 Krzysztof Piotr Michalak
Krzysztof Piotr Michalak Amelia Zofia Michalak2*†
Amelia Zofia Michalak2*†- 1Laboratory of Vision Science and Optometry, Physics and Astronomy Faculty, Adam Mickiewicz University in Poznań, Poznań, Poland
- 2Faculty of Medicine, Poznań University of Medical Sciences, Poznań, Poland
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has been recognized not only for its acute effects but also for its ability to cause LongCOVID Syndrome (LCS), a condition characterized by persistent symptoms affecting multiple organ systems. This review examines the molecular and immunological mechanisms underlying LCS, with a particular focus on autophagy inhibition, chronic inflammation, oxidative, nitrosative and calcium stress, viral persistence and autoimmunology. Potential pathophysiological mechanisms involved in LCS include (1) autoimmune activation, (2) latent viral persistence, where SARS-CoV-2 continues to influence host metabolism, (3) reactivation of latent pathogens such as Epstein-Barr virus (EBV) or cytomegalovirus (CMV), exacerbating immune and metabolic dysregulation, and (4) possible persistent metabolic and inflammatory dysregulation, where the body fails to restore post-infection homeostasis. The manipulation of cellular pathways by SARS-CoV-2 proteins is a critical aspect of the virus’ ability to evade immune clearance and establish long-term dysfunction. Viral proteins such as NSP13, ORF3a and ORF8 have been shown to disrupt autophagy, thereby impairing viral clearance and promoting immune evasion. In addition, mitochondrial dysfunction, dysregulated calcium signaling, oxidative stress, chronic HIF-1α activation and Nrf2 inhibition create a self-sustaining inflammatory feedback loop that contributes to tissue damage and persistent symptoms. Therefore understanding the molecular basis of LCS is critical for the development of effective therapeutic strategies. Targeting autophagy and Nrf2 activation, glycolysis inhibition, and restoration calcium homeostasis may provide novel strategies to mitigate the long-term consequences of SARS-CoV-2 infection. Future research should focus on personalized therapeutic interventions based on the dominant molecular perturbations in individual patients.
1 Introduction
Since its emergence in late 2019, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has posed an unprecedented challenge to global health. While the acute phase of Coronavirus Disease 2019 (COVID-19) is increasingly well understood, growing attention has turned toward long-term complications. LongCOVID Syndrome (LCS) affects millions of patients worldwide and manifests as a constellation of multisystem symptoms persisting months after viral clearance, with mechanisms that remain elusive (1, 2).
Recent studies have highlighted the ability of SARS-CoV-2 proteins to interfere with key intracellular pathways, including autophagy, mitochondrial dynamics, oxidative and nitrosative stress responses, calcium signaling, and the regulation of transcription factors such as HIF-1α and Nrf2 (3–11). These perturbations are believed to contribute to both viral persistence and immune dysregulation. However, no study to date has thoroughly examined how viral proteins simultaneously affect multiple cellular pathways, each controlled by its own feedback mechanisms, and how the disruption of these systems may interact to worsen or prolong disease. This lack of a systems-level perspective limits our understanding of how SARS-CoV-2 causes long-term cellular dysfunction.
This review aims to fill this gap by providing an integrative analysis of how viral proteins reprogram intracellular signaling, with particular focus on autophagy, Nrf2-mediated antioxidant defenses, calcium signaling, and mitochondrial function. We further examine how dysregulation of feedback mechanisms between these pathways may lead to sustained cellular stress and impaired resolution of inflammation. A better understanding of these interdependencies could inform novel therapeutic strategies for both acute and post-acute COVID-19.
2 LongCOVID syndrome
LongCOVID-19 Syndrome (LCS) is characterized by a variety of health complications affecting multiple organ systems, including the respiratory, cardiovascular, hematologic, genitourinary, gastrointestinal, and neurologic systems (1, 2). The underlying cause of these symptoms is thought to be the persistent activation of the immune system (1, 12). Various definitions have been proposed for LCS. Some authors propose to distinguish between Post Acute COVID-19 (4-12 weeks after infection), Post COVID-19 (more than 12 weeks) and Long COVID-19 which includes both of these terms (2). Another term used is Post-Acute Sequelae of SARS-CoV-2 Infection (PASC). However, it should be noted that this classification is somewhat artificial and has a negligible impact on the current analysis.
