 Perrine Cruaud1,2,3*
Perrine Cruaud1,2,3* Adrien Vigneron1,2,3
Adrien Vigneron1,2,3 Patricia Pignet1
Patricia Pignet1 Jean-Claude Caprais4Françoise Lesongeur1,2,3
Jean-Claude Caprais4Françoise Lesongeur1,2,3 Laurent Toffin1,2,3
Laurent Toffin1,2,3 Anne Godfroy1,2,3Marie-Anne Cambon-Bonavita1,2,3
Anne Godfroy1,2,3Marie-Anne Cambon-Bonavita1,2,3- 1UMR6197, Laboratoire de Microbiologie des Environnements Extrêmes, IFREMER, Technopôle Brest Iroise, Plouzané, France
- 2UMR6197, Laboratoire de Microbiologie des Environnements Extrêmes, Université de Bretagne Occidentale, Plouzané, France
- 3UMR6197, Laboratoire de Microbiologie des Environnements Extrêmes, Centre National de la Recherche Scientifique, Technopôle Brest Iroise, Plouzané, France
- 4Laboratoire ≪ Environnements Profonds ≫, IFREMER, EEP-LEP, Technopôle Brest Iroise, Plouzané, France
The Sonora Margin cold seeps present a seafloor mosaic pattern consisting of different faunal assemblages and microbial mats. To better understand if sedimentary microbial communities reflect this patchy distribution, all major habitats were investigated using four complementary approaches: 16S rRNA gene sequence 454 pyrosequencing, quantitative polymerase chain reaction, fluorescence in situ hybridization and geochemistry analyses. This study reveals that sediments populated by different surface assemblages show distinct porewater geochemistry features and are associated with distinct microbial communities. In the sediments underlying the microbial mat and the surrounding macrofauna, microbial communities were dominated by anaerobic methane oxidizers (archaeal anaerobic methanotroph ANME) and sulfate-reducing Deltaproteobacteria. In contrast, sediment-associated microbial communities underlying the megafauna habitats (vesicomyids and siboglinids) were characterized by a lower biomass and important proportions of the Marine Benthic Group D (MBG-D), Chloroflexi as well as filamentous Gammaproteobacteria and Deltaproteobacteria. Together, geochemical and microbial surveys indicate that porewater methane concentrations play an important role in the microbial community structure and subsequently in the establishment of the surface colonizers. Furthermore, presence and activity of the surface colonizers influence the underlying microbial communities probably because of modification of energy source availabilities.
Introduction
Seafloor emissions of hydrocarbon low-temperature fluids are a characteristic feature of cold seep ecosystems. These fluids are derived mainly from sedimented organic matter and support complex food webs based on microbial chemoautotrophic primary production (Van Dover et al., 2002). Diffusive and advective transports of potential substrates in sediment establish a succession of different redox zones from aerobic to anaerobic processes, based on the availability of electron donors (e.g., sulfide and methane) and acceptors (e.g., oxygen, nitrate, sulfate) (Jørgensen, 1977; Froelich et al., 1979; Engelen and Cypionka, 2009; Orcutt et al., 2011). For example, the sulfate-dependent Anaerobic Oxidation of Methane (AOM) occurs in anoxic zones of sediments, where upward diffusing methane meets sulfate from the seawater (Devol et al., 1984; Iversen and Jørgensen, 1985; Orcutt et al., 2011). This process appears to be mainly mediated by anaerobic methanotrophic Archaea (ANMEs), which are frequently observed in structured aggregates with sulfate-reducing bacteria mostly affiliated to Deltaproteobacteria. This process, as well as the activity of abundant sulfate-reducing bacteria, generates high concentrations of sulfide produced in porewater sediments which provides high fluxes of sulfide to the seafloor (Boetius et al., 2000; Orphan et al., 2001; Hinrichs and Boetius, 2002; Jørgensen and Boetius, 2007). These uprising sulfide-enriched fluids fuelled various surface assemblages such as mat-forming giant bacteria (Lloyd et al., 2010; Grünke et al., 2011; Mckay et al., 2012) or symbiont-bearing invertebrates (Sibuet and Olu, 1998; Dubilier et al., 2008; Cambon-Bonavita et al., 2009). These different types of communities are associated with specific seeps (characterized by different rates of fluid flow velocity and composition) depending on their ability to grow on available energy sources and to tolerate oxygen limitations and toxic sulfide concentrations (Barry et al., 1997; Sibuet and Olu, 1998; Sahling et al., 2002; Niemann et al., 2006; Lichtschlag et al., 2010; Pop Ristova et al., 2012; Ruff et al., 2013).
Sulfide-consuming bacterial mats live at the interface between opposed gradients of oxygen from the water column and sulfide from the sediments (Nelson et al., 1989; Schulz and Jorgensen, 2001; Lloyd et al., 2010; Grünke et al., 2011; Mckay et al., 2012). Some mobile organisms, such as gastropods, might feed on these mat-forming bacteria (Waren and Bouchet, 2009). At cold seeps with moderate fluid flow rates (Dubilier et al., 2008), benthic assemblages are dominated by chemosynthetic tubeworms (e.g., Siboglinidae) and bivalves (e.g., Vesicomyidae) harboring intracellular bacterial symbionts that consume sulfide and/or methane (Felbeck et al., 1981; Cavanaugh, 1983; Fisher, 1990). While vesicomyids live partially buried in sediments, capturing sulfide from sediments with their foot and pumping oxygen from seawater with their siphons (Cavanaugh, 1983; Arp et al., 1984; Fisher, 1990), siboglinids mainly use the posterior extension of their body (the “root”), to acquire sulfide from sediments and extend their branchial plumes in ambient seawater for oxygen uptake (Julian et al., 1999; Freytag et al., 2001; Duperron et al., 2014).
The hydrocarbon-rich cold seeps of the Guaymas Basin, located along a transform fault of the Sonora Margin, form a complex ecosystem showing various surface assemblages (Lonsdale, 1985; Simoneit et al., 1990; Paull et al., 2007; Vigneron et al., 2013). Microbial communities, especially those involved in AOM, occurring in sediments underlying microbial mats and the surrounding macrofauna habitats have been recently studied (Vigneron et al., 2013, 2014a). These works highlighted that ANMEs were metabolically active and AOM might represent a major microbial process at the Sonora Margin cold seeps. However, the microbial diversity and distribution in sediments underlying other dominant benthic cold seep assemblages and an overview of the microbial communities of the Sonora Margin sediments are still missing.
In this study, we investigated four cold seep sites in the Guaymas Basin, which exhibited contrasted surface assemblages (Figure 1, Table S1). Two sites (Ayala and Vasconcelos BIG13) were populated by chemosynthetic vesicomyid bivalves, one site (Juarez) was colonized by chemosynthetic siboglinid tubeworms while the fourth site (Vasconcelos BIG18) hosted a white microbial mat surrounded by a macrofaunal assemblage composed of ampharetid polychaetes and gastropods. Additionally, one site without observable seepage activity or microbial/faunal presence was analyzed as a “reference” sample.

Figure 1. Bathymetric map of the Sonora Margin and localization and pictures of the sampling sites.
At each site, we analyzed the diversity and distribution of microbial communities from the seawater/sediment interface down to 15 cm below the seafloor (cmbsf), using ARISA fingerprinting, 454-pyrosequencing, quantification of dominant microbial groups and microscopy, completed by porewater geochemistry. Our main objectives were (i) to characterize and compare microbial communities inhabiting these sediments, and (ii) to evaluate how biogeochemical factors shaped the microbial community structures. Doing so we tried to better understand (i) how the sedimentary microbial communities and their potential metabolisms may impact the surface colonizer distribution and therefore influence diversity patterns of chemosynthetic organisms at cold seeps and in return, (ii) how the surface chemosynthetic organisms may influence the sedimentary microbial communities.
Materials and Methods
Sampling Sites and Methods
Four cold seep sites situated at the Sonora Margin were investigated. Sediment samples were collected using push cores operated by the manned submersible Nautile during the “BIG” cruise (June 2010). Sampling sites were selected according to visual observations of the seafloor and methane plumes in the water column (H. Ondreas, pers. com.). The Ayala site, located at a water depth of 1560 m and spotted with the marker BIG14, was covered by abundant and scattered vesicomyid aggregates distributed on an estimated area of 15 by 20 meters (Figure 1, Table S1). The Vasconcelos BIG13 site, 1 km away from the Ayala site and located at a water depth of 1570 m of water depth, was colonized by vesicomyid populations surrounding a large and thin microbial mat (Figure 1, Table S1). The Juarez site, situated 160 m away from the Ayala site, was located at a water depth of 1560 m and spotted with the marker BIG12 (Figure 1, Table S1). This site was characterized by the occurrence of tubeworm bushes composed of specimens of Lamellibrachia barhami and Escarpia spp. siboglinids. Our last sampling site, the Vasconcelos BIG18 site, was located 30 meters away from the Vasconcelos BIG13 site. The Vasconcelos BIG18 site was colonized by a thick white Beggiatoa-like mat and a surrounding macrofaunal assemblage (called WM14 and EWM14 in Vigneron et al., 2013) (Figure 1, Table S1), mainly composed of Hyalogirina sp. gastropods (A. Waren and M. Portail pers. com.) and Ampharetidae (M. Rabiller pers. com.). Sediment cores were collected either from under the microbial mat or the assemblage of macrofauna. Finally, a reference site without seafloor manifestation of cold seeps was also sampled (called REF in Vigneron et al., 2013) (Table S1).
At each location, two replicates of sediment push cores were collected for microbiological and sulfate analyses and one sediment push core for methane, sulfate and sulfide analyses (Table S1). Porewater analyses were performed as described in Vigneron et al. (2013). Briefly, porewaters were obtained by spinning down 10 cm3 of sediments per horizon for sulfate analysis and using Rhizon samplers for hydrogen sulfide and methane analysis. Sulfate concentrations were measured by ion exchange chromatography, hydrogen sulfide concentrations were determined by colorimetry and methane concentrations were quantified by using the headspace technique and gas chromatograph equipped with flame-ionization detector. Oxygen profiles were measured using an oxygen minisensor OX 100 coupled to a picoammeter (PA 2000, UNISENSE) and a micro-manipulator using a profix data acquisition software (UNISENSE).
After recovering on board, sediment cores were immediately transferred to a cold room (~8°C) for sub-sampling. Sediment cores were cut into 2 cm thick layers and then frozen at −80°C for further nucleic acid extractions. Two cm3 of sediments were collected from each layer and fixed for 4 h in PBS (Phosphate Buffered Saline)/formaldehyde (3% final) at 4°C, washed twice with PBS and stored in an ethanol/PBS (1:1, vol/vol) buffer at −20°C for fluorescence in situ hybridization (FISH).
DNA Extraction
In order to increase the diversity of extracted lineages, we used two different extraction methods. For each sample, total nucleic acids were extracted from: 4 × 0.6 g of frozen sediments were extracted using FastDNA® SPIN Kit for Soil (Bio101 Systems, MP Biomedicals™) with some modifications (Webster et al., 2003; Roussel et al., 2009) and 3 × 2.5 g of frozen sediments were extracted using a modified method described in Zhou et al. (1996) and detailed in Cruaud et al. (2014). While the FastDNA Spin commercial kit is based on a mechanical lysis with ceramic and silica beads in a bead beater, the Zhou's method involves cycles of freeze-thawing in a high-salt extraction buffer and a phenol-chloroform extraction. Replicate of crude DNA extracts were then pooled and purified using the Wizard DNA clean-up kit (Promega, Madison, WI) according to the manufacturer instructions. Purified DNA extracts were stored at −20°C.
