 Gemma M. McLaughlin
Gemma M. McLaughlin- Independent Researcher, Christchurch, New Zealand
Introduction: Vespula vulgaris is an invasive wasp that causes considerable detriment to native birds and invertebrates in New Zealand. Reducing at least 80% of invasive wasp densities is necessary to manage the problems this species presents to its invaded range. To explore the function of target genes for the genetic management of V. vulgaris, screening of appropriate reference genes is crucial for conducting the reverse transcriptase-quantitative real-time PCR (RT-qPCR). The selection of appropriate reference genes is an important but often overlooked consideration when delving into RNA research. Many studies rely on one of two tried and trusted reference genes widely used in the literature, which may not be suitable for the normalization of data under particular variables.
Methods: Here, I selected six reference genes of V. vulgaris and evaluated their stability across two conditions: developmental stage and sex by using five different tools for analysis: the ΔCt method, geNorm, NormFinder, BestKeeper, and RefFinder.
Results: Differing appropriate reference genes for different research foci: TBP, EF1A, RPL18X3, and CAPZB for developmental stage treatment, and KTB, EF1A, and CAPZB amongst the sexes.
Discussion: My study further emphasizes that there is no “one size fits all” reference gene, and advocates for analysis of reference gene suitability when conducting gene quantification experiments.
1 Introduction
Vespula vulgaris is an invasive eusocial insect affecting suburban and beech forest areas throughout New Zealand (1). Vespula wasps exist at the highest global densities in New Zealand (2), to the detriment of the native avifauna and invertebrates, especially in beech forests (3–5). Extensive preliminary research is needed to lay a foundation for genetic research on Vespula control. The recent genome assembly and annotation of three Vespula genomes (V. vulgaris, V. gemanica, and V. pensylvanica; 6) has been a great advancement towards this, as it provides the actual sequence of genes of the organism of interest instead of relying on orthologs from closely related species from genetic databases such as NCBI. RNA interference (RNAi) approaches could be employed to silence genes pivotal to Vespula development. Identifying the desired target genes for silencing, then analysis of their abundance post-transcription will determine the adequacy of RNAi to control this pest species. Functional gene studies can identify the target genes ideal for silencing, and their transcript abundance can be demonstrated using quantitative real-time PCR (RT-qPCR).
RT-qPCR is an excellent method for accurately quantifying gene expression in various tissues. It evaluates the expression patterns of genes under a range of experimental conditions (7, 8) and is frequently used for interpretations of relative gene expression, providing exceptionally accurate and reproducible quantitation of gene copies with high throughput (9).
RT-qPCR is easy to use, fast, avoids the use of radioactivity, and requires a minimal amount of RNA. However, some issues reside in the use of RT-qPCR which include the variability of RNA and varying reverse transcription and PCR efficiencies (10). Unlike DNA in the genome which remains context-independent (i.e., normally cells contain the same DNA sequence), RNA in the transcriptome is context-dependent (i.e., the mRNA component level varies with pathology, physiology or development; 10), which can make it difficult to achieve an accurate result.
Ideally, such sample-to-sample variation could be normalized by measuring the levels of a single universal cellular RNA, which is present at all stages of development at constant levels, irrespective of experimental treatment or tissue used (11). These so-called reference genes are widely employed as an internal RNA reference for Northern blotting, RNase protection, and RT-qPCR practices. However, in many studies, only a single, usually popular, reference gene is used, usually without any investigation/justification for the choice of the reference gene or why they are only using one. Historically, a single reference gene has been used for RNA transcription analyses. Suzuki, Higgins and Crawford (10) found that over 90% of RNA transcription analyses published in high-impact journals in 1999 used only one reference gene. One gene in particular – glyceraldehyde-3-phosphate dehydrogenase (GAPDH) – was commonly used as the sole reference gene for RNA analysis involving human tissue biopsies, despite a lack of research supporting the suitability of this gene. GAPDH was not investigated as a suitable candidate until 2002, when GAPDH mRNA levels were found to be higher in some cancers but lower or unaltered in others in comparison to normal tissue (12) and, therefore, an unsuitable reference gene for RNA analysis. β-actin is also a frequently used control for RNA, however β-actin mRNA levels have been found to increase significantly in the adrenal glands of hypophysectomized rats exposed to adrenocorticotropin (13), increase in rat myocardium after abdominal aortic banding (14), and increases in the liver of rats with vitamin B6 deficiency (15), thus rendering it as an unsuitable reference gene when conducting such research, especially as the sole reference gene. Other papers also advocate for using more than one reference gene (see 10, 16).
Clearly, it is crucial to employ the appropriate reference genes for accurate gene expression analyses. As no experiments concerning gene expression in V. vulgaris currently exist, validation of suitable reference genes is necessary for accurate genetic analysis. This paper aimed to identify the most appropriate reference genes for genetic analysis in V. vulgaris via selection and validation of a suite of reference genes under the treatment conditions of sexes (male and female) and developmental stages (larvae, pupae, and adults). I selected 10 different reference genes as potential reference genes: dimethyladenosine transferase (DIMT), F-actin-capping protein subunit beta (CAPZB), 60s ribosomal protein (60s RP), peptidylprolyl isomerase domain and WD repeat-containing protein (PPWD), king tubby (KTUB), ribosomal protein L18 isoform X3 (RPL18X3), 18s ribosomal RNA (18s rRNA), adenosine kinase 1 (AK1), elongation factor 1-alpha-like (EF1A), and TATA binding (TBP). These genes were chosen as they have been used in previous reference gene studies in Hymenoptera (17, 18), and all are employed in the metabolic and physiological activities of cells (19, 20). The stability of these potential reference genes was initially analyzed using an RT-qPCR and evaluating standard curves. The top six genes were taken and underwent RT-qPCRs, and expression stabilities were analyzed using four different modes of software: BestKeeper (21), geNorm (16), NormFinder (22), and the ΔCt method (23). A final ranking from the cumulative data sets was also conferred via RefFinder (24). Based on my findings, the reference genes were ranked from most to least stable, and recommendations were made for RT-qPCR experiments in V. vulgaris. Finally, the DNA-dependent RNA polymerase II subunit protein RPB7 gene (RPB7) was used as the target gene to verify my findings. To my knowledge, this study is the first to identify stable RT-qPCR reference genes for V. vulgaris, which can aid in accurate gene expression analysis for V. vulgaris for further analysis of targeted genes for RNAi.
