 Tobias O. Apinjoh1,2*
Tobias O. Apinjoh1,2* Marcelus U. Ajonina3,4
Marcelus U. Ajonina3,4 Deriba Abera5
Deriba Abera5 Hanesh F. Chi3
Hanesh F. Chi3 Roland B. Tata1Regina N. Mugri3
Roland B. Tata1Regina N. Mugri3 Lemu Golassa6
Lemu Golassa6 Eric A. Achidi1
Eric A. Achidi1 Alfred Amambua-Ngwa7
Alfred Amambua-Ngwa7- 1Department of Biochemistry and Molecular Biology, University of Buea, Buea, Cameroon
- 2Department of Chemical and Biological Engineering, The University of Bamenda, Bambili, Cameroon
- 3Department of Microbiology and Parasitology, University of Buea, Buea, Cameroon
- 4Department of Public Health, The University of Bamenda, Bambili, Cameroon
- 5School of Medical Laboratory Sciences, Jimma University, Oromia, Ethiopia
- 6Aklilu Lemma Institute of Pathobiology, Addis Ababa University, Addis Ababa, Ethiopia
- 7Medical Research Council Unit The Gambia at London School of Hygiene and Tropical Medicine, Fajara, Gambia
Background: Malaria elimination is threatened by the emergence and rapid spread of drug resistance. Understanding the demographic history of Plasmodium falciparum and the genetic basis of adaptation to antimalarial treatment and host immunity is critical to elimination efforts. This study sought to characterize the diversity of P. falciparum isolates across different altitudes along the slope of Mount Cameroon.
Methodology: Using the Illumina next-generation sequencing platform P. falciparum isolates from low, intermediate and high altitudes along the slope of Mount Cameroon were studied to determine their genetic diversity, population structures, and signatures of selection in known drug resistance alleles.
Results: A total of 77,253 single-nucleotide polymorphisms were identified from 220 quality P. falciparum clinical isolates from high (24,214), intermediate (24,426), and low (28,613) altitude. About 49%, 48.2% and 30% of the parasite isolates from high, intermediate, and low altitudes, respectively had FWS values > 0.95 indicative of dominant mixed genotype infections and low population sub-structure with high potential for out-crossing. No significant difference was observed in within-host diversity while population structure analysis did not separate the isolates in the three major altitudinal groups by PCA, FST and admixtures, suggesting bidirectional gene flow among the populations. A total of 94 antigenic genes under balancing selection were detected in the area including vaccine candidate gene ama1, eba175, msp1, trap, dblmsp, and clag2. Moreover, 17 of these genes were identified to be under both recent positive directional and positive balancing selection including the prominent host immune target genes surfin 8.2, trap, and ama1. Recent directional selection analysis using integrated standardized haplotype score (iHS) did not detect any selection signatures in the Pfdhfr, Pfdhps, Pfmdr1, and PfK13 genes. Furthermore, no PfKelch13 validated mutation associated with artemisinin resistance was identified in this study and no structural divergence was noticed among the P. falciparum parasite populations across different altitudes around the Mount Cameroon region.
Discussion: Findings revealed high genetic diversity and low population structure suggesting that malaria transmission remain high in the study area and parasite populations circulating around the slope of Mount Cameroon are homogenous.
Introduction
Malaria due to Plasmodium falciparum remains the most significant cause of morbidity and death globally, with the number of cases and deaths increasing from 2019 to 2020, mostly owing to medical treatment interruptions during the COVID-19 epidemic (WHO, 2021). The global burden and economic impact of the disease are disproportionately high in sub-Saharan Africa, primarily among children under the age of 5 years (WHO, 2021), with an estimated direct cost of at least US$12 billion each year, with costs affecting both individuals and governments (CDC, 2021). In Cameroon, deaths caused by malaria constitute 30%–35% of the overall fatality and 67% of the yearly pediatric mortality figures (Mbako, 2017).
Despite the tremendous and concerted malaria control efforts made in recent years, with interventions resulting in a global decline in morbidity and mortality between 2000 and 2015 (WHO, 2016), the disease remains endemic in 87 countries and territories (CDC, 2021). The emergence and spread of antimalarial drug and insecticide resistance in the parasite, and among Anopheles mosquitoes, is thought to be a major impediment to the management of malaria especially in endemic regions (Apinjoh et al., 2016; Menard and Dondorp, 2017).
Plasmodium falciparum has developed resistance to practically all antimalarial medications, with Southeast Asia’s Greater Mekong subregion being the locus for most cases of antimalarial drug resistance. Resistance to conventional antimalarials, notably chloroquine (CQ), the backbone of malaria treatment in the 20th century, was first observed in Cambodia in the late 1950s and then spread internationally (Payne, 1987). Later, the same region’s pyrimethamine-resistant parasites also spread to Africa (Roper et al., 2004). Recently, resistance to the artemisinin and non-artemisinin components of artemisinin-based combination therapy (ACT), which is currently used as a first- and second-line treatment in the majority of endemic countries, has emerged in parts of Southeast Asia, threatening the efficacy of this important antimalarial treatment (Arya et al., 2021). A recent study in Uganda (Balikagala et al., 2021) suggests the emergence of artemisinin-resistant P. falciparum in Africa, amidst several documented treatment failures to ACT regimens in the African region (Ndiaye et al., 2008; Rosenthal, 2021; Tuedom et al., 2021).
Widespread chloroquine (CQ) resistance in Cameroon, as in many other malaria-endemic countries, and subsequent antimalarial treatment failures, led to the replacement of CQ, which was widely used from the 1970s (Sansonetti et al., 1985), with amodiaquine (AQ) combined with CQ or sulfadoxine–pyrimethamine (SP) (Basco et al., 2006). Artemisinin combination therapy (ACT) is currently recommended for the treatment of uncomplicated P. falciparum (Sayang et al., 2009; Whegang et al., 2019), and injectable artemether or quinine are recommended for severe malaria, whereas SP remains in use for the intermittent preventative treatment of malaria during pregnancy (IPTp) (PNLP, 2012).
Plasmodium falciparum resistance may occur due to drug pressure on the parasite, which induces spontaneous genetic changes, lowering its sensitivity to host immunity and therapeutic interventions (Kidgell et al., 2006). Genes encoding immunological and therapeutic targets are thus under natural selection and exhibit balanced or directional selection signatures (Duffy et al., 2015) to propagate and transmit “host immune-/drug”-adapted parasite strains. Such selection may be influenced by differences in innate human susceptibility and ecological transmission that alter acquired immunity, and/or drug pressure (Abera et al., 2021). Knowledge about genetic variation patterns due to parasite adaptation to host environments and drug interventions is required for effective malaria control.
Although previous studies on P. falciparum population genomics in East (Abera et al., 2021) and West (Amambua-Ngwa et al., 2012; Mobegi et al., 2014) Africa have revealed strong directional selection around known drug resistance genes and signatures of balancing selection on multiple candidate vaccine antigens, little is known about the genomic variations underlying local adaptive mechanisms of P. falciparum populations in Cameroon. A recent study in Ethiopia revealed that P. falciparum parasites have minimal genetic diversity and that these parasites were structurally divergent from Southeast Asian and other East African groups (Abera et al., 2021).
This study sought to investigate the genetic diversity and the population structure of the P. falciparum parasite at different altitudes around the slope of Mount Cameroon, assess signatures of selection, and determine the prevalence of markers of antimalarial resistance in the parasite population. This will provide actionable information for malaria epidemiology and control.
Materials and methods
Study area
The study was conducted in communities on the eastern slope of Mount Cameroon with varying malaria transmission profiles and topographical characteristics. The area has a forested equatorial climate, which is influenced by the ocean and mountains, with two distinct seasons: a brief dry season (from November to March) and a lengthy rainy season (from March to November) (Wanji et al, 2003). It is characterized by relatively steady temperatures that range from 18°C in August to 35°C in March, and the relative humidity (75%–80%), average annual rainfall (2,625 mm), and precipitation (2,000–10,000 mm) are relatively high (Wanji et al., 2003). Malaria transmission is widespread and perennial in the region, with parasitemia levels being highest during the rainy season and at lower altitudes (Achidi et al., 2008). Plasmodium falciparum is responsible for most infections, whereas Anopheles gambiae is the area’s dominant, most aggressive, and most active malarial vector. Although the Bakweri tribe, of the Bantu ethnic group, is indigenous to this area (Ngwa, 2000), the region’s fertile volcanic soils and large plantations have attracted individuals from other parts of the nation, mostly from the northwest’s Semi-Bantu ethnic group. There is a substantial level of human migration between localities, mainly for educational, recreational, and commercial purposes.
Sample collection and processing
Individuals with asymptomatic or uncomplicated malaria were prescreened using light microscopy and enrolled through cross-sectional surveys in 10 randomly selected rural, semirural, and urban communities between May 2013 and March 2014, as described previously by Apinjoh et al. (2015; 2017). Communities below 200 m, between 385 m and 575 m, and greater than 636 m above sea level were considered to be at low, intermediate, and high altitudes, respectively. DNA extracted (Qiagen, UK) from CF11 leukocyte-depleted (wwarn.org) venous blood was then sequenced on the Illumina HiSeq platform (Illumina, San Diego, CA, USA), and, subsequently, genotyped using well-established methods, as described previously by Amambua-Ngwa et al. (2019).
DNA sequencing
A total of 239 P. falciparum samples were sequenced on the Illumina Genome HiSeq platform. Library preparation and sequencing were undertaken by the Wellcome Trust Sanger Institute core teams, as described by Auburn et al. (2012), and as part of the MalariaGEN P. falciparum Community Project (https://www.malariagen.net/projects/p-falciparum-community-project). Short-sequence reads with 76 bp were generated, and variant discovery and genotype calling were carried out as described in the P. falciparum Community Project (https://www.malariagen.net/projects/p-falciparum-community-project). Briefly, after removing sequences that mapped to the human reference genome, the sequence reads were aligned to Pf3D7 reference version 3 (ftp://ftp.sanger.ac.uk/pub/project/pathogens/gff3/2016–07/Pfalciparum.genome.fasta.gz) using Burrows–Wheeler Alignment (BWA) mem tools version 0.7.15 (https://github.com/lh3/bwa.git) with -M parameter to mark shorter split hits as secondary.
The Picard version 2.6.0 (http://picard.sourceforge.net) tool was used to clean and mark duplicates of the aligned sequences, and default parameters of GATK base quality score recalibration, version 4.2.6.0, were applied to improve the quality of aligned bases using the pass variants from the Pf Genetic Crosses project (http://www.malariagen.net/data/pf-crosses-1.0). The stats utility from samtools version 1.2 (Li et al., 2009) was used to generate standard alignment metrics for each sample. GATK’s CallableLoci (Depristo et al., 2011), with default parameters, was used to determine the genomic positions callable in each sample.
Variant calling and genotyping were carried out following the GATK best practice pipeline (Auwera et al., 2013) using utilities HaplotypeCaller and GenotypeGCVFs of GATK version 4.2.6.0, respectively. Each variant was assigned a quality score with GATK’s VariantRecalibrator and ApplyRecalibration using the pass variants from the Pf Genetic Crosses project. This step assigned a quality score named VQSLOD to each variant. High values of VQSLOD indicated a higher quality. Variants were annotated using snpEff version 4.1 (Cingolani et al., 2012), with gene annotations downloaded from GeneDB (Logan-Klumpler et al., 2012) at the following URL: ftp://ftp.sanger.ac.uk/pub/project/pathogens/gff3/2016–10/Pfalciparum.noseq.gff3.gz . Each sample was genotyped for polymorphic-coding SNPs across the genome, ensuring that there was a minimum of 5 × paired-end coverage across each variant per sample. Polymorphic sites within hypervariable, telomeric, and repetitive sequence regions were excluded. Vcftools version 0.1.16 (Danecek et al., 2011) and bcftools version 1.10.2 (https://github.com/vcftools/vcftools.git) were used to filter the genomic data. Biallelic high-quality SNPs (VQSLOD ≥ 3) in the coding region loci with a minor allele frequency of at least 2%, individual samples, and SNP sites missing less than 10% across the isolates were extracted and used for downstream analysis as described (Amambua-Ngwa et al., 2019; Abera et al., 2021).
