
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 April 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1583886
This article is part of the Research Topic Integrative AI and Multi-Omics: Precision Medicine in Immuno-Inflammation View all articles
 Zhitao Li1
Zhitao Li1 Chunrong Gong2*
Chunrong Gong2*Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory impairment, and neuroinflammation, with no definitive cure currently available. The NLRP3 inflammasome, a key mediator of neuroinflammation, has emerged as a critical player in AD pathogenesis, contributing to the accumulation of β-amyloid (Aβ) plaques, tau hyperphosphorylation, and neuronal damage. This review explores the mechanisms by which the NLRP3 inflammasome is activated in AD, including its interactions with Aβ, tau, reactive oxygen species (ROS), and pyroptosis. Additionally, it highlights the role of the ubiquitin system, ion channels, autophagy, and gut microbiota in regulating NLRP3 activation. Therapeutic strategies targeting the NLRP3 inflammasome, such as IL-1β inhibitors, natural compounds, and novel small molecules, are discussed as promising approaches to mitigate neuroinflammation and slow AD progression. This review underscores the potential of NLRP3 inflammasome inhibition as a therapeutic avenue for AD.
Alzheimer’s disease (AD) is a prevalent, chronic, and progressive neurodegenerative disorder, often leading to significant cognitive decline and memory impairment in its early stages, with later phases causing substantial daily functioning challenges and psychiatric symptoms (1). Currently, there are no definitive strategies for the prevention or treatment of AD (2), and its incidence is escalating rapidly due to global aging trends (3). The pathogenesis of AD is primarily associated with the accumulation of β-amyloid (Aβ), hyperphosphorylation of tau proteins, and the formation of neurofibrillary tangles. Recent studies have further underscored the critical role of neuroinflammation in the progression of AD (4, 5).
Among the various neuroinflammatory mediators, the NLRP3 inflammasome has gained attention as a potential therapeutic target (6). First described by Martinon et al. (7), the inflammasome is a multi-protein complex that includes caspases, ASC, and cytoplasmic pattern recognition receptors (8). It recognizes pathogen- or danger-associated molecular patterns, activating Caspase-1, which processes IL-1β and IL-18 precursors into active cytokines (9–11). NLRP3, the most extensively studied inflammasome, has been implicated in the pathogenesis of AD (10, 12) This review summarizes the structure, activation mechanisms, and involvement of the NLRP3 inflammasome in AD, as well as potential therapeutic strategies targeting this inflammasome.
In brain regions affected by AD, microglia are frequently observed in proximity to Aβ plaques. Research has demonstrated that these microglial cells play a crucial role in eliminating Aβ through mechanisms such as phagocytosis and proteolysis (13). The activation of the NLRP3 inflammasome, however, is not solely triggered by fibrillar Aβ but also by smaller Aβ oligomers and protofibrils (14). Aβ induces microglial activation through multiple signaling pathways, including NF-κB, which upregulates the expression of NLRP3 and pro-IL-1β (15). Moreover, soluble Aβ disrupts lysosomal stability, leading to the release of cathepsins and subsequent activation of the NLRP3 inflammasome (16). Additionally, Aβ oligomers impair mitochondrial function, causing oxidative stress and the release of mitochondrial DNA, which further exacerbates NLRP3 inflammasome activation (17). As AD progresses, chronic NLRP3 activation leads to excessive microglial activation, diminishing their ability to clear Aβ and fostering its accumulation (18, 19). Notably, APP/PS1 transgenic mice lacking NLRP3 exhibit reduced Aβ deposition, suggesting that NLRP3 contributes to Aβ accumulation and accelerates disease progression (20).
Tau proteins activate the NLRP3 inflammasome in microglial cells via an ASC-dependent mechanism, triggering IL-1β release and neuroinflammation (16, 21). This activation also regulates tau phosphorylation by modulating kinases and phosphatases, leading to tau hyperphosphorylation and aggregation in neurons (22). Hyperphosphorylated tau aggregates (NFTs) are recognized by microglial TLRs, inducing NLRP3 inflammasome assembly through the NF-κB pathway. Additionally, tau uptake by microglial lysosomes can release cathepsin B, further activating the inflammasome. The resulting IL-1β and IL-18 release exacerbates neuroinflammation and accelerates AD (23). Tau precursors can also trigger neuroinflammation and impair memory through NLRP3 pathways (22). Thus, targeting NLRP3 activation may offer a potential AD therapy.
ROS are crucial regulators of the NLRP3 inflammasome, with their generation closely linked to enzymes such as peroxiredoxin and NADPH oxidase (24). In normal conditions, thioredoxin (TRX) and its binding partner thioredoxin-interacting protein (TXNIP) form a stable complex. However, under oxidative stress, an increase in ROS levels leads to the oxidation of TRX, which in turn neutralizes ROS and causes the dissociation of the TRX-TXNIP complex (25). This dissociation allows TXNIP to interact with NLRP3, thereby recruiting ASC and procaspase-1, which ultimately promotes the assembly and activation of the inflammasome (26). Furthermore, mitochondrial dysfunction can lead to the release of mitochondrial DNA, a potent activator of the NLRP3 inflammasome (27). Different ROS sources selectively modulate NLRP3 signaling in AD (28). Mitochondrial ROS from electron transport chain (ETC) dysfunction exacerbates oxidative damage, impairing neuronal energy metabolism and enhancing NLRP3 activation (29). Conversely, NADPH oxidase-derived ROS, particularly NOX2 in microglia, amplifies ROS production, potentiating NLRP3 signaling and neuroinflammation (30). Targeting these distinct ROS pathways is crucial for precise NLRP3 inhibition in AD.
Pyroptosis, an inflammatory programmed cell death, is activated by the NLRP3 inflammasome in AD (31). It promotes pro-inflammatory cytokine secretion, aiding Aβ plaque clearance but also inducing chronic neuroinflammation that accelerates AD progression (32). This process is mediated by gasdermin D (GSDMD) cleavage and caspase-1 activation (33). Pyroptosis occurs via two pathways: (1) The Classical Pathway, where caspase-1 cleaves IL-1β and IL-18 precursors, producing mature cytokines, and modifies GSDMD to form membrane pores, releasing cytokines and driving pyroptosis; and (2) The Non-classical Pathway, where caspase-11 (mice) or caspase-4/5 (humans) is activated by cytosolic LPS, triggering pyroptosis through specific cleavage (34, 35). In AD, Aβ-induced pyroptosis is mediated by the NLRP3-caspase-1-GSDMD axis, exacerbating neuroinflammation and neuronal damage (36–38). GSDMD knockout inhibits astrocyte pyroptosis, mitigating Aβ42-induced brain and vascular damage in APP/PS1 mice (39). These findings suggest that targeting inflammasome-driven pyroptosis holds potential as a novel therapeutic approach for AD.
