1
School of Biomedical Sciences, University of Nottingham Medical School, Queen’s Medical Centre, Nottingham, UK
2
Department of Anatomy and Neurobiology, University of Maryland School of Medicine, Baltimore, MD, USA
Cannabinoids have been shown to possess anticonvulsant properties in whole animal models of epilepsy. The present investigation sought to examine the effects of cannabinoid receptor activation on kainic acid (KA)-induced epileptiform neuronal excitability. Under urethane anesthesia, acute KA treatment (10 mg kg−1, i.p.) entrained the spiking mode of simultaneously recorded neurons from random firing to synchronous bursting (% change in burst rate). Injection of the high-affinity cannabinoid agonist (-)-11-hydroxy-8-tetrahydrocannabinol-dimethyl-heptyl (HU210, 100 μg kg−1, i.p.) following KA markedly reduced the burst frequency (% decrease in burst frequency) and reversed synchronized firing patterns back to baseline levels. Pre-treatment with the central cannabinoid receptor (CB1) antagonist N-piperidino-5-(4-clorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide (rimonabant, SR141716A 3 mg kg−1, i.p.) completely prevented the actions of HU210. The present results indicate that cannabinoids exert their antiepileptic effects by impeding pathological synchronization of neuronal networks in the hippocampus.
The appearance of epileptic seizures is commonly associated with aberrant hypersynchronization of large neuronal aggregates (Walker and Kullmann, 1999
). One of the most commonly affected brain areas is the hippocampus, where several mechanisms underlying pathological synchronization have been identified (Miles and Wong, 1983
). Such mechanisms include alterations in gap junctional signaling (Jefferys, 1995
), imbalances of excitatory/inhibitory currents (Cohen et al., 2002
) and an enhancement of inhibitory GABAergic feedback (Haas et al., 1996
). In the present study, hippocampal epileptiform activity was induced pharmacologically with kainic acid (KA). This excitotoxin produces an episode of status epilepticus (Sokal and Large, 2001
) and complex epileptiform EEG patterns (Ben-Ari, 1985
; Medvedev and Willoughby, 1999
) when administered acutely in vivo.
Available evidence from human and animal seizure models suggests that various cannabinoid compounds (derived from the marijuana plant, Cannabis sativa) have antiepileptic properties (Cunha et al., 1980
; Wallace et al., 2002
). Furthermore, anandamide has been shown to modulate seizure threshold and severity through CB1 receptors implicating the endogenous cannabinoid system in epilepsy (Karanian et al., 2007
). An earlier study by Hàjos et al. (2000)
provided a mechanistic explanation for the effects of the endogenous cannabinoid receptor ligand anandamide. Using hippocampal slices, the authors showed that bath application of cannabinoid receptor agonists reduce the power of KA-induced network oscillations, these actions were prevented by pre-treatment with the CB1 receptor antagonist rimonabant.
Population studies provide valuable information in terms of temporal associations among recorded neurons and of specific features of single-neuronal firing. As functional synaptic correlations between the neurons within the population (activity correlations) are created, single-unit fire in phase, whereas spikes from outside the correlated network are out of phase (Abeles et al., 1993
). The present study was aimed at investigating the effects of cannabinoid receptor activation on KA-induced bursting activity in the CA1 hippocampal layer of anesthetized rats. We employed multiple simultaneous extracellular single-neuron recordings to physiologically verify the antiepileptic actions of cannabinoids and to better delineate the dynamics of population discharge patterns underlying epilepsy.
Animals and Surgical Procedures
Male Sprague-Dawley rats (n = 13, Charles River Laboratories), weighing 300–350 g at the time of surgery were housed in groups of three in large plastic cages and maintained on a 12:12-h light–dark cycle (lights on at 7:00 AM). Water and food were available ad libitum. All experiments were performed in accordance to UK Home Office regulations under project license 40/1955 and with NIH guidelines (publication 86–23). Animals were anesthetized with urethane (25% w/v in water) at a dose of 1.4 g kg−1 (i.p.) and mounted on a modified Kopf 1730 stereotaxic apparatus.