2 Methods
2.1 Sample collection
V. vulgaris nests were collected from around the Dunedin/Mosgiel region after advertising free excavation services on social media in collaboration with Nichol’s Garden Centre. Excavated nests were stored in plastic buckets in a 4°C cold room overnight to induce stasis and inhibit function. Individual adults, pupae, larvae, and adults of both sexes were then selected and stored at −80°C for RNA extraction.
Developmental stages included in this study were third-instar larvae, pupae, and three-day-old adults. Developmental stages were identified by specific features: a grub-like appearance for larvae and visualised as the middle size of the larvae present; pupae were selected from capped cells and had clear eye definition; and adults were selected upon emergence from their cells and monitored for 72 hours before freezer storage.
For the sexes, adult workers and adult drones were used. Gynes/queens were excluded from this treatment as they are genetically identical to workers and were not as accessible as workers (e.g., 15 nests with queens would have had to be excavated, as opposed to a single nest for sufficient workers).
2.2 Sample preparation
The stability of different reference genes in V. vulgaris was assessed across (i) developmental stages and (ii) sexes. Wasps used for life stages were selected the day after nest collection. Larvae and pupae were collected from nest combs, and adults were collected after emerging from their silk caps and stored in foraging boxes for three days then placed in 1.5 mL Eppendorf tubes and stored at −80°C until RNA extraction. Three samples were used for each life stage/experiment, with each experiment conducted in triplicate. Under the sexes condition, three sets of five adult workers and drones were selected separately.
2.3 RNA extraction
The Qiagen RNeasy Mini Kit was used for RNA extraction. Whole samples were removed from -80°C storage, transferred to a 1.5 ml Eppendorf tube, and placed on dry ice. 300 µl of Trizol was added, and the sample was homogenized using a 1000 µl pipette tip for a minimum of three minutes. More Trizol was added to make up the final volume of 1 ml, along with 200 µl of chloroform. The tube was shaken vigorously by hand for 30 seconds and then incubated at room temperature for three minutes. Samples were centrifuged at maximum speed (10,000 xg) for 15 minutes. 450 µl of the upper aqueous upper phase was transferred to a new sterile 1.5 ml Eppendorf tube with the rest discarded. 1x volume of 100%, RNA-free ethanol was added and mixed by pipetting. Samples were transferred to a RNeasy Mini spin column placed in a 2 ml collection tube, and centrifuged at 12,000 xg for one minute. The flow-through was discarded, and samples were loaded with 350 µl of buffer RW1 and centrifuged at 13,000 xg for one minute. 10 µl DNase 1 stock solution was added to 70 µl of RDD buffer, mixed by gently inverting the tube, and centrifuged briefly. This was transferred directly to the RNeasy column membrane, and incubated at room temperature for 15 minutes. The column was washed with 350 µl RW1 buffer and centrifuged for one minute at 13,000 xg with flow-through discarded. 500 µl buffer RPE was added and centrifuged as above. An additional 500 µl buffer RPE was added and centrifuged for two minutes at 8,000 xg with flow-through discarded. A “dry” centrifuge run at full spin followed to dry out the membrane. The column was transferred to a new sterile Eppendorf, and 40 µl RNase-free water was added directly to the spin column membrane. This was then incubated at room temperature for one minute and centrifuged for one minute at 8,000 xg. This was repeated once, with 5 µl aliquoted for quality assessment, while the remainder of the final samples were stored at −80°C. The 5µl aliquots were assessed for RNA purity, with 1.5 µl used for concentration quantification by measuring the absorbance at a wavelength of 206 nm with a spectrophotometer (NanoDrop 2000C, Thermo Fisher Scientific, USA), with the remainder run on a 1% agarose gel electrophoresis with a 1 Kb+ ladder (Invitrogen) and 2 µl of gel pilot loading dye (Qiagen) to check for clean bands and therefore a lack of degradation.
2.4 RNA purification
Samples with a 260/230 ratio of <1.5 underwent a clean-up to improve purity. A 1/10 sample volume of NaAc and 2.5x volume of 100% ethanol was added to each sample and stored at −80°C overnight. Samples were spun at 12,000 g for 30 minutes at 4°C, and the supernatant was removed. 200µl of 70% ethanol was added and samples were spun at 12,000 g for 10 minutes at 4°C. The supernatant was removed, and samples were air-dried for 5–10 minutes. Samples were resuspended in 20 µl RNase-free water and rerun in the Nanodrop to check for altered purity and concentration.
2.5 Verification of candidate reference genes
Candidate reference genes were first selected from Polistes genomes available on NCBI. Genes were identified in the V. vulgaris genome (Harrop et al., 2020) with diamond v0.9.24.125 using the command “diamond blastp –query proteins_of_interest.faa –db Vespula_vulgaris”. Proteins were matched to Polistes genome protein models and hits were further classified using visual alignment. Samtools faidx version 1.13-5-gb188dd8 was used to extract the predicted genes from the V. vulgaris genome where the proteins matched. Gene-specific primers were then designed using Primer Wizard (Benchling) and used to clone the open reading frame (ORF) of each reference gene. Primer sequences used for this study are shown in Table 1.

Table 1. Primers used for studying reference gene expression in V. vulgaris by RT-qPCR.
