
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Malar., 03 April 2024
Sec. Antimalarial Drug Resistance
Volume 2 - 2024 | https://doi.org/10.3389/fmala.2024.1363969
This article is part of the Research TopicWomen in Malaria ResearchView all 14 articles
 Sonal Kale1†‡
Sonal Kale1†‡ Swapna M. Uplekar2‡Nabamita Bandyopadhyay1‡
Swapna M. Uplekar2‡Nabamita Bandyopadhyay1‡ Pavitra N. Rao2‡Syed Z. Ali3
Pavitra N. Rao2‡Syed Z. Ali3 Surya Kant Sharma3
Surya Kant Sharma3 Nikunj Tandel4†Ankita Patel4Ranvir Singh4Aaron Dank4Sangamithra Ravishankaran5G. Sri Lakshmi Priya5Aswin Asokan5
Nikunj Tandel4†Ankita Patel4Ranvir Singh4Aaron Dank4Sangamithra Ravishankaran5G. Sri Lakshmi Priya5Aswin Asokan5 Alex Eapen5
Alex Eapen5 Om. P. Singh1
Om. P. Singh1 Jane M. Carlton2*†
Jane M. Carlton2*† Prashant K. Mallick1*
Prashant K. Mallick1*Introduction: Tracking the emergence and spread of antimalarial drug resistance is critical for supporting progress towards the control and eventual elimination of malaria in South Asia, especially India. Plasmodium falciparum has evolved resistance to virtually every antimalarial drug, and significant progress has been made to identify the molecular genetic mechanisms involved in the most common types of resistance.
Methods: An amplicon sequencing protocol was used for molecular surveillance of antimalarial drug resistance in a total of 158 patient isolates collected from December 2012 to September 2015 from three sites in south, west and east India: Tamil Nadu, Gujarat, and Odisha respectively. Five full length Plasmodium falciparum genes whose mutant proteins are implicated in antimalarial drug resistance were investigated: Pfcrt for chloroquine, Pfdhfr for pyrimethamine, Pfdhps for sulfadoxine, Pfk13 for artemisinin and Pfmdr1 for resistance to multiple antimalarial drugs.
Results: We observed a high proportion of wild-type Pfcrt and Pfdhfr haplotypes from the P. falciparum-dominant site Rourkela, while mutant Pfcrt and Pfdhfr haplotypes were fixed at the P. vivax dominant sites Chennai and Nadiad. The wild-type Pfdhps haplotype was predominant across all study sites. We identified mutations in the propeller domain of Pfk13, although they are not associated with resistance to artemisinin. Finally, using samples taken from the same patient on day 2, day 7, and day 14 after artemisinin combination treatment, we were able to observe changes in allele frequency of drug resistance genes during the course of an infection.
Discussion: This is the first high-throughput deep sequencing study of five full-length P. falciparum genes in clinical isolates from three different study sites in India with varying transmission ecologies. Amplicon sequencing based on ion-torrent has the potential to track changes in the frequency of drug resistant alleles as a patient is undergoing drug therapy, and to identify new resistance mutations as they increase in frequency in the patient. This study showed possibility of whole gene sequencing, leads to in-depth molecular surveillance of multiple antimalarial resistant candidates and furthermore suggests investigations on reversal of resistant genotypes due to usage of artemisinin combination therapy in P. falciparum prevalent areas of the country.
Malaria remains a major cause of mortality and morbidity in tropical and sub-tropical regions worldwide. Despite a decrease in malaria cases in past two decades, many endemic nations, including India report cases of the disease regularly. Massive efforts by a worldwide malaria elimination program have reduced the number of malaria cases by more than 40% in 2021 compared with 2015(2023). However, the number of deaths has remained the same over the past three years and India accounted for about 83% of all malaria deaths in the WHO South-East Asia Region (2023). Over 60% of malaria cases in India are caused by Plasmodium falciparum. India has also joined malaria elimination efforts, allocating financial resources and infrastructure for the launch of a ‘National Framework for Malaria Elimination in India 2016-2030’. This effort includes all Indian states with varying levels of malaria transmission and outlines specific strategies to achieve the following milestones: 1) eliminate malaria throughout the entire country by 2030; and 2) maintain a malaria-free status in areas where transmission has been interrupted and prevent re-introduction of malaria.
The emergence and spread of parasite resistance against available antimalarials constitutes a major threat towards the efforts of the elimination program, as evidenced by the reversal of previous malaria eradication successes (Shah et al., 2011). Routine molecular surveillance of parasite strains is an essential tool for identifying and limiting the spread of emerging antimalarial resistance. Sanger sequencing is the gold standard for validation of genotypes identified in parasite clinical isolates, but length of time and scalability remain limiting factors; these may be overcome by deep sequencing that generates large amounts of genome sequence data in a short time by parallelization of sequencing reactions. The technology provides high sequencing coverage, which enables the detection of low-frequency mutations relevant to drug resistance, or low frequency wild-type parasites arising after reversion of mutant genotypes following antimalarial drug withdrawal from a region. Deep sequencing can also provide insight into the genetic diversity of loci within a single subject over the course of treatment (Mideo et al., 2013).
We previously described a novel amplicon sequencing protocol for molecular surveillance of P. falciparum drug resistance loci in field settings (Rao et al., 2016). This protocol enabled the identification of polymorphisms in five important genes implicated in P. falciparum resistance against the mainstay antimalarial drugs; Pfcrt for chloroquine resistance, Pfdhfr for pyrimethamine resistance, Pfdhps for sulfadoxine resistance, Pfk13 for artemisinin resistance, and Pfmdr1 for resistance to multiple antimalarials. CQ has remained the first-line therapy for P. vivax in India, even after its use against P. falciparum was discontinued. Following widespread CQ resistance, a combination of the antifolates sulfadoxine and pyrimethamine (SP) was introduced in India as the second-line treatment for P. falciparum in 1982. Resistance to sulfadoxine (SUL) and pyrimethamine (PYR) has been associated with mutations in the folate biosynthesis pathway enzymes PfDHPS and PfDHFR, respectively, that reduce binding of the antifolates to their targets. Artemisinin and its derivatives artesunate, artemether, and dihydroartemisinin, are the most effective antimalarials currently available, and administered in combination (artemisinin combination therapy, ACT) with partner drugs e.g., lumefantrine and piperaquine, due to their short half-lives. In India, artesunate is used in combination with SP (AS-SP), except in the Northeast region where artemether and lumefantrine (AL) is used due to widespread SP resistance (Anvikar et al., 2014). Artemisinin resistance was first identified in Cambodia and spread quickly through Southeast Asia, while also emerging simultaneously at different foci in South Asia. Mutations in the propeller domain of the kelch protein K13 are partially associated with delayed clearance of parasites after ACT treatment (Ariey et al., 2014; Straimer et al., 2015), although PfK13-independent resistance is also reported [reviewed in (Pandit et al., 2023)]. Evidence of artemisinin-resistant P. falciparum in West Bengal, India, was reported in 2018 (Das et al., 2018), although the finding is controversial (Rasmussen et al., 2019). A more recent study purports to have identified treatment failure in patients receiving ACT in Chhattisgarh, central India, in the absence of PfK13 polymorphisms (Das et al., 2021).
In the study presented here, we used our deep sequencing protocol to elucidate the landscape of drug resistance at three different field study sites in India, representing varying transmission ecologies, as part of ongoing epidemiology studies at the Center for the Study of Complex Malaria in India (Carlton et al., 2022a; Carlton et al., 2022b). We identified a high proportion of parasites from the P. falciparum-dominant site Rourkela carrying wild-type Pfcrt and Pfdhfr haplotypes, while mutant Pfcrt and Pfdhfr haplotypes were fixed at the P. vivax-dominant sites Chennai and Nadiad; the wild-type Pfdhps haplotype was predominant across all study sites. We identified mutations in the propeller domain of PfK13, although they are not associated with resistance to artemisinin. We were also able to track the change in frequency of drug resistant haplotypes during the course of infection in 14 subjects with samples collected on subsequent days following ACT treatment.
The three study sites have been described in detail previously (Das et al., 2012; van Eijk et al., 2019). Briefly: subjects were enrolled at (i) Besant Nagar catchment area in the city of Chennai, Tamil Nadu, a hypo-endemic malaria site with P. vivax dominant; (ii) Nadiad Civil Hospital/health center in the Vatva ward of Ahmedabad, Gujarat, hypo-endemic for P. vivax and P. falciparum; and (iii) a health center clinic in Rourkela and surrounding rural villages, Odisha, with meso-to-hyperendemic malaria, and P. falciparum dominant.
Blood samples were collected from patients enrolled at three study sites in India, namely Chennai, Nadiad and Rourkela after obtaining informed consent, and treated as per India’s national drug treatment guidelines (Anvikar et al., 2014). Approximately 3-5 ml blood were collected in EDTA vacutainers during clinic enrollment (day 0) and follow-up visits on day 2, day 7, or day 14 post-ACT treatment represented as v1, v2 and v3 respectively in the year 2012-2015. Thick and thin blood smear were prepared from these blood samples for microscopy identification and DNA extraction was performed using the QIAamp DNA Blood Mini or Midi Kit (Qiagen Inc., catalog nos. 51106, 51185). Blood Samples were collected from 141 patients (98 male and 43 female, age range 2-69 years old). A total of 158 P. falciparum isolates from 141 patients at the time of enrollment and follow up from December 2012 to September 2015. The erythrocytic P. falciparum parasites were detected by microscopy in 140 isolates (mean parasitemia 14,073.16 parasites/ul; 95%CI: 3,822.15- 24,324.18); 18 isolates were submicroscopic. The detailed description of collected samples is provided in Table 1. A total of 149 isolates were positive for P. falciparum by species-specific PCR, nine were PCR negative. Subsequent analysis excluded certain follow-up samples due to either the absence of P. falciparum malaria positivity post-treatment or insufficient parasite DNA yield for PCR amplification and sequencing. Poor-quality data were also removed to ensure robustness of the analysis.
