 Ji-Qing Duan
Ji-Qing Duan Hui Liu1
Hui Liu1 Jia-Qiao Wu
Jia-Qiao Wu- 1Department of Intensive Care Unit, The Third People's Hospital of Chengdu, Affiliated Hospital of Southwest Jiaotong University, Chengdu, China
- 2Department of Anesthesia, The Third People's Hospital of Chengdu, Affiliated Hospital of Southwest Jiaotong University, Chengdu, China
A 24-year-old man presented with insidious onset progressive gait disturbance and was finally diagnosed with autosomal recessive hereditary spastic paraplegia. Two novel mutations, including a frameshift mutation (c.5687_5691del) and a non-sense mutation (c.751C>T), were identified in the SPG11 gene of the patient through whole genome sequencing. The frameshift mutation of c.5687_5691del leads to a change in amino acid synthesis beginning with amino acid No. 1896 arginine and terminating at the 8th amino acid after the change (p. Arg1896MetfsTer8). The non-sense mutation (c.751C>T) causes the conversion of codon 251st encoding the amino acid Gln into a stop codon (p. Gln251Ter), resulting in premature termination of peptide synthesis. Although confirmation of compound-heterozygosity could not be performed, our findings enriched the phenotypic spectrum of SPG11 mutations related to hereditary spastic paraplegia.
Background
A 24-year-old man presented with insidious onset progressive gait disturbance and was finally diagnosed with autosomal recessive hereditary spastic paraplegia. Two novel mutations, including a frameshift mutation (c.5687_5691del) and a non-sense mutation (c.751C>T), were identified in the SPG11 gene of the patient through whole-genome sequencing. The frameshift mutation of c.5687_5691del leads to a change in amino acid synthesis beginning with amino acid No. 1896 arginine and terminating at the eighth amino acid after the change (p. Arg1896MetfsTer8). The non-sense mutation (c.751C>T) causes the conversion of codon 251, encoding the amino acid Gln into a stop codon (p. Gln251Ter), resulting in premature termination of peptide synthesis. Although the confirmation of compound-heterozygosity could not be performed, our findings enriched the phenotypic spectrum of SPG11 mutations related to hereditary spastic paraplegia.
Case presentation
A 24-year-old man born in a non-consanguineous family developed insidious onset progressive gait disturbance accompanied by weakness in the lower limbs for the past 4 years. The weakness of his lower extremities began distally and gradually progressed proximally. His performance at school was below average. Upon examination, his speech showed mild dysarthria, and his lower extremity strength was Medical Research Council (MRC) grade 4/5 with brisk reflexes and a bilateral extensor plantar response. He had pes cavus, normal muscle volume, bilateral foot drop, positive Babinski's sign, and scissor gait. There were no signs suggestive of upper limb problems. The scores of the mini-mental state examination (MMSE, normal range ≥27 points) and Montreal Cognitive Assessment (MoCA, normal range ≥26 points) were 29 points and 22 points, respectively, indicating mild cognitive impairment in the patient. Blood and cerebrospinal fluid (CSF) testing, including routine blood tests, serum immunologic tests, liver, and kidney function tests, as well as CSF biochemistry, immunologic tests, and oligoclonal bands, were all within the normal range. Magnetic resonance imaging (MRI) of the brain showed an extremely thin corpus callosum on sagittal views and symmetrical periventricular white matter lesions (Figures 1A, B). Thoracic MRI showed that the thoracic spinal cord was significantly thinner and reduced in volume (Figure 2). However, there was no obvious abnormal signal in the thoracic spinal cord on MRI images. No obvious abnormality was found in an electromyogram (EMG) or an electroencephalogram (EEG) of the patient. Somatosensory evoked potentials (SEPs) showed a significant decrease in the amplitude and central conduction velocity of the lower extremities. Whole-genome sequencing (WGS) was performed on the patient. A frameshift mutation (c.5687_5691del) and a non-sense mutation (c.751C>T) were identified in the SPG11 gene of the patient (Figure 3). The diagnosis of autosomal recessive HSP with a thin corpus callosum was finally confirmed according to the clinical manifestations and genetic test results of the patient (Patel et al., 2016; Zhang et al., 2016; Meyyazhagan et al., 2022). The Spastic Paraplegia Rating Scale (SPRS) was 16 points according to a previous study (Schüle et al., 2006). The patient received a daily dose of 60 mg edaravone and 0.5 g citicoline for ~2 weeks, and he was seen for follow-up visits once every 3 months. However, the weakness of his lower extremities and his gait disturbance was not effectively relieved.