Recent studies highlight the interconnected mechanisms of Aβ accumulation, tau hyperphosphorylation, ROS generation, and defective autophagy in driving AD progression (34). Aβ and tau aggregates activate microglia, increasing ROS and NLRP3 inflammasome activation, which exacerbates mitochondrial dysfunction and impairs autophagy (40). This creates a vicious cycle in which NLRP3 serves as a pivotal intersection point, amplifying neuroinflammation and neuronal damage (40). Targeting NLRP3 offers a promising therapeutic strategy to disrupt these cascades, addressing the complex interplay of Aβ, tau, ROS, and autophagy in AD (Figure 1).

Figure 1. The role of NLRP3 inflammasome in AD.
Ubiquitin, a small protein present in all eukaryotic cells, is capable of forming chains through enzymatic processes, which signal target proteins for degradation (41). This modification, referred to as ubiquitination, is carried out by a series of specialized enzymes that alter target proteins, marking them for proteasomal recognition and breakdown (42). In conjunction with autophagy, this system plays a critical role in maintaining cellular homeostasis (43). Disruptions in the ubiquitin-proteasome pathway have been linked to Alzheimer’s disease and other neurodegenerative conditions (44). Within the E1, E2, and E3 enzyme families, E3 ligases such as SCF-FBXL2 selectively target NLRP3 and its associated molecules, including ASC and caspase-1, promoting NLRP3 ubiquitination and proteasomal degradation, thereby regulating its activation (45). Additionally, Cullin1 associates with NLRP3, enhancing its ubiquitination but without triggering degradation, which serves to inhibit NLRP3 inflammasome activation (46).
Ion channels play a critical role in modulating the activation of the NLRP3 inflammasome, with K+ efflux identified as a pivotal signaling event (47). In AD, imbalances in the homeostasis of Na+ and K+ disrupt the electrophysiological properties of neurons, contributing to the pathophysiological change characteristic of the disease (48). Studies by Gritsenko A et al. (49) demonstrated that K+ efflux in human monocytes leads to the aggregation of ASC, cleavage of caspase-1, and subsequent processing of GSDMD. Inflammatory factors significantly influence the efficacy of therapies for inflammatory diseases (50–55). Inhibition of NLRP3 or genetic deletion of NLRP3 and GSDMD blocks the release of IL-18, highlighting the crucial role of early inflammasome assembly before IL-1β production (56). Additionally, extracellular ATP, released in response to bacterial toxins, activates P2X7 purinergic receptors, which disturb intracellular ion balance and promote K+ efflux (57). This perturbation enables the assembly of NLRP3 with ASC, forming an active inflammasome complex that cleaves procaspase-1 into active caspase-1. Caspase-1 then facilitates the secretion of IL-1β and IL-18 (58). Furthermore, GSDMD is cleaved by the inflammasome, creating membrane pores that allow the release of these cytokines (45).
Autophagy is a fundamental cellular process in eukaryotic cells that degrades and recycles cytoplasmic components, such as damaged organelles and protein aggregates, to maintain cellular homeostasis (59). Wang D et al. (60) revealed that excessive accumulation of manganese compromises lysosomal integrity by altering their structure and impairing their function. In manganese-induced NLRP3-caspase-1 inflammasome activation, the release of cathepsin B from lysosomes plays a critical role (61). Additionally, inflammatory stimuli such as alum, crystalline materials, and protein aggregates can trigger autophagy (62), leading to lysosomal destabilization and rupture (63). Following lysosomal rupture, cathepsin B is released and directly interacts with NLRP3, thereby promoting the activation of the NLRP3 inflammasome (62).
In AD pathogenesis, Aβ peptides compromise synaptic plasticity and inhibit long-term potentiation (64). Parajuli et al. showed that Aβ promotes the conversion of pro-IL-1β into its active form, IL-1β, thereby enhancing microglia-mediated neurotoxicity (65). This process is largely driven by increased caspase-1 activity and the activation of NOD-like receptors, specifically NLRP3, which features a pyrin domain (66). Mitochondrial-derived ROS, and to a lesser degree, ROS generated by NADPH oxidase, play a pivotal role in initiating NLRP3 activation (67). Elevated ROS levels activate TRPM2 channels, which subsequently activate NLRP3 and caspase-1, thereby increasing IL-1β production (68). Notably, the use of mitochondrial ROS inhibitors, such as DPI, significantly reduces both ROS and IL-1β levels, indicating a suppression of NLRP3 activation (69).
GM, a consortium of symbiotic microorganisms within the human intestinal tract, has been implicated in various diseases, including AD. Dysbiosis, or disruptions in microbial composition, is frequently observed in individuals with AD (70). The gut-brain axis, an increasingly studied area, highlights how microbiota influences brain function, with both probiotics and prebiotics playing roles in modulating microbial and immune systems (71). Dysbiosis impairs the intestinal barrier, allowing pathogen-associated molecular patterns (PAMPs) to trigger the release of pro-inflammatory cytokines. These cytokines can then travel to the brain, aggravating the progression of AD (72–74). Moreover, microbial activation of the NLRP3 inflammasome leads to the upregulation of caspase-1 in the AD brain, further advancing disease pathology (75). While short-chain fatty acids (SCFAs) from commensal bacteria bind GPCRs (GPR43, GPR41), inhibit HDACs, reduce cytokines, and downregulate NLRP3 in microglia/astrocytes (76). Thus, restoring a balanced gut microbiota may reduce neuroinflammation and enhance cognitive function in AD patients (77).
Early therapeutic strategies targeting the NLRP3 inflammasome pathway have largely focused on IL-1β modulation. Notable agents with proven efficacy include anakinra, a recombinant IL-1 receptor antagonist; canakinumab, a monoclonal antibody against IL-1β; and rilonacept, a soluble decoy receptor that binds IL-1β by incorporating IL-1R1 and IL-1RAcP domains (78). In preclinical experiments using the 3xTg AD model, anakinra was shown to reduce Aβ and tau accumulation, decrease IL-1β levels, and enhance cognitive function (79). Furthermore, both anakinra treatment and genetic deletion of IL-1R improved mitochondrial dysfunction and alleviated memory deficits associated with Aβ in in vivo and in vitro models (80). Despite these promising results, the challenge of the blood-brain barrier (BBB) has hindered further exploration of IL-1β-based therapies in AD (80). A Phase 2 clinical trial assessing the efficacy of canakinumab in AD patients is currently ongoing (NCT04795466). However, since IL-1β acts as a downstream effector, directly targeting NLRP3 or its inflammasome components could potentially offer more substantial therapeutic advantages (81).