Plasmodium falciparum samples with low-quality sequences and no altitude information were then discarded (19 samples), and 220 high-quality sequences from high (51), intermediate (139), and low altitudes (30) were retained for assessing the effect of altitude on the genetic diversity of parasites, and its contribution to drug resistance. The minimum of 30 samples from each population category in the study is sufficiently large to determine population genomic variation. Analyses of the optimum sample size for population genetic studies (Trask et al., 2011; Hale et al., 2012; Nazareno et al., 2017; Flesch et al., 2018) suggest that even eight samples could be sufficient to accurately report intrapopulation (within-population) and interpopulation (between-population) genetic diversity estimates.
Analysis of population genetic diversity and mixedness of infection
The genotype mixedness of infection (within host/sample diversity) was estimated using the inbreeding coefficient within a population (FWS) metric as described by Manske et al. (2012):
FWS = 1 − HW/HS, (1)
where HW represents the within-isolate expected heterozygosity, calculated from the relative allele frequencies for all genic SNPs, and HS represents the heterozygosity of the local population. The FWS values range from zero (high diversity of infection) to one (single infection within the sample). For this analysis, individual alleles with coverage < 5 reads and positions with total coverage < 20 reads were classified as missing data. Isolates with > 10% missing SNP data and SNPs with > 10% missing isolate data were discarded. Isolates with FWS scores > 0.95 were classified as having a single predominant genotype due to a low genome-wide diversity.
Analysis of population structure and admixture
Principal component analysis (PCA) was used to estimate the population structure. This method simplified the complexity of high-dimensional data by geometrically projecting them onto a limited number of lower dimensions called principal components (PCs). The first PC minimizes the total distance between the data, as it maximizes the variance between the projected points; PC1 thus has the greatest variance as compared with other PCs, followed by PC2, and so on. However, all the PCs are not typically useful because much of the variance in the patterns of the data was covered by the first few PCs (Lever et al., 2017). In addition, the glPCA function in the R version 3.6.3 software package (The R Foundation for Statistical Computing, Vienna, Austria) was used to calculate the first 10 principal component axes, and the first three PCA axes were retained to explain the genomic variation among P. falciparum populations. The data was thinned down by pruning SNPs with pairwise linkage disequilibrium (LD) R2 values greater than 0.05. An allele-sharing matrix was then calculated using the pruned SNP loci with the R glPCA function that uses between- and within-group variances to determine the population’s genetic structure. Discriminant analysis of principal components (DAPC), which determines ancestry proportions and membership probability, was carried out using the adegenet version 2.1.10 package in R, as described previously (Jombart et al., 2010). This method identified and described population genetic clusters and their relationships and modeled genetic variation/allele sharing across space (altitude) to determine admixture. The optimum PCs for DAPC analysis, which prevent overfitting and instability in the membership probabilities or exclude useful information from the analysis, were selected using the optim.a. score function (Jombart et al., 2010).
Population allele frequency differentiation analysis
Analyses of allele frequency distributions between population FST values (Weir & Cockerham, 1984) were calculated using vcftools version 0.1.16 after excluding SNPs missing more than 10% data, as described by Abera et al. (2021), correcting for differences due to population size variation. The FST values range from zero to one, and are indicative of there being no and complete population differentiation, respectively.
Detection of selection signatures in P. falciparum populations
Allele frequency distributions within the population were assessed by estimating Tajima’s D indices (Tajima, 1989) using vcftools, after removing missing data per SNP. Tajima’s D values were determined for each SNP, and the average value was calculated for each gene using the tapply function in R. Genes with at least five SNPs and Tajima’s D values greater than one were considered to be under positive balancing selection. Standardized integrated haplotype score (iHS) analysis was undertaken to identify positive directional selection signatures using SNP data with allele frequency > 5%. iHS was estimated using the rehh version 3.2.2 package in R with default parameters (Gautier & Vitalis, 2012), after imputing missing SNP data using beagle version 5.2 (Browning & Browning, 2007). An |iHS| > 2.5 (top 1% of the expected distribution) (Voight et al., 2006) was used as a cutoff value to report genes under recent directional selection as reported for genome analysis of West African P. falciparum (Oyebola et al., 2017).
Detection of selection signatures difference between P. falciparum populations
To fully capture evidence of potential selection signature differences in P. falciparum populations at high, intermediate, and low altitudes, the cross-population Rsb statistics of rehh version 3.2.2 package in R were used, as iHS may not be suitable for detecting positive selection for those SNPs that have reached or are near to fixation in the local P. falciparum population (Voight et al., 2006). An Rsb score of < −5 or > 5 indicates that there is strong evidence of potential selection signature differences among P. falciparum populations (Duffy et al., 2015). The Rsb metrics (Tang et al., 2007) in the rehh package (Gautier & Vitalis, 2012) were used to detect the directional selection signature difference between populations. The scan for population-specific directional selection was performed using SNPs with a minimum minor allele frequency of 0.05, and to assess the relative haplotype lengths between two populations standardized against the genome-wide average, as used previously (Mobegi et al., 2014; Duffy et al., 2015). Calculation of haplotype breakdown during Rsb scores was terminated if the gaps between adjacent SNPs were > 20 kb. For determining Rsb, putative selection windows were defined by calculating the distance over which the extended haplotypes decayed to a level of 0.05 in both directions. For the analysis of between-population differences using the Rsb metric, |Rsb| > 5 was used to identify high-scoring SNPs.
Results
Sequencing of P. falciparum isolates and SNPs allele frequency distributions
Of the 239 P. falciparum isolates sequenced, 19 were removed due to low-quality sequences or a lack of altitude information. High-quality sequence data obtained from 220 samples enabled the identification of 682,462 (51 samples), 681,773 (139 samples), and 681,909 (30 samples) biallelic SNPs at high, intermediate, and low altitudes, respectively, with less than 10% missing SNPs and missing individual isolate data. Sequences from the intergenic regions had a lower read coverage than the coding regions, as expected, because of extreme A + T nucleotide contents in the intergenic regions. Thus, 78.23% (534,085), 78.4% (533,716), and 78.3% (533,953) of all SNPs identified from high, intermediate, and low altitudes, respectively, were located within the genes. Approximately 3,608, 3,644, and 3,847 genes analyzed at high, intermediate, and low altitudes, respectively, had at least one SNP (Figure 1).
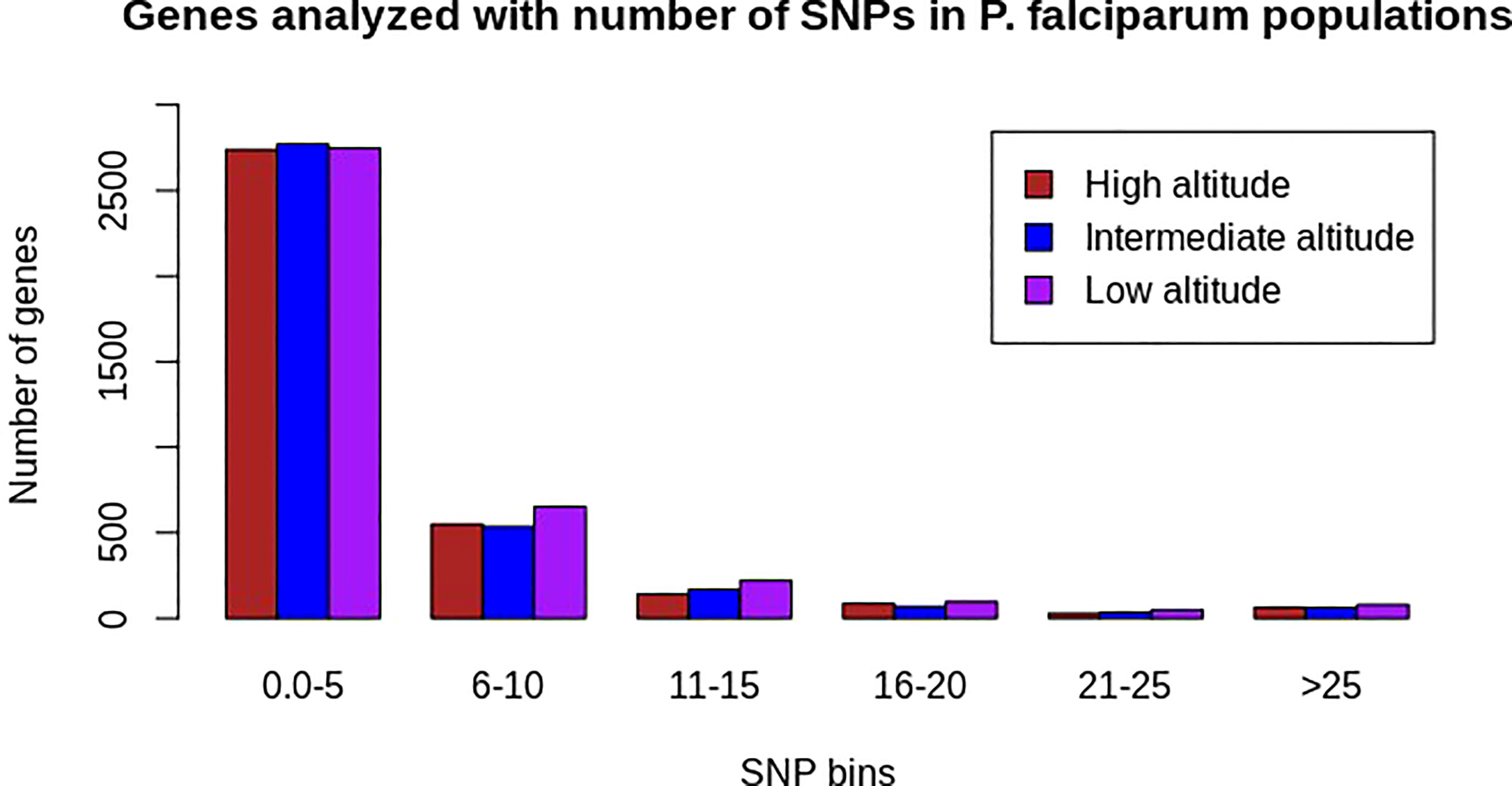
Figure 1 Distribution of the number of SNPs across all analyzed genes at high (N = 3607), intermediate (N = 3643), and low (N = 3846) altitudes in natural Plasmodium falciparum populations from southwestern Cameroon. The x-axis represents SNP bins and the y-axis represents the number of genes containing a range of SNPs bins for each population category (the colors brown, blue, and pink represent populations from high, intermediate, and low altitudes, respectively). Most genes from each population had SNP bin categories of 0–5 SNPs.
Approximately 24,214, 24,426, and 28,613 SNPs at high, intermediate, and low altitudes, respectively, were polymorphic in at least one sample of P. falciparum. Of these, 43.8%, 44.4%, and 45.4% were non-synonymous-coding SNPs; 20.9%, 20.7%, and 21.3% were synonymous-coding SNPs; 28.31%, 28.26%, and 27% were in intergenic regions; 3.3%, 3.04%, and 2.89% were in other intragenic regions; and 3.66%, 3.6%, and 3.46% were in intron regions at high, intermediate, and low altitudes, respectively (Figure 2; Supplementary Table S1).