Reverse transcription of samples into synthesized cDNA was conducted using the following reagents: 4 µl 5x VILO™ Reaction Mix, 2 µl 10x SuperScript® Enzyme Mix, 1 µg RNA, and nuclease-free water to a total volume of 20 µl. Samples were then incubated at 25°C for 10 minutes, 42°C for 60 minutes, and terminated at 85°C for 5 minutes. Samples were stored at −20°C until ready for further testing. Primers were checked for specificity via a PCR. Two controls were used: a water-only control and 100 ng of genomic DNA from a gyne sample to verify that amplification from genomic DNA results in a bigger fragment. Reagents consisted of 5 µl NEBNext® Ultra™ II Q5® Master Mix (Ipswich, MA), 1 µl of each of the forward and reverse primers (10 µM), 1 µl of cDNA or 3 µl of gDNA, and nuclease-free water to a total of 10 µl. Conditions of the PCR amplification comprised of 3 min at 94°C, 35 cycles of 94°C for 10 s, 58°C for 30 s, 72°C for 20 s, and a final 7 min at 72°C. Samples were visualised on a 1% agarose gel electrophoresis with a 1 Kb+ ladder (Invitrogen) and 2 µl of gel pilot loading dye (Qiagen).
To measure the efficiency of the reference genes I performed a PCR on a Light Cycler 480 thermocycler (LC480-II; Roche Diagnostics, Rotkreuz, Switzerland). An RNA sample underwent three reverse transcription reactions at 20 µl each to a pooled total volume of 60 µl. This was diluted with 240 µl of nuclease-free water to act as the undiluted initial sample. A serial dilution of cDNA (1:1, 1:10, 1:100, 1:1,000 and 1:10,000) was made with 5 µl of dilutions for each well and one water control. The qPCR reactions comprised of a master mix with 2x SYBR green mix (10 µl), 10 pmol/µl each forward and reverse primer (0.6 µl), and 3.8 µl RNase-free water to a final 15 µl volume. Each dilution was done in triplicate for each gene. he reaction cycle was as follows: 3 min at 94 °C, followed by 35 cycles of 30 s at 94°C, 30 s at 58°C, and 20 s at 72°C, and a final 7 min at 72°C. Each plate included a water-negative control for each primer pair. Analysis of the amplification plot and melting curve followed each reaction.
2.6 Validation and analyses of the stability of the candidate reference genes
All 10 primers underwent an initial RT-qPCR to test their amplification potential. This list was reduced to the six most effective primers, as four failed to produce stable curves across replicates.
2.7 Data analysis
Six reference genes with the most stable standard curves were selected for further analyses. The relative expression profiles of these genes were determined at three different developmental stages (larvae, pupae, and adults) and sexes (workers and drones). The stability of these six genes was assessed using Microsoft Excel-based software tools: BestKeeper, geNorm, NormFinder, and the ΔCt method. BestKeeper uses geometric means of Ct values to express the stability of reference genes. A standard deviation (SD) and stability value are used (SV); the lower SV represents more stable genes (21). geNorm automatically calculates the stability measure M by calculating the average pairwise variation of the reference gene against all other genes used in the analysis (16). NormFinder implements an ANOVA-derived model that calculates intra- and inter-group variation and then ranks the reference genes by an SV (22). The ΔCt method, where “pairs of genes” are compared and a rank order is determined based on mean delta Ct scores. In these analyses, genes with the lowest values are ranked as the most stably expressed for that given experimental condition. And finally, a comprehensive ranking of the stability of all candidate reference genes established from these four different statistical algorithms is employed using a Web-based analysis tool, RefFinder (https://www.heartcure.com.au/reffinder/; 22).
2.7.1 Validation of selected reference genes
To confirm the reliability of the reference genes, the relative expression profiles of RPB7 were determined at different development stages (larva, pupa and adult). These were normalised with the two most stable reference genes (TBP and EF1A) and the least stable reference gene (KTUB). Relative quantification of the target gene was calculated via the 2-ΔΔCt method (25).
3 Results
3.1 PCR amplification of candidate reference genes
The primer specificity was validated by using 1% agarose gels (Figure 1A). Primer specificity of all candidate reference genes was further tested via the amplification and dissociation curve analysis of every gene, visualized with a single peak and no detectable signal in the negative control (Figure 1B).

Figure 1. Confirmation of amplicon size and primer specificity. (A) Amplification results for Vespula vulgaris candidate reference genes from cDNA template. Columns 1–10 respectively represent 18s rRNA, 60s, AK1, CAPZB, DIMT, TBP, EF1A, KTUB, PPWD, RPL18X3, and M is the DNA ladder. (B) Melt curve analysis of all Vespula vulgaris candidate reference genes. All RT-qPCR products had a single melting curve suggesting the breakdown of only one PCR product.
Standard curves were generated for every gene based on a five-fold serial dilution of the pooled cDNA (Figure 1B). The correlation coefficient (R2) values ranged from 0.8–1, and the PCR efficiency values determined by the standard curve ranged from 81.41%–268.42% (Table 1). From this data, the six following genes were selected for further analyses: CAPZB, KTUB, RPL18X3, AK1, EF1A, and TBP.
3.2 Expression profiling of candidate reference genes
Expression levels were defined as the number of amplification cycles required to reach a fixed threshold in the exponential phase of the PCR reaction (26). Thresholds were automatically set by the instrument software in all genes to determine the Ct values. These expression levels were tested across two treatments in V. vulgaris, with variable Ct values highlighting a range of expression levels and varied expression patterns for these six reference genes (Figure 2).
