 Sara J. Bender
Sara J. Bender Colleen A. Durkin
Colleen A. Durkin Chris T. Berthiaume
Chris T. Berthiaume Rhonda L. Morales
Rhonda L. Morales
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 26 March 2014
Sec. Aquatic Microbiology
Volume 1 - 2014 | https://doi.org/10.3389/fmars.2014.00003
Diatoms are among the most diverse groups of phytoplankton in the ocean. Despite their widely recognized influence on ocean ecosystems and global biogeochemistry, little is known about the impact of this diversity on large-scale processes. Here, we examined the ramifications of between-species diversity by documenting the transcriptional response of three diatoms —Thalassiosira pseudonana, Fragilariopsis cylindrus, and Pseudo-nitzschia multiseries —to the onset of nitrate limitation of growth, a common limiting nutrient in the ocean. The three species shared 5583 clusters of orthologous genes based on OrthoMCL clustering of publically available diatom genomes. These clusters represent 30–54% of the predicted genes in each diatom genome. Less than 5% of genes within these core clusters displayed the same transcriptional responses across species when growth was limited by nitrate availability. Orthologs, such as those involved in nitrogen uptake and assimilation, as well as carbon metabolism, were differently expressed across the three species. The two pennate diatoms, F. cylindrus and P. multiseries, shared 3839 clusters without orthologs in the genome of the centric diatom T. pseudonana. A majority of these pennate-clustered genes, as well as the non-orthologous genes in each species, had minimal annotation information, but were often significantly differentially expressed under nitrate limitation, indicating their potential importance in the response to nitrogen availability. Despite these variations in the specific transcriptional response of each diatom, overall transcriptional patterns suggested that all three diatoms displayed a common physiological response to nitrate limitation that consisted of a general reduction in carbon fixation and carbohydrate and fatty acid metabolism and an increase in nitrogen recycling. Characterization of these finely tuned responses will help to better predict which types of diatoms will bloom under which sets of environmental factors.
Diatoms are a globally influential group of marine phytoplankton with an estimated 200,000 species on Earth (Kooistra et al., 2007). They first appeared in the fossil record about 190 million years ago (Sims et al., 2006) and subsequently diverged into four main groups—the radial and bi/multipolar centric diatoms and the more recently diverged raphid and araphid pennate diatoms (Kooistra et al., 2007). Globally, diatoms contribute an estimated 40% to marine primary production (Falkowski et al., 2004). Fueled by seasonal and episodic upwelling events, diatom production leads to extensive organic matter export as part of the biological pump and provides an important link between the carbon and nitrogen cycles (Longhurst and Harrison, 1989; Dugdale and Wilkerson, 1998; Wilkerson et al., 2000; Martin et al., 2011).
Nitrogen uptake and assimilation in diatoms is directly connected to their carbon metabolism. Nitrogen assimilation requires ATP and reductants derived from photosynthesis (Falkowski and Stone, 1975) and carbon skeletons derived from the TCA cycle (Turpin, 1991; Behrenfeld et al., 2008; Hockin et al., 2012). When diatom growth is limited by nitrate availability, protein levels and cellular nitrogen concentrations drop and carbon to nitrogen ratios increase (Harrison et al., 1990; LaRoche et al., 1993; Claquin et al., 2002), triggering increased intracellular nitrogen recycling and storage of carbon-rich compounds, such as lipids (Allen et al., 2011; Palmucci et al., 2011; Hockin et al., 2012). Ultimately, a decrease in chlorophyll pigments results in cellular chlorosis (Kolber et al., 1988) and leads to a reduction in photosynthetic capacity, which in turn negatively impacts energy conversion, carbon fixation, and cell growth (Behrenfeld et al., 2008).
Molecular insights into diatom carbon and nitrogen metabolism come largely from analysis of two distantly related model diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum, both with publicly available whole genome sequences (Armbrust et al., 2004; Bowler et al., 2008). Genomic differences between T. pseudonana and P. tricornutum include the predicted cellular localization and number of isoforms of carbonic anhydrases and bicarbonate transporters, both required for the enhanced delivery of carbon dioxide to the carbon fixation enzyme, ribulose bis-phosphate carboxylase (Kroth et al., 2008), and key genes involved in glycolysis and gluconeogenesis (Montsant et al., 2005; Smith et al., 2012). In general, transcriptional and metabolomic studies suggest that increased availability of energy and carbon stores from glycolysis and the TCA cycle fuels the urea cycle, with the subsequent production of compounds containing recycled nitrogen, such as urea and arginine (Allen et al., 2006, 2011; Bender et al., 2012; Hockin et al., 2012; Smith et al., 2012). Despite these general commonalities, relatively few othologs shared by the two diatoms display similar transcriptional patterns under nitrogen limitation (Maheswari et al., 2010). Some of the genes with shared transcriptional patterns are involved in nitrogen assimilation (e.g., nitrate/nitrite reductase) and the first step of the urea cycle (e.g., carbamoyl phosphate synthetase) (Mock et al., 2008; Allen et al., 2011; Hockin et al., 2012; Ashworth et al., 2013). However, the remainder of the urea cycle genes and genes encoding urease and nitrate and ammonium transporters, are transcribed differently by the two diatoms; e.g., urease gene expression is elevated in T. pseudonana under nitrate limitation vs. nutrient-replete conditions, whereas this gene exhibits constitutive expression in P. tricornutum (Allen et al., 2011; Hockin et al., 2012).
We chose to further explore the implications of diatom diversity by extending transcriptional analyses to Fragilariopsis cylindrus, and Pseudo-nitzschia multiseries, two widely distributed raphid diatoms, both with recently available whole genome sequences: F. cylindrus is present in polar waters and P. multiseries is common in coastal and estuarine waters. We compared these diatoms to the estuarine model bipolar centric species T. pseudonana. Physiological differences (e.g., temperature range, nutrient quotas, maximum growth rates) between these diatoms were chosen to reflect the physiological diversity found among diatoms. We conducted a comparative transcriptional analysis of the diatoms grown under a comparable transition from optimal growth conditions to growth limited by nitrogen availability. Our goal was to identify those transcriptional patterns induced by nitrate limitation that are uniform across diatoms and those aspects that are exclusive to a given species. Findings from this study have implications for how diatom metabolism is modeled in the marine environment and how we classify and generalize the metabolic response of this diverse group of marine phytoplankton.
Thalassiosira pseudonana (Hustedt) Hasle et Heimdal (Provasoli-Guillard National Center for Marine Algae and Microbiota, Maine, USA; CCMP 1335), Fragilariopsis cylindrus (Provasoli-Guillard National Center for Marine Algae and Microbiota, CCMP 1102), and Pseudo-nitzschia multiseries (S. Bates, Fisheries and Oceans Canada; CLNN-17) were maintained at their optimum growth temperatures without bubbling in semi-continuous batch cultures under continuous light (100 μmol photons m−2s−1) in modified artificial seawater (Berges et al., 2001) with f/2 concentrations of nutrients (882 μ M NaNO3, 106 μM Na2SiO3, 36.2 μ MNaH2PO4; Guillard and Ryther, 1962). Growth of non-axenic triplicate cultures of T. pseudonana at 20°C, F. cylindrus at 4°C and P. multiseries at 13°C were monitored with a 10-AU fluorometer (Turner, California, USA). Cultures were considered acclimated to these growth conditions when the growth rates of three consecutive transfers were not significantly different from one another (ANCOVA, Brand et al., 1981).
Each acclimated diatom culture was then transferred into a batch culture with nutrient-replete media (882 μ M NaNO3, 106 μ M Na2SiO3, 36.2 μ MNaH2PO4) and a batch culture with low nitrate media (55 μ MNaNO3, 212 μ MNa2SiO3, 72.4 μ M NaH2PO4) in artificial seawater, maintaining three biological replicates per condition and per diatom (Berges et al., 2001). The reduced amount of nitrate relative to other nutrients, including a doubling of Na2SiO3 and NaH2PO4 concentrations, ensured that the onset of stationary phase in the low nitrate media was caused by availability of nitrate. In total, there were triplicate biological replicates of the low-nitrate and the replete-media conditions for each diatom (N = 18). Experiments were conducted in 10 L (T. pseudonana and F. cylindrus) or 4 L (P. multiseries) bottles; all cultures were bubbled with sterile filtered air. Pseudo-nitzschia multiseries was also stirred continuously. Photosynthetic yield of photosystem II (Fv/Fm) was monitored with a PhytoPAM fluorometer (Walz, Germany). The onset of stationary phase was defined by when chlorophyll a fluorescence of a culture no longer increased exponentially and Fv/Fm values decreased. Nitrate limitation of growth in the low nitrate media was confirmed by removing two 50 mL aliquots from the remaining cultures and adding 1764 μ M NaNO3 to aliquot one, no nutrients to aliquot two, and 1764 μ M NaNO3 to the remaining post-harvest large-volume cultures. All samples were monitored for at least two more days to confirm that chlorophyll a fluorescence and Fv/Fm increased after the addition of nitrate to aliquot one and the large-volume cultures.
Between 4.5 and 1.5 L of the experimental cultures (approximately half of the volume of each culture) were filtered onto 0.8 μm polycarbonate filters (Millipore, Massachusetts, USA) during mid-exponential growth (nutrient-replete) or at the onset of stationary phase due to nitrate limitation. Filtered cells were immediately flash frozen in liquid nitrogen and stored at −80°C until later RNA extraction. Samples were also taken to determine concentrations of nutrients. Approximately 50 mL of sample from each experimental culture were syringe-filtered (0.2 μm) into Falcon tubes during mid-exponential growth or at the onset of stationary phase for each replicate and stored at −20°C for later quantification of dissolved nutrients. Dissolved nutrient concentrations in P. multiseries nutrient-replete treatments were taken from a separate but identical, set of experiments conducted 1 week prior. All nutrients, including the nitrate concentration, were analyzed at the University of Washington Marine Chemistry Laboratory on a Technicon AutoAnalyser II (Seal Analytical, Wisconsin, USA) following the methods of UNESCO (1994).
