 Suman Tamang
Suman Tamang Aditi Arya1
Aditi Arya1 Vineeta Singh
Vineeta Singh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Malar., 24 February 2025
Sec. Antimalarial Drug Resistance
Volume 3 - 2025 | https://doi.org/10.3389/fmala.2025.1530088
This article is part of the Research TopicWomen in Malaria ResearchView all 14 articles
Objectives: Plasmodium vivax is the most widespread Plasmodium spp. globally and the second most common cause of human malaria. However, very little is known about its biology due to the lack of continuous in vitro/ex vivo culture. In such circumstances, genomic studies provide an alternative for understanding the biology of P. vivax parasites. To date, most of the genomic studies on P. vivax have been based on Sal-I as a reference genome; however, with the recent release of the PvP01 and PvW1 reference genomes with higher quality and improved assemblies following continual improvements in annotation, the adoption of these genomes as a reference for genomic studies appears more advantageous. In this study, allelic differences in putative antimalarial drug resistance genes and vaccine candidate genes with assembly features of these three genomes are analyzed and summarized.
Methods: The nucleotide and amino acid sequences of five P. vivax putative antimalarial drug resistance genes and three vaccine candidate genes for all three reference genomes (i.e., Sal-I, PvP01, and PvW1) were retrieved from PlasmoDB and aligned together using MEGA11. Any differences in the nucleotide and codon sequences between the genomes were recorded.
Results: Various allelic differences in the putative antimalarial drug resistance and vaccine candidate genes between the three reference genomes were observed, which included the positions previously identified as candidate markers for these gene variants.
Conclusions: As antimalarial drug resistance genotyping studies rely on candidate genetic markers to classify resistant or sensitive parasites, knowledge of the allelic differences among references is important. Furthermore, the heterogeneity of the vaccine candidate genes should be taken into account when designing vaccines for P. vivax.
