 David R. Turner1*
David R. Turner1* Eric P. Achterberg2
Eric P. Achterberg2 Chen-Tung A. Chen3
Chen-Tung A. Chen3 Simon L. Clegg4
Simon L. Clegg4 Vanessa Hatje5
Vanessa Hatje5 Maria T. Maldonado6
Maria T. Maldonado6 Sylvia G. Sander7
Sylvia G. Sander7 Constant M. G. van den Berg8
Constant M. G. van den Berg8 Mona Wells9
Mona Wells9- 1Department of Marine Sciences, University of Gothenburg, Gothenburg, Sweden
- 2GEOMAR Helmholtz Centre for Ocean Research, Kiel, Germany
- 3Department of Oceanography, National Sun Yat-sen University, Kaohsiung, Taiwan
- 4School of Environmental Sciences, University of East Anglia, Norwich, UK
- 5Centro Interdisciplinar de Energia e Ambiente, Instituto de Química, Universidade Federal da Bahia, Salvador, Brazil
- 6Department of Earth Ocean and Atmospheric Sciences, University of British Columbia, Vancouver, BC, Canada
- 7Department of Chemistry, NIWA/University of Otago Research Centre for Oceanography, Dunedin, New Zealand
- 8Earth and Ocean and Ecological Sciences, University of Liverpool, Liverpool, UK
- 9Department of Environmental Science, Xi'an Jiaotong-Liverpool University, Suzhou, China
We elaborate the need for a quality-controlled chemical speciation model for seawater and related natural waters, work which forms the major focus of SCOR Working Group 145. Model development is based on Pitzer equations for the seawater electrolyte and trace components. These equations can be used to calculate activities of dissolved ions and molecules and, in combination with thermodynamic equilibrium constants, chemical speciation. The major tasks to be addressed are ensuring internal consistency of the Pitzer model parameters (expressing the interactions between pairs and triplets of species, which ultimately determines the calculated activities), assessing uncertainties, and identifying important data gaps that should be addressed by new measurements. It is recognized that natural organic matter plays an important role in many aquatic ecosystems, and options for including this material in a Pitzer-based model are discussed. The process of model development begins with the core components which include the seawater electrolyte and the weak acids controlling pH. This core model can then be expanded by incorporating additional chemical components, changing the standard seawater composition and/or broadening the range of temperature and pressure, without compromising its validity. Seven important areas of application are identified: open ocean acidification; micronutrient biogeochemistry and geochemical tracers; micronutrient behavior in laboratory studies; water quality in coastal and estuarine waters; cycling of nutrients and trace metals in pore waters; chemical equilibria in hydrothermal systems; brines and salt lakes.
1. Introduction
Ocean composition is changing at an unprecedented rate as a result of anthropogenic pressures, with important implications for the health of the oceans and for economic activities. In a recent survey of global ocean research priorities, ocean acidification and the effects of cumulative stressors were identified as two of the top three areas of concern (Rudd, 2014). Understanding the impacts of ocean acidification on areas of human concern, such as fisheries, is a matter of great urgency. Knowledge of chemical speciation, which describes the distribution of the elements between different chemical forms, is essential to understand how changes in ocean chemistry affect bioavailability of different elements (Tessier and Turner, 1995). For example, the speciation of both carbonate and (micronutrient) trace metals are complex functions of natural water composition, pH, temperature, and pressure. Changes in ocean chemistry may be small on an annual basis, but are modulated on short time and space scales by physical parameters, such as salinity, temperature, and irradiance, as well as variations in upwelling and ocean currents. Therefore, these chemical changes need to be measured accurately and consistently across the globe to monitor and understand contemporary processes. In addition, chemical changes need to be modeled accurately to evaluate future scenarios and remediation strategies. These requirements are linked by the need to understand chemical speciation—for example that of carbonate and trace metal micronutrients—both in natural waters and in the reference materials and solutions used for analytical method verification and instrument calibration.
Recognizing the importance of chemical speciation modeling, SCOR, the Scientific Committee on Ocean Research of the International Council for Science, created Working Group 145 tasked with establishing a reference seawater chemical speciation model that is user-friendly, and freely available to the marine science community. This paper, which constitutes the first report from the Working Group, describes the approach that will be used and identifies different applications to be supported by this chemical speciation model. These span a variety of aquatic environments, and include measurement, calibration and laboratory studies. Each application has its own requirements for the chemical species to be included, and the physical conditions (temperature and pressure) to be covered. To this end, we have proposed a set of core components that are essential for all marine science applications, followed by the specific requirements for each application. This approach is summarized in Figure 1, where in addition to the core components, seven different groups of application have been identified. The major tasks for the Working Group include: ensuring self-consistency of the chemical speciation model and traceability of its parameters to thermodynamic measurements, establishing quantitatively the uncertainties in the calculated speciation, and identifying important data gaps to be addressed by new measurements.
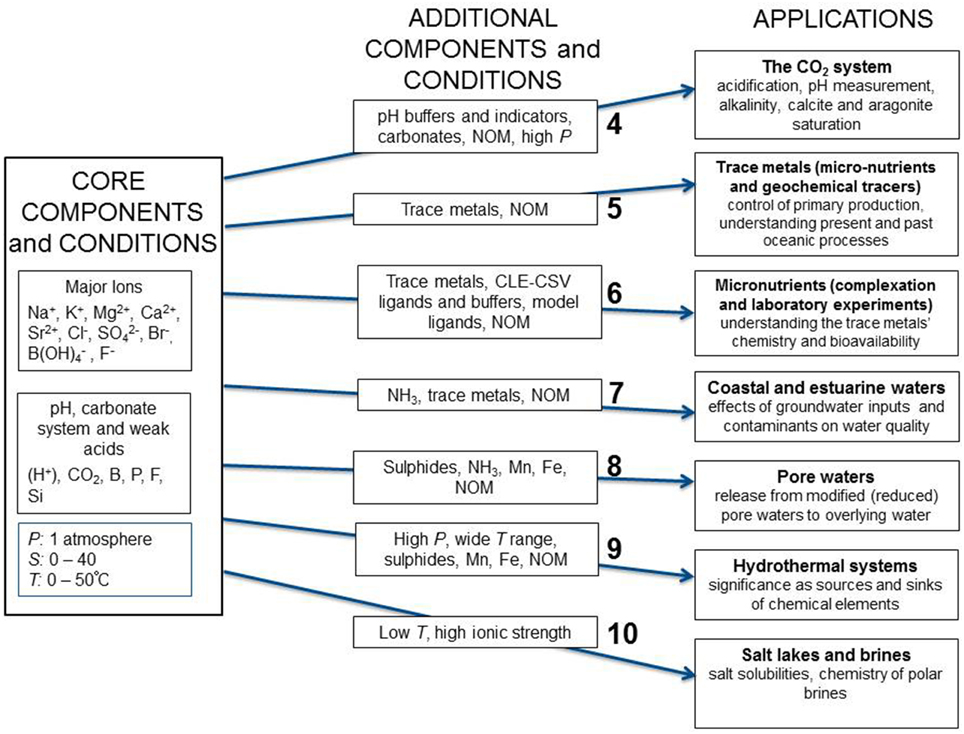
Figure 1. Schematic diagram showing the core components and conditions, and seven groups of additional components and conditions with associated applications for the Pitzer chemical speciation model proposed by the SCOR WG 145. These conditions and applications cover the majority of problems biogeochemists face in describing the chemical speciation of elements in marine and estuarine environments. Each set of additional components, conditions and applications is discussed in the text section indicated by the number shown in the diagram.
2. Theory
2.1. Pitzer Equations
The form in which a trace element or other component of seawater is present, and its tendency to react, depends on its activity (Clegg and Whitfield, 1991). This is the product of its concentration (usually mol per kg seawater), and an activity coefficient (γ) which is a complex function of temperature, pressure, and salinity (or, more generally, solution composition). Many of the important reactions in seawater involve acid-base equilibria, which introduces pH as a further variable.
The Pitzer model (Pitzer, 1991) is a set of equations to calculate activity coefficients (and hence all forms of chemical equilibria) in aqueous solutions as functions of composition and concentration, temperature, and pressure. The equations for water and solute activities, and thermal and volumetric properties, are derived from a single expression for the excess Gibbs energy of the solutions. The equations contain sets of parameters (which are functions of temperature and pressure) of two kinds: “pure” solution parameters whose values are determined by fitting to data for solutions containing single electrolytes (e.g., NaCl, MgSO4); and “mixture” parameters whose values are determined from measurements containing, typically, two different electrolytes with a common ion. (e.g., NaCl and Na2SO4).
The data needed to build a model of a complex mixture, such as seawater, include: solvent and solute activities, apparent molar enthalpies and heat capacities (yielding the variation of the model parameters with temperature), apparent molar volumes and compressibilities (the variation of the parameters with pressure), salt solubilities, liquid/liquid phase partitioning, equilibrium partial pressures of volatile solutes, and others. The model is described in detail by Pitzer (1991), and its application to the chemistry of natural waters by Clegg and Whitfield (1991). For any solution, the major effort in constructing a Pitzer model is the determination of the parameter values; a process that often includes the resolution of differences between inconsistent sets of data, and obtaining approximate values of parameters for which there are no data.