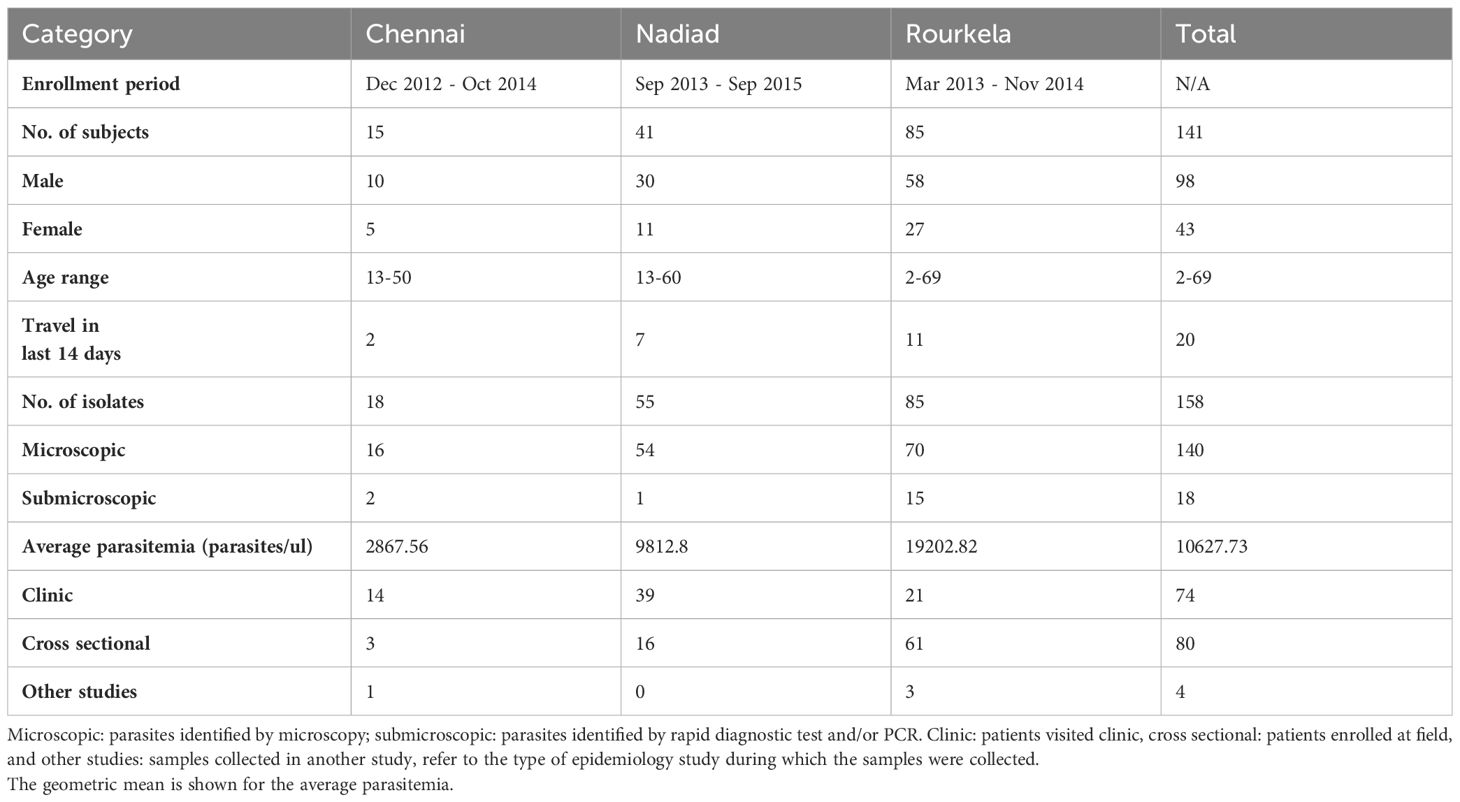
Table 1 Summary of samples collected during epidemiology studies at three sites in India and used in this study.
For library preparation primers and PCR conditions were as described (Rao et al., 2016). For each sample, 10 ul of each amplicon was pooled, the pooled mixture purified using a QIAquick PCR Purification Kit (Qiagen Inc., catalog no. 28106) and quantitated using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, catalog no. Q32866). Barcoded sequencing libraries were generated from 100-500 ng of purified pooled PCR product using ion express™ barcode adapters (Life Technologies, catalog no. 4471250 for barcodes 1-16; catalog no. 4474009 for barcodes 17-32) and library prep kit (New England Biolabs, catalog no. E6285L). The size and quality of libraries were determined using an Agilent bioanalyzer, and quantified by quantitative PCR using Ion Torrent Library Quantification kit (KAPA biosystems, KK4857), on a LightCyclerR 480 (Roche). For most experiments, 16 barcoded libraries were pooled for a single run on an Ion Torrent Personal Genome Machine using Ion PGM™ Hi-Q™ sequencing technology and Ion™ 314v2 type chips (one library pool was run on an Ion™ 318v2 chip). Genomic DNA from P. falciparum strains Dd2 (MRA-150G) and 7G8 (MRA-152G) was obtained from the Malaria Research and Reference Reagents Resource (MR4), and control libraries prepared with Dd2 or 7G8 DNA were included in each run. Ten P. falciparum reference strains (NF54, TM91C235, HB3, W2, K1, V1/S, D10, GB4, D6, FCB) were also sequenced on different runs to ensure that the variant calling results were in concordance with published genotypes for these isolates.
Sequencing data were analyzed using the Torrent Suite 5.0.2 software, with raw reads de-multiplexed and filtered using standard quality filtering parameters, and read quality assessed using the Torrent Suite FastQC plugin v0.10.1. A multi-FASTA reference file containing the P. falciparum 3D7 sequence and 300bp flanking regions for each target gene was generated [PlasmoDB (Warrenfeltz et al., 2018)], and high quality reads aligned to the reference file using Torrent Mapping Alignment Program v5.0.
Variant calling was performed with low stringency parameters using the Torrent Variant Caller plugin (v5.0). A list of all positions in all genes that were variable in at least one sample was generated; for samples where no variants were reported at these positions, the presence of the reference allele was confirmed by obtaining raw nucleotide counts and quality scores from corresponding BAM files using BEDTools (Quinlan and Hall, 2010). Variant filtering criteria were determined based on the quality of known SNPs in MR4 reference isolates sequenced in each run. Variant calls with SNP allele depth <10 reads and base quality score <10 were discarded. Calls with SNP allele frequency between 20% - 80% were marked as heterozygous, and used to classify isolates as single or multi-clonal infections. SNP calls were processed to determine codon changes based on the P. falciparum 3D7 sequence using custom Perl and MS Excel VBA scripts. In the case of multiple nucleotide polymorphisms (MNPs) affecting the same codon, if both SNP alleles occurred at a frequency >80%, they were merged to calculate the resulting codon change; variant calls with SNP allele frequency <20% were discarded. For the Pfcrt gene, MNPs affecting codon positions 72-76 were filtered using a higher SNP allele frequency cut-off of 30%. Variants in the Pfcrt gene were manually curated using the IGV genome browser (Thorvaldsdottir et al., 2013) due to a high proportion of homopolymer repeats and poor alignment of reads. Population structure was examined using a non-model-based multivariate analysis method implemented in the adegenet 2.0.1 package using the dudi.pca function. In the case of multi-allelic SNPs, the major allele was used to represent the dominant genotype at that position.
A panel of five P. falciparum genes implicated in antimalarial drug resistance: Pfk13, Pfcrt, Pfmdr1, Pfdhfr, and Pfdhps were amplified and sequenced. One of our previously published amplicon sequencing protocol that sequences the complete length of each gene (Rao et al., 2016) was used here and a summary of observed high quality single nucleotide polymorphisms in these five genes is described in Supplementary Table 1. Implications of various mutations associated with resistance identified at the three sites are described below in the context of each associated antimalarial drug. Evaluation on of these isolates yielded baseline genotype data on the antimalarial drug resistant candidates during initial years of artemisinin combination therapy usage.