RNA was extracted from frozen cells on filters using the ToTALLY RNA extraction kit (Life Technologies, New York, USA). The RNA was incubated with DNase I (Ambion, New York, USA) at 37°C for 2 h and purified by DNase inactivation reagent (Ambion). Messenger RNA was polyA-selected from total RNA using the Ambion Micro Poly(A)Purist Kit (Life Technologies, New York, USA) to minimize bacterial RNA sequencing. The mRNA was then prepared for sequencing according to the SOLiD Total RNA-Seq Kit protocol. For each sample, 700 million beads were loaded onto a slide and run on a Life Technologies (New York, USA) SOLiD sequencer version 4. Raw sequence reads and processed sequence reads for the three diatoms and two experimental conditions were deposited in the NCBI Gene Expression Omnibus (Accession # GSE56132: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE56132).
Predicted sequence read length was 50 base pairs. Sequence reads were converted from raw color-space into fastq format and then trimmed based on quality score using trimfastq (Iverson et al., 2012; SEAStAR, http://armbrustlab.ocean.washington.edu/seastar). The appropriate read error probability cutoff (“−p”) was determined based on a subsample of reads trimmed at 10 equally spaced cutoff values. The cutoff value that yielded the highest number of bases aligned was chosen. Trimmed reads with a length shorter than 30 colorspace transitions (“−l”) or an entropy value less than 3.0 (“−e”) were discarded. Trimmed reads were aligned with BWA (v.0.5.9) to predicted genes for Thalassiosira pseudonana (http://genome.jgi.doe.gov/Thaps3/Thaps3.home.html), Fragilariopsis cylindrus (http://genome.jgi.doe.gov/Fracy1/Fracy1.home.html) or Pseudo-nitzschia multiseries (http://genome.jgi.doe.gov/Psemu1/Psemu1.home.html). Parameters for “bwa aln” were “-n.001 -c -l 18 -k 2,” and parameters for “bwa samse” were “−n 500000” (Li and Durbin, 2009). Counts, the number of sequence reads that aligned to predicted genes, were calculated from the resulting SAM alignment files using ref_select as part of the SEAStAR package. Only those sequences that aligned to predicted genes were used for subsequent analyses.
Transcriptomes from biological triplicates for each condition and species (N = 18) were analyzed with the R software package edgeR from Bioconductor (Robinson et al., 2010). Alignment counts generated from ref_select in the SEAStAR package were normalized to library size for each replicate in edgeR. Then, an estimate of common dispersion of normalized counts for the three biological replicates within each treatment was calculated so that triplicates could be pooled for further analyses (Robinson and Smyth, 2007). Pairwise comparisons of pooled counts (i.e., transcript abundances) between treatments (nitrate-limited vs. nutrient-replete) were made with the exactTest function. A false discovery rate (fdr) of 0.01 was used to account for Type I errors; all p values provided have been fdr-corrected. P-values < 0.01 reflect statistically significant differences between nitrate-limited and nutrient-replete gene transcription. Up-regulated genes exhibit greater transcript abundances in response to nitrate-limited growth and down-regulated genes exhibit lower transcript abundances under nitrate-limited growth. For each diatom, the nitrate-limited transcript abundances were normalized to the nutrient-replete “control” abundances to give a log fold-change value, also referred to as the expression ratio.
In order to compare the transcriptomes among the diatoms, it was necessary to determine what genes had orthologs in the three diatoms genomes. Orthologous genes were clustered using the Identification of Ortholog Groups for Eukaryotic Genomes (OrthoMCL) software and default settings (Li et al., 2003). Potential function of predicted proteins in an orthologous cluster was based on a translated query sequence search of the three whole genomes (BLASTX v2.2.27+, e-value 1 × 10−5) to proteins within the Kyoto Encyclopedia of Genes and Genomes (KEGG) 2012-05-09 database (Altschul et al., 1990; Kanehisa and Goto, 2000; Kanehisa et al., 2012). The KEGG module annotation for the protein with the lowest e-value was assigned to all gene members of a cluster. Annotations were categorized as “multiple pathways” if the KEGG annotations differed between orthologs within a cluster. KEGG module annotations associated with genes that exhibited significant differential expression were hierarchically clustered using the heatmap.2 function in R on default settings to calculate the distance matrix (R gplots package). Transcribed genes that clustered within only one diatom or between the two pennate diatoms only were translated and compared to a protein database of P. tricornutum (http://genome.jgi-psf.org/Phatr2/Phatr2.home.html), and 21 publicly available transcriptome databases (Moore Foundation Marine Microbiology Initiative-supported Marine Microbial Eukaryote Transcriptome Sequencing Project, National Center for Genome Resources) using BLASTX with an e-value cutoff of 1 x 10−5 (Supplementary Table S1).
A total of 49,170 genes are predicted to be encoded by the three diatom genomes (T. pseudonana, 11,390 predicted genes; F. cylindrus, 18,077 predicted genes; P. multiseries, 19,703 predicted genes). Of this total, 18,077 orthologous genes were shared amongst the three diatoms and grouped into 5583 clusters (Figure 1) and were categorized as the core clusters. A minority of the core orthologous clusters (629) was composed of multi-copy gene families; 4954 clusters contained single copy gene families (Table 1). The two pennate diatoms, F. cylindrus and P. multiseries, shared 3839 clusters (4639 and 5105 genes, respectively) with no orthologs in the T. pseudonana genome and were categorized as the pennate clusters. A majority of these pennate clusters (3481) were composed of single copy genes (Table 1). Forty percent of the genes in each diatom did not cluster with genes from the two other diatoms and were defined as the non-orthologous clusters (Figure 1). A majority of these genes were present as single copies and did not cluster with any other non-orthologous genes (Table 1).
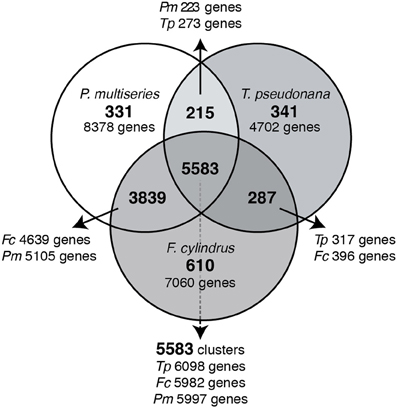
Figure 1. Venn diagram of predicted genes from T. pseudonana, F. cylindrus and P. multiseries. The numbers of OrthoMCL-identified homologous gene clusters are written in bold and the numbers of genes within the OrthoMCL clusters are written below or adjacent.
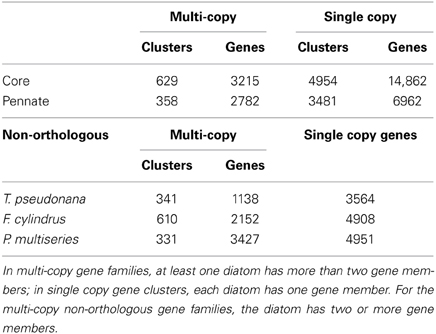
Table 1. Number of multi-copy and single copy gene clusters and genes based on OrthoMCL analysis for the core, pennate, and non-orthologous groups.
To identify the transcriptional response to the onset of stationary phase due to nitrate-limitation, the three diatom species were harvested while in exponential growth and once cell division ceased due to depletion of nitrate from the growth media. Exponential growth rates under optimal growth temperatures for each diatom were 0.87 ± 0.19 day−1 for T. pseudonana, 0.34 ± 0.04 day−1 for F. cylindrus, and 0.60 ± 0.12 day−1 for P. multiseries (Table 2). The photochemical yield of photosystem II (Fv/Fm) was 0.68 for T. pseudonana and P. multiseries and 0.52 for F. cylindrus at time of harvest under nutrient-replete growth. The low Fv/Fm values for F. cylindrus are common for this diatom during growth at an irradiance of 100 μmol photons m−2 s−1 (Arrigo et al., 2010).
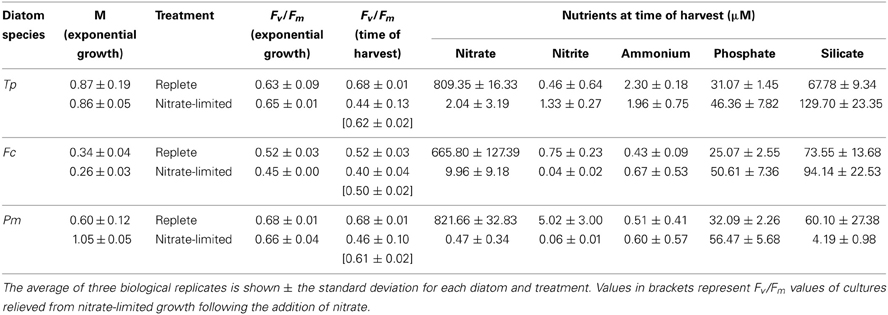
Table 2. Exponential growth rates in replete and nitrate-limited media prior to growth limitation, Fv/Fm, and nutrient concentrations for cells harvested under nutrient-replete or nitrate-limited growth.