Among the five major human Plasmodium species, Plasmodium vivax remains one of the most important malaria parasite species in malaria-endemic regions outside Africa, contributing approximately 9.2 million malaria cases in 2023 (World Health Organization (WHO), 2024; Schneider and Escalante, 2013). The recent increase in the proportion of P. vivax cases observed in areas such as the Greater Mekong Subregion (GMS) suggests that P. vivax malaria could pose a major obstruction to the elimination of malaria (World Health Organization (WHO), 2024; World Health Organization (WHO), 2022). The development of resistance by malaria parasites against the currently available antimalarial drugs is one of the major issues that impede malaria control. Surveillance of these resistant parasites to track the emergence and spread of resistance is of enormous importance in order to effectively respond to the threat of antimalarial drug resistance. Among the various methods available for the genetic surveillance of drug-resistant parasites, targeted genotyping of the loci associated with drug resistance is the most widely employed because it is rapid and cost-effective (Buyon et al., 2021; Gen et al., 2021). Despite its importance, the molecular mechanism behind antimalarial drug resistance in P. vivax remains undetermined. Moreover, thus far, there are no validated molecular markers available for the monitoring of P. vivax drug resistance, even though various polymorphisms have been reported in putative drug resistance genes, viz., dihydrofolate reductase (Pvdhfr), dihydropteroate synthase (Pvdhps), multidrug resistance gene 1 (Pvmdr1), chloroquine resistance transporter (Pvcrt-o), and kelch 12 (Pvk12) (World Health Organization, 2020; Buyon et al., 2021). Although these mutations have not been linked to a drug sensitivity phenotype, they serve as candidate molecular markers for antimalarial drug resistance. In the case of P. vivax, most of the candidate molecular markers thought to be associated with antimalarial