Mutations encoding amino acid substitutions in the chloroquine resistance transporter PfCRT (PF3D7_0709000) at positions 72, 74, 75, 76, 97, 220, 271, 326, 356 and 371 have been associated with increased resistance to chloroquine (CQ), with the K76T substitution being the primary mediator of resistance (Fidock et al., 2000; Djimdé et al., 2001; Sidhu et al., 2002). A total of 147 of 158 isolates sequenced carried only one haplotype at the PfCRT locus; one isolate had multiple Pfcrt haplotypes, and 10 isolates had partial or missing PfCRT haplotype data (Table 2). The CQ-resistant Pfcrt haplotype S72V73M74N75T76, purported to be the most prevalent Pfcrt haplotype in India (Vathsala et al., 2004), was present in 71/147 isolates, including all isolates from Chennai and most isolates from Nadiad, but only two isolates from Rourkela. Contrastingly, the CQ-sensitive wild-type haplotype C72V73M74N75K76 was detected in 46 isolates from Rourkela but absent from Nadiad and Chennai. The CQ-resistant haplotype C72V73I74E75T76 was present in 30/147 isolates: 28 isolates from Rourkela and 2 isolates from Nadiad. Both copy number variation and mutations encoding substitutions in the multidrug resistance protein PfMDR1 (PF3D7_0523000) codons 86, 184, 1034, 1042 and 1246 can modulate resistance to various antimalarial drugs (Menard and Dondorp, 2017). While our protocol does not permit gene copy number assessment, we were able to identify nucleotide polymorphisms associated with resistance to CQ. A total of 129 of 158 isolates sequenced carried only one Pfmdr1 haplotype, while 10 isolates possessed more than one Pfmdr1 haplotype, which may be attributed to increased Pfmdr1 copy number (Table 2). A total of 19 isolates had partial or missing haplotype data. The single-mutant Pfmdr1 haplotype N86F184S1034N1042D1246 was predominant (57/129) among isolates in our study, and the wild-type Pfmdr1 haplotype N86Y184S1034N1042D1246 was also prevalent in several (45/129) isolates. Double-mutant Pfmdr1 haplotypes were rare, with only 5/129 isolates containing the haplotypes Y86F184S1034N1042D1246 or N86F184S1034D1042D1246. The single mutation-bearing haplotype Y86Y184S1034N1042D1246 was present in 22/129 isolates, eleven each from Nadiad and Rourkela.
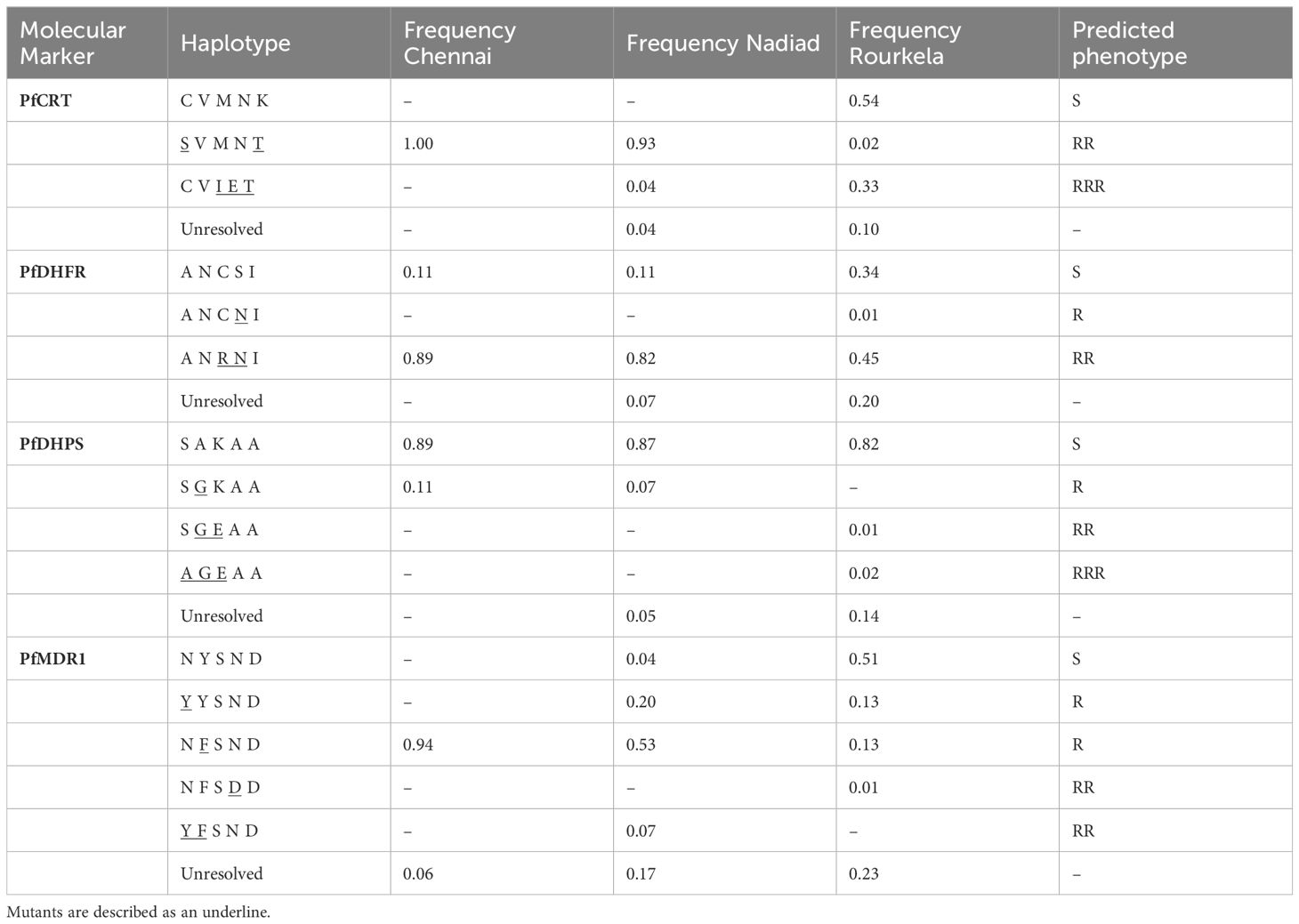
Table 2 Frequency of haplotypes for four drug resistance markers at the three field sites Chennai, Nadiad and Rourkela.
A total of 12 haplotype combinations between PfCRT and PfMDR1 were observed in 122 isolates with complete sequence information for these genes across the three sites. Rourkela had the largest diversity of haplotypes, with ten haplotype combinations at these two loci, and Chennai had the least diversity (Table 3), with only one haplotype combination. The Pfcrt and Pfmdr1 haplotype combination of S72V73M74N75T76 + N86F184S1034N1042D1246 was most common among isolates surveyed in our study (47/122), but was seen primarily in Chennai and Nadiad and not in Rourkela. The combination of wild-type haplotypes at both loci (C72V73M74N75K76 + N86Y184S1034N1042D1246) was seen only at Rourkela, and present in the highest proportion at that site.
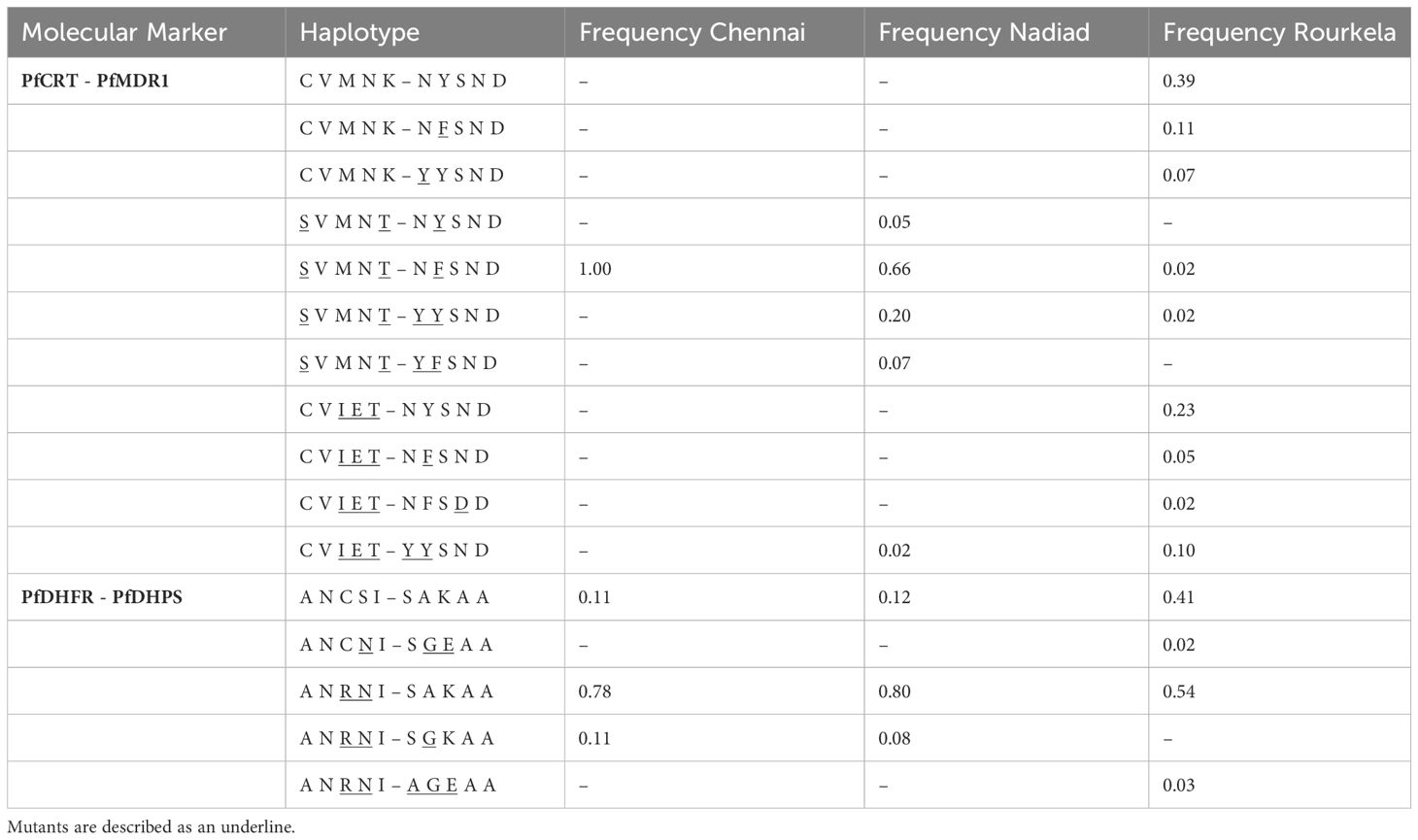
Table 3 Frequency of haplotypes for combinations of two drug resistance markers (PfCRT-PfMDR1 and PfDHDR-PfDHPS) at the three field sites Chennai, Nadiad and Rourkela.