ARISA
An Automated fingerprinting method of Ribosomal Intergenic Spacer Analyses (ARISA) was carried out for rapid monitoring of microbial community composition in all sediment samples (Fisher and Triplett, 1999) using DNA 7500® Chip on an Agilent 2100 Bioanalyzer® (Agilent Technology, Santa Clara, USA). ARISA-PCR was performed with primers targeting the archaeal 16S-23S intergenic spacer region (Table 1). PCR conditions were as described in Vigneron et al. (2013). All data were recovered using the 2100 Expert® software (Agilent Technology). Comparisons between samples were allowed by normalization of the data by the raw signal intensity before statistical analyses. Non-metric multidimensional scaling (NMDS) (Kruskal, 1964) was carried out based on the Bray-Curtis similarity index (Bray and Curtis, 1957) using the software package Vegan (Oksanen et al., 2012) based on the software environment R (v. 3.0.2).
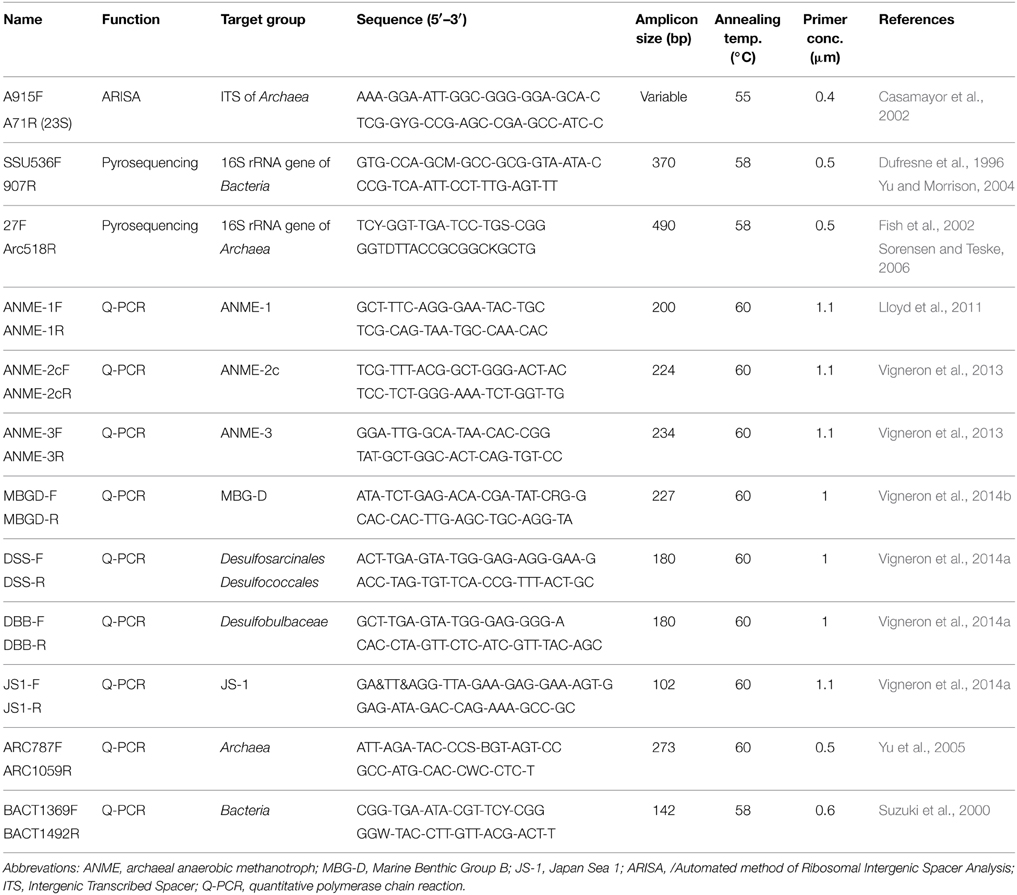
Table 1. PCR primers used for ARISA, 454-pyrosequencing and real-time PCR of 16S rRNA genes.
PCR Amplifications and Pyrosequencing
The 16S rRNA gene was amplified by PCR (from one sediment push core replicate per sampling location) using archaeal and bacterial targeted primers. The primer set for Bacteria amplified the V4-V5 hypervariable regions with SSU536F (Dufresne et al., 1996) and 907R (Yu and Morrison, 2004), while the archaeal primer set amplified the V1-V2-V3 hypervariable regions with 27F (Fish et al., 2002) and Arc518R (Sorensen and Teske, 2006) (Table 1). The primers were fused to 5–10-nucleotide key tags and to the 454 GS-FLX sequencing adaptor using the Lib-A chemistry. To allow multiplex sequencing (6 sampling locations with the different sediment sections), 23 such primers associated to Roche adaptor A and 23 associated to Roche adaptor B were synthesized for both primer sets, each with a different key tag (Table S2). These fusion primers were designed based on MIDs recommended by Roche and to minimize secondary structures as predicted by the OligoAnalyzer software (Owczarzy et al., 2008). The amplifications of the 16S rRNA genes were performed under the following conditions: 10 min at 95°C, then 30 cycles for Bacteria and 35 cycles for Archaea including 30 s at 95°C, 45 s at 58°C and 45 s at 72°C and a final step of 6 min at 72°C. PCR amplifications were performed in independent replicates (X and Y duplicates) using the following reagents in a 25 μL reaction volume: 1X Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Agilent Technologies, Palo Alto, CA), 0.5 μM of each primer (Eurofins MWG Operon, Ebersberg, Germany) and 1 μL of purified DNA template. The final volume was adjusted to 25 μL with sterile water. Absence of contaminations was checked by negative controls. PCR products were purified on TAE agarose gel (1.2%) and using PCR clean-up Gel extraction Nucleospin® Gel and PCR clean-up kit (Macherey-Nagel, Düren, Germany).
Microfluidic digital PCR (Fluidigm Corporation, San Francisco, CA) was used to quantify nucleic acids in purified amplicons. Amplicon products obtained for each PCR replicate were mixed equimolarly (108 molecules per microliter of each amplicon) and separately. A total of 4 mixes were obtained (Bacteria replicate X, Bacteria replicate Y, Archaea replicate X and Archaea replicate Y). Emulsion PCR and sequencing were then performed independently for these 4 mixes on a 454 Life Sciences Genome Sequencer GS-FLX (PicoTiterPlate divided in 4 regions) (Roche Diagnostics, Indianapolis, IN). Quantification, emulsion PCR, and sequencing were performed by the Biogenouest platform (Rennes, France).
Pyrosequencing Data Analyses
To minimize the effect of random sequencing errors (Huse et al., 2007; Kunin et al., 2010), pyrosequencing reads analyses and filtering were performed from the sff file using the Mothur pipeline (Schloss et al., 2009). We allowed for 1 mismatch to the barcode and 2 mismatches to the primer. We also removed sequences (i) shorter than 200 bp, (ii) containing homopolymers longer than 8 bp (iii) that aligned to the incorrect region within the 16S rRNA gene (Silva release 115, www.arb-silva.de), (iv) identified as chimeras using Uchime algorithm (Edgar et al., 2011; Schloss et al., 2011), and (v) affiliated to Archaea when Bacteria was targeted and inversely.
Using the tag combinations, sequences were assigned to their respective sample. In order to minimize the impact of higher number of sequences per sample, all the following analyzes were performed on the same number of sequences for the bacterial (1013 sequences) and the archaeal (686 sequences) regions.
Taxonomic assignments of the reads were performed using the Mothur version of the “Bayesian” classifier (Schloss et al., 2009) on both SILVA database (release 115, 418,497 sequences of Bacteria and 17,530 sequences of Archaea, www.arb-silva.de) and a personal database composed of deep-sea marine sediment sequences from reference publications (1677 bacterial and 786 archaeal sequences, as described in Cruaud et al., 2014). Results obtained from the two databases were then compared. If the results obtained for some microbial taxa were different depending on the database used, sequences affiliated to these taxa were extracted and BLAST analyzed against GenBank (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Consistencies of SILVA taxonomic affiliations were then checked by phylogenetic reconstructions with our reference database sequences and BLAST hits obtained. Phylogenetic trees were estimated using a distance method. Pairwise nucleotide sequence divergences were calculated using a Kimura-2-parameter model of substitution (Kimura, 1980) and Neighbor-Joining trees (Saitou and Nei, 1987) were reconstructed using MEGA 4.0.2 (Tamura et al., 2007). Robustness of topologies was assessed by bootstrap procedures using 100 replicates (Felsenstein, 1985). Then, our reference database was updated and supplemented according to the results obtained. Taxonomic assignments of the reads were finally performed on our updated and supplemented database (2549 bacterial and 803 archaeal sequences, Table S3). Only microbial groups retrieved in both PCR duplicates (X and Y) were conserved for further analyses.
Non-metric multidimensional scaling (NMDS) (Kruskal, 1964) was carried out based on the Bray-Curtis similarity measure (Bray and Curtis, 1957). Analysis of similarity (ANOSIM) was used to determine significant differences between sampling locations based on a Bray-Curtis similarity measure. SIMPER (Similarity Percentages) analyses were used to determine the contribution of each microbial groups to the Bray-Curtis dissimilarity (Clarke, 1993). These statistical analyses were performed using the software package Vegan (Oksanen et al., 2012) based on the software environment R (v. 3.0.2).
The raw sequencing data have been submitted to the NCBI database under SRA accession number SRP056234.
Quantitative Real-time PCR
Archaeal and bacterial abundances were estimated by quantitative PCR. Amplifications were performed with a Step One Plus instrument (Life technologies, Gaithersburg, MD) in a final volume of 25 μL using PerfeCTa® SYBR® Green SuperMix ROX (Quanta Bioscience), 1 ng of crude DNA template (extracted with the Zhou's method) and primers with appropriate concentrations and annealing temperatures (Table 1) according to the manufacturer's instructions. Standard curves were obtained in triplicate with 10-fold serial dilution (105–109 copies per μL) of plasmids containing environmental 16S rRNA genes of selected microbial lineages. The efficiencies of the reactions were above 85% and R2 of standard curves were close to 0.99. Samples were diluted until the crossing point decreased log-linearly with sample dilutions, indicating the absence of inhibition effect. Q-PCR results were expressed in copy number per gram of sediment.
Fluorescence In situ Hybridization (FISH)
Twenty microliters of a 100-fold dilution of fixed sediment subsamples were immobilized on 0.22 μm GTTP polycarbonate filters (Merck Millipore, Darmstadt, Germany). Hybridization conditions were optimized to maximize fluorescence signal and probe specificity (Probes listed in Table S4). Filters were hybridized in a reaction mix containing 0.5 μM of each probe in a formamide hybridization buffer [0.9M NaCl, 0.02M Tris-HCl, 0.01% sodium dodecyl sulfate (SDS), X% deionized formamide, Table S4] for 3 h at 46°C. Samples were washed at 48°C for 15 min in a washing buffer (NaCl, 0.02 M Tris-HCl, 0.004 M EDTA, 0.01% SDS) and rinsed briefly with cold water. Finally, filters were fixed on slides and treated with Slow Fade® Gold antifading reagent containing 4′-6-diamidino-2-phenylindole (DAPI) (Invitrogen, Life technologies, CA, USA). Observations and images were performed with a Zeiss Imager Z2 microscope (Zeiss, Göttingen, Germany) equipped with the slider module ApoTome® (Zeiss), the Colibri light technology (Zeiss) and using an AxioCam MRm camera (Zeiss). Epifluorescence acquisitions were treated using the ZEN software (Zeiss). A minimum of three filters was fully explored for each sediment layer.