drug resistance so far have been largely based on P. vivax Salvador I (Sal-I), the first complete P. vivax reference genome (Carlton et al., 2008). The development of the P. vivax Sal-I reference genome and its release in 2006 marked a significant milestone in the genomics of P. vivax and served as a foundational resource for the generation of subsequent reference genomes. Recently, with the release of the PvP01 (PNG isolate-2016) and PvW1 (Thailand isolate-2021) reference genomes that have improved assemblies and continual improvements with annotation thereafter, it appears a prudent decision for researchers to also utilize these high-quality genomes as reference for molecular studies (Auburn et al., 2016; Minassian et al., 2021; Goo, 2022). As pointed out by the WHO—”molecular markers are an asset for confirming resistance, in the analysis of trends and as an early warning signal”—it is important to understand the allelic variations between the Sal-I, PvP01, and PvW1 reference genomes with respect to the previously reported candidate molecular markers for the P. vivax antimalarial drug resistance genes (World Health Organization, 2020). In addition, compared with the well-researched Plasmodium falciparum, there is a massive lack of knowledge with regard to vaccine development in P. vivax (da Veiga et al., 2023). One of the most significant obstacles encountered in the development of vaccines against P. vivax is the high genetic diversity of the candidate vaccine antigens, resulting in allele- or strain-specific immune response, suggesting that a detailed understanding of genetic diversity is crucial for the selection of ideal candidates for vaccine development (Draper et al., 2018; da Veiga et al., 2023). Currently, different P. vivax vaccines at various stages of development utilize the Sal-I sequence; therefore, it is important to point out the allelic differences of these vaccine candidates with respect to other reference genomes that have recently been made available (Payne et al., 2017; Singh et al., 2018). In this brief overview, the allelic differences in