Mutations encoding amino acid substitutions at positions 51, 59, 108, 164 in the enzyme dihydrofolate reductase PfDHFR (PF3D7_0417200) are associated with increased resistance to pyrimethamine (PYR), with the S108N mutation primarily responsible for resistance (Cowman et al., 1988; Sirawaraporn et al., 1997). 137 of 158 isolates sequenced carried only a single haplotype at the PfDHFR locus; 12 isolates carried multiple Pfdhfr haplotypes and 9 isolates had partial or missing Pfdhps haplotype data (Table 2). The double-mutant Pfdhfr haplotype A16N51R59N108I164 was most common among isolates (99/137) in our study, while the single-mutant A16N51C59 N108I164 haplotype was extremely rare (1/137); triple (A16I51R59N108I164) or quadruple (A16I51R59N108L164) mutant haplotypes could not be detected. Several isolates (37/137) had only the PYR-sensitive wild-type Pfdhfr haplotype A16N51C59S108I164. Resistance to sulfadoxine (SUL) has been associated with mutations encoding amino acid substitutions at positions 436, 437, 540, 581 and 613 in the enzyme dihydropteroate synthetase (PfDHPS), with the A437G substitution arising first (Brooks et al., 1994; Triglia et al., 1998). A total of 143 of 158 isolates sequenced carried only a single haplotype at the PfDHPS locus; 15 isolates had partial or missing Pfdhps haplotype data. The SUL-sensitive wild-type Pfdhps haplotype S436A437K540A581A613 predominated in our study, with 134/143 isolates carrying only this haplotype. Two isolates from Rourkela possessed the triple mutant Pfdhps haplotype A436G437E540A581A613, while the double mutant haplotype S436G437E540A581A613 was only detected in a single isolate. Six isolates carried the single-mutant S436G437K540A581A613 haplotype.
A total of five haplotype combinations between the PfDHFR and PfDHPS loci were seen in 129 isolates with complete sequence information for these two genes across the three sites (Table 3). The combination of double-mutant PfDHFR with wild-type PfDHPS (A16N51R59N108I164 + S436A437K540A581A613) was predominant among isolates in our study (87/129). Two samples from Rourkela had the strongest predictors of treatment failure in our dataset- the combination of double-mutant PfDHFR with triple-mutant PfDHPS (A16N51R59N108I164 + A436G437E540A581A613). Only 33 isolates, primarily from Rourkela, carried wild-type haplotypes at both loci. Two other haplotype combinations were observed in the population: (A16N51R59N108I164 + S436G437K540A581A613), seen in six isolates and (A16N51C59N108I164 + S436G437E540A581A613) seen in one isolate.
Mutations in the propeller domain of the kelch protein coded by the Pfk13 gene (PF3D7_1343700) have been shown to be associated with resistance to artemisinin derivatives (Ariey et al., 2014; Straimer et al., 2015), which is defined as a delayed clearance of parasites from the blood following artemisinin administration (Dondorp et al., 2009). The Pfk13 gene was completely conserved among 92/158 isolates that were successfully sequenced at this locus, and a majority of the SNPs observed in the remaining isolates led to synonymous changes (60/92 SNPs). Only two isolates had substitutions in the propeller domain: V555L in N0319-V1, and A578S in R5025. We did not identify any of the four validated SNPs (C580Y, R539T, I543T or Y493H) implicated in artemisinin resistance in our isolates (Straimer et al., 2015). The K189T mutation was seen in 17 isolates in our study, all from Rourkela. Mutations encoding amino acid substitutions in PfCRT (such as I356T) are part of the genetic background associated with the emergence of artemisinin resistance (Miotto et al., 2015), and were present in 20 isolates from our study (18 from Rourkela, two from Nadiad).
Resistance to the artemisinin combination therapy (ACT) partner drugs lumefantrine, mefloquine and piperaquine, has also been linked to certain Pfcrt and Pfmdr1 haplotypes. The wild type PfCRT K76 allele, associated with lumefantrine tolerance (Sisowath et al., 2009), was present in 46/129 isolates, all from Rourkela. The wild-type N86 allele of PfMDR1, associated with decreased susceptibility to lumefantrine (Sisowath et al., 2005), was observed in 103 of a total of 129 isolates in our study bearing only one Pfmdr1 haplotype. The N86F184D1246 haplotype, selected for after artemether-lumefantrine (AL) treatment in Africa (Dokomajilar et al., 2006; Humphreys et al., 2007), was observed in 58/129 isolates.
For 14 subjects (three from Chennai and eleven from Nadiad), drug resistance genes were sequenced from parasite isolates collected on day 2, or day 7, or day 14 following ACT treatment. A majority of the post-treatment follow-up samples had drug resistance haplotypes that were identical to samples collected from the same patient at day 0 (Supplementary Table 2). However, subject N0319 had a mixture of wild-type (N86Y184S1034N1042D1246) and mutant (Y86F184S1034N1042D1246) PfMDR1 haplotypes on day 0, but only the mutant (Y86F184S1034N1042D1246) haplotype could be detected on day 2 (Supplementary Table 2). Most subjects did not have parasitemia detectable by microscopy or PCR day 2 onwards following ACT treatment.
In order to identify possible multi-clonal infections among the clinical isolates, we ranked the isolates based on the total number of high-quality heterozygous variants (i.e., positions containing more than one allele) as described earlier (Rao et al., 2016) (Figure 1). Isolates containing more than twice the median number of heterozygous variants in the dataset were classified as potential multi-clonal infections. This included 30 isolates in total, eight from Nadiad and 22 from Rourkela (Supplementary Table 3). We did not identify any multiclonal infections in Chennai.
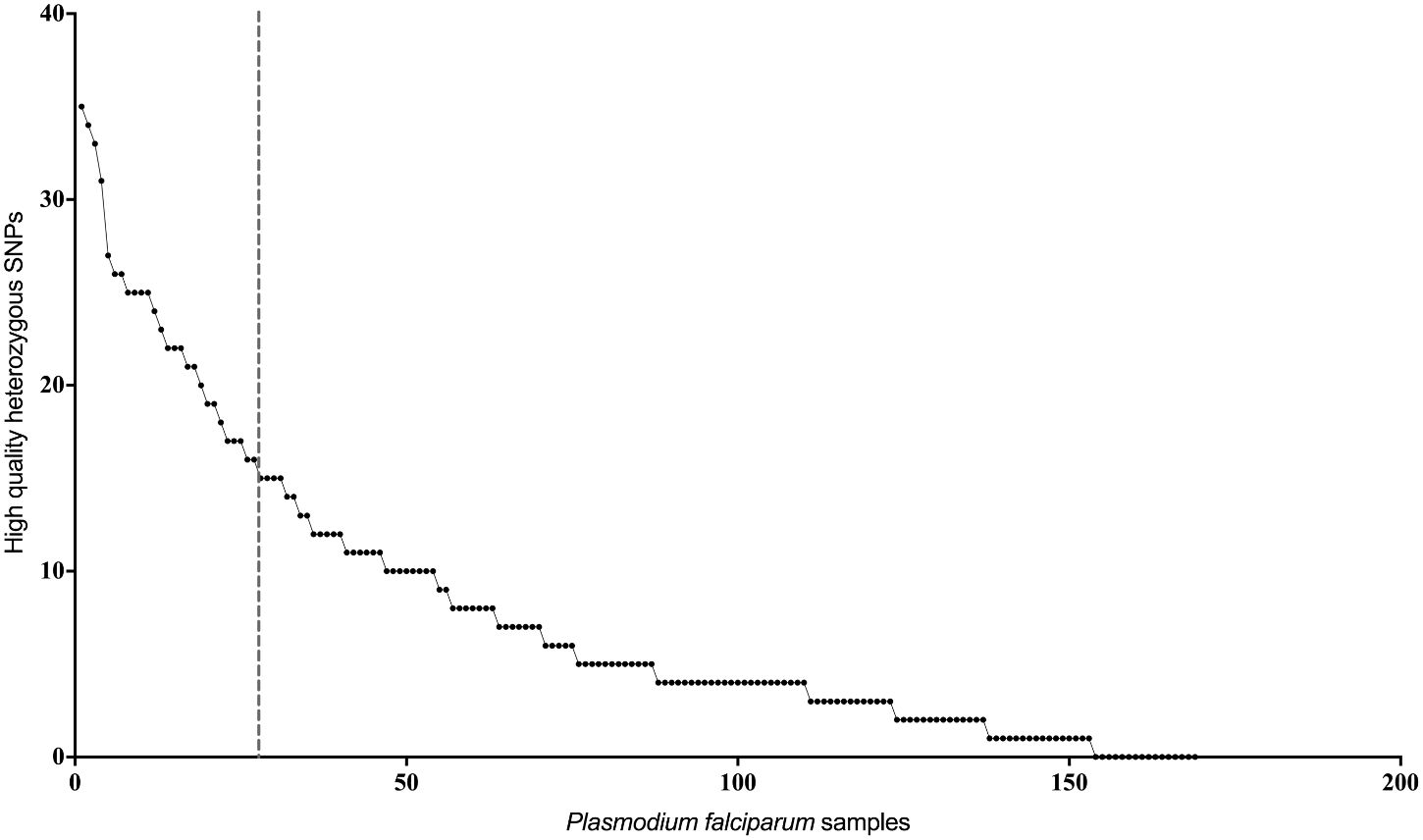
Figure 1 Estimation of mixed genotypes using heterozygous variant calls. Plot showing number of high quality heterozygous variant calls (y-axis) for each P. falciparum clinical isolate. Isolates on the left of the vertical dotted line, which represents twice the median number of heterozygous variants observed in the entire dataset, were classified as potential mixed genotype infections.