Recording Procedures
Recordings were made with a bundle of eight microwire electrodes (Formvar-coated stainless steel, 50-μm diameter; NBLabs, TX, USA), with a stainless steel ground electrode connected to a unity-gain 8 channel J-type field effect transistor headstage (NBLabs). Electrodes were stereotaxically positioned via fine micromanipulator control over the hippocampus (−5.5 mm AP, 3.0 L relative to bregma and −2.2 DV from the brain surface) according to Paxinos and Watson (1986)
. The temperature of the animal was maintained at 37.7°C with the aid of a rectal probe connected to a homeothermic control unit (Harvard Apparatus, MA, USA). The second level of amplification occurred at a 16-channel differential preamplifier (fixed gain ×100; Plexon Inc, TX, USA) after which the signal was transferred to a Multi-Channel Acquisition Processor (Plexon Inc, TX, USA). This allowed for computer-controlled amplification, filtering, switching and digital signal processing of the signals originating from the microwires. The impedance of the electrodes was ∼300 kΩ measured at 1 kHz. Commercially available software (RASPUTIN; Plexon Inc, TX, USA) allowed on-line isolation and discrimination (using dual voltage–time widows or principal component analysis) of neuronal spikes from background noise. Typically, one to four action potential waveforms were observed and discriminated per microwire during any given experiment. Activity was also displayed on a Tektronix D11 5000 series dual-beam oscilloscope and corresponding action potential waveforms were isolated with the aid of a Gould 1425 Digital Oscilloscope and a Gould Type 125 waveform processor. Firing rate was also monitored aurally with the aid of a loudspeaker. Signals were filtered using a second order low cut-off (500 Hz) and high cut-off (5 kHz), with a 50-Hz notch filter. The gain block was a programmable gain multiplier with gain steps 1–30 (total gain: ×1000 to ×32,000); typically the gain was set at ×10,000 for the experiments.
Pharmacological Manipulations
HU210 was obtained from Tocris Cookson (UK, USA), KA was obtained from Sigma Chemical (UK, USA) and rimonabant (SR141716A) was a generous gift from Sanofi-Synthelabo. HU210 and rimonabant were resuspended to stock concentration (10−2 M) in ethanol and refrigerated at −20°C. Drugs were dissolved in a physiological 1.0-ml saline (0.9% NaCl)/10-μl Tween 80 (1%) vehicle solution for injection on the day of the experiment. KA was resuspended to stock concentration (10−2 M) in distilled water and frozen (−20°C) in 200-μl aliquots. When required, the aliquots were thawed and diluted in 0.2-ml saline and for each experiment an aliquot was thawed and diluted in 0.2-ml saline to the desired dose. All drugs were injected intraperitoneally (i.p.).
Electrophysiological Analyses
Discriminated single-unit data was initially visualized off-line as spike rasters; the spike raster representing action potential spikes, viewed as vertical ticks, corresponding to the temporal (time stamped) occurrence of the spikes. Data was subsequently analyzed as integrated firing rate histograms (accumulated over 5 or 10 s epochs); burst analyses were calculated for epochs of 15 s during periods of bursting activity and cross-correlation histograms (CCHs; Gerstein, 1970
), between pairs of hippocampal neurons, were computed using NeuroExplorer (Plexon Inc, TX, USA). Basal activity was calculated as the mean ± SEM of firing during a selected 10-min pre-drug period for overall firing analyses and over 15 s for burst analyses. Visual evaluation of bursting discharge activity, induced by KA injection, in the spike raster displays revealed two types of neuronal discharge – “micro-bursts” and “macro-bursts”. Micro-bursts were empirically defined as follows: maximum burst start interval of 170 ms, maximum interspike interval within the burst 300 ms; minimum inter-burst interval 200 ms; minimum duration for a burst 100 ms and minimum number of spikes within a burst, minimum of three spikes. Macro-bursts were much slower oscillations in overall firing (clearly visible over longer time frames – see Figure 3
A) with a periodicity of 180–220 s. CCH analysis (Figure 1
B) shows the conditional probability of the occurrence of a spike in the spike train at time t on the condition that there is a reference event (or reference spike; in this case spikes from neuron 1) at time 0 (Gerstein, 1970
). Therefore, if the graph shows a flat profile, neurons are not firing synchronously with respect to a given reference neuron. Correlated discharge activity (i.e., functional firing synchronicity) between pairs of neurons were revealed in CCHs as a central peak (or trough) centered on the zero time bin, while a flat cross-correlation indicated non-correlated discharge activity between neuronal pairs (Eblen-Zajjur and Sandkuhler, 1997
). Conventional analyses of variance (ANOVA) were used to study treatment–response differences. The Scheffé’s post-hoc test was used to assess differences between and within treatments. The criterion of significance for the ANOVAs and post-hoc tests was set at p < 0.05. All data are expressed mean ± SEM and statistical analyses were computed using Statistica (Statsoft Inc, OK, USA).