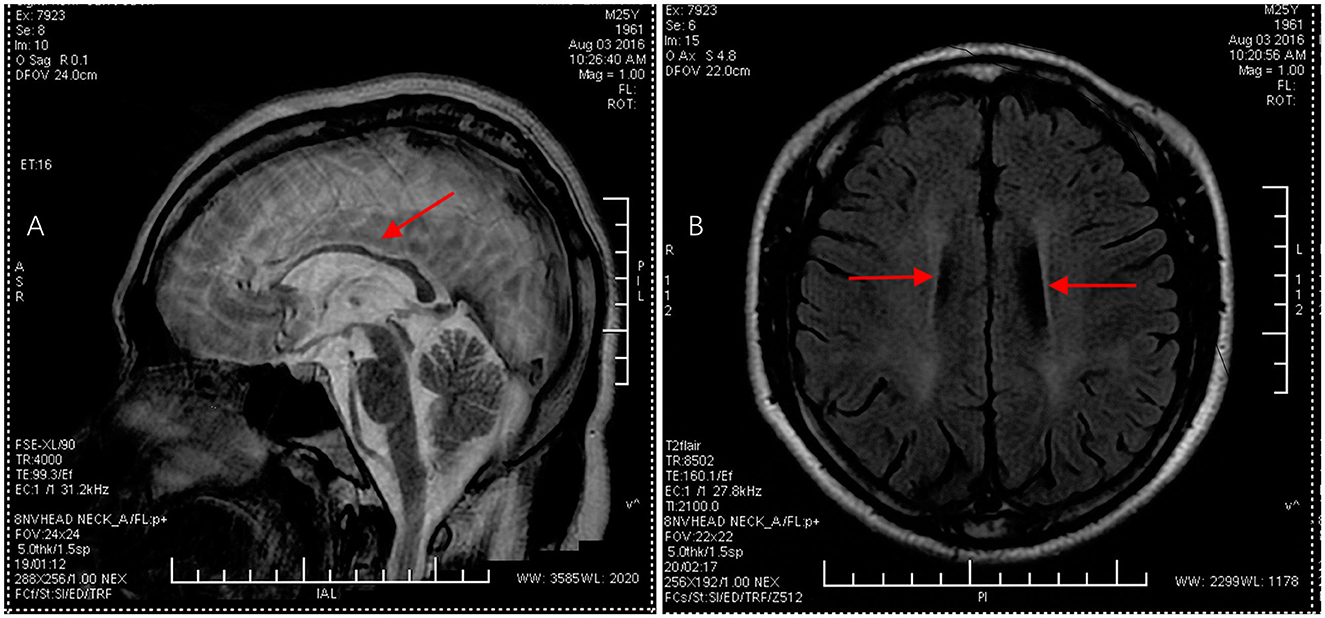
Figure 1. Brain MRI images in the proband. (A) Sagittal T2-weighted image displayed thinning of the corpus callosum, as indicated by the red arrow. (B) Axial T1-weighted image showed symmetrical periventricular white matter lesions, as indicated by the red arrow.