GB has demonstrated protective effects against ischemic brain injury and neurotoxicity induced by Aβ (82). In models of hypoxic-ischemic brain damage in rats, GB diminishes NLRP3 inflammasome activation, thereby alleviating neuroinflammation and mitigating AD-related pathology in BV2 cells (83). Additionally, GB treatment has been reported to reduce Aβ-induced pathological alterations and inhibit NLRP3 inflammasome activation (84). Furthermore, GB promotes the upregulation of anti-inflammatory markers in M2 microglia, while concurrently suppressing the release of pro-inflammatory cytokines in M1 microglia (85). Through autophagy-dependent pathways, GB also curbs NLRP3 inflammasome activation, ultimately safeguarding cognitive function in SAMP8 mice (84, 86).
SFN exhibits notable anti-inflammatory, antioxidant, and neuroprotective properties (87). Studies suggest that SFN effectively diminishes the release of IL-1β and IL-18 in LPS-activated microglia, while also inhibiting the overexpression of NLRP3 and caspase-1 proteins (88). Furthermore, SFN prevents pyroptosis in microglia by inhibiting caspase-1 activity (89), and attenuates NLRP3 inflammasome activation via the downregulation of NF-κB (88), thus reducing inflammatory responses (90).
OLT1177, an orally available and selective inhibitor of the NLRP3 inflammasome, has demonstrated considerable therapeutic promise (91). This compound binds directly to NLRP3, blocking its ATPase function and disrupting several inflammasome activation pathways. In APP/PS1 transgenic mice, OLT1177 treatment partially alleviated cognitive impairments as assessed by the Morris water maze test. It also decreased microglial activation and lowered cortical plaque accumulation (92). Although there is limited research on its application in neurodegenerative disorders, OLT1177’s excellent safety profile, favorable pharmacokinetic characteristics, and minimal side effects underscore its potential as a therapeutic agent for AD (93).
MCC950 is a potent anti-inflammatory compound that selectively inhibits NLRP3 inflammasome activation by targeting its NACHT domain (94). In models of Alzheimer’s disease, MCC950 effectively dampens the inflammasome activation triggered by Aβ or tau, preventing the cleavage and release of caspase-1 and IL-1β. Additionally, it completely halts immune responses induced by Aβ aggregates and low-molecular-weight oligomers (95). In vitro experiments using Aβ-stimulated human primary neurons pre-treated with MCC950 demonstrated its capacity to inhibit pyroptosis, thereby significantly reducing Aβ-induced neuronal toxicity. Other NLRP3 inhibitors currently under exploration include IFM-514 (96), CY-09 (97), DFV890 (98), Tranilast (99), Oridonin (100), Selnolast (101), and Inzomelid (102).
Nonsteroidal anti-inflammatory drugs (NSAIDs) have shown potential in delaying the onset of Alzheimer’s disease (AD) or reducing its risk, likely through their modulation of the NLRP3 inflammasome pathway (103). For instance, indomethacin, a well-known NSAID, has been demonstrated to inhibit both NLRC4 and NLRP3 inflammasomes. This inhibition leads to a reduction in the expression of IL-1β and caspase-1, thereby alleviating neuroinflammation and mitigating memory deficits associated with AD (104). MicroRNAs have been shown to suppress NLRP3 expression, leading to improved cognitive function in rodent models of Alzheimer’s disease (105, 106). In AD patients, reduced miR-22 levels in AD patients are associated with increased NLRP3 activation, while overexpression of miR-22 inhibits GSDMD-mediated pyroptosis, thereby reducing neuroinflammation and cognitive decline in AD mice (107). These findings highlight the potential of targeting miRNAs to modulate NLRP3 activity as a novel therapeutic strategy for AD.
In addition to miRNAs, autophagy and mitophagy also modulate NLRP3 activation. For example, IIIM-941 has been shown to induce autophagy via AMPK, inhibiting NLRP3 activity (108). A-68930 activates dopamine D1 receptors, promoting NLRP3 degradation through AMPK/autophagy, reducing IL-1β/IL-18 secretion, and mitigating Aβ1-42-induced neuroinflammation (109). Ginkgo biloba extract EGb 761 enhances microglial autophagy, downregulates NLRP3, and attenuates Aβ-induced IL-1β/caspase-1 activation in TgCRND8 mice (110). Furthermore, Quercetin stimulates mitophagy, suppressing mtROS-driven NLRP3 activation and protecting against neuronal damage (111). These studies suggest that enhancing autophagy and mitophagy in microglia may offer a promising therapeutic approach for AD.
A recent study has identified cornuside as a promising anti-AD agent. Cornuside has been shown to restores mitophagic flux, enabling the efficient the removal of damaged mitochondria and the recovery of mitochondrial function. These mechanisms contribute to the inhibition of NLRP3 inflammasome activation, thereby reducing neuronal and synaptic damage and improving cognitive function (112). Structurally unique diterpenoids, isolated from the mangrove plant Excoecaria agallocha L., have emerged as promising anti-neuroinflammatory agents. These compounds exert their effects by inhibiting macrophage polarization and suppressing the activation of the NLRP3 inflammasome, highlighting their potential in mitigating neuroinflammation (113). Additionally, Thonningianin A (ThA) has demonstrated the ability to suppress NLRP3 inflammasome-driven inflammation and curb the overactivation of microglia and astrocytes through the induction of autophagy (114). Moreover, autophagy has been implicated in mitigating neuroinflammation in AD by modulating NLRP3 inflammasome activity (115). Consequently, targeting the autophagy-NLRP3 inflammasome axis using ThA holds potential as a novel therapeutic approach for AD (23). Additionally, research suggests that Eriodictyol exerts beneficial effects on AD by inhibiting NLRP3 activation. Eriodictyol can cross the BBB and significantly reduce the expression of NLRP3, caspase-1, and ASC proteins in brain tissue, while also decreasing the inflammatory cytokines IL-1β and IL-18. These effects improve cognitive function and memory (116), as well as attenuate AD pathology (23, 117). The ongoing phase 3 trial TRAILBLAZER-ALZ 2 (NCT04437511) (118), phase 2 clinical trial (NCT04795466) (81) and other AD clinical trials will offer a more comprehensive strategy for AD treatment (119) (Table 1).