On completion of experiments, the location of the recording electrode tips was marked by passing a 60-μA current for 30 s through electrode pairs in the array. Animals were then transcardially perfused with a 4% paraformaldehyde/5% potassium ferrocyanide solution. Ferrocyanide creates a blue-green stain by reacting with deposited iron ions at the mark site. The brains were then removed and stored at 4°C in the same fixative solution overnight. Vibratome sections were then cut (80 μm) and transferred to slides with the aid of mounting medium (Vectashield). Photographic images were made using a low power objective without further counter-staining of the sections.
A total of 58 neurons (n = 6 rats) were recorded showing action potential signal-to-noise amplitude ratios of at least a 3:1. KA (10 mg kg−1) produced a significant increase in the number of bursts over pre-drug activity in 46 of 58 units from 4.1 ± 1.06 to 10.5 ± 1.18 bursts (Figures 1
A and 2
; F(1,57) = 15.74, p < 0.0008). Every neuron studied was significantly (p < 0.05) excited from pre-drug basal firing following KA injection (Figure 3
; 6.89 ± 1.18 to 13.05 ± 2.04 Hz, for the entire population). The onset latency for KA observable effects was ∼6 min after injection.
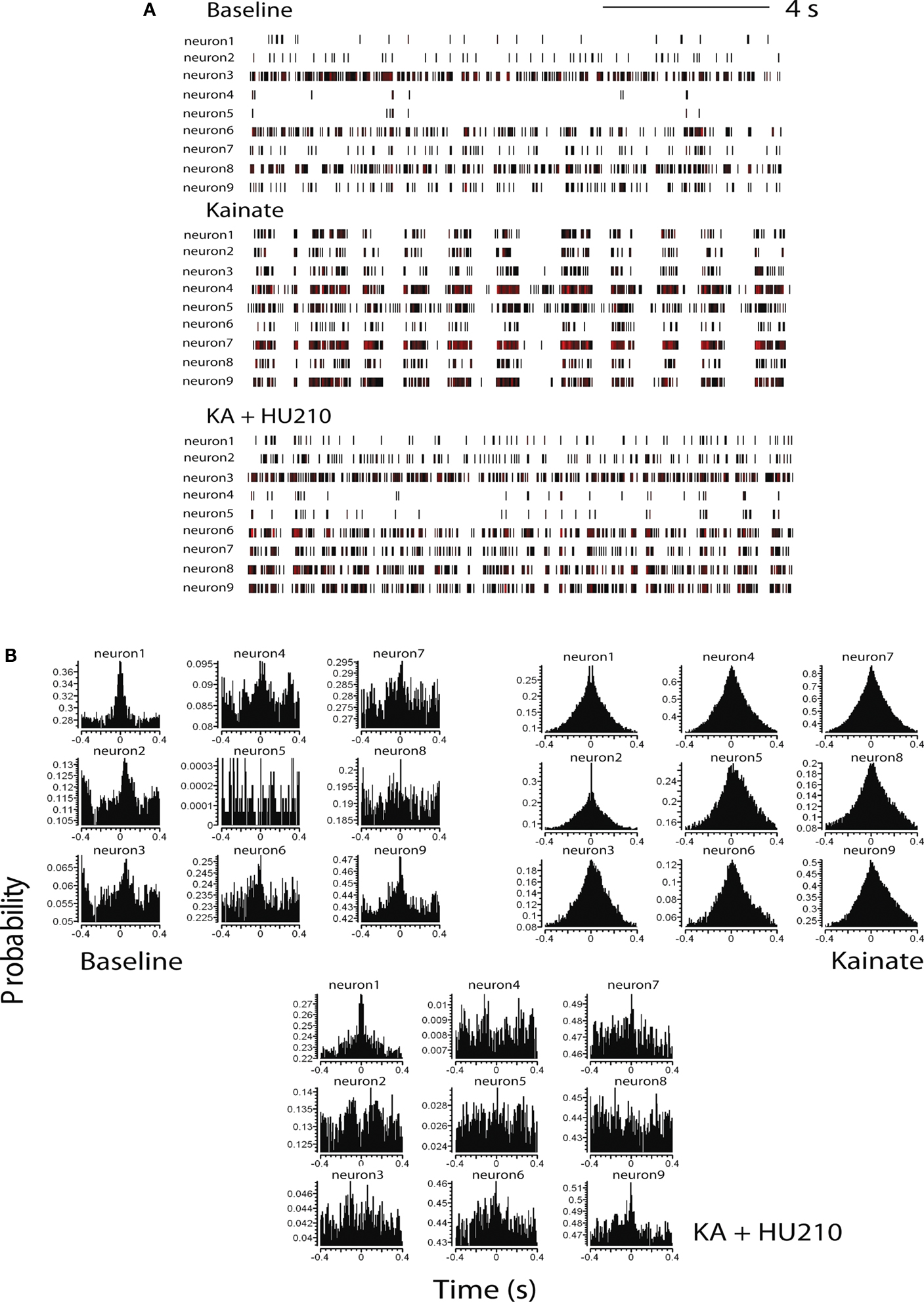
Figure 1. Cannabinoids reverse kainate-induced epileptiform-like activity in the hippocampus. (A) Representative spike rasters for nine simultaneously recorded neurons showing activity during pre-drug basal firing (upper panel) and the induction of synchronous burst firing induced by kainate (KA; 10 mg kg−1; middle panel). HU210 (100 μg kg−1) reversed the KA-induced synchronous activity to that seen during basal activity. (B) Representative cross-correlation histogram (CCH) analyses (with auto-correlogram for neuron 1) for the same neurones shown in the raster displays above. CCHs again illustrate KA-induce synchronous activity and its reversal by HU210. CCH reference was neuron 1 (time 0); bin width 1 ms. All doses i.p.
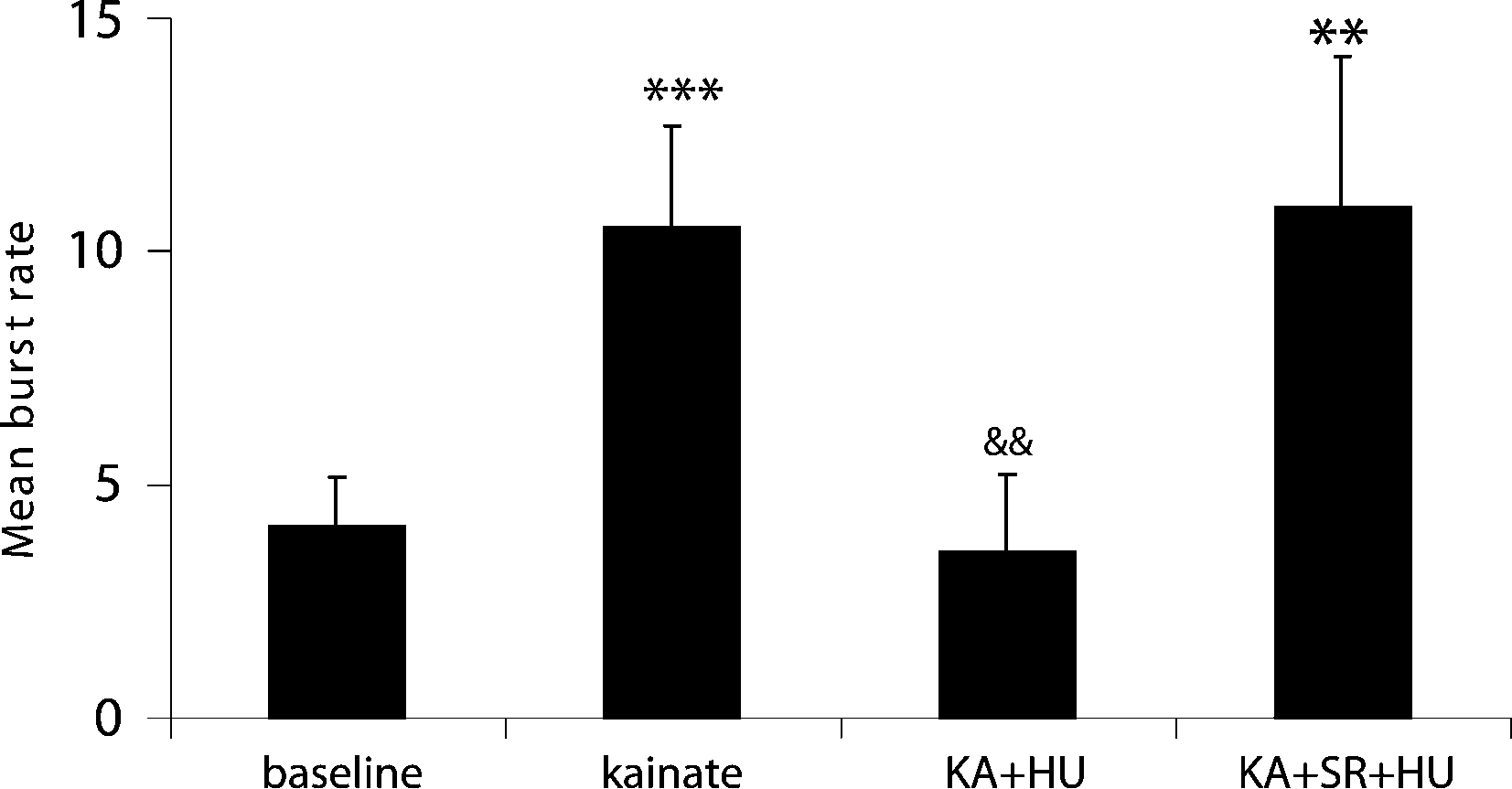
Figure 2. Incidence of mean burst rate (bursts per analysis epoch ± SEM) as a function of drug treatment. KA significantly increased the burst frequency from pre-drug basal activity and entrained the network to burst synchronously. Co-application of the CB1 receptor agonist HU210 (HU) significantly decreased burst rate and disrupted the synchronicity in firing induced by KA. When injected