In order to identify population-specific variation in the drug resistance genes we performed principal component analysis (PCA) using sequence data at 230 polymorphic positions. The first two principal components accounted for 49.1% of the variation in the SNP data (PC1 = 30.74%, PC2 = 18.36%). The PCA showed isolates from Chennai and Nadiad clustering together, while isolates from Rourkela were split into two separate clusters (Figure 2A). Further analysis revealed that the three clusters correspond to distinct Pfcrt haplotypes (codons 72-76). The two clusters of isolates from Rourkela were associated with haplotypes C72V73I74E75T7 and C72V73M74N75K76, whereas the cluster containing isolates from Chennai and Nadiad was associated with the S72V73M74N75T76 haplotype. Depending on the specific Pfcrt haplotype, MR4 reference strains were placed across all three clusters (Figure 2B).
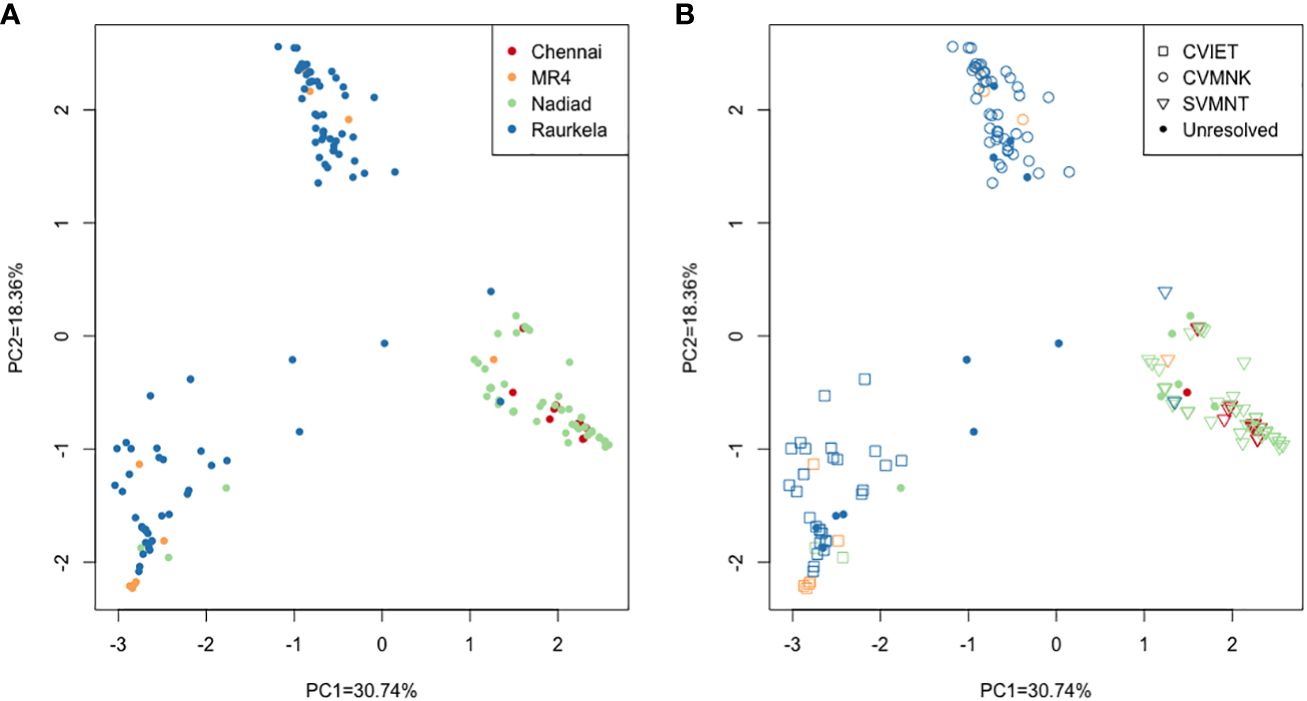
Figure 2 Principal component analysis using high quality SNP data from all P. falciparum isolates. (A) Each isolate is colored by site; (B) Each isolate is colored by site and shape denotes haplotype observed at PfCRT 72-76 locus. Unresolved refers to missing or partial data and mixed infection samples.
Using our NGS amplicon-sequencing protocol to genotype 158 isolates at five full length drug resistance loci, we surveyed antimalarial drug resistance allele frequencies of P. falciparum infections at three field sites. We observed a diverse distribution of three PfCRT and five PfMDR1 haplotypes across the sites: the CQ resistance-associated PfCRT S72V73M74N75T76 haplotype (Mehlotra et al., 2001; Mixson-Hayden et al., 2010) likely introduced to South India through Sri Lanka or the Andaman and Nicobar Islands from Papua New Guinea (Mallick et al., 2013) was fixed in Chennai and Nadiad, while in Rourkela either the CQ resistance-associated PfCRT C72V73I74E75T76 haplotype or the CQ-sensitive wild-type PfCRT C72V73M74N75K76 haplotype was found. Since Rourkela has the highest prevalence of P. falciparum, and the prevalence of P. vivax is higher in Chennai and Nadiad, resulting in presumably greater CQ usage and resulting drug pressure, this may explain the higher PfCRT S72V73M74N75T76 haplotype frequency at the latter two field sites. This is also borne out by PCA, where the Rourkela parasites group into two distinct clusters, corresponding to CQ-resistant (C72V73I74E75T76) and CQ-sensitive (C72V73M74N75K76) haplotypes. While the S72V73M74N75T76 haplotype is predominant in most of India and Odisha showed a higher prevalence of the C72V73I74E75T76 haplotype (Mallick et al., 2012). This haplotype may have been introduced to Rourkela through Northeast India, following the well-established migratory route of Southeast Asian P. falciparum strains into India. We propose that in the absence of CQ pressure in Rourkela, there has been reversion to the wild-type C72V73M74N75K76 haplotype from the mutant C72V73I74E75T76 haplotype as the fitness cost associated with the C72V73I74E75T76 haplotype may be greater than that of the S72V73M74N75T76 haplotype (Sá et al., 2009).
CQ resistance can also be modulated by mutations in PfMDR1 when they occur in the background of resistance-conferring mutations in PfCRT (Sá et al., 2009). The PfMDR1 substitution N86Y contributes to CQ-resistance but has been associated with sensitization of P. falciparum parasites to dihydroartemisinin (DHA) and lumefantrine (LMF) (Veiga et al., 2016). We observed a predominance of the PfMDR1 N86 allele and PfCRT S72V73M74N75T76 haplotype in Chennai and Nadiad, which corroborates previous observations in regions of low P. falciparum transmission in India (Mallick et al., 2012).
In this study, the wild-type SUL-sensitive Pfdhps haplotype S436A437K540A581A613 was predominant across all three sites, occurring primarily in combination with the PYR-resistant, double-mutant Pfdhfr haplotype A16N51R59N108I164 Nadiad and Chennai. However, in Rourkela, there were comparable numbers of isolates with the wild type A16N51C59S108I164 and double-mutant A16N51R59N108I164 DHFR haplotypes. The double mutant A16N51R59N108I164 Pfdhfr haplotype was predominant at each site, corroborating previous studies from India (Ahmed et al., 2004; Kar et al., 2016). We primarily detect wild-type Pfdhps and double-mutant Pfdhfr haplotypes, that confer no resistance or low-level resistance to PYR and SUL, respectively. This observation coupled with the absence of quadruple or quintuple mutant haplotypes at all three sites suggest that P. falciparum resistance to SP therapy at these three sites is low (Ahmed et al., 2004). Recent studies also showed a similar observation of high frequency of wildtype Pfdhps in various parts of India (Pathak et al., 2020; Jacob et al., 2021; Rana et al., 2022) and wildtype Pfdhfr haplotype in Odisha (Rana et al., 2022). However, double mutant Pfdhfr haplotypes prevail in central (Pathak et al., 2020), western (Ozarkar et al., 2021) and northeastern part of India (Jacob et al., 2021).