Results
Sampling Sites
For easier reading, the following codes are used throughout text to identify the studied sediment cores. The codes refer to the geographic location of the sampling site and the organisms living on the sediment surface.
Ayala—vesicomyids (Figure 1, top right corner); Juarez—siboglinids (Figure 1, bottom right corner); Vasconcelos BIG18—macrofauna (Figure 1, bottom left corner, macrofauna composed of ampharetid polychaetes and gastropods); Vasconcelos BIG18—white mat (Figure 1, bottom left corner, microbial mat) and Vasconcelos BIG13—vesicomyids (Figure 1, top left corner). Taken as a whole, sites colonized by vesicomyids and siboglinids are named as “megafauna habitats.”
Geochemical Characterization
Methane was detected in all sediments but absent in the reference site (Figure 2). The highest methane concentrations (almost 1.2 mM) were measured throughout the sediments underlying the Vasconcelos BIG18—white mat site (Figure 2F). Methane concentrations were also high, though two fold lower in the sediments of the Vasconcelos BIG18—macrofauna site (peak of 606.5 μM at 7 cmbsf, Figure 2E). Methane concentrations were two orders of magnitude lower for habitats colonized by megafauna (vesicomyids and siboglinids) reaching maximum concentrations of 13 μM at 8 cmbsf for the Ayala—vesicomyids site (Figure 2B), 4.60 μM for the Juarez—siboglinids site (Figure 2C) and 3.40 μM for the Vasconcelos BIG13—vesicomyids (Figure 2D). It is noteworthy that methane concentrations might have been underestimated due to outgassing during core retrieval.
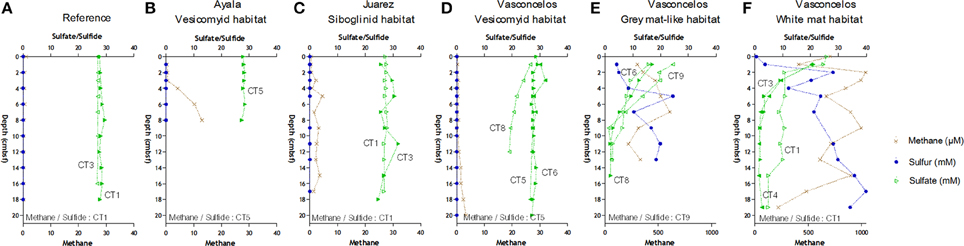
Figure 2. Geochemical profiles in cold seep sediments of the Sonora Margin, Guaymas Basin. Dissolved methane (cross), sulfate (full and empty triangle) and sulfide (full and empty circle) concentrations in porewater sediments. Sediment cores were from (A) Reference, (B) Ayala—vesicomyids, (C) Juarez—siboglinids, (D) Vasconcelos BIG13—vesicomyids, (E) Vasconcelos BIG18—macrofauna and (F) Vasconcelos BIG18—white mat sites.
Sulfide porewater concentrations increased quickly with depth and reached 40 and 20 mM at the deepest layers of the Vasconcelos BIG18—white mat and macrofauna sites respectively (Figures 2E,F). Sulfide porewater concentrations were below the technical detection limit [10 μM] for the sediments associated with habitats colonized by megafauna and reference site (Figures 2A–D).
In cores collected in the Vasconcelos BIG18—white mat and macrofauna sites, porewater sulfate concentrations decreased quickly from about 22 mM in surface layer to about 7 mM at 7 cmbsf. Sulfate concentrations at other habitats remained relatively constant to 28 mM with depth except for CT8 in the Vasconcelos BIG13—vesicomyids where sulfate concentrations decreased until 19 mM at 12 cmbsf.
Cores collected from all seeps had similar oxygen depth penetration ranging from 2.2 to 3.6 mm.
ARISA Fingerprints
ARISA was used as a fast method to compare archaeal community structure across our samples. Statistical analyses of the archaeal-community fingerprints showed a clear difference between microbial communities from the reference sediments and those from the cold seep sediments. For cold seep samples, the NMDS ordination plot indicated that microbial structures from a given sampling location clustered together (Figure S1). The archaeal community structure of the Vasconcelos BIG18 sites (white mat and macrofauna) appeared to be the most divergent from the archaeal community structure of the reference sediments. Archaeal community structures in the sediments colonized by megafauna assemblages appeared a little more similar to those of the reference sediments, though still closer to those of the the Vasconcelos BIG18 sites.
Archaeal Diversity
Pyrosequencing was used to describe microbial community diversity and distribution in the selected sites. 32.4% of total sequences were removed during quality control procedures (primer and barcode mismatches, long homopolymers, short sequences: 15%, sequences not aligned with the correct region: 15.8% and chimeras: 1.6%). After filtering, a total of 31,556 partial 16S rRNA gene sequences were used for archaeal diversity analyses. Taxonomic affiliation of these sequences highlighted various community structures according to sampling sites. Bray-Curtis similarities between samples are shown in Figure 3A and percentages of the different archaeal lineages are reported on Figure S2 (Results obtained with SILVA database are presented on Table S5).
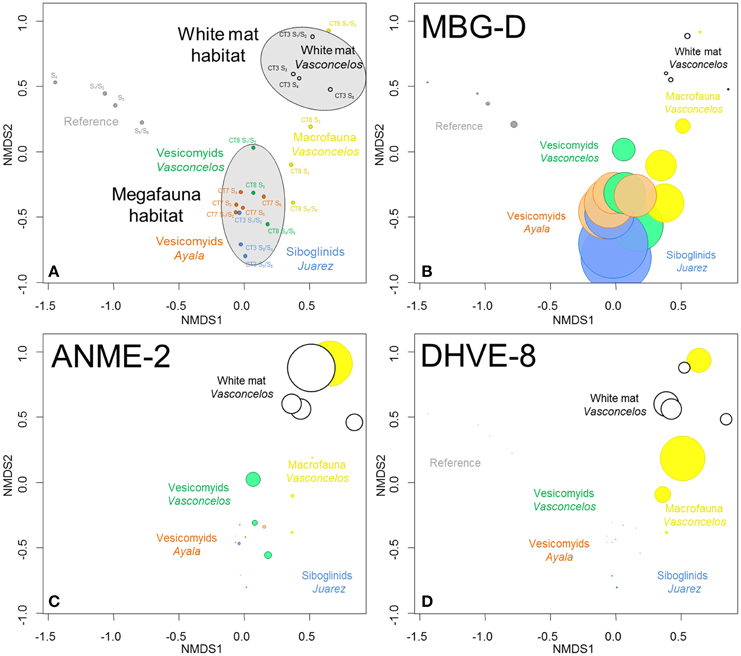
Figure 3. (A) NMDS ordination plots derived from the Bray-Curtis similarity measure between samples based on taxonomic affiliation of pyrosequencing reads for Archaea. Stress = 0.07. (B–D) Size of colored circles (gray: Reference, orange: Ayala—vesicomyids, blue: Juarez—siboglinids, green: Vasconcelos BIG13—vesicomyids, yellow: Vasconcelos BIG18—macrofauna and black: Vasconcelos BIG18—white mat sites) indicates microbial group proportions in each sample (B) MBG-D, (C) ANME-2 and (D) DHVE-8 groups.
Statistical analyses suggested that microbial communities from the reference sediments were significantly distinct from those observed in cold seep sediments (ANOSIM, p < 0.001) (Figure 3A). Similarity percentage analyses (SIMPER) (Clarke, 1993) highlighted that sequences affiliated to the Marine Group I (MG-I, 37.8% contribution), the Marine Benthic Group D (MBG-D, 22.2% contribution, Figure 3B) and the ANME-2 (Anaerobic Methanotroph, 7.3% contribution, Figure 3C) were mainly responsible for the differences between these sites. It is noteworthy that MG-I represented the majority of the detected sequences in the reference sediments while ANME-2 and MBG-D were mainly detected in cold seep sediments (Figure S2). Other archaeal groups such as the Marine Benthic Group E (MBG-E) and the Marine Benthic Group A (MBG-A) were only detected in the reference sediments (Figure S2).
Archaeal communities observed in cold seep sediments colonized with siboglinid and vesicomyid assemblages (megafauna habitats) were significantly different from those occurring in sediment underlying the Vasconcelos BIG18—white mat (ANOSIM, p < 0.002, Figure 3A). SIMPER analyses highlighted that MBG-D (37.8% contribution, Figure 3B), ANME-2 (18.6% contribution, Figure 3C) and DHVE-8 (Deep Sea Hydrothermal Vent Euryarchaeota group 8, 13.4% contribution, Figure 3D) were mainly responsible for the differences between these habitats. While sequences affiliated to MBG-D were largely dominant among sequences from samples collected from megafauna habitats (Figure 3B and Figure S2), ANME-2, ANME-1, and DHVE-8 were the main archaeal groups detected among the sequences from the Vasconcelos BIG18—white mat site (Figures 3B,C and Figure S2).
In shallow sediments (0–6 cm), archaeal community compositions associated with sediments colonized by gastropods and polychaetes (Vasconcelos BIG18—macrofauna habitat) appeared more similar to those associated with the Vasconcelos BIG18—white mat. In contrast, in deeper horizons, these compositions were more similar to those associated with megafauna assemblages (Figure 3A). Indeed sequences affiliated to ANME-2 and DHVE-8 were mainly detected in shallow sediments of the Vasconcelos BIG18—macrofauna habitat (Figures 3C,D and Figure S2), while sequences detected in deeper horizons were mainly affiliated to MBG-D (Figure 3B and Figure S2). ANME-1 sequences mainly detected in deeper sediment samples collected from under the Vasconcelos BIG18—white mat were also retrieved in significant proportions in deeper sediment samples collected from the Vasconcelos BIG18—macrofauna habitat (Figure S2).
Archaeal communities associated with the two vesicomyid habitats clustered together and appeared different from those associated with the siboglinid habitat (ANOSIM, p < 0.049). SIMPER analyses highlighted that MBG-D (36.8% contribution, detected in larger proportion in the Juarez—siboglinids habitat, Figure 3B and Figure S2) and AMOS1A (from “AMIS” Anaerobic Methane Incubation System, Girguis et al., 2003; Girguis et al., 9.5% contribution, detected in larger proportion in the vesicomyid habitats, Figure S2) were mainly responsible for the differences between these habitats.
Bacterial Diversity
24.7% of total sequences were removed during quality control procedures (primer and barcode mismatches, long homopolymers, short sequences: 11.7%, sequences not aligned with the correct region: 5% and chimeras: 8%). After filtering, a total of 46,598 partial sequences for the bacterial 16S rRNA gene were used for diversity analyses. Bray-Curtis similarities in bacterial diversity are shown in Figure 4A and proportions for the different bacterial groups are reported on Figure S3 (Results obtained with SILVA database are presented on Table S5).