in the presence of the CB1 receptor antagonist rimonabant (SR), HU210 did not alter KA-induced synchronous bursting activity (**p < 0.01; ***p < 0.005 vs baseline and &&p < 0.01 vs KA-induced bursting).
Inspection of firing rate histograms showed that KA treatment elicited two types of bursts: micro-bursts (as defined in “Materials and methods”) and macro-bursts that lasted ∼202 ± 21.2 s in duration (Figure 3
A). The number of KA-induced micro-burst events was significantly reduced by the high-affinity cannabinoid agonist HU210 from 10.5 ± 1.18 to 3.55 ± 2.07 bursts during each analysis period (Figures 1 and 2
; F(1,57) = 8.89, p < 0.05). The KA-induced elevation in firing rate was only marginally reduced by concurrent administration of HU210 (100 μg kg−1), decreasing the firing rate of simultaneously recorded neurons from 13.05 ± 2.04 to 10.74 ± 1.56 Hz on this short time scale (F(2,85) = 1.1, p < 0.38, data not shown). The frequency of macro-bursts was similarly inhibited (Figure 3
) by HU210 from 3.7 ± 1.1 to 1.1 ± 0.3 bursts (F(1,57) = 3.2, p < 0.05, data not shown); however on this longer time scale, overall firing rate was significantly reduced 11.15 ± 3.04 to 4.84 ± 2.26 (F(2,85) = 8.4, p < 0.05).