Previous analyses of the LCS problem have focused on describing the immunologic, molecular, and morphologic changes in the organs or tissues that are damaged or dysregulated (1, 2). The criteria for the diagnosis of LCS have also been discussed in the context of different symptoms and affected organs (1, 2). The features of hyperinflammation, coagulation disorders, oxidative stress and tissue-dependent damage are presented as major endpoint features of LCS. However, little attention has been paid to the initial pathobiochemical and molecular changes in cellular metabolism that lead to the observed end-effects. Assuming a large variety of different LCS types, the question arises as to what is the reason for the prolonged duration of symptoms in patients after SARS-CoV-2 infection and to what extent the molecular and immunological changes observed during and after COVD-19 are the reason for the symptoms of LCS.
Four main alternatives can be considered as the reason for LCS:
1. Induction of a type of autoimmunity [29,30]. The autoantibodies, after binding to some receptors on the surface of the cells, induce its metabolic reprogramming causing regulatory dysfunctions.
2. COVID-19 infection induces other chronic pathogens that were already present in the host cells (e.g. Epstein-Barr virus, cytomegalovirus) (13, 14) to go to the “higher level” of their body infiltration causing the magnification of their earlier latent symptoms. The development of intracellular infections can be attributed to intracellular metabolic or immunological changes that facilitate pathogen propagation. Autophagy inhibition induced by SARS-CoV-2 proteins (15–18) appears to be a possible facilitator of disease progression.
3. The SARS-CoV-2 virus remains in the cells after recovery, it is not completely eliminated and enters the chronic active state (19, 20). The patient’s symptoms are to a high degree the direct metabolic consequences of the presence of the virus in the host cells.
4. Neither SARS-CoV-2 nor other pathogens or autoimmunity are present in the host cell as an activating stimulus. The presence of SARS-CoV-2, other intracellular pathogens and autoimmunity have been excluded as initiating elements of LCS. The metabolic and immunological alterations have been shown to be a consequence of the pathological state (e.g. chronic mitochondrial dysfunction, chronic microcirculatory disturbances), and the organism is unable to leave this state and return to a healthy states characterized by proper activity of all metabolic pathways and transcription factors. The only possible explanation for LCS is the inability of host cells to exit the deregulated state.
It is important to note that the first three proposed causes of LCS may overlap in individual patients, and the fourth cause can only be considered after the first three have been ruled out.
Autoimmune disorders and the induction of other pathogens will be discussed in a basic outline, as it is dedicated to a separate review. However, a detailed analysis will focus on the impact of individual viral proteins on metabolism, assuming that LongCovid-19 syndrome is generated by an active chronic viral infection and interference of viral proteins with host metabolism, as is the case with other viruses that enter a state of chronic active infection, such as EBV (21–26), CMV (27), HCV (28, 29), HSV (30, 31). In this case, the key to Long Covid syndrome therapy is to understand the interaction of virus proteins with the host’s metabolism in order to reverse the disorders and restore the immune system’s ability to remove virus-laden cells or restore the ability of host cells to clear the virus.
2.1 COVID-19 activates multiple autoimmune diseases
Autoimmunity appears to be a important component of the symptoms associated with LCS, as evidenced by the presence of various autoantibodies in a significant proportion of patients with both LCS and COVD-19 [29,30]. Numerous reports have indicated the presence of various autoantibodies following infection with SARS-CoV-2 (32). These autoantibodies associated with the SARS-CoV-2 infection include autoantibodies against type I interferons, lupus anticoagulant (LAC), antinuclear antibodies (ANA), rheumatoid factor (RF), p- and c-antineutrophil cytoplasmic antibodies (pANCA, cANCA), anticardiolipin antibodies (ACL), anti-Ro52 antibodies and anti-phosphatidylserine antibodies (32). Antiphospholipid antibodies were mainly represented by IgG ACL (48%), followed by IgM ACL (21%), especially in COVID-19 positive patients (32). As reported by Zhou et al. (33), autoantibodies were found to be directed against 12 different host antigens. These immune molecular elements included antinuclear antibodies (ANA), anti-SSA/Ro antibodies, anti-Scl-70 antibodies, and anti-U1-RNP antibodies.
Vojdani et al. (34) demonstrated by ELISA that certain monoclonal antibodies directed against the spike protein of SARS-CoV-2 showed reactivity with several self-antigens, including glutamate decarboxylase-65 (GAD-65), mitochondrial proteins, phospholipids, and hepatocyte microsomes.