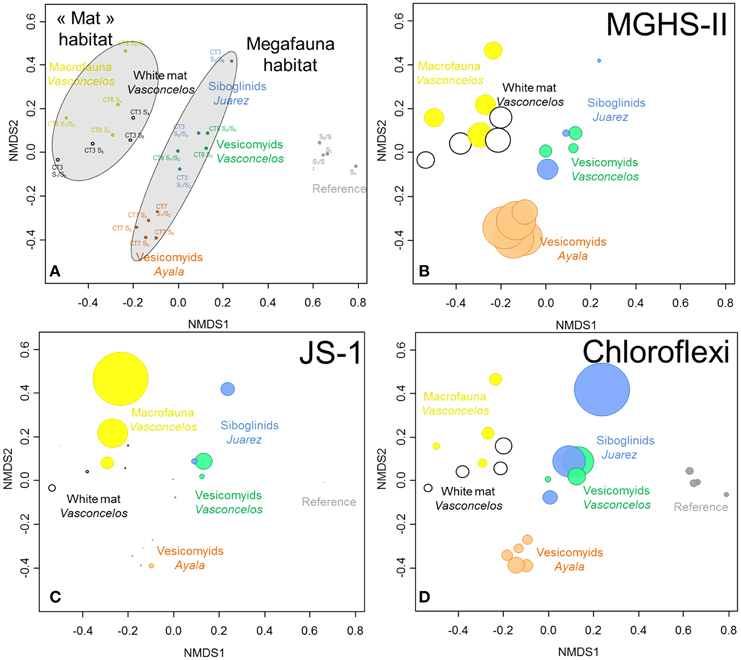
Figure 4. (A) NMDS ordination plots derived from the Bray-Curtis similarity measure between samples based on taxonomic affiliation of pyrosequencing reads for Bacteria. Stress = 0.09. (B–D) Size of colored circles (gray: Reference, orange: Ayala—vesicomyids, blue: Juarez—siboglinids, green: Vasconcelos BIG13—vesicomyids, yellow: Vasconcelos BIG18—macrofauna and black: Vasconcelos BIG18—white mat sites) indicates microbial group proportions in each sample (B) MHGS-II, (C) candidate division JS-1 and (D) Chloroflexi groups.
Statistical analyses showed a clear dissimilarity between bacterial community compositions from the reference sediments and all other sites (ANOSIM, p < 0.001, Figure 4A). SIMPER analyses highlighted that number of reads affiliated to specific lineages of Gammaproteobacteria were mainly responsible for these differences. In reference sediments, the JTB255 marine benthic group (Japan Trench Bacteria clone 255, Li et al., 1999) represented 12% of the detected sequences in average while only a few sequences from this group were detected in sediments from cold seep habitats. In contrast microbial communities identified in cold seep sediments were mainly affiliated to lineages of Deltaproteobacteria, especially Desulfobacteraceae, Epsilonproteobacteria, MHGS-II relatives group (sequences close to Maorithyas hadalis gill thioautotrophic symbiont phylotype II Fujiwara et al., 2001, Figure 4B), candidate division JS-1 (Japan Sea 1, Figure 4C), CFB (Bacteroidetes group) and Chloroflexi (Figure 4D).
In cold seep sites, bacterial communities underlying megafauna habitats appeared to cluster together compared to those associated with the microbial mat and macrofauna habitats (ANOSIM, p < 0.001, Figure 4A). SIMPER analyses highlighted that Desulfobacteraceae (8.9% contribution), candidate division JS-1 (6.5%) and Sulfurovum relatives (5.2%) were mainly responsible for the differences between these habitats. These bacterial groups were mainly detected in the sediments underlying the white mat and the macrofauna assemblage (Vasconcelos BIG18). On the contrary, larger percentages of Chloroflexi Division 1, including Caldilineales and Anaerolineales lineages, were globally detected in the sediments associated with megafauna habitats (Figure S3).
Bacterial communities associated with the bottom layer of the Vasconcelos BIG18—macrofauna habitat (20.9% sequences affiliated to the candidate division JS-1 and 1.2% affiliated to Sulfurovum relatives) differed from those associated with the surface layer of the Vasconcelos BIG18—white mat habitat (2.6% for the candidate division JS-1 and 17.3% for the Sulfurovum relative group).
In megafauna habitats, bacterial communities associated with the Juarez—siboglinids and the Vasconcelos BIG13—vesicomyids sediments appeared more similar to each other than to those associated with the Ayala—vesicomyids sediments (ANOSIM, p < 0.004). SIMPER analyses highlighted that Gammaproteobacteria and particularly MHGS-II relatives group (9.5% contribution), were responsible for the differences between these sites.
Quantitative Real-time PCR
In order to get a more comprehensive picture of microbial communities inhabiting the Sonora Margin sediments, we chose to combine our NGS results with two complementary approaches: FISH microscopy and Q-PCR analysis, as already suggested (Cruaud et al., 2014). Then, microbial groups highlighted by pyrosequencing and SIMPER analyses were also analyzed by Q-PCR. Thereby, relative abundances of ANME-1, ANME-2, ANME-3, and MBG-D for Archaea and DSS (Desulfosarcina-Desulfococcus group), DBB (Desulfobulbus group) and candidate division JS-1 for Bacteria were estimated by Q-PCR (Figure 5). Due to the very few data available on microorganisms from cold seeps, the number of 16S rRNA gene copy per cell remains uncertain. We thus considered a single 16S rRNA gene copy for each quantified populations, as previously suggested (Vigneron et al., 2013). Overall, relative abundances were higher for the Vasconcelos BIG18—macrofauna and white mat habitats for each microbial group except for the MBG-D group for which higher relative abundances were found for the Juarez—siboglinids and the Vasconcelos BIG13—vesicomyids habitats (Figure 5).
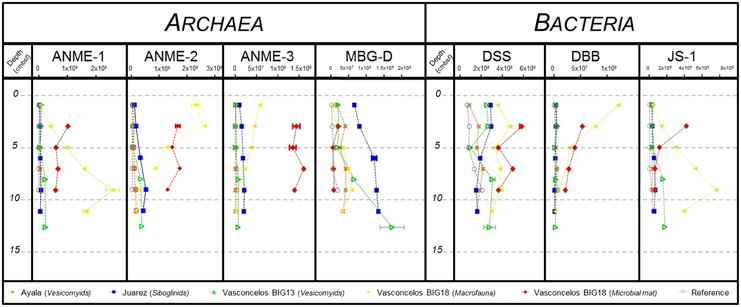
Figure 5. DNA copy numbers of the 16S rRNA gene per gram of sediment for ANME-1, ANME-2, ANME-3, MBG-D, Desulfosarcina/Desulfococcus group (DSS), Desulfobulbus group (DBB) and candidate division JS-1, from cores of (gray) Reference, (orange) Ayala—vesicomyids, (blue) Juarez—siboglinids, (green) Vasconcelos BIG13—vesicomyids, (yellow) Vasconcelos BIG18—macrofauna and (red) Vasconcelos BIG18—white mat sites in function of depth in cold seep sediments of the Sonora Margin.
Regarding the Archaea, ANME-1 relative abundances increased with depth, except for the Vasconcelos BIG18—white mat habitat. Between surface and bottom layer, we observed a 2–3 times increase for the megafauna habitats and a 60 times increase for the Vasconcelos BIG18—macrofauna habitat. Maximum abundances reached 6.30 × 107 16S rRNA gene copies for the Ayala—vesicomyids habitat, 7.50 × 107 for the Juarez—siboglinids habitat, 2.32 × 108 for the Vasconcelos BIG13—vesicomyids habitat. The highest value was found for the Vasconcelos BIG18—macrofauna habitat with 2.60 × 109 copies of 16S rRNA gene per gram of sediments. ANME-1 relative abundances slightly decreased from 1.03 × 109 between 2 and 4 cmbsf to 5.93 × 108 between 8 and 10 cmbsf for the Vasconcelos BIG18—white mat habitat. ANME-2 relative abundances increased slightly with depth for the megafauna habitats. We observed a 5–10 times increase for the megafauna habitats (up to 1.48 × 108 gene copies at the Ayala—vesicomyids habitat, 5.01 × 108 for the Juarez—siboglinids habitat and 3.56 × 108 for the Vasconcelos BIG13—vesicomyids habitat). On the contrary, ANME-2 relative abundances decreased with depth but were globally higher for the Vasconcelos BIG18—macrofauna habitat (from 2.65 × 109 to 1.25 × 108 copies of 16S rRNA gene per gram of sediments) and were fluctuating for the Vasconcelos BIG18—white mat habitat (around 1.54 × 109). ANME-3 relative abundances slightly increased with depth for the Juarez—siboglinids and the Vasconcelos BIG13—vesicomyids habitats (max. 2.12 × 107 and 7.7 × 106 respectively), while they decreased for the Vasconcelos BIG18—macrofauna habitat (from 5.79 × 107 to 1.35 × 106) and were close to the detection limit for the Ayala—vesicomyids habitat (max. 3.8 × 105). ANME-3 relative abundances were higher and relatively fluctuant for the Vasconcelos BIG18—white mat habitat (around 1.45 × 108). MBG-D relative abundances increased with depth and reached the highest quantities for the Vasconcelos BIG13—vesicomyids, the Juarez—siboglinids, and the Vasconcelos BIG18—macrofauna habitats. Between the surface and bottom layer, relative abundances were 9 times higher for the Vasconcelos BIG13—vesicomyids and the Vasconcelos BIG18—macrofauna habitats and 2 times higher for the Juarez—siboglinids habitat (max. 1.73 × 108, 1.34 × 108, and 5.90 × 107 respectively). MBG-D relative abundances were relatively constant with depth for the Ayala—vesicomyids and the Vasconcelos BIG18—white mat habitats (around 3.36 × 107 and 1.03 × 107 respectively).
Regarding the Bacteria, DSS relative abundances slightly decreased with depth for the Juarez—siboglinids and the Vasconcelos BIG18—white mat habitats (from 2.82 × 108 to 1.57 × 108 and 5.76 × 108 to 3.56 × 108 copies of 16S rRNA gene per gram of sediments respectively). They were relatively fluctuant for other habitats (1.58 × 108 for the Ayala—vesicomyids, 2.34 × 108 for the Vasconcelos BIG13—vesicomyids, 3.63 × 108 for Vasconcelos BIG18—macrofauna and 4.45 × 108 for the Vasconcelos BIG18—white mat habitats). DBB relative abundances decreased with depth for the Vasconcelos BIG18—macrofauna and the white mat habitats. We observed a 44 times decrease for the Vasconcelos BIG18—macrofauna habitat (max 1.22 × 108 at the surface layer) and a 2.5 times decrease for the Vasconcelos BIG18—white mat habitat (max 5.44 × 107 at the surface layer). DBB relative abundances were relatively constant and lower for other habitats (4.76 × 106 for Ayala—vesicomyids, 3.4 × 106 for Juarez—siboglinids and 4.41 × 106 for Vasconcelos BIG13—vesicomyids). Finally, JS-1 relative abundances increased with depth for the Vasconcelos BIG13—vesicomyids and the Vasconcelos BIG18—macrofauna habitats (5 times higher (max 1.80 × 108) and 21 times higher (max 7.55 × 108) respectively between surface and bottom layer). On the opposite, JS-1 relative abundances decreased for the Vasconcelos BIG18—white mat habitat (divided by 6.57, max 4.23 × 108), while they were relatively constant though lower at other habitats (3.58 × 107 for the Ayala—vesicomyids habitat and 5.04 × 107 for the Juarez—siboglinids habitat).
Fluorescence In situ Hybridization (FISH)
FISH observations using probes described in Table S4 were carried out to visualize likely active microbial cells (Teske, 2005). Overall higher cell density and morphological diversity were observed in sediments underlying the Vasconcelos BIG18—macrofauna and the white mat habitats (Figure 6).