five P. vivax putative antimalarial drug resistance genes and three most promising vaccine candidate genes between the three reference genomes are analyzed. In addition, the assembly features of the three major P. vivax reference genomes (i.e., Sal-I, PvP01, and PvW1) are summarized (Tables 1, 2; Supplementary Table S1).
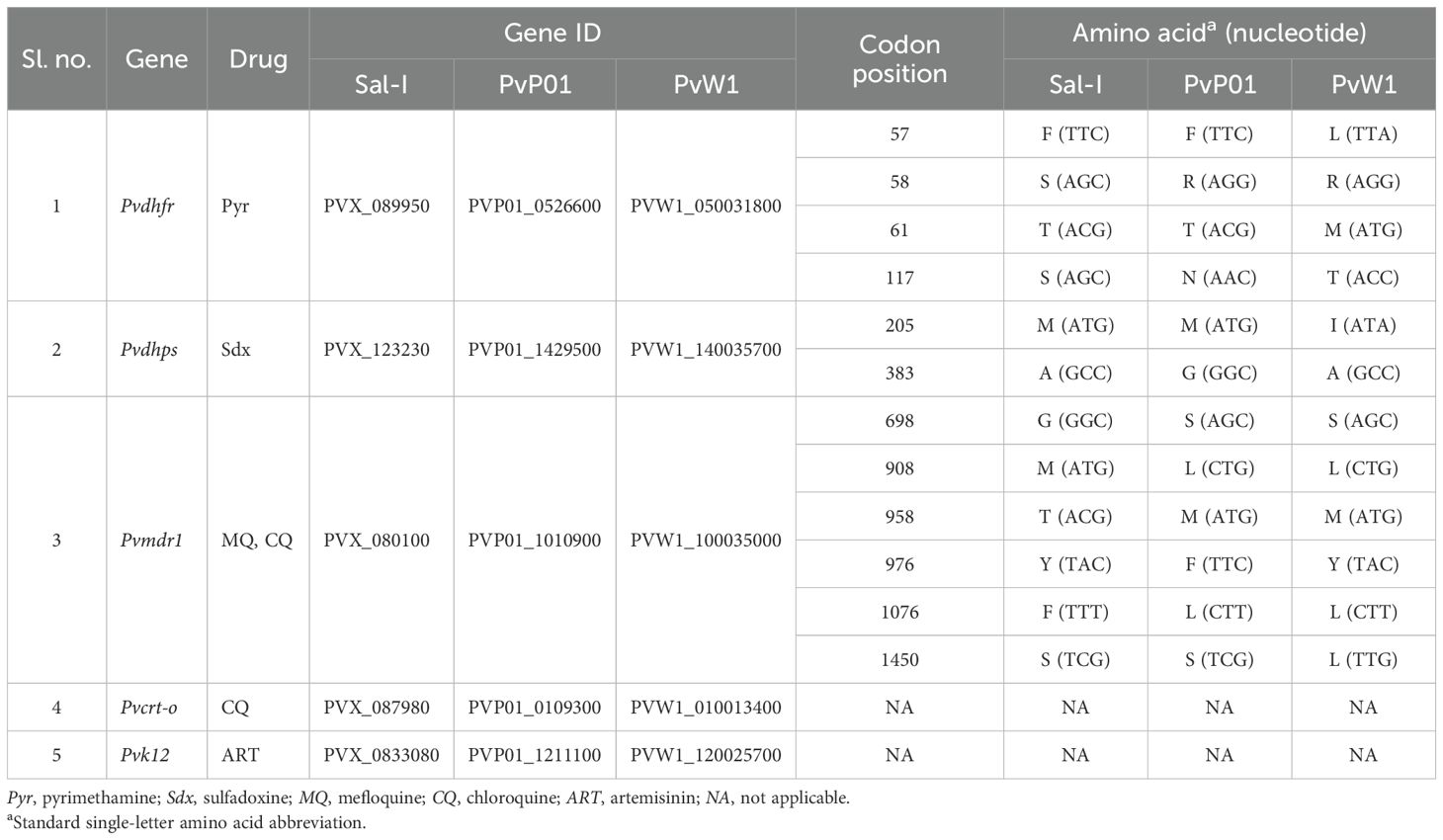
Table 1. Allelic variations in the Plasmodium vivax antimalarial drug resistance genes between the Sal-I, PvP01, and PvW1 reference genomes with codon positions and gene IDs.
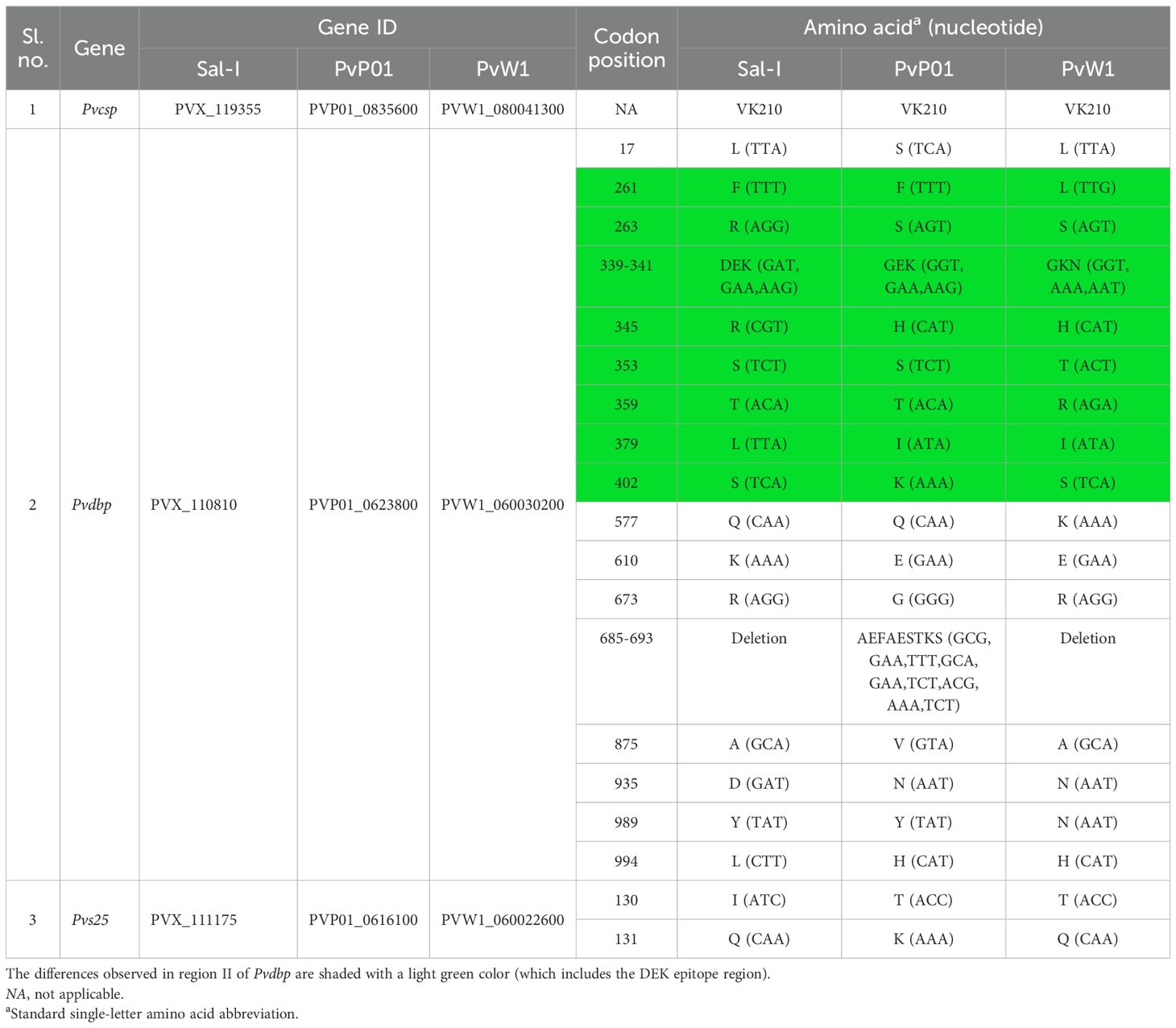
Table 2. Allelic variations in the vaccine candidate genes between the Sal-I, PvP01, and PvW1 reference genomes with codon positions and gene IDs.
The nucleotide and amino acid sequences of the P. vivax putative drug resistance genes (Pvdhps, Pvdhfr, Pvmdr1, Pvcrt-o, and Pvk12) and three vaccine candidate genes, viz., circumsporozoite surface protein (Pvcsp), duffy binding protein (Pvdbp), and ookinete surface protein (Pvs25), for all three reference genomes (Sal-I, PvP01, and PvW1) were obtained from PlasmoDB, 68th release, May 7, 2024 (https://plasmodb.org/) (Amos et al., 2022). The study utilized the following genome versions: Sal-I (GCA_000002415.2), PvP01 (GCA_900093555.2), and PvW1 (GCA_914969965.1). The nucleotide sequences of each gene from the three reference genomes were aligned together using the ClustalW multiple sequence alignment in MEGA11 with a BLOSUM62 scoring matrix. Differences in the nucleotide positions were determined (Tamura et al., 2021). Similarly, the amino acid sequences from the three reference genomes were aligned, and any differences in codon were noted. Quality control measures were implemented during the alignment analysis process, including manual inspection of alignment for consistency and accuracy and the exclusion of poorly aligned regions, particularly in the terminal ends of some gene sequences. The results obtained are summarized in Tables 1, 2. Furthermore, the assembly features of all three reference genomes are collated and listed in Supplementary Table S1.