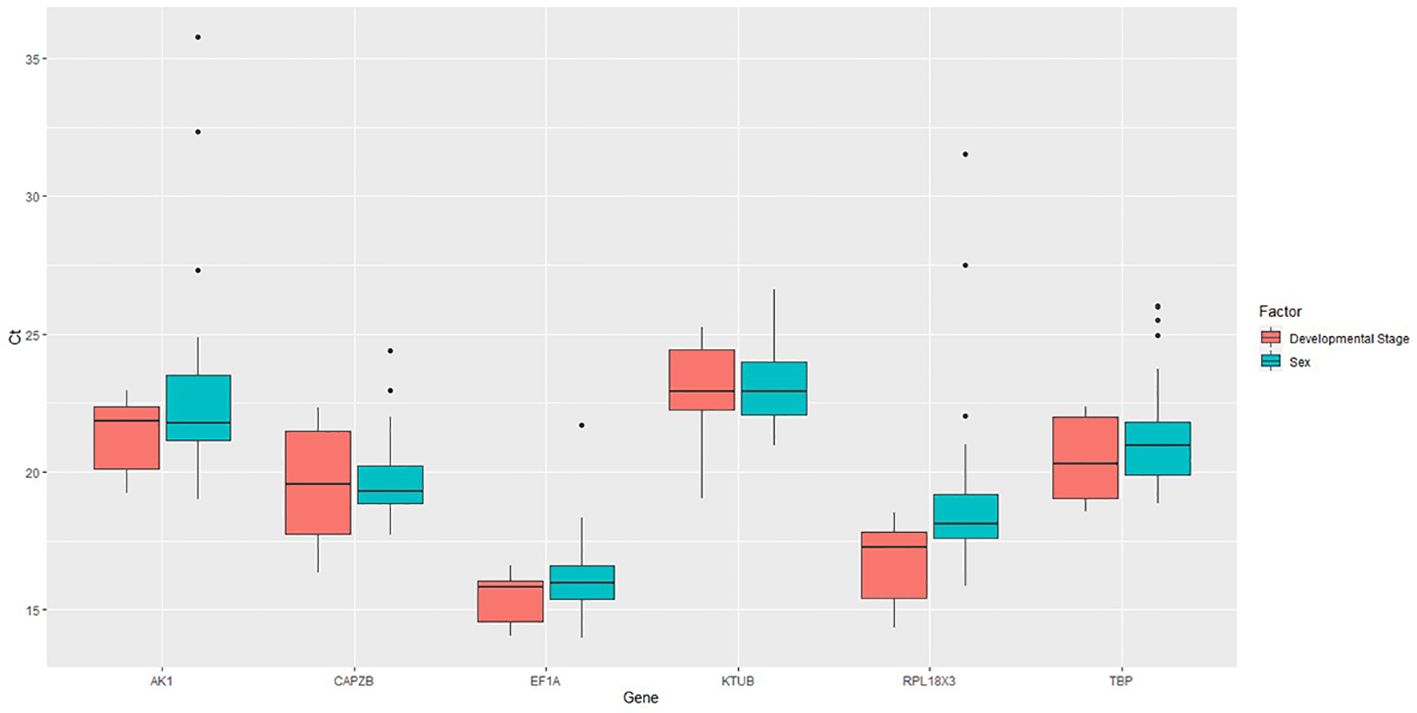
Figure 2. Expression profiles of candidate reference genes in both treatments. Box plot analysis showing the raw Ct values of candidate reference genes in V. vulgaris.
The expression levels of CAPZB, RPL18X3, and TBP showed greater variability within the developmental stage, while the Ct values for AK, KTUB, and TBP were the most varied in the sexes. EF1A and KTUB showed the narrowest range of variability between factors, suggesting a relatively stable expression at developmental stages and sexes (Figure 2).
3.3 Expression stability and ranking of candidate genes
All candidate reference genes were ranked based on the stability of gene expression for both treatments via four statistical algorithms.
BestKeeper ranked the candidate reference genes AK1 as the most stably expressed candidate reference gene in both developmental stage and sex, and CAPZB as the least stably expressed candidate reference gene in both conditions. Based on the ΔCt method ranking system, the candidate reference genes TBP and CAPZB were the most stably expressed genes for developmental stage and sex, respectively. Analysis of candidate reference genes ranked EF1A as the most stable gene based on both developmental stage and sex expression profiling. Using the analysis tool NormFinder, genes TBP and KTUB were ranked as the most stable genes across developmental stages and sexes, respectively. The overall values of the stability of the six selected candidate reference genes can be viewed in Table 2.
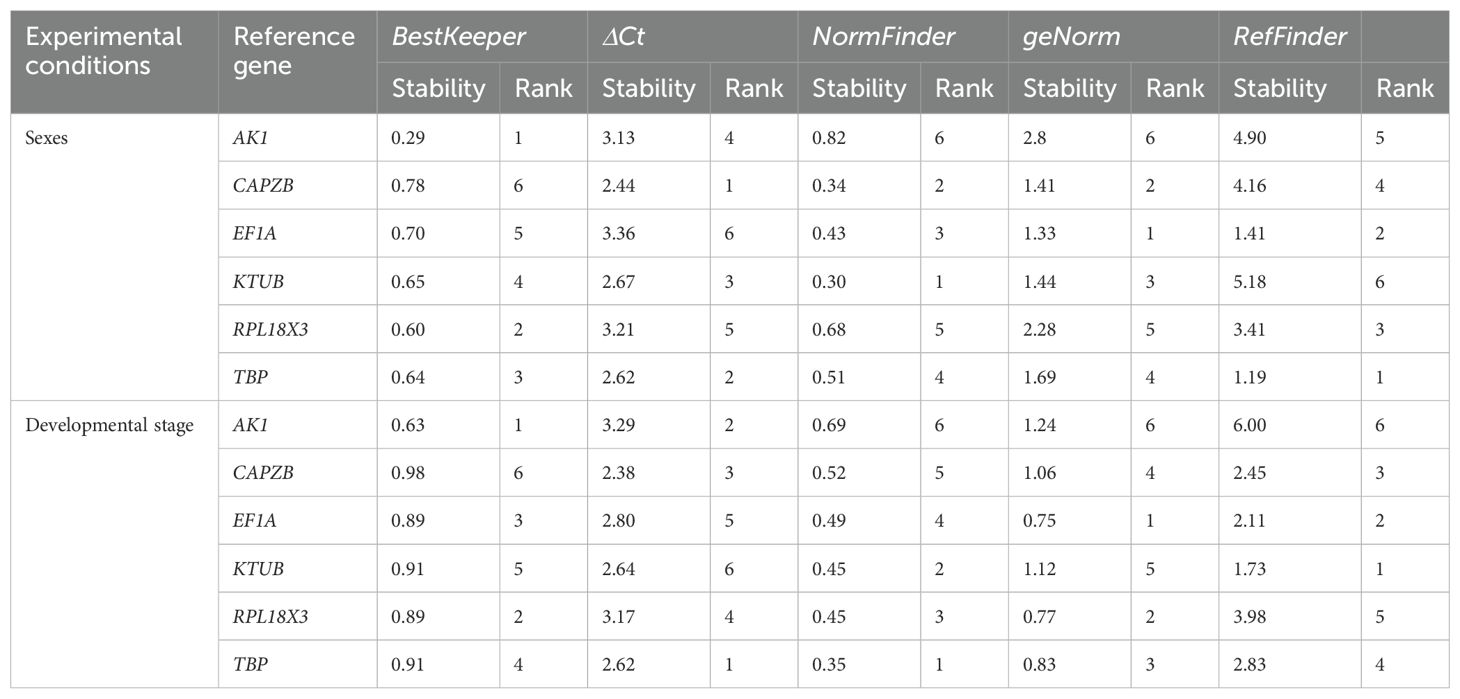
Table 2. Stability of the top six reference genes from four different variables.