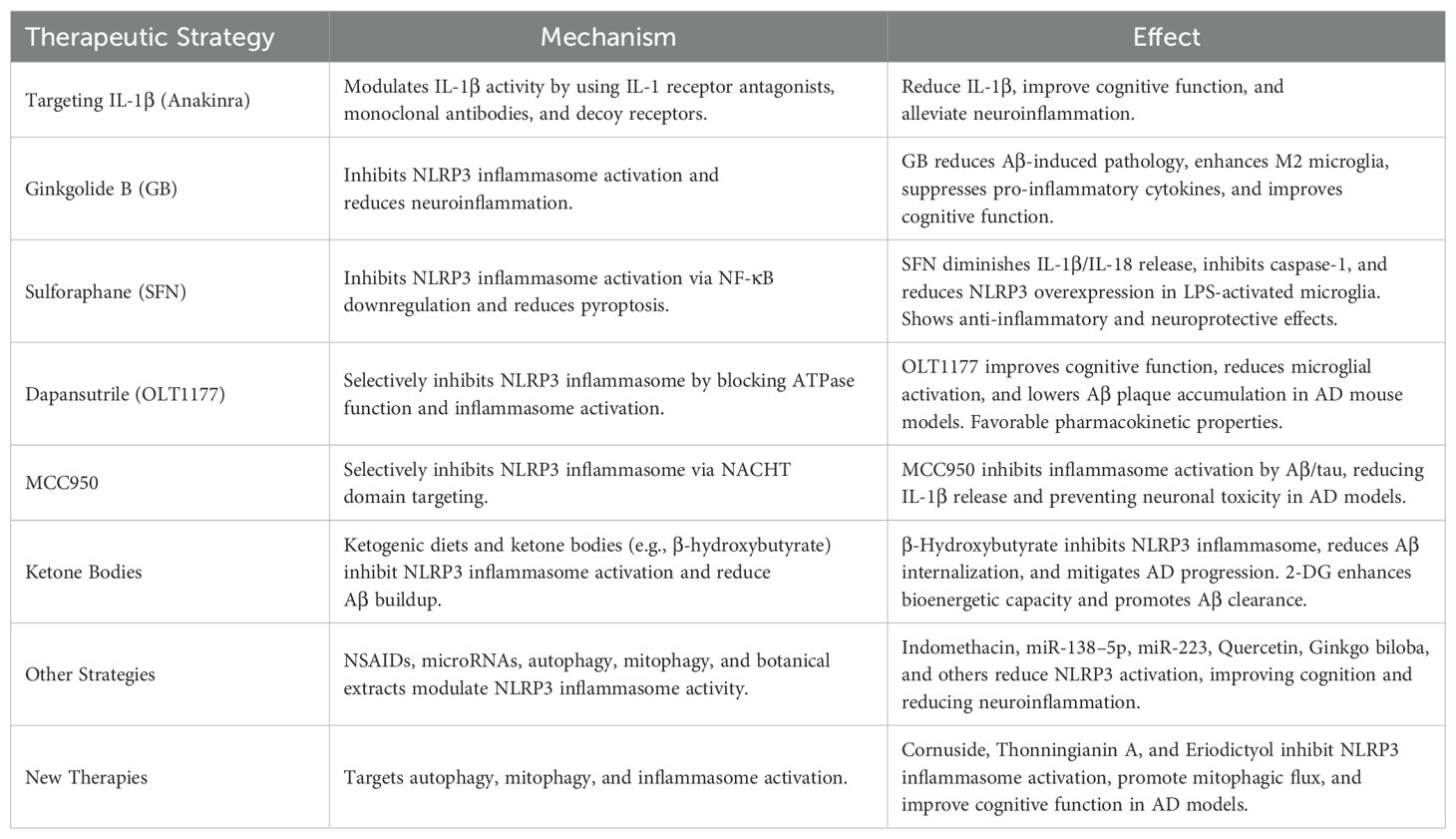
Table 1. The therapies targeting NLRP3 Inflammasome in AD.
The NLRP3 inflammasome plays a pivotal role in the pathogenesis of Alzheimer’s disease by driving neuroinflammation, Aβ accumulation, tau pathology, and neuronal damage. Its activation is influenced by multiple factors, including ROS, mitochondrial dysfunction, and gut microbiota dysbiosis. Targeting the NLRP3 inflammasome through various therapeutic strategies, such as IL-1β modulation, natural compounds, and small-molecule inhibitors, offers promising potential to alleviate neuroinflammation and slow disease progression. Future research should focus on developing NLRP3-targeted therapies that can effectively cross the blood-brain barrier and provide long-term benefits in AD patients. Moreover, integrating biomarker identification and precision-targeted drug design into clinical research could expedite the transition from bench to bedside, ultimately offering earlier intervention and better protection against neurodegeneration in individuals at risk for or diagnosed with AD. By addressing the central role of the NLRP3 inflammasome in AD, these therapeutic approaches may pave the way for more effective treatments for this debilitating disease.