The principal Pitzer chemical speciation model of seawater is that of Millero and co-workers at the University of Miami (Millero and Roy, 1997; Millero and Pierrot, 1998; Waters and Millero, 2013). Using many of the same data sources, Clegg and Whitfield (1995) also developed a seawater model, including the protonation of dissolved ammonia. The model of Millero and co-workers is applicable primarily to major ions in seawater (from 0 to 50°C, and 0 to >40 salinity) containing the species H+, Na+, K+, Mg2+, Ca2+, Sr2+, Cl−, Br−, OH−, , , , , , CO2, B(OH)3, and H2O. The use of the model to calculate pH, and some of the problems concerning pH scales that still need to be addressed, are discussed by Waters and Millero (2013). There is, as yet, no comprehensive evaluation of the uncertainties in model-calculated speciation arising from uncertainties in the thermodynamic measurements upon which they are based. The data for trace metal activities and complexation in seawater—which are important for understanding their behavior and fate—are best described as patchy, and the effects of pressure on activities are only well characterized for aqueous solutions of some of the major seawater electrolytes (e.g., NaCl). This limits the accuracy of calculations of chemical speciation in the subsurface ocean.
The parameters that comprise the Pitzer models developed for seawater are drawn from many sources, include data up to a century old, and are often not optimized for solutions of seawater but rather for modeling brines, e.g., Harvie et al. (1984). To our knowledge, the number of new studies yielding the activities and other data needed to improve the model has been in decline for many years, though the need to quantitatively understand ocean biogeochemistry, especially carbonate chemistry, has increased significantly. Furthermore, there is no comprehensive evaluation relating the capabilities of speciation models, and the measurements upon which they are based, to current and future needs in chemical oceanography as exemplified in current programs, such as the international GEOTRACES Program (Henderson et al., 2007).
The matrix of Pitzer parameters for major ions in seawater—of the composition noted above but excluding Sr2+ and boric acid—is considerable: 36 sets of cation-anion interactions, and potentially 210 ternary or “mixture” parameters that express the interactions between two dissimilar ions of one charge type, and one of the opposite charge type. Some of these can be neglected if all the interacting species are at very low concentration. Nevertheless, the large numbers of interactions and the fact that they can vary with both temperature and pressure emphasizes the need to (1) assess the completeness and reliability of the model, (2) validate its basis in measured thermodynamic properties (including analyses to relate the uncertainties in the output quantities of the model to those of the fundamental data—for each interaction—upon which it is based); and (3) establish the sets of measurements still needed to adequately characterize the behavior of seawater, estuarine and other natural waters encountered worldwide. The treatment of uncertainties could adopt the methods applied to the IAPWS 1995 equation of state for water (Feistel et al., 2016).
When Pitzer equations are used to calculate activity coefficients, the stability constants used for the chemical equilibria are thermodynamic constants K, which are functions of temperature and pressure only. However, many practical applications make use of stoichiometric constants K* (sometimes also called conditional constants), which are expressed in terms of concentrations and are thus functions of temperature, pressure and solution composition. This dependence on solution composition—via the activity coefficients γ—limits the practical value of stoichiometric constants. Taking the dissociation of HF as an example, and using square brackets to represent concentrations:
Values of the stoichiometric constant K* measured in normal seawater can be used only for solutions of seawater composition and for the salinity, or salinities, for which K* has been determined. Any variations from seawater composition—such as might be found in enclosed seas, pore waters, and some estuaries—will lead to changes in the values of the three activity coefficients in the equation above and therefore a change in K*.
Models based on the Pitzer equations are used to calculate chemical speciation at equilibrium, although there is of course no guarantee that a particular system is at equilibrium. In marine environments, the different redox couples that are present are, in general, not in equilibrium with each other. The Pitzer equations can be used to examine this phenomenon by calculating the equilibrium speciation in the two oxidation states of a redox couple: combining measured total concentrations and the standard potential for the couple allows the degree of disequilibrium to be established. Such calculations can also be used to establish the relative oxidizing or reducing power of different redox couples at in situ conditions. A knowledge of the equilibrium speciation may also contribute to the analysis of rate processes in places where complexation of a metal ion affects the rate of oxidation (Santana-Casiano et al., 2005).
2.2. Natural Organic Matter (NOM)
The Pitzer equations are applicable to reactions involving relatively simple chemical species whose chemical composition and structure are well defined. Natural organic matter (NOM) in natural waters, including seawater, falls outside this definition since it is a polydisperse material comprising a mixture of many different molecular structures (Koch et al., 2005). Modeling of NOM in seawater is currently restricted to its complexation of trace metals; the available information is derived from competitive ligand exchange cathodic stripping voltammetry (CLE-CSV) titrations (section 6.1), which are summarized as the “concentrations” and “stability constants” of one or more ligand classes. This operational summary cannot be applied in conditions that depart from those used in the titration, although in many cases a broad agreement between different studies has been observed. An alternative strategy worthy of investigation is the use of modeling approaches for freshwater NOM, following extensive laboratory studies of extracted material. Three modeling approaches have been developed: the Windermere Humic Acid Model (WHAM) (Tipping et al., 2011); the Stockholm Humic Model (SHM) (Gustafsson, 2001); and the NICA-Donnan model (Koopal et al., 2005). All three approaches explicitly recognize the polydisperse nature of NOM, and also explicitly treat the development of charge due to ionization and complexation reactions. The model codes developed for these approaches in fresh waters use simple extended Debye-Hückel equations for the calculation of activity coefficients. These equations are not adequate for use in seawater. However, the WHAM formulation has recently been combined with a Pitzer model (Ulfsbo et al., 2015), providing the basis to test this approach for NOM modeling in seawater.
3. Core Components and Conditions
3.1. The Seawater Electrolyte
The basis of any speciation model that aims to predict activity coefficients accurately is the background electrolyte that determines the physicochemical properties of seawater, and the chemical environment experienced by trace species. On a molar concentration basis, seven chemical elements account for 99.9% of the dissolved species in seawater. Models of the seawater electrolyte normally include the 11 elements whose concentrations exceed 1 μmol kg−1 and constitute a constant or near-constant proportion of salinity. These are, in order of descending concentration, chlorine (Cl), sodium (Na), magnesium (Mg), sulfur (S), calcium (Ca), potassium (K), (inorganic) carbon (C), bromine (Br), boron (B), strontium (Sr), and fluorine (F).
The definition of the core speciation model also includes the ranges of the three master variables temperature, salinity, and pressure. A temperature range of 0–50°C and a salinity range of 0–50 correspond to the ranges of many speciation models, although the most complete data collection is usually at 25°C. Information on pressure dependence is often limited, so the core speciation model that the Working Group will consider is initially for 1 atmosphere pressure.
3.2. pH
The practical scales used by chemical oceanographers to measure pH are summarized by Waters and Millero (2013). The complexities of these scales, and the difficulty of defining pH, arise because neither the concentration nor the activity of H+ can be measured directly and independently of other quantities (Dickson, 1984). On the total scale, the H+ associated with (as ) is included in the pH, so that:
The seawater pH scale also includes the H+ bound to F− (as HF), so that,
In the above equations the stoichiometric dissociation constants K*HSO4 (for reaction ↔ H+ + ) and K*HF (HF ↔ H+ + F−) vary with temperature, pressure, and salinity. More generally, they vary with chemical composition which affects pH in natural waters whose composition differs from that of normal seawater.
The total and seawater pH scales have been adopted for the following practical reason: the artificial seawater solutions used for the characterization of pH buffers and pH indicators must contain a relatively high concentration of sulfate in order to match the major ionic composition of natural seawater. However, glass and hydrogen pH electrodes respond only to the free H+ in solution, while analytical hydrogen ion concentrations obtained from measurements on these scales include both H+ and . This difference is significant: −log10[H+] and pHT at salinity 35 and 25°C differ by 0.128 pH units, while the difference between pHT and pHSWS is only 0.005 units. The accuracy with which the free H+ concentration can be determined from pHT and pHSWS is currently limited by the accuracy with which K*HSO4 is known (Waters and Millero, 2013). Waters and Millero recommend further work to resolve discrepancies between measured and modeled activity coefficients in seawater-like solutions containing sulfate.
Figure 2 shows concentrations of the principal species controlling pH in a salinity 35 seawater at 25°C, calculated using an ion-interaction speciation model (Clegg and Whitfield, 1995). It is clear, from the relatively high concentrations of carbonate and borate, that seawater pH is largely controlled by equilibria involving these species.