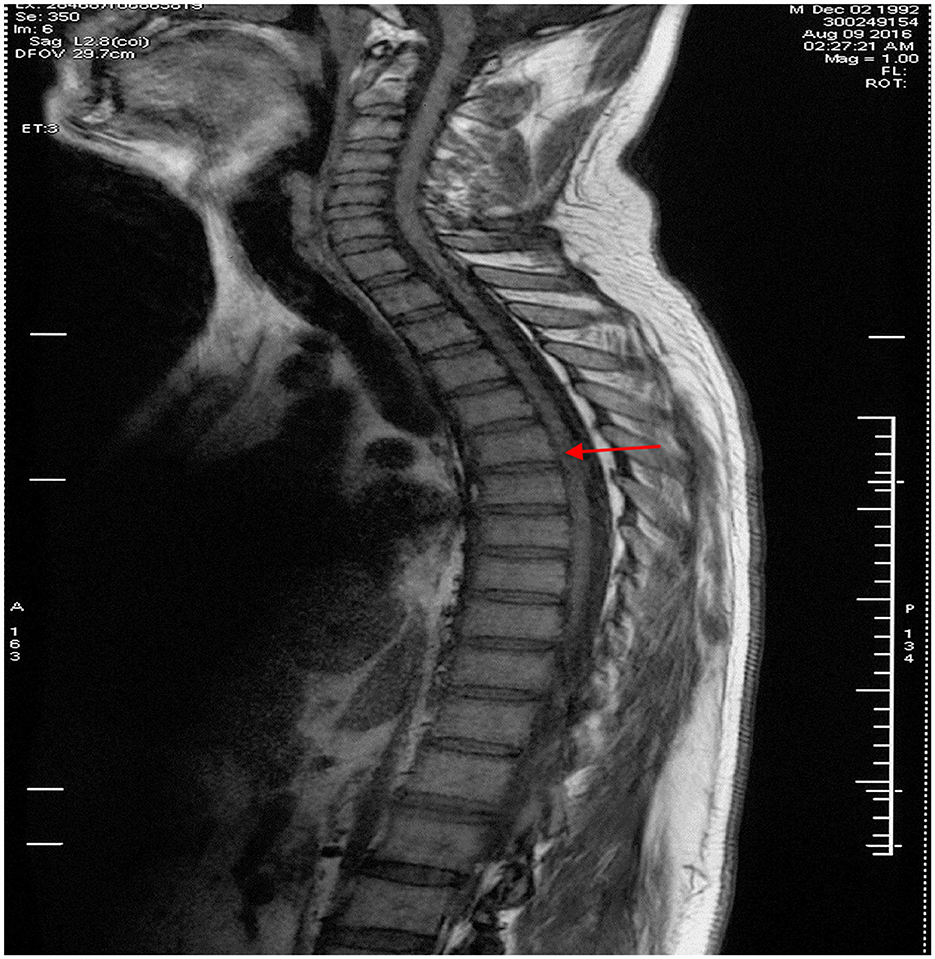
Figure 2. Thoracic spinal MRI image in the proband. Thoracic spinal MRI revealed thinning of the thoracic spinal cord with volume loss but no abnormal signal in the cord on sagittal T1-weighted imaging, as indicated by the red arrow.
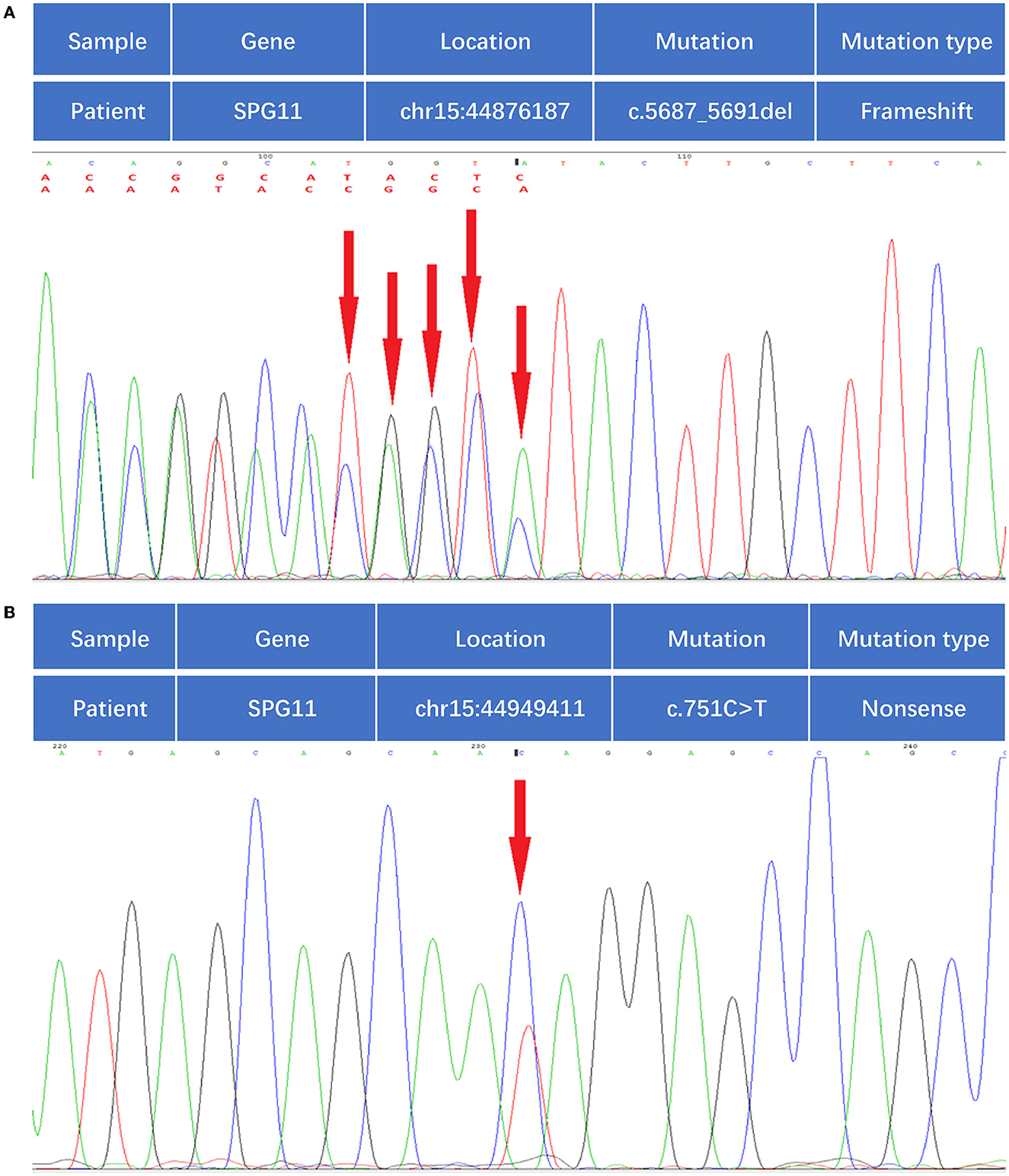
Figure 3. Whole-genome sequencing identified compound heterozygous mutations in the SPG11 gene. (A) Mutation of c.5687-5691del, as indicated by the red arrow. (B) Mutation of c.751C>T, as indicated by the red arrow.
Discussion
HSP is a genetically heterogeneous group of neurodegenerative disorders characterized by progressive spasticity and lower extremity weakness as a result of retrograde axonal degeneration of the corticospinal tracts and posterior columns (Samaranch et al., 2008; Stevanin et al., 2008). HSP with a thin corpus callosum (HSP-TCC) is a distinguished subgroup of complicated forms and is often inherited as a recessive trait (Sperfeld et al., 2005; Hourani et al., 2009; Riverol et al., 2009). HSP-TCC has a high rate of disability, and most of the affected patients will become wheelchair-bound one or two decades after the onset of the disease (Stevanin et al., 2007). The clinical features of HSP begin primarily before the second decade of life. The estimated prevalence for HSP of all types ranges from 1:100,000 to 10:100,000, affecting diverse ethnic groups. The diagnosis of HSP is mainly based on clinical characteristics, neuroimaging, and gene testing (Shribman et al., 2019). The pathogenic mechanism, associated clinical