As reported by Chang et al. (35), multiple IgG autoantibodies were found in hospitalized patients with confirmed cases of COVID-19. Some of these autoantibodies have been shown to contribute to the formation of proinflammatory immune complexes, primarily on endothelial cell surfaces. This has the potential to lead to vascular inflammation and thrombosis. For example, increased formation of neutrophil extracellular traps (NETs) was found in patients with COVID-19 who also had vasculitis (35). This phenomenon was found to be associated with significant neutrophil activation and production of proinflammatory NETs, which were observed to contain nucleic acids, histones, and various inflammatory peptides or proteins (35). Autoantibodies identified in this study included anti-C1q antibodies, previously observed in systemic lupus erythematosus, as well as anti-β2GP1, anti-bactericidal/permeability-increasing protein (BPI), and anti-ACE-2 antibodies. In addition, 60-80% of all hospitalized patients with confirmed COVID-19 expressed at least one anti-centromere antibody (ACA), a finding also reported in other autoimmune diseases, most commonly in CREST syndrome (35).
As also found by Wang et al. (36), IgG isotypes from patients with anti-GM-CSF, anti-CXCL-1 or anti-CXCL-7 autoantibodies have the potential to block the signaling of these proteins. In addition, increased antibody-dependent cellular phagocytosis was observed in Raji B cells or Jurkat T cells due to the presence of anti-CD38 or anti-CD3ε autoantibodies (36). Furthermore, the study established a correlation between autoantigens such as NXPH-1, PCSK-1, SLC2A10 and DCD and markers of COVID-19 severity, including D-dimer, ferritin, C-reactive protein and lactate, which were observed to increase in cases of severe COVID-19 (36). In addition, there have been reports of an association between viral-induced autoimmunity and COVID-19 based on the mechanism of molecular mimicry. Certain proteins on the surface of SARS-CoV-2 have been shown to be biochemically similar to those found in host cells (37, 38). Therefore, it is plausible that some patients with COVD-19 may manifest cross-reactive immunologic responses analogous to those observed in other pathologic conditions, such as acute post-streptococcal glomerulonephritis or rheumatic endocarditis.
The research on the existence of autoimmunity in LCS patients is less numerous, but significant. According to Ampudia et al. (39), there is persistent autoimmune activation and a proinflammatory state in LCS. For example, the frequency of β2-glycoprotein-1 (β2-GP1) IgM autoantibodies, classically described in antiphospholipid syndrome, was higher in LCS patients compared to pre-pandemic controls (39). The same study also found that 19 of 33 acute cases of COVID-19 and 21 of 33 LCS patients expressed at least one autoantibody. According to Ampudia et al., LCS patients have higher levels of circulating naive B cells, which are a known source of autoantibodies. Furthermore, patients with LCS had very high levels of circulating proinflammatory cytokines such as IFN-α, TNF-α, G-CSF, IL17A, IL-6, IL1-β, and IL-13, but also a decrease in interferon-γ-induced protein-10 (IP-10) (39). Persistent dysregulation of IL-6 (one of the major proinflammatory cytokines in COVID-19) was found to be associated with generalized fatigue, sleep disturbance, depression, and anxiety and is one of the major proinflammatory molecules associated with the development of autoinflammatory responses and autoimmunity via pre-existing B lymphocytes (39). The clinical presentation of LCS has been found to be influenced by other molecular elements, including IL-1β, TNF-α, IFN-γ, IL-10, IL-2, C-reactive protein, MCP-1, serum amyloid-A and metabolites of the kynurenine pathway (39).
In conclusion, the autoimmunity that develops during and after SARS-CoV-2 infection is a significant problem that needs to be analyzed independently for different types of autoantibodies. Further research is needed to determine which antibodies are “silent” (i.e., do not react with any proteins or receptors in host cells) and which are active, i.e., react with some proteins and thereby induce or inhibit specific molecular pathways. The issue of autoimmunity in LCS requires dedicated research and analysis, with a focus on the inhibition of persistent induced inflammation. Treatment should aim to gradually reduce the levels of inflammatory antibodies, thereby facilitating recovery.