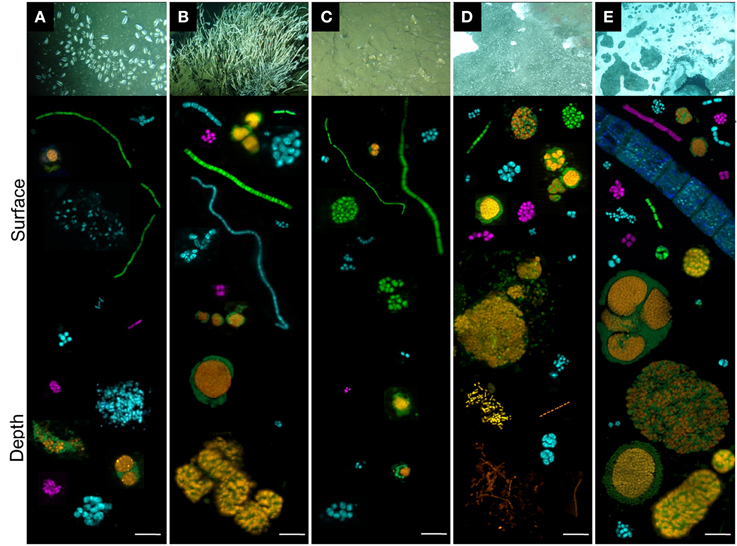
Figure 6. Individual cells and cell aggregates of ANME (orange), Deltaproteobacteria (green), Gammaproteobacteria (blue) and Epsilonproteobacteria (purple) visualized with fluorescent-labeled oligonucleotide probes. Each aggregate, single cell or heaps were taken from different pictures. Sediments were from (A) Ayala—vesicomyids, (B) Juarez—siboglinids, (C) Vasconcelos BIG13—vesicomyids, (D) Vasconcelos BIG18—macrofauna and (E) Vasconcelos BIG18—white mat sites, according to depth (surface and depth). Scale is 10 μm.
Regarding the Archaea, ANME-2 labeled cells were detected in each habitat and at each analyzed depth. They were always observed in structured consortia with bacterial cells labeled with Deltaproteobacteria probe (orange and green aggregates in Figure 6). The most abundant and the largest ANME-2/Deltaprotebacteria aggregates (diameter up to 100 μm) were observed in deep sediments underlying the Vasconcelos BIG18—white mat habitat (Figure 6E). Abundant but smaller ANME-2/Deltaproteobacteria aggregates were also observed in the surface sediments of the Vasconcelos BIG18—macrofauna habitat (Figure 6D) and in a minor proportion in the deepest sediments of the Juarez—siboglinids habitat (Figure 6B). ANME-2/Deltaproteobacteria aggregates were scarce and small (2–5 μm diameter) throughout the core of sediments colonized by vesicomyids (Figures 6A,C). Numerous ANME-1 labeled archaeal cells were observed as rod chains without detectable bacterial partner in the deep sediments of the Vasconcelos BIG18—macrofauna (Figure 6D) and the white mat habitats. They were observed as scarce rod-shaped cells in the other habitats. Unfortunately MBG-D and Thermoplasmatales were not detected using TPM666 probe but the majority of the observed cells labeled with the archaeal probe (Arch915) were also labeled with ANME probes.
Regarding the Bacteria, Deltaproteobacteria, Gammaproteobacteria and Epsilonprocteobacteria labeled cells were detected in each cold seep sediment core. Within each habitat, morphological diversity of Deltaproteobacteria cells (green cells in Figure 6) was greater in surface sediments than in deep sediments. Deltaproteobacteria were observed as tetrads, multicellular filaments, cocci monospecific clusters and associated with ANME-2 labeled cells in shallow sediments and almost exclusively within ANME consortia in the deepest layers (Figure 6). Whatever the sample, Gammaproteobacteria (blue cells in Figure 6) were observed as diplococci, tetrads, streptobacilli, and cluster of small cocci or large shapeless cocci. Gammaproteobacteria cells were more frequently observed in deeper horizons of the Ayala—vesicomyids habitat (Figure 6A), than in deeper horizons of the Juarez—siboglinids, the Vasconcelos BIG18—macrofauna and the white mat habitats (Figures 6B,D,E). They seemed steadier with depth in the Vasconcelos BIG13—vesicomyids habitat (Figure 6C). Epsilonproteobacteria labeled cells (purple in Figure 6) were less detected than Deltaprotebacteria and Gammaproteobacteria labeled cells, except for the surface sediments of the Vasconcelos BIG18—white mat habitat (Figure 6E). Most of them were observed as compact monospecific aggregates of small cocci. Epsilonproteobacteria cells were more numerous in deep sediments than in surface of the Ayala—vesicomyids habitat (Figure 6A), while it was the opposite situation for the Juarez—siboglinids, the Vasconcelos BIG18—macrofauna and white mat habitats (Figures 6B,D,E). Interestingly, multicellular bacterial filaments (mainly Delta- and Gammaproteobacteria) up to 0.7 cm long were recorded only in the surface sediments of the megafauna habitats (Figures 6A–C) and rare giant filamentous Gammaproteobacteria, probably corresponding to mat-forming bacteria, were observed in the surface sediments of the Vasconcelos BIG18—white mat habitat (Figure 6E).
Discussion
Cold seep ecosystems of the Sonora Margin in the Guaymas Basin show a high spatial heterogeneity with non-overlapping habitats dominated by different chemosynthetic organisms colonizing the surface, as previously observed in other cold seep ecosystems (Niemann et al., 2006; Fischer et al., 2012; Ruff et al., 2013). On the Sonora Margin seafloor, dense communities of bivalve Vesicomyidae, tubeworm Siboglinidae and microbial mats interspersed visually with large area of bare sediments.
Microorganisms occurring in the non-seep area (e.g., MG-I, MBG-E, MBG-A, specific lineages of Gammaproteobateria) were different from the microbial community identified in the cold seep sediments (e.g., ANME, MBG-D, candidate division JS-1) (Figures S2, S3). Previous study on the same samples (Vigneron et al., 2013) failed to amplified RNA for the reference non-seep site with the same primers, suggesting that these microbial communities were probably not very active in these sediments. Indeed, the DNA detected in our study could be due to the persistence of nucleic acids after cell death (Keer and Birch, 2003). Furthermore, MG-I account for a major portion of prokaryotic picoplankton (Delong et al., 1994; Fuhrman and Campbell, 1998; Karner et al., 2001). Thus sequences detected in our study in the reference site may in part result from the amplification of the 16S rRNA genes of some decaying cells. Likewise MBG-E and MBG-A are frequently detected in deep-sea sediments located outside any active area (Vetriani et al., 1999; Suzuki et al., 2004; Teske and Sorensen, 2008; Wang et al., 2010). In contrast, the detected microbial communities of the cold seep sediments were comparable to communities previously identified in other methane-rich environments (e.g., ANME groups, MBG-D, sulfate-reducers, candidate division JS1…) (Inagaki et al., 2006; Lösekann et al., 2007; Cambon-Bonavita et al., 2009; Lloyd et al., 2010; Pachiadaki et al., 2010, 2011).
In cold seep sediments, fluid emissions and more particularly methane fluxes appear to be important factors shaping the cold seep microbial communities (Boetius et al., 2000; Constan, 2009; Pachiadaki et al., 2011). The spatial variation of porewater sulfate, sulfide and methane concentrations measured in our study, potentially creates niche partitioning among microbial and animal communities. Indeed, we clearly observed that differences of surface colonizers reflected different microbial populations and distinct porewater geochemistry. The microbial mat habitat, which is fuelled by the highest methane porewater concentrations (Figure 2F) as previously observed in other cold seep ecosystems (Sahling et al., 2002; Knittel et al., 2003, 2005; Niemann et al., 2006; Fischer et al., 2012), harbored the most abundant and potentially active microorganisms (Figures 5, 6). These microorganisms, mainly ANME-2 observed in large consortia (diameter up to 100 μm) with Deltaproteobacteria (Figures 3C, 5, 6, Figures S2, S3), could oxidize the methane via AOM and produce the high concentrations of sulfide measured in the porewater (Figure 2F) (Boetius et al., 2000; Orphan et al., 2002; Lloyd et al., 2011; Milucka et al., 2012). These high sulfide concentrations could then allow the development of thiotrophic microbial mats at the surface sediment (Debeer et al., 2006; Fischer et al., 2012), potential sulfide oxidizers such as some Gamma- (MHGS-II Fujiwara et al., 2001) and Epsilonproteobacteria (Sulfurovum relatives Inagaki et al., 2004) detected by both pyrosequencing and FISH microscopy. Nevertheless, these high sulfide concentrations could exclude the megafauna from these areas since hydrogen sulfide is usually toxic to metazoans (Vetter et al., 1991). While many ANMEs were detected in these sediments, porewater methane concentrations remained elevated even at the top layer (Figure 2). This suggests that methane fluxes were too high to be entirely consumed by microbial communities. This also suggests a potential methane production in these sediments which might be mediated by the minority of methanogens identified by pyrosequencing (Figure S2) or by potentially versatile ANMEs such as. ANME-1 (Lloyd et al., 2011; Underwood et al., 2015), detected in these sediments by molecular analyses and observed as single rod cells in chain without bacterial partners (Figure 6). Additionally, a high percentage of sequences related to DHVE-8 were detected in this habitat and around (Figures 3D, 4), as previously observed in cold seep and hydrothermal vent ecosystems (Nercessian et al., 2004; Lloyd et al., 2010). Important proportions of sequences affiliated to DHVE-8 group are unusual in cold seep ecosystems but have already been highlighted in sediments underlying a microbial mat from a Gulf of Mexico hydrocarbon seep (Lloyd et al., 2010). This might reveal a potential link between the occurrences of both microbial mat at the surface and DHVE-8 in sediments.
In the edge of the microbial mat, colonized by gastropods and polychaetes (called macrofauna), methane porewater concentrations were two fold lower than in microbial mat sediments (Figures 2E,F). This feature leads likely to the restricted abundance of ANMEs and Deltaproteobacteria detected by Q-PCR and FISH (Figures 5, 6D) and therefore to the lower sulfide concentrations detected in porewater (Figure 2E). These moderate sulfide concentrations might then allow colonization by the macrofauna living at the seafloor. However, sulfide concentrations measured in the sediments were probably too high (25 mM at 5 cmbsf) for siboglinids and vesicomyids, which live half-buried in sediments (Arp et al., 1984; Julian et al., 1999).
In megafauna habitats, methane porewater concentrations were 100-fold lower than in microbial mat sediments (Figures 2B–D). Moreover, relative quantification of the dominant microbial groups and FISH observations indicated that microbial biomass in sediments underlying the megafauna was lower than in other habitats (Figures 5, 6), suggesting that methane and sulfur cycles might be less active than in the former environments. These low methane concentrations might limit the ANME biomass and potential AOM activity (Constan, 2009; Pachiadaki et al., 2011), as revealed by pyrosequencing, FISH and Q-PCR analyses of these sediments (Figures 3B, 5, 6, Figures S2, S3). Nevertheless, since ANME-2 and potential sulfate-reducer aggregates were detected, sulfide might likely be produced in these sediments but would remain below our technical detection limit [10 μM] (Figures 2B,C,D). Lack of sulfide observed in the surface sediment is a common feature for clam and tubeworm habitats at cold seeps (Barry et al., 1997; Sahling et al., 2002; Levin et al., 2003; Niemann et al., 2006; Fischer et al., 2012). Without elevated and toxic sulfide concentrations, vesicomyids, and tubeworms can colonize the surface. They dwell down in the sediments to reach the buried sulfide pool probably produced by deeper ANME/Deltaproteobacteria consortia as detected in the bottom sediment layer of the tubeworm habitat. Sulfide is then used by their symbionts (Childress et al., 1991; Freytag et al., 2001; Fischer et al., 2012).