All nitrate-limited cultures were harvested when chlorophyll a plateaued to ensure that the physiological responses detected among the diatoms reflected the same change in condition, despite the inherent physiological differences between the species. When nitrate-limited cells were harvested, Fv/Fm ranged between 0.46 and 0.40, depending on the diatom. When nitrate was added back to the nitrate-limited cultures, Fv/Fm increased to nutrient-replete values confirming that the ceased growth of the limited cultures was due to nitrogen availability (Table 2). Nitrate concentrations in the nitrate-limited cultures were 2-3 orders of magnitude less than the nitrate concentrations of the nutrient-replete cultures and an order of magnitude less than the phosphate and silicic acid concentrations of either the nutrient-replete or nitrate-limited cultures (Table 2). At time of harvest, F. cylindrus nitrate concentrations in growth-limited cultures were higher (9 μ M) than the other two growth-limited diatoms (<2 μ M; Table 2). Previous work has shown that F. cylindrus experiences the beginning of growth limited by nitrate at this concentration (Sommer, 1986); differences in nitrate concentrations at time of harvest reflect known differences in nitrogen uptake kinetics for each diatom. Silicate concentrations for P. multiseries nitrate-limited cultures were 4 μ M, indicating that these cultures may have been silicate uptake limited (Pan et al., 1996); however, upon the re-addition of nitrate, the relative fluorescence for each P. multiseries nitrate-limited biological replicate doubled over the course of 2-3 days (data not shown), and the Fv/Fm values recovered to levels observed during nutrient-replete growth (Table 2).
The vast majority of predicted genes in each diatom was transcribed either during exponential growth and/or when limited for nitrate (Table 3). About half the genes that displayed significantly different transcript abundances under nitrate limitation relative to exponential conditions (defined here as differentially expressed) were part of the orthologous core group (1016–2106 genes) (Table 3; Supplementary Table S2), with 1.5–2.5 times more of these genes displaying fewer transcripts (down-regulated) than more transcripts (up-regulated) relative to control, replete conditions (Table 3; Figure 2). Thalassiosira pseudonana displayed the greatest range of differential expression followed by P. multiseries, whereas differential expression of genes encoded by F. cylindrus fell within a smaller range of values (Figure 2). Core genes with significant differential expression in one diatom did not necessarily exhibit the same differential expression patterns in the other diatoms, contributing to the observed variation.
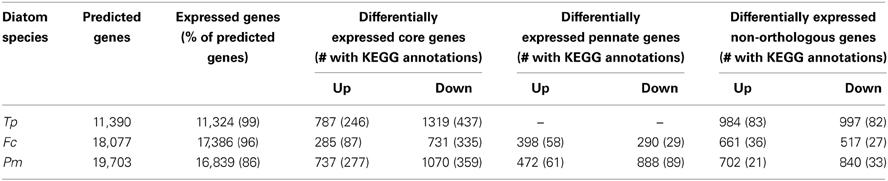
Table 3. Genomic and transcriptomic comparisons of orthology and expression among T. pseudonana (Tp), F. cylindrus (Fc), and P. multiseries (Pm), including genes with differential expression under nitrate limitation (p < 0.01) for the core, pennate and non-orthologous gene subsets.
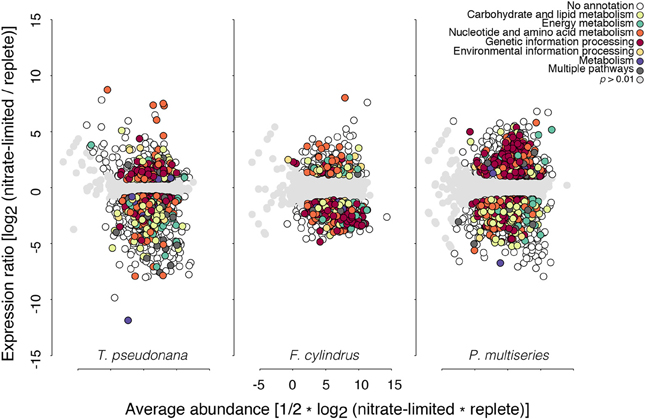
Figure 2. Ratio-average (RA) plots of expressed core orthologous genes among T. pseudonana (left), F. cylindrus (center) and P. multiseries (right) under nitrate limitation relative to nutrient-replete control conditions. White circles indicate genes with significant differential expression (p < 0.01); gray circles indicate genes where expression does not differ significantly from the nutrient-replete control (p > 0.01). Genes with significant differential expression that mapped to a KEGG module are highlighted in color. Genes where expression was only detected under one condition and not under both conditions fall on the separate diagonal line of the plot.
About a third (32-42%) of the differentially expressed orthologous core genes had a KEGG module annotation (Table 3). When the presence and abundance of transcripts with KEGG annotations were compared across the normalized diatom transcriptomes, three groups emerged. One group contained KEGG annotations for transcripts up-regulated during nitrate limitation in all three diatoms. The other two groups contained KEGG annotations for transcripts down-regulated during nitrate limitation: one consisted of T. pseudonana and P. multiseries annotations (Figure 3) and the other consisted of F. cylindrus annotations. RNA processing transcripts constituted the largest portion of annotations in the up-regulated subset (10%) and cofactor and vitamin biosynthesis transcripts constituted the largest portion in the down-regulated subset (11%). Nitrogen-related annotations (e.g., nitrogen metabolism and polyamine biosynthesis) were more abundant (28%) in the up-regulated subset of transcripts than in the down-regulated subset (19% down-regulated). Carbon-related annotations (e.g., central carbohydrate metabolism and carbon fixation) were more abundant (23%) in the down-regulated than up-regulated (17%) subset of transcripts. Additional KEGG annotations in the up-regulated group were for the spliceosome (8%), protein processing (4%), and branched-chain amino acid metabolism (4%), all with potential links to cell repair and nitrogen recycling. Abundant KEGG annotations in the down-regulated groups were for the ribosome (9%), central carbohydrate metabolism (8%), fatty acid metabolism (7%), and carbon fixation (4%). Overall, there were twice as many KEGG annotations associated within the down-regulated group.
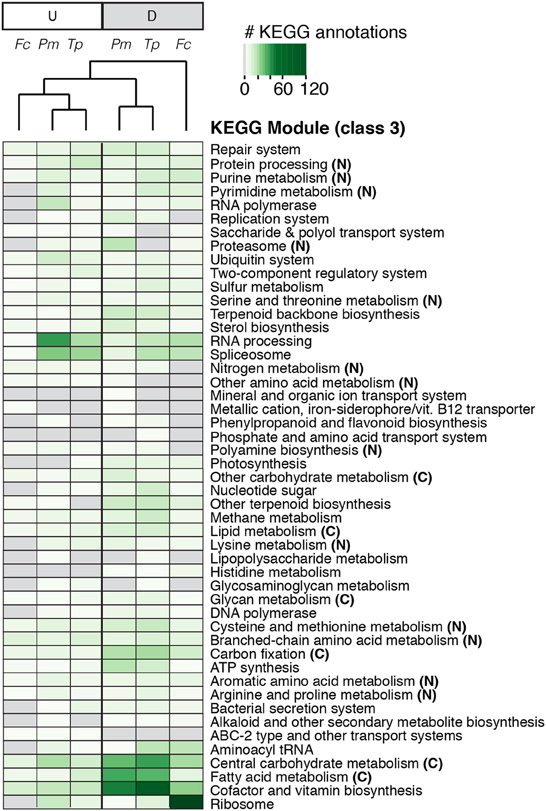
Figure 3. Heatmap of KEGG annotations for differentially expressed orthologous genes based on presence, absence, and abundance of annotations. The darker green colors indicate a greater number of differentially expressed genes with KEGG annotations; gray boxes, no differentially expressed genes had KEGG annotations. KEGG modules associated with carbon (C) and nitrogen (N) metabolism are labeled accordingly. Treatments and KEGG modules are clustered hierarchically based on similarities and differences in KEGG annotation abundances. Abbreviations: Tp, T. pseudonana; Fc, F. cylindrus; Pm, P. multiseries; up-regulated (U) and down-regulated (D).
A small subset (< 5%) of the orthologous genes that were present in the three diatom genomes shared the same differential expression patterns across all three diatoms, such that all transcribed genes within a given cluster were significantly up- or down- regulated in response to nitrate limitation (Figure 4). We define the expression patterns of these genes as the shared core response to nitrate limitation. Genes within 24 orthologous clusters were all up-regulated in response to nitrate limitation. Eleven of these clusters had associated KEGG module annotations that suggested involvement in the formation, degradation and transport of nitrogen-rich compounds including genes involved in serine biosynthesis, leucine degradation, cell repair and protein processing (Figure 4). Genes with shared differential expression included a putative ABC transport system (Figure 4). Genes within 160 orthologous clusters were all down-regulated in response to nitrate limitation. Sixty-three of these clusters had KEGG annotations that suggested involvement in porphyrin and chlorophyll metabolism, glycolysis, fatty acid biosynthesis, terpenoid backbone biosynthesis, the pentose phosphate pathway, photosynthesis and carbon fixation. The largest annotated gene cluster in the down-regulated core subset contained six ferredoxin genes required for electron transport (Energy metabolism module, Figure 4). These results indicate a general enhancement of transcription of genes involved in nitrogen processing and a reduction of transcription of genes involved in carbon metabolism and electron transport in response to nitrate limitation. Four genes in the shared core response were identified as nitrate-only in a separate study of T. pseudonana by (Mock et al., 2008), who included in their comparison iron limitation, low temperature and high pH conditions. Three of the gene clusters encode hypothetical proteins with no obvious annotations (Tp268343, Fc164230, Pm303116; Tp4888, Fc206263, Pm284060; Tp6551, Fc227588, Pm315644); the fourth gene cluster is connected to carotenoid biosynthesis (Tp264039, Fc169705, Pm242952).