2.2 Activation of other active chronic infections
A recent reports describe that SARS-CoV-2-induced infection may lead to reactivation of EBV (40–42). Other viruses that belong to the Herpesvirus family: HHV-6 and HHV-7 were also shown to be reactivated in patients infected with SARS-CoV-2 (42). The effect of reactivation of pathogens is in line with studies indicating that the SARS-CoV-2 virus disrupts various immune functions, including the blocking of autophagy in numerous molecular mechanisms (16). Another mechanism observed for the SARS-CoV-2 NSP15 protein is the inhibition of type 1 interferon production (43). It has been observed that a virus with a mutated NSP15 induced normal interferon production, while the wild type induced a broad immune response including the induction of ER stress and upregulation of over 2,800 genes, including networks associated with activating the unfolded protein response and the proinflammatory response associated with viral pathogenesis (43). It indicates the important role of this viral protein in weakening antiviral immunity.
It is important to acknowledge that the chronic active viremia is not exclusive to the SARS-CoV-2. Various other viruses, including Epstein-Barr virus (EBV) (21–26), cytomegalovirus (27), Ebola virus (44), Zika virus (45), enteroviruses (46), Coxsackie (47–49) and measles virus (50), have also been documented to enter the active chronic state and induce various health complications. EBV, a common virus in the general population, often progresses to active chronic infection. Studies employing blood PCR in healthy individuals by various authors suggest a population burden of 18-30% (51–54) and up to 80% in the older adult population in Qatar (55). Population analyses for CMV are similar. A study by Huifen Li et al. (56) demonstrates that active chronic CMV infection can persist for decades in older individuals and leads to a threefold increase in blood levels of the pro-inflammatory IL-6. The study further suggests that the virus occupies immune cells, including monocytes, CD4 and CD8 lymphocytes. In a group of subjects aged 70-79 years, 46% had CMV DNA detected in monocytes. As in the case with SARS-CoV-2, both viruses have been postulated to induce autoimmunity and hypercoagulation (57–64) including disseminated intravascular coagulation (DIC) (65–67).
EBV, CMV and Coxsackie B3 have also been reported to inhibit autophagy (16, 68, 69), suggesting a potential for these two viruses and SARS-CoV-2 to facilitate each other’s spread within the body. Other viruses that chronically block autophagy may also contribute to this process. Naendrup et al. (14) reported that in a cohort of 117 critically ill COVID-19 patients, EBV reactivation was detected in 16% and CMV in 9%. Reactivations were more frequent in patients receiving corticosteroids (58% for EBV, 55% for CMV) (14). While targeted treatment with ganciclovir improved survival in CMV patients (83% vs. 0% without treatment), rituximab did not show a significant effect on EBV outcomes. These findings suggest that while viral reactivations are common in severe cases of COVID-19, further study is necessary to determine their clinical impact and the benefits of targeted treatment.
2.3 Sars-Cov-2 still persisted in the cells despite negative tests
The mounting body of evidence indicates the presence of SARS-CoV-2 RNA and protein in a broad spectrum of tissue types, collected weeks or months after the onset of acute SARS-CoV-2 infection. The viral RNA or protein has been identified in the majority of organs and tissues, including the liver, stomach, tonsils, gallbladder, and lungs (20, 70–74). The preponderance of evidence for the existence of a SARS-CoV-2 reservoir in individuals with LCS stems from three sources: tissue biopsy studies, studies of SARS-CoV-2 proteins in plasma and studies using features of the adaptive immune response to infer the presence of a SARS-CoV-2 reservoir in tissues.
One of the earliest reports concerning the prolonged positive PCR tests at recovery patients was published by Lan et al. (19). Four members of the hospital staff were prescribed antiviral treatment (75 mg of oseltamivir taken orally every 12 hours) due to a positive diagnosis of SARS-CoV-2 infection. Subsequently, two consecutive negative RT-PCR test results were obtained. The time from the onset of symptoms to recovery ranged from 12 to 32 days. Thereafter, the subjects were instructed to adhere to a five-day home quarantine protocol. Subsequent RT-PCR tests, conducted between 5 and 18 days later using two distinct PCR kits, yielded positive results, despite the absence of symptoms as determined by clinical examination and chest CT findings.
Cheung et al. (20) detected nucleocapsid protein (NP) of SARS-CoV-2 in various organs including the liver, colon, lymph nodes, appendix, ileum, haemorrhoids and gallbladder, in five cancer patients who had recovered from the disease. These patients had tested negative for SARS-CoV-2 between 9 to 180 days prior. Notably, viral antigen was detected in all tissues of two patients, suggesting widespread multi-organ involvement from the viral infection. It is noteworthy that in the colon, viral antigen was detected exclusively in normal colonic crypts and polyps, while it was absent from neoplastic tissues.
