 David M. Feliciano
David M. Feliciano
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mamm. Sci. , 10 August 2023
Sec. Nervous System and Cognate Behaviors
Volume 2 - 2023 | https://doi.org/10.3389/fmamm.2023.1231778
This article is part of the Research Topic Editors' Showcase: Nervous System and Cognate Behaviors View all 9 articles
The capacity to integrate complex sensory cues and to coordinate an adequate behavioral response often requires integration of information within the outermost part of the mammalian brain called the cerebral cortex. The laminar and columnar cytoarchitecture of the cerebral cortex contains neurons that establish proximal and distal connections. Genetically encoded transcription factors ensure the generation of the appropriate number, types, locations, and connections of cortical neurons. However, somatic mutations that alter cortical development provide evidence that post-transcriptional regulation is equally important. An example is that somatic mutations in regulators and substrates of mammalian target of rapamycin (mTOR) are associated with neuropsychiatric and neurological manifestations. mTOR is a protein kinase that phosphorylates substrates that control mRNA translation and anabolic processes. Numerous challenges remain in uncovering the mechanisms by which mutations in regulators and substrates of mTOR impact behavior. Here, evidence is provided that somatic mosaicism can be modeled in the developing murine cerebral cortex which may have clinical significance.
A singular genome which is present within a fertilized zygote is responsible for generating the genomic and cellular architecture of an entire organism. In diploid organisms, half of the alleles are paternally derived, and half are maternally derived. When an allele is inherited, the frequency of each variant allele (VAF) is estimated to be ~50%. Genetic testing currently relies on this notion. For example, to determine the genetic cause of a disease, a patient may have DNA from blood, saliva, hair, or skin collected for genetic analysis. The expectation is that the patient has two inherited alleles with a VAF of 50% and deviations from this expectation may underlie disease. It is well accepted that only certain tissues or cell types may be changed in many diseases. One possible mechanism for why only specific tissues or cell types are affected in some diseases is that cellular genomes within an organism can change. Indeed, studies on Zea mays and later in patients having hemophilia demonstrated that cellular genomes may be altered by endogenous genomic modifiers called transposons (McCLINTOCK, 1950; Kazazian et al., 1988). Seminal studies on oncoviruses, oncogenes, and tumor suppressors also highlighted that the somatic genome was mutable (Varmus et al., 1972; Nigro et al., 1989; Ballester et al., 1990; Li et al., 1997; Kwon et al., 2006). Thus, postzygotic de novo mutations may cause two cells within an organism to be genetically different. This genetic mosaicism can have significant effects leading to neurological diseases, the causes of which have been undetectable until recently. Low VAF in regions of the brain are now realized to underlie some neurodevelopmental disorders which can be detected because of advances in DNA sequencing and computational analyses. Somatic genomic instability was also statistically modeled for sporadically arising diseases which were hypothesized to arise from de novo mutations (Knudson, 1971). This model, called the Two-hit hypothesis extends to numerous neurocutaneous disorders, including proteus syndrome with hypomelanosis of ito, for which patients have visually recognizable cutaneous somatic changes. Rudolf Happle argued that somatic mosaicism underlies the development of these disorders (Happle, 1987). Contemporaneously, evidence for somatic mosaicism was found in the growths of neurocutaneous syndromes Neurofibromatosis (NF) and Tuberous Sclerosis Complex (TSC) which are also characterized by neurocutaneous lesions and benign growths in the brain (Nigro et al., 1989; Verhoef et al., 1995). However, the role of somatic mutations, ranging from transposition events to point mutations, has only recently been recognized as a normal part of brain development (Bizzotto and Walsh, 2022). In some cases, somatic mosaicism that alters select pathways can cause malformations of cortical development that lead to a range of neurological manifestations and are the cause of numerous neurodevelopmental disorders. A fundamental neuroscientific question that remains then, is why such a mechanism for genomic heterogeneity exists. As more patients, tissues, and cells are sequenced and as sequencing technologies advance, the prevalence of somatic variants is sure to grow. Hopefully, so too will the appreciation for the capacity of variants to shape neurological function. This review will highlight the prevailing evidence that somatic mutations that alter mTOR function change neuronal circuitry and behavior and that this can be modeled in rodents which may be relevant for understanding the importance and role of genetic mosaicism in shaping normal physiological function. What follows is a description of the mTOR complex 1 pathway (Figure 1).
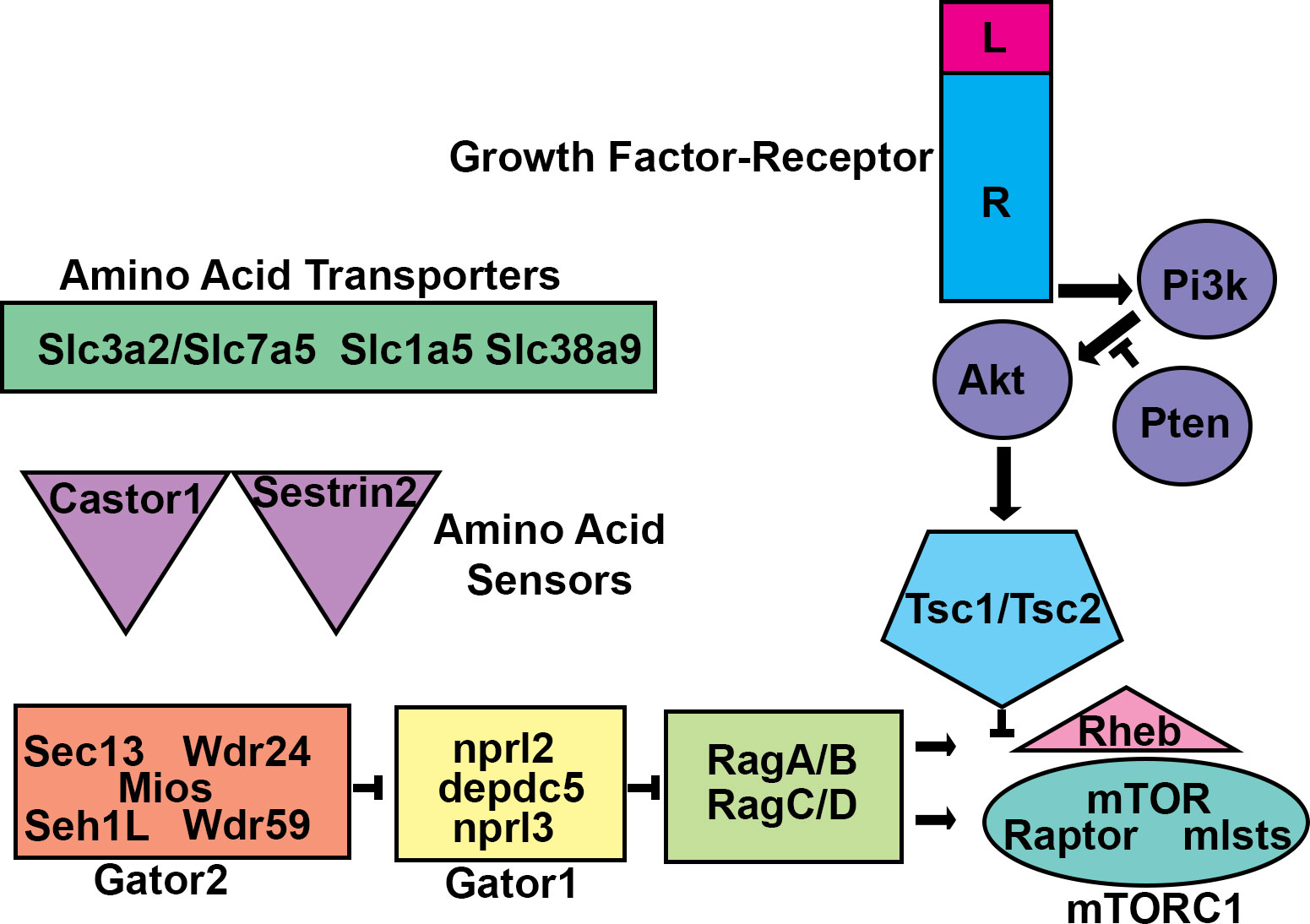
Figure 1 mTORC1 Regulation. Schematic diagram of the mTORC1 circuit. Amino acid transporters allow amino acids into cells and/or subcellular compartments. Amino acids are sensed by proteins such as castor or sestrin2. The sensor proteins then work through GATOR2 to prevent GATOR1, a GAP, from inhibiting the RAG heteromeric GTPases. RAG-GTP allows for mTOR Complex 1 (mTORC1) to interact with and become activated by the monomeric GTPase RHEB at the lysosome membrane. RHEB activation of mTORC1 is inhibited by the TSC1/TSC2 heteromeric GAP. TSC1/TSC2 can be inhibited by phosphorylation by protein kinases including AKT. AKT is activated by growth factor signaling initiated by transmembrane receptors which activate the lipid kinase PI3K. PI3K signaling is balanced by the lipid phosphatase PTEN. Mutations that alter these components can lead to malformations of cortical development.
mTORC1 and mTORC2 are complexes that utilize the mechanistic target of rapamycin (mTOR) as a core catalytic kinase (Laplante and Sabatini, 2012). mTORC1 is inhibited by rapamycin, a molecule synthesized by Streptomyces hygroscopicus (Vézina et al., 1975). Rapamycin acutely inhibits mTORC1 but sustained exposure can also prevent the assembly of mTORC2 (Chung et al., 1992; Brown et al., 1994; Sabatini et al., 1994; Sabers et al., 1995; Burnett et al., 1998; Sarbassov et al., 2006; Lamming et al., 2012). Rapamycin works by causing FK506-binding protein (FKBP12) to bind to and inhibit mTORC1 (Sabatini et al., 1994). mTORCs contain homodimeric circular catalytic mTOR kinase loops held together by mTORC specific proteins (Yip et al., 2010; Aylett et al., 2016; Saxton and Sabatini, 2017). These proteins are Raptor and Rictor which stabilize mTORC1 and mTORC2, respectively (Hara et al., 2002; Dos et al., 2004).
mTORC1 is activated by the monomeric GTPase, RHEB (Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). RHEB-GTP activates mTORC1. RHEB can hydrolyze GTP to GDP (Yamagata et al., 1994). When this happens, mTORC1 is turned off. RHEB dependent GTP hydrolysis is tightly controlled by hamartin and tuberin which are encoded by the TSC1 and TSC2 genes (Beugnet et al., 2003; Garami et al., 2003; Inoki et al., 2003; Zhang et al., 2003). Hamartin and tuberin form a GTPase activating protein (GAP) complex that prevents RHEB-mTORC1 activation. Tuberin exerts GAP activity towards RHEB. Hamartin stabilizes tuberin along with TBC1D7 (Dibble et al., 2012). mTORC1 activity is elevated in the absence of hamartin, tuberin, and TBC1D7. In addition, hamartin and tuberin are targeted by signaling pathways that titrate mTORC1 activity. For example, AKT phosphorylates and inhibits tuberin (Cai et al., 2006).
The amino acids leucine, arginine, and glutamine facilitate RHEB-mTORC1 activation (Hara et al., 1998). Activation is promoted by mTORC1 translocation to the lysosome where RHEB is located (Sancak et al., 2010; Bar-Peled et al., 2012; Efeyan et al., 2012). Translocation is mediated by heterodimeric RAG GTPases (RagA or B bound to RagC or D) (Bar-Peled et al., 2012). RAG-GTP facilitates interaction of and activates RHEB-MTORC1. RAG-RHEB-MTORC1 interaction is opposed by a GAP called GATOR1 (Bar-Peled et al., 2013). GATOR1 is comprised of DEPDC5, Nprl2, and Nprl3. GATOR1 turns off RAG-RHEB-mTORC1. A second complex called GATOR2 counters GATOR1(Bar-Peled et al., 2013). GATOR2 (Mios, WDR24, WDR59, Seh1L, Sec13) inhibits GATOR1 thereby turning on RAG-RHEB-mTORC1. GATOR2 inhibits GATOR1 when leucine and arginine sensors are engaged and allow for RAG-RHEB-mTORC1 activation (Chantranupong et al., 2014; Chantranupong et al., 2016; Wolfson et al., 2016).
mTORC1 controls translation of select mRNAs by phosphorylating the eukaryotic initiation factor 4E (eIF4E) binding protein (4EBP), which is an inhibitor of eif4E (Burnett et al., 1998; Gingras et al., 1999). mTORC1 also phosphorylates p70S6 kinase (p70S6K) which catalyzes ribosomal S6 subunit (S6) phosphorylation (Chung et al., 1992; Price et al., 1992). mTORC1 dependent 4EBP inhibition and S6 activation stimulates translation of anabolic mRNAs involved in processes including ribosome biogenesis (Choo et al., 2008; Thoreen et al., 2012). mTOR also regulates catabolic cellular processes (Ganley et al., 2009; Jung et al., 2009; Kim et al., 2011; Zhang and Manning, 2015).
Somatic mutations occurring along the mTOR pathway alter brain development. What follows is a description of case examples of mutations and the commensurate abnormalities that occur in the CNS. Next is a discussion of pathogenic variants organized by genes (Figure 2) and reported mutations (Table 1) starting from the catalytic subunit MTOR.

Figure 2 Pathogenic Variants. Variants of pathogenic or likely pathogenic significance as well as uncertain significance are listed for (A) MTOR (B) DEPDC5 (C) TSC2 from gnomAD browser (Karczewski et al., 2020). Note that both ClinVar variants and gnomAD variants are provided. Synonymous variants are not demonstrated due to space constraints. Despite a significant number of pathogenic variants for each gene that have been discovered, many more of uncertain significance have been identified and warrant further investigation.