3.4 Comprehensive ranking of selected reference genes via RefFinder
After the expression stabilities for developmental stage and sex were analysed by the above four methods, RefFinder was employed to calculate an overall stability ranking and therefore the most appropriate reference genes for these experimental conditions.
For developmental stages, analyses using the ΔCt method and NormFinder both ranked the candidate reference gene TBP as the most stable, while BestKeeper and geNorm ranked AK1 and EF1A as the most stable genes, respectively. In sex, the most stable gene varied for each form of analysis, with AK1 for BestKeeper, CAPZB for the ΔCt method, KTUB when ranked by NormFinder, and EF1A when using geNorm (Table 1). These algorithms varied in the ranking of the most unstable genes, putting AK1 as the least stable in sex for all measurements bar BestKeeper, while NormFinder and geNorm consistently ranked AK1 as the least stable gene under the sex condition. RefFinder ranked the stability of these six genes from most to least stable in the developmental stage TBP>EF1A>RPL18X3>CAPZB> AK1>KTUB (Figure 3A) and as KTUB>EF1A>CAPZB>TBP>RPL18X3>AK1 under the treatment for sex (Figure 3B). geNorm attempted to determine an optimal number of reference genes required to normalise target gene data, but could not recommend an amount as the variability between sequential normalisation factors (based on the n and n+1 least variable reference targets) is relatively high (geNorm V >0.15). Additional candidate reference genes were recommended, or if not possible, to use the four reference targets with the lowest M value when running an experiment under the developmental stage condition, or three reference targets with the lowest M value for experiments concerning sex. This was recommended as using multiple (in this case, non-optimal) reference targets would result in more accurate normalization compared to using a single nonvalidated reference gene.
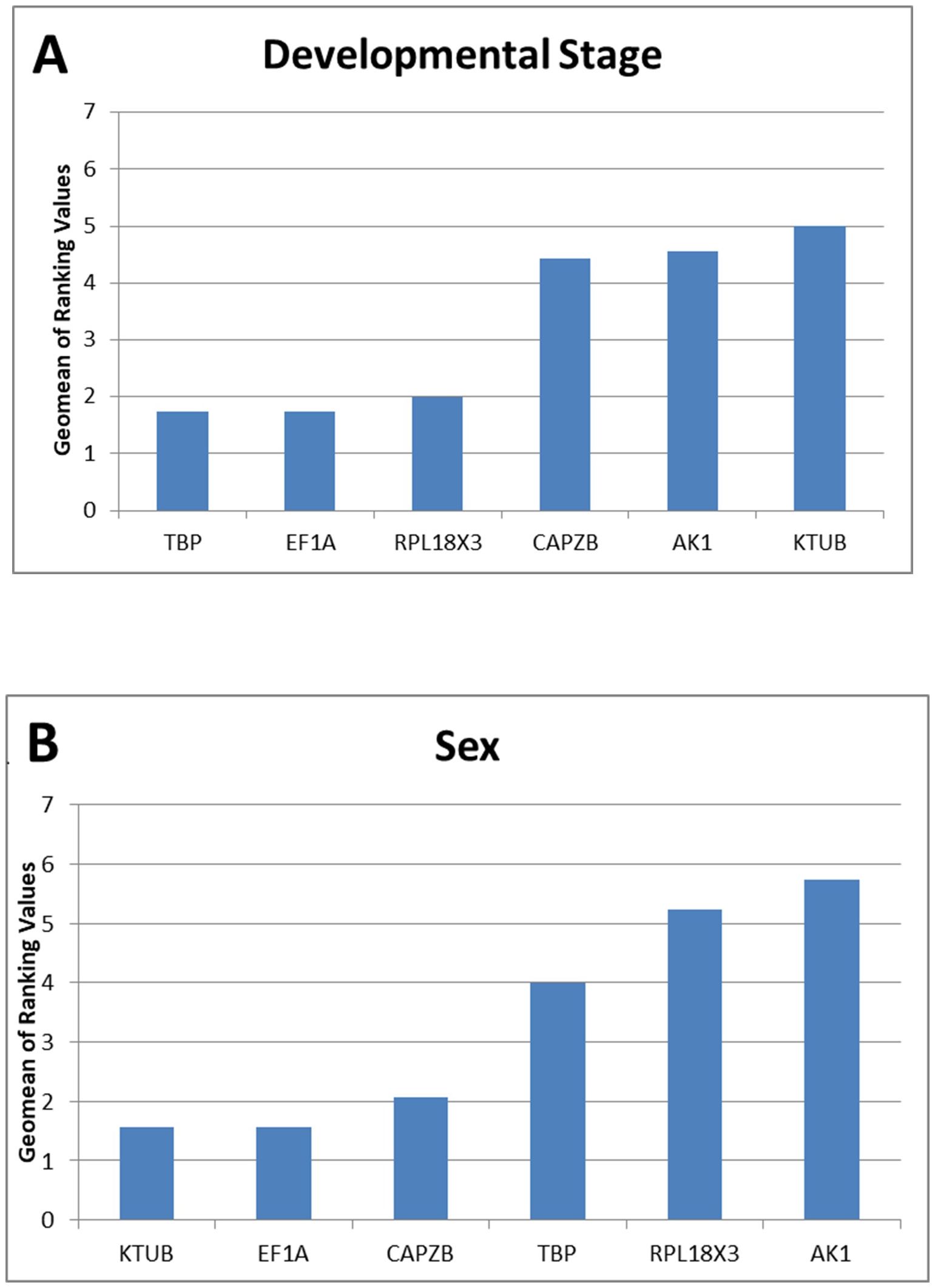
Figure 3. Stability of candidate reference gene expression under developmental stage (A) and sex (B) treatments. Lower Geomean values indicate more stable expression based on RefFinder analyses.