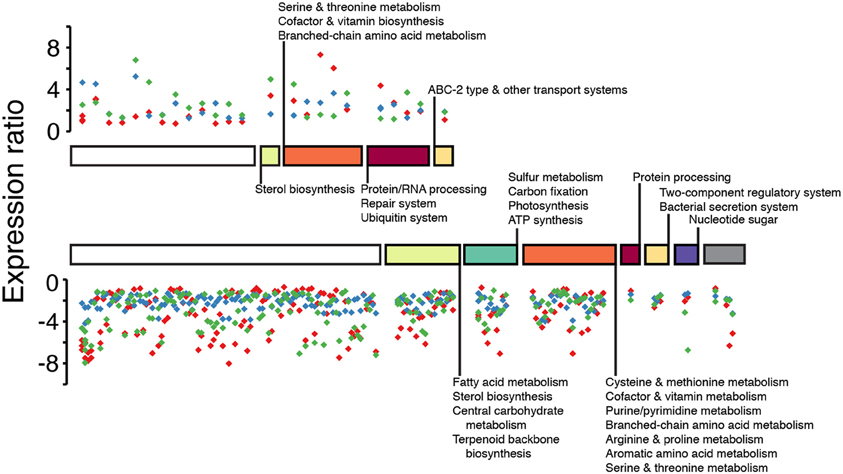
Figure 4. Scatterplot of core orthologous genes with shared significant differential patterns among the three diatoms. Differentially expressed orthologous genes from the same cluster are stacked vertically based on their gene expression ratio; colored diamonds correspond to each species (red, T. pseudonana; blue, F. cylindrus; green, P. multiseries). Stacked clusters are sorted according to their KEGG module ID (x-axis; colored bars). Clusters with no KEGG module ID are represented by the white bars. From left to right: yellow bar, carbohydrate and lipid metabolism; green bar, energy metabolism; orange bar, nucleotide and amino acid metabolism; red bar, genetic information processing; gold bar, environmental information processing; purple bar, metabolism; gray bar, multiple pathways. The length of colored bars along the x-axis is proportional to the number of differentially expressed orthologous gene clusters represented within a KEGG module. Y-axis, expression ratio: Log2 (nitrate-limited / replete) treatments.
We examined transcriptional patterns of the genes shared between only the pennate diatoms to further explore potential group-specific responses to nitrate limitation. A minority of the 3839 pennate gene clusters was significantly differentially expressed, and those genes with the greatest differential expression had no KEGG module annotations. When the small fraction of differentially expressed genes with a KEGG module annotation (Table 3) were clustered hierarchically, three groups formed (Figure 5A). The up-regulated subsets for both diatoms contained more annotations associated with RNA processing/polymerase, as well as cysteine and methionine metabolism. The down-regulated subset contained more annotations associated with central carbohydrate metabolism and carbon fixation. More P. multiseries down-regulated genes were associated with lipid and fatty acid metabolism compared to the other subsets. About 160 genes in both diatoms displayed a shared response to nitrate limitation (all expressed genes were up- or down- regulated in a given cluster). Nearly half of the up-regulated genes (8 genes out of 20) with an annotation were involved in nucleotide and amino acid metabolism (e.g., polyamine synthesis, lysine metabolism). In the down-regulated fraction, 13 out of 19 genes with KEGG annotations were associated with carbohydrate and lipid metabolism. This subset included genes connected to glycolysis, gluconeogenesis, the Calvin Cycle, and isoprenoid biosynthesis.
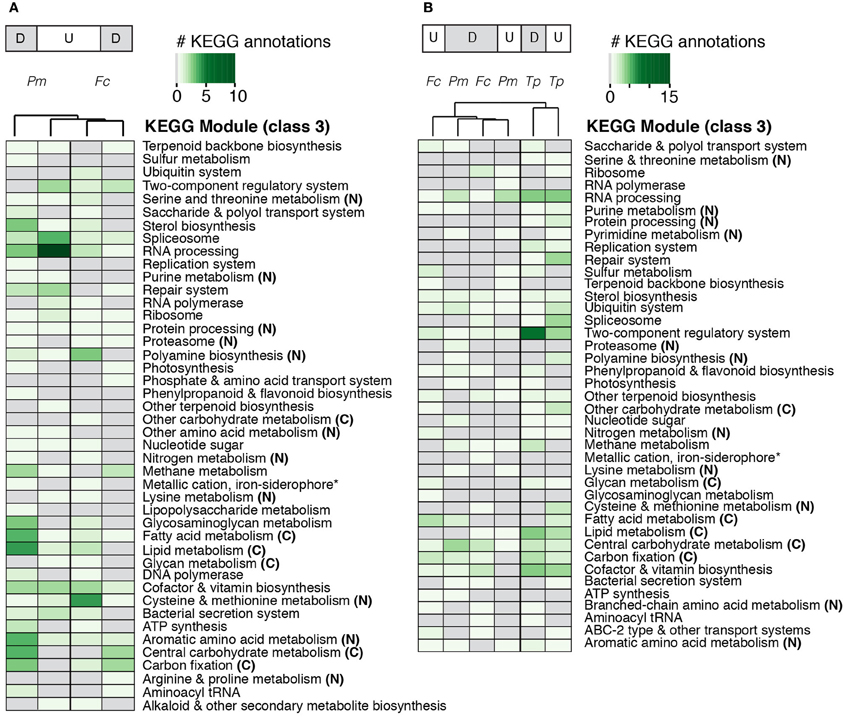
Figure 5. Heatmaps of KEGG annotations for differentially expressed (A) pennate genes and (B) non-orthologous genes based on presence, absence, and abundance of annotations. The darker green colors indicate a greater number of differentially expressed genes with KEGG annotations; gray boxes, no differentially expressed genes had KEGG annotations. KEGG modules associated with carbon (C) and nitrogen (N) metabolism are labeled accordingly. Treatments and KEGG modules are clustered hierarchically based on similarities and differences in KEGG annotation abundances. Abbreviations: Tp, T. pseudonana; Fc, F. cylindrus; Pm, P. multiseries; up-regulated (U); down-regulated (D). Asterisks, abbreviation for Metallic cation, iron-siderophore and Vitamin B12 transporter.
Forty-six percent (1981 genes; T. pseudonana), 40% (1178 genes; F. cylindrus), and 32% (1542 genes; P. multiseries) of the significantly differentially expressed genes were non-orthologous (Table 3). Tens of these genes in each diatom were transcribed only under nitrate limitation, with no detected transcription under nutrient-replete conditions. Few of the non-orthologous genes (8%, T. pseudonana; 5%, F. cylindrus; 4%, P. multiseries) had an assigned KEGG module annotation. When the presence and abundance of KEGG annotations of differentially expressed non-orthologous genes were compared across the normalized diatom transcriptomes, the pennate diatoms (F. cylindrus and P. multiseries) clustered separately from T. pseudonana (Figure 5B). In T. pseudonana, up-regulated genes were involved in RNA processing, two-component regulatory systems, spliceosome, repair system, and cofactor and vitamin biosynthesis. The greatest number of nitrate-limited annotated genes in F. cylindrus were involved in carbon fixation and fatty acid metabolism. Pseudo-nitzschia multiseries had the fewest KEGG module annotations in the up- and down-regulated subsets; the most annotated genes in the up-regulated subset encoded proteins involved in RNA processing. Among all down-regulated genes, a higher abundance of genes involved in carbon metabolism (glycolysis, lipid metabolism, and the pentose phosphate pathway) were observed. None of the most differentially expressed genes had KEGG annotations.
Overall, transcription patterns differed between the centric and the pennate non-orthologous gene subsets: T. pseudonana differentially transcribed a greater number of genes involved in lipid metabolism, two-component regulatory systems, and general cellular repair and information processing (e.g., RNA processing, spliceosome), whereas the pennate diatoms differentially transcribed a greater number of genes involved in central carbohydrate metabolism and fatty acid metabolism.
We examined in more detail the transcription patterns of genes known to be required for carbon metabolism, focusing on those genes involved in glycolysis (glucose to pyruvate) and gluconeogenesis (pyruvate to glucose) (Figure 6). Of the 13 genes examined, seven are shared between glycolysis/gluconeogenesis; all of these genes had at least two orthologs in each diatom except the gene that encodes for enolase and in several instances, expression patterns for a given diatom differed between gene copies. For glucose 6-phosphate isomerase, two T. pseudonana copies (of four) were down-regulated and one P. multiseries copy (of four) was up-regulated. No significant differential expression was observed in F. cylindrus. There are two isoforms for phosphofructokinase, an ATP-dependent isoform and a pyrophosphate-dependent isoform (Smith et al., 2012). Transcription of the two T. pseudonana gene copies differed from another, whereas F. cylindrus and P. multiseries each up-regulated one of two gene copies. In the next two shared steps that involve fructose bisphosphate aldose and glyceraldehyde phosphate dehydrogenase, all three diatoms down-regulated expression in at least two gene copies per diatom. In addition, F. cylindrus up-regulated expression for at least one copy for both genes, and P. multiseries up-regulated expression for one copy for glyceraldehyde phosphate dehydrogenase. For one of the fructose bisphosphate adolase clusters (cluster 4, Figure 6) and two of the clusters for glyceraldehyde phosphate dehydrogenase (clusters 3 and 5, Figure 6), expression was uniform across gene orthologs. For phosphoglycerate kinase, three copies (of four) for T. pseudonana and one copy (of two) for P. multiseries were down-regulated; F. cylindrus had an up-regulated copy and a down-regulated copy. Contrasting expression patterns were observed across gene copies for T. pseudonana (one up-regulated, one down-regulated) and P. multiseries (two up-regulated and one down-regulated) for phosphoglycerate mutase. In F. cylindrus one gene copy was down-regulated. No significant differences in expression were detected for enolase.