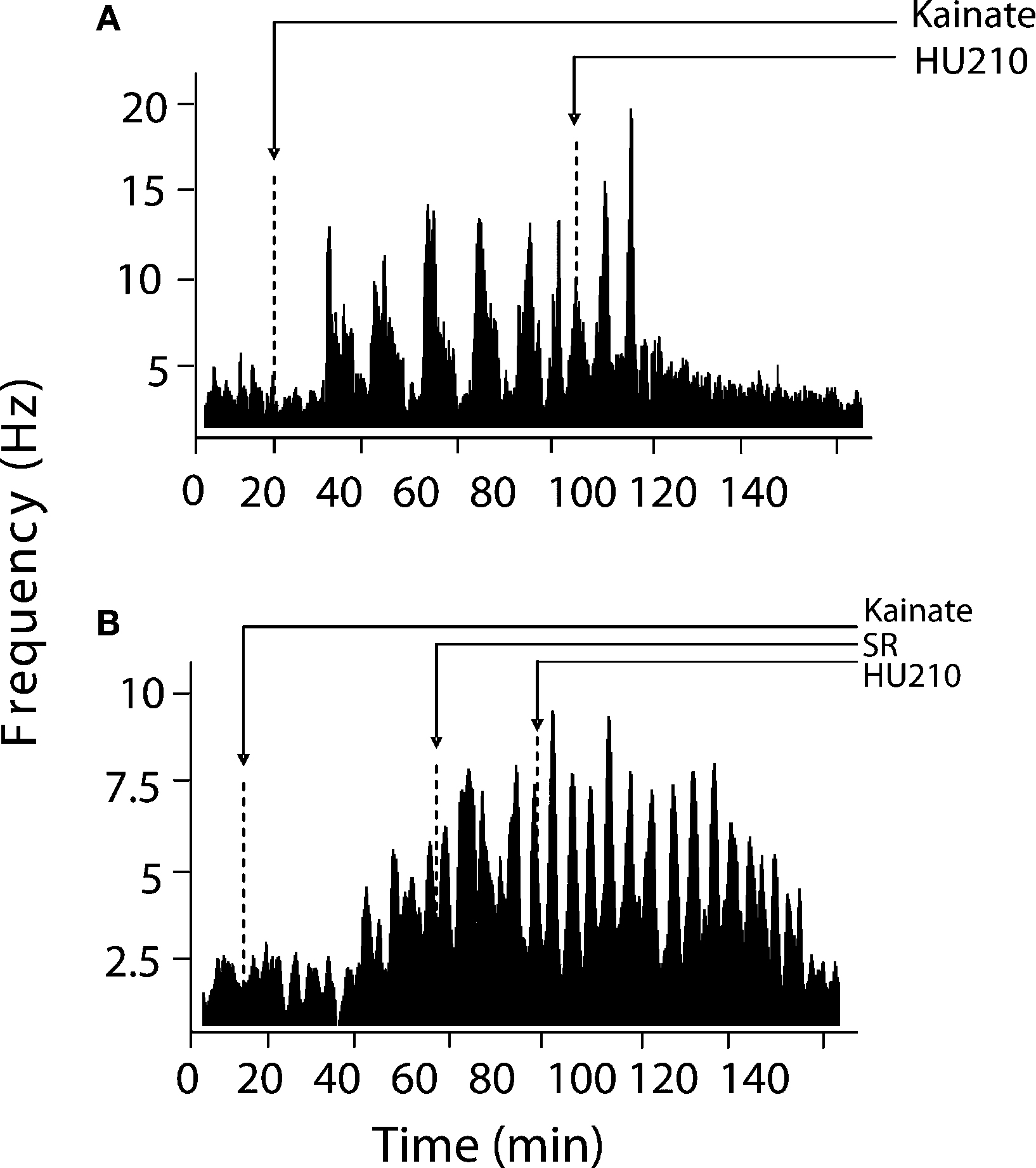
Figure 3. Modulation of KA-induced slow oscillations in firing by CB1 receptors. (A) Representative integrated firing rate histograms (bin width 10 s) illustrating KA-induced macro-burst firing pattern (periodicity ∼200 s). Injection of HU210 abolished this slow oscillatory firing activity. (B) Pre-treatment with the CB1 antagonist rimonabant (SR) completely blocked the actions of HU210. Arrow heads indicate the time of HU210 (100 μg kg−1) or rimonabant (3 mg kg−1) injections during KA (10 mg kg−1). All doses i.p.
Cross-correlation histogram analyses confirmed the visual inspection of the raster displays that there is limited or no synchronous activity between hippocampal neurons during pre-drug basal activity, at least under the recording conditions of the present experiments (Figure 1
B). Following KA injection, corresponding with the period of KA-induced micro-burst and macro-burst activity, there was development of synchronous activity, revealed by peaks in the CCHs around time bin 0 (Figure 1
B). This was reversed by injection of HU210, resulting in CCHs showing a flat profile similar to that expressed under basal conditions (Figure 1
B), indicative of non-synchronous activity.