These results suggested that porewater geochemistry and fluid flows would shape directly the microbial community structure in the sediments, as previously observed (Niemann et al., 2006; Lösekann et al., 2007). Fluid flows could also exert a selective pressure and therefore influence the faunal distribution at cold seep seafloor through microbial activities.
If the nature of the dominant surface colonizers reflects the underlying chemistry profiles and microbial activities, colonizers can also have an impact on the microbial community structures and modify the sediment geochemistry (Cordes et al., 2005; Fischer et al., 2012). Previous studies have demonstrated that chemosynthetic megafauna and microbial mat influence their local environment by bioturbation, bioirrigation, burrowing, and by exudates, altering the local seafloor biogeochemistry (Barry et al., 1997; Schulz and Jorgensen, 2001; Levin et al., 2003; Fischer et al., 2012). For example, Epsilonproteobacteria affiliated to sulfur-oxidizing Sulfurovum sp. (Inagaki et al., 2004; Pachiadaki et al., 2011; Vigneron et al., 2014a; Jones et al., 2015), representing 17.3% of the pyrosequencing reads in the surface sediments of the microbial mat (Figure S3), might be favored by the high sulfide concentrations potentially generated by AOM and by the micro-oxygenation of the sediments generated by the gliding mobility of giant filamentous bacteria (Schulz and Jorgensen, 2001). Likewise other aerobic groups detected by pyrosequencing in the surface layers might benefit from the microbial mat movements. In contrast, the detection of strictly anaerobic ANME-2 aggregates near the sediment surface (Figures 3C, 5, 6 and Figure S2) and the elevated abundance of ANME-1 (Figures 5, 6 and Figure S2) and candidate division JS-1, usually detected in totally anoxic sediments (Webster et al., 2004; Biddle et al., 2012) could be a consequence of the heterotrophic gastropods and polychaetes lifestyle. Indeed these macrofauna do not dig into the sediments but are supposed to graze the surface microbial communities and/or use a part of sedimented organic matter (Fauchald and Jumars, 1979; Sahling et al., 2002; Waren and Bouchet, 2009; Ruff et al., 2013). Thus micro-oxygenation in sediment underlying the macrofauna was probably low or nonexistent allowing the development of anaerobic lineages in the surface layers. Finally, in megafauna assemblages, sulfide was not detected (Figures 2B–D) and the observed AOM aggregates were reduced and localized in the deepest sediment layers (Figures 6A–C). Sulfide consumption and bioirrigation by vesicomyids and siboglinids could generate a deeper sulfate transition zone (SMTZ). AOM aggregates would then migrate more deeply in sediments as previously suggested (Cambon-Bonavita et al., 2009). This confirms that megafauna can modify porewater and microbial community structures in deeper sediments with their root or foot (Sahling et al., 2002; Cordes et al., 2005, 2010; Fischer et al., 2012). The observation of Deltaproteobacteria and Gammaproteobacteria multicellular filaments, exclusively in surface sediments of megafauna assemblages (Figures 6A–C) could also be explain by the occurrence of gaps between oxic and sulfidic zones detected in these sediments. These multicellular microbial filaments could actively seek out microenvironments where they find an optimal nutrient supply and couple oxygen consumption, theoretically possible only near the seafloor, at the very thin oxygenated zone (few millimeters), to hydrogen sulfide oxidation buried deeper in sediments (Nielsen et al., 2010; Pfeffer et al., 2012). The detection by pyrosequencing and Q-PCR of other microbial groups under faunal assemblages, such as MBG-D and Chloroflexi (Figures 3B, 4D, 5, Figures S2, S3), could also be explained by the occurrence of surface animals. Indeed, MBG-D, also known as MG-III (Delong, 1998), and Chloroflexi with potential metabolisms based on protein, amino acid and fatty acid degradations (Hugenholtz et al., 1998; Sekiguchi and Kamagata, 2004; Yamada et al., 2006; Lloyd et al., 2013) could be favored by exudates or metabolic wastes from fauna. For example, recent studies highlighted a local excretion of metabolites by the bivalves (Joye et al., 2010; Pop Ristova et al., 2012). Tubeworm and vesicomyid exudates could provide substrates available for MBG-D and Chloroflexi relatives detected in sediments underlying the mega- and macro-fauna and thereby explain their frequent detection in periphery of other cold seeps (Knittel et al., 2005; Lazar et al., 2010) and hydrothermal sediments (Biddle et al., 2012). The release of sulfate by tubeworm roots (Julian et al., 1999; Freytag et al., 2001; Cordes et al., 2005) could also increase microbial hydrocarbon degradation in the vicinity of tubeworm aggregations (Joye et al., 1999; Boetius, 2005). This might explain the presence of other Deltaproteobacteria and candidate division JS-1 in the siboglinid sediments, which could be possible partners in hydrocarbon-degrading sulfate reducing consortia (Phelps et al., 1998).
Conclusion
In this study we used three complementary approaches (454-pyrosequencing, Q-PCR and FISH), to characterize unexplored sediments of the Sonora Margin. Sediments underlying microbial mats, surrounding macrofauna as well as vesicomyid and siboglinid assemblages, were colonized by microbial communities typically found in other methane-rich ecosystems. However, differences in microbial community structure and lineage abundances were detected according to the sampled habitat. Our results indicated strong relationships between porewater geochemistry, microbial communities and surface colonizers. Thus, the occurrence of chemosynthetic communities at the seafloor might depend on the intensity and composition of the fluid seepages, which also depend on the abundance and activity of microbial communities involved in sulfur and methane cycles. In return, surface colonizers could impact microbial communities by modifying the spatial localization of the different energy sources and by probably releasing organic substrates.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to the crews of the research vessel L'Atalante and the submersible Nautile of the cruise “BIG” and the scientific team for their work on board. This cruise was funded by IFREMER (France) and has benefited from a work permit in Mexican waters (DAPA/2/281009/3803, 28 Octobre 2009). We thank Maxime Galan, Maria-Cristina Ciobanu, Jean-Yves Rasplus, Astrid Cruaud, Philippe Vandenkoornhuyse, Alexandra Dheilly and Sophie Coudouel for helpful scientific discussions. This study was supported by a Carnot Institute funding and by an IFREMER/Brittany region PhD grant for PC.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2015.00053
Figure S1. NMDS ordination plot derived from the Bray Curtis similarity measure between samples based on archaeal ARISA fingerprinting. Stress = 0.1. Each sample is represented by a colored and annotated dot. Sediment cores from (gray dots) Reference, (orange dots) Ayala—vesicomyids, (blue dots) Juarez—siboglinids, (green dots) Vasconcelos BIG13—vesicomyids, (yellow dots) Vasconcelos BIG18—macrofauna and (black and white dots) Vasconcelos BIG18—white mat sites.
Figure S2. Taxonomic affiliation of archaeal pyrosequencing reads after filtering for each sample. Higher percentages are highlighted in darker blue cells.
Figure S3. Taxonomic affiliation of bacterial pyrosequencing reads after filtering for each sample. Higher percentages are highlighted in darker green cells.
Table S1. Overview of some site features: localization, depth, sampled push cores, corresponding habitat, and analyses realized on each push core.
Table S2. Fusion primers used for pyrosequencing analyses.
Table S3. List of sequences included in our updated and supplemented reference database and associated details (NCBI accession numbers, reference publications, associated affiliations, and affiliations used in our database).
Table S4. Oligonucleotide probes used for fluorescence in situ hybridization.
Table S5. Taxonomic affiliation of pyrosequencing reads after filtering, using SILVA database (release 115). Results for Bacteria and Archaea according to samples.
References
Arp, A. J., Childress, J. J., and Fisher, C. R. (1984). Metabolic and blood-gas transport characteristics of the hydrothermal vent bivalve Calyptogena magnifica. Physiol. Zool. 57, 648–662.
Barry, J. P., Kochevar, R. E., and Baxter, C. H. (1997). The influence of pore-water chemistry and physiology on the distribution of vesicomyid clams at cold seeps in Monterey Bay: implications for patterns of chemosynthetic community organization. Limnol. Oceanogr. 42, 318–328. doi: 10.4319/lo.1997.42.2.0318
Biddle, J. F., Cardman, Z., Mendlovitz, H., Albert, D. B., Lloyd, K. G., Boetius, A., et al. (2012). Anaerobic oxidation of methane at different temperature regimes in Guaymas Basin hydrothermal sediments. ISME J. 6, 1018–1031. doi: 10.1038/ismej.2011.164
Boetius, A. (2005). Microfauna–macrofauna interaction in the seafloor: lessons from the tubeworm. PLoS Biol. 3:e102. doi: 10.1371/journal.pbio.0030102
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi: 10.1038/35036572
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Cambon-Bonavita, M. A., Nadalig, T., Roussel, E., Delage, E., Duperron, S., Caprais, J. C., et al. (2009). Diversity and distribution of methane-oxidizing microbial communities associated with different faunal assemblages in a giant pockmark of the Gabon continental margin. Deep Sea Res 56, 2248–2258. doi: 10.1016/j.dsr2.2009.04.007
Casamayor, E. O., Massana, R., Benlloch, S., Øvreås, L., Diez, B., Goddard, V. J., et al. (2002). Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4, 338–348. doi: 10.1046/j.1462-2920.2002.00297.x
Cavanaugh, C. M. (1983). Symbiotic chemoautotrophic bacteria in marine-invertebrates from sulphid-rich habitats. Nature 302, 58–61. doi: 10.1038/302058a0
Childress, J., Fisher, C., Favuzzi, J., Kochevar, R., Sanders, N., and Alayse, A. (1991). Sulfide-driven autotrophic balance in the bacterial symbiont-containing hydrothermal vent tubeworm, Riftia pachyptila Jones. Biol. Bull. Mar. Biol. Lab. Woods Hole 180, 135–153.
Clarke, K. R. (1993). Non−parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143.
Constan, L. (2009). A Correlation of Anaerobic Methane Oxidizing Archaea with Geochemical Gradients in Coastal Californian Marine Sediments. Ph.D. thesis, University of British Columbia, Vancouver.
Cordes, E. E., Arthur, M. A., Shea, K., Arvidson, R. S., and Fisher, C. R. (2005). Modeling the mutualistic interactions between tubeworms and microbial consortia. PLoS Biol. 3:e77. doi: 10.1371/journal.pbio.0030077
Cordes, E. E., Cunha, M. R., Galeron, J., Mora, C., Roy, O. L., Sibuet, M., et al. (2010). The influence of geological, geochemical, and biogenic habitat heterogeneity on seep biodiversity. Mar. Ecol. 31, 51–65. doi: 10.1111/j.1439-0485.2009.00334.x
Cruaud, P., Vigneron, A., Lucchetti-Miganeh, C., Ciron, P. E., Godfroy, A., and Cambon-Bonavita, M.-A. (2014). Influence of DNA extraction methods, 16S rRNA targeted hypervariable regions and sample origins on the microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl. Environ. Microbiol. 80, 4626–4639. doi: 10.1128/aem.00592-14
Debeer, D., Sauter, E., Niemann, H., Kaul, N., Foucher, J. P., and Witte, U. (2006). In situ fluxes and zonation of microbial activity in surface sediments of the Haakon Mosby mud volcano. Limnol. Oceanogr. 51, 1315–1331. doi: 10.4319/lo.2006.51.3.1315
Delong, E. F. (1998). Everything in moderation: archaea as ‘non-extremophiles’. Curr. Opin. Genet. Dev. 8, 649–654.