This study also generated sequence data for the full length pfk13 gene from isolates at our three sites Rourkela, Chennai and Nadiad. Our study identified two mutations in the propeller region, one from an isolate in Nadiad (V555L in sample N0319-V1) and the other from Rourkela (A578S in sample R5025). The V555L mutation has been detected in Senegal at a very low frequency (Talundzic et al., 2017), while A578S has been detected in several isolates from Africa, India and Bangladesh (Mohon et al., 2014; Mishra et al., 2015; Menard et al., 2016; Mishra et al., 2016). Neither has been associated with resistance among field isolates or in in vitro assays. The K189T mutation outside the propellor domain observed in our study in a subset of isolates from Rourkela has been reported from Senegal, Guyana, the China-Myanmar border, and Northeast India (Torrentino-Madamet et al., 2014; Wang et al., 2015; Mishra et al., 2016; Rahman et al., 2016). These rare and random pfk13 gene mutations in Indian parasites observed in various geographic areas suggests that no selection pressure is occurring in the field to enhance expansion of these genotypes.
Continued surveillance of pfk13 haplotypes in India is vital and pressing. The World Health Organization has classified 13 mutations as validated molecular markers for artemisinin resistance and nine mutations are listed as ‘candidate or associated’ markers (WHO, 2022). Since one of the validated mutations F446I is commonly observed in Myanmar and also reported from the eastern border of India (Tun et al., 2015; Kagoro et al., 2022), there has been concern about the spread of artemisinin resistance into India in a manner similar to that seen for the spread of CQ and SP resistance. In addition, independent emergence of these artemisinin resistance mutations has been observed in various countries in Africa (Musyoka et al., 2020; Kayiba et al., 2021; Owoloye et al., 2021), Papua New Guinea (Miotto et al., 2020), South America (Chenet et al., 2016), and Southeast Asia (Takala-Harrison et al., 2015), indicating that emergence could occur in any endemic country. Currently, 13 of the mutations in the propellor domain of pfk13 gene have been reported from areas in India in low frequencies (Chatterjee et al., 2015; Mishra et al., 2015; Mishra et al., 2016; Manuj et al., 2017; Mishra et al., 2017; Das et al., 2018; Choubey et al., 2023), with only three of the W.H.O. validated mutations having been observed, and mutations occurring rarely and randomly across different geographic areas (Mishra et al., 2015; Mishra et al., 2016; Das et al., 2018). Only three of the ‘associated’ mutations were observed in more than one geographic area (Mishra et al., 2016; Manuj et al., 2017; Mishra et al., 2017). Only one study has reported a novel mutation, G625R, in the pfk13 propellor domain in parasites from West Bengal which showed association with artemisinin resistance (Das et al., 2018). However, a recent analysis of 53 whole genome sequences of parasites from West Bengal found no known mutations associated with artemisinin resistance in the pfk13 gene (Choubey et al., 2023).
Our parasite isolates from Rourkela showed greater diversity than isolates from Chennai and Nadiad at all drug resistance loci. PCA clustering revealed that parasites from Chennai and Nadiad were more closely related, whereas the parasites in Rourkela separated into two populations that were associated with distinct Pfcrt haplotypes. There was a near-equal distribution of wild-type and mutant haplotypes at the Pfcrt, Pfdhfr and Pfmdr1 loci in Rourkela. The eight isolates that possessed wild-type haplotypes at all the drug resistance loci were also from Rourkela. More than 70% of the possible multi-clonal infections identified were from Rourkela, the remainder from Nadiad, and none were from Chennai. More infections are likely to be multi-clonal in isolates from Rourkela because of the higher prevalence and transmission of P. falciparum there, compared to Nadiad or Chennai, where the prevalence of P. falciparum is much lower.
Another application of our amplicon-seq assay is to study the variation in within-host diversity in response to chemotherapy. This is particularly interesting for multi-clonal infections as it can enable us to track changes in the frequency of individual alleles before and after drug treatment (Mideo et al., 2013). Our analysis of parasite from follow-up (collected after ACT treatment) samples was limited to a small number of isolates from Chennai and Nadiad. For all pairs of isolates collected from the same individual, the key drug resistance SNPs were identical at enrollment and follow-up isolates, except in the case of one patient isolate from Nadiad that possessed a mixture of CQ-resistant and CQ-sensitive haplotypes at the Pfmdr1 locus at enrollment but retained only the CQ-resistant haplotype following drug treatment. Deep sequencing methods provide a means to identify changes in frequency of drug resistance alleles as well as identify new mutations as they are selected during drug treatment. However, performing deep sequencing require a technical expertise on library preparation and bioinformatic analysis, which may limit such means of molecular surveillance to few central labs in the country. In contrast, routine sanger sequencing is easy to perform and analyze, but associated with higher cost when performed for full-length multiple genes.
There are two key limitations of our study. Firstly, the relatively small size of follow-up samples post-ACT treatment constrains the depth of our analysis concerning alterations in drug resistance allele frequencies after treatment. Future longitudinal investigations with larger samples are essential for a more thorough understanding of the dynamics of drug resistance evolution. Secondly, the study was limited to three specific field sites in India, and findings may not be applicable to other regions within the country. Future studies should aim to incorporate a wider geographic scope to capture the full diversity of drug resistance profiles in India. In addition, study was not able to capture the copy number variation in Pfmdr1 gene, which could have inferred signature of multidrug resistance in P. falciparum.
In conclusion, our study presents a comprehensive analysis of antimalarial drug resistance allele frequencies in P. falciparum isolates across three diverse field sites in India. The observed heterogeneity in drug resistance haplotypes underscores the complex dynamics of drug pressure and genetic selection within parasite populations. Our findings demonstrate the fixation of the CQ resistance-associated PfCRT S72V73M74N75T76 haplotype in Chennai and Nadiad, contrasting with the mixed haplotype distribution in Rourkela, suggestive of varying levels of CQ pressure. Furthermore, we identify a predominance of the PfMDR1 N86 allele and PfCRT S72V73M74N75T76 haplotype in low transmission settings, indicating potential targets for focused surveillance and intervention efforts. While variability in PfDHFR haplotypes was observed across sites, the predominance of the double mutant A16N51R59N108I164 haplotype underscores the importance of continued monitoring for SP resistance. In pfk13 rare mutations were identified, suggesting limited selection pressure for artemisinin resistance in Indian parasites. Our study highlights the significance of considering within-host diversity and multi-clonal infections in malaria transmission dynamics, providing insights essential for guiding evidence-based control strategies. India has evidenced the decline in malaria cases with the usage of artemisinin combination therapy, which in turn may have declined the resistant genotypes for antimalarials. The observations of high frequency of wild type genotypes for Pfcrt gene suggest further molecular surveillance for the evidence of reversal of chloroquine resistance in the P. falciparum prevalent areas. Low number of mutations observed in Pfdhfr gene may have similar implication. This study contributes to the ongoing efforts towards malaria elimination by informing targeted surveillance, treatment strategies, and the development of novel interventions aimed at mitigating the spread of drug-resistant malaria in India.
The original contributions presented in the study are publicly available. This data can be found here: NCBI SRA, accession PRJNA1062426. All data are also available through the WorldWide Antimalarial Research Network (WWARN) website http://www.wwarn.org/tracking-resistance with WWARN unique identifier DINZU.
The studies involving humans were approved by New York University Institutional Review Board (Study #i10-00173) and the Ethics Committee of the National Institute of Malaria Research, India (ICMR). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
SK: Formal Analysis, Writing – original draft. SU: Methodology, Writing – original draft. NB: Formal analysis, Writing – original draft. PR: Methodology, Writing – original draft. SA: Investigation, Writing – review & editing. SS: Project administration, Writing – review & editing. NT: Investigation, Writing – review & editing. AP: Investigation, Writing – review & editing. RS: Project administration, Writing – review & editing. AD: Formal analysis, Writing – original draft. SR: Methodology, Writing – review & editing. GL: Methodology, Writing – review & editing. AA: Methodology, Writing – review & editing. AE: Project administration, Writing – original draft. OS: Project administration, Supervision, Writing – original draft. JC: Conceptualization, Funding acquisition, Writing – review & editing, Writing – original draft. PM: Supervision, Writing – review & editing, Writing – original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number U19AI089676 as part of the International Centers for Excellence in Malaria Research.
We thank the Malaria Reference and Resource Reagent repository MR4 for providing P. falciparum strains. We thank Drs. Lalitha Ramanathapuram, Patrick Sutton, Anna Maria van Eijk and CSCMi teams in Delhi, Nadiad, Rourkela and Chennai for their work, and Dr. Martina Bradic at New York University for linkage analysis. We also thank Director and staff of the National institute of Malaria Research in Delhi, India. This manuscript bears the National Institute of Malaria Research publication screening committee approval number 56/2019.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmala.2024.1363969/full#supplementary-material
Ahmed A., Bararia D., Vinayak S., Yameen M., Biswas S., Dev V., et al. (2004). Plasmodium falciparum isolates in India exhibit a progressive increase in mutations associated with sulfadoxine-pyrimethamine resistance. Antimicrob. Agents Chemother. 48, 879–889. doi: 10.1128/AAC.48.3.879-889.2004
Anvikar A. R., Arora U., Sonal G., Mishra N., Shahi B., Savargaonkar D., et al. (2014). Antimalarial drug policy in India: Past, present & future. Indian J. Med. Res. 139, 205–215.
Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A. C., Khim N., et al. (2014). A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55. doi: 10.1038/nature12876
Brooks D. R., Wang P., Read M., Watkins W. M., Sims P. F., Hyde J. E. (1994). Sequence variation of the hydroxymethyldihydropterin pyrophosphokinase: dihydropteroate synthase gene in lines of the human malaria parasite, Plasmodium falciparum, with differing resistance to sulfadoxine. Eur. J. Biochem. 224, 397–405. doi: 10.1111/j.1432-1033.1994.00397.x
Carlton J. M., Eapen A., Kessler A., Anvikar A. R., Hoffmann A., Singh O. P., et al. (2022a). Advances in basic and translational research as part of the center for the study of complex malaria in India. Am. J. Trop. Med. Hyg 107, 97–106. doi: 10.4269/ajtmh.21-1333
Carlton J. M., Sahu P. K., Wassmer S. C., Mohanty S., Kessler A., Eapen A., et al. (2022b). The impact, emerging needs, and new research questions arising from 12 years of the center for the study of complex malaria in India. Am. J. Trop. Med. Hyg 107, 90–96. doi: 10.4269/ajtmh.21-1277
Chatterjee M., Ganguly S., Saha P., Bankura B., Basu N., Das M., et al. (2015). No polymorphism in plasmodium falciparum K13 propeller gene in clinical isolates from kolkata, India. J. Pathog. 2015, 374354. doi: 10.1155/2015/374354
Chenet S. M., Akinyi Okoth S., Huber C. S., Chandrabose J., Lucchi N. W., Talundzic E., et al. (2016). Independent emergence of the plasmodium falciparum kelch propeller domain mutant allele C580Y in Guyana. J. Infect. Dis. 213, 1472–1475. doi: 10.1093/infdis/jiv752
Choubey D., Deshmukh B., Rao A. G., Kanyal A., Hati A. K., Roy S., et al. (2023). Genomic analysis of Indian isolates of Plasmodium falciparum: Implications for drug resistance and virulence factors. Int. J. Parasitol. Drugs Drug Resist. 22, 52–60. doi: 10.1016/j.ijpddr.2023.05.003
Cowman A. F., Morry M. J., Biggs B. A., Cross G. A., Foote S. J. (1988). Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc. Natl. Acad. Sci. U S A. 85, 9109–9113. doi: 10.1073/pnas.85.23.9109
Das A., Anvikar A. R., Cator L. J., Dhiman R. C., Eapen A., Mishra N., et al. (2012). Malaria in India: the center for the study of complex malaria in India. Acta tropica. 121, 267–273. doi: 10.1016/j.actatropica.2011.11.008
Das S., Kar A., Manna S., Mandal S., Mandal S., Das S., et al. (2021). Artemisinin combination therapy fails even in the absence of Plasmodium falciparum kelch13 gene polymorphism in Central India. Sci. Rep. 11, 9946. doi: 10.1038/s41598-021-89295-0
Das S., Saha B., Hati A. K., Roy S. (2018). Evidence of artemisinin-resistant plasmodium falciparum malaria in eastern India. N Engl. J. Med. 379, 1962–1964. doi: 10.1056/NEJMc1713777
Djimdé A., Doumbo O. K., Steketee R. W., Plowe C. V. (2001). Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet 358, 890–891. doi: 10.1016/S0140-6736(01)06040-8
Dokomajilar C., Nsobya S. L., Greenhouse B., Rosenthal P. J., Dorsey G. (2006). Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob. Agents Chemother. 50, 1893–1895. doi: 10.1128/AAC.50.5.1893-1895.2006
Dondorp A. M., Nosten F., Yi P., Das D., Phyo A. P., Tarning J., et al. (2009). Artemisinin resistance in Plasmodium falciparum malaria. N Engl. J. Med. 361, 455–467. doi: 10.1056/NEJMoa0808859
Fidock D. A., Nomura T., Talley A. K., Cooper R. A., Dzekunov S. M., Ferdig M. T., et al. (2000). Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 6, 861–871. doi: 10.1016/S1097-2765(05)00077-8
Humphreys G. S., Merinopoulos I., Ahmed J., Whitty C. J., Mutabingwa T. K., Sutherland C. J., et al. (2007). Amodiaquine and artemether-lumefantrine select distinct alleles of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for uncomplicated malaria. Antimicrob. Agents Chemother. 51, 991–997. doi: 10.1128/AAC.00875-06
Jacob C. G., Thuy-Nhien N., Mayxay M., Maude R. J., Quang H. H., Hongvanthong B., et al. (2021). Genetic surveillance in the Greater Mekong subregion and South Asia to support malaria control and elimination. Elife 10. doi: 10.7554/eLife.62997.sa2
Kagoro F. M., Barnes K. I., Marsh K., Ekapirat N., Mercado C. E. G., Sinha I., et al. (2022). Mapping genetic markers of artemisinin resistance in Plasmodium falciparum malaria in Asia: a systematic review and spatiotemporal analysis. Lancet Microbe 3, e184–ee92. doi: 10.1016/S2666-5247(21)00249-4
Kar N. P., Chauhan K., Nanda N., Kumar A., Carlton J. M., Das A. (2016). Comparative assessment on the prevalence of mutations in the Plasmodium falciparum drug-resistant genes in two different ecotypes of Odisha state, India. Infect. Genet. Evol. 41, 47–55. doi: 10.1016/j.meegid.2016.03.014
Kayiba N. K., Yobi D. M., Tshibangu-Kabamba E., Tuan V. P., Yamaoka Y., Devleesschauwer B., et al. (2021). Spatial and molecular mapping of Pfkelch13 gene polymorphism in Africa in the era of emerging Plasmodium falciparum resistance to artemisinin: a systematic review. Lancet Infect. Dis. 21, e82–e92. doi: 10.1016/S1473-3099(20)30493-X
Mallick P. K., Joshi H., Valecha N., Sharma S. K., Eapen A., Bhatt R. M., et al. (2012). Mutant pfcrt “SVMNT” haplotype and wild type pfmdr1 “N86” are endemic in Plasmodium vivax dominated areas of India under high chloroquine exposure. Malar J. 11, 16. doi: 10.1186/1475-2875-11-16
Mallick P. K., Sutton P. L., Singh R., Singh O. P., Dash A. P., Singh A. K., et al. (2013). Microsatellite analysis of chloroquine resistance associated alleles and neutral loci reveal genetic structure of Indian Plasmodium falciparum. Infect. Genet. Evol. 19, 164–175. doi: 10.1016/j.meegid.2013.07.009
Manuj K., Das M. C. K., Chetry S., Dutta P. (2017). PfK13 kelch propeller domain and Pfmdr1 sequence polymorphism in Plasmodium falciparum field isolates from Northeast region, India. Hum. Parasitic Dis. 2017, 9 1–9 9.