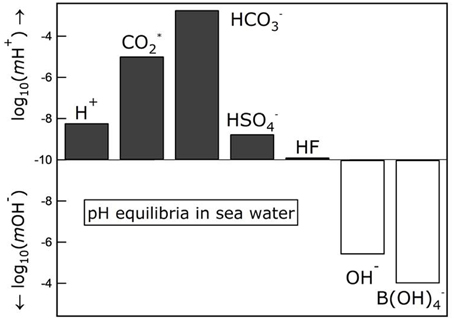
Figure 2. Concentrations of the principal species controlling pH in salinity 35 seawater at 25°C, calculated using an ion-interaction speciation model (Clegg and Whitfield, 1995). refers to the sum of dissolved CO2 and H2CO3.
3.3. Weak Acids
The chemical species in the model include the weak acids that are involved in defining the pH and alkalinity of seawater. These are clearly illustrated in the open-ended definition of total alkalinity AT (Dickson, 1981):
The core chemical species in the model have been selected based on a significant contribution (>1 μmol kg−1) to AT in the open ocean. This includes all the species in Equation (4), with the exception of the weak base ammonia.
4. The CO2 System
4.1. Limitations of Current Calculation Programs
A number of software packages are freely available for carrying out calculations on the CO2 system, and are based on stoichiometric constants parameterized as a function of salinity, temperature, and pressure (Orr et al., 2015). However, the use of salinity as a master variable carries the drawback that these calculations are not applicable to waters of different compositions. This is a particular problem at lower salinities where major ion composition of the seawater/freshwater mixture does not correspond simply to diluted seawater. Compared with seawater, river waters generally have high Mg, Ca, and borate concentrations relative to Na, Cl, and SO4. Furthermore, river waters are typically high in organic ligands of various molecular sizes. This material can contribute to alkalinity, as noted above, and its decomposition in estuaries can cause substantial changes in the CO2 system (Cai, 2011; Chen et al., 2013; Yang et al., 2013).
The interpretation of measured alkalinity, and its use in calculations of CO2 equilibria in natural waters, requires identification of all the relevant weak acids (i.e., those that contribute more than ca. 1 μmol kg−1 to the alkalinity), and their degrees of dissociation at the temperature, salinity, and pressure of interest. This presents an analytical challenge in many estuarine and brackish waters where natural organic matter can make a significant contribution to alkalinity (Hernandez-Ayon et al., 2007; Kulinski et al., 2014). Complete modeling of the CO2 system in such waters will also require a treatment of the acid-base chemistry of this material. Overall, there is a great deal to be gained by integrating calculations of the carbon dioxide system, and pH, into a Pitzer model of chemical speciation.
4.2. Calcium Carbonate Solubility
An aspect of ocean acidification that has received a great deal of attention is the concomitant increase in the solubility of calcium carbonate, and its consequences for calcifying marine organisms. A large number of experiments have been carried out exposing different calcifying (and non-calcifying) marine organisms to reduced pH. These experiments show a range of results, and that, at least in the short term, not all calcifying organisms are negatively affected (Dupont and Poertner, 2013). It is, however, clear that oceanic pH is falling and that organisms that precipitate the more soluble forms of calcium carbonate will be greatly affected. Corals, for example, precipitate aragonite, which is the most soluble pure form of calcium carbonate, although high magnesium calcites can have even higher solubilities (Haese et al., 2014). Projections show that aragonite will become undersaturated in large parts of the ocean during the next century. As for the CO2 system equilibria in solution, the solubility products of calcite and aragonite are currently available as functions of salinity, temperature, and pressure (Millero, 2007). Incorporation of these equilibria into a Pitzer framework would enable improved calculations of the solubility products of calcite and aragonite in waters whose composition differs from standard seawater.
4.3. Buffers for pH Measurement
The recognition of ocean acidification as “the other CO2 problem” has highlighted the need for precise and accurate pH measurements in seawater. Two methods are currently recognized as meeting these requirements: glass electrodes calibrated in artificial seawater buffers; and indicator spectrophotometry (Dickson et al., 2007), also calibrated against buffer solutions. The most widely used buffer for this purpose is “tris” (2-amino-2-hydroxymethyl-1,3-propanediol), although alternative buffers, such as 2-aminopyridine, morpholine (tetrahydro-l,4-isoxazine) and bis (2-amino-2-methyl-l.3-propanediol) can be used when the pH departs significantly from the normal seawater range (Dickson, 1993). The pH values assigned to these buffers are available as functions of salinity and temperature, and have been derived by least-squares fitting pH values determined from a large number of potentiometric measurements in cells without liquid junctions (DelValls and Dickson, 1998). As yet, these measurements cover a limited salinity range (20–40). An alternative approach to fitting the buffer pH values to semi-empirical functions of salinity and temperature, is to derive Pitzer parameters for the interactions of these two buffers with the components of seawater. This would then allow these important pH buffers to be incorporated into the planned unified model of seawater, and also facilitate the treatment of low salinity buffer solutions.
4.4. Sulphonephthalein Indicators
Indicator spectrophotometry has gained wide acceptance as the method of choice for measurements of seawater pH. These measurements require the stoichiometric stability constant of the indicator dye at the salinity and temperature of the measurement. This is currently achieved by laboratory measurements of these constants under a range of conditions, followed by least squares fitting of the data to semi-empirical functions of salinity and temperature (Liu et al., 2011). As with the pH buffers, an alternative approach to fitting the indicator pH values to semi-empirical functions of salinity and temperature is to derive Pitzer parameters for the interactions of the relevant indicators with the components of seawater. This would allow these indicators to be incorporated into the planned unified seawater model. The most important indicators are m-cresol purple, phenol red and thymol blue, which are used for pH measurements; and also bromocresol green and bromocresol purple which are used for the determination of alkalinity by the single point method.
5. Trace Metals: Micronutrients and Geochemical Tracers
The key role of trace metals in ocean science has been recognized in the development of the GEOTRACES programme (Henderson et al., 2007), which is coordinating a global survey of trace elements and isotopes in the ocean. GEOTRACES focuses on the behavior of trace metals as micronutrients and as tracers for ocean processes; this division of roles is not exclusive: some trace metals, such as Cd and Mn, fall into both categories.
5.1. Micronutrients
It is now recognized that the bioavailability of iron is a key factor in determining primary production and/or phytoplankton community structure in large areas of the ocean (Turner and Hunter, 2001). The thermodynamically stable oxidation state in oxic waters, Fe(III), is poorly soluble with a solubility of the order of 10−11 mol L−1 in seawater in the absence of organic ligands (Liu and Millero, 2002). However, strong organic complexation can maintain significantly higher concentrations. While organic complexation by (as yet) not fully characterized ligands dominates the speciation of Fe(III), there is continuing interest in defining the inorganic complexation and solubility as essential supporting information. There is also considerable interest in Fe(II), both as a component of anoxic waters, and as metastable Fe(II) produced by photochemical reduction in surface waters. The speciation of this metastable Fe(II) and its effects on Fe oxidation rates are key factors determining the bioavailability of Fe in sunlit surface waters.
While much attention has been focused on Fe in recent years, several other metals are essential for biological processes. A recent review gives the generalized ranking of trace metal content in phytoplankton as Fe ≈ Zn > Mn ≈ Ni ≈ Cu >> Co ≈ Cd (Twining and Baines, 2013). While all these metals act as micronutrients, some can also have toxic effects at elevated concentrations (Cd, Ni, Cu, Zn, see also section 7.2). Four of these metals (Fe, Zn, Cu, and Cd) are obligatory core parameters on GEOTRACES cruise sections, which are providing extensive new information on these metals, both in terms of dissolved (<0.2 μm) concentrations and CLE-CSV titration data (see section 6.1). Chemical speciation modeling will provide an excellent tool to help extract full value from these unique global datasets.
5.2. Tracers of Ocean Processes
Manganese is present as insoluble MnO2 at equilibrium in oxic seawater. However, relatively slow oxidation rates result in dissolved Mn concentrations at the nM level. This slow oxidation has led to the use of Mn as a tracer for releases from anoxic environments, where Mn is present as dissolved Mn(II), and from hydrothermal systems where significant amounts of reduced Mn are released into the surrounding waters (German et al., 1999). The speciation of dissolved Mn has been considered to be dominated by weakly complexed Mn(II), but recent work indicates that Mn(III) may also be present at significant concentrations (Luther et al., 2015).
In surface waters, Al has been used as a tracer for dust input, which is a major source of Fe to the ocean. The strong correlation between dissolved Al and silicic acid has prompted suggestions of biological control of Al distributions (van Hulten et al., 2014). However, for example in the Mediterranean Sea, the correlation between Al and Si could be explained purely by vertical transport of biogenic particles (Rolison et al., 2015).
The lanthanides series are a powerful set of tracers because of their coherent and predictable behavior. Lanthanides have been used in many studies to investigate redox conditions (Liu et al., 1988; Sholkovitz and Schneider, 1991), particulate exchange and scavenging processes (Andersson et al., 2008), water mass transport (Scher and Martin, 2004; Haley et al., 2008) and identification of benthic sources of pore fluid (Abbott et al., 2015). More recently, the lanthanides' widespread use in high-technology processes and products is increasing their environmental occurrence. For example, lanthanides are now being used as tracers of wastewater-derived contaminants in the marine environment (Hatje et al., 2014, 2016).