In separate study Tao et al. (75) demonstrated that gastrointestinal (GI) symptoms were exhibited by a considerable proportion of patients with COVID-19. Furthermore, the study revealed that seven out of fifteen patients with confirmed cases of SARS-CoV-2 RNA positivity in their stool samples exhibited negative results in their respiratory samples and did not manifest any gastrointestinal symptoms. In vitro transcriptional analysis employing a SARS-CoV-2-infected cell model revealed that the 3’ end of the viral genome was significantly more covered compared to the 5’ end, suggesting an indication of active viral replication and infection (75). Furthermore, the presence of opportunistic pathogens, including Collinsella aerofaciens, Collinsella anakaei, Streptococcus infantis, and Morganella morganii, was observed in higher concentrations in SARS-CoV-2-positive fecal samples (75). Additionally, these samples exhibited an augmented capacity for nucleotide biosynthesis and amino acid and carbohydrate metabolism (glycolysis). However, the level of short-chain fatty acid-producing bacteria was diminished (75). These findings suggest a potential significant role for GI dysbiosis in SARS-CoV-2 patients; however, the direction of causality remains uncertain, as the study does not elucidate whether the dysbiosis is a consequence or a cause of the infection.
In a separate study, Tejerina et al. (76) utilized RT-PCR to detect SARS-CoV-2 in plasma, urine, and stool samples from patients with LCS. The patients were examined 39 to 67 days after their initial diagnosis of COVD-19. The study’s participants included 29 patients who had reported symptoms such as fatigue, myalgia, dyspnea, inappropriate tachycardia, and low-grade fever for more than 4 weeks after their initial diagnosis of COVD-19. The disease manifested as mild in 55% of cases, while 13 patients (45%) exhibited positive plasma RT-PCR results and 51% were positive in at least one RT-PCR sample (plasma, urine, or stool). Notably, 18 patients (62%) were undergoing antiviral treatment using lopinavir/ritonavir and hydroxychloroquine, which led to a substantial improvement in their health status (76). The findings indicate a pattern of persistent or recurrent/intermittent SARS-CoV-2 viremia in approximately 50% of patients with non-specific symptoms, which could be interpreted as LCS.
Further research on viral persistence in the human body can be found in (77). The analyses presented herein demonstrate that the probability of long-term viral persistence in the human body subsequent to a diagnosis of COVD-19 infection is relatively high, irrespective of the severity of the disease. Consequently, this possibility should be addressed with caution during the analysis of patients exhibiting LCS symptoms. A comprehensive review of the literature reveals that persistent viral infection may be responsible for approximately 50-70% of LCS patients.
3 Molecular basis for the LongCOVID syndrome caused by SARS-CoV-2 persistence
To understand the mechanisms underlying the metabolic effects of SARS-CoV-2 on host cell metabolism, it is imperative to elucidate the mechanisms of inflammation regulation, which consist of feedbacks between cytokines, oxidative stress, nitrosative stress, calcium stress, reticuloendoplasmic (ER) stress, HIF-1α, autophagy, and Nrf2. These feedbacks have been described by Michalak et al. (78). The disruption of these feedback loops by SARS-CoV-2 is a critical factor in understanding the mechanisms by which it induces LCS syndrome. A particular focus is placed on autophagy, as its disruption has been identified as a key factor in the development of both acute hyperinflammatory and chronic active COVID-19 (16, 18, 79–84) as well as many other diseases such as e.g. autoimmune and neurodegenerative diseases (85–89). A diagram of the couplings in inflammation control between the mentioned elements based mainly on the review by Michalak et al. (78), is shown in Figure 1. The analysis of the interaction of individual viral proteins on the metabolism in the context of the above-mentioned feedbacks allows us to understand the multifaceted mechanisms of the development of LongCovid-19 syndrome, as well as the reason for the therapeutic difficulties of this condition. This multiple coupling system will therefore be briefly discussed.