On analysis, various candidate single nucleotide polymorphisms (SNPs) of antimalarial drug resistance were observed in the Pvdhfr, Pvdhps, and Pvmdr-1 genes of the PvP01 and PvW1 reference genomes (Table 1) (Pornthanakasem et al., 2016; Chaturvedi et al., 2021; Ferreira et al., 2021). In the Pvdhfr gene, an allelic difference was observed between Sal-I, PvP01, and PW1 at four codon positions, i.e., 57, 58, 61, and 117 (Table 1). In Sal-I and PvP01, the Pvdhfr gene at codon position 57 translates into F, whereas the same position translates into L in PvW1. Similarly, codon position 58 translates into S in Sal-I, but into R in both PvP01 and PvW1. Allelic differences at codon positions 61 and 117 in the Pvdhfr gene were also observed. The mutations at codon positions 57, 58, 61, and 117 in the Pvdhfr gene, identified as F57L, S58R, T61M, and S117N/T, respectively, are considered key candidate markers for pyrimethamine resistance in P. vivax due to their high prevalence in regions such as Southeast Asia, where pyrimethamine resistance has been extensively reported, indicating their significance in genetic surveillance and resistance monitoring efforts (Chaturvedi et al., 2021). Allelic variations were observed at two codon positions, i.e., 205 and 383, in the Pvdhps gene. Codon 205 translates into M in Sal-I and PvP01, but into I in PvW1. Similarly, the codon at position 383 codes for A in Sal-I and PvW1, whereas it codes for G in PvP01. Mutations M205I and A383G in the Pvdhps gene are commonly observed in areas where sulfadoxine resistance is prevalent and are important candidate mutations for sulfadoxine resistance in P. vivax (Benavente et al., 2021; Chaturvedi et al., 2021). Allelic differences at five codon positions were identified, viz., 698, 908, 958, 976, 1076, and 1450, in the Pvmdr1 gene between Sal-I, PvP01, and PvW1 (Table 1). Mutations at corresponding gene positions are commonly reported in the Pvmdr1 gene (i.e., G698S, M908L, T958M, Y976F/V, F1076L/I/T, and S1450L), with the most prevalent being Y976F/V and F1076L/I/T (Ferreira et al., 2021). For Pvcrt-o and Pvk12, no allelic differences were observed among the three reference genomes (Table 1).
Comparison of the three most promising vaccine candidate genes (i.e., Pvcsp, Pvdbp, and Pvs25) between the three reference genomes revealed varying levels of heterogeneity (Draper et al., 2018; da Veiga et al., 2023). Pvcsp is a key pre-erythrocytic stage vaccine candidate, with two formulations—VMP001 and peptides N, R, and C—currently undergoing early-phase clinical trials (da Veiga et al., 2023). The pre-erythrocytic stage represents a critical bottleneck in the Plasmodium life cycle, with minimal parasitemia levels, and csp, the major sporozoite surface protein, being directly exposed to host antibodies during migration to the liver, positioning Pvcsp-based vaccines at the forefront of P. vivax research (De et al., 2021). However, the highly polymorphic nature of Pvcsp, which leads to strain-specific immune responses, presents a significant challenge, making the heterogeneity of Pvcsp a crucial factor in vaccine design. Based on the repetitive sequences at the central region, three different variants of the gene have been described, viz., VK210, VK247, and P. vivax-like (Gimenez et al., 2021). Upon comparison, the VK210-type repeats were observed to be present in all three reference genomes (Sal-I, PvP01, and PvW1) (Table 2). An essential consideration in the development of an effective Pvcsp-based P. vivax vaccine is the inclusion of all three repeat types present. Pvdbp is an important molecule that plays a role in reticulocyte invasion by the parasite, making it a crucial candidate for blood-stage vaccine (da Veiga et al., 2023). The cysteine-rich region II of Pvdbp, which is responsible for erythrocyte binding, has been utilized in the development of various Pvdbp-based vaccines. Two such vaccines, PvDBPII/GLA-SE and ChAd63/MVA PvDBP RII, have completed phase I clinical trials (da Veiga et al., 2023). It is critical to acknowledge that both vaccines are based on Sal-I alleles, with strain-specific immune responses having been observed (Cole-Tobian et al., 2009; De et al., 2021; da Veiga et al., 2023). This underscores the necessity to incorporate multiple Pvdbp variant alleles in the design of a vaccine with broader specificity, capable of inducing strain-transcending immunity. Amino acid differences at various positions of the gene, including the DEK epitope in region II of Pvdbp, were observed (Table 2) (da Veiga et al., 2023). The DEK epitope is a key B-cell site in the polymorphic region II of Pvdbp, which is crucial as it is targeted by human inhibitory antibodies that block the invasion of P. vivax into red blood cells. However, its polymorphism poses a challenge as it enables immune evasion, necessitating vaccine strategies that address this variability (Chootong et al., 2009). Upon analysis of the transmission blocking vaccine candidate Pvs25, high conservation was observed between the three reference genomes, with only two amino acid differences at codon positions 130 and 131 (Table 2) (Miyata et al., 2011). Phase I clinical trials of Pvs25-based vaccines yielded promising results, indicating the potential of the Pvs25 protein to elicit an immune response. However, a clinical trial involving Pvs25 formulated with the adjuvant Montanide ISA 51 resulted in notable reactogenicity (Malkin et al., 2005; Wu et al., 2008; da Veiga et al., 2023). Therefore, the identification of an optimal adjuvant combination that enhances vaccine efficacy while minimizing adverse reactogenicity remains a critical area of focus for the future development of Pvs25-based vaccines.