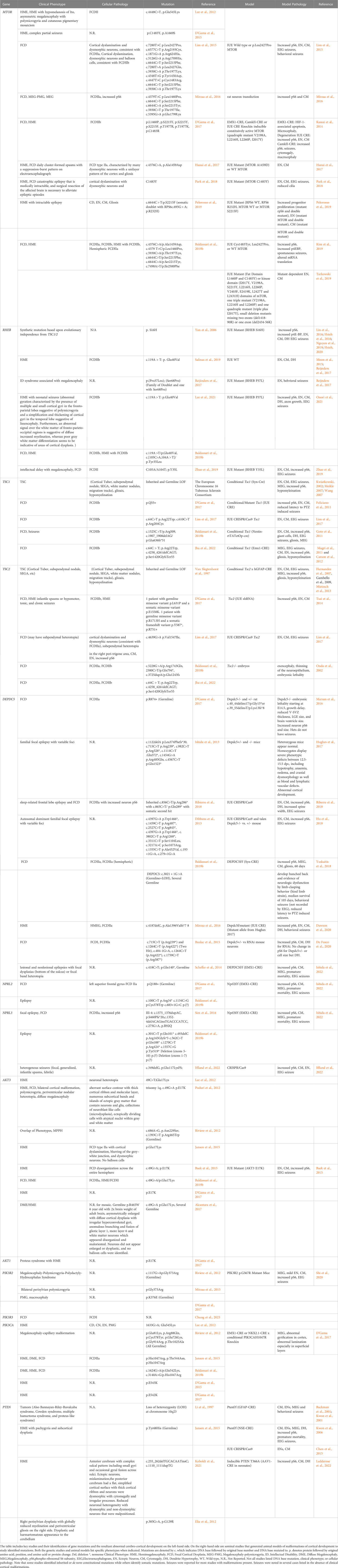
Table 1 Somatic Mosaicism.
A 5 year old patient having focal skin discoloration (hypomelanosis of ito) and hemimegalencephaly (HME) was discovered to harbor a somatic mutation in MTOR (c.4448C>T; p.Cys1483Tyr) at a 8-36% variant allele frequency (VAF) in tissue removed from affected cortex (Lee et al., 2012). HME is an enlargement of one side of the brain, typically involving the cerebral cortex and often contains improper lamination of the cortex with dysmorphic cytomegalic neurons. The patient had cortical dyslamination with ectopic and cytomegalic neurons. This patient had hypomelanosis of ito too which is interesting given the low allele frequency within the brain and the fact that these mutations were not found in any blood sample (Lee et al., 2012). These results suggest that the mutation might have arisen in cells that give rise to both the skin and cortex or that one of the inherited alleles is a mutant allele. It would be useful to know whether skin lesions also contained the same MTOR mutation. Nevertheless, this patient had elevated mTORC1 activity indicated by increased pS6 in brain tissue. Whether MTORC2 signaling was also elevated was not examined. This could be critical for developing potential patient therapies since rapalogs only target MTORC1. A second patient with HME and complex partial seizures was discovered that had a 14% VAF for p.Cys1483Tyr (D’Gama et al., 2015). D’Gama and colleagues subsequently reported 8 patients with mosaic MTOR mutations (D’Gama et al., 2017). The presence of this same mutation and another (MTOR c.5005G>T, p.A1669S) was later reported in a study examining focal cortical dysplasia (FCD) (Mirzaa et al., 2016). FCD is reminiscent of HME with the exception that FCD is confined to a smaller cortical region. FCD can be classified into type I, type IIa or type IIb and type III. FCDIIa and FCDIIb have focal mislamination, ectopically positioned cytomegalic, and dysmorphic neurons and differ based on the presence of balloon cells found in FCDIIb.
MTOR mutations were also found in ~15% of all patients diagnosed with FCDII (Lim et al., 2015). Over-expression of plasmids encoding representative MTOR mutations in HEK293T cells increased mTORC1 signaling which mirrored immunohistochemical quantification of mTORC1 activity in resected FCD. Several groups have introduced DNA plasmids into neural stem cells (NSCs) that generate the different types of cells in the brain using a technique called in utero electroporation (Figure 3). In utero electroporation of mutant MTOR plasmids into the developing cerebral cortex of mice induced rapamycin sensitive mTORC1 activity and seizures (Lim et al., 2015). These results demonstrate that the developing rodent cerebral cortex may allow for screening the veracity of claims that a somatic variant is pathogenic. A heroic study further examined 16 MTOR mutants in culture and performed in utero electroporations of select mutants having the greatest effects on MTORC1 pathway activity thereby demonstrating the utility of this methodological pipeline for assessing pathogenic variants (Tarkowski et al., 2019). Moreover, therapies may also be tested in these models.
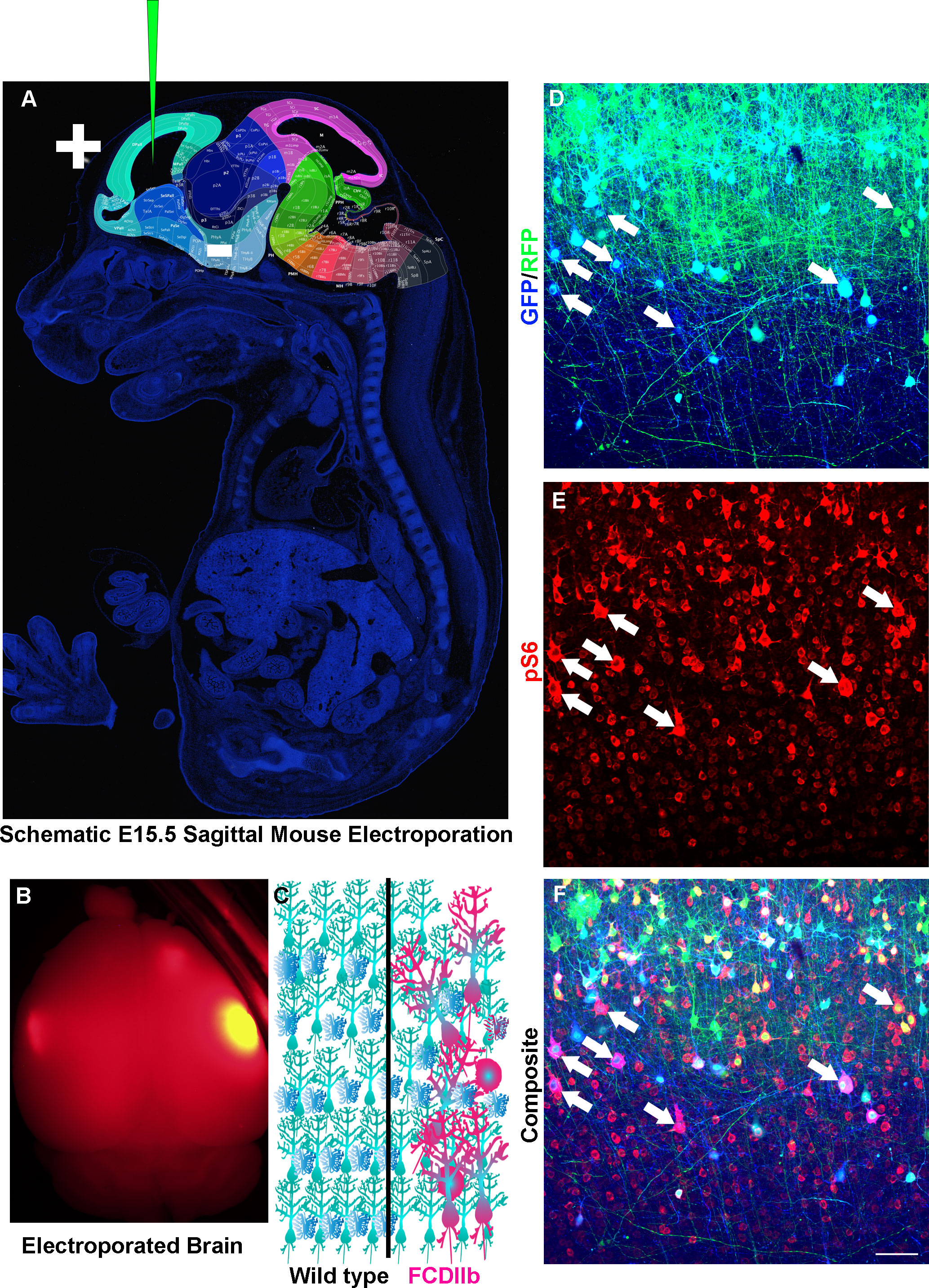
Figure 3 Modeling Somatic Mosaicism. (A) Sagittal section of an E15.5 mouse with regions of the brain highlighted and labeled. Modified from Allen Institute for Brain Science. Depicted is a glass capillary tube with plasmids (Green) inserted into the developing embryo. Injection of plasmid is followed by injection of short electrical pulses that facilitate the entry of plasmid into target cells. In utero electroporation as discussed here is typically targeted to radial glia which are neural stem cells that generate neurons and astrocytes. (B) An example of an adult mouse brain from an e15-15 electroporation. (C) Schematic diagram of wild-type (left) and Focal cortical dysplasia IIb (FCDIIb, right) brains. Note the cytomegalic and dysmorphic neurons along with balloon cells that are strewn throughout the normally hexalaminar cerebral cortex. (D–F) In utero electroporation of an e15-16 mouse with CRE recombinase and GFP (Blue) into RFP (Green) inducible mice having conditional and mutant TSC genes and stained for phospho-S6 (Red). Arrows point to cytomegalic neurons and balloon cells.
VAF correlates with the clinical presentation of patients that have mTOR pathway mutations and cortical malformations. Patients with a low MTOR VAF have less severe malformations (FCD IIa) whereas those with a higher MTOR VAF had severe or diffuse HME (including with polymicrogyria) (Mirzaa et al., 2016). VAF or the presence of additional mutations was not examined in HME brain tissue which prevented direct comparisons, however. Nevertheless, tissue with greater changes to cytoarchitecture also had greater mTORC1 activity. Importantly, over-expression of mutant MTOR enhanced mTORC1 pathway activity and cell size in rat neurons which could be rescued with the mTORC1 inhibitor RAD001 (Mirzaa et al., 2016). Thus, lissencephalic mice and gyrencephalic rat brains both have neurons that can be altered by MTOR mutants and screened using drug therapies.
An additional 18 cases of somatic MTOR mutations were identified in a group of 76 out of 283 patients for which mutations were detectable (Chung et al., 2023). Although additional somatic mosaic MTOR mutations were identified and could phenocopy the FCD aspects of HME, why such changes occur was unclear (Hanai et al., 2017). In utero electroporation of plasmids encoding p.Cys1483Tyr and p.Leu-2427Pro mutations found in patients also causes abnormal development leading to FCD-like changes (Kim et al., 2019). Importantly, the rodent cortex again demonstrated utility for screening cellular and neuroanatomical changes caused by variants (Kim et al., 2019). FACS sorted neurons had translational changes associated with ribonucleoside metabolic pathways, RNA, processing, and regulation of organelle and cilia assembly (Kim et al., 2019). The link to ciliagenesis was solidified by the demonstration that activating MTOR mutations prevent autophagy causing a buildup of OFD1 (Park et al., 2018). OFD1 buildup prevented the generation of non-motile cilia and thereby abrogated WNT mediated morphological polarization (Park et al., 2018). These results indicate that reactivation of autophagy could be a useful treatment to treat patients. Rapamycin has a well-established role in activating autophagy in yeast and has some effect within mammalian brain (Noda and Ohsumi, 1998). However, other treatments, for example, those that activate pro-autophagic pathways such as AMPK activation of Ulk1 could also be useful (Egan et al., 2011). Moreover, autophagy plays a critical role in synaptic pruning (Tang et al., 2014). Altered mTOR pathway activity in ASD patients has revealed mTOR-dependent inhibition of autophagy may mediate hyperconnectivity (Tang et al., 2014). Not surprisingly, many patients with FCDs or HME also have ASDs. One surprising finding comes from a CRE-inducible transgenic mouse model harboring 4-point mutations in MTOR making it constitutively active (Kassai et al., 2014). This study utilized an EMX1-CRE mouse to turn on MTOR in excitatory neuron stem cells in the embryonic brain (Kassai et al., 2014). The result was massive cortical neuron apoptosis and microcephaly which is not reported in in utero electroporation models. However, assessing such effects following electroporation is a challenge since electroporation efficiency and success rate of electroporation are not easily assessable. It is however plausible that mutations originate in a different neural stem cell pool and produce different results. Alternatively, there may be limited nutrients or extracellular factors that limit the utility of using this global model. Nevertheless, the generation of a transgenic mouse demonstrates an interesting conundrum for electroporation. Plasmids are not genomically integrated at an efficient rate and therefore electroporation seldom allows for the consistent assessment on certain cell types. For example, astrocytes and neural stem cells are challenging to target using plasmid electroporation. However, the use of transposon technology during electroporation overcomes some of these limitations (Chen et al., 2015).