3.5 Validation of selected reference genes in V. vulgaris
The findings from this study show a striking difference in the stability of candidate reference genes from one condition to the other in V. vulgaris. While a comprehensive ranking by RefFinder showed that KTUB expressed the highest stability between sexes, it also indicated that the same gene had the most varied stability under the developmental stages condition. TBP and EF1A were considered the most stably expressed gene for the developmental stages condition. The expression patterns of RPB7 across various life stages were inconsistent when normalized with these two most stable reference genes (Figure 4). Transcripts of RPB7 Ct values showed some variation across all developmental stages, while normalization of transcripts using TBP and CAPZB highlighted peak RPB7 expression in pupae (Figure 4). Overall, RPB7 had varied expression patterns, with over 400-fold more expression when normalized with the least stable KTUB gene, compared to a less than four-fold change in expression when normalized with EF1A (Figure 4).
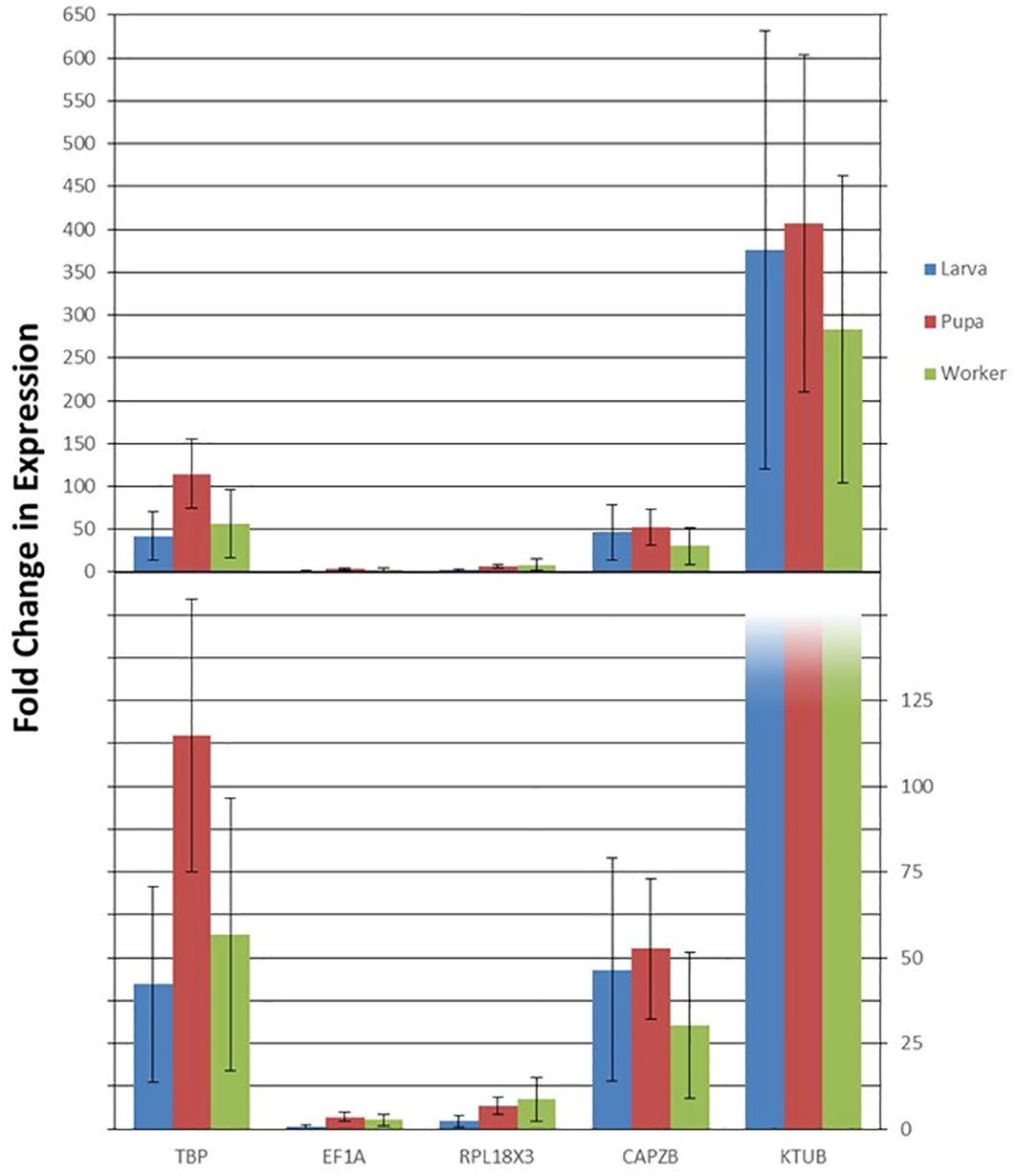
Figure 4. Validation of the candidate reference genes. Relative expression levels of the target gene RPB7 in different samples using different normalization factors (the four most and one least stable genes). Error bars represent the means and standard deviation of three biological replicates.
4 Discussion
To further investigate the biological function of a gene in an organism, quantification of that gene is crucial and requires experiments run in conjunction with reference genes of high stability. There have been several recent publications on selecting suitable reference genes for Hymenoptera (27–32), all highlighting the variability of such reference genes across a suite of experimental conditions. This paper aimed to find suitable reference genes for RNAi experiments in V. vulgaris, using five different algorithms to identify the stability of six reference genes. To date, there has been no investigation into appropriate reference genes for gene expression research in this social wasp. The genes selected for this study are regularly used as reference genes in other insect gene expression papers and were thus selected for this study. I chose six reference genes as candidate genes to examine their stability under different developmental stages and sexes. In an effort to reduce errors in analysis caused by selecting co-regulated transcripts, the candidate genes were ranked according to four statistical models (BestKeeper, ΔCt method, geNorm, and NormFinder).