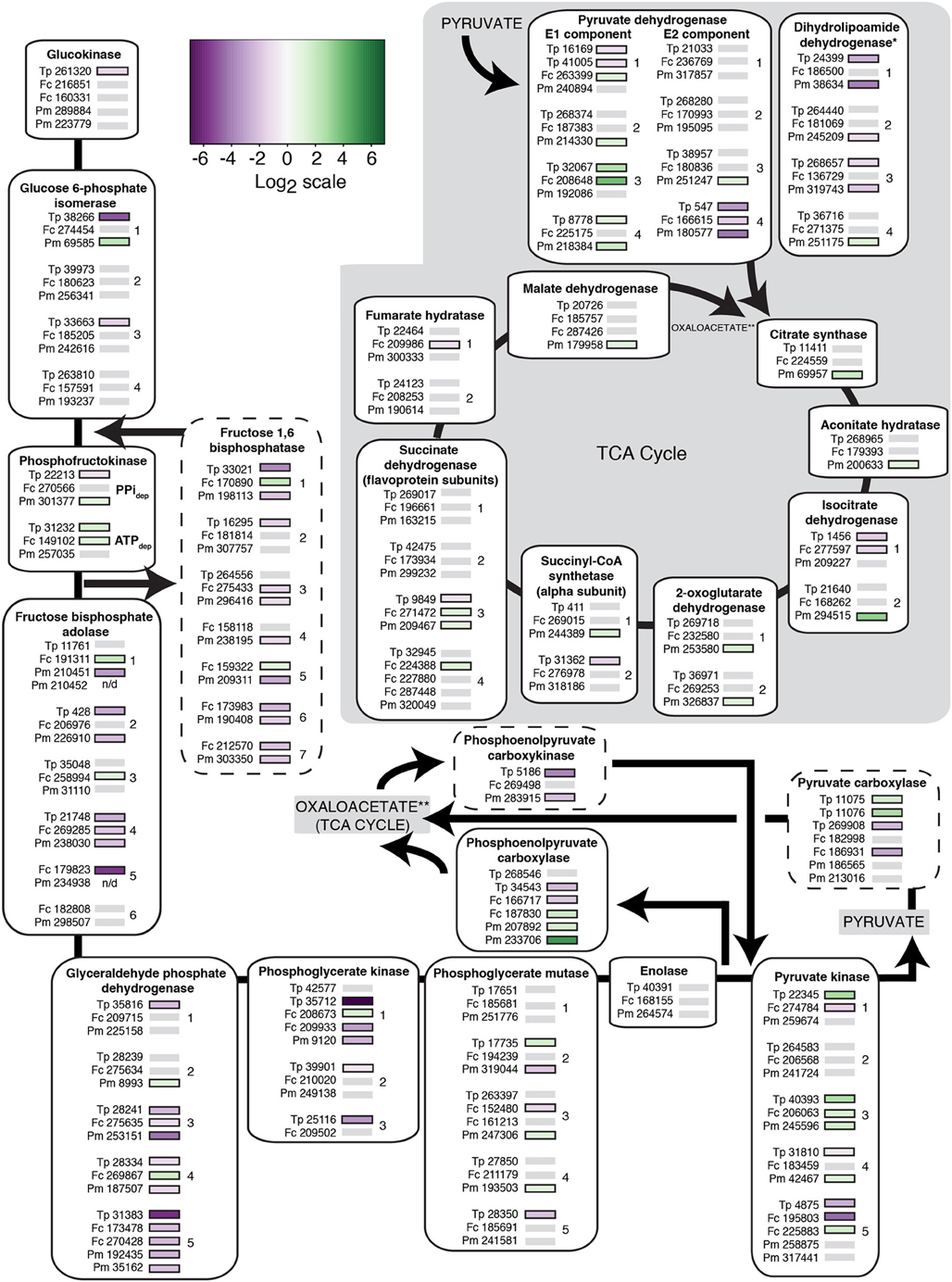
Figure 6. Heat map of the transcriptional response to nitrate limitation for known carbon-related genes in each diatom connected to glycolysis/gluconeogensis and the TCA cycle. The protein ID of genes encoded by each species (Tp, T. pseudonana; Fc, F. cylindrus; Pm, P. multiseries) is next to a color-coded bar of significant differential expression relative to a control treatment (p < 0.05). Gray bars indicate genes without significant differential expression; “n/d” indicates that gene transcription was not detected. Genes are grouped according to their respective isoforms and according to their OrthoMCL clusters (indicated with a number, when applicable). Only genes that grouped with at least one other diatom ortholog are shown; single copy genes are not shown. Predicted localizations of gene products may differ among orthologs, but are not shown. PPidep, pyrophosphate dependent; ATPdep, ATP dependent. Single asterisks, dihydrolipoamide dehydrogenase catalyzes two steps in the TCA cycle; double asterisks, TCA cycle location where reaction products are channeled to and from. The dashed lines highlight steps in gluconeogenesis. Gray shading, TCA cycle. Genes were chosen based on gene annotations from Smith et al. (2012) and Hockin et al. (2012).
Three genes specific to gluconeogenesis also exhibited a variety of expression patterns. In the first steps of this pathway, pyruvate is converted to phosphoenolpyruvate via pyruvate carboxylase and phosphoenolpyruvate carboxykinase (Figure 6). Two (of three) T. pseudonana pyruvate carboxylase gene copies were up-regulated and the third gene copy was down-regulated in response to nitrate-limited growth. One F. cylindrus copy was down-regulated and no significant differential expression was present for the two P. multiseries copies. Each diatom had one gene representative for phosphoenolpyruvate carboxykinase. Thalassiosira pseudonana and P. multiseries down-regulated their copy, whereas the F. cylindrus gene copy exhibited no significant differential expression. The gene that encodes for fructose 1,6 bisphosphatase, one of the final steps of gluconeogenesis, was differentially expressed in each diatom (Smith et al., 2012). The majority of genes with differential expression were down-regulated, including all of the differentially expressed T. pseudonana and P. multiseries genes. Fragilariopsis cylindrus up-regulated two of its gene copies. To date, the gene that encodes for the final step of gluconeogenesis, glucose 6-phosphatase, has not been identified in a diatom genome (Smith et al., 2012).
Glucokinase, phosphoenolpyruvate carboxylase, and pyruvate kinase are specific to glycolysis. For the first step of glycolysis, the T. pseudonana gene copy encoding glucokinase was down-regulated and no significant differential expression was observed for the two F. cylindrus and two P. multiseries copies under nitrate limitation (p < 0.05). Phosphoenolpyruvate carboxylase, which catalyzes the formation of oxaloacetate, exhibited a variety of differential expression patterns. One (of two) T. pseudonana gene copies was down-regulated, both P. multiseries gene copies were up-regulated, and F. cylindrus had one up-regulated and one down-regulated gene copy. The most up-regulated gene copies encode the final step of glycolysis- the production of pyruvate: two copies (of five) for T. pseudonana, two copies (of six) for F. cylindrus, and two copies (of six) for P. multiseries were all up-regulated. Up-regulated expression patterns were uniform across the three diatoms in one orthologous cluster (cluster 3, Figure 6).
After pyruvate is produced as part of glycolysis, it is channeled into the TCA cycle where it is oxidized as part of ATP production. A majority of genes related to the TCA cycle exhibited no significant differential expression. Genes encoding pyruvate dehydrogenase and dihydrolipoamide dehydrogenase, required for the conversion of pyruvate to acetyl-CoA, displayed mixed expression patterns (Figure 6). For pyruvate dehydrogenase, each diatom had at least one up-regulated and one down-regulated gene copy. Thalassiosira pseudonana down-regulated two (of four) gene copies and P. multiseries down-regulated 3 (of four) gene copies for dihydrolipoamide dehydrogenase. Pseudo-nitzschia multiseries also up-regulated one gene copy for this enzyme and F. cylindrus exhibited no significant differential expression. Dihydrolipoamide dehydrogenase is also involved in the conversion of 2-oxoglutarate to succinyl-CoA. Pseudo-nitzschia multiseries was distinctive in that it up-regulated one copy for each gene in the TCA cycle pathway with the exception of the gene that encodes fumarate hydratase. In contrast, T. pseudonana down-regulated one copy (of two) for isocitrate dehydrogenase and succinyl-CoA synthetase and one copy (of four) for succinate dehydrogenase. Fragilariopsis cylindrus down-regulated one copy (of two) for isocitrate dehydrogenase and fumarate hydratase, and up-regulated two copies (of six) for succinate dehydrogenase.
Genes connected to nitrogen uptake and assimilation were also examined. Each diatom has multiple copies of each type of transporter gene, except for the gene encoding the nitrite transporter (Figure 7). Under nitrate limitation, T. pseudonana down-regulated transcription of three (of six) ammonium transporter genes, whereas F. cylindrus and P. multiseries up-regulated transcription of three (of five) and two (of four) genes, respectively (p < 0.05; Figure 7). Nitrate-limited cultures of T. pseudonana and F. cylindrus up-regulated one (of three) and two (of four) urea transporter genes, respectively; these genes were not differentially expressed by nitrate-limited cultures of P. multiseries. The nitrate transporter genes were all up-regulated in response to nitrate limitation, except for one down-regulated copy in P. multiseries. The nitrate reductase gene in T. pseudonana (one copy) and P. multiseries (one copy) were both up-regulated, whereas the two F. cylindrus copies did not exhibit significant differential expression. Only the F. cylindrus nitrite transporter gene was down-regulated in response to nitrate limitation. Both types of nitrite reductase genes encoded by T. pseudonana and P. multiseries were up-regulated in response to nitrate limitation and one (of three) copies encoded by F. cylindrus (Figure 7).