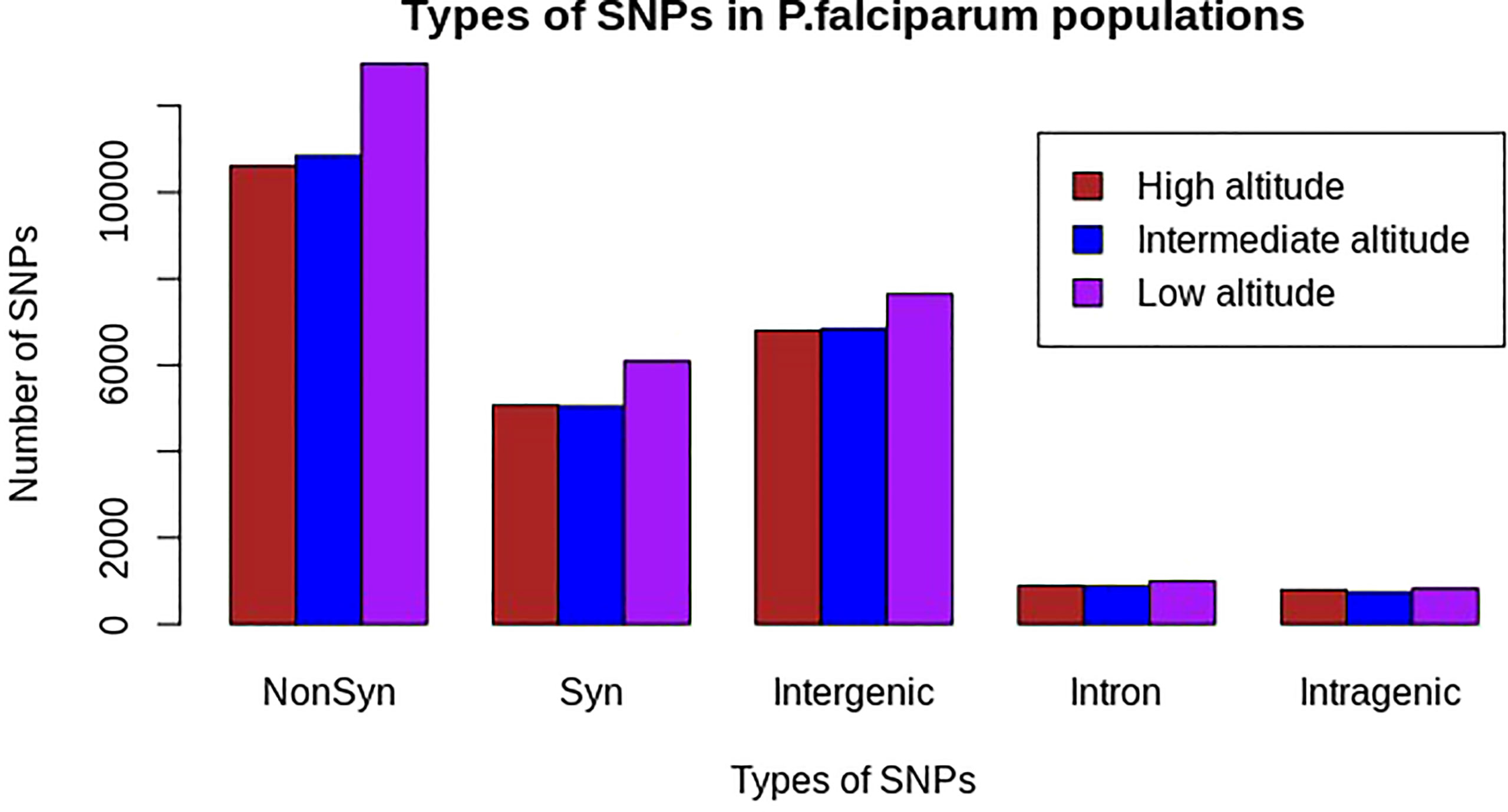
Figure 2 Distribution of polymorphic SNP types in natural Plasmodium falciparum populations at different altitudes from southwestern Cameroon. NonSyn, non-synonymous; Syn, synonymous; Intergenic, SNPs found in intergenic regions; Intron, SNPs found in intron region; Intragenic, other SNPs in coding regions other than non-synonymous and synonymous mutations. The x-axis represents types of mutations based on SNP effect and the y-axis represents the frequency of each SNP in the respective populations. The colors brown, blue, and pink represent populations from high, intermediate, and low altitudes, respectively. Non-synonymous mutations were high in all populations, as compared with other mutations, and relatively the highest in the population from low altitude, whereas other intragenic SNPs were the lowest in all populations.
Single-nucleotide polymorphisms with a minor allele frequency (MAF) < 5% were common in all analyzed P. falciparum populations following the exclusion of monomorphic SNPs. Furthermore, SNPs with minor alleles frequency < 5% occurred more frequently in samples from low altitudes than in isolates from high and intermediate altitudes (Figure 3).
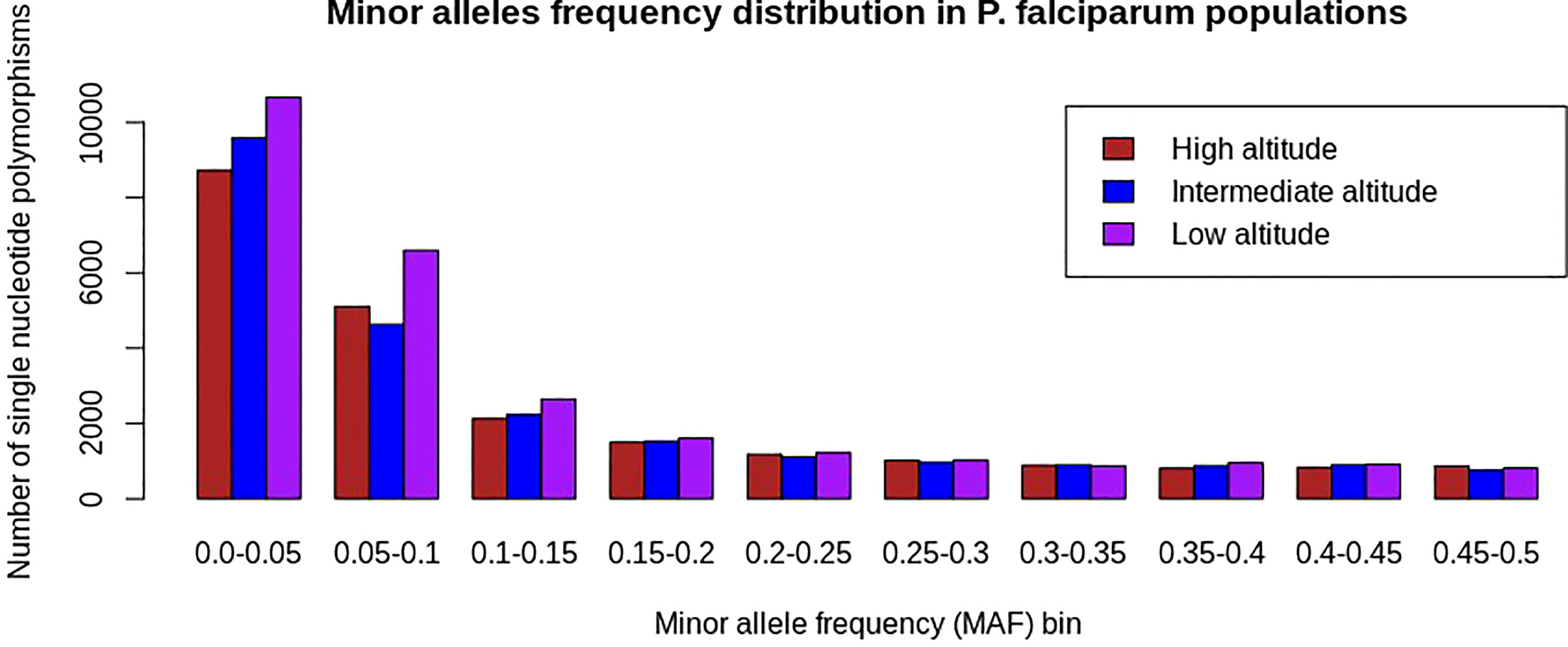
Figure 3 Minor allele frequency distribution of SNPs in Plasmodium falciparum populations at different altitudes in southwestern Cameroon. The x-axis represents SNPs binned into 10 equal sizes of 0.05 and the y-axis represents the frequency of SNPs with respective minor allele frequency bins. In all P. falciparum populations, there is an overabundance of low-frequency SNPs (MAF < 5%), which means rare alleles were common in each population and is consistent with population expansion, bottlenecks, or under directional selections as the result of pressure from drugs, human immunity, or other environmental factors. The colors brown, blue, and pink represent P. falciparum populations from high, intermediate, and low altitudes, respectively. SNPs with a minor allele frequency of less than 15% were relatively high in the population from the low altitude, indicating that rare alleles were the most common in this population. This might show that the population from the low altitude was under intense adaptation pressure as compared with others.
Genomic diversity of P. falciparum infections
The distribution of FWS scores was similar across isolates in each population, ranging from 0.363 to 0.997 (mean = 0.829, median = 0.937), 0.275 to 0.998 (mean = 0.831, median = 0.941), and 0.333 to 0.997 (mean = 0.772, median = 0.820) for P. falciparum infections at high, intermediate, and low attitudes, respectively (Figure 4; Supplementary Table S2). The proportion of isolates with FWS > 0.95, indicative of single genotype-dominated infections, was 25 (49%), 67 (48.2%), and 9 (30%) at high, intermediate, and low altitudes, respectively. No significant difference was observed in the mean FWS scores between P. falciparum populations from high and intermediate altitudes (p = 0.61), high and low altitudes (p = 0.93), and intermediate and low attitudes (p = 0.76). The majority of the samples had multiple infections or high levels of genetic diversity in all populations, which was consistent with high malaria prevalence and transmission rates reported throughout the years in the study area.
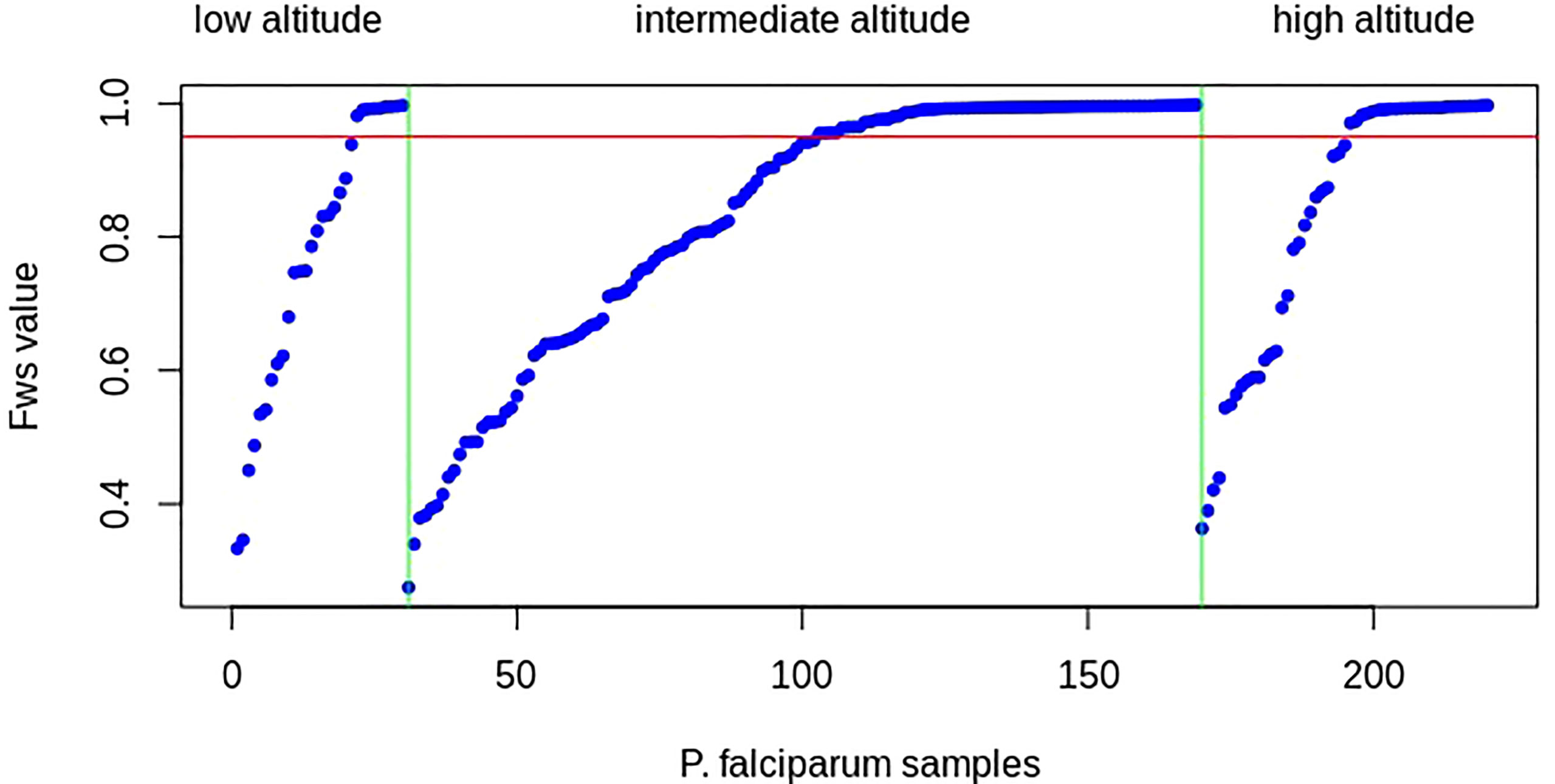
Figure 4 Within-infection FWS for Plasmodium falciparum sampled at high, intermediate, and low altitudes (listed above the figure) in southwestern Cameroon, ordered by increasing FWS within samples of each population. The x-axis represents P. falciparum samples (blue dots) and the y-axis represents Wright’s fixation index (FWS) for each clinical sample. The red line marks FWS = 0.95, above which an isolate is considered a single predominant genotype. The vertical green lines separate the P. falciparum population based on their altitude of origin. More than 50% of samples from all populations had FWS values of less than 0.95, indicating that most samples had two or more than two P. falciparum strains (high multiplicity of infections and high genetic diversity in each population), which is consistent with high P. falciparum malaria transmission or a high probability of population genetic outcrossing in the study areas.