Mehlotra R. K., Fujioka H., Roepe P. D., Janneh O., Ursos L. M., Jacobs-Lorena V., et al. (2001). Evolution of a unique Plasmodium falciparum chloroquine-resistance phenotype in association with pfcrt polymorphism in Papua New Guinea and South America. Proc. Natl. Acad. Sci. U S A. 98, 12689–12694. doi: 10.1073/pnas.221440898
Menard D., Dondorp A. (2017). Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. doi: 10.1101/cshperspect.a025619
Menard D., Khim N., Beghain J., Adegnika A. A., Shafiul-Alam M., Amodu O., et al. (2016). A worldwide map of plasmodium falciparum K13-propeller polymorphisms. N Engl. J. Med. 374, 2453–2464. doi: 10.1056/NEJMoa1513137
Mideo N., Kennedy D. A., Carlton J. M., Bailey J. A., Juliano J. J., Read A. F. (2013). Ahead of the curve: next generation estimators of drug resistance in malaria infections. Trends Parasitol. 29, 321–328. doi: 10.1016/j.pt.2013.05.004
Miotto O., Amato R., Ashley E. A., MacInnis B., Almagro-Garcia J., Amaratunga C., et al. (2015). Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234. doi: 10.1038/ng.3189
Miotto O., Sekihara M., Tachibana S. I., Yamauchi M., Pearson R. D., Amato R., et al. (2020). Emergence of artemisinin-resistant Plasmodium falciparum with kelch13 C580Y mutations on the island of New Guinea. PloS Pathog. 16, e1009133. doi: 10.1371/journal.ppat.1009133
Mishra N., Bharti R. S., Mallick P., Singh O. P., Srivastava B., Rana R., et al. (2016). Emerging polymorphisms in falciparum Kelch 13 gene in Northeastern region of India. Malar J. 15, 583. doi: 10.1186/s12936-016-1636-4
Mishra S., Bharti P. K., Shukla M. M., Ali N. A., Kashyotia S. S., Kumar A., et al. (2017). Clinical and molecular monitoring of Plasmodium falciparum resistance to antimalarial drug (artesunate+sulphadoxine-pyrimethamine) in two highly malarious district of Madhya Pradesh, Central India from 2012-2014. Pathog. Glob Health 111, 186–194. doi: 10.1080/20477724.2017.1331875
Mishra N., Prajapati S. K., Kaitholia K., Bharti R. S., Srivastava B., Phookan S., et al. (2015). Surveillance of artemisinin resistance in Plasmodium falciparum in India using the kelch13 molecular marker. Antimicrob. Agents Chemother. 59, 2548–2553. doi: 10.1128/AAC.04632-14
Mixson-Hayden T., Jain V., McCollum A. M., Poe A., Nagpal A. C., Dash A. P., et al. (2010). Evidence of selective sweeps in genes conferring resistance to chloroquine and pyrimethamine in Plasmodium falciparum isolates in India. Antimicrob. Agents Chemother. 54, 997–1006. doi: 10.1128/AAC.00846-09
Mohon A. N., Alam M. S., Bayih A. G., Folefoc A., Shahinas D., Haque R., et al. (2014). Mutations in Plasmodium falciparum K13 propeller gene from Bangladesh (2009-2013). Malar J. 13, 431. doi: 10.1186/1475-2875-13-431
Musyoka K. B., Kiiru J. N., Aluvaala E., Omondi P., Chege W. K., Judah T., et al. (2020). Prevalence of mutations in Plasmodium falciparum genes associated with resistance to different antimalarial drugs in Nyando, Kisumu County in Kenya. Infect. Genet. Evol. 78, 104121. doi: 10.1016/j.meegid.2019.104121
Owoloye A., Olufemi M., Idowu E. T., Oyebola K. M. (2021). Prevalence of potential mediators of artemisinin resistance in African isolates of Plasmodium falciparum. Malar J. 20, 451. doi: 10.1186/s12936-021-03987-6
Ozarkar A., Kanyal A., Dass S., Deshpande P., Deobagkar D., Karmodiya K. (2021). Analysis of drug resistance marker genes of Plasmodium falciparum after implementation of artemisinin-based combination therapy in Pune district, India. J. Biosci. 46. doi: 10.1007/s12038-021-00200-3
Pandit K., Surolia N., Bhattacharjee S., Karmodiya K. (2023). The many paths to artemisinin resistance in Plasmodium falciparum. Trends Parasitol. 39, 1060–1073. doi: 10.1016/j.pt.2023.09.011
Pathak A., Mårtensson A., Gawariker S., Sharma A., Diwan V., Purohit M., et al. (2020). Stable high frequencies of sulfadoxine-pyrimethamine resistance associated mutations and absence of K13 mutations in Plasmodium falciparum 3 and 4 years after the introduction of artesunate plus sulfadoxine-pyrimethamine in Ujjain, Madhya Pradesh, India. Malar J. 19, 290. doi: 10.1186/s12936-020-03274-w
Quinlan A. R., Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rahman R., Martin M. J., Persaud S., Ceron N., Kellman D., Musset L., et al. (2016). Continued sensitivity of plasmodium falciparum to artemisinin in Guyana, with absence of kelch propeller domain mutant alleles. Open Forum Infect. Dis. 3, ofw185. doi: 10.1093/ofid/ofw185
Rana R., Khan N., Sandeepta S., Pati S., Das A., Bal M., et al. (2022). Molecular surveillance of anti-malarial drug resistance genes in Plasmodium falciparum isolates in Odisha, India. Malar J. 21, 394. doi: 10.1186/s12936-022-04403-3
Rao P. N., Uplekar S., Kayal S., Mallick P. K., Bandyopadhyay N., Kale S., et al. (2016). A method for amplicon deep sequencing of drug resistance genes in plasmodium falciparum clinical isolates from India. J. Clin. Microbiol. 54, 1500–1511. doi: 10.1128/JCM.00235-16
Rasmussen C., Valecha N., Ringwald P. (2019). Lack of convincing evidence of artemisinin resistance in India. Clin. Infect. Dis. 69, 1461–1462. doi: 10.1093/cid/ciz166
Sá J. M., Twu O., Hayton K., Reyes S., Fay M. P., Ringwald P., et al. (2009). Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc. Natl. Acad. Sci. U S A. 106, 18883–18889. doi: 10.1073/pnas.0911317106
Shah N. K., Dhillon G. P., Dash A. P., Arora U., Meshnick S. R., Valecha N. (2011). Antimalarial drug resistance of Plasmodium falciparum in India: changes over time and space. Lancet Infect. Dis. 11, 57–64. doi: 10.1016/S1473-3099(10)70214-0
Sidhu A. B., Verdier-Pinard D., Fidock D. A. (2002). Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298, 210–213. doi: 10.1126/science.1074045
Sirawaraporn W., Sathitkul T., Sirawaraporn R., Yuthavong Y., Santi D. V. (1997). Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc. Natl. Acad. Sci. U S A. 94, 1124–1129. doi: 10.1073/pnas.94.4.1124
Sisowath C., Petersen I., Veiga M. I., Mårtensson A., Premji Z., Björkman A., et al. (2009). In vivo selection of Plasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J. Infect. Dis. 199, 750–757. doi: 10.1086/596738
Sisowath C., Strömberg J., Mårtensson A., Msellem M., Obondo C., Björkman A., et al. (2005). In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by artemether-lumefantrine (Coartem). J. Infect. Dis. 191, 1014–1017. doi: 10.1086/427997
Straimer J., Gnädig N. F., Witkowski B., Amaratunga C., Duru V., Ramadani A. P., et al. (2015). Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347, 428–431. doi: 10.1126/science.1260867
Takala-Harrison S., Jacob C. G., Arze C., Cummings M. P., Silva J. C., Dondorp A. M., et al. (2015). Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 211, 670–679. doi: 10.1093/infdis/jiu491
Talundzic E., Ndiaye Y. D., Deme A. B., Olsen C., Patel D. S., Biliya S., et al. (2017). Molecular Epidemiology of Plasmodium falciparum kelch13 Mutations in Senegal Determined by Using Targeted Amplicon Deep Sequencing. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.02116-16
Thorvaldsdottir H., Robinson J. T., Mesirov J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Torrentino-Madamet M., Fall B., Benoit N., Camara C., Amalvict R., Fall M., et al. (2014). Limited polymorphisms in k13 gene in Plasmodium falciparum isolates from Dakar, Senegal in 2012-2013. Malar J. 13, 472. doi: 10.1186/1475-2875-13-472
Triglia T., Wang P., Sims P. F., Hyde J. E., Cowman A. F. (1998). Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J. 17, 3807–3815. doi: 10.1093/emboj/17.14.3807
Tun K. M., Imwong M., Lwin K. M., Win A. A., Hlaing T. M., Hlaing T., et al. (2015). Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect. Dis. 15, 415–421. doi: 10.1016/S1473-3099(15)70032-0
van Eijk A. M., Sutton P. L., Ramanathapuram L., Sullivan S. A., Kanagaraj D., Priya G. S. L., et al. (2019). The burden of submicroscopic and asymptomatic malaria in India revealed from epidemiology studies at three varied transmission sites in India. Sci. Rep. 9, 17095. doi: 10.1038/s41598-019-53386-w
Vathsala P. G., Pramanik A., Dhanasekaran S., Devi C. U., Pillai C. R., Subbarao S. K., et al. (2004). Widespread occurrence of the Plasmodium falciparum chloroquine resistance transporter (Pfcrt) gene haplotype SVMNT in P. falciparum malaria in India. Am. J. Trop. Med. Hyg. 70, 256–259. doi: 10.4269/ajtmh.2004.70.256
Veiga M. I., Dhingra S. K., Henrich P. P., Straimer J., Gnädig N., Uhlemann A. C., et al. (2016). Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat. Commun. 7, 11553. doi: 10.1038/ncomms11553
Wang Z., Shrestha S., Li X., Miao J., Yuan L., Cabrera M., et al. (2015). Prevalence of K13-propeller polymorphisms in Plasmodium falciparum from China-Myanmar border in 2007-2012. Malar J. 14, 168. doi: 10.1186/s12936-015-0672-9
Warrenfeltz S., Basenko E. Y., Crouch K., Harb O. S., Kissinger J. C., Roos D. S., et al. (2018). EuPathDB: the eukaryotic pathogen genomics database resource. Methods Mol. Biol. 1757, 69–113.
WHO (2022). Available online at: https://www.who.int/news-room/questions-and-answers/item/artemisinin-resistance.
Keywords: malaria, Plasmodium falciparum, antimalarial drug resistance, genomic surveillance, India
Citation: Kale S, Uplekar SM, Bandyopadhyay N, Rao PN, Ali SZ, Sharma SK, Tandel N, Patel A, Singh R, Dank A, Ravishankaran S, Priya GSL, Asokan A, Eapen A, Singh OP, Carlton JM and Mallick PK (2024) Antimalarial drug resistance profiling of Plasmodium falciparum infections in India using Ion Torrent deep sequencing. Front. Malar. 2:1363969. doi: 10.3389/fmala.2024.1363969
Received: 31 December 2023; Accepted: 12 March 2024;
Published: 03 April 2024.
Edited by:
Alena Pance, University of Hertfordshire, United KingdomReviewed by:
Ariel H. Magallon-Tejada, Gorgas Memorial Institute of Health Studies, PanamaCopyright © 2024 Kale, Uplekar, Bandyopadhyay, Rao, Ali, Sharma, Tandel, Patel, Singh, Dank, Ravishankaran, Priya, Asokan, Eapen, Singh, Carlton and Mallick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane M. Carlton, SmFuZUNhcmx0b25Aamh1LmVkdQ==; Prashant K. Mallick, cGttbXJjMDFAZ21haWwuY29t
†Present addresses: Sonal Kale, Laboratory of Malaria Immunology and Vaccinology, National Institute of Allergy and Infectious Disease, NIH, Bethesda, MD, United States
Jane M. Carlton, Department of Molecular Microbiology and Immunology, Johns Hopkins Malaria Research Institute, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, United States
Nikunj Tandel, Institute of Science, Nirma University, Ahmedabad, Gujarat, India
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.