The close relationship between the depth profiles of Cd and phosphate concentrations makes Cd an attractive candidate as a proxy for phosphate concentrations in palaeoceanography, although the relationship shows significant variation (de Baar et al., 1994).
6. Micronutrients: Complexation and Laboratory Experiments
6.1. CLE-CSV Titrations
The most widely used method for characterizing the organic complexation of trace metals in seawater is currently Competitive Ligand Exchange (CLE), although direct electrochemical measurements by Anodic Stripping Voltammetry (ASV) are also used, e.g., Sinoir et al. (2016). CLE involves the addition of a competing ligand which has two properties: it forms a complex that competes for trace metals with the natural unknown ligands present in the sample; and it forms a complex with the target trace metal that adsorbs on a mercury electrode and can thus be detected by Cathodic Stripping Voltammetry (CSV). The methods of fitting the CLE/CSV and ASV titration data accurately have been improved significantly in recent times (Pizeta et al., 2015). However, to enable accurate CLE characterization of the complexation of the trace metal with unknown ligands, it is essential to quantify the complexation of the metal by the added competing ligand, as well as competition reactions with other metals.
For CLE measurements, the most commonly used added ligands are SA (salicylaldoxime), which complexes with Cu and Fe (Campos and van den Berg, 1994; Buck et al., 2007; Abualhaija and van den Berg, 2014); NN (1-nitroso-2-naphthol), which complexes with Fe (Aldrich and van den Berg, 1998); APDC (aminopyrrolidinedithiocarbamate), which complexes with Zn (van den Berg, 1985); and DHN (dihydroxynaphthalene), which complexes with Fe (van den Berg, 2006). Other relevant ligands are TAC [2-(2-thiazolylazo)-p-cresol], which has been used as competing ligand for the determination of Fe organic complexation in seawater (Croot and Johansson, 2000; Sander et al., 2015); and DMG (dimethyglyoxime), which has been used for determinations of Co and Ni complexation (van den Berg et al., 1991; Ellwood and van den Berg, 2001).
In addition to modeling the chemistry of the added ligands, attention needs to be paid to the buffers added to control pH in the CSV measurements. Optimal buffers are those near the natural pH of the water. While the carbonate system acts as the major pH buffer in seawater, sample deaeration undertaken during the CSV analysis disrupts this function, so that an additional buffer is needed. As well as buffering close to the natural pH of seawater, the added buffer must have only weak interactions with the trace metals being studied, so that it does not act as an additional competing ligand. The two buffers that are most commonly used are EPPS [4-(2-hydroxyethyl)-1-piperazinepropanesulphonic acid] and HEPES [4-(2-hydroxyethyl) piperazine-1-ethanesulphonic acid], however phosphate buffer is becoming more popular as it can be cleaned very efficiently to remove any traces of organic ligands and trace metals. A quantitative understanding of these buffers' chemistry in seawater would allow the buffering effect to be calculated more accurately, thus providing a better definition of the chemical conditions of the complexation titrations.
6.2. Model Ligands for Laboratory Experiments
Model organic chelators are used in culture media to ensure that the inorganic trace metal concentrations stay constant during culturing. Furthermore, the use of these chelators, in conjunction with chemical equilibrium modeling, allows the researcher to manipulate the inorganic concentration of a given trace metal while maintaining the inorganic concentrations of the remaining bioactive trace elements unchanged. This design is especially suitable for controlled physiological studies investigating the response of phytoplankton to limiting or toxic trace metal concentrations. The most common general model chelator in culture studies is EDTA (ethylenediaminetetraacetic acid; Price et al., 1988; Sunda et al., 2005). However, the chelator of choice may vary depending on the research question. For example, if Cu is the metal of interest, one might use DTPA (diethylene triamine pentaacetic acid) instead of EDTA, given that DTPA is a stronger chelator of Cu than EDTA.
Limiting growth in cultures by manipulation of a specific trace metal is often challenging, and requires the use of well-chosen metal chelators. For example, siderophores, such as DFB (desferrioxamine B), and DFE (desferrioxamine E), are often used to significantly reduce the concentrations of inorganic Fe in the culture media, and thus induce Fe limitation in phytoplankton with extremely low Fe requirements (Strzepek et al., 2011). Similarly, Cu specific chelators, such as Cyclam (1,4,8,11-tetraazacyclotetradecane) and Cyclen (1,4,7,10-tetraazacyclododecane) are frequently used to induce Cu limitation in phytoplankton cultures (Semeniuk, 2014). In addition, these organic ligands are frequently used in uptake experiments (Semeniuk et al., 2015), as they are believed to mimic naturally occurring strong organic ligands of Fe and Cu in seawater. Naturally occurring thiols, such as glutathione, cysteine and thiourea are known to strongly bind Cu and other chalcophilic metals (Laglera and van den Berg, 2003). Chemical modeling that includes these organic chelators will greatly enhance our understanding of the chemical speciation of the trace metals in laboratory growth and uptake media, as well as in situ oceanic conditions.
7. Coastal and Estuarine Systems
7.1. Estuaries and Groundwater Discharge
Estuaries are where the river water meets seawater, and where the mixture interacts with the land, oceans, atmosphere, sediments, and biota. The freshwater end members are typically richer in major nutrients (nitrate, nitrite, ammonia, phosphate, and silicate) compared with ocean waters, hence biological productivities are generally high in estuaries. River waters subject to anthropogenic influence frequently have high concentrations of micronutrients and toxic elements (Liu et al., 2010), while permafrost degradation affects the chemistry of Arctic rivers and coastal seas through the mobilization of organic matter (Semiletov et al., 2016). Furthermore, submarine groundwaters with unique chemical compositions discharge into many estuaries and coastal waters (Zhang and Mandal, 2012). In the case of eutrophication, the pH and dissolved oxygen concentration are both lowered following organic matter breakdown, with consequences for acid-base and redox reactions. Moreover, when riparian tidal freshwater lands and oligohaline marshes are inundated by seawater, competition by major cations, such as Mg and Ca can result in the release of trace metals from particulate matter (Wang et al., 2012; Zhang and Mandal, 2012).
7.2. Contaminant Discharge
The United Nations Environmental Programme (UNEP, 2013) has highlighted the phenomenon of “global chemical intensification”: in many parts of the world, the anthropogenic inputs of metals to the ocean are estimated to be far greater than non-anthropogenic inputs, and the way in which this affects global biogeochemical cycles and ecosystem diversity is as yet poorly understood. Islam and Tanaka (2004) and Davies (1978) noted that the 10 most significant contaminant elements in marine waters are, in order of decreasing toxicity Hg, Cd, Ag, Ni, Se, Pb, Cu, Cr, As, and Zn. Four of these (Cd, Ni, Cu, and Zn) are classified as key micronutrients (Twining and Baines, 2013), and Se and Cr can in addition act as micronutrients.
Specific examples of contaminant discharge include run-off from metal mines, their associated mine tailings, and also naturally occurring acid rock drainage springs. The metals that are commonly found at elevated concentrations (micromolar to millimolar) in mine runoff include Fe, Cu, Zn (Brown et al., 2005; Braungardt et al., 2007), and depending on the ore formations, may also include Cd, Ni, Ag, Au, As. High acidity (pH < 1) is associated with the oxidation of iron pyrites and the subsequent formation of sulfuric acid. High acidity also mobilizes metals and prevents their removal by precipitation and scavenging.
Industrial discharges are also potential sources of inorganic contaminants, including those from metallurgical industry (metals), fertilizer industry (N, P), and desalination plants (metals). Furthermore, domestic waste water treatment operations discharge inorganic contaminants into receiving waters, and the growth of aquaculture in coastal zones is associated with increasing pollution by inorganic compounds.
8. Pore Waters
Pore waters are aqueous solutions that occupy the pore spaces between particles in sediments and can often be considered in equilibrium with the sediments. Diagenetic reactions within sediments and at the sediment-pore water interface control, among others, the recycling of nutrients, carbon, trace elements and contaminants, the dissolution of carbonates, the flux of organic matter to benthic communities and the burial of several elements (see for example Berner, 1980; Klinkhammer, 1980; Klinkhammer et al., 1982). Pore water studies have provided a link between water column transport processes and sedimentary accumulation by showing evidence for the release of metals associated with the degradation of organic matter (Sholkovitz et al., 1989).
Chemical changes across redox interfaces in marine systems can have a profound impact on metal solubility and bioavailability. At these oxic-anoxic interfaces, changes in oxidation state, as well as complexation with reduced sulfur species and formation of insoluble sulfides, are common. In coastal areas, redox interfaces are found in anoxic sediments, in anoxic basins and fjords, as well as in sewage outfalls. In the open ocean, oxic-anoxic interfaces are less common, but changes in trace metal solubility and speciation are often observed in oxygen minimum zones. Thus, water columns with oxic-anoxic interfaces have an impact on the cycling of trace metals in the global ocean.