Population structure, admixtures, and allele frequency differentiation analysis
The principal component analysis did not clearly distinguish the parasite isolates into their respective geographical origins/altitudinal groups from where they were sampled (Figure 5). Admixture analyses were consistent with the outcome of PCA, with no separation between isolates from the three altitudes, as there was gene flow among the populations.
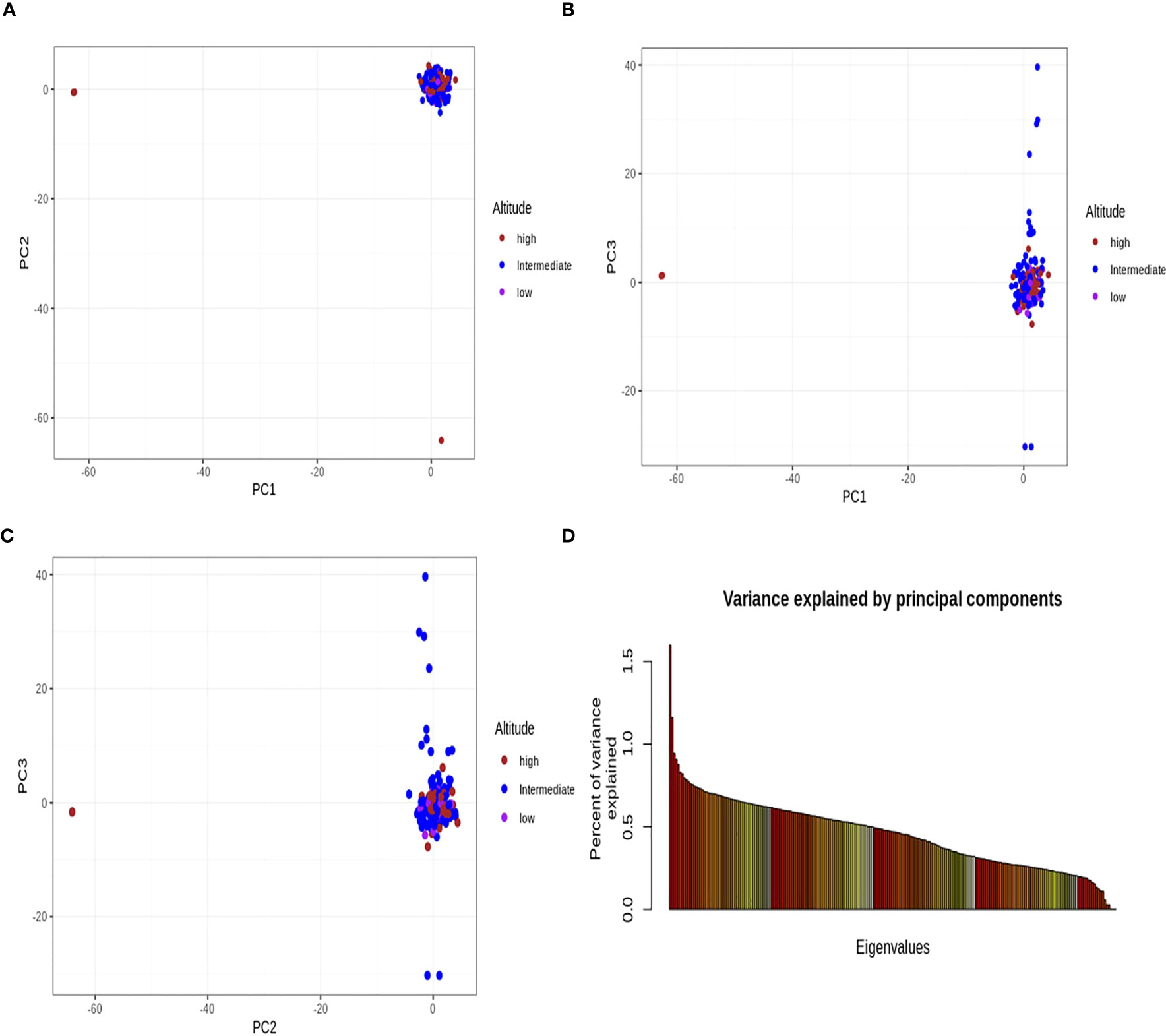
Figure 5 Principal component analysis. All the principal components (A–C) did not identify the Plasmodium falciparum populations from the three altitudinal zones of southwestern Cameroon. (D) Percentage of variance explained by each principal component axis. The first three principal component axes further explained the variance found in the data. PC1 explained 1.63%, PC2 explained 1.16%, and PC3 explained 0.97% of the variance in the analyzed data. The altitude stands for the origin of each population. The brown, blue, and pink dots represent the P. falciparum samples from high, intermediate, and low altitudes, respectively. This principal component analysis did not separate the populations from one another and was consistent with gene flow (i.e., population gene-sharing) among these populations.
This admixture analysis showed that none of the three components were differentiated according to the optimized cluster value of K = 3. Multiple parasite subpopulations were detected in parasites from all altitudes of Mount Cameroon (Figure 6). There was bidirectional gene flow among the isolates from high, intermediate, and low altitudes of Mount Cameroon, which is consistent with other studies carried out in West Africa and across the continent (Duffy et al., 2015; Amambua-Ngwa et al., 2019). These studies reported there were high probabilities of gene flow within or between the P. falciparum populations of Middle and West African countries.
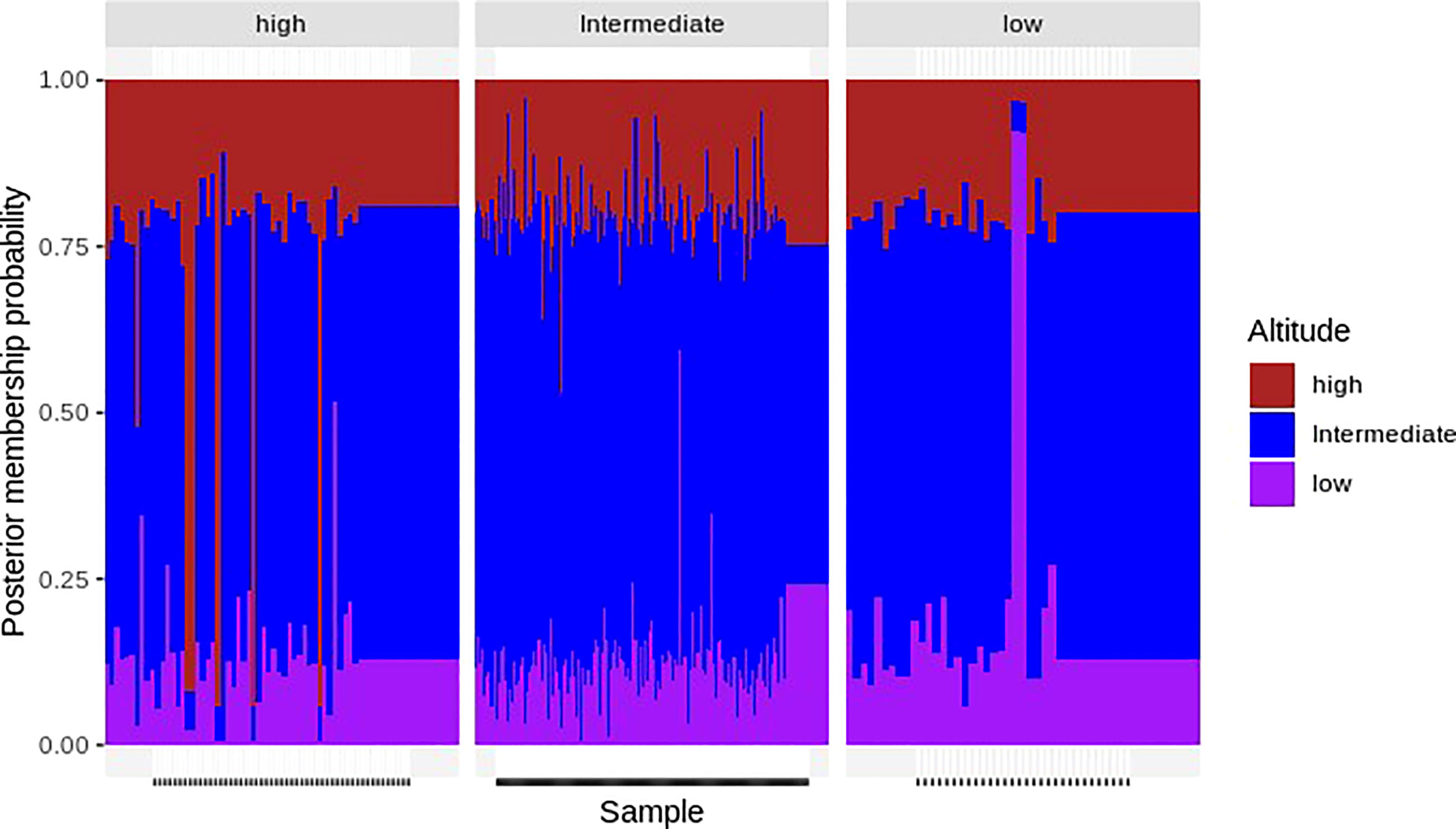
Figure 6 An admixture analysis—the x-axis represents the assigned samples to the altitude area listed above the figure after modeling allele sharing probability, and the y-axis represents the membership probability of the samples. Color-coded altitudes (the colors brown, blue, and pink represent Plasmodium falciparum parasite populations from high, intermediate, and low altitudes, respectively) listed at the right of the figure indicate the origin of the samples. Analysis of admixture did not differentiate any of the three components based on an optimized cluster value of K = 3, and multiple subpopulations of P. falciparum were found in all locations. There was a bidirectional gene flow among parasite populations.
The clustering of all parasite isolates was consistent with the fixation index (FST) values with or without correcting for sample size. The FST values of isolates from high versus intermediate altitude were 0.0054 (p < 2.2 × 10–16), 0.0087 (p < 2.2 × 10–16) for high versus low altitude, and 0.0075 (p < 2.2 × 10–16) for intermediate versus low altitude (Table 1) This suggests a low population differentiation among P. falciparum isolates from different altitudes along the slopes of Mount Cameroon.

Table 1 Pairwise population divergence (as measured by FST) among Plasmodium falciparum populations from southwestern Cameroon.
Detection of selection signatures in P. falciparum populations
The average Tajima’s D values for high, intermediate, and low altitude isolates were 0.01 (p = 0.16), 0.12 (p < 2.2 × 10–16), and −0.02 (p = 0.004), respectively, across the entire genome. Approximately 117, 134, and 98 genes in the P. falciparum population at high (Supplementary Table S3), intermediate (Supplementary Table S4), and low (Supplementary Table S5) altitudes, respectively, had at least five SNPs with Tajima’s D values > 1, suggesting that they were most likely under balancing selection. It is notable that 93 of the 98 genes with a Tajima’s D value > 1 at a low altitude also had a value > 1 at intermediate and high altitudes (Supplementary Table S6). Among these genes, crt, ama1, eba175, msp1, trap, dblmsp, clag2, and clag8 have been previously reported to be under balancing or mixed selections. Of these genes, clag8, ama1, and trap encode surface proteins that undergo balancing selection and have previously been identified as potential vaccine candidates. The Duffy-binding-like merozoite surface protein 2 gene (pfdblmsp2) has been implicated in parasite resistance to halofantrine, mefloquine, and lumefantrine.
Genome-wide analysis for evidence of recent positive directional selection due to drug pressure, the impact of the immune response, or other mechanisms identified 36, 49, and 39 genes at high (Supplementary Figure S1, Supplementary Table S7), intermediate (Supplementary Figure S2, Supplementary Table S8), and low (Supplementary Figure S3, Supplementary Table S9) altitudes, respectively, with at least two SNPs that could be under significant positive selection. Interestingly, 22 consistent sweeps were present in populations at all altitudes in the study area (Table 2). The genes surfin8.2 on chromosome 8 and trap on chromosome 13, previously reported as erythrocytic and pre-erythrocytic vaccine candidates, respectively, were detected with higher SNPs at elevated |iHSs|, indicating continuous selection on these antigenic loci. In addition, other vaccine candidates such as the celtos, clag8, and ama1 genes, were also detected, as was the chloroquine-resistance transporter gene crt, although only at low and intermediate altitudes. No signatures around the multidrug resistance mdr1 gene and the antifolate drug target genes dhfr and dhps were detected.