MTORC1 must associate with RHEB-GTP to be activated. Electroporation of a constitutively active (CA) RHEB (S16H) mutant into neonatal and subsequently, embryonic mice, revealed phenotypes associated with hyperactivation of MTORC1 including MTORC1 pathway activation and ectopically positioned cytomegalic neurons (Yan et al., 2006; Lin et al., 2016). In utero electroporation of CA-RHEB led to seizures which could be initiated independent of lamination defects (Hsieh et al., 2016). Moreover, the amount of plasmid can be titrated to mimic the variable levels of MTORC1 activity that occur in patients (Nguyen et al., 2019). It could also be useful to vary promoters for electroporation studies since dilution of plasmids has resulted in a gradient of change which has not yet been quantified. Nevertheless, RHEB effects can be reversed by expressing a constitutively active form of the translation regulatory protein EIF4E-BP (Lin et al., 2016). These results indicated that RHEB mutations could theoretically cause cortical malformations. This hypothesis was soon realized in the discovery of patients with RHEB mutations that had intellectual delay and megalencephaly (Reijnders et al., 2017). Indeed, electroporation of mutant or wild-type RHEB expression is sufficient to induce most cellular phenotypes (Moon et al., 2015; Sokolov et al., 2018). A somatic RHEB mutation (c.119A > T: p. Glu40Val) was subsequently identified in an HME patient with an FCDIIb histopathology (Salinas et al., 2019). An additional FCD patient having somatic RHEB mutations in two adjacent nucleotides (A104T, C105A) resulted in a p.Y35L mutation that enhanced GTP binding (Zhao et al., 2019). In utero electroporation of this mutant into mice phenocopied previous mutant RHEB experiments and created another FCDII model. A similar RHEB p.Y37L mutant which produces stronger and TSC1/2 resistant activation of MTORC1 activity in comparison to the RHEB S16H mutant also induces seizures that are independent of heterotopias that are formed (Onori et al., 2021). In addition to enhanced dendrite arbors, mutant neurons also have faster growing axons with extensive ramifications (Onori et al., 2021). Mutant axons had a broader and ectopic (lower layer) targeting in the contralateral hemisphere. Although the neurons had enhanced excitability, blockade of axon vesicle release could reverse the spontaneous seizures in this model (Onori et al., 2021). RHEB pY35L mutations have since been identified in other patients with FCDIIB and HME (Lee et al., 2021). Although RHEB mutations occur infrequently this work builds a critical foundation for understanding how somatic mutations that influence the MTORC1 pathway can cause cytoarchitectonic errors and lead to neurological manifestations including seizures (Chung et al., 2023). There are several major questions about RHEB mutations which exist. For example, are there mTORC1 independent effects? Another surprising fact is that mTOR requires signals to become localized to the lysosome. Do RHEB mutations only enhance the level of mTORC1 signaling or is the duration of mTORC1 signaling also increased? If the latter, how can this occur if the subcellular localization of MTORC1 is also under stringent control? Could this indicate that some amount of MTORC1 is always in proximity to RHEB, and that the level of RHEB-GTP is more important for MTORC1 activation than subcellular localization of MTORC1? Finally, does the subcellular distribution or kinetics of localization of mutant RHEB change? Answering these questions may help to generate better mechanistic insight into pathogenesis and to developing therapeutic strategies.
RHEB is inhibited by a GAP comprised of proteins encoded by the TSC1 and TSC2 genes (Garami et al., 2003; Inoki et al., 2003; Zhang et al., 2003). Inactivating TSC1/TSC2 mutations promote mTORC1 pathway activity (Feliciano, 2020). Inactivating mutations in TSC1 or TSC2 cause TSC (The European Chromosome 16 Tuberous Sclerosis Consortium, 1993; Van Slegtenhorst et al., 1997). TSC is a multi-system disorder characterized by prominent focal dysplastic regions including within the brain (Feliciano et al., 2013; Hasbani and Crino, 2018; Feliciano, 2020). The majority of TSC patients have seizures (Kingswood et al., 2014; Kingswood et al., 2017; Nabbout et al., 2019). Seizures called infantile spasm frequently occur in infants with focal seizures developing later on (Lux and Osborne, 2004; Kelley and Knupp, 2018; Nabbout et al., 2019). Seizure manifestations are heterogenous (Nabbout et al., 2019). Seizures are likely caused by focal dysplastic regions called cortical tubers that are present in the majority of patients (Krueger et al., 2013b; Kingswood et al., 2017). Seizure foci often overlap with cortical tubers (Mohamed et al., 2012). Electrophysiological analysis of tubers demonstrated that tubers have an extensive contribution to cortical hyperexcitability (Despouy et al., 2019). In agreement, cortical tuber removal prevents seizures (Fallah et al., 2013; Fallah et al., 2015). Surgical removal of peri-tuber regions enhances outcomes which indicates that regions outside of the tuber may also contribute to hyperexcitability (Fallah et al., 2015). Cellular and molecular changes within and surrounding cortical tubers may influence their electrophysiological properties, spread of hyperexcitability, and clinical presentation (Doherty et al., 2005; Boer et al., 2008; Kaczorowska et al., 2011; Canevini et al., 2018; Nabbout et al., 2019). There are however case reports of TSC patients without tubers that exhibit seizures which indicates additional mechanisms of epileptogenesis may exist in TSC (Wang et al., 2007). Moreover, removal of conditional Tsc1 from postmitotic neurons in mice induces seizures in the absence of some tuber features and supports this hypothesis (Meikle et al., 2007; Wang et al., 2007). Tubers are histologically heterogenous (Chu-Shore et al., 2009; Gallagher et al., 2010; Zhang et al., 2018). They can vary in cellular composition (Mühlebner et al., 2016). Similar to FCD, tubers are regions for which the cortex loses laminar cytoarchitecture and have dysmorphic cytomegalic neurons and balloon cells (Ferrer et al., 1984; Huttenlocher and Heydemann, 1984; Mühlebner et al., 2016). However, tubers contain giant-cells (Yamanouchi et al., 1997a; Yamanouchi et al., 1997b; Mizuguchi and Takashima, 2001; Mizuguchi et al., 2002).
Somatic mosaicism has long been recognized in TSC (Verhoef et al., 1995; Verhoef et al., 1999; Sampson et al., 1997). There are multiple types of mosaicism that can occur. It is important to keep in context how this might be relevant to defining which variants are pathogenic in the related disorders discussed in this review. The first type of mosaicism is germline mosaicism (Dabora et al., 2001). This occurs when a non-carrier parent produces gametes that may carry a mutation that is subsequently inherited. The second is classical mosaicism, which occurs when a de novo mutation arises during development (Verhoef et al., 1999; Qin et al., 2010b; Tyburczy et al., 2015; Lim et al., 2017; Klonowska et al., 2023). Although it is feasible to ascertain whether a parent carries a variant, it is challenging to determine the precise time of development when the de novo variant arises. However, groups have made compelling progress in this arena which might be clinically applicable (Bizzotto et al., 2021). Nevertheless, a late occurring mutation would affect a minor fraction of cells. The third type of mosaicism is that which Knudson proposed, which is the manifestation of the two-hit hypothesis (Knudson, 1971). In this model, a mutant allele and wild-type allele are inherited, followed by conversion of the wild-type allele into a mutant allele. Knudson later used the Eker rat, which carries a Tsc2 mutation, to test this model (Eker and Mossige, 1961). They demonstrated that the tumors have Tsc2 loss of heterozygosity (LOH) (Hino et al., 1993; Yeung et al., 1994; Kubo et al., 1995). While most TSC phenotypes require LOH, there are many reports that have used heterozygous rodents to suggest haploinsufficiency. One question not yet answered is how often these models have LOH. Thus, routine examination of tissue from heterozygous models should also examine conversion of the second allele.
A human induced pluripotent stem cell (IPSC) culture model was created that has conditional TSC1 alleles to determine the effect of losing TSC genes on neurodevelopment(Blair et al., 2018). IPSCs were used to generate NSCs that were subsequently differentiated. Loss of both TSC1 alleles was required for cortical tuber phenotypes (Blair et al., 2018). Winden et al. however demonstrated that heterozygous and homozygous TSC2 mutant human IPSC derived NSCs exhibit significant changes that mirrored the conditional TSC1 NSC model. Surprisingly, heterozygous and homozygous TSC2 mutant NSCs had the same amount of tuberin (c.5238_5255del p.H1746_R1751del, TSC2+/-) (Winden et al., 2019). The difference between homozygous and heterozygous requirements in these models could be caused by the mutation, NSC type, culture condition, or LOH in the TSC2+/- cells. Detection of LOH in patient tubers while detectable, may be limited by VAF detection in low abundance cell types (Crino et al., 2010; Qin et al., 2010a). On the other hand, either mechanism may be sufficient to generate tubers depending on the mutation. Another human IPSC-NSC model demonstrated that TSC2 mutant heterozygous IPSCs undergo LOH during culture (Eichmüller et al., 2022). These analyses challenged the proposition for LOH but were specific for a specialized caudal late inhibitory progenitor (CLIP) which has low tuberin expression. SEGAs and tubers are not symmetrically (bilaterally) localized in patients. Since both brain hemispheres have CLIP cells and both hemispheres would be proposed to harbor the same pathogenic variant, why only 5-20% of TSC patients develop SEGAs is unclear. Moreover, TSC patients’ cortical tubers are present in anatomically and cellularly distinct regions. In fact, TSC patients have many types of lesions outside of the brain which seldom occur symmetrically. In contradiction to a cell-type specific function of TSC genes, loss of both copies of Tsc1 or Tsc2 in many different neural cell types causes overlapping phenotypes in rodents. Finally, as discussed below, transgenic mice with conditional alleles subject to electroporation or crossed to CRE driver mice have more severe manifestations than heterozygous models. Nevertheless, the specificity of SEGAs occurring most frequently along the lateral ventricles near the foramen of Monroe is compelling. It is also of interest to consider that while no differential methylation pattern is currently recognized for maternal vs. paternal TSC1 or TSC2, there may be additional mechanisms that factor into the penetrance of phenotypes.
An inherited homozygous dominant negative mutation or two recessive mutations appear incompatible with mammalian viability. No cases of TSC patients that have inherited two mutant alleles has been reported. This is supported by the fact that Tsc1 and Tsc2 homozygous deletion is embryonic lethal in mice and rats (Rennebeck et al., 1998; Onda et al., 1999; Kobayashi et al., 2001). Moreover, Tsc2 heterozygous neuroepithelial cells have no differences whereas homozygous null cells recapitulate key aspects of those seen in TSC (Onda et al., 2002). Mice having conditional TSC alleles have circumvented this limitation and demonstrated that phenotypes most often can be detected following loss of both TSC alelles (Kwiatkowski, 2002; Hernandez et al., 2007). For example, homozygous Tsc1 deletion from NSCs generates mislamination, macrocephaly, cytomegaly, hypomyelination, and reactive gliosis with seizures (Goto et al., 2011; Magri et al., 2011; Carson et al., 2012; Mietzsch et al., 2013). Homozygous Tsc2 deletion also generates macrocephalic mice having cytomegaly, hypomyelination, reactive gliosis and seizures (Way et al., 2009; Mietzsch et al., 2013). Tsc1 removal from neurons using synapsin I promoter driven CRE also caused mTORC1 hyperactivation, cytomegalic and dysmorphic neurons, megalencephaly, severe seizures and premature mortality (Meikle et al., 2007; Wang et al., 2007). The absence of a discrete cortical tuber in TSC conditional mice has provoked discussions that loss of TSC genes in large cellular populations in the cortex might cause the entire cortex to represent a cortical tuber (Wong, 2012). To recapitulate LOH and the focal nature of cortical tubers, in utero electroporation of CRE recombinase into mice carrying a mutant and conditional Tsc1 allele was performed (Feliciano et al., 2011). The in utero model generated focal mosaic patterning, cytomegaly, and hyper-active mTORC1 similar to that seen in patient tubers (Feliciano et al., 2011). Two conditional Tsc1 alleles appears sufficient to allow modeling of this phenotype (Robens et al., 2016; Cox et al., 2018). In utero Tsc1/2 knockdown also increased the number of axons in developing cortical neurons (Choi et al., 2008). Importantly, recent reports suggest that loss of TSC genes through a Two Hit mechanism can also cause FCD (D’Gama et al., 2017; Lim et al., 2017; Baldassari et al., 2019b; Jha et al., 2022; Chung et al., 2023). In utero electroporation of CRISPR-Cas9 system targeting TSC genes was used to model the FCD which produced cortical dyslamination, cytomegalic neurons, and induced seizures (Lim et al., 2017).Taken together, TSC1/TSC2 somatic mutations within TSC or FCD patients can cause malformations in the cortex that often underlie seizures. It remains to be seen whether such somatic mutations underlie the many other neuropsychiatric manifestations in these patients.
The role of mTORC1 in the pathogenesis and clinical manifestations in TSC is underscored by the utility of clinically approved mTORC1 inhibitors. mTORC1 inhibitors include everolimus and sirolimous which are derived from the macrolide antifungal compound rapamycin which is synthesized by the bacteria Streptomyces hygroscopicus (Vézina et al., 1975). Rapamycin causes FK506-binding protein (FKBP12) to inhibit mTORC1 phosphorylation of select substrates including the p70 ribosomal S6 kinase (Chung et al., 1992; Brown et al., 1994; Sabatini et al., 1994; Sabers et al., 1995). mTORC1 phosphorylation of other substrates such as Ulk1 and 4EBP is however, incompletely inhibited by rapamycin (Choo et al., 2008; Thoreen et al., 2009; Kang et al., 2013). Despite these critical limitations, rapamycin analogs (rapalogs) are effective in treating TSC and FCD models. Rapamycin treatment of the Synapsin-CRE x Tsc1 model reverses elevated mTORC1 pathway activity, neuron size, cortical thickness, and increased survival (Meikle et al., 2008). Similar results were achieved in NSC Tsc1 deletion models (Anderl et al., 2011; Magri et al., 2011). The importance of mTORC1 in lesion formation was confirmed in focal models which revealed specific windows of time for which rapamycin reverses select cellular phenotypes (Tsai et al., 2014; Lim et al., 2017; Cox et al., 2018). SEGAs are TSC associated subcortical growths that occur within the brain of TSC patients. The first study to use mTORC1 inhibitors to treat these growths found that rapamycin was highly effective and caused SEGAs to regress (Franz et al., 2006). Everolimus treatment caused TSC SEGA regression by 30-50% within 6 months and coincidentally reduced seizure burden (Krueger et al., 2013a). 17 of 20 patients had a reduction in seizures with a 73% median decrease (Krueger et al., 2013c). Extension of the EXIST (EXamining everolimus In a Study of TSC) phase III clinical trials demonstrated long-term everolimus decreased seizure frequency and was particularly impressive at a high 9-15 ng/mL exposure (Curatolo et al., 2018). Early MRI and EEG fingerprints predict neurological outcome in TSC children with severity and age of seizure onset being a predictor of intellectual disability (De Ridder et al., 2021; Hulshof et al., 2022). Moreover, many neonates that have normal EEG before 2 months of age develop aberrant activity later and can represent an opportunity to prevent encephalopathy (Kingswood et al., 2014; Słowińska et al., 2018; De Ridder et al., 2021). Indeed, early intervention with vigabatrin improves neurological outcome with adjuvant everolimus further reducing seizures (Curatolo et al., 2018; Nabbout et al., 2021; Śmiałek et al., 2023). These results indicate that early treatment may facilitate proper brain development which could have lifelong implications and sets the stage for treating patients that have other mTORC1 pathway variants that cause cortical malformations.