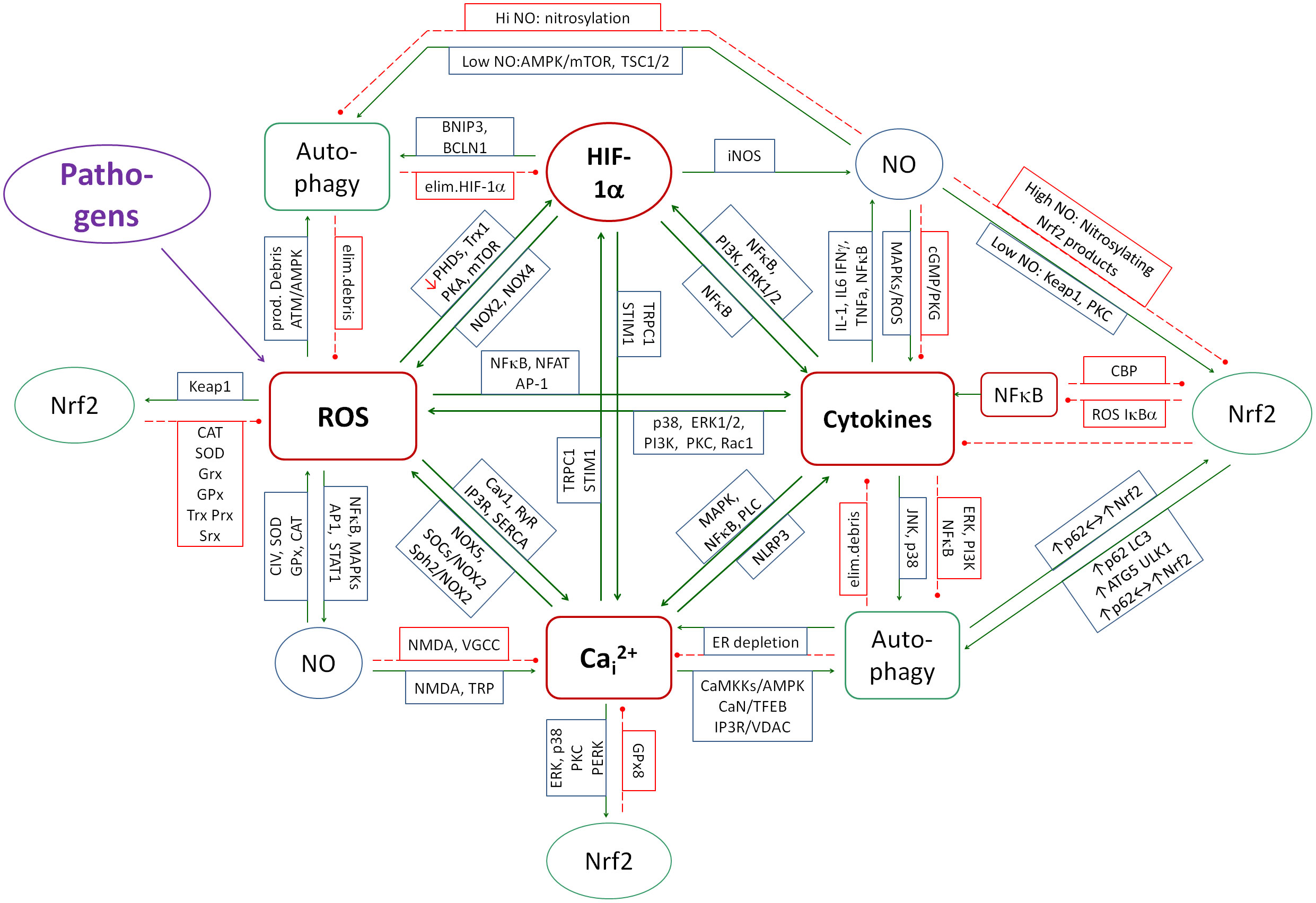
Figure 1. A diagram of the couplings that contribute to development of inflammation. The system of mainly positive couplings between inflammation (cytokines), ROS, Cai2+ and HIF-1α forms the core of inflammation induction. NO is partly positively and partly negatively coupled to the core elements and contributes at low concentrations to the control of inflammation and at high concentrations to its amplification. Nrf2 and autophagy create the system of mainly negative couplings that reduce and control inflammation, ROS production, Cai2+ and HIF-1α. Green solid arrows - activation, red dashed lines - inhibition.
3.1 Couplings between cytokines, NOX, NO, Cai2+ and HIF-1α
From the point of view of control theory, it is not easy to design a system that, when exposed to a variety of pathogens, excites itself to levels high enough to fight the pathogen, but not so high as to exceed the threshold of self-destruction. Evolution has programmed a system in which cytokines, NOXs, HIF-1α and Cai2+ form a mutual positive feedback system. When induced by a pathogen, they reinforce each other to fight the pathogen. On the other hand, autophagy and Nrf2 are regulators of this spiral, turning on mechanisms that inhibit this coupling spiral. HIF-1α and NO have a dual effect, as their action reinforces the inflammatory spiral in some conditions and inhibits it in others. When analyzing the effect of SARS-CoV-2 proteins on metabolism, one must keep this regulatory system in mind, since the activities of all these elements are disrupted by the viral proteins. The total effect of the virus on metabolism must take into account not only the sum of the individual interactions but also the mutual feedbacks between these elements, which amplifies the total effect.