Delong, E. F., Wu, K. Y., Prézelin, B. B., and Jovine, R. V. (1994). High abundance of Archaea in Antarctic marine picoplankton. Nature 371, 695–697. doi: 10.1038/371695a0
Devol, A., Anderson, J., Kuivila, K., and Murray, J. (1984). A model for coupled sulfate reduction and methane oxidation in the sediments of Saanich Inlet. Geochim. Cosmochim. Acta 48, 993–1004. doi: 10.1016/0016-7037(84)90191-1
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Dufresne, S., Bousquet, J., Boissinot, M., and Guay, R. (1996). Sulfobacillus disulfidooxidans sp. nov., a new acidophilic, disulfide-oxidizing, gram-positive, spore-forming bacterium. Int. J. Syst. Bacteriol. 46, 1056–1064. doi: 10.1099/00207713-46-4-1056
Duperron, S., Gaudron, S. M., Lemaitre, N., and Bayon, G. (2014). A microbiological and biogeochemical investigation of the cold seep tubeworm Escarpia southwardae (Annelida: Siboglinidae): Symbiosis and trace element composition of the tube. Deep Sea Res. 90, 105–114. doi: 10.1016/j.dsr.2014.05.006
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engelen, B., and Cypionka, H. (2009). The subsurface of tidal-flat sediments as a model for the deep biosphere. Ocean Dynamics 59, 385–391. doi: 10.1007/s10236-008-0166-1
Fauchald, K., and Jumars, P. A. (1979). The diet of worms: a study of polychaete feeding guilds. Oceanogr. Mar. Biol. Ann. Rev. 17, 193–284.
Felbeck, H., Childress, J. J., and Somero, G. N. (1981). Calvin-Benson cycle and sulfide oxidation enzymes in animals from sulfide-rich habitats. Nature 293, 291–293. doi: 10.1038/293291a0
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Fischer, D., Sahling, H., Nöthen, K., Bohrmann, G., Zabel, M., and Kasten, S. (2012). Interaction between hydrocarbon seepage, chemosynthetic communities, and bottom water redox at cold seeps of the Makran accretionary prism: insights from habitat-specific pore water sampling and modeling. Biogeosciences 9, 2013–2031. doi: 10.5194/bg-9-2013-2012
Fish, S. A., Shepherd, T. J., Mcgenity, T. J., and Grant, W. D. (2002). Recovery of 16S ribosomal RNA gene fragments from ancient halite. Nature 417, 432–436. doi: 10.1038/417432a
Fisher, C. (1990). Chemoautotrophic and methanotrophic symbioses in marine invertebrates. Rev. Aquat. Sci. 2, 399–436.
Fisher, M. M., and Triplett, E. W. (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 65, 4630–4636.
Freytag, J. K., Girguis, P. R., Bergquist, D. C., Andras, J. P., Childress, J. J., and Fisher, C. R. (2001). A paradox resolved: sulfide acquisition by roots of seep tubeworms sustains net chemoautotrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 13408–13413. doi: 10.1073/pnas.231589498
Froelich, P. N., Klinkhammer, G., Bender, M. A. A., Luedtke, N., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090. doi: 10.1016/0016-7037(79)90095-4
Fuhrman, J. A., and Campbell, L. (1998). Marine ecology: microbial microdiversity. Nature 393, 410–411. doi: 10.1038/30839
Fujiwara, Y., Kato, C., Masui, N., Fujikura, K., and Kojima, S. (2001). Dual symbiosis in the cold-seep thyasirid clam Maorithyas hadalis from the hadal zone in the Japan Trench, western Pacific. Mar. Ecol. Prog. Ser. 214, 151–159. doi: 10.3354/meps214151
Girguis, P. R., Orphan, V. J., Hallam, S. J., and Delong, E. F. (2003). Growth and methane oxidation rates of anaerobic methanotrophic archaea in a continuous-flow bioreactor. Appl. Environ. Microbiol. 69, 5472–5482. doi: 10.1128/AEM.69.9.5472-5482.2003
Grünke, S., Felden, J., Lichtschlag, A., Girnth, A. C., De Beer, D., Wenzhöefer, F., et al. (2011). Niche differentiation among mat-forming, sulfide-oxidizing bacteria at cold seeps of the Nile Deep Sea Fan (Eastern Mediterranean Sea). Geobiology 9, 330–348. doi: 10.1111/j.1472-4669.2011.00281.x
Hinrichs, K., and Boetius, A. (2002). The anaerobic oxidation of methane: new insights in microbial ecology and biogeochemistry. Ocean Margin Syst. 457–477. doi: 10.1007/978-3-662-05127-6_28
Hugenholtz, P., Goebel, B. M., and Pace, N. R. (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180, 4765–4774.
Huse, S. M., Huber, J. A., Morrison, H. G., Sogin, M. L., and Welch, D. M. (2007). Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 8, 1–9. doi: 10.1186/gb-2007-8-7-r143
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., et al. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments, on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820. doi: 10.1073/pnas.0511033103
Inagaki, F., Takai, K., Nealson, K. H., and Horikoshi, K. (2004). Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 54, 1477–1482. doi: 10.1099/ijs.0.03042-0
Iversen, N., and Jørgensen, B. (1985). Anaerobic methane oxidation rates at the sulfate-methane transition in marine sediments from Kattegat and Skagerrak (Denmark). Limnol. Oceanogr. 30, 944–955. doi: 10.4319/lo.1985.30.5.0944
Jones, D. S., Flood, B. E., and Bailey, J. V. (2015). Metatranscriptomic analysis of diminutive Thiomargarita-like bacteria (“Candidatus Thiopilula spp.”) from abyssal cold seeps of the Barbados Accretionary Prism. Appl. Environ. Microbiol. 81, 3142–3156. doi: 10.1128/aem.00039-15
Jørgensen, B. (1977). Bacterial sulfate reduction within reduced microniches of oxidized marine sediments. Mar. Biol. 41, 7–17. doi: 10.1007/BF00390576
Jørgensen, B. B., and Boetius, A. (2007). Feast and famine - microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/nrmicro1745
Joye, S. B., Bowles, M. W., Samarkin, V. A., Hunter, K. S., and Niemann, H. (2010). Biogeochemical signatures and microbial activity of different cold-seep habitats along the Gulf of Mexico deep slope. Deep Sea Res. 57, 1990–2001. doi: 10.1016/j.dsr2.2010.06.001
Joye, S. B., Connell, T. L., Miller, L. G., Oremland, R. S., and Jellison, R. S. (1999). Oxidation of ammonia and methane in an alkaline, saline lake. Limnol. Oceanogr. 44, 178–188. doi: 10.4319/lo.1999.44.1.0178
Julian, D., Gaill, F., Wood, E., Arp, A. J., and Fisher, C. R. (1999). Roots as a site of hydrogen sulfide uptake in the hydrocarbon seep vestimentiferan Lamellibrachia sp. J. Exp. Biol. 202, 2245–2257.
Karner, M. B., Delong, E. F., and Karl, D. M. (2001). Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409, 507–510. doi: 10.1038/35054051
Keer, J. T., and Birch, L. (2003). Molecular methods for the assessment of bacterial viability. J. Microbiol. Methods 53, 175–183. doi: 10.1016/S0167-7012(03)00025-3
Kimura, M. (1980). A simple method for estimating evolutionnary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Knittel, K., Boetius, A., Lemke, A., Eilers, H., Lochte, K., and Pfannkuche, O. (2003). Activity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia margin, Oregon). Geomicrobiol. J. 20, 269–294. doi: 10.1080/01490450303896
Knittel, K., Lösekann, T., Boetius, A., Kort, R., and Amann, R. (2005). Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 71, 467–479. doi: 10.1128/aem.71.1.467-479.2005
Kruskal, J. B. (1964). Nonmetric multidimensional scaling: a numerical method. Psychometrika 29, 115–129. doi: 10.1007/BF02289694
Kunin, V., Engelbrektson, A., Ochman, H., and Hugenholtz, P. (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123. doi: 10.1111/j.1462-2920.2009.02051.x
Lazar, C. S., Dinasquet, J., Pignet, P., Prieur, D., and Toffin, L. (2010). Active archaeal communities at cold seep sediments populated by siboglinidae tubeworms from the storegga slide. Microb. Ecol. 60, 516–527. doi: 10.1007/s00248-010-9654-1
Levin, L. A., Ziebis, W., Mendoza, G. F., Growney, V. A., Tryon, M. D., Brown, K. M., et al. (2003). Spatial heterogeneity of macrofauna at northern California methane seeps: influence of sulfide concentration and fluid flow. Mar. Ecol. Prog. Ser. 265, 123–139. doi: 10.3354/meps265123
Li, L., Kato, C., and Horikoshi, K. (1999). Microbial diversity in sediments collected from the deepest cold-seep area, the Japan Trench. Mar. Biotechnol. 1, 391–400. doi: 10.1007/pl00011793
Lichtschlag, A., Felden, J., Bruchert, V., Boetius, A., and De Beer, D. (2010). Geochemical processes and chemosynthetic primary production in different thiotrophic mats of the Hakon Mosby Mud Volcano (Barents Sea). Limnol. Oceanogr. 55, 931–949. doi: 10.4319/lo.2009.55.2.0931
Lloyd, K. G., Albert, D. B., Biddle, J. F., Chanton, J. P., Pizarro, O., and Teske, A. (2010). Spatial structure and activity of sedimentary microbial communities underlying a Beggiatoa spp. mat in a Gulf of Mexico hydrocarbon seep. PLoS ONE 5:e8738. doi: 10.1371/journal.pone.0008738
Lloyd, K. G., Alperin, M. J., and Teske, A. (2011). Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ. Microbiol. 13, 2548–2564. doi: 10.1111/j.1462-2920.2011.02526.x
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Lonsdale, P. F. (1985). A transform continental margin rich in hydrocarbons, Gulf of California. AAPG Bull. 69, 1160–1180.
Lösekann, T., Knittel, K., Nadalig, T., Fuchs, B., Niemann, H., Boetius, A., et al. (2007). Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl. Environ. Microbiol. 73, 3348–3362. doi: 10.1128/aem.00016-07
Mckay, L. J., Macgregor, B. J., Biddle, J. F., Albert, D. B., Mendlovitz, H. P., Hoer, D. R., et al. (2012). Spatial heterogeneity and underlying geochemistry of phylogenetically diverse orange and white Beggiatoa mats in Guaymas Basin hydrothermal sediments. Deep Sea Res. 67, 21–31. doi: 10.1016/j.dsr.2012.04.011
Milucka, J., Ferdelman, T. G., Polerecky, L., Franzke, D., Wegener, G., Schmid, M., et al. (2012). Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 491, 541–546. doi: 10.1038/nature11656
Nelson, D. C., Wirsen, C. O., and Jannasch, H. W. (1989). Characterization of large, autotrophic beggiatoa spp abundant at hydrothermal vents of the Guaymas basin. Appl. Environ. Microbiol. 55, 2909–2917.