Amino acids facilitate RHEB-mTORC1 activation (Hara et al., 1998). Amino acids allow GATOR2 to inhibit GATOR1 (Bar-Peled et al., 2013; Parmigiani et al., 2014; Chantranupong et al., 2016; Kimball et al., 2016). GATOR1 is the GAP for the heterodimeric RAG GTPase (RagA or B bound to RagC or D) at the lysosome (Bar-Peled et al., 2013). Mutations in the GATOR1 components, DEPDC5, NPRL2, and NPRL3 cause cortical malformations. Two seminal genetics studies were published in 2013.
One study described 19 families with autosomal dominant focal epilepsy were used for linkage analysis and identified 22q12 association Familial DEPDC5 mutations were initially described as a cause of familial focal epilepsy with multiple foci. Further sequencing confirmed mutations including nonsense and missense mutations demonstrating loss of function of DEPDC5 as the likely cause (Ishida et al., 2013). The other study demonstrated that DEPDC5 mutations occur in >10% of families with non-lesional focal epilepsy (Dibbens et al., 2013). Patients from these families can have focal dysplasias (bottom of the sulcus) or focal band heterotopia (Scheffer et al., 2014). To provide mechanistic studies on DEPDC5, knockout rats were generated, but died embryonically (Marsan et al., 2016). Heterozygous DEPDC5 rats still had dysmorphic cytomegalic neurons with pS6 and reduced firing rates, but do not exhibit seizures (Marsan et al., 2016). DEPDC5 heterozygous e12.5 cultured neurons also have similar morphological changes (De Fusco et al., 2020). These results demonstrate that loss of a single allele could be sufficient for cellular phenotypes. However, the heterozygous mouse neurons do not have detectably higher pS6 in vitro as measured by western blot (De Fusco et al., 2020). In contrast, DEPDC5 null e12.5 mouse neuron cultures as well as DEPDC5 knockdown neurons have a more severe phenotype than heterozygotes including increased mTORC1 pathway activity and enhanced dendrite arbors (De Fusco et al., 2020). A requirement for loss of both alleles is consistent with a genome engineering approach using CRISPR/Cas9 and Talen technology to remove DEPDC5 from mice (Hughes et al., 2017). Thus, one might consider Knudson’s two hit hypothesis as a potential mechanism for DEPDC5 phenotypes. Indeed, using matched blood/brain lesion samples, Knudson’s two hit hypothesis again came to fruition (Baulac et al., 2015; Baldassari et al., 2019b). This result was further solidified by in utero electroporation of DEPDC5 targeted gRNAs using CRISPR/Cas9 (Ribierre et al., 2018). Interestingly, these mice also develop seizures. In agreement, in utero electroporation and mutation of rat DEPDC5 recapitulates this phenotype (Hu et al., 2018). However, a neuron selective CRE driver (synapsin-I promoter) mouse crossed to those having conditional DEPDC5 exhibited nearly all phenotypes of patients (Yuskaitis et al., 2018). Mice also had spontaneous seizures associated with premature mortality which could be rescued by rapamycin (Yuskaitis et al., 2019). Depdc5 conditional mice were crossed to Depdc5 mice and subject to in utero electroporation of CRE recombinase, similar to the Tsc1 tuber model, which was sufficient to model nearly all FCD phenotypes seen in FCD patients with DEPDC5 mutations (Dawson et al., 2020). Taken together, it appears that loss of both alleles of DEPDC5 can recapitulate the phenomenon seen in patients.
Linkage analysis of first cousins with focal epilepsy and later a cohort of FCD patients associated mutations in NPRL3 with seizures (Sim et al., 2016). Another 5 mutations in NPRL3 and 5 in NPRL2 were identified in a cohort of 404 focal epilepsy patients (Ricos et al., 2016). Interestingly, the penetrance of mutations appeared low for some families (Korenke et al., 2016). Additional mutations in NPRL2 and NPRL3 continue to be identified in a range of epilepsy patients (Baldassari et al., 2019a). Using Emx1-CRE to drive recombination in the dorsal telencephalon led to a nearly overlapping phenotype of Nprl3 and or Nprl2 conditional mice (Ishida et al., 2022). Importantly, the same manuscript compared both models as well as conditional Depdc5 mice (Ishida et al., 2022). These results confirmed that loss of both alleles of any GATOR1 component is sufficient to cause hyperactivation of the mTORC1 pathway and induce seizures (Ishida et al., 2022). The effect of Nprl3 on neuron mTORC1 activity and neuron morphology was further validated using in utero electroporation CRISPR/Cas9 mediated mutation (Ishida et al., 2022).
The presence of PIP3 recruits AKT to the cell membrane where it is phosphorylated by phosphoinositide-dependent protein kinase 1 (PDK1) T308 (Alessi et al., 1997). Subsequent phosphorylation of AKT T473 leads to complete activation (Sarbassov et al., 2005). AKT phosphorylates numerous substrates that regulate cell survival (BAD), metabolism (GSK3a/b), and proliferation (p27 Kip1 and p21 Cip1). AKT is highly expressed within the developing brain (Owada et al., 1997). It is highly expressed in a proportion of embryonic NSCs and over-expression increases NSC proliferation including in the V-SVZ and OB (Sinor and Lillien, 2004).
Tuberin is a substrate of AKT (Cai et al., 2006). Tuberin along with hamartin, which are encoded by TSC1 and TSC2, form a GTPase activating protein (GAP) that inhibit RHEB (Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). Tuberin has a GAP domain that causes the monomeric GTPase RHEB to hydrolyze GTP (Tee et al., 2003). Hamartin and TBC1D7 together appear to stabilize Tuberin (Dibble et al., 2012). RHEB activates mammalian target of rapamycin mTOR (Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). mTOR can also phosphorylate AKT 473. Specifically, an mTOR heteromer called mTOR Complex 2 that associates with a protein called rictor phosphorylates AKT phosphorylation (Sarbassov et al., 2005). Rheb activates a distinct mTOR complex (mTORC1) that contains the protein raptor (Hara et al., 2002; Kim et al., 2002; Dos et al., 2004).
Somatic AKT1 mutations had been uncovered in proteus syndrome (Lindhurst et al., 2011). However, examination of AKT1 in 20 patients with HME failed to reveal somatic mutations. However, somatic mutations in AKT3 were discovered (Lee et al., 2012). A contemporaneous report of an AKT3 mutation in an HME patient was described by another group that indicated AKT3 activation normally occurs within dividing apical NSCs near the lumen of the ventricles (Poduri et al., 2012). However, an AKT1 E17K pathogenic mutation was identified in a patient with HME and Proteous syndrome (D’Gama et al., 2017). AKT3 E17K mutations were also identified in another HME patient in the same study (D’Gama et al., 2017). The recurrence of the AKT3 E17K mutation in studies of HME and FCD or related syndromes is exceptionally impressive and demonstrates the pathogenicity of this mutation (Baek et al., 2015; Jansen et al., 2015; Alcantara et al., 2017; D’Gama et al., 2017; Baldassari et al., 2019b). In utero electroporation of a plasmid encoding an AKT3 E17K mutant which mimicked a mutation found HME patients, prevented excitatory cortical neuron lamination leading to ectopic positioning, dysmorphic phenotypes, and cytomegaly (Baek et al., 2015). Interestingly, AKT3 constitutive activity altered transcriptomes leading to expression of reelin which was responsible for luring and entrapping non-electroporated cells. Somatic duplication of a region containing AKT3 have also been reported (Conti et al., 2015). Taken together, AKT3 mutations are a cause of abnormal cortical development. It is unclear to what extent the abnormal activity of AKT3 during development as opposed to after development, within postmitotic neurons causes neurological manifestations such as epilepsy. It would appear however, that hyperactivation caused by mutations to AKT3 or upstream regulators of AKT3 could be treated with AKT3 inhibitors and is of further clinical exploration. Given that AKT3 is a protein kinase, a major question that remains is which substrates are responsible for the changes seen in patients with AKT. The AKT substrate that rises above all others is tuberin, the inactivation of which causes a range of neurological manifestations.
Information conveyed by growth factors are transmitted by receptors having tyrosine kinase activity. The ability of somatic alterations that effect integral membrane proteins to change cells was demonstrated by oncogenic viruses that induce cellular transformation (Frykberg et al., 1983; Downward et al., 1984). Such growth factor receptor kinases exert effects by acting on intracellular signaling proteins. Later it was demonstrated that an avian sarcoma virus that encoded oncoviral proteins had similar transforming activity but acts as an intracellular signaling kinase (Macara et al., 1984; Sugimoto et al., 1984; Whitman et al., 1985; Kaplan et al., 1986). This transforming activity was identified to be mediated by a lipid kinase (Whitman et al., 1988). However, this lipid kinase phosphorylated the 3-OH group of phosphatidylinositols and generated phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (Auger et al., 1989; Ruderman et al., 1990). PIP3 subsequently acts as a second messenger to help recruit and activate AKT. The oncoviral studies support the idea that somatic changes in lipid signaling can also alter the balance of cell division. A hint that mutations in PI3K could disrupt brain development came from studies of a girl that had megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome (MPPH) which revealed a mutation in PIK3R2 (Nakamura et al., 2014). The PIK3R2 mutation was detected in blood leukocytes at a VAF of 47.7%. LOH or de novo mutations within the brain were not examined, but the authors concluded based on VAF, that the mutation was likely a de novo germline mutation (Nakamura et al., 2014). The first report that somatic mutations may result in hemimegalencephaly included identification of several patients have PI3KCA mutations. One mutation, PIK3CA c.1633G>A (p.Glu545Lys) was recurrent (Lee et al., 2012). This mutation was also identified by other groups (D’Gama et al., 2015; D’Gama et al., 2017; Baldassari et al., 2019b). However, a different mutation, PIK3CA pH1047R which is associated with diverse cancers inspired D’Gama and colleagues to cross a previously generated inducible form of this mutation to different CRE driver mice. EMX1-CRE dependent expression of the PIK3CA mutant caused MEG, abnormal cortical gyrification, and abnormal lamination thereby recapitulating many HME/FCD-like phenotypes (D’Gama et al., 2017). A larger study later built off of this finding and uncovered additional patients with PIK3CA mutations that had HME (Chung et al., 2023). Single cell RNA sequencing of two of these patients uncovered changes in the cellular composition and revealed altered transcript abundance that indicated altered lipid biosynthesis and metabolism in astrocytes.
Somatic mutations in PIK3R2 encoding regulatory subunit of PI3K have also been discovered in numerous patients. Notably, mosaic mutations in children with bilateral perisylvian polymicrogyria (Mirzaa et al., 2015). This mutation was further modeled in mice which had extensive megencephaly, mild ectopic neurons, cytomegaly, increased pS6 and EEG seizures (Shi et al., 2020). In addition, PI3K regulatory subunit (PIK3R3) mutations have also been described (Chung et al., 2023). Taken together, somatic mutations that increase PI3K activity, also disturb brain development. These studies generate many provocative questions, namely whether there are convergent mechanisms by which PI3K is working, could PI3K inhibition be used to treat these patients, is it a specific lipid product such as PIP3 that is altering brain development or a non-physiological product at fault, and finally, do all cells affected by these mutations cause the anatomical and clinical presentations or is there any specificity of how the mutations change brain development.
Mechanisms for reducing PIP3 also exist to prevent excessive downstream signaling. Evidence of such a system came from studies on somatic mutations that occur in brain tumors. LOH on chromosome ten q23 encoding a lipid phosphatase with homology to chicken tensin, and therefore called PTEN, is a common occurrence in cancers including glioblastoma (Li et al., 1997). PTEN is a lipid phosphatase, and like tensin, has a Src Homology 2 (SH2) domain allows for docking at phospho-tyrosine residues. PTEN removes the 3’ phosphate of the inositol ring of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), resulting in phosphatidylinositol (4, 5)-bisphosphate (PIP2) (Parsons, 2020). Inactivating mutations in PTEN therefore lead to a buildup of PIP3, a special lipid messenger. PTEN appears to function as a tumor suppressor, as its loss is readily detected in glioblastoma. Bannayan-Riley-Ruvalcaba syndrome, Cowden syndrome, multiple hamartoma syndrome, and proteus-like syndrome are now covered by the umbrella term PTEN Hamartoma syndrome (Parsons, 2020).