Knowledge of biogeochemical processes in marine sediments and benthic fluxes of pore water is essential for understanding the global carbon cycle and climate (Berner, 1980; Siegenthaler and Sarmiento, 1993; Ridgwell and Hargreaves, 2007). Moreover, pore water is a key exposure route for metal contaminants to organisms associated with the sediments, in particular infauna (Chapman et al., 2002). Knowledge of metal speciation in pore waters is necessary for the development of predictions of bioavailability and for reliable risk assessment strategies. This knowledge could be incorporated in biotic ligand models to derive better-founded quality criteria for marine environments.
8.1. Sulfides
The need to model chemistry in anoxic pore waters, and to understand the chemical processes occurring at the oxic/anoxic boundary, focuses attention on redox reactions, and on the chemistry of reduced oxidation states. The most abundant of these are NH3, Fe(II), Mn(II), and sulfide. The most challenging of these species for chemical modeling is sulfide. In the last 2 decades, metal sulfide speciation in aquatic systems has become of great interest, due to the discovery of nanomolar levels of sulfide in oxic seawater (Luther and Tsamakis, 1989). Sulfides become stable in oxic conditions by forming complexes with trace elements, especially with Hg, Cu (Dyrssen, 1988; Luther and Tsamakis, 1989); and Pb (Bura-Nakic et al., 2007). These metal (M)—sulfide species in oxic waters include simple MHS+ (or complexes, but also higher-order unprotonated clusters (multinuclear oligomers) with high stability constants (Rozan et al., 2000). In the case of Cu and Zn, the resulting metal-sulfide species are resistant to oxidation in oxic waters and include a mixture of dissolved metal-sulfide complexes and active metal-sulfide nanoparticles (Rozan et al., 1999; Sukola et al., 2005).
9. Hydrothermal Systems
Hydrothermal venting occurs in two forms: as hot (up to 450°C), or diffuse venting (5–100°C). Hot vents are mainly found close to mid ocean ridges, subduction zones, and arcs; whilst diffuse venting also occurs off axis and in areas with mild tectonic activity. Recently, it has been argued that seafloor venting may provide a significant source of the bio-essential Fe, Cu, and Zn, as well as other metals, due to their stabilization with organic ligands and nanoparticulate sulfides (Sander and Koschinsky, 2011; Yucel et al., 2011; Nishioka et al., 2013). These results place new constraints on submarine metal vent fluxes worldwide, including an indication that the majority of Fe supplied to hydrothermal plumes should come from entrainment of diffuse flow (German et al., 2015). Submarine hydrothermal venting has recently been suggested to have the potential to impact ocean biogeochemistry at the global scale (Tagliabue et al., 2010; Sander and Koschinsky, 2011; Wu et al., 2011; Fitzsimmons et al., 2014). This is the case because processes active in hydrothermal plumes are so vigorous that the residence time of seawater cycling through hydrothermal plumes is comparable to the residence time of deep-water mixing by thermohaline circulation.
9.1. High Temperature Venting (>100°C)
Since hot hydrothermal vents expel fluid at temperatures between 100 and 450°C, depending on vent activity and depth, these fluids are highly reactive and far from thermodynamic equilibrium with the surrounding seawater. Besides their high temperature, they exhibit extreme pH values (<3 or >9), are highly reduced and may have a very different ionic composition to that of standard seawater. In fact, the fluid composition is defined by the underlying bedrock, pressure and temperature, and may reflect phase separation. Once the venting fluids encounter cold oxygenated seawater, precipitation reactions occur instantly, resulting in the commonly seen black (or white) smokers. While the chemical signature of hot hydrothermal vents can be followed in the hydrothermal plume over thousands of kilometers, the temperature will only be >300°C for seconds or millimeters after discharge. Most chemical reactions with the seawater will occur at temperatures between 4 and 300°C. Taking samples that are representative of in-situ conditions is almost impossible as samples will undergo spontaneous degassing upon the release of pressure, and cooling. Thus, to fully understand the reactions and processes occurring at depth in the presence of high temperature and pressure, it is essential to model the speciation of the fluid at in situ conditions.
9.2. Low Temperature Venting (<100°C)
Shallow vents are of great importance for the supply of micro- and macronutrients to the photic zone. In contrast to hot vents, the majority of shallow vents are characterized by diffuse venting, making them an interesting field of research and a natural laboratory to link speciation with biological uptake and toxicity (Klevenz et al., 2012; Kleint et al., 2015). For the vast majority of chemical tracers enriched in vent fluids, net fluxes to the oceans are modified as these tracers are incorporated into Fe-rich polymetallic sulfide and oxyhydroxide particles that sink to the seafloor at or close to mid ocean ridges (Mottl and McConachy, 1990; German et al., 1991; Kadko, 1993). Since diffuse vent fluids have undergone modification in the sub-seafloor and are composed of more than 90% seawater, they have had time to partially equilibrate with ambient seawater. However, often they are still exposed to reducing conditions. The mixing zone for diffuse venting is in the order of meters. Beyond that mixing zone, minor ions still undergo reactions that need to be modeled to understand the chemistry and biological uptake in the vicinity of these diffuse vents.
10. Salt Lakes and Brines
Brines, i.e., natural waters with substantially higher ionic strengths than seawater, are formed in two ways: during ice formation in polar waters where salt rejection increases the salt content of the remaining water; and during evaporation in salt lakes.
10.1. Polar Brines
Polar regions are subject to intensive research activities, as they are particularly sensitive to rising temperatures and increasing atmospheric CO2 concentrations, whilst at the same time playing a key role in global biogeochemical cycles and climate. Ocean acidification adds another stressor to these rapidly changing ecosystems (Orr et al., 2005). Polar regions experience extremes in primary productivity. In the Southern Ocean, low productivity is common in extensive regions with low iron supply, while high productivity is observed in regions with substantial iron supply from sediments (South Georgia) or ice melt (Nielsdottir et al., 2012). Whereas, iron supply over a large part of the Arctic is considered sufficient to sustain primary productivity (Klunder et al., 2012), the macro-nutrient concentrations may become exhausted following ice retreat. The freezing of sea ice results in brines with high salinity (100 or more), with freezing points well below that of normal seawater (ca. −1.8°C). These are conditions that are not included in chemical speciation models for seawater. In particular, the carbonate system in the polar oceans is not adequately described at sub-zero temperatures and salinities >50. In order to improve our mechanistic understanding of the dynamics of polar carbonate chemistry, and to allow quantification of CO2 fluxes across the atmosphere–ice–seawater interfaces, Pitzer based chemical speciation models covering these conditions will be valuable.
10.2. Salt Lakes
Salt lakes are systems with very high ionic strength, the record being held by Don Juan Pond (Antarctica), which contains 3.72 mol kg−1 CaCl2 and 0.5 mol kg−1 NaCl (Marion, 1997). The major focus in modeling of salt lakes and brines is the accurate prediction of precipitation equilibria as a function of temperature and composition (e.g., Harvie et al., 1984; Vanèina et al., 1986, 1997; Greenberg and Møller, 1989), with two major areas of application. The first is in understanding the evolution of past environments. An example is the modeling of evaporite sequences (layers of different salts accumulated over time) to infer the temperatures, concentrations, and compositions in the water body at the time of deposition. This type of work can be used to link the evolution of the water body to long term climate variations. The second area concerns the modeling of future scenarios. Interest here is focused on water bodies that are saturated, or close to saturation, by one or more salts in response to changes in inflow/outflow and evaporation. Such changes can be due to human activities. The Dead Sea, for example, is decreasing in volume, resulting in salt precipitation.
11. Concluding Remarks
This paper outlines the programme of work for the development of a quality-controlled chemical speciation model for seawater and related systems, including descriptions of the different applications that can benefit from the model. Ensuring accessibility by the marine science community will be addressed in a future publication. SCOR Working Group 145 welcomes comments on the proposed programme of work. Comments can be sent to the corresponding author.
Author Contributions
DT developed the overall structure of the paper. All authors contributed sections of text to this paper, and approved the submitted version.
Funding
This work was partially supported by Award #1243377 from the U.S. National Science Foundation to the Scientific Committee on Oceanic Research (SCOR), and is a contribution from SCOR Working Group 145. SGS received funding from MBIE contract C01X1005.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, A. N., Haley, B. A., McManus, J., and Reimers, C. E. (2015). The sedimentary flux of dissolved rare earth elements to the ocean. Geochim. Cosmochim. Acta 154, 186–200. doi: 10.1016/j.gca.2015.01.010
Abualhaija, M. M., and van den Berg, C. M. G. (2014). Chemical speciation of iron in seawater using catalytic cathodic stripping voltammetry with ligand competition against salicylaldoxime. Mar. Chem. 164, 60–74. doi: 10.1016/j.marchem.2014.06.005
Aldrich, A. P., and van den Berg, C. M. G. (1998). Determination of iron and its redox speciation in seawater using catalytic cathodic stripping voltammetry. Electroanalysis 10, 369–373. doi: 10.1002/(sici)1521-4109(199805)10:6<369::aid-elan369>3.3.co;2-n
Andersson, P. S., Porcelli, D., Frank, M., Bjork, G., Dahlqvist, R., and Gustafsson, O. (2008). Neodymium isotopes in seawater from the Barents Sea and Fram Strait Arctic-Atlantic gateways. Geochim. Cosmochim. Acta 72, 2854–2867. doi: 10.1016/j.gca.2008.04.008
Berner, R. A. (1980). Early Diagenesis, a Theoretical Approach. Princeton, NJ: Princeton University Press.