Nercessian, O., Prokofeva, M., Lebedinski, A., L'haridon, S., Cary, C., Prieur, D., et al. (2004). Design of 16S rRNA−targeted oligonucleotide probes for detecting cultured and uncultured archaeal lineages in high−temperature environments. Environ. Microbiol. 6, 170–182. doi: 10.1046/j.1462-2920.2004.00560.x
Nielsen, L. P., Risgaard-Petersen, N., Fossing, H., Christensen, P. B., and Sayama, M. (2010). Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature 463, 1071–1074. doi: 10.1038/nature08790
Niemann, H., Losekann, T., De Beer, D., Elvert, M., Nadalig, T., Knittel, K., et al. (2006). Novel microbial communities of the Haakon Mosby mud volcano and their role as a methane sink. Nature 443, 854–858. doi: 10.1038/nature05227
Oksanen, J., Blanchet, F., Kindt, R., Legendre, P., Minchin, P., O'hara, R., et al. (2012). Vegan: Community Ecology Package. R package version 2.1–14/r2120. Available online at: http://R-Forge.R-project.org/projects/vegan/
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/MMBR.00039-10
Orphan, V. J., Hinrichs, K. U., Ussler, W., Paull, C. K., Taylor, L. T., Sylva, S. P., et al. (2001). Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl. Environ. Microbiol. 67, 1922–1934. doi: 10.1128/aem.67.4.1922-1934.2001
Orphan, V. J., House, C. H., Hinrichs, K.-U., Mckeegan, K. D., and Delong, E. F. (2002). Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc. Natl. Acad. Sci. U.S.A. 99, 7663–7668. doi: 10.1073/pnas.072210299
Owczarzy, R., Tataurov, A. V., Wu, Y., Manthey, J. A., Mcquisten, K. A., Almabrazi, H. G., et al. (2008). IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 36, W163–W169. doi: 10.1093/nar/gkn198
Pachiadaki, M. G., Lykousis, V., Stefanou, E. G., and Kormas, K. A. (2010). Prokaryotic community structure and diversity in the sediments of an active submarine mud volcano (Kazan mud volcano, East Mediterranean Sea). FEMS Microbiol. Ecol. 72, 429–444. doi: 10.1111/j.1574-6941.2010.00857.x
Pachiadaki, M., Kallionaki, A., Dählmann, A., De Lange, G., and Kormas, K. (2011). Diversity and spatial distribution of prokaryotic communities along a sediment vertical profile of a deep-sea mud volcano. Microb. Ecol. 62, 655–668. doi: 10.1007/s00248-011-9855-2
Paull, C. K., Ussler, W. III. Peltzer, E. T., Brewer, P. G., Keaten, R., Mitts, P. J., et al. (2007). Authigenic carbon entombed in methane-soaked sediments from the northeastern transform margin of the Guaymas Basin, Gulf of California. Deep Sea Res. 54, 1240–1267. doi: 10.1016/j.dsr2.2007.04.009
Pfeffer, C., Larsen, S., Song, J., Dong, M., Besenbacher, F., Meyer, R. L., et al. (2012). Filamentous bacteria transport electrons over centimetre distances. Nature 491, 218–221. doi: 10.1038/nature11586
Phelps, C. D., Kerkhof, L. J., and Young, L. Y. (1998). Molecular characterization of a sulfate−reducing consortium which mineralizes benzene. FEMS Microbiol. Ecol. 27, 269–279. doi: 10.1111/j.1574-6941.1998.tb00543.x
Pop Ristova, P., Wenzhöfer, F., Ramette, A., Zabel, M., Fischer, D., Kasten, S., et al. (2012). Bacterial diversity and biogeochemistry of different chemosynthetic habitats of the REGAB cold seep (West African margin, 3160 m water depth). Biogeosci. Disc. 9, 8337–8385. doi: 10.5194/bg-9-5031-2012
Roussel, E. G., Sauvadet, A.-L., Allard, J., Chaduteau, C., Richard, P., Bonavita, M.-A. C., et al. (2009). Archaeal methane cycling communities associated with gassy subsurface sediments of Marennes-Oléron Bay (France). Geomicrobiol. J. 26, 31–43. doi: 10.1080/01490450802599284
Ruff, S. E., Arnds, J., Knittel, K., Amann, R., Wegener, G., Ramette, A., et al. (2013). Microbial communities of deep-sea methane seeps at hikurangi continental margin (New Zealand). PLoS ONE 8:e72627. doi: 10.1371/journal.pone.0072627
Sahling, H., Rickert, D., Lee, R. W., Linke, P., and Suess, E. (2002). Macrofaunal community structure and sulfide flux at gas hydrate deposits from the Cascadia convergent margin, NE Pacific. Mar. Ecol. Prog. Ser. 231, 121–138. doi: 10.3354/meps231121
Saitou, N., and Nei, M. (1987). The neighbor-joining method - A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310. doi: 10.1371/journal.pone.0027310
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schulz, H. N., and Jorgensen, B. B. (2001). Big bacteria. Annu. Rev. Microbiol. 55, 105–137. doi: 10.1146/annurev.micro.55.1.105
Sekiguchi, Y., and Kamagata, Y. (2004). “Microbial community structure and functions in methane fermentation technology for wastewater treatment,” in Strict and Facultative Anaerobes: Medical and Environmental Aspects, eds M. M. Nakano and P. Zuber (Norfolk: Horizon Scientific Press), 361–384.
Sibuet, M., and Olu, K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. 45, 517–567. doi: 10.1016/S0967-0645(97)00074-X
Simoneit, B. R. T., Lonsdale, P. F., Edmond, J. M., and Shanks, W. C. (1990). Deep-water hydrocarbon seeps in Guaymas basin, Gulf of California. Appl. Geochem. 5, 41–49. doi: 10.1016/0883-2927(90)90034-3
Sorensen, K. B., and Teske, A. (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl. Environ. Microbiol. 72, 4596–4603. doi: 10.1128/aem.00562-06
Suzuki, M. T., Taylor, L. T., and DeLong, E. F. (2000). Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl. Environ. Microbiol. 66, 4605–4614. doi: 10.1128/AEM.66.11.4605-4614.2000
Suzuki, Y., Inagaki, F., Takai, K., Nealson, K., and Horikoshi, K. (2004). Microbial diversity in inactive chimney structures from deep-sea hydrothermal systems. Microb. Ecol. 47, 186–196. doi: 10.1007/s00248-003-1014-y
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Teske, A. P. (2005). The deep subsurface biosphere is alive and well. Trends Microbiol. 13, 402–404. doi: 10.1016/j.tim.2005.07.004
Teske, A., and Sorensen, K. B. (2008). Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J. 2, 3–18. doi: 10.1038/ismej.2007.90
Underwood, S., Lapham, L., Teske, A., and Lloyd, K. G. (2015). Microbial community structure and methane-cycling activity of subsurface sediments at Mississippi Canyon 118 before the Deepwater Horizon disaster. Deep Sea Res. doi: 10.1016/j.dsr2.2015.01.011 (in press).
Van Dover, C. L., German, C. R., Speer, K. G., Parson, L. M., and Vrijenhoek, R. C. (2002). Evolution and Biogeography of Deep-Sea Vent and Seep Invertebrates. Science 295, 1253–1257. doi: 10.1126/science.1067361
Vetriani, C., Jannasch, H. W., Macgregor, B. J., Stahl, D. A., and Reysenbach, A. L. (1999). Population structure and phylogenetic characterization of marine benthic archaea in deep-sea sediments. Appl. Environ. Microbiol. 65, 4375–4384.
Vetter, R. D., Powell, M. A., and Somero, G. N. (1991). “Metazoan adaptations to hydrogen sulphide,” in Metazoan Life Without Oxygen, ed C. Bryant (London: Chapman and Ha/I), 109–128.
Vigneron, A., Cruaud, P., Pignet, P., Caprais, J.-C., Cambon-Bonavita, M.-A., Godfroy, A., et al. (2013). Archaeal and anaerobic methane oxidizer communities in the Sonora Margin cold seeps, Guaymas Basin (Gulf of California). ISME J. 7, 1595–1608. doi: 10.1038/ismej.2013.18
Vigneron, A., Cruaud, P., Pignet, P., Caprais, J. C., Gayet, N., Cambon−Bonavita, M. A., et al. (2014a). Bacterial communities and syntrophic associations involved in anaerobic oxidation of methane process of the Sonora Margin cold seeps, Guaymas Basin. Environ. Microbiol. 16, 2777–2790. doi: 10.1111/1462-2920.12324
Vigneron, A., Cruaud, P., Roussel, E. G., Pignet, P., Caprais, J. C., Callac, N., et al. (2014b). Phylogenetic and functional diversity of microbial communities associated with subsurface sediments of the Sonora Margin, Guaymas Basin. PLoS ONE 9:e104427. doi: 10.1371/journal.pone.0104427
Wang, P., Li, T., Hu, A., Wei, Y., Guo, W., Jiao, N., et al. (2010). Community structure of archaea from deep-sea sediments of the South China Sea. Microb. Ecol. 60, 796–806. doi: 10.1007/s00248-010-9746-y
Waren, A., and Bouchet, P. (2009). New gastropods from deep-sea hydrocarbon seeps off West Africa. Deep Sea Res. 56, 2326–2349. doi: 10.1016/j.dsr2.2009.04.013
Webster, G., Newberry, C. J., Fry, J. C., and Weightman, A. J. (2003). Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55, 155–164. doi: 10.1016/S0167-7012(03)00140-4
Webster, G., Parkes, R. J., Fry, J. C., and Weightman, A. J. (2004). Widespread occurrence of a novel division of bacteria identified by 16S rRNA gene sequences originally found in deep marine sediments. Appl. Environ. Microbiol. 70, 5708–5713. doi: 10.1128/AEM.70.9.5708-5713.2004
Yamada, T., Sekiguchi, Y., Hanada, S., Imachi, H., Ohashi, A., Harada, H., et al. (2006). Anaerolinea thermolimosa sp. nov., Levilinea saccharolytica gen. nov., sp. nov. and Leptolinea tardivitalis gen. nov., sp. nov., novel filamentous anaerobes, and description of the new classes Anaerolineae classis nov. and Caldilineae classis nov. in the bacterial phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 56, 1331–1340. doi: 10.1099/ijs.0.64169-0
Yu, Y., Lee, C., Kim, J., and Hwang, S. (2005). Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89, 670–679. doi: 10.1002/bit.20347
Yu, Z. T., and Morrison, M. (2004). Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 70, 4800–4806. doi: 10.1128/aem.70.8.4800-4806.2004
Keywords: microbial communities, pyrosequencing, MBG-D, ANME, faunal assemblages, Vesicomyidae, Siboglinidae, microbial mat
Citation: Cruaud P, Vigneron A, Pignet P, Caprais J-C, Lesongeur F, Toffin L, Godfroy A and Cambon-Bonavita M-A (2015) Microbial communities associated with benthic faunal assemblages at cold seep sediments of the Sonora Margin, Guaymas Basin. Front. Mar. Sci. 2:53. doi: 10.3389/fmars.2015.00053
Received: 04 May 2015; Accepted: 15 July 2015;
Published: 03 August 2015.
Edited by:
Hongyue Dang, Xiamen University, ChinaReviewed by:
J. Michael Beman, University of California, Merced, USAOlivia U. Mason, Florida State University, USA
Copyright © 2015 Cruaud, Vigneron, Pignet, Caprais, Lesongeur, Toffin, Godfroy and Cambon-Bonavita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Perrine Cruaud, EEP/LM2E, UMR6197, Laboratoire de Microbiologie des Environnements Extremes, IFREMER, Technopole Brest Iroise, BP70, Plouzane 29280, France, perrine.cruaud@gmail.com