Based on the importance of growth factor receptors in NSCs, it is not surprising that conditional deletion of PTEN using a nestin promoter CRE enhanced proliferation of tripotent neural progenitors (Groszer et al., 2001). Likewise, loss of PTEN from e14.5 NSCs enhances neurosphere self-renewal and cell cycle reentry (Groszer et al., 2006). Removal of PTEN using GFAP-CRE which is expressed in NSCs causes abnormal cytoarchitecture including in the hippocampal dentate gyrus where NSCs persist (Backman et al., 2001; Kwon et al., 2001). But many of the alterations are due to changes in neurons born from NSCs where PTEN loss increases cell size. Regardless, PTEN deletion in the adult V-SVZ also enhances neurogenesis (Gregorian et al., 2009). Likewise, removal of PTEN from the postnatal V-SVZ using tamoxifen inducible CRE expressed in nestin positive NSCs increases the number of neuroblasts in the V-SVZ. PTEN null neuroblasts likely have cell autonomous precocious differentiation since ectopically positioned neurons were present along the rostral migratory stream and in the SVZ (Zhu et al., 2012) PTEN deletion from human progenitors also alters cerebral organoids, causing a transient delay in differentiation, and promoting increased organoid size and gyrification (Li et al., 2017).
In combination with loss of other tumor suppressors such as NF1 and p53, PTEN deletion induces glioblastoma in mice (Llaguno et al., 2008). Interestingly, as reported for NF1 heterozygous mice, combined NF1/PTEN heterozygous deletion is insufficient for tumor formation (Llaguno et al., 2008). In contrast removal of one PTEN allele with both p53 alleles causes highly penetrant astrocytoma (Zheng et al., 2008). Whether LOH of PTEN occurred in this manuscript was unclear. Nevertheless, these studies point to the fact that the NSCs collected from the V-SVZ are likely tripotent progenitors since they have the capacity to generate neurons, astrocytes, and oligodendrocytes. These results could mean, that some NSC populations could be more susceptible to oncogenic transformation than others. In agreement, PTEN deletion in astrocytes appeared to have minimal effects on cytomegaly, proliferation, or activation of downstream pathways but combined PTEN and p53 deletion generated tumors that more frequently associated with the V-SVZ (Chow et al., 2011). PTEN deletion along with constitutive activation of PI3K does however cause high grade brain tumors that protrude from the V-SVZ into the lateral ventricles (Daniel et al., 2018). On the other hand, expression of constitutively active PI3K is sufficient to generate oligodendrogliomas indicating that there may be additional lipid phosphatases that might substitute for PTEN or that insufficient PIP3 builds up to form tumors in PTEN null NSCs (Daniel et al., 2018).
PTEN deletion also alters neuron morphology throughout the brain. One particularly striking example is that PTEN deletion using an NSE-CRE driver caused widespread cortical and hippocampal alterations (Kwon et al., 2006). These animals were macrocephalic, had neuron cytomegaly, and increased dendrite arborization and axon growth with enhanced synaptogenesis. These animals also revealed sensory hyperactivity with altered social responses. Neuron morphology is reproducibly altered in cortical pyramidal neurons (Chow et al., 2009). In utero electroporation of conditional PTEN mice has also confirmed a prominent role in regulating cortical neuron dendrite growth (Hsia et al., 2014). The use of CRISPR/Cas9 to remove PTEN has also confirmed that cortical neurons have cytomegaly and revealed that these neurons have altered excitatory post-synaptic currents (Chen et al., 2015).
Somatic mutations in PTEN are widespread in brain tumors. In addition, mutations are reported in a range of non-cancer syndromes which include Cowden Syndrome, Lhermitte-Dulcos disease (cerebellar dysplastic gangliocytoma), Bannayan-Riley-Ruvalcaba syndrome (BRRS), and autism spectrum disorder with macrocephaly. Cerebellar dysplastic gangliocytoma are dysplastic growths within the cerebellum. The prevalence of somatic mosaicism or LOH in these lesions is largely unknown, however most have inherited at least one mutant allele and at least one study has detected LOH (Zhou et al., 2003). One notable example was reported by Janesen et al. whom described a germline mutation in PTEN (Tyr68His) whom had right cortical macrocephaly with pachygyria and subcortical dysplasia into the V-SVZ (Jansen et al., 2015). These results are supported by a study that generated a knock-in mutation of threonine 366 to alanine in PTEN which demonstrated prevalent neuron cytomegaly, enhanced dendrite arborization, and behavioral changes (Ledderose et al., 2022). Thus, a single nucleotide change in PTEN has extensive neural effects. Therefore, detecting and delineating de novo somatic variants that are pathogenic, but account for a small variant allele frequency, may represent key challenges. One should also note that there are extensive challenges to detecting technical vs. biological variants and short read, long read, and single cell sequencing can all influence the capacity to detect mutations. Nevertheless, the prevalence of focal abnormalities does hint at plausible somatic mutation events. Subtle manifestations include focal dysplasia seen in patients with Cowden Syndrome or BRRS. An examples is that a case of a one-year-old that had HME subject to hemispherectomy had an indel and a 2-base duplication (Koboldt et al., 2021). Taken together, both PTEN and PI3K generation of lipids must be carefully controlled and mutations in these genes can lead to altered cortical development.
The overlapping clinical and anatomical phenotypes that have been long recognized in neurodevelopmental disorders has hinted at convergent pathogenic mechanisms. The fact that variants frequently influence the mTORC1 pathway components is perhaps not surprising given the highly evolutionarily conserved functions. Modeling somatic mosaicism has allowed for developing mechanistic insight into brain development and has yielded enthusiasm about potential therapies that target the mTORC1 pathway. In some cases, these therapies have been brought to clinic. Challenges that remain include developing non-invasive techniques to identify variant allele frequency in patients. In addition, long read technology with higher fidelity must be developed. In many patients, no mutations have been identified. It is unclear as to the frequency that abnormal cells cease to survive through apoptosis or clearance by immune cells such as microglia. The large number of non-pathogenic mutations may also help mold brain development in more subtle ways. Therefore, future experiments are needed to develop robust and high throughput examination of how a single variant in a gene, multiple changes in the same gene, or multiple changes among multiple genes may help to mold cerebrocortical cytoarchitecture and neural function.
DF wrote the manuscript in its entirety. All authors contributed to the article and approved the submitted version.
DF is supported by United States of America Department of Defense U.S. Army Medical Research Activity Award Congressionally Directed Medical Research Program Tuberous Sclerosis Complex Research Program W81XWH2010447 and National Institutes of Health 5P20GM139769-02.
DF acknowledges Angelique Bordey for reagents and helpful comments.
The author DF declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alcantara D., Timms A. E., Gripp K., Baker L., Park K., Collins S., et al. (2017). Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain 140, 2610–2622. doi: 10.1093/brain/awx203
Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R. J., Reese C. B., et al. (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269. doi: 10.1016/s0960-9822(06)00122-9
Anderl S., Freeland M., Kwiatkowski D. J., Goto J. (2011). Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum. Mol. Genet. 20, 4597–4604. doi: 10.1093/hmg/ddr393
Auger K. R., Serunian L. A., Soltoff S. P., Libby P., Cantley L. C. (1989). PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57, 167–175. doi: 10.1016/0092-8674(89)90182-7
Aylett C. H. S., Sauer E., Imseng S., Boehringer D., Hall M. N., Ban N., et al. (2016). Architecture of human mTOR complex 1. Science 351, 48–52. doi: 10.1126/science.aaa3870
Backman S. A., Stambolic V., Suzuki A., Haight J., Elia A., Pretorius J., et al. (2001). Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 29, 396–403. doi: 10.1038/ng782
Baek S. T., Copeland B., Yun E. J., Kwon S. K., Guemez-Gamboa A., Schaffer A. E., et al. (2015). An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat. Med. 21, 1445–1454. doi: 10.1038/nm.3982
Baldassari S., Picard F., Verbeek N. E., van Kempen M., Brilstra E. H., Lesca G., et al. (2019a). The landscape of epilepsy-related GATOR1 variants. Genet. Med. 21, 398–408. doi: 10.1038/s41436-018-0060-2
Baldassari S., Ribierre T., Marsan E., Adle-Biassette H., Ferrand-Sorbets S., Bulteau C., et al. (2019b). Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 138, 885–900. doi: 10.1007/s00401-019-02061-5
Ballester R., Marchuk D., Boguski M., Saulino A., Letcher R., Wigler M., et al. (1990). The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63, 851–859. doi: 10.1016/0092-8674(90)90151-4
Bar-Peled L., Chantranupong L., Cherniack A. D., Chen W. W., Ottina K. A., Grabiner B. C., et al. (2013). A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. doi: 10.1126/science.1232044
Bar-Peled L., Schweitzer L. D., Zoncu R., Sabatini D. M. (2012). Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208. doi: 10.1016/j.cell.2012.07.032
Baulac S., Ishida S., Marsan E., Miquel C., Biraben A., Nguyen D. K., et al. (2015). Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol. 77, 675–683. doi: 10.1002/ana.24368
Beugnet A., Tee A. R., Taylor P. M., Proud C. G. (2003). Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem. J. 372, 555–566. doi: 10.1042/BJ20021266
Bizzotto S., Dou Y., Ganz J., Doan R. N., Kwon M., Bohrson C. L., et al. (2021). Landmarks of human embryonic development inscribed in somatic mutations. Science 371, 1249–1253. doi: 10.1126/science.abe1544
Bizzotto S., Walsh C. A. (2022). Genetic mosaicism in the human brain: from lineage tracing to neuropsychiatric disorders. Nat. Rev. Neurosci. 23, 275–286. doi: 10.1038/s41583-022-00572-x
Blair J. D., Hockemeyer D., Bateup H. S. (2018). Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 24, 1568–1578. doi: 10.1038/s41591-018-0139-y
Boer K., Jansen F., Nellist M., Redeker S., van den Ouweland A. M. W., Spliet W. G. M., et al. (2008). Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res. 78, 7–21. doi: 10.1016/j.eplepsyres.2007.10.002
Brown E. J., Albers M. W., Bum Shin T., Ichikawa K., Keith C. T., Lane W. S., et al. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758. doi: 10.1038/369756a0
Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M. (1998). RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U. S. A. 95, 1432–1437. doi: 10.1073/pnas.95.4.1432
Cai S. L., Tee A. R., Short J. D., Bergeron J. M., Kim J., Shen J., et al. (2006). Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 173, 279–289. doi: 10.1083/jcb.200507119
Canevini M. P., Kotulska-Jozwiak K., Curatolo P., La Briola F., Peron A., Słowińska M., et al. (2018). Current concepts on epilepsy management in tuberous sclerosis complex. Am. J. Med. Genet. Part C Semin. Med. Genet. 178 (3), 299–308. doi: 10.1002/ajmg.c.31652
Carson R. P., Van Nielen D. L., Winzenburger P. A., Ess K. C. (2012). Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 45, 369–380. doi: 10.1016/j.nbd.2011.08.024
Chantranupong L., Scaria S. M., Saxton R. A., Gygi M. P., Shen K., Wyant G. A., et al. (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164. doi: 10.1016/j.cell.2016.02.035
Chantranupong L., Wolfson R. L., Orozco J. M., Saxton R. A., Scaria S. M., Bar-Peled L., et al. (2014). The sestrins interact with gator2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 9, 1–8. doi: 10.1016/j.celrep.2014.09.014
Chen F., Rosiene J., Che A., Becker A., Loturco J. (2015). Tracking and transforming neocortical progenitors by crispr/cas9 gene targeting and piggybac transposase lineage labeling. Dev 142, 3601–3611. doi: 10.1242/dev.118836
Choi Y. J., Di Nardo A., Kramvis I., Meikle L., Kwiatkowski D. J., Sahin M., et al. (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495. doi: 10.1101/gad.1685008
Choo A. Y., Yoon S. O., Sang G. K., Roux P. P., Blenis J. (2008). Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U. S. A. 105, 17414–17419. doi: 10.1073/pnas.0809136105
Chow L. M. L., Endersby R., Zhu X., Rankin S., Qu C., Zhang J., et al. (2011). Cooperativity within and among Pten, p53, and Rb Pathways Induces High-Grade Astrocytoma in Adult Brain. Cancer Cell 19, 305–316. doi: 10.1016/j.ccr.2011.01.039
Chow D. K., Groszer M., Pribadi M., Machniki M., Carmichael S. T., Liu X., et al. (2009). Laminar and compartmental regulation of dendritic growth in mature cortex. Nat. Neurosci. 12, 116–118. doi: 10.1038/nn.2255
Chung J., Kuo C. J., Crabtree G. R., Blenis J. (1992). Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236. doi: 10.1016/0092-8674(92)90643-Q
Chung C., Yang X., Bae T., Vong K. I., Mittal S., Donkels C., et al. (2023). Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development. Nat. Genet. 55, 209–220. doi: 10.1038/s41588-022-01276-9
Chu-Shore C. J., Major P., Montenegro M., Thiele E. (2009). Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology 72, 1165–1169. doi: 10.1212/01.wnl.0000345365.92821.86
Conti V., Pantaleo M., Barba C., Baroni G., Mei D., Buccoliero A. M., et al. (2015). Focal dysplasia of the cerebral cortex and infantile spasms associated with somatic 1q21.1-q44 duplication including the AKT3 gene. Clin. Genet. 88, 241–247. doi: 10.1111/cge.12476
Cox R. L., de Anda F. C., Mangoubi T., Yoshii A. (2018). Multiple critical periods for rapamycin treatment to correct structural defects in Tsc-1-suppressed brain. Front. Mol. Neurosci 11, 409. doi: 10.3389/fnmol.2018.00409
Crino P. B., Aronica E., Baltuch G., Nathanson K. L. (2010). Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 74, 1716–1723. doi: 10.1212/WNL.0b013e3181e04325
Curatolo P., Franz D. N., Lawson J. A., Yapici Z., Ikeda H., Polster T., et al. (2018). Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc. Heal. 2, 495–504. doi: 10.1016/S2352-4642(18)30099-3
Dabora S. L., Kwiatkowski D. J., Franz D. N., Roberts P. S., Nieto A., Chung J., et al. (2001). Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80. doi: 10.1086/316951
Daniel P. M., Filiz G., Brown D. V., Christie M., Waring P. M., Zhang Y., et al. (2018). PI3K activation in neural stem cells drives tumorigenesis which can be ameliorated by targeting the cAMP response element binding protein. Neuro. Oncol. 20, 1344–1355. doi: 10.1093/neuonc/noy068