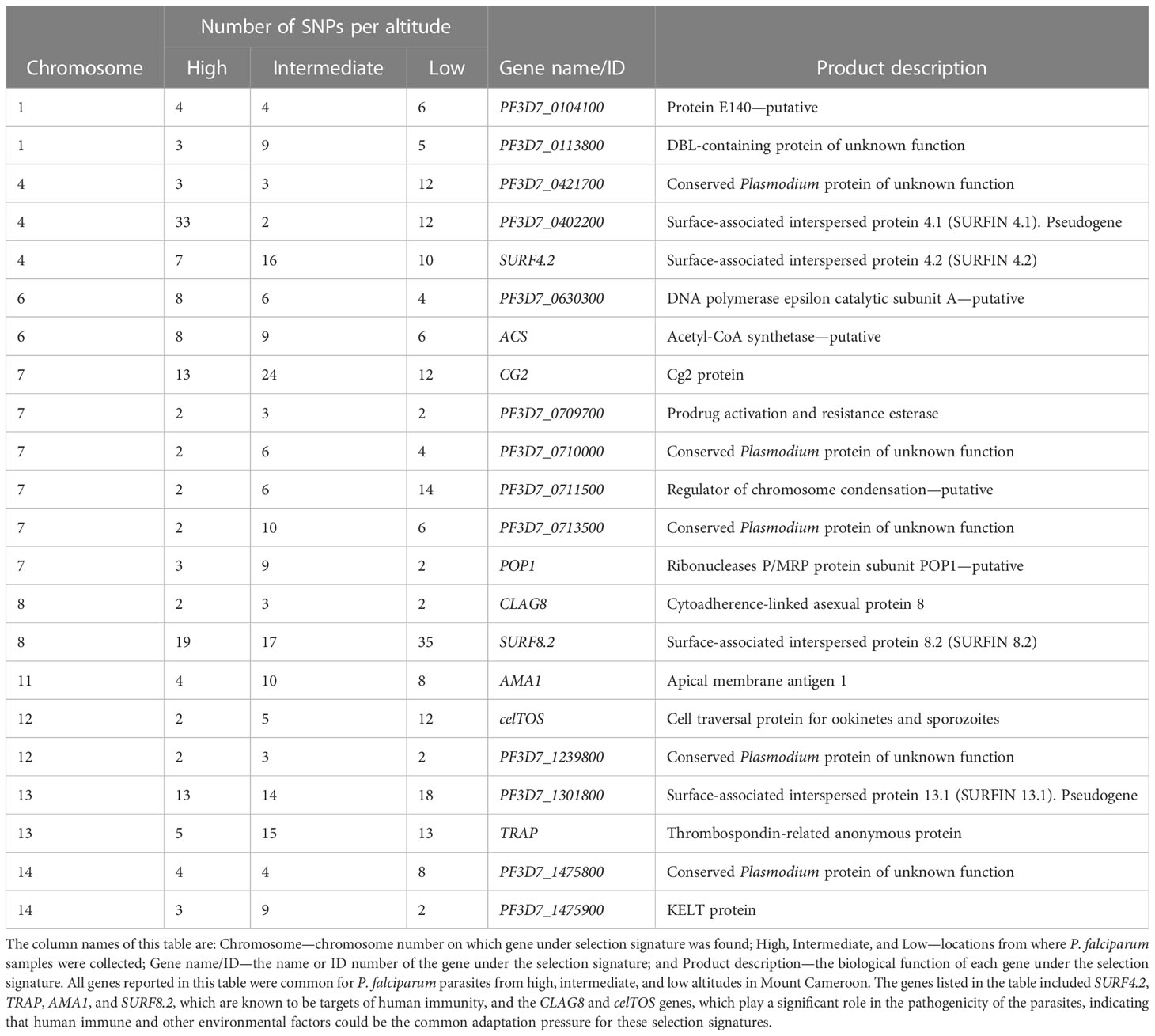
Table 2 Genes with at least two SNPs with recent positive directional selection in Plasmodium falciparum populations present at different altitudes in southwestern Cameroon, which were identified using the integrated haplotype score at a significance threshold of p < 0.01.
Of the genes under positive directional selection, 26 of the 36 at high (Supplementary Table S10), 30 of the 49 at intermediate (Supplementary Table S11), and 24 of the 39 at low altitudes (Supplementary Table S12), showed both positive directional and balancing selections.
Overall, 17 of the 22 genes under positive directional selection from the three altitudinal zones in the study area showed both positive balancing and directional selections (Table 3), among which are SURF4.2 on chromosome 4, cytoadherence-linked asexual protein 8 (CLAG8) on chromosome 8, and thrombospondin-related anonymous protein (TRAP) on chromosome 13. There was no detectable signature around the multidrug resistance mdr1 gene, and the antifolate drug target genes dhfr and dhps showed both positive balancing and directional selections.
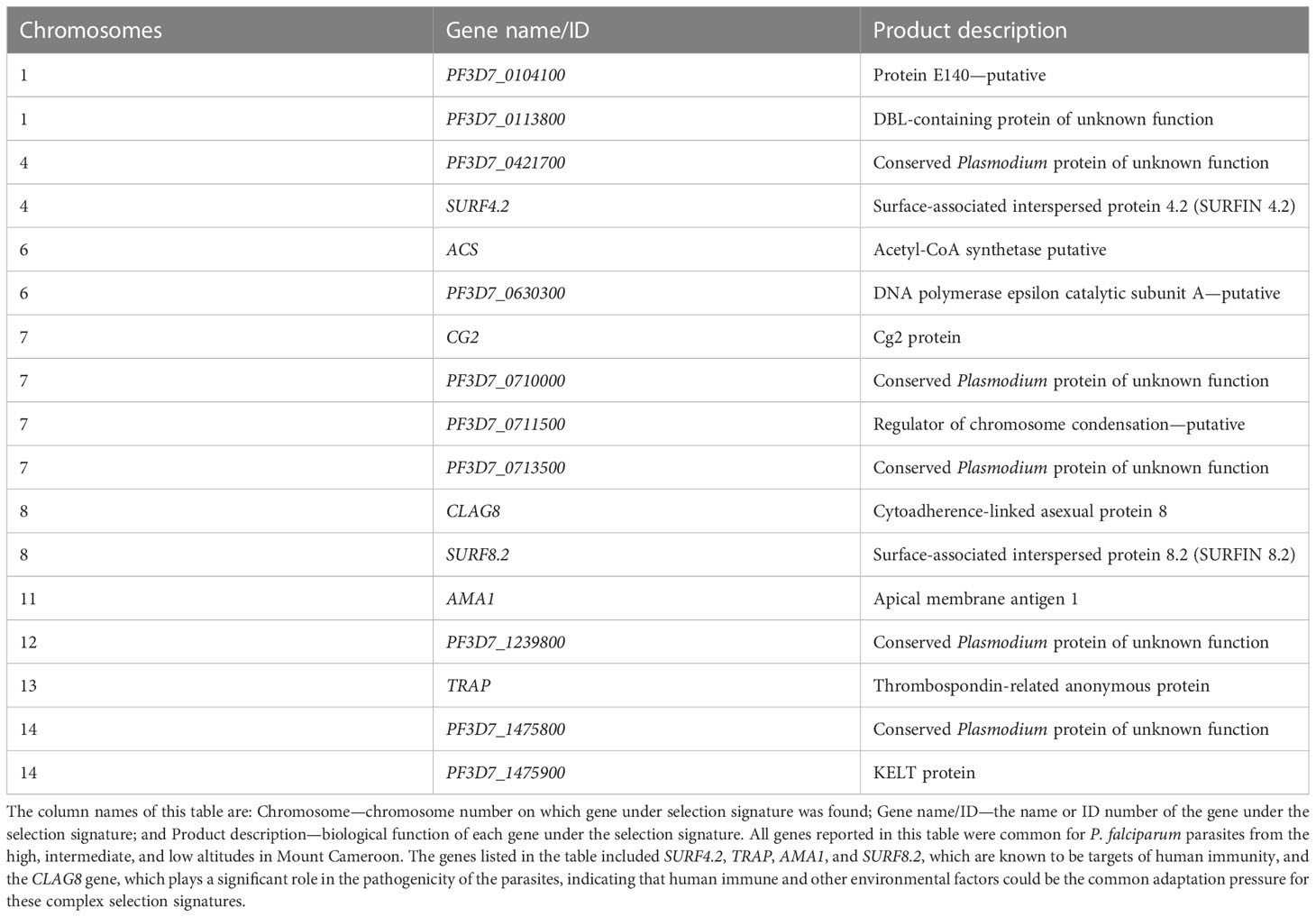
Table 3 Genes under both recent positive directional selection and positive balancing selections in Plasmodium falciparum populations at different altitudes in southwestern Cameroon.
Detection of selection signature differences between P. falciparum populations
Chromosomal loci with population-specific evidence of directional selection between high and intermediate altitudes were identified: five at a high altitude and four at an intermediate altitude (Figure 7, Supplementary Table S13). Selection signatures were observed on chromosome 4 for both high and low altitudes. Similarly, six chromosomal loci with population-specific evidence of directional selection between high and low altitudes were identified: three each at high and low altitudes (Figure 7, Supplementary Table S14). In addition, 11 chromosomal loci with evidence of directional selection between intermediate and low altitudes were detected: four at intermediate and seven at low altitudes (Figure 7, Supplementary Table S15)
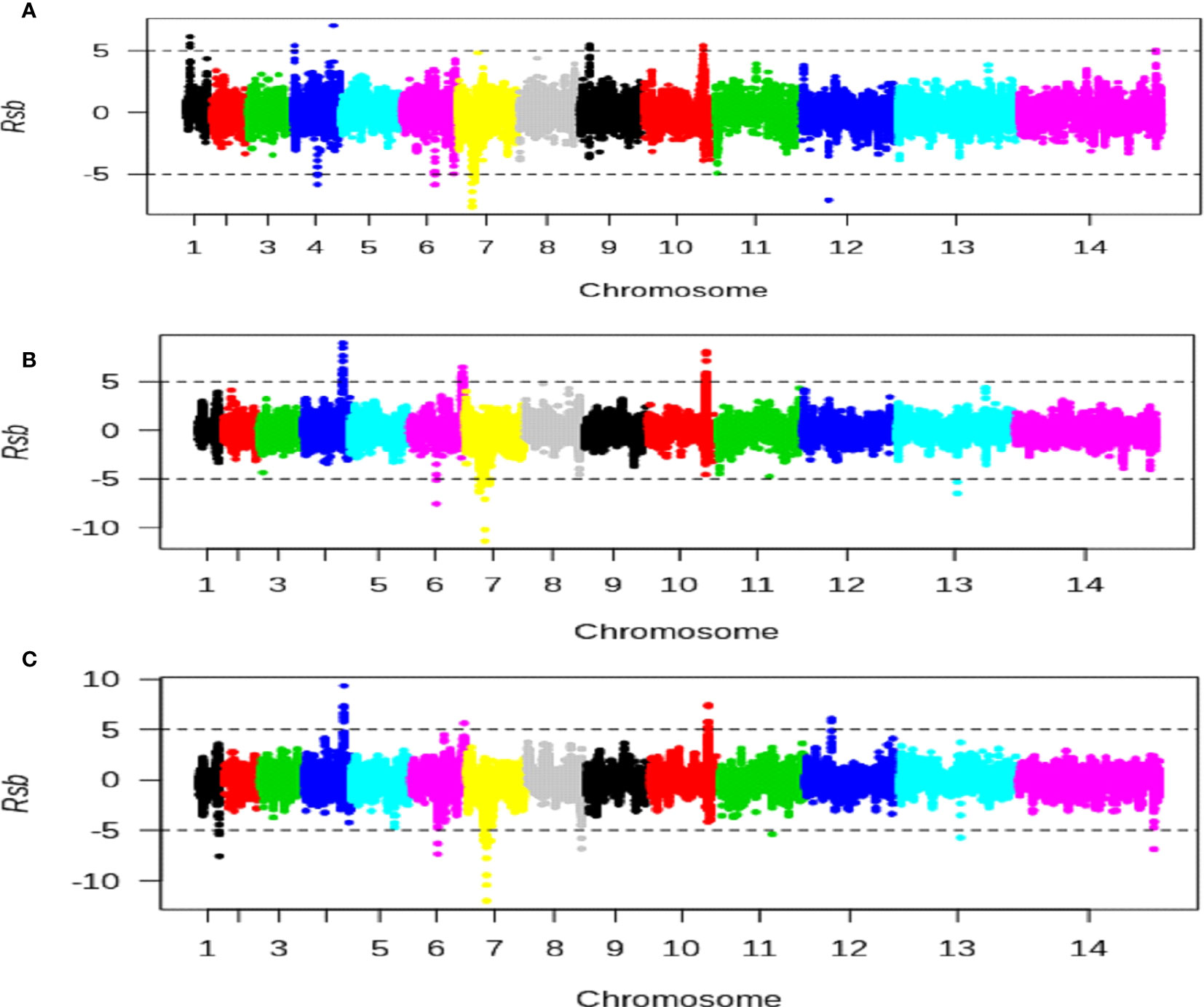
Figure 7 Scan for evidence of population-specific directional selection strength using the Rsb cross-population test for extended haplotypes. The x-axis represents Plasmodium falciparum chromosomes and the y-axis represents cross-population test Rsb metrics values. Positive Rsb metrics values above 5 and less than −5 (broken horizontal lines) indicate SNPs with specific stronger selection signatures in high versus intermediate (A), high versus low (B), or intermediate versus low (C) populations, respectively. Each region containing a gene with at least two SNPs having Rsb values above 5 or less than −5 indicated genes under population-specific strong directional selection signatures.