ZL: Writing – original draft. CG: Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as potential conflicts of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. (2017) 23:174–84. doi: 10.1038/nm.4267
2. Bali J, Gheinani AH, Zurbriggen S, Rajendran L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of beta-amyloid peptides. Proc Natl Acad Sci U.S.A. (2012) 109:15307–11. doi: 10.1073/pnas.1201632109
3. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. (2019) 15:501–18. doi: 10.1038/s41582-019-0228-7
4. Pallo SP, Johnson GV. Tau facilitates Abeta-induced loss of mitochondrial membrane potential independent of cytosolic calcium fluxes in mouse cortical neurons. Neurosci Lett. (2015) 597:32–7. doi: 10.1016/j.neulet.2015.04.021
5. Hansen DV, Hanson JE, Sheng M. Microglia in alzheimer’s disease. J Cell Biol. (2018) 217:459–72. doi: 10.1083/jcb.201709069
6. Pereira CF, Santos AE, Moreira PI, Pereira AC, Sousa FJ, Cardoso SM, et al. Is Alzheimer’s disease an inflammasomopathy? Ageing Res Rev. (2019) 56:100966. doi: 10.1016/j.arr.2019.100966
7. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
8. Xue Y, Enosi Tuipulotu, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. (2019) 40:1035–52. doi: 10.1016/j.it.2019.09.005
9. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
10. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflammation Res. (2015) 8:15–27. doi: 10.2147/JIR.S51250
11. Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Arindam C, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. (2014) 193:4214–22. doi: 10.4049/jimmunol.1400582
12. Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. (2018) 19:610–21. doi: 10.1038/s41583-018-0055-7
13. Merighi S, Nigro M, Travagli A, Gessi S. Microglia and alzheimer’s disease. Int J Mol Sci. (2022) 23:12990. doi: 10.3390/ijms232112990
14. Bai H, Zhang Q. Activation of NLRP3 inflammasome and onset of alzheimer’s disease. Front Immunol. (2021) 12:701282. doi: 10.3389/fimmu.2021.701282
15. Nopparat C, Boontor A, Kutpruek S, Govitrapong P. The role of melatonin in amyloid beta-induced inflammation mediated by inflammasome signaling in neuronal cell lines. Sci Rep. (2023) 13:17841. doi: 10.1038/s41598-023-45220-1
16. Van Zeller M, Dias D, Sebastiao AM, Valente CA. NLRP3 inflammasome: A starring role in amyloid-beta- and tau-driven pathological events in alzheimer’s disease. J Alzheimers Dis. (2021) 83:939–61. doi: 10.3233/JAD-210268
17. Li X, Jin Y, Ding X, Zhu T, Wei C, Yao L. Long-term exercise training inhibits inflammation by suppressing hippocampal NLRP3 in APP/PS1 mice. Sports Med Health Sci. (2023) 5:329–35. doi: 10.1016/j.smhs.2023.09.009
18. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC. specks cross-seed amyloid-beta Alzheimer’s disease. Nat. (2017) 552:355–61. doi: 10.1038/nature25158
19. Friker LL, Scheiblich H, Hochheiser IV, Brinkschulte R, Riedel D, Latz E, et al. beta-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep. (2020) 30:3743–3754 e6. doi: 10.1016/j.celrep.2020.02.025
20. Barczuk J, Siwecka N, Lusa W, Rozpędek-Kamińska W, Kucharska E, Majsterek I. Targeting NLRP3-mediated neuroinflammation in alzheimer’s disease treatment. Int J Mol Sci. (2022) 23:8979. doi: 10.3390/ijms23168979
21. Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. (2019) 137:599–617. doi: 10.1007/s00401-018-01957-y
22. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
23. Zu R, Lu H, Liu W, Shao S, Zheng J, Ying X, et al. Research progress in the molecular mechanism of NLRP3 inflammasome in alzheimer’s disease and regulation by natural plant products. Mol Neurobiol. (2025). doi: 10.1007/s12035-025-04715-w
24. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. (2009) 10:241–7. doi: 10.1038/ni.1703
25. Qayyum N, Haseeb M, Kim MS, Choi S. Role of thioredoxin-interacting protein in diseases and its therapeutic outlook. Int J Mol Sci. (2021) 22:2754. doi: 10.3390/ijms22052754
26. Zhang Y, Xing CJ, Liu X, Li YH, Jia J, Feng JG, et al. Thioredoxin-interacting protein (TXNIP) knockdown protects against sepsis-induced brain injury and cognitive decline in mice by suppressing oxidative stress and neuroinflammation. Oxid Med Cell Longev 2022. (2022) p:8645714. doi: 10.1155/2022/8645714
27. Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. (2016) 39:1503–18. doi: 10.1007/s12272-016-0827-4
28. Wei P, Yang F, Zheng Q, Tang W, Li JJFiCN. The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front Cell Neurosci. (2019) 13:73. doi: 10.3389/fncel.2019.00073
29. Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. (2022) 23:692–704. doi: 10.1038/s41590-022-01185-3
30. McCarty MF, DiNicolantonio JJ, Lerner A. A fundamental role for oxidants and intracellular calcium signals in Alzheimer’s pathogenesis—and how a comprehensive antioxidant strategy may aid prevention of this disorder. Int J Mol Sci. (2021) 22:2140. doi: 10.3390/ijms22042140
31. Moonen S, Koper MJ, Van Schoor E, Schaeverbeke JM, Vandenberghe R, von Arnim CAF, et al. Pyroptosis in Alzheimer’s disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol. (2023) 145:175–95. doi: 10.1007/s00401-022-02528-y
32. Lee S, Cho HJ, Ryu JH. Innate immunity and cell death in alzheimer’s disease. ASN Neuro. (2021) 13:17590914211051908. doi: 10.1177/17590914211051908
33. Xue W, Cui D, Qiu Y. Research progress of pyroptosis in alzheimer’s disease. Front Mol Neurosci. (2022) 15:872471. doi: 10.3389/fnmol.2022.872471
34. Liang T, Zhang Y, Wu S, Chen Q, Wang L. The role of NLRP3 inflammasome in alzheimer’s disease and potential therapeutic targets. Front Pharmacol. (2022) 13:845185. doi: 10.3389/fphar.2022.845185
35. Mangalmurti A, Lukens JR. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr Opin Neurobiol. (2022) 75:102575. doi: 10.1016/j.conb.2022.102575