The observed antiepileptic effects on hippocampal cell activity are mediated by CB1 receptors as the number of KA-induced micro-bursts was not significantly different from KA alone (9.98 ± 3.21; F(2,77) = 4.3, p < 0.69) when HU210 was injected in the presence of the CB1 receptor antagonist rimonabant (21 neurons, n = 3 rats, 3 mg kg−1, Figure 2
). Pre-treatment with rimonabant (3 mg kg−1) also prevented the blockade of KA-induced changes in firing rate and bursting activity patterns by HU210, whilst being devoid of effects of its own (5.81 ± 2.38 Hz during KA/SR and 7.01 ± 3.13 Hz during KA/SR/HU210, and 9.1 ± 1.13 total bursts during KA/SR and 11.24 ± 4.28 total bursts during KA/SR/HU210, F(2,77) = 8.1, p < 0.34, Figure 3
).
The present in vivo multiple single-unit study has generated two main findings. First, functionally cross-correlated epileptiform-like population activity can be evoked by the non-NMDA receptor agonist KA in the hippocampal CA1 pyramidal layer of urethane-anesthetized rats, in a way reminiscent of epileptiform-like activity observed in hippocampal dissociated and organotypic cultures (Jagger et al., 2001
, 2002
; Roe and Mason, 2002
; Sokal et al., 2000)
. The excitotoxin injection increased the firing rate of all the recorded neurons and also increased the burst frequency in the majority of simultaneously recorded cells whilst entraining them to a synchronized firing mode (no KA-induced inhibitions of firing were observed). Secondly, when injected in the presence of KA, the potent CB1 receptor agonist HU210 significantly reduced the elevation in burst rate (accompanied by a modest reduction in overall firing rate) elicited by KA treatment accompanied by a reversal of synchronized burst firing to random activity.
The hippocampal formation is one of the most seizure-prone structures in the brain due to its defining characteristic; the presence of a tri-synaptic circuit of fibers. The reverberation of impulse activity in the hippocampal loop is suggested to cause and maintain epileptiform-like activity (Stringer and Lothman, 1992
). Hippocampal cells are endowed with several voltage and ligand-gated conductances, which play a major role in the cells’ excitability. Even under normal basal conditions cells produce burst discharges that are qualitatively similar to epileptiform-like activity. Blockade of GABAergic transmission has been consistently proven to precipitate seizures. It has thus been assumed that abnormal GABAergic neurotransmission may be related to epilepsy (Prince, 1978
). Accordingly, the most common pharmacological manipulation to cause neuronal disinhibition is the use of the convulsant alkaloid bicuculline; a GABAA receptor antagonist (Margineanu and Wülfert, 1999
). However, in vivo epilepsy can also be induced with exposure to toxins or direct brain electrical stimulation, in a manner that may not be directly related to GABAergic mechanisms. One such agent is the excitotoxin KA, an AMPA/kainate receptor agonist. KA-induced seizures are a well-established model of temporal lobe epilepsy and epileptic seizures reliably occur at the chosen systemic dose of 10 mg kg−1 used in this study, a non-toxic dose (Sperk, 1994
). Interestingly, Khazipov and Holmes (2003)
elegantly implicated GABA inhibitory mechanisms in the emergence of synchronized epileptiform-like activity elicited by KA. In their in vivo superfused hippocampus preparation, GABAA receptor-mediated currents coincided with the inhibition of neuronal firing as assessed by the cross-correlogram of spikes vs inhibitory postsynaptic currents. The authors found that when inhibitory currents ceased, the probability of firing of pyramidal cells increased. Mediation of GABAA receptors was further confirmed by the finding that bicuculline perfusion suppressed KA-induced oscillations.