The comparative analyses showed some consistency across algorithms under both experimental conditions. NormFinder and geNorm produced highly similar results, while BestKeeper had the greatest discrepancies in comparison to the other algorithms; this finding was consistent with those of Gao et al. (17). Such fluctuations in ranking orders from these programs can make it difficult for researchers to select the optimal reference genes. Therefore, I used RefFinder for a final ranking, as this algorithm incorporates all of the above-mentioned algorithms for a final overall rank of candidate reference genes.
The final analysis conducted by RefFinder was used to rank the overall stability of the selected candidate reference genes and demonstrated that TBP, EF1A, RPL18X3, and CAPZB were the most stable genes under the developmental stage treatment, and KTB, EF1A, and CAPZB exhibited the highest stability values among sexes.
The findings here emphasize the instability of such reference genes in different conditions, and that we should not expect to have perfect reference genes across all conditions, and further reinforce that there should never be a single universal reference gene employed for expression analysis across all experimental conditions and species. It should be said that this does not mean that reference genes are pointless, but that they need to be explored so that the right ones can be implemented for appropriate experiments. In my study, EF1A expressed the most stability in both developmental stage and sex conditions, consistent with findings of reference gene analysis in the Sugarcane Stem Borer Chilo sacchariphagus (33), Tamarixia radiate (31) and Glenea cantor (34). This study showed that the expression of TBP was highly stable across developmental stages, but should not be used in studies involved with different sexes. KTUB had the most extreme levels of expression, as it was the least stable gene across developmental stage experiments, but then the most stable when tested under the condition of sex. CAPZB remained in the middle range for both conditions, while RPL18X3 flanked the upper and lower levels of stability in developmental stage and sex, respectively. AK1 was a consistently poor performer and should not be used for either condition.
One limitation of the algorithms used is that NormFinder, geNorm, and RefFinder cannot conduct analyses with missing data points, thus whole samples have to be discarded (16, 22, 24). BestKeeper and the ΔCt method do not have this issue, however, BestKeeper is limited in that it can only analyze a maximum of ten genes (21). Ideally, I would have liked to include additional conditions to test the reference genes on, such as tissues, temperature, and diet. However, limited samples, time constraints, and reagent costs made this unfeasible. Future work would greatly benefit from including these parameters in their analyses.
The gene RPB7 is a catalyst for transcription of DNA into RNA using four ribonucleoside triphosphates as substrates. The protein comprises of mobile elements that synthesis mRNA precursors and bind single-stranded DNA and RNA (35). The four most stable and one least stable reference gene underwent validation by determining the expression profiles of the target gene, RPB7. The expression level of RPB7 after normalization drastically varied across all five of these genes, suggesting they could not honestly reflect its expression level (Figure 4). These results are inconsistent with the gene normalizer concept, implying that using these reference genes will not normalize gene expression data in V. vulgaris.
Overall, this qPCR study of suitable reference genes for expression research in V. vulgaris has provided the first building blocks necessary. A lack of stability has been found, rendering many of these candidate reference genes unsuitable. Potentially, it may not be possible to identify suitable reference genes for qPCR analysis, thus the two most stable reference genes under the condition developmental stage would be preferable to no species-specific reference at all. More research into additional reference genes, in combination with more treatment conditions such as tissues and diet, would not go amiss for additional insights.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
GM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author would like to thank the Biological Heritage National Science Challenge for funding.
Conflict of interest
The author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. MacIntyre P, Hellstrom J. An evaluation of the costs of pest wasps (Vespula species) in. Int Pest Control. (2015) 57:162–3.
2. Thomas CD, Moller H, Plunkett GM, Harris RJ. The prevalence of introduced Vespula vulgaris wasps in a New Zealand beech forest community. N Z J Ecol. (1990) 13:63–72.
3. Beggs JR, Wilson PR. The kaka Nestor meridionalis, a New Zealand parrot endangered by introduced wasps and mammals. Biol Conserv. (1991) 56:23–38. doi: 10.1016/0006-3207(91)90086-O
4. Toft RJ, Rees JS. Reducing predation of orb-web spiders by controlling common wasps (Vespula vulgaris) in a New Zealand beech forest. Ecol Entomol. (1998) 23:90–5. doi: 10.1046/j.1365-2311.1998.00100.x
5. Beggs JR, Rees JS. Restructuring of Lepidoptera communities by introduced Vespula wasps in a New Zealand beech forest. Oecologia. (1999) 119:565–71. doi: 10.1007/s004420050820
6. Harrop TWR, Guhlin J, McLaughlin GM, Permina E, Stockwell P, Gilligan J, et al. High-Quality assemblies for three invasive social wasps from the vespula genus. G3 Genes Genomes Genet. (2020) 10:3479–88. doi: 10.1534/g3.120.401579
7. Gibson UEM, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res. (1996) 6:995–1001. doi: 10.1101/gr.6.10.995
8. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. (1996) 6:986–94. doi: 10.1101/gr.6.10.986
9. Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. (2005) 6:279–84. doi: 10.1038/sj.gene.6364190