Dawson R. E., Nieto Guil A. F., Robertson L. J., Piltz S. G., Hughes J. N., Thomas P. Q. (2020). Functional screening of GATOR1 complex variants reveals a role for mTORC1 deregulation in FCD and focal epilepsy. Neurobiol. Dis. 134, 1–15. doi: 10.1016/j.nbd.2019.104640
De Fusco A., Cerullo M. S., Marte A., Michetti C., Romei A., Castroflorio E., et al. (2020). Acute knockdown of Depdc5 leads to synaptic defects in mTOR-related epileptogenesis. Neurobiol. Dis. 139, 104822. doi: 10.1016/j.nbd.2020.104822
De Ridder J., Verhelle B., Vervisch J., Lemmens K., Kotulska K., Moavero R., et al. (2021). Early epileptiform EEG activity in infants with tuberous sclerosis complex predicts epilepsy and neurodevelopmental outcomes. Epilepsia 62, 1208–1219. doi: 10.1111/epi.16892
Despouy E., Curot J., Denuelle M., Deudon M., Sol J. C., Lotterie J. A., et al. (2019). Neuronal spiking activity highlights a gradient of epileptogenicity in human tuberous sclerosis lesions. Clin. Neurophysiol. 130, 537–547. doi: 10.1016/j.clinph.2018.12.013
D’Gama A. M., Geng Y., Couto J. A., Martin B., Boyle E. A., Lacoursiere C. M., et al. (2015). Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann. Neurol. 77, 720–725. doi: 10.1002/ana.24357
D’Gama A. M., Woodworth M. B., Hossain A. A., Bizzotto S., Hatem N. E., LaCoursiere C. M., et al. (2017). Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 21, 3754–3766. doi: 10.1016/j.celrep.2017.11.106
Dibbens L. M., De Vries B., Donatello S., Heron S. E., Hodgson B. L., Chintawar S., et al. (2013). Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet. 45, 546–551. doi: 10.1038/ng.2599
Dibble C. C., Elis W., Menon S., Qin W., Klekota J., Asara J. M., et al. (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546. doi: 10.1016/j.molcel.2012.06.009
Doherty C., Goh S., Poussaint T. Y., Erdag N., Thiele E. A. (2005). Prognostic significance of tuber count and location in tuberous sclerosis complex. J. Child Neurol. 20, 837–841. doi: 10.1177/08830738050200101301
Dos D. S., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., et al. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302. doi: 10.1016/j.cub.2004.06.054
Downward J., Yarden Y., Mayes E., Scrace G., Totty N., Stockwell P., et al. (1984). Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 307, 521–527. doi: 10.1038/307521a0
Efeyan A., Zoncu R., Sabatini D. M. (2012). Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 18, 524–533. doi: 10.1016/j.molmed.2012.05.007
Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Eichmüller O. L., Corsini N. S., Vértesy Á., Morassut I., Scholl T., Gruber V. E., et al. (2022). Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 375, 1–10. doi: 10.1126/science.abf5546
Eker R., Mossige J. (1961). A dominant gene for renal adenomas in the rat. Nature 189, 858–859. doi: 10.1038/189858b0
Elia M., Amato C., Bottitta M., Grillo L., Calabrese G., Esposito M., et al. (2012). An atypical patient with Cowden syndrome and PTEN gene mutation presenting with cortical malformation and focal epilepsy. Brain. Dev. 34 (10), 873–876. doi: 10.1016/j.braindev.2012.03.005
Fallah A., Guyatt G. H., Snead O. C., Ebrahim S., Ibrahim G. M., Mansouri A., et al. (2013). Predictors of seizure outcomes in children with tuberous sclerosis complex and intractable epilepsy undergoing resective epilepsy surgery: An individual participant data meta-analysis. PloS One 8, 1–10. doi: 10.1371/journal.pone.0053565
Fallah A., Rodgers S. D., Weil A. G., Vadera S., Mansouri A., Connolly M. B., et al. (2015). Resective epilepsy surgery for tuberous sclerosis in children: determining predictors of seizure outcomes in a multicenter retrospective cohort study. Neurosurgery 77, 517–524. doi: 10.1227/NEU.0000000000000875
Feliciano D. M. (2020). The neurodevelopmental pathogenesis of tuberous sclerosis complex (TSC). Front. Neuroanat. 14, 227–238. doi: 10.3389/fnana.2020.00039
Feliciano D. M., Lin T. V., Hartman N. W., Bartley C. M., Kubera C., Hsieh L., et al. (2013). A circuitry and biochemical basis for tuberous sclerosis symptoms: From epilepsy to neurocognitive deficits. Int. J. Dev. Neurosci. 31, 667–678. doi: 10.1016/j.ijdevneu.2013.02.008
Feliciano D. M., Su T., Lopez J., Platel J. C., Bordey A. (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest. 121, 1596–1607. doi: 10.1172/JCI44909
Ferrer I., Fabregues I., Coll J., Ribalta T., Rives A. (1984). Tuberous sclerosis: A Golgi study of cortical tuber. Clin. Neuropathol. 3, 47–51.
Franz D. N., Leonard J., Tudor C., Chuck G., Care M., Sethuraman G., et al. (2006). Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol. 59, 490–498. doi: 10.1002/ana.20784
Frykberg L., Palmieri S., Beug H., Graf T., Hayman M. J., Vennström B. (1983). Transforming capacities of avian erythroblastosis virus mutants deleted in the erbA or erbB oncogenes. Cell 32, 1070–1079. doi: 10.1016/0092-8674(83)90513-5
Gallagher A., Madan N., Stemmer-Rachamimov A., Thiele E. A. (2010). Progressive calcified tuber in a young male with tuberous sclerosis complex. Dev. Med. Child Neurol. 52, 1062–1065. doi: 10.1111/j.1469-8749.2010.03792.x
Ganley I. G., Lam D. H., Wang J., Ding X., Chen S., Jiang X. (2009). ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi: 10.1074/jbc.M900573200
Garami A., Zwartkruis F. J. T., Nobukuni T., Joaquin M., Roccio M., Stocker H., et al. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. doi: 10.1016/S1097-2765(03)00220-X
Gingras A. C., Gygi S. P., Raught B., Polakiewicz R. D., Abraham R. T., Hoekstra M. F., et al. (1999). Regulation of 4E-BP1 phosphorylation: A novel two step mechanism. Genes Dev. 13, 1422–1437. doi: 10.1101/gad.13.11.1422
Goto J., Talos D. M., Klein P., Qin W., Chekaluk Y. I., Anderl S., et al. (2011). Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc. Natl. Acad. Sci. U. S. A. 108, 1874–1886. doi: 10.1073/pnas.1106454108
Gregorian C., Nakashima J., Le Belle J., Ohab J., Kim R., Liu A., et al. (2009). Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J. Neurosci. 29, 1874–1886. doi: 10.1523/JNEUROSCI.3095-08.2009
Groszer M., Erickson R., Scripture-Adams D. D., Dougherty J. D., Le Belle J., Zack J. A., et al. (2006). PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. U. S. A. 103, 111–6. doi: 10.1073/pnas.0509939103
Groszer M., Erickson R., Scripture-Adams D. D., Lesche R., Trumpp A., Zack J. A., et al. (2001). Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186–9. doi: 10.1126/science.1065518
Hanai S., Sukigara S., Dai H., Owa T., Horike S., Otsuki T., et al. (2017). Pathologic active mTOR mutation in brain malformation with intractable epilepsy leads to cell-autonomous migration delay. Am. J. Pathol. 187, 1177–1185. doi: 10.1016/j.ajpath.2017.01.015
Happle R. (1987). Lethal genes surviving by mosaicism: A possible explanation for sporadic birth defects involving the skin. J. Am. Acad. Dermatol. 16, 899–906. doi: 10.1016/S0190-9622(87)80249-9
Hara K., Maruki Y., Long X., Yoshino K., Oshiro N., Hidayat S., et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. doi: 10.1016/S0092-8674(02)00833-4
Hara K., Yonezawa K., Weng Q. P., Kozlowski M. T., Belham C., Avruch J. (1998). Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273, 14484–14494. doi: 10.1074/jbc.273.23.14484
Hasbani D. M., Crino P. B. (2018). Tuberous sclerosis complex. Handb Clin Neurol 148, 813–822. doi: 10.1016/B978-0-444-64076-5.00052-1
Hernandez O., Way S., McKenna J., Gambello M. J. (2007). Generation of a conditional disruption of the Tsc2 gene. Genesis 45, 101–106. doi: 10.1002/dvg.20271
Hino O., Klein-Szanto A. J. P., Freed J. J., Testa J. R., Brown D. Q., Vilensky M., et al. (1993). Spontaneous and radiation-induced renal tumors in the Eker rat model of dominantly inherited cancer. Proc. Natl. Acad. Sci. U. S. A. 90, 327–331. doi: 10.1073/pnas.90.1.327
Hsia H. E., Kumar R., Luca R., Takeda M., Courchet J., Nakashima J., et al. (2014). Ubiquitin E3 ligase Nedd4-1 acts as a downstream target of PI3K/PTEN-mTORC1 signaling to promote neurite growth. Proc. Natl. Acad. Sci. U. S. A. 111, 13205–13210. doi: 10.1073/pnas.1400737111
Hsieh L. S., Wen J. H., Claycomb K., Huang Y., Harrsch F. A., Naegele J. R., et al. (2016). Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat. Commun. 7, 140–146. doi: 10.1038/ncomms11753
Hsieh L. S., Wen J. H., Nguyen L. H., Zhang L., Getz S. A., Torres-Reveron J., et al. (2020). Ectopic HCN4 expression drives mTOR-dependent epilepsy in mice. Sci Transl Med. 12(570), eabc1492. doi: 10.1126/scitranslmed.abc149
Hu S., Knowlton R. C., Watson B. O., Glanowska K. M., Murphy G. G., Parent J. M., et al. (2018). Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol. 84, 140–146. doi: 10.1002/ana.25272
Hughes J., Dawson R., Tea M., McAninch D., Piltz S., Jackson D., et al. (2017). Knockout of the epilepsy gene Depdc5 in mice causes severe embryonic dysmorphology with hyperactivity of mTORC1 signalling. Sci. Rep. 7, 1–15. doi: 10.1038/s41598-017-12574-2
Hulshof H. M., Kuijf H. J., Kotulska K., Curatolo P., Weschke B., Riney K., et al. (2022). Association of early MRI characteristics with subsequent epilepsy and neurodevelopmental outcomes in children with tuberous sclerosis complex. Neurology 98, e1216–e1225. doi: 10.1212/WNL.0000000000200027
Huttenlocher P. R., Heydemann P. T. (1984). Fine structure of cortical tubers in tuberous sclerosis: A Golgi study. Ann. Neurol. 16, 595–602. doi: 10.1002/ana.410160511
Iffland P. H., Everett M. E., Cobb-Pitstick K. M., Bowser L. E., Barnes A. E., Babus J. K., et al. (2022). NPRL3 loss alters neuronal morphology, mTOR localization, cortical lamination and seizure threshold. Brain. 145 (11), 3872–3885. doi: 10.1093/brain/awac044
Inoki K., Li Y., Xu T., Guan K. L. (2003). Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834. doi: 10.1101/gad.1110003
Ishida S., Picard F., Rudolf G., Noé E., Achaz G., Thomas P., et al. (2013). Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat. Genet. 45, 552–555. doi: 10.1038/ng.2601
Ishida S., Zhao D., Sawada Y., Hiraoka Y., Mashimo T., Tanaka K. (2022). Dorsal telencephalon-specific Nprl2- A nd Nprl3-knockout mice: Novel mouse models for GATORopathy. Hum. Mol. Genet. 31, 1519–1530. doi: 10.1093/hmg/ddab337
Jansen L. A., Mirzaa G. M., Ishak G. E., O’Roak B. J., Hiatt J. B., Roden W. H., et al. (2015). PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138, 1613–1628. doi: 10.1093/brain/awv045
Jha R., Kurup A., Kovilapu U. B., Ranjan R., Sondhi V. (2022). Somatic mutations involving TSC 1 and TSC2 genes in two children with focal cortical dysplasia. Brain Dev. 44, 166–172. doi: 10.1016/j.braindev.2021.10.002
Jung C. H., Jun C. B., Ro S. H., Kim Y. M., Otto N. M., Cao J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. doi: 10.1091/mbc.E08-12-1249
Kaczorowska M., Jurkiewicz E., Domańska-Pakiěa D., Syczewska M., Łojszczyk B., Chmielewski D., et al. (2011). Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia 52, 22–27. doi: 10.1111/j.1528-1167.2010.02892.x
Kang S. A., Pacold M. E., Cervantes C. L., Lim D., Lou H. J., Ottina K., et al. (2013). mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 364–373. doi: 10.1126/science.1236566
Kaplan D. R., Whitman M., Schaffhausen B., Raptis L., Garcea R. L., Pallas D., et al. (1986). Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc. Natl. Acad. Sci. U. S. A. 83, 3624–3628. doi: 10.1073/pnas.83.11.3624
Karczewski K. J., Francioli L. C., Tiao G., Cummings B. B., Alföldi J., Wang Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–444. doi: 10.1038/s41586-020-2308-7
Kassai H., Sugaya Y., Noda S., Nakao K., Maeda T., Kano M., et al. (2014). Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 7, 1626–1639. doi: 10.1016/j.celrep.2014.04.048
Kazazian H. H., Wong C., Youssoufian H., Scott A. F., Phillips D. G., Antonarakis S. E. (1988). Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332, 164–166. doi: 10.1038/332164a0
Kelley S. A., Knupp K. G. (2018). Infantile spasms—Have we made progress? Curr. Neurol. Neurosci. Rep. 18, 1–8. doi: 10.1007/s11910-018-0832-8
Kim J. K., Cho J., Kim S. H., Kang H. C., Kim D. S., Kim V. N., et al. (2019). Brain somatic mutations in MTOR reveal translational dysregulations underlying intractable focal epilepsy. J. Clin. Invest. 129, 4207–4223. doi: 10.1172/JCI127032
Kim J., Kundu M., Viollet B., Guan K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. doi: 10.1016/S0092-8674(02)00808-5
Kimball S. R., Gordon B. S., Moyer J. E., Dennis M. D., Jefferson L. S. (2016). Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell. Signal. 28, 896–906. doi: 10.1016/j.cellsig.2016.03.008
Kingswood J. C., Bruzzi P., Curatolo P., de Vries P. J., Fladrowski C., Hertzberg C., et al. (2014). TOSCA - first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet J. Rare Dis. 9, 182. doi: 10.1186/s13023-014-0182-9
Kingswood J. C., D’Augères G. B., Belousova E., Ferreira J. C., Carter T., Castellana R., et al. (2017). TuberOus SClerosis registry to increase disease Awareness (TOSCA) - Baseline data on 2093 patients. Orphanet J. Rare Dis. 12, 1–13. doi: 10.1186/s13023-016-0553-5
Klonowska K., Giannikou K., Grevelink J. M., Boeszoermenyi B., Thorner A. R., Herbert Z. T., et al. (2023). Comprehensive genetic and phenotype analysis of 95 individuals with mosaic tuberous sclerosis complex. Am. J. Hum. Genet. 110 (6), 979–988. doi: 10.1016/j.ajhg.2023.04.002
Knudson A. G. (1971). Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A. 68, 820–823. doi: 10.1073/pnas.68.4.820
Kobayashi T., Minowa O., Sugitani Y., Takai S., Mitani H., Kobayashi E., et al. (2001). A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. U. S. A. 98, 8762–8767. doi: 10.1073/pnas.151033798
Koboldt D. C., Miller K. E., Miller A. R., Bush J. M., McGrath S., Leraas K., et al. (2021). PTEN somatic mutations contribute to spectrum of cerebral overgrowth. Brain 144, 2971–2978. doi: 10.1093/brain/awab173
Korenke G. C., Eggert M., Thiele H., Nürnberg P., Sander T., Steinlein O. K. (2016). Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia 57, e60–e63. doi: 10.1111/epi.13307
Krueger D. A., Care M. M., Agricola K., Tudor C., Mays M., Franz D. N. (2013a). Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology 80, 574–580. doi: 10.1212/WNL.0b013e3182815428
Krueger D. A., Northrup H., Krueger D. A., Roberds S., Smith K., Sampson J., et al. (2013b). Tuberous sclerosis complex surveillance and management: Recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr. Neurol. 49, 255–265. doi: 10.1016/j.pediatrneurol.2013.08.002
Krueger D. A., Wilfong A. A., Holland-Bouley K., Anderson A. E., Agricola K., Tudor C., et al. (2013c). Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann. Neurol. 74, 679–687. doi: 10.1002/ana.23960
Kubo Y., Klimek F., Kikuchi Y., Bannasch P., Hino O. (1995). Early detection of knudson’s two-hits in preneoplastic renal cells of the eker rat model by the laser microdissection procedure. Cancer Res. 55, 989–990.