characteristics, and neuroimaging abnormalities vary greatly, owing to different disease-causing genes, making it challenging to distinguish HSP from other genetic disorders associated with spasticity. Gene testing is now widely available, but there are limitations, and molecular diagnosis cannot be performed in most suspected cases. With a further understanding of the pathophysiological basis of individual HSP subtypes, opportunities are emerging to provide targeted molecular therapies and personalized medications (Shribman et al., 2019).
To date, 15 distinct loci associated with HSP-TCC have been identified: SPG 1, SPG 11, SPG 15, SPG 18, SPG 21, SPG 44, SPG 45 (65), SPG 46, SPG 47, SPG 49, SPG 54, SPG 56, SPG 63, SPG 67, and SPG 71 (Boukhris et al., 2010; Finsterer et al., 2012). In the current study, we identified two novel SPG 11 gene mutations: a frameshift mutation (c.5687_5691del) and a non-sense mutation (c.751C>T). SPG11-associated HSP is supposed to be the most frequent type of complicated autosomal recessive HSP, especially for patients with a thin corpus callosum and intelligence disorder (Paisanruiz et al., 2008; Meyyazhagan et al., 2022). The SPG 11 gene, which is located at chromosome 15q13-15, encodes the spatacsin protein that is primarily expressed in the pineal gland, cerebral cortex, cerebellum, and hippocampus (Paisanruiz et al., 2008; Samaranch et al., 2008; Panza et al., 2022). The genetic analysis of SPG11 revealed frequent truncating mutations altering the protein structure and function in the KIAA1840 gene (Lossos et al., 2006; Fink, 2013).