Population-specific strong recent directional selections were also observed at genomic regions of chromosomes 1 and 4 in a P. falciparum population from high altitude, which includes two genes (PF3D7_0104100 and ACS6). Similarly, specific strong recent directional selections were found at genomic regions of chromosomes 4 (containing one gene, SMC3), 6 (containing one gene, ACS12), 7 (containing one gene, PF3D7_0709700), 12 (containing one gene, CelTOS) and chromosomes 1 (containing one gene, PF3D7_0114500), 6 (containing one gene, PF3D7_0616400), 7 (containing four genes, CG2, PF3D7_0710000, PF3D7_0711500, and PF3D7_0713200), 8 (SURF8.2), 11 (containing one gene, AMA1), 13 (containing one gene, TRAP) of P. falciparum populations from intermediate and low altitudes, respectively (Supplementary Table S16). These genes include immune-targeted genes such as AMA1, TRAP, SURF8.2, and CelTOS. This observation was consistent with reports of there being specific strong recent directional selections in immune-targeted genes of West African P. falciparum populations (Duffy et al., 2015).
Prevalence of mutations conferring antimalarial drug resistance in P. falciparum
Non-synonymous SNPs in resistance-associated genes were identified in isolates from high, intermediate, and low altitudes (Table 4). Four CQ-resistant alleles (pfcrt-K76T, pfcrt-A220S, pfcrt-Q271E pfcrt-I356T, and Pfmdr1-N86Y) and SP resistance-associated mutations in pfdhfr and pfdhfs genes were detected in isolates at high, intermediate, and low attitudes. The prevalence of pfdhr-N51I and pfdhr-C59R, the major pyrimethamine resistance-conferring alleles, was remarkably high at all attitudes (above 98%). Moreover, there was a variation in the prevalence of the drug resistance-conferring alleles Pfdhps-K142, Pfdhps-I431V, pfdhs-G437A, and Pfdhps-A581G, with pfdhps-I431V and pfdhps-A581G being the most prevalent (Table 4).
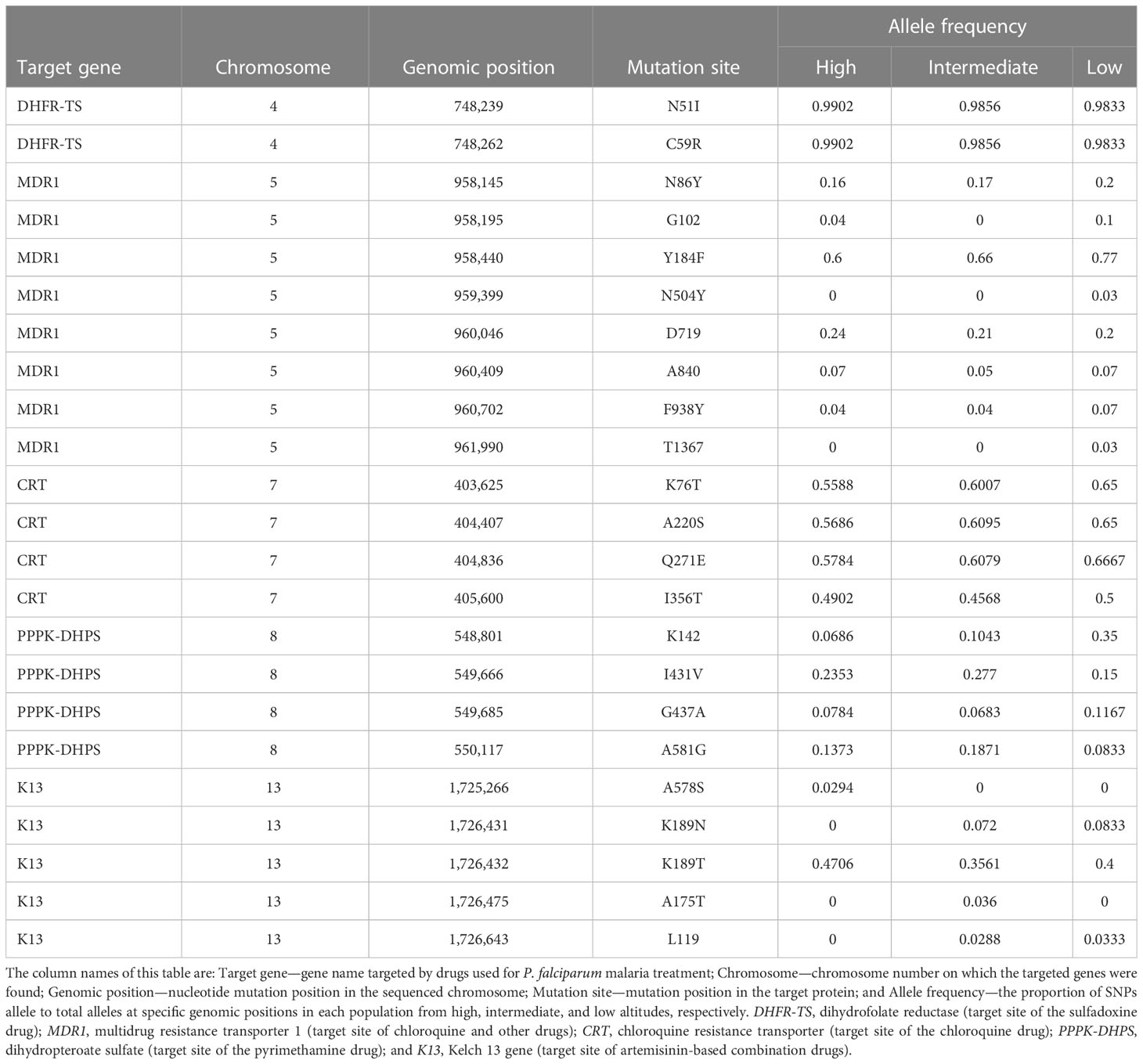
Table 4 Drug resistance-conferring alleles’ frequency in Plasmodium falciparum populations from high, intermediate, and low altitudes along the slope of Mount Cameroon.
Pfk13 mutations associated with artemisinin resistance showed a variation in all analyzed P. falciparum populations. The Pfk13-K189T mutation, previously identified in African P. falciparum populations (Verity et al., 2020; Abera et al., 2021), was observed at higher frequencies (> 35%) more than its pfk13-K189N (<7%), pfk13-A578S (<3%), pfk13-A175T (<3%) and pfk13-L119 (<3%) counterparts. In addition, Pfk13-K189T was observed in 47%, 36%, and 40% of isolates at high, intermediate, and low altitudes, respectively.
Discussion
Malaria parasites are under continual evolutionary selection from host immunity and antimalarial drug pressure, with the latter resulting in the emergence of resistant strains within the parasite population. Spontaneous changes may lead to lower drug sensitivity and allow parasites to circumvent host immunity and therapeutic interventions (Kidgell et al., 2006). As malaria endemic settings make progress toward eliminating the disease, genomic data can help identify relationships between parasite population, their structure, and transmission intensity, and, ultimately, generate actionable output to assess the effectiveness of control strategies (Wesolowski et al, 2018; Sy et al, 2022). Continuous monitoring of the parasite populations will therefore permit the identification of genetic changes to public health response and malaria elimination strategies. Studies in Cameroon have mainly focused on the prevalence of drug resistance mutations in P. falciparum isolates (Apinjoh et al., 2017), with limited reports on further characterization of the deadly P. falciparum using novel technologies. This study used Illumina next-generation sequencing to investigate the genetic diversity and population structure of the P. falciparum parasite at different altitudes around the slope of Mount Cameroon, as well as assessed signatures of selection and determined the prevalence of markers of antimalarial resistance in the parasite population. This will provide actionable information for malaria epidemiology and control.
Consistent with previous studies (Oyebola et al., 2017; Abera et al., 2021), there was a high percentage of non-synonymous- to synonymous-coding SNPs found at polymorphic loci across all P. falciparum populations from different altitudes in this study, suggesting that the parasites in the area were under selection pressure. Moreover, similar MAF of parasite populations across all altitudes was observed, with an over-representation of low-frequency (< 5%) variants, as previously reported (Abera et al., 2021). The low proportion of samples with high FWS scores is indicative of dominant mixed-genotype infections and a low-population substructure among the parasites (Auburn et al., 2012). Moreover, the risk of parasite outcrossing, and consequent threat to malaria control efforts, is moderately high in these populations.
Tajima’s D values and iHSs were used to explore the mechanisms of natural selection in the study area. Multiple (and almost 100 identical) genes in the P. falciparum population across all three altitudes had at least five SNPs with Tajima’s D values > 1, suggesting that these genes were most likely under balancing selection (Amambua-Ngwa et al., 2012; Ochola-Oyier et al., 2016). Among these genes, crt, ama1, eba175, msp1, trap, dblmsp, clag2, and clag 8 were previously reported to be under balancing or mixed selection (Amambua-Ngwa et al., 2012; Ye et al., 2019). The genes clag8, ama1, and trap which encodes surface proteins have previously been identified as potential vaccine candidates (Ochola-Oyier et al., 2016; Toenhake et al., 2018; Abera et al., 2021). The Duffy-binding-like merozoite surface protein 2 gene (pfdblmsp2) has been implicated in parasite resistance to halofantrine, mefloquine, and lumefantrine (Van Tyne et al., 2011). In all, 17 antigenic genes were identified as being under both recent positive directional and positive balancing selection, including the prominent host immune target genes surfin 8.2, trap, and ama1, indicating that antimalarial drugs and host immunity have been major selective agents. This is consistent with previous studies conducted in West Africa (Mobegi et al, 2014).
Interestingly, 22 consistent sweeps with |iHSs| > 2.5, which could be under significant positive directional selection, were present in populations at all altitudes in the study area. The genes surfin8.2 on chromosome 8 and trap on chromosome 13, previously reported as erythrocytic and pre-erythrocytic vaccine candidates, respectively (Amambua-Ngwa et al., 2018; Ajibola et al., 2021), were detected with higher SNPs at elevated |iHSs|, suggesting continuous selection on these antigenic loci. In addition, other vaccine candidates, such as the celtos, clag8, and ama1 genes, were also detected, as was the chloroquine resistance transporter gene crt, although only at low and intermediate altitudes. No signature around the multidrug-resistant mdr1 gene and antifolate drug target genes dhfr and dhps was detected. Overall, 17 of the 22 genes under positive directional selection from the three altitudinal zones in the study showed both positive balancing and directional selections, among which are SURF4.2 on chromosome 4, cytoadherence-linked asexual protein 8 (CLAG8) on chromosome 8, and thrombospondin-related anonymous protein (TRAP) on chromosome 13 (Amambua-Ngwa et al., 2018; Abera et al., 2021).