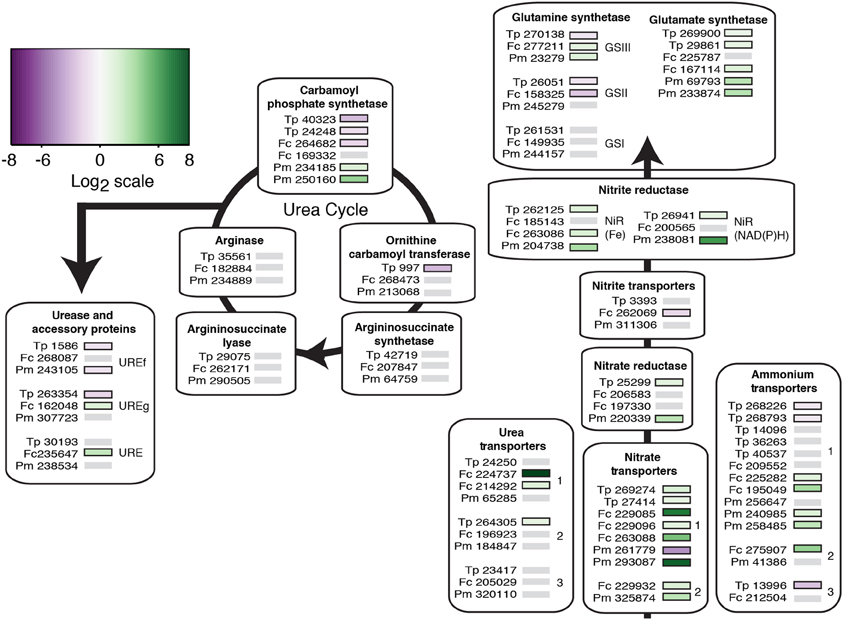
Figure 7. Heat map of the transcriptional response to nitrate limitation for known nitrogen—related genes in each diatom connected to uptake, assimilation, and the urea cycle. The protein ID of genes encoded by each species (Tp, T. pseudonana; Fc, F. cylindrus; Pm, P. multiseries) is next to a color-coded bar of significant differential expression relative to a control treatment (p < 0.05). Gray bars indicate genes without significant differential expression. Genes are grouped according to their respective isoforms and according to their OrthoMCL clusters (indicated with a number, when applicable). Only genes that grouped with at least one other diatom ortholog are shown; single copy genes are not shown. Predicted localizations of gene products may differ among orthologs, but are not shown. Nitrogen cycle genes were chosen based on gene annotations from Bender et al. (2012).
Once in a reduced form, nitrogen is assimilated into the cell through the GS-GOGAT (glutamine synthetase and glutamate synthetase) cycle. Each diatom has two gene copies for glutamate synthetase that form a single cluster; at least one gene copy per diatom was up-regulated. The glutamine synthetase genes grouped into GSI, GSII, and GSIII orthologous gene clusters. None of genes within the GSI cluster were differentially transcribed in response to nitrate limitation. The GSII genes were down-regulated by nitrate-limited T. pseudonana and F. cylindrus cells, and were not differentially transcribed by nitrate-limited P. multiseries cells. The GSIII gene encoded by T. pseudonana was down-regulated and the GSIII genes encoded by F. cylindrus and P. multiseries were up-regulated.
We also explored the possible role of the urea cycle and urea degradation in the diatom in response to nitrate limitation. A single cluster of carbamoyl phosphate synthetase (CPS) genes included the isoform involved in pyrimidine synthesis (pgCPSII) and the isoform required for the urea cycle (unCPS). Both CPS isoforms were down-regulated by nitrate-limited T. pseudonana, and F. cylindrus down-regulated the pgCPSII isoform (protein ID 264682). Nitrate-limited P. multiseries up-regulated both CPS isoforms. In subsequent urea cycle steps, the only gene with differential expression (down-regulation) was the ornithine carbamoyltransferase gene in T. pseudonana. Mixed expression patterns were observed for urease (URE) and its accessory genes (UREf and UREg).
More than 80% of the transcribed pennate-clustered genes were detected in at least one queried (bipolar/multipolar) centric diatom other than T. pseudonana (Supplementary Table S1) suggesting these genes were present in the bipolar/multipolar ancestor and were lost during the emergence of T. pseudonana. The remaining subset of pennate-specific genes that exhibited significant differential expression had minimal annotation information. Of the 258 gene subset, six genes had KEGG annotations that included a P. multiseries amine oxidase and a photosystem I accessory protein found in both F. cylindrus and P. multiseries. All of the F. cylindrus and P. multiseries non-orthologous transcribed genes displayed sequence similarities (e-value 1 × 10−5) to transcripts from at least one other queried centric diatom. About 1600 of the transcribed T. pseudonana non-orthologous genes were not detected in the eight queried transcriptome datasets of pennate diatoms (Supplementary Table S1). From this subset of 1600 genes, 185 non-orthologous genes were detected in T. pseudonana only (Supplementary Table S1). One non-orthologous gene identified in T. pseudonana (Tp1457) and encoding a hypothetical protein, was present in all of the queried centric diatoms but in none of the pennate diatoms, suggesting that this gene is of centric diatom origin, and was lost during the emergence of pennate lineages.
We compared transcriptional profiles of three phylogenetically distant diatoms—T. pseudonana, F. cylindrus, and P. multiseries—at the onset of nitrate limitation, a condition commonly associated with the end of diatom blooms when further cell division is limited by the depletion of nitrate. We confirmed that the experimental conditions induced nitrate limitation for each of the diatoms through the addition of nitrate at the onset of stationary phase and the measured resumption of cell division. Despite evolved differences in optimal light, temperature, and nutrient uptake rates required for growth, similarities in transcriptional patterns of a subset of the annotated core set of orthologous genes shared by the three diatoms indicate that the diatoms share pathway-level responses to changes in nitrogen availability. We observed an overall increase in potential nitrogen recycling pathways under nitrate limitation, and a general decrease in cellular metabolism, including a down-regulation of photosynthesis and carbon-related pathways, reiterating the known connection between carbon and nitrogen metabolism in the diatoms (Turpin, 1991; Hockin et al., 2012). A number of up-regulated genes were also connected to central carbohydrate metabolism, supporting the hypothesis that carbon skeletons supplied as part of the TCA cycle are used to re-assimilate nitrogen generated by protein degradation; this metabolic response allows the cell to shift cellular efforts toward the generation of essential nitrogen compounds to combat cellular stress (Harrison et al., 1990; LaRoche et al., 1993; Claquin et al., 2002; Tolonen et al., 2006; Hockin et al., 2012). This metabolic shift is most evident with the number of up-regulated genes connected to the final synthesis of pyruvate and the conversion of pyruvate to acetyl-CoA. These shared transcriptional similarities may reflect a diatom-wide response to changes in nitrogen availability.
The shared pennate-specific response involved a majority of transcripts with minimal functional annotations. The minority with annotations included up-regulated transcripts connected to nucleotide and amino acid metabolism and down-regulated genes connected to carbon metabolism. The largest difference in expression of shared pennate genes between F. cylindrus and P. multiseries was observed in the down-regulated subset. Greater transcriptional and functional similarities between the two pennate diatoms compared to the bipolar centric diatom reflect key differences in how each group has evolved to respond to its environment. Centric and pennate diatoms maintain fundamentally different physiologies from one another in how, for example, each group undergoes sexual reproduction (Armbrust, 2009) and responds to micronutrient availability and nutrient storage in open ocean environments (Strzepek and Harrison, 2004; Peers and Price, 2006; Sims et al., 2006).
Large variations in the underlying transcriptional patterns were detected across the three diatoms with less than 5% of the shared orthologous gene clusters similarly transcribed under nitrate starvation. The observed transcriptional diversity appears to result from four general mechanisms. First, a small percentage of proteins within each diatom belong to multi-copy gene families that commonly displayed different transcriptional patterns both within individual diatoms, as well as among the different diatoms. The extent of the genetic flexibility created by gene families may affect where and when nutrients are moved through the cell, ultimately influencing the flow of carbon and nitrogen (Lommer et al., 2012; Smith et al., 2012). For example, each diatom encodes multiple ammonium transporters, at least twice as many as the urea and nitrate transporters (Allen, 2005). Interestingly, under nitrate limitation, the three significantly expressed ammonium transporter genes in T. pseudonana were down-regulated, whereas the three significantly expressed by F. cylindrus and the two significantly expressed by P. multiseries were all up-regulated. Up-regulation of ammonium transporter gene expression has also been observed with two additional pennate diatoms, Cylindrotheca fusiformis and Phaeodactylum tricornutum (Hildebrand, 2005; Maheswari et al., 2010). Combined, these results suggest that pennate diatoms try to access ammonium when limited for nitrate, regardless of whether ammonia is actually available. A second example is fructose 1,6 bisphosphatase (FBP), which is linked to gluconeogenesis and the Calvin cycle and is also encoded by a multi-copy gene family (Smith et al., 2012). Fragilariopsis cylindrus and P. multiseries exhibited shared expression patterns distinct from T. pseudonana in two of three core orthologous clusters. Additionally, transcription of the F. cylindrus and P. multiseries pennate-clustered FBP orthologs was down-regulated during nitrate limitation in two (of four) orthologous pennate clusters. These additional orthologs have been hypothesized to confer a cellular advantage by providing the pennate diatoms with more flexibility (via more genes) in how they can react to environmental triggers (Martens et al., 2008; Smith et al., 2012). A similar examination of the more diverged araphid pennate diatoms would indicate whether these shared transcriptional patterns are a more generalized feature of pennate diatoms.
A second source of variation comes from the observation that about 40% of the genetic repertoire in each diatom is missing from the other two diatoms. About 32–46% of these genes are differentially expressed under nitrate limitation and most encode proteins of unknown function. Several genes were highly expressed under nitrate limitation, but not detected during the nutrient-replete conditions and vice versa, several genes highly expressed during nutrient-replete conditions were not detected during nitrate limitation. Genes specific to a given diatom lineage appear to play a critical role in how the cells respond to nitrogen availability. As an example, species within the Pseudo-nitzschia genus produce the nitrogen-containing toxin domoic acid, but the genetic basis of its synthesis is unknown (Auro and Cochlan, 2013). Additionally, diatom cells contain a vacuole for storing nitrate that can be used during periods of low environmental nitrogen, but the genes connected to the formation and maintenance of this vacuole have not been identified within the group (Raven, 1987; Armbrust et al., 2004; Katz et al., 2004; Allen et al., 2006). Distantly related genes may also encode diverged proteins that nonetheless carry out similar metabolic functions in the diatoms. This hypothesis was first proposed by Thompson et al. (2011), who conducted comparative transcriptomics on two closely-related ecotypes of Prochloroccocus and observed similar functional responses, but different transcriptional responses. Given the human health concerns associated with domoic acid and the ecological importance of the nitrate storage vacuole, targeted studies of nitrogen-responsive genes with unknown function could lead to discoveries about diatom nitrogen metabolism of both social and ecological importance.