The primary coupling is between cytokines and NADPH oxidases, which produce H2O2. These couplings form the main flywheel of inflammation in the fight against pathogens (see Figure 2). NOX-derived ROS stimulate the transcription of pro-inflammatory genes via factors such as NF-κB (90–92) or NFAT (93). Conversely, priming of NOXs occurs in response to a variety of cytokines such as TNF-α (94–96), IL-1β (97), IL-6 (98), IL-4 (99), IFN-γ (100), IL-8 (101), IL-12 (102), IL-15 (103), IL-17 (104), IL-23 (105), TGF-β (106–110). ROS also amplify inflammatory signals by activating kinase pathways such as p38 MAPK or ERK1/2, creating a complex network of interdependencies. Chronic maintenance of such couplings can lead to uncontrolled chronic inflammation and potential tissue damage. Figure 2 shows a simplified diagram of these couplings and the mediating role of transcription factors and kinase pathways.
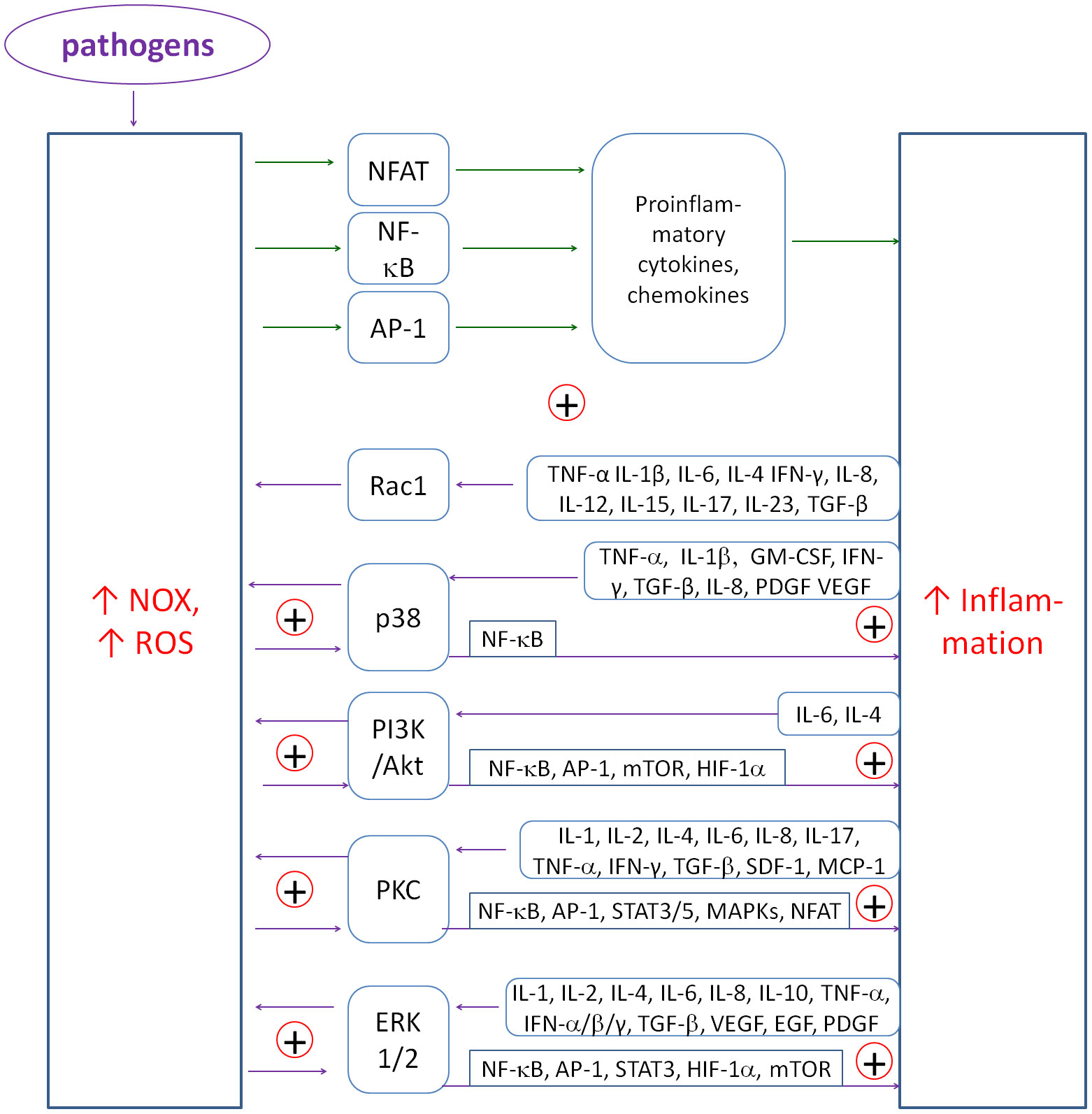
Figure 2. The diagram of positive couplings between NOX-derived ROS and inflammatory cytokines. The multiplicity of positive couplings forms the flywheel of inflammatory induction. ROS induce NF-κB, NFAT and AP-1 to produce mainly proinflammatory cytokines and chemokines. Conversely, cytokines induce NOXs to produce ROS mainly through p38, PI3K, PKC and ERK1/2 kinase pathways.