The Sal-I genome, which was derived from a monkey-adapted strain, serves as a foundational framework for the understanding of P. vivax, but may not fully represent the genetic diversity observed in wild isolates due to its adapted nature. PvP01, a reference genome derived from a Southeast Asian isolate, captures regional genetic variations, offering insights into the biology of P. vivax in populations with specific ecological and immunological pressures. Along with PvP01, PvW1 represents a wild parasite isolate with minimal adaptation and might provide a closer reflection of natural P. vivax populations. Supplementary Table S1 presents a comprehensive comparison of the genome assembly statistics across the three reference genomes, including their assembly features, their origin, the release year, and the sequencing platforms used. It highlights the progression in the reference genomes of P. vivax from the historical Sal-I to PvP01 and PvW1, showcasing significant improvements in sequencing technologies and genome assembly over time. PvP01 and PvW1 feature higher assembly completeness, fewer unassigned scaffolds, and richer gene annotations compared with the older Sal-I genome. The fewer unassigned scaffolds in the PvW1 assembly highlight the advantages of using a hybrid sequencing approach that combines long reads (PacBio) and short reads (Illumina) for the generation of more complete reference assemblies.
Molecular markers of antimalarial drug resistance are of immense importance as they strengthen the pillars of drug resistance monitoring. Allelic variations influence gene expression, antigenic variation, and host–pathogen interactions, highlighting the importance of using diverse genomes to study host specificity, immune evasion, and other biological traits. The present study summarizes the existing allelic differences between the Sal-I, PvP01, and PvW1 reference genomes with respect to the major putative antimalarial drug resistance and vaccine candidate genes, aiming at raising awareness of the polymorphisms observed as drug resistance mechanisms and vaccine efficacy are highly influenced by genomic variability. Hitherto, the phenotype of any P. vivax reference strains is unknown, and any reported mutations in its putative antimalarial drug resistance genes have not been clearly linked to resistance, primarily due to the lack of long-term in vitro culture of these parasites. Nonetheless, candidate SNPs that are commonly observed during the genotyping of putative genes are of immense significance in the current scenario as they serve as an important genetic surveillance tool in the absence of any other cost-effective approach. Furthermore, during the design of vaccines, it is crucial to take into account the heterogeneity of the candidate vaccine antigen as variations in the antigen-encoding genes could result in allele- or strain-specific immune response, reducing the overall efficacy of the vaccine. Future research should focus on the expansion of the P. vivax reference genome by incorporating a broader array of isolates from diverse geographical regions in order to create a more comprehensive pan-genome. This effort could significantly enhance the precision of genomic analyses, providing a thorough understanding of the global genetic variability of the parasite. Consequently, it would enable more accurate interpretations of the population structure, the evolutionary dynamics, and the identification of potential markers for drug resistance and vaccine development.