Among the mutations that have been reported in HSP, non-sense and frameshift mutations are the most frequent types described in SPG 11 (Liao et al., 2008; Cao et al., 2013; Tian et al., 2016; Du et al., 2018). To our knowledge, the mutation of SPG11 as seen in our case has never been reported in the literature. In our case, a frameshift mutation of c.5687_5691del (deletion of nucleotide number 5687_5691 in the coding region) was found in the SPG11 gene of the subject. The mutation resulted in a change in amino acid synthesis beginning with amino acid No. 1896 arginine and terminating at the eighth amino acid after the change (p. Arg1896MetfsTer8). A non-sense mutation of c.751C>T (nucleotide 751 in the coding region changes from C to T) was also noted, which causes the conversion of codon 251, encoding the amino acid Gln into a stop codon (p. Gln251Ter), resulting in premature termination of peptide synthesis. Such a mutation would bring about a truncated protein, resulting in a loss of the physiological function of the spatacsin protein (Stevanin et al., 2008). Both mutations can be pathogenic according to a joint consensus recommendation (Richards et al., 2015).
HSP induced by mutations of SPG 11 is characterized by early-onset progressive spasticity and weakness, a thin corpus callosum, and cognitive dysfunction. Approximately 79% of patients with HSP-TCC initially suffered from walking difficulties, and ~16% of patients first presented with symptoms of intellectual disability (Stevanin et al., 2008; Panza et al., 2022). In our case, the patient who presented with insidious onset progressive gait disturbance also had weakness in the lower limbs. A mild impairment of the cognitive function of the patient was also observed. Other symptoms of the patient included scissor gait, bilateral foot drop, and mild dysarthria. All the symptoms were consistent with previous studies (Liao et al., 2008; Cao et al., 2013; Tian et al., 2016; Du et al., 2018). Regrettably, the patient's father died in a car accident, his mother refused to undergo in-hospital genetic tests, and we failed to obtain his family pedigree. Although family segregation studies were not possible as explained, the phenotype and the characteristics of the variants detected indeed support their pathogenicity.
Conclusion
We reported a rare case of HSP-TCC accompanied by two novel mutations of SPG11, and our findings enriched the phenotypic spectrum of SPG11 mutations. For patients presenting with HSP-TCC, we recommend direct testing for the SPG11 gene to establish the role of SPG11 mutation in these rare disorders.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
J-QD wrote the initial draft (including substantive translation). HL revised the manuscript. J-QW approved the final version. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
HSP, hereditary spastic paraplegia; HSP-TCC, HSP with thin corpus callosum; PG11, Spastic paraplegia 11 = S; MRC, Medical Research Council; MMSE, mini-mental state examination; MoCA, Montreal Cognitive Assessment; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; EMG, electromyogram; EEG, electroencephalogram; WGS, whole-genome sequencing.
References
Boukhris, A., Feki, I., Elleuch, N., Miladi, M. I., Boland-Augé, A., Truchetto, J., et al. (2010). A new locus (SPG46) maps to 9p21.2-q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum. Neurogenetics 11, 441–448. doi: 10.1007/s10048-010-0249-2
Cao, L., Rong, T. Y., Huang, X. J., Fang, R., Wu, Z. Y., Tang, H. D., et al. (2013). Novel SPG11 mutations in Chinese families with hereditary spastic paraplegia with thin corpus callosum. Parkinsonism Relat. Disord. 19, 367–370. doi: 10.1016/j.parkreldis.2012.10.007
Du, J., Hu, Y. C., Tang, B. S., Jiang, H., and Shen, L. (2018). Identification of novel SPG11 mutations in a cohort of Chinese families with hereditary spastic paraplegia. Int. J. Neurosci. 128, 146–150. doi: 10.1080/00207454.2017.1378878
Fink, J. K. (2013). Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328. doi: 10.1007/s00401-013-1115-8
Finsterer, J., Löscher, W., Quasthoff, S., Wanschitz, J., Auer-Grumbach, M., and Stevanin, G. (2012). Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 318, 1–18. doi: 10.1016/j.jns.2012.03.025