The genomic regions on chromosomes 1, 4, 6, 7, 8, 11, 12, 13, and 14 with high iHSs in P. falciparum populations from high, intermediate, and low altitudes (Table 2) were marked as having stronger evidence of extended haplotypes in one or other of the populations using Rsb cross-population analysis (Supplementary Table S16). These regions contain vaccine target genes and others that might be subject to population-specific selection differences including ama1, trap, Surf8.2, and celtos, which code for vaccine candidates, Cg2 (mediator of RNA polymerase II transcription subunit 14), ACS12 (acylCoA synthetase), and SMC3 (structural maintenance of chromosomes protein 3). Strong selection signatures have previously been identified in genes linked to chloroquine resistance (crt), antifolate drugs SP (dhfr/dhps), and vaccine candidates of West African P. falciparum populations using haplotypes tests (Duffy et al., 2015). This selection signatures decay has been rapid following drug usage and malaria transmission intensity change over time because of rapid recombination in high malaria transmission areas. The results obtained here show local selection differences in P. falciparum populations, representing ongoing or recent selection events.
The within-isolate fixation index FWS had a similar range of values in each population at various altitudes. FWS is a genome-wide metric that averages heterozygosity across the genome in comparison with heterozygosity within the local parasite population (Manske et al., 2012), which is inversely related to the level of genomic complexity per infection. Although there was no significant difference in the mean FWS scores between P. falciparum isolates across all attitudes in the study area, FWS values tended to be higher at high altitudes where malaria transmission is low, and lower at low altitudes where malaria transmission is high. This is consistent with the findings of studies conducted in other African countries (Burkina Faso, Ghana, Mali), which associated high and low FWS values to settings with low and high malaria transmission, respectively (Auburn et al., 2012; Manske et al., 2012; Duffy et al., 2015).
An analysis of parasite population structure, PCA, and admixture revealed no clear distinction in parasites from the three altitude zones in the area. This suggests there was no separation between the isolates as there is gene flow among the populations under study. These observations are corroborated by several studies that report regional- and intercontinental-level structure in global P. falciparum parasite populations (Manske et al., 2012), indicating gene flow among populations at various altitudes. This bidirectional gene flow is highly likely across altitudinal zones in the study area, as they share the same ecological climate and malaria transmission intensity (Achidi et al., 2008), and there is a high degree of human mobility. The genome-wide FST values between the populations were also very low, suggesting a low population differentiation among P. falciparum from different altitudes (Duffy et al., 2015).
The prevalence of non-synonymous SNPs in the Pfdhfr gene (especially N51I and C59R) associated with SP resistance remains high, suggesting that this gene is highly conserved in this population. This may likely be due to the continuous use of SP in the country as part of intermittent preventative treatment in pregnancy (PNLP, 2012). This is consistent with previous studies in Kenya (Osborne et al., 2021) that found that the pfdhps gene was in the top 5% of identity-by-descent (IBD) positions in island isolates from Lake Victoria where SP is still being used as an IPTp. However, there was a variation in the prevalence of the resistance markers for SP-conferring alleles to Pfdhps (K142, I431V, G437A, and A581G). Furthermore, the proportion of mutant SNPs in the parasite population at high (24% pfdhps-I431V and 13% pfdhps-A581G), intermediate (28% pfdhps-I431V and 19% pfdhps-A581G), and low (15% pfdhps-I431V and 8% pfdhps-A581G) altitudes is consistent with previous articles on non-synonymous mutations in SP-associated genes in the area (Apinjoh et al., 2017) and elsewhere in the country (Chauvin et al., 2015). The continued use of SP as an IPTp is likely providing enough driving pressure to maintain these polymorphisms in the parasite populations in the region/country and across the continent. The pfcrt-K76T and Pfmdr1-N86Y alleles, which are strongly associated with chloroquine resistance (Chauvin et al., 2015), were detected in the study area as previously reported (Ndam et al., 2017). Moreover, three other mutations in CQ-resistant genes (pfcrt-A220S, pfcrt-Q271E, and pfcrt-I356T) were detected in isolates across all altitudes, where malaria transmission is widespread and persistent (Achidi et al., 2008).
None of the non-synonymous polymorphisms at N458Y, Y493H, R539T, I543T, R561H, and C580Y in the Kelch repeat region of the K13 propeller domain associated with artemisinin resistance (Ariey et al., 2014) was detected in any of the P. falciparum populations analyzed. However, other mutations in the gene, which have not been associated with partial artemisinin resistance (WHO, 2021), including the Pfk13-K189T mutation previously identified in African P. falciparum populations (Verity et al., 2020; Abera et al., 2021; Ocholla et al, 2014), were observed at higher frequencies (>35%), as were their pfk13-K189N (<7%), pfk13-A578S (<3%), pfk13-A175T (<3%), and pfk13-L119 (<3%) counterparts. Furthermore, non-synonymous mutations in the pfK13-A578S were recorded only in isolates at high altitudes, consistent with similar studies conducted in Africa (Kamau et al., 2014; de Laurent et al., 2018) and elsewhere in Cameroon (Safeukui et al., 2017). This study also reports the presence of the pfk13-K189T mutation in at least 35% of samples at all altitudes, with some non-synonymous SNPs (K189T and K189N) being found in samples taken from intermediate and low altitudes. The pfk13-K189T mutation has also been reported in previous studies in the region and some African countries (Apinjoh et al., 2017; Verity et al., 2020; Abera et al., 2021), although its role in artemisinin resistance is unknown. Other k13 mutant alleles of unknown function (A175T and L119) were also detected at low frequencies among isolates at intermediate and low altitudes; these need to be further characterized, and their role in artemisinin resistance identified.
Conclusion
This whole-genome analysis reveals high levels of genetic diversity (> 50% of analyzed samples in each category had Fws values less than 0.95) in P. falciparum parasite populations at different altitudes around the slopes of Mount Cameroon. There was no clear distinction/structure in parasites from the three altitude zones, which is indicative of there being bidirectional gene flow within parasites in the three zones. A total of 93 antigenic genes were observed to be under balance selection, including vaccine candidate genes ama1, eba175, msp1, trap, dblmsp, and clag2, with 22 under positive directional selection, among which are surfin 8.2 and trap, previously reported as erythrocytic and pre-erythrocytic vaccine candidates, respectively, and the ama1 gene, previously reported to encode an important merozoite target of acquired immunity. Although the prevalence of non-synonymous SNPs in the Pfdhfr gene (especially N51I and C59R) associated with SP resistance remains high, suggesting that this gene is highly conserved in this population, no PfKelch13-validated mutations were observed. The high level of genetic diversity and low-population structure suggests that malaria transmission remained high in the study area and parasite populations circulating around the slope of Mount Cameroon are homogeneous. Thus, more effort is neededto control malaria transmission and prevent the emergence of resistant alleles around the slope of Mount Cameroon.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the Faculty of Health Sciences, University of Buea ethics review board. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
TA: conceptualization; methodology; investigation; data curation; project administration; writing; review and editing; and funding acquisition. MA: investigation; data curation and analysis; and writing. DA: data curation and analysis; and writing. HC, RT, and RM: investigation; and data curation. LG and EA: review and editing. AA-N: funding acquisition; supervision; and review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Developing Excellence in Leadership and Genetics Training for Malaria Elimination in sub-Saharan Africa (DELGEME) program (Grant 107740/Z/15/Z) sponsored by the Developing Excellence in Leadership, Training, and Science (DELTAS) Africa initiative.
Acknowledgments
This work was supported through the DELTAS Africa Initiative (DELGEME grant 107740/Z/15/Z). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and was supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (DELGEME grant 107740/Z/15/Z) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors AA-N, EA, and TA declared that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer-review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmala.2023.1075755/full#supplementary-material
References
Abera D., Kibet C. K., Degefa T., Amenga-Etego L., Bargul J. L., Golassa L. (2021). Genomic analysis reveals independent evolution of Plasmodium falciparum populations in Ethiopia. Malaria J. 20 (1), 129. doi: 10.1186/s12936-021-03660-y
Achidi E. A., Apinjoh T. O., Mbunwe E., Besingi R., Yafi C., Wenjighe Awah N., et al. (2008). Febrile status, malarial parasitaemia and gastro-intestinal helminthiases in schoolchildren resident at different altitudes, in south-western Cameroon. Ann. Trop. Med. Parasitol. 102 (2), 103–118. doi: 10.1179/136485908X252287
Ajibola O., Diop M. F., Ghansah A., Amenga-Etego L., Golassa L., Apinjoh T., et al. (2021). In silico characterisation of putative Plasmodium falciparum vaccine candidates in African malaria populations. Sci. Rep. 11 (1), 16215. doi: 10.1038/s41598-021-95442-4
Amambua-Ngwa A., Amenga-Etego L., Kamau E., Amato R., Ghansah A., Golassa L., et al. (2019). Major subpopulations of Plasmodium falciparum in sub-Saharan Africa. Sci. (New York N.Y.) 365 (6455), 813–816. doi: 10.1126/science.aav5427
Amambua-Ngwa A., Jeffries D., Amato R., Worwui A., Karim M., Ceesay S., et al. (2018). Consistent signatures of selection from genomic analysis of pairs of temporal and spatial Plasmodium falciparum populations from The Gambia. Sci. Rep. 8 (1), 9687. doi: 10.1038/s41598-018-28017-5
Amambua-Ngwa A., Tetteh K. K. A., Manske M., Gomez-Escobar N., Stewart L. B., Deerhake M. E., et al. (2012). Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PloS Genet. 8 (11), e1002992. doi: 10.1371/journal.pgen.1002992
Apinjoh T., Anchang-Kimbi J., Ajonina M., Njonguo E., Njua-Yafi C., Ngwai A., et al. (2016). In Vivo Efficacy of Artesunate/Sulphadoxine-Pyrimethamine versus Artesunate/Amodiaquine in the Treatment of Uncomplicated P. falciparium Malaria in Children around the Slope of Mount Cameroon: A Randomized Controlled Trial. Biomedicines 4 (1), 5. doi: 10.3390/biomedicines4010005
Apinjoh T., Tata R. B., Anchang-Kimbi J., Chi H. F., Fon E. M., Mugri R. N., et al. (2015). Plasmodium falciparum merozoite surface protein 1 block 2 gene polymorphism in field isolates along the slope of mount Cameroon: a cross – sectional study. BMC Infect Dis 15, 309. doi: 10.1186/s12879-015-1066-x
Apinjoh T. O., Mugri R. N., Miotto O., Chi H. F., Tata R. B., Anchang-Kimbi J. K., et al. (2017). Molecular markers for artemisinin and partner drug resistance in natural Plasmodium falciparum populations following increased insecticide treated net coverage along the slope of mount Cameroon: Cross-sectional study. Infect. Dis. Poverty 6 (1), 136. doi: 10.1186/s40249-017-0350-y
Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A.-C., Khim N., et al. (2014). A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505 (7481), 50–55. doi: 10.1038/nature12876
Arya A., Kojom Foko L. P., Chaudhry S., Sharma A., Singh V. (2021). Artemisinin-based combination therapy (ACT) and drug resistance molecular markers: A systematic review of clinical studies from two malaria endemic regions – India and sub-Saharan Africa. Int. J. Parasitology: Drugs Drug Resistance 15, 43–56. doi: 10.1016/j.ijpddr.2020.11.006
Auburn S., Campino S., Miotto O., Djimde A. A., Zongo I., Manske M., et al. (2012). Characterization of within-host plasmodium falciparum diversity using next-generation sequence data. PloS One 7 (2), e32891. doi: 10.1371/journal.pone.0032891
Auwera G. A., Carneiro M. O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., et al. (2013). From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinf. 43 (1), 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
Balikagala B., Fukuda N., Ikeda M., Katuro O. T., Tachibana S.-I., Yamauchi M., et al. (2021). Evidence of artemisinin-resistant malaria in africa. New Engl. J. Med. 385 (13), 1163–1171. doi: 10.1056/NEJMoa2101746
Basco L. K., Ngane V. F., Ndounga M., Same-Ekobo A., Youmba J.-C., Abodo R. T. O., et al. (2006). Molecular epidemiology of malaria in Cameroon. XXI. Baseline therapeutic efficacy of chloroquine, amodiaquine, and sulfadoxine-pyrimethamine monotherapies in children before national drug policy change. Am. J. Trop. Med. Hygiene 75 (3), 388–395.
Browning S. R., Browning B. L. (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81 (5), 1084–1097. doi: 10.1086/521987
CDC (2021) CDC - malaria—Malaria worldwide—Impact of malaria. Available at: https://www.cdc.gov/malaria/malaria_worldwide/impact.html.