Although the utility of a different reference genome varies with the research context, with each offering distinct advantages and limitations, it is important to be aware of the genomic architecture of the reference strains under use as it makes the base for all omics studies. It is important to be cautious when using PvP01 and PvW1 as a reference for genotyping studies as alignment software programs might identify mutations at the abovementioned codon positions as the wild type will result in an incorrect interpretation of the results.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
VS: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. ST: Data curation, Formal analysis, Methodology, Software, Validation, Writing – original draft. AA: Methodology, Software, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors are grateful to the ICMR-National Institute of Malaria Research (NIMR) for providing infrastructure to carry out the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmala.2025.1530088/full#supplementary-material
Amos B., Aurrecoechea C., Barba M., Barreto A., Basenko E. Y., Bażant W., et al. (2022). VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50, D898. doi: 10.1093/NAR/GKAB929
Auburn S., Böhme U., Steinbiss S., Trimarsanto H., Hostetler J., Sanders M., et al. (2016). A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 1. doi: 10.12688/WELLCOMEOPENRES.9876.1
Benavente E. D., Manko E., Phelan J., Campos M., Nolder D., Fernandez D., et al. (2021). Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat. Commun. 12. doi: 10.1038/S41467-021-23422-3
Buyon L. E., Elsworth B., Duraisingh M. T. (2021). The molecular basis of antimalarial drug resistance in Plasmodium vivax. Int. J. Parasitol. Drugs Drug Resist. 16, 23–37. doi: 10.1016/J.IJPDDR.2021.04.002
Carlton J. M., Adams J. H., Silva J. C., Bidwell S. L., Lorenzi H., Caler E., et al. (2008). Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763. doi: 10.1038/NATURE07327
Chaturvedi R., Chhibber-Goel J., Verma I., Gopinathan S., Parvez S., Sharma A. (2021). Geographical spread and structural basis of sulfadoxine-pyrimethamine drug-resistant malaria parasites. Int. J. Parasitol. 51, 505–525. doi: 10.1016/J.IJPARA.2020.12.011
Chootong P., Ntumngia F. B., VanBuskirk K. M., Xainli J., Cole-Tobian J. L., Campbell C. O., et al. (2009). Mapping epitopes of the Plasmodium vivax duffy binding protein with naturally acquired inhibitory antibodies. Infect. Immun. 78, 1089. doi: 10.1128/IAI.01036-09
Cole-Tobian J. L., Michon P., Biasor M., Richards J. S., Beeson J. G., Mueller I., et al. (2009). Strain-specific duffy binding protein antibodies correlate with protection against infection with homologous compared to heterologous Plasmodium vivax strains in Papua New Guinean children. Infect. Immun. 77, 4009–4017. doi: 10.1128/IAI.00158-09
da Veiga G. T. S., Moriggi M. R., Vettorazzi J. F., Müller-Santos M., Albrecht L. (2023). Plasmodium vivax vaccine: What is the best way to go? Front. Immunol. 13. doi: 10.3389/FIMMU.2022.910236
De S. L., Ntumngia F. B., Nicholas J., Adams J. H. (2021). Progress towards the development of a P. vivax vaccine. Expert Rev. Vaccines 20, 97–112. doi: 10.1080/14760584.2021.1880898
Draper S. J., Sack B. K., King C. R., Nielsen C. M., Rayner J. C., Higgins M. K., et al. (2018). Malaria vaccines: recent advances and new horizons. Cell Host Microbe 24, 43. doi: 10.1016/J.CHOM.2018.06.008
Ferreira M. U., Nobrega de Sousa T., Rangel G. W., Johansen I. C., Corder R. M., Ladeia-Andrade S., et al. (2021). Monitoring Plasmodium vivax resistance to antimalarials: Persisting challenges and future directions. Int. J. Parasitol. Drugs Drug Resist. 15, 9–24. doi: 10.1016/J.IJPDDR.2020.12.001