Hourani, R., El-Hajj, T., Barada, W. H., Hourani, M., and Yamout, B. I. (2009). MR imaging findings in autosomal recessive hereditary spastic paraplegia. AJNR Am. J. Neuroradiol. 30, 936–940. doi: 10.3174/ajnr.A1483
Liao, S. S., Shen, L., Du, J., Zhao, G. H., Wang, X. Y., Yang, Y., et al. (2008). Novel mutations of the SPG11 gene in hereditary spastic paraplegia with thin corpus callosum. J. Neurol. Sci. 275, 92–99. doi: 10.1016/j.jns.2008.07.038
Lossos, A., Stevanin, G., Meiner, V., Argov, Z., Bouslam, N., Newman, J. P., et al. (2006). Hereditary spastic paraplegia with thin corpus callosum: reduction of the SPG11 interval and evidence for further genetic heterogeneity. Arch. Neurol. 63, 756–760. doi: 10.1001/archneur.63.5.756
Meyyazhagan, A., Kuchi Bhotla, H., Pappuswamy, M., and Orlacchio, A. (2022). The puzzle of hereditary spastic paraplegia: from epidemiology to treatment. Int. J. Mol. Sci. 23, 7665. doi: 10.3390/ijms23147665
Paisanruiz, C., Dogu, O., Yilmaz, A., Houlden, H., and Singleton, A. (2008). SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia (HSP). Neurology 70, 1384–1389. doi: 10.1212/01.wnl.0000294327.66106.3d
Panza, E., Meyyazhagan, A., and Orlacchio, A. (2022). Hereditary spastic paraplegia: genetic heterogeneity and common pathways. Exp. Neurol. 357, 114203. doi: 10.1016/j.expneurol.2022.114203
Patel, S., Sethi, P. K., Anand, I., Batra, A., and Gupta, P. (2016). Hereditary spastic paraplegia with a thin corpus callosum due to SPG11 mutation. Neurol. India 64, 171. doi: 10.4103/0028-3886.173660
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riverol, M., Samaranch, L., Pascual, B., Pastor, P., Irigoyen, J., Pastor, M. A., et al. (2009). Forceps minor region signal abnormality “ears of the lynx”: an early MRI finding in spastic paraparesis with thin corpus callosum and mutations in the spatacsin gene (SPG11) on chromosome 15. J. Neuroimaging 19, 52–60. doi: 10.1111/j.1552-6569.2008.00327.x
Samaranch, L., Riverol, M., Masdeu, J. C., Lorenzo, E., Vidal-Taboada, J. M., Irigoyen, J., et al. (2008). SPG11 compound mutations in spastic paraparesis with thin corpus callosum. Neurology 71, 332–336. doi: 10.1212/01.wnl.0000319646.23052.d1
Schüle, R., Holland-Letz, T., Klimpe, S., Kassubek, J., Klopstock, T., Mall, V., et al. (2006). The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology 67, 430–434. doi: 10.1212/01.wnl.0000228242.53336.90
Shribman, S., Reid, E., Crosby, A. H., Houlden, H., and Warner, T. T. (2019). Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 18, 1136–1146. doi: 10.1016/S1474-4422(19)30235-2
Sperfeld, A. D., Baumgartner, A., and Kassubek, J. (2005). Magnetic resonance investigation of the upper spinal cord in pure and complicated hereditary spastic paraparesis. Eur. Neurol. 54, 181–185. doi: 10.1159/000090294
Stevanin, G., Azzedine, H., Denora, P., Boukhris, A., Tazir, M., Lossos, A., et al. (2008). Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131, 772–784. doi: 10.1093/brain/awm293
Stevanin, G., Santorelli, F. M., Azzedine, H., Coutinho, P., Chomilier, J., Denora, P. S., et al. (2007). Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 39, 366. doi: 10.1038/ng1980
Tian, X., Wang, M., Zhang, K., and Zhang, X. (2016). Novel SPG 11 mutations in hereditary spastic paraplegia with thin corpus callosum in a chinese family. Can. J. Neurol. Sci. 43, 833–840. doi: 10.1017/cjn.2016.17
Keywords: hereditary spastic paraplegia, thin corpus, SPG11, gene mutation, case report
Citation: Duan J-Q, Liu H and Wu J-Q (2023) Case report: Novel mutations in the SPG11 gene in a case of autosomal recessive hereditary spastic paraplegia with a thin corpus callosum. Front. Integr. Neurosci. 17:1117617. doi: 10.3389/fnint.2023.1117617
Received: 06 December 2022; Accepted: 06 March 2023;
Published: 24 March 2023.
Edited by:
Antonio Orlacchio, Santa Lucia Foundation (IRCCS), ItalyReviewed by:
Martin Regensburger, University of Erlangen Nuremberg, GermanyXiangzhen Yuan, Shanghai Jiao Tong University, China
Copyright © 2023 Duan, Liu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-Qiao Wu, d3VqaWFxaWFvOTNAMTYzLmNvbQ==