Kwiatkowski D. J. (2002). A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11, 525–534. doi: 10.1093/hmg/11.5.525
Kwon C. H., Luikart B. W., Powell C. M., Zhou J., Matheny S. A., Zhang W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Kwon C. H., Zhu X., Zhang J., Knoop L. L., Tharp R., Smeyne R. J., et al. (2001). Pten regulates neuronal soma size: A mouse model of Lhermitte-Duclos disease. Nat. Genet. 29, 404–11. doi: 10.1038/ng781
Lamming D. W., Ye L., Katajisto P., Goncalves M. D., Saitoh M., Stevens D. M., et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. doi: 10.1126/science.1215135
Laplante M., Sabatini D. M. (2012). mTOR signaling. Cold Spring Harb. Perspect. Biol. 4, 1–3. doi: 10.1101/cshperspect.a011593
Ledderose J. M. T., Benitez J. A., Roberts A. J., Reed R., Bintig W., Larkum M. E., et al. (2022). The impact of phosphorylated PTEN at threonine 366 on cortical connectivity and behaviour. Brain 145, 3608–3621. doi: 10.1093/brain/awac188
Lee W. S., Baldassari S., Chipaux M., Adle-Biassette H., Stephenson S. E. M., Maixner W., et al. (2021). Gradient of brain mosaic RHEB variants causes a continuum of cortical dysplasia. Ann. Clin. Transl. Neurol. 8, 485–490. doi: 10.1002/acn3.51286
Lee J. H., Huynh M., Silhavy J. L., Kim S., Dixon-Salazar T., Heiberg A., et al. (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 44, 941–945. doi: 10.1038/ng.2329
Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. doi: 10.1126/science.275.5308.1943
Li Y., Muffat J., Omer A., Bosch I., Lancaster M. A., Sur M., et al (2017). Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell. 20 (3), 385–396.e3. doi: 10.1016/j.stem.2016.11.017
Lim J. S., Gopalappa R., Kim S. H., Ramakrishna S., Lee M., Kim W., et al. (2017). Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am. J. Hum. Genet. 100, 454–472. doi: 10.1016/j.ajhg.2017.01.030
Lim J. S., Kim W., Kang H. C., Kim S. H., Park A. H., Park E. K., et al. (2015). Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 21, 395–400. doi: 10.1038/nm.3824
Lin T. V., Hsieh L., Kimura T., Malone T. J., Bordey A. (2016). Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc. Natl. Acad. Sci. U. S. A. 113, 11330–11335. doi: 10.1073/pnas.1605740113
Lindhurst M. J., Sapp J. C., Teer J. K., Johnston J. J., Finn E. M., Peters K., et al. (2011). A mosaic activating mutation in AKT1 associated with the proteus syndrome. N. Engl. J. Med. 365, 611–619. doi: 10.1056/nejmoa1104017
Llaguno S. A., Chen J., Kwon C. H., Parada L. F. (2008). Neural and cancer stem cells in tumor suppressor mouse models of malignant astrocytoma. Cold Spring Harb Symp Quant Biol. 73, 421–6. doi: 10.1101/sqb.2008.73.005
Lux A. L., Osborne J. P. (2004). A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: Consensus statement of the West Delphi Group. Epilepsia 45, 1416–1428. doi: 10.1111/j.0013-9580.2004.02404.x
Macara I. G., Marinetti G. V., Balduzzi P. C. (1984). Transforming protein of avian sarcoma virus UR2 is associated with phosphatidylinositol kinase activity: Possible role in tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 81, 2728–2732. doi: 10.1073/pnas.81.9.2728
Magri L., Cambiaghi M., Cominelli M., Alfaro-Cervello C., Cursi M., Pala M., et al. (2011). Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462. doi: 10.1016/j.stem.2011.09.008
Marsan E., Ishida S., Schramm A., Weckhuysen S., Muraca G., Lecas S., et al. (2016). Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol. Dis. 89, 180–189. doi: 10.1016/j.nbd.2016.02.010
McCLINTOCK B. (1950). The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. U. S. A. 36, 344–55. doi: 10.1073/pnas.36.6.344
Meikle L., Pollizzi K., Egnor A., Kramvis I., Lane H., Sahin M., et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: Effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008
Meikle L., Talos D. M., Onda H., Pollizzi K., Rotenberg A., Sahin M., et al. (2007). A mouse model of tuberous sclerosis: Neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007
Mietzsch U., McKenna J., Reith R. M., Way S. W., Gambello M. J. (2013). Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J. Comp. Neurol. 521, 3817–3831. doi: 10.1002/cne.23380
Mirzaa G. M., Campbell C. D., Solovieff N., Goold C. P., Jansen L. A., Menon S., et al. (2016). Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 73 (7), 836–845. doi: 10.1001/jamaneurol.2016.0363
Mirzaa G. M., Conti V., Timms A. E., Smyser C. D., Ahmed S., Carter M., et al. (2015). Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: A next-generation sequencing study. Lancet Neurol. 14, 1182–95. doi: 10.1016/S1474-4422(15)00278-1
Mizuguchi M., Takashima S. (2001). Neuropathology of tuberous sclerosis. Brain Dev. 23, 508–515. doi: 10.1016/S0387-7604(01)00304-7
Mizuguchi M., Yamanouchi H., Becker L. E., Itoh M., Takashima S. (2002). Doublecortin immunoreactivity in giant cells of tuberous sclerosis and focal cortical dysplasia. Acta Neuropathol. 104, 418–424. doi: 10.1007/s00401-002-0575-z
Mohamed A. R., Bailey C. A., Freeman J. L., Maixner W., Jackson G. D., Harvey A. S. (2012). Intrinsic epileptogenicity of cortical tubers revealed by intracranial EEG monitoring. Neurology 79, 2249–2257. doi: 10.1212/WNL.0b013e3182768923
Moon U. Y., Park J. Y., Park R., Cho J. Y., Hughes L. J., McKenna J., et al. (2015). Impaired Reelin-Dab1 signaling contributes to neuronal migration deficits of tuberous sclerosis complex. Cell Rep. 12, 965–978. doi: 10.1016/j.celrep.2015.07.013
Mühlebner A., Van Scheppingen J., Hulshof H. M., Scholl T., Iyer A. M., Anink J. J., et al. (2016). Novel histopathological patterns in cortical tubers of epilepsy surgery patients with tuberous sclerosis complex. PloS One 11, 1–15. doi: 10.1371/journal.pone.0157396
Nabbout R., Belousova E., Benedik M. P., Carter T., Cottin V., Curatolo P., et al. (2019). Epilepsy in tuberous sclerosis complex: Findings from the TOSCA Study. Epilepsia Open 4, 73–84. doi: 10.1002/epi4.12286
Nabbout R., Belousova E., Benedik M. P., Carter T., Cottin V., Curatolo P., et al. (2021). Historical patterns of diagnosis, treatments, and outcome of epilepsy associated with tuberous sclerosis complex: results from TOSCA registry. Front. Neurol. 12, 396–398. doi: 10.3389/fneur.2021.697467
Nakamura K., Kato M., Tohyama J., Shiohama T., Hayasaka K., Nishiyama K., et al. (2014). AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin. Genet. 85, 396–398. doi: 10.1111/cge.12188
Nguyen L. H., Mahadeo T., Bordey A. (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci. 39, 2762–2773. doi: 10.1523/JNEUROSCI.2260-18.2019
Nigro J. M., Baker S. J., Preisinger A. C., Jessup J. M., Hosteller R., Cleary K., et al. (1989). Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708. doi: 10.1038/342705a0
Noda T., Ohsumi Y. (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273, 3963–3966. doi: 10.1074/jbc.273.7.3963
Onda H., Crino P. B., Zhang H., Murphey R. D., Rastelli L., Rothberg B. E. G., et al. (2002). Tsc2 null murine neuroepithelial cells are a model for human tuber giant cells, and show activation of an mTOR pathway. Mol. Cell. Neurosci. 21, 561–574. doi: 10.1006/mcne.2002.1184
Onda H., Lueck A., Marks P. W., Warren H. B., Kwiatkowski D. J. (1999). Tsc2(+/-) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J. Clin. Invest. 104, 687–695. doi: 10.1172/JCI7319
Onori M. P., Koene L. M. C., Schafer C. B., Nellist M., De Brito Van Velze M., Gao Z., et al. (2021). RHEB/mTOR hyperactivity causes cortical malformations and epileptic seizures through increased axonal connectivity. PloS Biol. 19, 1–34. doi: 10.1371/journal.pbio.3001279
Owada Y., Utsunomiya A., Yoshimoto T., Kondo H. (1997). Expression of mRNA for Akt, Serine-Threonine Protein Kinase, in the Brain during Development and Its Transient Enhancement Following Axotomy of Hypoglossal Nerve. J. Mol. Neurosci. 9, 27–33. doi: 10.1007/BF02789392
Park S. M., Lim J. S., Ramakrishina S., Kim S. H., Kim W. K., Lee J., et al. (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99, 83–97.e7. doi: 10.1016/j.neuron.2018.05.039
Parmigiani A., Nourbakhsh A., Ding B., Wang W., Kim Y. C., Akopiants K., et al. (2014). Sestrins Inhibit mTORC1 Kinase Activation through the GATOR Complex. Cell Rep. 9, 1281–1291. doi: 10.1016/j.celrep.2014.10.019
Parsons R. (2020). Discovery of the pten tumor suppressor and its connection to the pi3k and akt oncogenes. Cold Spring Harb. Perspect. Med. 10, 1–11. doi: 10.1101/cshperspect.a036129
Pelorosso C., Watrin F., Conti V., Buhler E., Gelot A., Yang X., et al. (2019). Somatic double-hit in MTOR and RPS6 in hemimegalencephaly with intractable epilepsy. Hum. Mol. Genet. 28 (22), 3755–3765. doi: 10.1093/hmg/ddz194
Poduri A., Evrony G. D., Cai X., Elhosary P. C., Beroukhim R., Lehtinen M. K., et al. (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. doi: 10.1016/j.neuron.2012.03.010
Price D. J., Grove J. R., Calvo V., Avruch J., Bierer B. E. (1992). Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 257, 973–977. doi: 10.1126/science.1380182
Qin W., Chan J. A., Vinters H. V., Mathern G. W., Franz D. N., Taillon B. E., et al. (2010a). Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 20, 1096–1105. doi: 10.1111/j.1750-3639.2010.00416.x
Qin W., Kozlowski P., Taillon B. E., Bouffard P., Holmes A. J., Janne P., et al. (2010b). Ultra deep sequencing detects a low rate of mosaic mutations in tuberous sclerosis complex. Hum. Genet. 127, 573–582. doi: 10.1007/s00439-010-0801-z
Reijnders M. R. F., Kousi M., Van Woerden G. M., Klein M., Bralten J., Mancini G. M. S., et al. (2017). Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 8, 1052. doi: 10.1038/s41467-017-00933-6
Rennebeck G., Kleymenova E. V., Anderson R., Yeung R. S., Artzt K., Walker C. L. (1998). Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc. Natl. Acad. Sci. U. S. A. 95, 15629–15634. doi: 10.1073/pnas.95.26.15629
Ribierre T., Deleuze C., Bacq A., Baldassari S., Marsan E., Chipaux M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest. 128, 2452–2458. doi: 10.1172/JCI99384
Rivière J. B., Mirzaa G. M., O'Roak B. J., Beddaoui M., Alcantara D., Conway R. L., et al. (2012). De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 44 (8), 934–940. doi: 10.1038/ng.2331
Ricos M. G., Hodgson B. L., Pippucci T., Saidin A., Ong Y. S., Heron S. E., et al. (2016). Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann. Neurol. 79, 120–131. doi: 10.1002/ana.24547