10. Bustin SA, Nolan T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech. (2004) 15:155–66.
11. Suzuki T, Higgins PJ, Crawford DR. Control selection for RNA quantitation. Biotechniques. (2000) 29:332–7. doi: 10.2144/00292rv02
12. Tricarico C, Pinzani P, Bianchi S, Paglierani M, Distante V, Pazzagli M, et al. Quantitative real-time reverse transcription polymerase chain reaction: Normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal Biochem. (2002) 309:293–300. doi: 10.1016/S0003-2697(02)00311-1
13. Imai T, Seo H, Murata Y, Ohno M, Satoh Y, Funahashi H, et al. Adrenocorticotropin increases expression of c-fos and β-actin genes in the rat adrenals. Endocrinology. (1990) 127:1742–7. doi: 10.1210/endo-127-4-1742
14. Chapman D, Weber KT, Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res. (1990) 67:787–94. doi: 10.1161/01.RES.67.4.787
15. Oka T, Komori N, Kuwahata M, Sassa T, Suzuki I, Okada M, et al. Vitamin B6 deficiency causes activation of RNA polymerase and general enhancement of gene expression in rat liver. FEEBS Lett. (1993) 331:162–4. doi: 10.1016/0014-5793(93)80318-O
16. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. (2002) 3:1–12. doi: 10.1186/gb-2002-3-7-research0034
17. Gao XK, Zhang S, Luo JY, Wang CY, Lü LM, Zhang LJ, et al. Comprehensive evaluation of candidate reference genes for gene expression studies in Lysiphlebia japonica (Hymenoptera : Aphidiidae) using RT-qPCR. Gene. (2017) 637:211–8. doi: 10.1016/j.gene.2017.09.057
18. Liu Z, Xiao J, Xia Y, Wu Q, Zhao C, Li D. Selection and validation of reference genes for RT-qPCR-based analyses of Anastatus japonicus Ashmead (Hymenoptera: Helicopteridae). Front Physiol. (2022) 13:1–13. doi: 10.3389/fphys.2022.1046204
19. He Y, Zhong Y, Bao Z, Wang W, Xu X, Gai Y, et al. Evaluation of Angelica decursiva reference genes under various stimuli for RT-qPCR data normalization. Sci Rep. (2021) 11:1–10. doi: 10.1038/s41598-021-98434-6
20. Guo C, Peng X, Wang H, Zheng X, Hu P, Zhou J, et al. Bacterial diversity of Leptocybe invasa Fisher & La Salle (Hymenoptera: Eulophidae) from different geographical conditions in China. Arch Insect Biochem Physiol. (2021) 108:e21847. doi: 10.1002/arch.v108.4
21. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper - Excel-based tool using pair-wise correlations. Biotechnol Lett. (2004) 26:509–15. doi: 10.1023/B:BILE.0000019559.84305.47
22. Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. (2004) 64:5245–50. doi: 10.1158/0008-5472.CAN-04-0496
23. Silver N, Best S, Jiang J, Thein SL. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol. (2006) 7:1–9. doi: 10.1186/1471-2199-7-33
24. Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. (2012) 80:75–84. doi: 10.1007/s11103-012-9885-2
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
27. Liu Y, Zhou J, Qiu Z, Hu P, Chen X, Yang Z. Identification and validation of reference genes for expression analysis using RT-qPCR in leptocybe invasa fisher and la salle (Hymenoptera: eulophidae). Insets. (2023) 14:1–17. doi: 10.3390/insects14050456
28. Gao Y, Wang S, Xu D, Yu H, Wu L, Lin Q, et al. GeoChip as a metagenomics tool to analyze the microbial gene diversity along an elevation gradient. Genomics Data. (2014) 2:132–4. doi: 10.1016/j.gdata.2014.06.003
29. Freitas FCP, Depintor TS, Agostini LT, Luna-lucena D, Nunes FMF, Bitondi MMG, et al. Evaluation of reference genes for gene expression analysis by real- time quantitative PCR (qPCR) in three stingless bee species (Hymenoptera : Apidae : Meliponini). Sci Rep. (2019) 9:1–13. doi: 10.1038/s41598-019-53544-0
30. Deng Y, Zhao H, Yang S, Zhang L, Zhang L. Screening and validation of reference genes for RT-qPCR under different Honeybee viral infections and dsRNA treatment. Front Microbiol. (2020) 11:1–12. doi: 10.3389/fmicb.2020.01715
31. Guo CF, Pan HP, Zhang LH, Ou D, Lu ZT, Khan MM, et al. Comprehensive assessment of candidate reference genes for gene expression studies using RT-qPCR in Tamarixia radiata, a predominant parasitoid of Diaphorina citri. Genes (Basel). (2020) 10:1–17. doi: 10.3390/genes11101178
32. Huo LX, Bai XP, Che WN, Ning SF, Lv L, Zhou JC, et al. Selection and evaluation of qPCR reference genes for expression analysis in the tiny egg parasitoid wasp, (Hymenoptera: Trichogrammatidae). bioRxiv. (2021), 2021–7.
33. Wang Z, Shang X, Wei J, Tian X, Liu Y, Zhang G. Evaluation and Validation of Reference Genes for Gene Expression Analysis Using qRT-PCR in the Sugarcane Stem Borer Chilo sacchariphagus (Lepidoptera: Pyralidae). Insects. (2024) 15:594. doi: 10.3390/insects15080594
34. Ahmadzada T, Reid G, McKenzie DR. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys Rev. (2018) 10:69–86. doi: 10.1007/s12551-017-0392-1
35. Kershnar E, Wu SY, Chiang CM. Immunoaffinity purification and functional characterization of human transcription factor IIH and RNA polymerase II from clonal cell lines that conditionally express epitope-tagged subunits of the multiprotein complexes. J Biol Chem. (1998) 273:34444–53. doi: 10.1074/jbc.273.51.34444
Keywords: Vespula vulgaris, reference gene, RT-qPCR, gene expression, invasive wasp
Citation: McLaughlin GM (2025) Evaluation of candidate reference genes for gene expression research in Vespula vulgaris. Front. Insect Sci. 5:1495626. doi: 10.3389/finsc.2025.1495626
Received: 13 September 2024; Accepted: 20 January 2025;
Published: 13 February 2025.
Edited by:
Sudeshna Mazumdar-Leighton, University of Delhi, IndiaReviewed by:
Suneet Pandher, Punjab Agricultural University, IndiaKanakachari Mogilicherla, Indian Institute of Rice Research (ICAR), India
Copyright © 2025 McLaughlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gemma M. McLaughlin, Z2VtbWEubS5tY2xhdWdobGluQGdtYWlsLmNvbQ==