The hippocampus is one of the areas with the highest cannabinoid receptor binding densities in the rat brain (Herkenham et al., 1991
). This structure also exhibits the highest concentrations of both anandamide (Felder et al., 1996
) and 2-arachidonylglycerol (Stella et al., 1997
), the two most abundant endogenous cannabinoid ligands. It is now well accepted that presynaptic CB1 receptors are located on the terminals of cholecystokinin (CCK+)-containing GABAergic interneurons to inhibit Ca2+ dependent GABA release via N-type channels (for review see Freund et al., 2003
). Cannabinoid receptors are also reported to be situated on glutamatergic presynaptic terminals inhibiting glutamate release via terminal N-type and P/Q type Ca2+ channels (Twitchell et al., 1997
) in autaptic circuits as well as in hippocampal slices (Kawamura et al., 2006
). Thus, at least in the hippocampus, CB1 receptors are located on (CCK+) basket and dendritically projecting cells (Freund, 2003
; Hàjos et al., 2000)
as well as on excitatory afferents to pyramidal cells (Takahashi and Castillo, 2006
). We are therefore left with the view that mixed actions of HU210 on inhibitory and excitatory neurotransmission are responsible for reversing synchronized burst firing. Since intact GABAergic inhibition is necessary for the maintenance of KA-induced bursting, the activation of CB1 receptors by HU210 on the terminals of different populations of GABAergic neurons in the hippocampus and the corresponding inhibition of GABAA synaptic inhibition (Hoffman and Lupica, 2000
) could be sufficient to shunt synchronous population bursting. However, direct effects of CB1 agonists in suppressing glutamatergic excitatory neurotransmission have been observed in a KA model of seizure (Monory et al., 2006
). This suggests CB1 receptors are also critically involved in the control of excitatory glutamatergic neurotransmission. Thus, activation of CB1 receptors may therefore confer inhibitory effects via suppression of excitatory synapses under hyperexcitable conditions. Nevertheless, because of the systemic nature of the dosing regimen employed in our study, contributions from outside the hippocampal circuitry cannot be ruled out (microinjection studies are currently ongoing in our laboratory to address this issue).
The finding that the CB1 receptor antagonist rimonabant was devoid of effect on KA-induced synchronized bursting is puzzling (but see Robbe et al., 2006
) since the present KA manipulations represent the ideal conditions for the on-demand release of endocannabinoids hippocampal slices. This may indicate that the basal level of endocannabinoid binding to CB1 receptors is not sufficient to effectively modulate epileptiform-like activity in anesthetized animals by virtue of achieving a ceiling effect wit the dose of KA used here. It would be interesting to determine whether an effect of rimonabant would be uncovered when recording are performed in awake, freely-moving rats. It would also be important to determine whether injection of rimonabant prevents the induction of epileptiform-like activity by KA, since it does not reverse KA effects on its own. Nevertheless, the present study replicates and builds upon previous findings that KA-induced increases in firing and burst firing rates are attenuated by inhibition of anandamide (an endocannabinoid) hydrolysis (Coomber et al., 2008
). However, under those conditions rimonabant only partly diminished the effects of anandamide hydrolysis blockade (in that particular study functional connectivity was not assessed). The appearance of “macro-bursts” composed of “micro-bursts” in the present report could be associated with metabolic recovery following intense increases in firing rate. Alternatively, the extensive calcium influx during periods of sustained elevated activity may sequentially trigger downregulation of inhibitory and/or excitatory processing (see Hàjos and Freund, 2002
) and the generation of the subsequent volley of intense firing.
These previously unseen findings suggest that the reduction in spike timing coordination and the associated decrease in hippocampal population activity are important in the well-documented antiepileptic actions of cannabinoid compounds (Smith, 2005
). A major implication of these data is that the synchrony of spike timing in neuronal assemblies is a critical component of epileptiform-like activity in vivo. The effect of cannabinoids on network dynamics is noteworthy because neurons form assemblies defined by synchronous action potential firing and these changes in population synchronicity are thought to accompany status epilepticus (Williams et al., 2007
).
In conclusion we have shown that cannabinoid receptors modulate the rhythmicity and synchronization generated by inhibitory neuronal networks in the rat hippocampus. Further investigations are required to determine the clinical relevance and the site of action of the cannabinoid agonist, since drugs that reduce synchronization may represent novel anticonvulsants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks are due to Clare Spicer for technical support. This research was funded in part by NIH grant DA022340 to JFC.
Felder, C. C., Nielson, A., Briley, E. M., Palkovits, A., Priller, J., Axelrod, J., Richardson, J. M., Riggin, R. M., Paul, G. A., and Becker, G. W. (1996). Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS. Lett. 393, 231–235.
Monory, K., Massa, F., Egertová, M., Eder, M., Blaudzun, H., Westenbroek, R., Kelsch, W., Jacob, W., Marsch, R., Ekker, M., Long, J., Rubenstein, J. L., Goebbels, S., Nave, K. A., During, M., Klugmann, M., Wölfel, B., Dodt, H. U., Zieglgänsberger, W., Wotjak, C. T., Mackie, K., Elphick, M. R., Marsicano, G., and Lutz, B. (2006). The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51, 455–466.