Braungardt, C. B., Achterberg, E. P., Gledhill, M., Nimmo, M., Elbaz-Poulichet, F., Cruzado, A., et al. (2007). Chemical speciation of dissolved Cu, Ni, and Co in a contaminated estuary in southwest Spain and its influence on plankton communities. Environ. Sci. Technol. 41, 4214–4220. doi: 10.1021/es063042h
Brown, J., Sander, S., Craw, D., and Hunter, K. (2005). Measurement of labile metals in acid rock drainage springs, New Zealand: field application of anodic stripping voltammetry. Appl. Geochem. 20, 1533–1545. doi: 10.1016/j.apgeochem.2005.04.014
Buck, K. N., Lohan, M. C., Berger, C. J. M., and Bruland, K. W. (2007). Dissolved iron speciation in two distinct river plumes and an estuary: implications for riverine iron supply. Limnol. Oceanogr. 52, 843–855. doi: 10.4319/lo.2007.52.2.0843
Bura-Nakic, E., Krznaric, D., Jurasin, D., Helz, G. R., and Ciglenecki, I. (2007). Voltammetric characterization of metal sulfide particles and nanoparticles in model solutions and natural waters. Anal. Chim. Acta 594, 44–51. doi: 10.1016/j.aca.2007.04.065
Cai, W. J. (2011). Estuarine and coastal ocean carbon paradox: CO2 sinks or sites of terrestrial carbon incineration? Ann. Rev. Mar. Sci. 3, 123–145. doi: 10.1146/annurev-marine-120709-142723
Campos, M. L. A. M., and van den Berg, C. M. G. (1994). Determination of copper complexation in sea-water by cathodic stripping voltammetry and ligand competition with salicylaldoxime. Anal. Chim. Acta 284, 481–496.
Chapman, P. M., Wang, F., Germano, J. D., and Batley, G. (2002). Pore water testing and analysis: the good, the bad, and the ugly. Mar. Pollut. Bull. 44, 359–366. doi: 10.1016/s0025-326x(01)00243-0
Chen, C. T. A., Huang, T. H., Chen, Y. C., Bai, Y., He, X., and Kang, Y. (2013). Air-sea exchanges of CO2 in the world's coastal seas. Biogeosciences 10, 6509–6544. doi: 10.5194/bg-10-6509-2013
Clegg, S. L., and Whitfield, M. (1995). A chemical model of seawater including dissolved ammonia and the stoichiometric dissociation constant of ammonia in estuarine water and seawater from –2°C to 40°C. Geochim. Cosmochim. Acta 59, 2403–2421. doi: 10.1016/0016-7037(95)00135-2
Clegg, S., and Whitfield, M. (1991). “Activity coefficients in natural waters,” in Activity Coefficients in Electrolyte Solutions, 2nd Edn., ed K. S. Pitzer (Boca Raton, FL: CRC Press), 279–434.
Croot, P. L., and Johansson, M. (2000). Determination of iron speciation by cathodic stripping voltammetry in seawater using the competing ligand 2-(2-thiazolylazo)-p-cresol (TAC). Electroanalysis 12, 565–576. doi: 10.1002/(sici)1521-4109(200005)12:8<565::aid-elan565>3.3.co;2-c
Davies, A. G. (1978). Pollution studies with marine plankton; Part II. Heavy metals. Adv. Mar. Biol. 15, 381–508.
de Baar, H. J. W., Saager, P. M., Nolting, R. F., and Vandermeer, J. (1994). Cadmium versus phosphate in the world ocean. Mar. Chem. 46, 261–281. doi: 10.1016/0304-4203(94)90082-5
DelValls, T. A., and Dickson, A. G. (1998). The pH of buffers based on 2-amino-2-hydroxymethyl-1,3-propanediol (‘tris’) in synthetic sea water. Deep Sea Res. 45, 1541–1554. doi: 10.1016/s0967-0637(98)00019-3
Dickson, A. G. (1981). An exact definition of total alkalinity and a procedure for the estimation of alkalinity and total inorganic carbon from titration data. Deep Sea Res. 28A, 609–623.
Dickson, A. G. (1984). pH scales and proton transfer reactions in saline media such as seawater. Geochim. Cosmochim. Acta 48, 2299–2308.
Dickson, A. G. (1993). pH buffers for sea-water media based on the total hydrogen-ion concentration scale. Deep Sea Res. 40, 107–118. doi: 10.1016/0967-0637(93)90055-8
Dickson, A., Sabine, C., and Christian, J. (eds.). (2007). Guide to Best Practices for Ocean CO2 Measurements. Sidney, BC: PICES Special Publication 3.
Dupont, S., and Poertner, H. O. (2013). A snapshot of ocean acidification research. Mar. Biol. 160, 1765–1771. doi: 10.1007/s00227-013-2282-9
Ellwood, M. J., and van den Berg, C. M. G. (2001). Determination of organic complexation of cobalt in seawater by cathodic stripping voltammetry. Mar. Chem. 75, 33–47. doi: 10.1016/s0304-4203(01)00024-x
Feistel, R., Lovell-Smith, J. W., Saunders, P., and Seitz, S. (2016). Uncertainty of empirical correlation equations. Metrologia 53, 1079–1090. doi: 10.1088/0026-1394/53/4/1079
Fitzsimmons, J. N., Boyle, E. A., and Jenkins, W. J. (2014). Distal transport of dissolved hydrothermal iron in the deep South Pacific Ocean. Proc. Natl. Acad. Sci. U.S.A. 111, 16654–16661. doi: 10.1073/pnas.1418778111
German, C. R., Campbell, A. C., and Edmond, J. M. (1991). Hydrothermal scavenging at the mid-atlantic ridge - modification of trace-element dissolved fluxes. Earth Planet. Sci. Lett. 107, 101–114. doi: 10.1016/0012-821x(91)90047-l
German, C. R., Legendre, L. L., Sander, S. G., Niquil, N., Luther, G. W. III Bharati, L., et al. (2015). Hydrothermal Fe cycling and deep ocean organic carbon scavenging: model-based evidence for significant POC supply to seafloor sediments. Earth Planet. Sci. Lett. 419, 143–153. doi: 10.1016/j.epsl.2015.03.012
German, C. R., Rudnicki, M. D., and Klinkhammer, G. P. (1999). A segment-scale survey of the Broken Spur hydrothermal plume. Deep Sea Res. 46, 701–714. doi: 10.1016/s0967-0637(98)00078-8
Greenberg, J. P., and Møller, N. (1989). The prediction of mineral solubilities in natural waters - a chemical equilibrium model for the Na-K-Ca-Cl-SO4-H2O system to high concentration from 0°C to 250°C. Geochim. Cosmochim. Acta 53, 2503–2518. doi: 10.1016/0016-7037(89)90124-5
Gustafsson, J. P. (2001). Modeling the acid-base properties and metal complexation of humic substances with the stockholm humic model. J. Colloid Interface Sci. 244, 102–112. doi: 10.1006/jcis.2001.7871
Haese, R. R., Smith, J., Weber, R., and Trafford, J. (2014). High-Magnesium calcite dissolution in tropical continental shelf sediments controlled by ocean acidification. Environ. Sci. Technol. 48, 8522–8528. doi: 10.1021/es501564q
Haley, B. A., Frank, M., Spielhagen, R. F., and Eisenhauer, A. (2008). Influence of brine formation on Arctic Ocean circulation over the past 15 million years. Nat. Geosci. 1, 68–72. doi: 10.1038/ngeo.2007.5
Harvie, C. E., Møller, N., and Weare, J. H. (1984). Prediction of mineral solubilities in natural waters: the Na-K-Mg-Ca-H-Cl-SO4-OH-HCO3-CO3-H2O system to high ionic strength at 25°C. Geochim. Cosmochim. Acta 48, 723–751.