Chauvin P., Menard S., Iriart X., Nsango S. E., Tchioffo M. T., Abate L., et al. (2015). Prevalence of Plasmodium falciparum parasites resistant to sulfadoxine/pyrimethamine in pregnant women in Yaoundé, Cameroon: Emergence of highly resistant pfdhfr/pfdhps alleles. J. Antimicrobial Chemotherapy 70 (9), 2566–2571. doi: 10.1093/jac/dkv160
Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) , 80–92. doi: 10.4161/fly.19695
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi: 10.1093/bioinformatics/btr330
de Laurent Z. R., Chebon L. J., Ingasia L. A., Akala H. M., Andagalu B., Ochola-Oyier L. I., et al. (2018). Polymorphisms in the K13 gene in plasmodium falciparum from different malaria transmission areas of kenya. Am. J. Trop. Med. Hygiene 98 (5), 1360–1366. doi: 10.4269/ajtmh.17-0505
DePristo M., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. doi: 10.1038/ng.806
Duffy C. W., Assefa S. A., Abugri J., Amoako N., Owusu-Agyei S., Anyorigiya T., et al. (2015). Comparison of genomic signatures of selection on Plasmodium falciparum between different regions of a country with high malaria endemicity. BMC Genomics 16 (1), 527. doi: 10.1186/s12864-015-1746-3
Flesch E. P., Rotella J. J., Thomson J. M., Graves T. A., Garrott R. A. (2018). Evaluating sample size to estimate genetic management metrics in the genomics era. Mol. Ecol. Resour. 18 (5), 1077–1091. doi: 10.1111/1755-0998.12898
Gautier M., Vitalis R. (2012). rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28 (8), 1176–1177. doi: 10.1093/bioinformatics/bts115
Hale M. L., Burg T. M., Steeves T. E. (2012). Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PloS One 7 (9), e45170. doi: 10.1371/journal.pone.0045170
Jombart T., Devillard S., Balloux F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 11 (1), 94. doi: 10.1186/1471-2156-11-94
Kamau E., Campino S., Amenga-Etego L., Drury E., Ishengoma D., Johnson K., et al. (2014). K13-propeller polymorphisms in plasmodium falciparum parasites from sub-saharan africa. J. Infect. Dis. jiu608 211 (8), 1352–1355. doi: 10.1093/infdis/jiu608
Kidgell C., Volkman S. K., Daily J., Borevitz J. O., Plouffe D., Zhou Y., et al. (2006). A systematic map of genetic variation in plasmodium falciparum. PloS Pathog. 2 (6), e57. doi: 10.1371/journal.ppat.0020057
Lever J., Krzywinski M., Altman N. (2017). Principal component analysis. Nat. Methods 14 (7), 641–642. doi: 10.1038/nmeth.4346
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi: 10.1093/bioinformatics/btp352
Logan-Klumpler F. J., De Silva N., Boehme U., Rogers M. B., Velarde G., McQuillan J. A., et al. (2012). GeneDB—an annotation database for pathogens. Nucleic Acids Research 40 (Issue D1), D98–D108. doi: 10.1093/nar/gkr1032
Manske M., Miotto O., Campino S., Auburn S., Almagro-Garcia J., Maslen G., et al. (2012). Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487 (7407), 375–379. doi: 10.1038/nature11174
Mbako D. J. (2017). Enhancing malaria prevention in cameroon through community participation: an in-depth review. Cent. Afr. J. Public Health 3 (6), 97. doi: 10.11648/j.cajph.20170306.12
Menard D., Dondorp A. (2017). Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harbor Perspect. Med. 7 (7), a025619. doi: 10.1101/cshperspect.a025619
Mobegi V. A., Duffy C. W., Amambua-Ngwa A., Loua K. M., Laman E., Nwakanma D. C., et al. (2014). Genome-wide analysis of selection on the malaria parasite plasmodium falciparum in west african populations of differing infection endemicity. Mol. Biol. Evol. 31 (6), 1490–1499. doi: 10.1093/molbev/msu106
Nazareno A. G., Bemmels J. B., Dick C. W., Lohmann L. G. (2017). Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 17 (6), 1136–1147. doi: 10.1111/1755-0998.12654
Ndam N. T., Basco L. K., Ngane V. F., Ayouba A., Ngolle E. M., Deloron P., et al. (2017). Reemergence of chloroquine-sensitive pfcrt K76 Plasmodium falciparum genotype in southeastern Cameroon. Malaria J. 16 (1), 130. doi: 10.1186/s12936-017-1783-2
Ndiaye J. L. A., Faye B., Diouf A. M., Kuété T., Cisse M., Seck P. A., et al. (2008). Randomized, comparative study of the efficacy and safety of artesunate plus amodiaquine, administered as a single daily intake versus two daily intakes in the treatment of uncomplicated falciparum malaria. Malaria J. 7 (1), 16. doi: 10.1186/1475-2875-7-16
Ochola-Oyier L. I., Okombo J., Wagatua N., Ochieng J., Tetteh K. K., Fegan G., et al. (2016). Comparison of allele frequencies of Plasmodium falciparum merozoite antigens in malaria infections sampled in different years in a Kenyan population. Malaria J. 15 (1), 261. doi: 10.1186/s12936-016-1304-8
Osborne A., Manko E., Takeda M., Kaneko A., Kagaya W., Chan C., et al. (2021). Characterizing the genomic variation and population dynamics of Plasmodium falciparum malaria parasites in and around Lake Victoria, Kenya. Sci. Rep. 11 (1), 19809. doi: 10.1038/s41598-021-99192-1
Oyebola K. M., Idowu E. T., Olukosi Y. A., Awolola T. S., Amambua-Ngwa A. (2017). Pooled-DNA sequencing identifies genomic regions of selection in Nigerian isolates of Plasmodium falciparum. Parasites Vectors 10 (1), 320. doi: 10.1186/s13071-017-2260-z
Payne D. (1987). Spread of chloroquine resistance in Plasmodium falciparum. Parasitol. Today 3 (8), 241–246. doi: 10.1016/0169-4758(87)90147-5
PNLP (2012). Plan Strategique nationale de lutte contre le paludisme 2011–2015 (Rapport Minsante Cameroun). Retrieved October 11, 2022 Available at: https://www.minsante.cm
Roper C., Pearce R., Nair S., Sharp B., Nosten F., Anderson T. (2004). Intercontinental spread of pyrimethamine-resistant malaria. Science 305 (5687), 1124–1124. doi: 10.1126/science.1098876
Rosenthal P. J. (2021). Are artemisinin-based combination therapies for malaria beginning to fail in africa? Am. J. Trop. Med. Hygiene 105 (4), 857–858. doi: 10.4269/ajtmh.21-0797
Safeukui I., Fru-Cho J., Mbengue A., Suresh N., Njimoh D. L., Bumah V. V. (2017). Investigation of polymorphisms in the P. falciparum artemisinin resistance marker kelch13 in asymptomatic infections in a rural area of Cameroon. bioRxiv 148999. doi: 10.1101/148999
Sansonetti P. J., Lebras C., Verdier F., Charmot G., Dupont B., Lapresle C. (1985). CHLOROQUINE-RESISTANT PLASMODIUM FALCIPARUM IN CAMEROON. Lancet 325 (8438), 1154–1155. doi: 10.1016/S0140-6736(85)92453-5
Sayang C., Gausseres M., Vernazza-Licht N., Malvy D., Bley D., Millet P. (2009). Treatment of malaria from monotherapy to artemisinin-based combination therapy by health professionals in urban health facilities in Yaoundé, central province, Cameroon. Malaria J. 8, 176. doi: 10.1186/1475-2875-8-176
Sy M., Deme A. B., Warren J. L. (2022). et al. Plasmodium falciparum genomic surveillance reveals spatial and temporal trends, association of genetic and physical distance, and household clustering. Sci. Rep. 12, 938. doi: 10.1038/s41598-021-04572-2
Tajima F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 (3), 585–595. doi: 10.1093/genetics/123.3.585
Tang K., Thornton K. R., Stoneking M. (2007). A new approach for using genome scans to detect recent positive selection in the human genome. PloS Biol. 5 (7), e, 171. doi: 10.1371/journal.pbio.0050171
Toenhake C. G., Fraschka S. A.-K., Vijayabaskar M. S., Westhead D. R., van Heeringen S. J., Bártfai R. (2018). Chromatin accessibility-based characterization of the gene regulatory network underlying plasmodium falciparum blood-stage development. Cell Host Microbe 23 (4), 557–569.e9. doi: 10.1016/j.chom.2018.03.007
Trask J. A. S., Malhi R. S., Kanthaswamy S., Johnson J., Garnica W. T., Malladi V. S., et al. (2011). The effect of SNP discovery method and sample size on estimation of population genetic data for Chinese and Indian rhesus macaques (Macaca mulatta). Primates 52 (2), 129–138. doi: 10.1007/s10329-010-0232-4
Tuedom A. G. B., Sarah-Matio E. M., Moukoko C. E. E., Feufack-Donfack B. L., Maffo C. N., Bayibeki A. N., et al. (2021). Antimalarial drug resistance in the Central and Adamawa regions of Cameroon: Prevalence of mutations in P. falciparum crt, Pfmdr1, Pfdhfr and Pfdhps genes. PloS One 16 (8), e0256343. doi: 10.1371/journal.pone.0256343
Van Tyne D., Park D. J., Schaffner S. F., Neafsey D. E., Angelino E., Cortese J. F., et al. (2011). Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in plasmodium falciparum. PloS Genet. 7 (4), e1001383. doi: 10.1371/journal.pgen.1001383
Verity R., Aydemir O., Brazeau N. F., Watson O. J., Hathaway N. J., Mwandagalirwa M. K., et al. (2020). The impact of antimalarial resistance on the genetic structure of Plasmodium falciparum in the DRC. Nat. Commun. 11 (1), 2107. doi: 10.1038/s41467-020-15779-8
Voight B. F., Kudaravalli S., Wen X., Pritchard J. K. (2006). A map of recent positive selection in the human genome. PloS Biol. 4 (3), 0446–0458. doi: 10.1371/journal.pbio.0040072
Wanji S., Tanke T., Atanga S. N., Ajonina C., Nicholas T., Fontenille D. (2003). Anopheles species of the mount Cameroon region: Biting habits, feeding behaviour and entomological inoculation rates. Trop. Med. Int. Health 8 (7), 643–649. doi: 10.1046/j.1365-3156.2003.01070.x
Weir B. S., Cockerham C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38 (6), 1358. doi: 10.2307/2408641
Wesolowski A., Taylor A. R., Chang H. H., Verity R., Tessema S., Bailey J. A., et al. (2018). Mapping malaria by combining parasite genomic and epidemiologic data. BMC Med. 16 (1), 190. doi: 10.1186/s12916-018-1181-9
Whegang Y. S., Chiabi A., Basco L. K. (2019). Monitoring the efficacy and safety of artemisinin-based combination therapies: A review and network meta-analysis of antimalarial therapeutic efficacy trials in cameroon. Drugs R&D 19 (1), 1–14. doi: 10.1007/s40268-018-0259-3
WHO (2021). World malaria report 2021 (World Health Organization). Available at: https://apps.who.int/iris/handle/10665/350147.
World Health Organization. (2016). World health statistics 2016: monitoring health for the SDGs, sustainable development goals. World Health Organization. Available at: https://apps.who.int/iris/handle/10665/206498.
Keywords: Plasmodium falciparum, genomic diversity, population structure, signatures of selection, drug resistance
Citation: Apinjoh TO, Ajonina MU, Abera D, Chi HF, Tata RB, Mugri RN, Golassa L, Achidi EA and Amambua-Ngwa A (2023) Genomic analysis of Plasmodium falciparum isolates across different altitudinal zones along the slope of Mount Cameroon. Front. Malar. 1:1075755. doi: 10.3389/fmala.2023.1075755
Received: 20 October 2022; Accepted: 10 July 2023;
Published: 31 July 2023.
Edited by:
Anita Ghansah, Noguchi Memorial Institute for Medical Research, GhanaReviewed by:
Valentina Dianora Mangano, University of Pisa, ItalyRachel Daniels, University of Massachusetts Medical School, United States
Copyright © 2023 Apinjoh, Ajonina, Abera, Chi, Tata, Mugri, Golassa, Achidi and Amambua-Ngwa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias O. Apinjoh, YXBpbmpvaC50b2JpYXNAdWJ1ZWEuY20=