Gen M., Ahouidi A., Ali M., Almagro-Garcia J., Amambua-Ngwa A., Amaratunga C., et al. (2021). An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 6, 1–31. doi: 10.12688/WELLCOMEOPENRES.16168.2
Gimenez A. M., Salman A. M., Marques R. F., López-Camacho C., Harrison K., Kim Y. C., et al. (2021). A universal vaccine candidate against Plasmodium vivax malaria confers protective immunity against the three PvCSP alleles. Sci. Rep. 11. doi: 10.1038/S41598-021-96986-1
Goo Y. K. (2022). Vivax malaria and the potential role of the subtelomeric multigene vir superfamily. Microorganisms 10. doi: 10.3390/MICROORGANISMS10061083
Malkin E. M., Durbin A. P., Diemert D. J., Sattabongkot J., Wu Y., Miura K., et al. (2005). Phase 1 vaccine trial of Pvs25H: a transmission blocking vaccine for Plasmodium vivax malaria. Vaccine 23, 3131–3138. doi: 10.1016/J.VACCINE.2004.12.019
Minassian A. M., Themistocleous Y., Silk S. E., Barrett J. R., Kemp A., Quinkert D., et al. (2021). Controlled human malaria infection with a clone of Plasmodium vivax with high-quality genome assembly. JCI Insight 6. doi: 10.1172/JCI.INSIGHT.152465
Miyata T., Harakuni T., Sugawa H., Sattabongkot J., Kato A., Tachibana M., et al. (2011). Adenovirus-vectored Plasmodium vivax ookinete surface protein, Pvs25, as a potential transmission-blocking vaccine. Vaccine 29, 2720–2726. doi: 10.1016/J.VACCINE.2011.01.083
Payne R. O., Silk S. E., Elias S. C., Milne K. H., Rawlinson T. A., Llewellyn D., et al. (2017). Human vaccination against Plasmodium vivax Duffy-binding protein induces strain-transcending antibodies. JCI Insight 2. doi: 10.1172/JCI.INSIGHT.93683
Pornthanakasem W., Riangrungroj P., Chitnumsub P., Ittarat W., Kongkasuriyachai D., Uthaipibull C., et al. (2016). Role of Plasmodium vivax dihydropteroate synthase polymorphisms in sulfa drug resistance. Antimicrob. Agents Chemother. 60, 4453. doi: 10.1128/AAC.01835-15
Schneider K. A., Escalante A. A. (2013). Fitness components and natural selection: Why are there different patterns on the emergence of drug resistance in Plasmodium falciparum and Plasmodium vivax? Malar. J. 12, 1–11. doi: 10.1186/1475-2875-12-15/FIGURES/2
Singh K., Mukherjee P., Shakri A. R., Singh A., Pandey G., Bakshi M., et al. (2018). Malaria vaccine candidate based on Duffy-binding protein elicits strain transcending functional antibodies in a Phase I trial. NPJ Vaccines 3, 1–10. doi: 10.1038/s41541-018-0083-3
Tamura K., Stecher G., Kumar S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/MOLBEV/MSAB120
World Health Organization (2020). Tackling antimalarial drug resistance Launch of the WHO Report on antimalarial drug efficacy, resistance and response: 10 years of surveillance. Geneva: World Health Organization. p. 64.
World Health Organization (WHO). World malaria report 2024. Available online at: https://www.who.int/publications/i/item/9789240104440 (Accessed January 1, 2025).
World Health Organization (WHO). (2022). World malaria report 2022. Geneva: World Health Organization.
Keywords: antimalarial drug resistance, malaria vaccine, Plasmodium vivax, PvP01, PvW1, reference genome, Sal-I
Citation: Tamang S, Arya A and Singh V (2025) Allelic variations in putative antimalarial drug resistance and vaccine candidate genes among Plasmodium vivax reference genomes. Front. Malar. 3:1530088. doi: 10.3389/fmala.2025.1530088
Received: 18 November 2024; Accepted: 13 January 2025;
Published: 24 February 2025.
Edited by:
Alena Pance, University of Hertfordshire, United KingdomReviewed by:
Ariel H. Magallon-Tejada, Gorgas Memorial Institute of Health Studies, PanamaCopyright © 2025 Tamang, Arya and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vineeta Singh, dmluZWV0YXNfMjAwMEB5YWhvby5jb20=
†ORCID: Suman Tamang, orcid.org/0000-0002-2892-2862
Vineeta Singh, orcid.org/0000-0003-0113-7394
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.