A third source of transcriptional variability among the three diatoms involves the differential regulation of separate components of the same metabolic pathway as evidenced by the over-representation of carbon-related pathways in both the up- and down- regulated core, pennate, and non-orthologous subsets. This phenomenon has been shown previously for TCA cycle and urea cycle genes in T. pseudonana (Hockin et al., 2012). Transcriptional and protein abundance for the gene that encodes carbamoyl phosphate synthetase (first reaction of the urea cycle) was lower under nitrate limitation, yet protein concentrations for subsequent urea cycle enzymes were elevated under nitrate limitation (Hockin et al., 2012). A similar decoupling among urea cycle genes was observed in P. tricornutum (Allen et al., 2011). We also observed this decoupling for TCA cycle genes; more differentially expressed genes were detected at the beginning of the cycle rather than in subsequent steps. Differential regulation within a metabolic pathway may be the result of post-transcriptional and post-translational modification for select genes or proteins, respectively, or it may be attributed to a lag in how nitrogen and carbon are partitioned within the cell, with the first enzymatic reaction of a pathway responding to changes in nutrient availability earlier than downstream reactions (Poulsen et al., 2006; Hockin et al., 2012).
Last, the observed transcriptional diversity may reflect variations in the regulatory networks that differentially control gene expression among the different diatoms, particularly between the centric and the two pennate diatoms. Gene methylation/demethylation could provide one mechanism by which centric and pennate diatoms coordinate their transcriptional responses. Veluchamy et al. (2013) found that DNA methylation affected the expression of over 300 genes in the pennate diatom P. tricornutum and suppressed the differential expression of two key nitrogen metabolism genes (i.e., carbamoyl phosphate synthetase and nitrite reductase) in cultures grown under nitrate-replete growth. These findings suggest that hypomethylation may play an important role in regulating the expression of sets of genes (e.g., transporters) under a given set of environmental conditions (Veluchamy et al., 2013). Additionally, Ashworth et al. (2013) identified transcription factors that putatively control different regulatory networks in T. pseudonana, resulting in co-expression of thousands of genes in response to different environmental conditions including the day/night cycle and the transition from nutrient-replete growth to nitrate limitation. Koester et al. (2012) found evidence that transcription factors within T. pseudonana are under positive selection, implying that mutations within the genes encoding transcription factors convey adaptive phenotypes. The different transcriptional profiles of orthologous single copy genes observed in the different species may reflect, in part, genetic differences between transcription factors and thus which genes are incorporated into a given network. A nitrogen network proposed in Prochlorococcus involves the transcriptional regulator NtcA, which has been shown to regulate expression not only of key nitrogen genes, but also genes related to photosynthesis (Su et al., 2005; Tolonen et al., 2006).
In this study, the three model diatoms represent phylogenetically and physiologically distinct organisms. Given these inherent differences, our findings provide a conservative survey of shared genes that are differentially expressed in response to changes in nitrate availability and they highlight the genetic and transcriptional diversity within the diatom group. Additional transcriptional studies that investigate the similarities and differences of diatoms to long-term nitrate limitation, as well as to other nutrient stressors, will help to determine whether or not the marked diversity in gene-specific responses translates directly to significantly different ecological consequences in the field.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. All research was funded through the Gordon and Betty Moore Foundation Marine Microbiology Investigator Award to E. Virginia Armbrust.
The authors acknowledge Franziska Lutz and Tiffany Truong for assistance with laboratory culturing, Betsy Welsh for assistance with analyses conducted in R, and Jody W. Deming, Anitra Ingalls, and Micaela S. Parker for input on the manuscript. The authors also thank the two reviewers for their insightful comments and suggestions. This research was funded by a Gordon and Betty Moore Foundation Marine Microbiology Investigator Award to E. Virginia Armbrust. MMETSP Samples in Supplementary Table S1 were sequenced at the National Center for Genome Resources through Grant #2637 from the Gordon and Betty Moore Foundation.
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmars.2014.00003/abstract
Supplementary Table S1. List of queried Joint Genome Institute (JGI) or Marine Microbial Eukaryotic Transcriptome Sequencing Project (MMETSP) diatoms for BLASTX search.
Supplementary Table S2. Significant differential expression information (p < 0.01) for all T. pseudonana (Tp), F. cylindrus (Fc), and P. multiseries (Pm) genes under nitrate limitation (relative to the nutrient-replete control) that were present in the core, pennate, or non-orthologous groups. Expression ratios represent the expression for three biological replicates for the nitrate-limited treatment normalized to the expression for three biological replicates for the nutrient-replete control. Species shared (column 8) values indicate how many species shared an ortholog; cluster IDs (column 9) are based on OrthoMCL analysis (NA = single copy gene); KEGG module annotation information is given in columns 10–17 (NA = no annotation information). Column 16 lists merged KEGG module class 2 annotations for instances where there were multiple class 2 module annotations for shared orthologs (see Materials and Methods). A gene may occupy multiple lines when it is associated with multiple KEGG module annotations. Expression information, including the adjusted p-value is given for three pooled replicates per treatment (see Materials and Methods).
Allen, A. E. (2005). Beyond sequence homology: redundant ammonium transporters in a marine diatom are not functionally equivalent. J. Phycol. 41, 4–6. doi: 10.1111/j.0022-3646.2005.41101_2.x
Allen, A. E., Dupont, C. L., Obornik, M., Horak, A., Nunes-Nesi, A., McCrow, J. P., et al. (2011). Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature 473, 203–209. doi: 10.1038/nature10074
Allen, A. E., Vardi, A., and Bowler, C. (2006). An ecological and evolutionary context for integrated nitrogen metabolism and related signaling pathways in marine diatoms. Curr. Opin. Plant Biol. 9, 264–273. doi: 10.1016/j.pbi.2006.03.013
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Armbrust, E. V. (2009). The life of diatoms in the world's oceans. Nature 459, 185–192. doi: 10.1038/nature08057
Armbrust, E. V., Berges, J. A. J., Bowler, C. C., Green, B. R. B., Martinez, D. D., Putnam, N. H., et al. (2004). The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86. doi: 10.1126/science.1101156
Arrigo, K. R., Mills, M. M., Kropuenske, L. R., van Dijken, G. L., Alderkamp, A.-C., and Robinson, D. H. (2010). Photophysiology in two major southern ocean phytoplankton taxa: photosynthesis and growth of Phaeocystis antarctica and Fragilariopsis cylindrus under different irradiance levels. Integr. Comp. Biol. 50, 950–966. doi: 10.1093/icb/icq021
Ashworth, J., Coesel, S., Lee, A., Armbrust, E. V., Orellana, M. V., and Baliga, N. S. (2013). Genome-wide diel growth state transitions in the diatom Thalassiosira pseudonana. Proc. Natl. Acad. Sci. U.S.A. 110, 7518–7523. doi: 10.1073/pnas.1300962110
Auro, M. E., and Cochlan, W. P. (2013). Nitrogen utilization and toxin production by two diatoms of the Pseudo-nitzschia pseudodelicatissima complex: P. cuspidata and P. fryxelliana. J. Phycol. 49, 156–169. doi: 10.1111/jpy.12033
Behrenfeld, M. J., Halsey, K. H., and Milligan, A. J. (2008). Evolved physiological responses of phytoplankton to their integrated growth environment. Philos. Trans. R. Soc. B 363, 2687–2703. doi: 10.1098/rstb.2008.0019
Bender, S. J., Parker, M. S., and Armbrust, E. V. (2012). Coupled effects of light and nitrogen source on the urea cycle and nitrogen metabolism over a diel cycle in the marine diatom Thalassiosira pseudonana. Protist 163, 232–251. doi: 10.1016/j.protis.2011.07.008
Berges, J. A., Franklin, D. J., and Harrison, P. J. (2001). Evolution of an artificial seawater medium: improvements in enriched seawater, artificial water over the last two decades. J. Phycol. 37, 1138–1145. doi: 10.1046/j.1529-8817.2001.01052.x
Bowler, C. C., Allen, A. E. A., Badger, J. H. J., Grimwood, J. J., Jabbari, K. K., Kuo, A. A., et al. (2008). The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239–244. doi: 10.1038/nature07410
Brand, L. E., Guillard, R. R. L., and Murphy, L. S. (1981). A method for the rapid and precise determination of acclimated phytoplankton reproduction rates. J. Plankton Res. 3, 193–201. doi: 10.1093/plankt/3.2.193
Claquin, P., Martin-Jezequel, V., Kromkamp, J., Veldhuis, M., and Kraay, G. (2002). Uncoupling of silicon compared with carbon and nitrogen metabolisms and the role of the cell cycle in continuous cultures of Thalassiosira pseudonana (Bacillariophyceae) under light, nitrogen, and phosphorus control. J. Phycol. 38, 922–930. doi: 10.1046/j.1529-8817.2002.t01-1-01220.x
Dugdale, R., and Wilkerson, F. (1998). Silicate regulation of new production in the equatorial Pacific upwelling. Nature 391, 270–273. doi: 10.1038/34630
Falkowski, P. G., Katz, M. E., Knoll, A. H., Quigg, A., Raven, J. A., Schofield, O. M., et al. (2004). The evolution of modern eukaryotic phytoplankton. Science 305, 354–360. doi: 10.1126/science.1095964
Falkowski, P. G., and Stone, D. (1975). Nitrate uptake in marine phytoplankton- Energy sources and interaction with carbon fixation. Mar. Biol. 32, 77–84. doi: 10.1007/BF00395161
Guillard, R. R., and Ryther, J. H. (1962). Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt, and Detonula confervacea (cleve) Gran. Can. J. Microbiol. 8, 229–239. doi: 10.1139/m62-029
Harrison, P. J., Thompson, P., and Calderwood, G. A. (1990). Effects of nutrient and light limitation on the biochemical composition of phytoplankton. J. Appl. Phycol. 2, 45–56. doi: 10.1007/BF02179768
Hildebrand, M. (2005). Cloning and functional characterization of ammonium transporters from the marine diatom Cylindrotheca fusiformis (Bacillariophyceae). J. Phycol. 41, 105–113. doi: 10.1111/j.1529-8817.2005.04108.x
Hockin, N. L., Mock, T., Mulholland, F., Kopriva, S., and Malin, G. (2012). The response of diatom central carbon metabolism to nitrogen starvation is different from that of green algae and higher plants. Plant Physiol. 158, 299–312. doi: 10.1104/pp.111.184333
Iverson, V., Morris, R. M., Frazar, C. D., Berthiaume, C. T., Morales, R. L., and Armbrust, E. V. (2012). Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335, 587–590. doi: 10.1126/science.1212665
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi: 10.1093/nar/gkr988
Katz, M. E., Finkel, Z. V., Grzebyk, D., Knoll, A. H., and Falkowski, P. G. (2004). Evolutionary trajectories and biogeochemical impacts of marine eukaryotic phytoplankton. Annu. Rev. Ecol. Syst. 35, 523–556. doi: 10.1146/annurev.ecolsys.35.112202.130137
Koester, J. A., Swanson, W. J., and Armbrust, E. V. (2012). Positive selection within a diatom species acts on putative protein interactions and transcriptional regulation. Mol. Biol. Evol. 30, 422–434. doi: 10.1093/molbev/mss242
Kolber, Z. S., Zehr, J. P., and Falkowski, P. G. (1988). Effects of growth, irradiance and nitrogen limitation on photosynthetic energy conversion in photosystem II. Plant Physiol. 88, 923–929. doi: 10.1104/pp.88.3.923
Kooistra, W., Gersonde, R., Medlin, L. K., and Mann, D. G. (2007). “The origin and evolution of the diatoms: their adaptation to a planktonic existence,” in Evolution of Primary Producers in the Sea, eds P. G. Falkowski and A. H. Knoll (Amsterdam: Elsevier Academic Press), 210–225.