36. Han C, Yang Y, Guan Q, Zhang X, Shen H, Sheng Y, et al. New mechanism of nerve injury in Alzheimer’s disease: beta-amyloid-induced neuronal pyroptosis. J Cell Mol Med. (2020) 24:8078–90. doi: 10.1111/jcmm.v24.14
37. Jia J, Zhang X, Xu G, Zeng X, Li L. Thioredoxin-1 inhibits amyloid-beta(25-35)-induced activation of NLRP1/caspase-1/GSDMD pyroptotic pathway in PC12 cells. Mol Biol Rep. (2022) 49:3445–52. doi: 10.1007/s11033-022-07177-8
38. Huang Y, Li X, Luo G, Wang J, Li R, Zhou C, et al. Pyroptosis as a candidate therapeutic target for Alzheimer’s disease. Front Aging Neurosci. (2022) 14:996646. doi: 10.3389/fnagi.2022.996646
39. Hong W, Hu C, Wang C, Zhu B, Tian M, Qin H. Effects of amyloid beta (Abeta)42 and Gasdermin D on the progression of Alzheimer’s disease in vitro and in vivo through the regulation of astrocyte pyroptosis. Aging (Albany NY). (2023) 15:12209–24. doi: 10.18632/aging.205174
40. Ayyubova G, Madhu LN. Microglial NLRP3 inflammasomes in alzheimer’s disease pathogenesis: from interaction with autophagy/mitophagy to therapeutics. Mol Neurobiol. (2025) p:1–20. doi: 10.1007/s12035-025-04758-z
41. Bialek W, Collawn JF, Bartoszewski R. Ubiquitin-dependent and independent proteasomal degradation in host-pathogen interactions. Molecules. (2023) 28:6740. doi: 10.3390/molecules28186740
42. Finley D, Ciechanover A, Varshavsky A. Ubiquitin as a central cellular regulator. Cell. (2004) 116:S29–32. doi: 10.1016/S0092-8674(03)00971-1
43. Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. (2017) 40:441–9. doi: 10.14348/molcells.2017.0115
44. Hegde AN, Smith SG, Duke LM, Pourquoi A, Vaz S. Perturbations of ubiquitin-proteasome-mediated proteolysis in aging and alzheimer’s disease. Front Aging Neurosci. (2019) 11:324. doi: 10.3389/fnagi.2019.00324
45. Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. FEBS J. (2020) 287:11–26. doi: 10.1111/febs.v287.1
46. Wan P, Zhang Q, Liu W, Jia Y, Ai S, Wang T, et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. (2019) 33:5793–807. doi: 10.1096/fj.201801681R
47. Koumangoye R. The role of Cl(-) and K(+) efflux in NLRP3 inflammasome and innate immune response activation. Am J Physiol Cell Physiol. (2022) 322:C645–52. doi: 10.1152/ajpcell.00421.2021
48. Graham SF, Nasarauddin MB, Carey M, McGuinness B, Holscher C, Kehoe PG, et al. Quantitative measurement of [Na+] and [K+] in postmortem human brain tissue indicates disturbances in subjects with Alzheimer’s disease and dementia with Lewy bodies. J Alzheimers Dis. (2015) 44:851–7. doi: 10.3233/JAD-141869
49. Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols EM, Davis DM, et al. Priming is dispensable for NLRP3 inflammasome activation in human monocytes in vitro. Front Immunol. (2020) 11:565924. doi: 10.3389/fimmu.2020.565924
50. Zhai X, Zhang H, Xia Z, Liu M, Du G, Jiang Z, et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. (2024) 6:101032. doi: 10.1016/j.jhepr.2024.101032
51. Xiao J, Lin H, Liu B, Xia Z, Zhang J, Jin J. Decreased S1P and SPHK2 are involved in pancreatic acinar cell injury. biomark Med. (2019) 13:627–37. doi: 10.2217/bmm-2018-0404
52. Xiao J, Huang K, Lin H, Xia Z, Zhang J, Li D, et al. Mogroside II(E) inhibits digestive enzymes via suppression of interleukin 9/interleukin 9 receptor signalling in acute pancreatitis. Front Pharmacol. (2020) 11:859. doi: 10.3389/fphar.2020.00859
53. Zhang H, Xia T, Xia Z, Zhou H, Li Z, Wang W, et al. KIF18A inactivates hepatic stellate cells and alleviates liver fibrosis through the TTC3/Akt/mTOR pathway. Cell Mol Life Sci. (2024) 81:96. doi: 10.1007/s00018-024-05114-5
54. Zhang JJ, Rizk R, Li X, Lee BG, Matthies ML, Bietz KA, et al. Interleukin-10 exhibit dose-dependent effects on macrophage phenotypes and cardiac remodeling after myocardial infarction. Front Physiol. (2024) 15:1481460. doi: 10.3389/fphys.2024.1481460
55. Zhang H, Ni M, Wang H, Zhang J, Jin D, Busuttil RW, et al. Gsk3beta regulates the resolution of liver ischemia/reperfusion injury via MerTK. JCI Insight. (2023) 8:e151819. doi: 10.1172/jci.insight.151819
56. Vande Walle L, Stowe IB, Sacha P, Lee BL, Demon D, Fossoul A, et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PloS Biol. (2019) 17:e3000354. doi: 10.1371/journal.pbio.3000354
57. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. (2018) 19:1222. doi: 10.3390/ijms19041222
58. Savio LEB, de Andrade Mello P, da Silva CG, Coutinho-Silva R. The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol. (2018) 9:52. doi: 10.3389/fphar.2018.00052
59. Xia Y, Liu N, Xie X, Bi G, Ba H, Li L, et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy. (2019) 15:960–75. doi: 10.1080/15548627.2019.1569916
60. Wang D, Zhang J, Jiang W, Cao Z, Zhao F, Cai T, et al. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy. (2017) 13:914–27. doi: 10.1080/15548627.2017.1293766
61. Tinkov AA, Paoliello MMB, Mazilina AN, Skalny AV, Martins AC, Voskresenskaya ON, et al. Molecular targets of manganese-induced neurotoxicity: A five-year update. Int J Mol Sci. (2021) 22:4646. doi: 10.3390/ijms22094646
62. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
63. Zhou J, Li C, Lu M, Jiang G, Chen S, Li H, et al. Pharmacological induction of autophagy reduces inflammation in macrophages by degrading immunoproteasome subunits. PloS Biol. (2024) 22:e3002537. doi: 10.1371/journal.pbio.3002537
64. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. (2011) 31:6627–38. doi: 10.1523/JNEUROSCI.0203-11.2011
65. Parajuli B, Sonobe Y, Horiuchi H, Takeuchi H, Mizuno T, Suzumura A. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis. (2013) 4:e975. doi: 10.1038/cddis.2013.503
66. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