Robens B. K., Grote A., Pitsch J., Schoch S., Cardoso C., Becker A. J. (2016). Minute amounts of hamartin wildtype rescue the emergence of tuber-like lesions in conditional Tsc1 ablated mice. Neurobiol. Dis. 95, 134–44. doi: 10.1016/j.nbd.2016.07.006
Ruderman N. B., Kapeller R., White M. F., Cantley L. C. (1990). Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl. Acad. Sci. U. S. A. 87, 1411–5. doi: 10.1073/pnas.87.4.1411
Sabatini D. M., Erdjument-Bromage H., Lui M., Tempst P., Snyder S. H. (1994). RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43. doi: 10.1016/0092-8674(94)90570-3
Sabers C. J., Martin M. M., Brunn G. J., Williams J. M., Dumont F. J., Wiederrecht G., et al. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270, 815–822. doi: 10.1074/jbc.270.2.815
Salinas V., Vega P., Piccirilli M. V., Chicco C., Ciraolo C., Christiansen S., et al. (2019). Identification of a somatic mutation in the RHEB gene through high depth and ultra-high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur. J. Med. Genet. 62, 1–5. doi: 10.1016/j.ejmg.2018.11.005
Sampson J. R., Maheshwar M. M., Aspinwall R., Thompson P., Cheadle J. P., Ravine D., et al (1997). Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet 61 (4), 843–51. doi: 10.1086/514888
Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010). Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. doi: 10.1016/j.cell.2010.02.024
Sarbassov D. D., Ali S. M., Sengupta S., Sheen J. H., Hsu P. P., Bagley A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Saxton R. A., Sabatini D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Scheffer I. E., Heron S. E., Regan B. M., Mandelstam S., Crompton D. E., Hodgson B. L., et al. (2014). Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann. Neurol. 75, 782–787. doi: 10.1002/ana.24126
Shi X., Lim Y., Myers A. K., Stallings B. L., Mccoy A., Zeiger J., et al. (2020). PIK3R2/pik3r2 activating mutations result in brain overgrowth and EEG changes. Ann. Neurol. 88, 1077–1094. doi: 10.1002/ana.25890
Sim J. C., Scerri T., Fanjul-Fernández M., Riseley J. R., Gillies G., Pope K., et al. (2016). Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann. Neurol. 79, 132–137. doi: 10.1002/ana.24502
Sinor A. D., Lillien L. (2004). Akt-1 expression level regulates CNS precursors. J. Neurosci. 24, 8531–8541. doi: 10.1523/JNEUROSCI.1470-04.2004
Słowińska M., Jóźwiak S., Peron A., Borkowska J., Chmielewski D., Sadowski K., et al. (2018). Early diagnosis of tuberous sclerosis complex: A race against time. How to make the diagnosis before seizures? Orphanet J. Rare Dis. 13, 1–10. doi: 10.1186/s13023-018-0764-z
Śmiałek D., Kotulska K., Duda A., Jóźwiak S. (2023). Effect of mTOR inhibitors in epilepsy treatment in children with tuberous sclerosis complex under 2 years of age. Neurol. Ther. 12, 931–946. doi: 10.1007/s40120-023-00476-7
Sokolov A. M., Seluzicki C. M., Morton M. C., Feliciano D. M. (2018). Dendrite growth and the effect of ectopic Rheb expression on cortical neurons. Neurosci. Lett. 671, 140–147. doi: 10.1016/j.neulet.2018.02.021
Sugimoto Y., Whitman M., Cantley L. C., Erikson R. L. (1984). Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglyercol. Proc. Natl. Acad. Sci. U. S. A. 81, 1–5. doi: 10.1073/pnas.81.7.2117
Tang G., Gudsnuk K., Kuo S. H., Cotrina M. L., Rosoklija G., Sosunov A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tarkowski B., Kuchcinska K., Blazejczyk M., Jaworski J. (2019). Pathological mTOR mutations impact cortical development. Hum. Mol. Genet. 28, 1098–1101. doi: 10.1093/hmg/ddz042
Tee A. R., Manning B. D., Roux P. P., Cantley L. C., Blenis J. (2003). Tuberous Sclerosis Complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. doi: 10.1016/S0960-9822(03)00506-2
The European Chromosome 16 Tuberous Sclerosis Consortium (1993). Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315. doi: 10.1016/0092-8674(93)90618-Z
Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M. (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. doi: 10.1038/nature11083
Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., et al. (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032. doi: 10.1074/jbc.M900301200
Tsai V., Parker W. E., Orlova K. A., Baybis M., Chi A. W. S., Berg B. D., et al. (2014). Fetal brain mTOR signaling activation in tuberous sclerosis complex. Cereb. Cortex 24, 315–327. doi: 10.1093/cercor/bhs310
Tyburczy M. E., Dies K. A., Glass J., Camposano S., Chekaluk Y., Thorner A. R., et al. (2015). Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PloS Genet. 11, 1–17. doi: 10.1371/journal.pgen.1005637
Van Slegtenhorst M., De Hoogt R., Hermans C., Nellist M., Janssen B., Verhoef S., et al. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. doi: 10.1126/science.277.5327.805
Varmus H. E., Weiss R. A., Friis R. R., Levinson W., Bishop J. M. (1972). Detection of avian tumor virus-specific nucleotide sequences in avian cell DNAs. Proc. Natl. Acad. Sci. 69, 20–4. doi: 10.1073/pnas.69.1.20
Verhoef S., Bakker L., Tempelaars A. M. P., Hesseling-Janssen A. L. W., Mazurczak T., Jozwiak S., et al. (1999). High rate of mosaicism in tuberous sclerosis complex. Am. J. Hum. Genet. 64, 1632–1637. doi: 10.1086/302412
Verhoef S., Vrtel R., van Essen T., Bakker L., Sikkens E., Halley D., et al. (1995). Somatic mosaicism and clinical variation in tuberous sclerosis complex. Lancet 345 (8943), 202. doi: 10.1016/S0140-6736(95)90213-9
Vézina C., Kudelski A., Sehgal S. N. (1975). Rapamycin (AY-22, 989) a new antifungal antibiotic. J. Antibiot. (Tokyo). 28, 721–726. doi: 10.7164/antibiotics.28.721
Wang Y., Greenwood J. S. F., Calcagnotto M. E., Kirsch H. E., Barbaro N. M., Baraban S. C. (2007). Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann. Neurol. 61 (2), 139–52. doi: 10.1002/ana.21058
Way S. W., Mckenna J., Mietzsch U., Reith R. M., Wu H. C. J., Gambello M. J. (2009). Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum. Mol. Genet. 18, 1252–1265. doi: 10.1093/hmg/ddp025
Whitman M., Downes C. P., Keeler M., Keller T., Cantley L. (1988). Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332, 644–6466. doi: 10.1038/332644a0
Whitman M., Kaplan D. R., Schaffhausen B., Cantley L., Roberts T. M. (1985). Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315, 239–42. doi: 10.1038/315239a0
Winden K. D., Sundberg M., Yang C., Wafa S. M. A., Dwyer S., Chen P. F., et al. (2019). Biallelic mutations in TSC2 lead to abnormalities associated with cortical tubers in human ipsc-derived neurons. J. Neurosci. 39, 9294–9305. doi: 10.1523/JNEUROSCI.0642-19.2019
Wolfson R. L., Chantranupong L., Saxton R. A., Shen K., Scaria S. M., Cantor J. R., et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. doi: 10.1126/science.aab2674
Wong M. (2012). A tuber-ful animal model of tuberous sclerosis at last? Epilepsy Curr. 12, 15–16. doi: 10.5698/1535-7511-12.1.15
Yamagata K., Sanders L. K., Kaufmann W. E., Yee W., Barnes C. A., Nathans D., et al. (1994). rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel ras-related protein. J. Biol. Chem. 269, 16333–1639. doi: 10.1016/s0021-9258(17)34012-7
Yamanouchi H., Ho M., Jay V., Becker L. E. (1997a). Giant cells in cortical tubers in tuberous sclerosis showing synaptophysin-immunoreactive halos. Brain Dev. 19, 21–24. doi: 10.1016/S0387-7604(96)00079-4
Yamanouchi H., Jay V., Rutka J. T., Takashima S., Becker L. E. (1997b). Evidence of abnormal differentiation in giant cells of tuberous sclerosis. Pediatr. Neurol. 17, 49–53. doi: 10.1016/S0887-8994(97)00036-2
Yan L., Findlay G. M., Jones R., Procter J., Cao Y., Lamb R. F. (2006). Hyperactivation of mammalian target of rapamycin (mTOR) signaling by a gain-of-function mutant of the Rheb GTPase. J. Biol. Chem. 281, 19793–19797. doi: 10.1074/jbc.C600028200
Yeung R. S., Xiao G. H., Jin F., Lee W. C., Testa J. R., Knudson A. G. (1994). Predisposition to renal carcinoma in the Eker rat is determined by germ- line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc. Natl. Acad. Sci. U. S. A. 91, 11413–11416. doi: 10.1073/pnas.91.24.11413
Yip C. K., Murata K., Walz T., Sabatini D. M., Kang S. A. (2010). Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38, 768–774. doi: 10.1016/j.molcel.2010.05.017
Yuskaitis C. J., Jones B. M., Wolfson R. L., Super C. E., Dhamne S. C., Rotenberg A., et al. (2018). A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 111, 91–101. doi: 10.1016/j.nbd.2017.12.010
Yuskaitis C. J., Rossitto L. A., Gurnani S., Bainbridge E., Poduri A., Sahin M. (2019). Chronic mTORC1 inhibition rescues behavioral and biochemical deficits resulting from neuronal Depdc5 loss in mice. Hum. Mol. Genet. 28, 2952–2964. doi: 10.1093/hmg/ddz123
Zhang Y., Gao X., Saucedo L. J., Ru B., Edgar B. A., Pan D. (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578–581. doi: 10.1038/ncb999
Zhang Y., Manning B. D. (2015). mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 14, 2011–2017. doi: 10.1080/15384101.2015.1044188
Zhang M. N., Zou L. P., Wang Y. Y., Pang L. Y., Ma S. F., Huang L. L., et al. (2018). Calcification in cerebral parenchyma affects pharmacoresistant epilepsy in tuberous sclerosis. Seizure 60, 86–90. doi: 10.1016/j.seizure.2018.06.011
Zhao S., Li Z., Zhang M., Zhang L., Zheng H., Ning J., et al. (2019). A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp. Mol. Med. 51, 1–11. doi: 10.1038/s12276-019-0277-4
Zheng H., Ying H., Yan H., Kimmelman A. C., Hiller D. J., Chen A. J., et al. (2008). p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133. doi: 10.1038/nature07443
Zhou X. P., Marsh D. J., Morrison C. D., Chaudhury A. R., Maxwell M., Reifenberger G., et al. (2003). Germline inactivation of PTEN and dysregulation of the phosphoinositol-3- kinase/akt pathway cause human lhermitte-duclos disease in adults. Am. J. Hum. Genet. 73, 1191–1198. doi: 10.1086/379382
Keywords: somatic mosaicism, focal cortical dysplasia, tuberous sclerosis complex, mTORC1, malformations of cortical development
Citation: Feliciano DM (2023) Modeling genetic mosaicism of the mammalian target of rapamycin pathway in the cerebral cortex. Front. Mamm. Sci. 2:1231778. doi: 10.3389/fmamm.2023.1231778
Received: 30 May 2023; Accepted: 11 July 2023;
Published: 10 August 2023.
Edited by:
Ruth Benavides-Piccione, Spanish National Research Council (CSIC), SpainReviewed by:
Carlos Lorenzo, University of Rovira i Virgili, SpainCopyright © 2023 Feliciano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Feliciano, ZGZlbGljaUBjbGVtc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.