Hatje, V., Bruland, K. W., and Flegal, A. R. (2014). Determination of rare earth elements after pre-concentration using NOBIAS-chelate PA-1 (R) resin: method development and application in the San Francisco Bay plume. Mar. Chem. 160, 34–41. doi: 10.1016/j.marchem.2014.01.006
Hatje, V., Bruland, K. W., and Flegal, A. R. (2016). Increases in anthropogenic gadolinium anomalies and rare earth element concentrations in San Francisco Bay over a 20 year record. Environ. Sci. Technol. 50, 4159–4168. doi: 10.1021/acs.est.5b04322
Henderson, G. M., Anderson, R. F., Adkins, J., Andersson, P., Boyle, E. A., Cutter, G., et al. (2007). GEOTRACES - An international study of the global marine biogeochemical cycles of trace elements and their isotopes. Chem. Erde 67, 85–131. doi: 10.1016/j.chemer.2007.02.001
Hernandez-Ayon, J. M., Zirino, A., Dickson, A. G., Camiro-Vargas, T., and Valenzuela-Espinoza, E. (2007). Estimating the contribution of organic bases from microalgae to the titration alkalinity in coastal seawaters. Limnol. Oceanogr. 5, 225–232. doi: 10.4319/lom.2007.5.225
Kadko, D. (1993). Excess Po-210 and nutrient recycling within the california coastal transition zone. J. Geophys. Res. 98, 857–864. doi: 10.1029/92jc01932
Kleint, C., Kuzmanovski, S., Powell, Z., Buehring, S. I., Sander, S. G., and Koschinsky, A. (2015). Organic Cu-complexation at the shallow marine hydrothermal vent fields off the coast of Milos (Greece), Dominica (Lesser Antilles) and the Bay of Plenty (New Zealand). Mar. Chem. 173, 244–252. doi: 10.1016/j.marchem.2014.10.012
Klevenz, V., Sander, S. G., Perner, M., and Koschinsky, A. (2012). Amelioration of free copper by hydrothermal vent microbes as a response to high copper concentrations. Chem. Ecol. 28, 405–420. doi: 10.1080/02757540.2012.666531
Klinkhammer, G., Heggie, D. T., and Graham, D. W. (1982). Metal diagenesis in oxic marine-sediments. Earth Planet. Sci. Lett. 61, 211–219. doi: 10.1016/0012-821x(82)90054-1
Klinkhammer, G. P. (1980). Early diagenesis in sediments from the eastern Equatorial Pacific. 2. Pore water metal results. Earth Planet. Sci. Lett. 49, 81–101. doi: 10.1016/0012-821x(80)90151-x
Klunder, M. B., Bauch, D., Laan, P., de Baar, H. J. W., van Heuven, S., and Ober, S. (2012). Dissolved iron in the Arctic shelf seas and surface waters of the central Arctic Ocean: impact of Arctic river water and ice-melt. J. Geophys. Res. 117:C01027. doi: 10.1029/2011jc007133
Koch, B. P., Witt, M. R., Engbrodt, R., Dittmar, T., and Kattner, G. (2005). Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochim. Cosmochim. Acta 69, 3299–3308. doi: 10.1016/j.gca.2005.02.027
Koopal, L. K., Saito, T., Pinheiro, J. P., and van Riemsdijk, W. H. (2005). Ion binding to natural organic matter: general considerations and the NICA-Donnan model. Colloids Surf. A 265, 40–54. doi: 10.1016/j.colsurfa.2004.11.050
Kulinski, K., Schneider, B., Hammer, K., Machulik, U., and Schulz-Bull, D. (2014). The influence of dissolved organic matter on the acid-base system of the Baltic Sea. J. Mar. Syst. 132, 106–115. doi: 10.1016/j.jmarsys.2014.01.011
Laglera, L. M., and van den Berg, C. M. G. (2003). Copper complexation by thiol compounds in estuarine waters. Mar. Chem. 82, 71–89. doi: 10.1016/s0304-4203(03)00053-7
Liu, K.-K., Atkinson, L., Quinones, R., and Talaue-McManus, L. (eds.). (2010). Carbon and Nutrient Fluxes in Continental Margins: A Global Synthesis. Berlin: Springer-Verlag.
Liu, X., Patsavas, M. C., and Byrne, R. H. (2011). Purification and characterization of meta-cresol purple for spectrophotometric seawater pH measurements. Environ. Sci. Technol. 45, 4862–4868. doi: 10.1021/es200665d
Liu, X. W., and Millero, F. J. (2002). The solubility of iron in seawater. Mar. Chem. 77, 43–54. doi: 10.1016/s0304-4203(01)00074-3
Liu, Y. G., Miah, M. R. U., and Schmitt, R. A. (1988). Cerium - a chemical tracer for paleo-oceanic redox conditions. Geochim. Cosmochim. Acta 52, 1361–1371. doi: 10.1016/0016-7037(88)90207-4
Luther, G. W. III Madison, A. S., Mucci, A., Sundby, B., and Oldham, V. E. (2015). A kinetic approach to assess the strengths of ligands bound to soluble Mn(III). Mar. Chem. 173, 93–99. doi: 10.1016/j.marchem.2014.09.006
Luther, G. W., and Tsamakis, E. (1989). Concentration and form of dissolved sulfide in the oxic water column of the ocean. Mar. Chem. 27, 165–177. doi: 10.1016/0304-4203(89)90046-7
Marion, G. M. (1997). A theoretical evaluation of mineral stability in Don Juan Pond, Wright Valley, Victoria Land. Antarct. Sci. 9, 92–99.
Millero, F. J. (2007). The marine inorganic carbon cycle. Chem. Rev. 107, 308–341. doi: 10.1021/cr0503557
Millero, F. J., and Pierrot, D. (1998). A chemical equilibrium model for natural waters. Aquat. Geochem. 4, 153–199. doi: 10.1023/a:1009656023546
Millero, F. J., and Roy, R. N. (1997). A chemical equilibrium model for the carbonate system in natural waters. Croatica Chemica Acta 70, 1–38.
Mottl, M. J., and McConachy, T. F. (1990). Chemical processes in buoyant hydrothermal plumes on the East Pacific Rise near 21°N. Geochim. Cosmochim. Acta 54, 1911–1927. doi: 10.1016/0016-7037(90)90261-i
Nielsdottir, M. C., Bibby, T. S., Moore, C. M., Hinz, D. J., Sanders, R., Whitehouse, M., et al. (2012). Seasonal and spatial dynamics of iron availability in the Scotia Sea. Mar. Chem. 130, 62–72. doi: 10.1016/j.marchem.2011.12.004
Nishioka, J., Obata, H., and Tsumune, D. (2013). Evidence of an extensive spread of hydrothermal dissolved iron in the Indian Ocean. Earth Planet. Sci. Lett. 361, 26–33. doi: 10.1016/j.epsl.2012.11.040
Orr, J. C., Epitalon, J. M., and Gattuso, J. P. (2015). Comparison of ten packages that compute ocean carbonate chemistry. Biogeosciences 12, 1483–1510. doi: 10.5194/bg-12-1483-2015
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., et al. (2005). Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms. Nature 437, 681–686. doi: 10.1038/nature04095
Pitzer, K. S. (ed.). (1991). Activity Coefficients in Electrolyte Solutions. Boca Raton, FL: CRC Press.
Pizeta, I., Sander, S. G., Hudson, R. J. M., Omanovic, D., Baars, O., Barbeau, K. A., et al. (2015). Interpretation of complexometric titration data: an intercomparison of methods for estimating models of trace metal complexation by natural organic ligands. Mar. Chem. 173, 3–24. doi: 10.1016/j.marchem.2015.03.006
Price, N. M., Harrison, G. I., Hering, J. G., Robert, J. M. H., Nirel, P. M. V., Palenik, B., et al. (1988). Preparation and chemistry of the artificial algal culture medium aquil. Biol. Oceanogr. 6, 443–461.
Ridgwell, A., and Hargreaves, J. C. (2007). Regulation of atmospheric CO2 by deep-sea sediments in an Earth system model. Global Biogeochem. Cycles 21:GB2008. doi: 10.1029/2006gb002764
Rolison, J. M., Middag, R., Stirling, C. H., Rijkenberg, M. J. A., and de Baar, H. J. W. (2015). Zonal distribution of dissolved aluminium in the Mediterranean Sea. Mar. Chem. 177, 87–100. doi: 10.1016/j.marchem.2015.05.001
Rozan, T. F., Benoit, G., and Luther, G. W. (1999). Measuring metal sulfide complexes in oxic river waters with square wave voltammetry. Environ. Sci. Technol. 33, 3021–3026. doi: 10.1021/es981206r
Rozan, T. F., Lassman, M. E., Ridge, D. P., and Luther, G. W. (2000). Evidence for iron, copper and zinc complexation as multinuclear sulphide clusters in oxic rivers. Nature 406, 879–882. doi: 10.1038/35022561
Rudd, M. A. (2014). Scientists' perspectives on global ocean research priorities. Front. Mar. Sci. 1:36. doi: 10.3389/fmars.2014.00036
Sander, S. G., and Koschinsky, A. (2011). Metal flux from hydrothermal vents increased by organic complexation. Nat. Geosci. 4, 145–150. doi: 10.1038/ngeo1088
Sander, S. G., Tian, F., Ibisanmi, E. B., Currie, K. I., Hunter, K. A., and Frew, R. D. (2015). Spatial and seasonal variations of iron speciation in surface waters of the Subantarctic front and the Otago Continental Shelf. Mar. Chem. 173, 114–124. doi: 10.1016/j.marchem.2014.09.001
Santana-Casiano, J. M., González-Dávila, M., and Millero, F. J. (2005). Oxidation of nanomolar levels of Fe(II) with oxygen in natural waters. Environ. Sci. Technol. 39, 2073–2079. doi: 10.1021/es049748y
Scher, H. D., and Martin, E. E. (2004). Circulation in the Southern Ocean during the Paleogene inferred from neodymium isotopes. Earth Planet. Sci. Lett. 228, 391–405. doi: 10.1016/j.epsl.2004.10.016
Semeniuk, D. M. (2014). Copper Nutrition and Transport Mechanisms in Plankton Communities in the Northeast Pacific Ocean. Ph.D. University of British Columbia.