67. Lee H, Jose PA. Coordinated contribution of NADPH oxidase- and mitochondria-derived reactive oxygen species in metabolic syndrome and its implication in renal dysfunction. Front Pharmacol. (2021) 12:670076. doi: 10.3389/fphar.2021.670076
68. Zheng Q, Tan Q, Ren Y, Reinach PS, Li L, Ge C, et al. Hyperosmotic stress-induced TRPM2 channel activation stimulates NLRP3 inflammasome activity in primary human corneal epithelial cells. Invest Ophthalmol Vis Sci. (2018) 59:3259–68. doi: 10.1167/iovs.18-23965
69. Aminzadeh M, Roghani M, Sarfallah A, Riazi GH. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int Immunopharmacol. (2018) 54:78–85. doi: 10.1016/j.intimp.2017.10.024
70. Mancuso C, Santangelo R. Alzheimer’s disease and gut microbiota modifications: The long way between preclinical studies and clinical evidence. Pharmacol Res. (2018) 129:329–36. doi: 10.1016/j.phrs.2017.12.009
71. Suganya K, Koo BS. Gut-brain axis: role of gut microbiota on neurological disorders and how probiotics/prebiotics beneficially modulate microbial and immune pathways to improve brain functions. Int J Mol Sci. (2020) 21:7551. doi: 10.3390/ijms21207551
72. Jie Z, Xia H, Zhong SL, Feng Q, Li S, Liang S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. (2017) 8:845. doi: 10.1038/s41467-017-00900-1
73. Askarova S, Umbayev B, Masoud AR, Kaiyrlykyzy A, Safarova Y, Tsoy A, et al. The links between the gut microbiome, aging, modern lifestyle and alzheimer’s disease. Front Cell Infect Microbiol. (2020) 10:104. doi: 10.3389/fcimb.2020.00104
74. Yamazaki Y, Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of alzheimer’s disease. Int J Mol Sci. (2017) 18:1965. doi: 10.3390/ijms18091965
75. Pennisi M, Crupi R, Di Paola R, Ontario ML, Bella R, Calabrese EJ, et al. Inflammasomes, hormesis, and antioxidants in neuroinflammation: Role of NRLP3 in Alzheimer disease. J Neurosci Res. (2017) 95:1360–72. doi: 10.1002/jnr.23986
76. Qian X-h, Xie R-Y, Liu X-l, Tang H-dJA. Mechanisms of short-chain fatty acids derived from gut microbiota in Alzheimer’s disease. Aging Dis. (2022) 13:1252–66. doi: 10.14336/AD.2021.1215
77. Shen H, Guan Q, Zhang X, Yuan C, Tan Z, Zhai L, et al. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 100:109884. doi: 10.1016/j.pnpbp.2020.109884
78. Dubois EA, Rissmann R, Cohen AF. Rilonacept and canakinumab. Br J Clin Pharmacol. (2011) 71:639–41. doi: 10.1111/j.1365-2125.2011.03958.x
79. Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. (2011) 187:6539–49. doi: 10.4049/jimmunol.1100620
80. Batista AF, Rody T, Forny-Germano L, Cerdeiro S, Bellio M, Ferreira ST, et al. Interleukin-1beta mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-beta oligomers. J Neuroinflamm. (2021) 18:54. doi: 10.1186/s12974-021-02099-x
81. McManus RM, Latz E. NLRP3 inflammasome signalling in Alzheimer’s disease. Neuropharmacology. (2024) 252:109941. doi: 10.1016/j.neuropharm.2024.109941
82. Wang J, Ding Y, Zhuang L, Wang Z, Xiao W, Zhu J. Ginkgolide B−induced AMPK pathway activation protects astrocytes by regulating endoplasmic reticulum stress, oxidative stress and energy metabolism induced by Abeta1−42. Mol Med Rep. (2021) 23:457. doi: 10.3892/mmr.2021.12096
83. Liu GZ, Niu TT, Yu Q, Xu BL, Li XQ, Yuan BY, et al. Ginkgolide attenuates memory impairment and neuroinflammation by suppressing the NLRP3/caspase-1 pathway in Alzheimer’s disease. Aging (Albany NY). (2023) 15:10237–52. doi: 10.18632/aging.205072
84. Shao L, Dong C, Geng D, He Q, Shi Y. Ginkgolide B inactivates the NLRP3 inflammasome by promoting autophagic degradation to improve learning and memory impairment in Alzheimer’s disease. Metab Brain Dis. (2022) 37:329–41. doi: 10.1007/s11011-021-00886-2
85. You JE, Kim Ej, Kim HW, Kim JS, Kim K, Kim PH. Exploring the role of guanylate-binding protein-2 in activated microglia-mediated neuroinflammation and neuronal damage. Biomedicines. (2024) 12:1130. doi: 10.3390/biomedicines12051130
86. Zhang Y, Zhao Y, Zhang J, Gao Y, Li S, Chang C, et al. Ginkgolide B inhibits NLRP3 inflammasome activation and promotes microglial M2 polarization in Abeta(1-42)-induced microglia cells. Neurosci Lett. (2021) 764:136206. doi: 10.1016/j.neulet.2021.136206
87. Kiser C, Gonul CP, Olcum M, Genc S. Inhibitory effects of sulforaphane on NLRP3 inflammasome activation. Mol Immunol. (2021) 140:175–85. doi: 10.1016/j.molimm.2021.10.014
88. Brandenburg LO, Kipp M, Lucius R, Pufe T, Wruck CJ. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflammation Res. (2010) 59:443–50. doi: 10.1007/s00011-009-0116-5
89. Greaney AJ, Maier NK, Leppla SH, Moayeri M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J Leukoc Biol. (2016) 99:189–99. doi: 10.1189/jlb.3A0415-155RR
90. Tufekci KU, Ercan I, Isci KB, Olcum M, Tastan B, Gonul CP, et al. Sulforaphane inhibits NLRP3 inflammasome activation in microglia through Nrf2-mediated miRNA alteration. Immunol Lett. (2021) 233:20–30. doi: 10.1016/j.imlet.2021.03.004
91. Kluck V, Jansen T, Janssen M, Comarniceanu A, Efde M, Tengesdal IW, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. (2020) 2:e270–80. doi: 10.1016/S2665-9913(20)30065-5
92. Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D'Alessandro A, et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U.S.A. (2020) 117:32145–54. doi: 10.1073/pnas.2009680117
93. Cho H, Balaji S, Hone NL, Moles CM, Sheikh AQ, Crombleholme TM, et al. Diabetic wound healing in a MMP9-/- mouse model. Wound Repair Regener. (2016) 24:829–40. doi: 10.1111/wrr.2016.24.issue-5
94. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
95. Luciunaite A, McManus RM, Jankunec M, Racz I, Dansokho C, Dalgediene I, et al. Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem. (2020) 155:650–61. doi: 10.1111/jnc.14945