Kroth, P. G., Chiovitti, A., Gruber, A., Martin-Jezequel, V., Mock, T., Parker, M. S., et al. (2008). A model for carbohydrate metabolism in the diatom Phaeodactylum tricornutum deduced from comparative whole genome analysis. PLoS ONE 3:e1426. doi: 10.1371/journal.pone.0001426
LaRoche, J., Geider, R. J., Graziano, L. M., Murray, H., and Lewis, K. (1993). Induction of specific proteins in eukaryotic algae grown under iron-deficient, phosphorus-deficient, or nitrogen-deficient conditions. J. Phycol. 29, 767–777. doi: 10.1111/j.0022-3646.1993.00767.x
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoockert, C., and Roos, D. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lommer, M., Specht, M., Roy, A.-S., Kraemer, L., Andreson, R., Gutowska, M. A., et al. (2012). Genome and low-iron response of an oceanic diatom adapted to chronic iron limitation. Genome Biol. 13:R66. doi: 10.1186/gb-2012-13-7-r66
Longhurst, A. R., and Harrison, W. G. (1989). The biological pump - profiles of plankton production and consumption in the upper ocean. Prog. Oceanogr. 22, 47–123. doi: 10.1016/0079-6611(89)90010-4
Maheswari, U., Jabbari, K., Petit, J.-L., Porcel, B. M., Allen, A. E., Cadoret, J.-P., et al. (2010). Digital expression profiling of novel diatom transcripts provides insight into their biological functions. Genome Biol. 11:R85. doi: 10.1186/gb-2010-11-8-r85
Martens, C., Vandepoele, K., and Van de Peer, Y. Y. (2008). Whole-genome analysis reveals molecular innovations and evolutionary transitions in chromalveolate species. Proc. Natl. Acad. Sci. U.S.A. 105, 3427–3432. doi: 10.1073/pnas.0712248105
Martin, P., Lampitt, R. S., Jane Perry, M., Sanders, R., Lee, C., and D'Asaro, E. (2011). Export and mesopelagic particle flux during a North Atlantic spring diatom bloom. Deep-Sea Res. Pt. I 58, 338–349. doi: 10.1016/j.dsr.2011.01.006
Mock, T., Samanta, M. P., Iverson, V., Berthiaume, C., Robison, M. M., Holtermann, K., et al. (2008). Whole-genome expression profiling of the marine diatom Thalassiosira pseudonana identifies genes involved in silicon bioprocesses. Proc. Natl. Acad. Sci. U.S.A. 105, 1579–1584. doi: 10.1073/pnas.0707946105
Montsant, A., Jabbari, K., Maheswari, U., and Bowler, C. (2005). Comparative genomics of the pennate diatom Phaeodactylum tricornutum. Plant Physiol. 137, 500–513. doi: 10.1104/pp.104.052829
Palmucci, M., Ratti, S., and Giordano, M. (2011). Ecological and evolutionary implications of carbon allocation in marine phytoplankton as a function of nitrogen availability: a fourier transform infrared spectroscopy approach. J. Phycol. 47, 313–323. doi: 10.1111/j.1529-8817.2011.00963.x
Pan, Y., Rao, D., Mann, K. H., Brown, R. G., and Pocklington, R. (1996). Effects of silicate limitation on production of domoic acid, a neurotoxin, by the diatom Pseudo-nitzschia multiseries. I. Batch culture studies. Mar. Ecol. Prog. Ser. 131, 225–233. doi: 10.3354/meps131225
Peers, G., and Price, N. M. (2006). Copper-containing plastocyanin used for electron transport by an oceanic diatom. Nature 441, 341–344. doi: 10.1038/nature04630
Poulsen, N. N., Chesley, P. M., and Kroger, N. (2006). Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J. Phycol. 42, 1059–1065. doi: 10.1111/j.1529-8817.2006.00269.x
Raven, J. A. (1987). The role of vacuoles. New Phytol. 106, 357–422. doi: 10.1111/j.1469-8137.1987.tb00149.x
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., and Smyth, G. K. (2007). Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23, 2881–2887. doi: 10.1093/bioinformatics/btm453
Sims, P. A., Mann, D. G., and Medlin, L. K. (2006). Evolution of the diatoms: insights from fossil, biological and molecular data. Phycologia 45, 361–402. doi: 10.2216/05-22.1
Smith, S. R., Abbriano, R. M., and Hildebrand, M. (2012). Comparative analysis of diatom genomes reveals substantial differences in the organization of carbon partitioning pathways. Algal Res. 1, 2–16. doi: 10.1016/j.algal.2012.04.003
Sommer, U. (1986). Nitrate- and silicate-competition among antarctic phytoplankton. Mar. Biol. 91, 345–351. doi: 10.1007/BF00428628
Strzepek, R. F., and Harrison, P. J. (2004). Photosynthetic architecture differs in coastal and oceanic diatoms. Nature 431, 689–692. doi: 10.1038/nature02954
Su, Z., Olman, V., Mao, F., and Xu, Y. (2005). Comparative genomics analysis of NtcA regulons in cyanobacteria: regulation of nitrogen assimilation and its coupling to photosynthesis. Nucleic Acids Res 33, 5156–5171. doi: 10.1093/nar/gki817
Thompson, A. W., Huang, K., Saito, M. A., and Chisholm, S. W. (2011). Transcriptome response of high- and low-light-adapted Prochlorococcus strains to changing iron availability. ISME J. 5, 1580–1594. doi: 10.1038/ismej.2011.49
Tolonen, A. C., Aach, J., Lindell, D., Johnson, Z. I., Rector, T., Steen, R., et al. (2006). Global gene expression of Prochlorococcus ecotypes in response to changes in nitrogen availability. Mol. Syst. Biol. 2, 53–53. doi: 10.1038/msb4100087
Turpin, D. (1991). Effects of inorganic N availability on algal photosynthesis and carbon metabolism. J. Phycol. 27, 14–20. doi: 10.1111/j.0022-3646.1991.00014.x
UNESCO. (1994). “Protocols for the Joint Global Ocean Flux Study (JGOFS) core measurements,” in IOC Manual and Guides 29, ed A. H. Knap (Paris: Intergovernmental Oceanographic Commission), 170.
Veluchamy, A., Lin, X., Maumus, F., Rivarola, M., Bhavsar, J., Creasy, T., et al. (2013). Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat. Commun. 4, 2091. doi: 10.1038/ncomms3091
Keywords: nitrate metabolism, algal physiology, transcriptomics, orthologous genes, Thalassiosira pseudonana, Fragilariopsis cylindrus, Pseudo-nitzschia multiseries
Citation: Bender SJ, Durkin CA, Berthiaume CT, Morales RL and Armbrust EV (2014) Transcriptional responses of three model diatoms to nitrate limitation of growth. Front. Mar. Sci. 1:3. doi: 10.3389/fmars.2014.00003
Received: 13 January 2014; Paper pending published: 15 February 2014;
Accepted: 11 March 2014; Published online: 26 March 2014.
Edited by:
Karla B. Heidelberg, University of Southern California, USAReviewed by:
Byron Crump, Oregon State University, USACopyright © 2014 Bender, Durkin, Berthiaume, Morales and Armbrust. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: E. Virginia Armbrust, Center for Environmental Genomics, School of Oceanography, University of Washington, Box 357940, Seattle, WA 98195, USA e-mail:YXJtYnJ1c3RAdXcuZWR1
†Present address: Sara J. Bender and Colleen A. Durkin, Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution, Woods Hole, USA
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.