Semeniuk, D. M., Bundy, R. M., Payne, C. D., Barbeau, K. A., and Maldonado, M. T. (2015). Acquisition of organically complexed copper by marine phytoplankton and bacteria in the northeast subarctic Pacific Ocean. Mar. Chem. 173, 222–233. doi: 10.1016/j.marchem.2015.01.005
Semiletov, I., Pipko, I., Gustafsson, O., Anderson, L. G., Sergienko, V., Pugach, S., et al. (2016). Acidification of East Siberian Arctic Shelf waters through addition of freshwater and terrestrial carbon. Nat. Geosci. 9, 361–365. doi: 10.1038/nego2695
Shahidul Islam, M., and Tanaka, M. (2004). Impacts of pollution on coastal and marine ecosystems including coastal and marine fisheries and approach for management: a review and synthesis. Mar. Pollut. Bull. 48, 624–649. doi: 10.1016/j.marpolbul.2003.12.004
Sholkovitz, E. R., Piepgras, D. J., and Jacobsen, S. B. (1989). The pore water chemistry of rare-earth elements in Buzzards Bay sediments. Geochim. Cosmochim. Acta 53, 2847–2856. doi: 10.1016/0016-7037(89)90162-2
Sholkovitz, E. R., and Schneider, D. L. (1991). Cerium redox cycles and rare-earth elements in the Sargasso Sea. Geochim. Cosmochim. Acta 55, 2737–2743. doi: 10.1016/0016-7037(91)90440-g
Siegenthaler, U., and Sarmiento, J. L. (1993). Atmospheric carbon dioxide and the ocean. Nature 365, 119–125.
Sinoir, M., Ellwood, M. J., Butler, E. C. V., Bowie, A. R., Mongin, M., and Hassler, C. S. (2016). Zinc cycling in the Tasman Sea: Distribution, speciation and relation to phytoplankton community. Mar. Chem. 182, 25–37. doi: 10.1016/j.marchem.2016.03.006
Strzepek, R. F., Maldonado, M. T., Hunter, K. A., Frew, R. D., and Boyd, P. W. (2011). Adaptive strategies by Southern Ocean phytoplankton to lessen iron limitation: uptake of organically complexed iron and reduced cellular iron requirements. Limnol. Oceanogr. 56, 1983–2002. doi: 10.4319/lo.2011.56.6.1983
Sukola, K., Wang, F. Y., and Tessier, A. (2005). Metal-sulfide species in oxic waters. Anal. Chim. Acta 528, 183–195. doi: 10.1016/j.aca.2004.10.009
Sunda, W., Price, N. M., and Morel, F. M. M. (2005). “Trace metal ion buffers and their use in culture studies,” in Algal Culturing Techniques, ed R. Anderson (Burlington, NC: Academic Press), 35–65.
Tagliabue, A., Bopp, L., Dutay, J. C., Bowie, A. R., Chever, F., Jean-Baptiste, P., et al. (2010). Hydrothermal contribution to the oceanic dissolved iron inventory. Nat. Geosci. 3, 252–256. doi: 10.1038/ngeo818
Tessier, A., and Turner, D. R. (eds.) (1995). Metal Speciation and Bioavailability in Aquatic Systems. Chichester: John Wiley.
Tipping, E., Lofts, S., and Sonke, J. (2011). Humic ion-binding model VII: a revised parameterisation of cation-binding by humic substances. Environ. Chem. 8, 225–235. doi: 10.1071/EN11016
Turner, D. R., and Hunter, K. A. (eds.) (2001). The Biogeochemistry of Iron in Seawater. Chichester: John Wiley.
Twining, B. S., and Baines, S. B. (2013). “The trace metal composition of marine phytoplankton,” in Annual Review of Marine Science, Vol. 5, eds C. A. Carlson and S. J. Giovannoni (Palo Alto, CA: Annual Reviews), 191–215.
Ulfsbo, A., Kuliñski, K., Anderson, L. G., and Turner, D. R. (2015). Modelling organic alkalinity in the Baltic Sea using a Humic-Pitzer approach. Mar. Chem. 168, 18–26. doi: 10.1016/j.marchem.2014.10.013
UNEP (2013). Global Chemicals Outlook: Towards Sound Management of Chemicals. Nairobi: United Nations Environment Programme.
Vanèina, V., Kester, D. R., and Bilinski, H. (1997). Application of the Pitzer model to solar salt brine chemistry. Croatica Chemica Acta 70, 55–69.
Vanèina, V., Plavšiæ, M., Bilinski, H., Branica, M., and Millero, F. J. (1986). Preparation and solubility of northupite from brine and its adsorption properties for Cu(II) and Cd(II) in seawater. Geochim. Cosmochim. Acta 50, 1329–1336. doi: 10.1016/0016-7037(86)90309-1
van den Berg, C. M. G. (1985). Determination of the zinc complexing capacity in seawater by cathodic stripping voltammetry of zinc-APDC complex ions. Mar. Chem. 16, 121–130.
van den Berg, C. M. G. (2006). Chemical speciation of iron in seawater by cathodic stripping voltammetry with dihydroxynaphthalene. Anal. Chem. 78, 156–163. doi: 10.1021/ac051441+
van den Berg, C. M. G., Khan, S. H., Daly, P. J., Riley, J. P., and Turner, D. R. (1991). An electrochemical study of Ni, Sb, Se, Sn, U and V in the estuary of the Tamar. Estuarine Coast. Shelf Sci. 33, 309–322.
van Hulten, M. M. P., Sterl, A., Middag, R., de Baar, H. J. W., Gehlen, M., Dutay, J. C., et al. (2014). On the effects of circulation, sediment resuspension and biological incorporation by diatoms in an ocean model of aluminium. Biogeosciences 11, 3757–3779. doi: 10.5194/bg-11-3757-2014
Wang, D., Lin, W., Yang, X., Zhai, W., Dai, M., and Chen, C. T. A. (2012). Occurrences of dissolved trace metals (Cu, Cd, and Mn) in the Pearl River Estuary (China), a large river-groundwater-estuary system. Cont. Shelf Res. 50, 54–63. doi: 10.1016/j.csr.2012.10.009
Waters, J. F., and Millero, F. J. (2013). The free proton concentration scale for seawater pH. Mar. Chem. 149, 8–22. doi: 10.1016/j.marchem.2012.11.003
Wu, J., Wells, M. L., and Rember, R. (2011). Dissolved iron anomaly in the deep tropical-subtropical Pacific: evidence for long-range transport of hydrothermal iron. Geochim. Cosmochim. Acta 75, 460–468. doi: 10.1016/j.gca.2010.10.024
Yang, L., Hong, H., Chen, C. T. A., Guo, W., and Huang, T. H. (2013). Chromophoric dissolved organic matter in the estuaries of populated and mountainous Taiwan. Mar. Chem. 157, 12–23. doi: 10.1016/j.marchem.2013.07.002
Yucel, M., Gartman, A., Chan, C. S., and Luther, G. W. III (2011). Hydrothermal vents as a kinetically stable source of iron-sulphide-bearing nanoparticles to the ocean. Nat. Geosci. 4, 367–371. doi: 10.1038/ngeo1148
Keywords: chemical speciation, seawater, modeling, equilibria, pH, trace metals, biogeochemical cycles
Citation: Turner DR, Achterberg EP, Chen C-TA, Clegg SL, Hatje V, Maldonado MT, Sander SG, van den Berg CMG and Wells M (2016) Toward a Quality-Controlled and Accessible Pitzer Model for Seawater and Related Systems. Front. Mar. Sci. 3:139. doi: 10.3389/fmars.2016.00139
Received: 31 May 2016; Accepted: 26 July 2016;
Published: 16 September 2016.
Edited by:
Marta Álvarez, Instituto Español de Oceanografía, SpainReviewed by:
Marta Plavsic, Rudjer Boskovic Institute, CroatiaWei-Dong Zhai, Shandong University, China
Copyright © 2016 Turner, Achterberg, Chen, Clegg, Hatje, Maldonado, Sander, van den Berg and Wells. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Turner, ZGF2aWQudHVybmVyQG1hcmluZS5ndS5zZQ==