96. Torres S, Brol MJ, Magdaleno F, Schierwagen R, Uschner FE, Klein S, et al. The specific NLRP3 antagonist IFM-514 decreases fibrosis and inflammation in experimental murine non-alcoholic steatohepatitis. Front Mol Biosci. (2021) 8:715765. doi: 10.3389/fmolb.2021.715765
97. Wang X, Sun K, Zhou Y, Wang H, Zhou Y, Liu S, et al. NLRP3 inflammasome inhibitor CY-09 reduces hepatic steatosis in experimental NAFLD mice. Biochem Biophys Res Commun. (2021) 534:734–9. doi: 10.1016/j.bbrc.2020.11.009
98. Madurka I, Vishnevsky A, Soriano JB, Gans SJ, Ore DJS, Rendon A, et al. DFV890: a new oral NLRP3 inhibitor-tested in an early phase 2a randomised clinical trial in patients with COVID-19 pneumonia and impaired respiratory function. Infection. (2023) 51:641–54. doi: 10.1007/s15010-022-01904-w
99. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. (2018) 10:e8689. doi: 10.15252/emmm.201708689
100. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. (2018) 9:2550. doi: 10.1038/s41467-018-04947-6
101. McFarthing K, Buff S, Rafaloff G, Fiske B, Mursaleen L, Fuest R, et al. Parkinson’s disease drug therapies in the clinical trial pipeline: 2023 update. J Parkinsons Dis. (2023) 13:427–39. doi: 10.3233/JPD-239901
102. Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. (2022) 43:653–68. doi: 10.1016/j.tips.2022.04.003
103. Deardorff WJ, Grossberg GT. Targeting neuroinflammation in Alzheimer’s disease: evidence for NSAIDs and novel therapeutics. Expert Rev Neurother. (2017) 17:17–32. doi: 10.1080/14737175.2016.1200972
104. Karkhah A, Saadi M, Pourabdolhossein F, Saleki K, Nouri HR. Indomethacin attenuates neuroinflammation and memory impairment in an STZ-induced model of Alzheimer’s like disease. Immunopharmacol Immunotoxicol. (2021) 43:758–66. doi: 10.1080/08923973.2021.1981374
105. Feng X, Hu J, Zhan F, Luo D, Hua F, Xu G. MicroRNA-138-5p regulates hippocampal neuroinflammation and cognitive impairment by NLRP3/caspase-1 signaling pathway in rats. J Inflammation Res. (2021) 14:1125–43. doi: 10.2147/JIR.S304461
106. Zhang M, Wang L, Huang S, He X. MicroRNA-223 targets NLRP3 to relieve inflammation and alleviate spinal cord injury. Life Sci. (2020) 254:117796. doi: 10.1016/j.lfs.2020.117796
107. Han C, Guo L, Yang Y, Guan Q, Shen H, Sheng Y, et al. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav. (2020) 10:e01627. doi: 10.1002/brb3.v10.6
108. Ali M, Gupta M, Wani A, Sharma A, Abdullaha M, Kour D, et al. IIIM-941, a stilbene derivative inhibits NLRP3 inflammasome activation by inducing autophagy. Front Pharmacol. (2021) 12:695712. doi: 10.3389/fphar.2021.695712
109. Cheng ZY, Xia QP, Hu YH, Wang C, He L. Dopamine D1 receptor agonist A-68930 ameliorates Abeta(1-42)-induced cognitive impairment and neuroinflammation in mice. Int Immunopharmacol. (2020) 88:106963. doi: 10.1016/j.intimp.2020.106963
110. Liu X, Hao W, Qin Y, Decker Y, Wang X, Burkart M, et al. Long-term treatment with Ginkgo biloba extract EGb 761 improves symptoms and pathology in a transgenic mouse model of Alzheimer’s disease. Brain Behav Immun. (2015) 46:121–31. doi: 10.1016/j.bbi.2015.01.011
111. Han X, Xu T, Fang Q, Zhang H, Yue L, Hu G, et al. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. (2021) 44:102010. doi: 10.1016/j.redox.2021.102010
112. Zhou F, Lian W, Yuan X, Wang Z, Xia C, Yan Y, et al. Cornuside alleviates cognitive impairments induced by Abeta(1-42) through attenuating NLRP3-mediated neurotoxicity by promoting mitophagy. Alzheimers Res Ther. (2025) 17:47. doi: 10.1186/s13195-025-01695-w
113. Li JY, Chen MB, Fu TT, Liu FF, Liu J, Xu CJ, et al. Discovery of structurally intriguing diterpenoids as anti-neuroinflammatory agents from mangrove plant Excoecaria agallocha L. via inhibiting macrophage polarization and inflammasome. Phytochemistry. (2025) 234:114440. doi: 10.1016/j.phytochem.2025.114440
114. Zhang C, Qiu WQ, Yu L, Pan R, Teng JF, Sang ZP, et al. Development of cancer-associated fibroblasts subtype and prognostic model in gastric cancer and the landscape of tumor microenvironment. Int J Biochem Cell Biol. (2022) 152:106309. doi: 10.1016/j.biocel.2022.106309
115. Qiu WQ, Pan R, Tang Y, Zhou XG, Wu JM, Yu L, et al. Lychee seed polyphenol inhibits Abeta-induced activation of NLRP3 inflammasome via the LRP1/AMPK mediated autophagy induction. BioMed Pharmacother. (2020) 130:110575. doi: 10.1016/j.biopha.2020.110575
116. Li N, Li M, Guo M, Meng T, Mu B, Yu J, et al. Eriodictyol improves cognitive function in 5xFAD mice of Alzheimer’s disease by inhibiting the microglia NLRP3 inflammasome signaling pathway. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. (2023) 39:973–80.
117. Li L, Li WJ, Zheng XR, Liu QL, Du Q, Lai YJ, et al. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol Med. (2022) 28:11. doi: 10.1186/s10020-022-00442-3
118. Melchiorri D, Merlo S, Micallef B, Borg J-J, Dráfi FJFiP. Alzheimer’s disease and neuroinflammation: will new drugs in clinical trials pave the way to a multi-target therapy? Front Pharmacol. (2023) 14:1196413. doi: 10.3389/fphar.2023.1196413
Keywords: NLRP3, neuro-inflammation, IL-1β, inflammasome, Alzheimer’s disease, microglia, inhibitor
Citation: Li Z and Gong C (2025) NLRP3 inflammasome in Alzheimer’s disease: molecular mechanisms and emerging therapies. Front. Immunol. 16:1583886. doi: 10.3389/fimmu.2025.1583886
Received: 26 February 2025; Accepted: 19 March 2025;
Published: 07 April 2025.
Edited by:
Jing Zhang, University of South Dakota, United StatesReviewed by:
Shengshan Xu, Jiangmen Central Hospital, ChinaCopyright © 2025 Li and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunrong Gong, ODk1ODkwMTQ4QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.