Cai2+ and HIF-1α are additional elements that drive the inflammatory spiral (see Figure 1). ROS-induced increases in Cai2+ levels activate the NLRP3 inflammasome, leading to IL-1β production and enhanced inflammation (111). In turn, inflammatory cytokines increase Ca²+ influx into cells via MAPK, PLC (phospholipase C), and NF-κB, establishing a positive feedback loop. PLC produces IP3 and DAG. IP3 binds to IP3 receptors in the endoplasmic reticulum, resulting in the release of Ca2+ ions from the ER into the cytoplasm (112), whereas DAG activates transient receptor potential canonical (TRPC) channels, resulting in the influx of Ca2+ ions from the extracellular space (113).
HIF-1α activity depends on multiple feedbacks with ROS, NO, cytokines and Ca2+. It has a dual function: it is protective for mitochondria under hypoxic conditions and pro-inflammatory in response to stressors. The protective effect is mainly due to inhibition of PDH (pyruvate dehydrogenase) and pyruvate influx into mitochondria, thereby reducing Krebs cycle turnover and mito-ROS production (114). The positive feedback between HIF-1α and succinate/fumarate acts as an amplifier of this relationship (115) as succinate and fumarate contribute to the stabilization of HIF-1α, while activation of HIF-1α leads to an increase in their concentration in mitochondria by blocking PDH, activating glycolysis (GLL), gluconeogenesis (GNG), and the glucose transporter GLUT-1. ROS stabilize HIF-1α by inhibiting its degradation and increasing its stability (116, 117). Stabilization of HIF-1α, in turn, induces NOX expression (118), which further increases ROS production. In addition, ROS activate NF-κB, which increases HIF-1α transcription (119). In turn, HIF-1α activates inflammation mainly through NF-κB (120). Proinflammatory cytokines such as TNF-α and IL-1β induce HIF-1α through activation of the PI3K/Akt and NF-κB pathways (121–124). In addition, HIF-1α modulates the expression of calcium channels such as TRPC1 and STIM1, which increase Ca2+ influx into the cytoplasm (125–128). The increase in Ca2+ can activate calmodulin-dependent proteins that increase HIF-1α activity, closing the positive feedback loop (129). In summary, HIF-1α exerts a protective effect against mito-stress. However, under conditions of chronic inflammation, its interactions with ROS, Cai2+ and cytokines promote the perpetuation of inflammation.
3.2 The role of kinase pathways
Kinase pathways, including MAPK (p38, ERK1/2, JNK), PI3K/Akt, JAK/STAT, AMPK and cAMP/PKA, have been identified as central regulators of inflammation and autophagy processes. The p38, ERK1/2, JNK and PI3K/Akt pathways are mainly proinflammatory, while AMPK and cAMP/PKA are anti-iflammatory. The JAK/STAT pathway is actually a series of pathways depending on the type of STAT protein. Some STATs are pro-inflammatory and some are anti-inflammatory. A diagram of the action of individual signaling pathways and selected metabolites on autophagy is shown in Figure 3. This diagram shows the complexity of the autophagy regulation. Even a small imbalance of this control can disrupt the process leading to cellular pathology. It is worth noting that in the case of a severe inflammation, NO changes its effect from pro-autophagic to anti-autophagic, which can have an important impact on the final balance of this process.
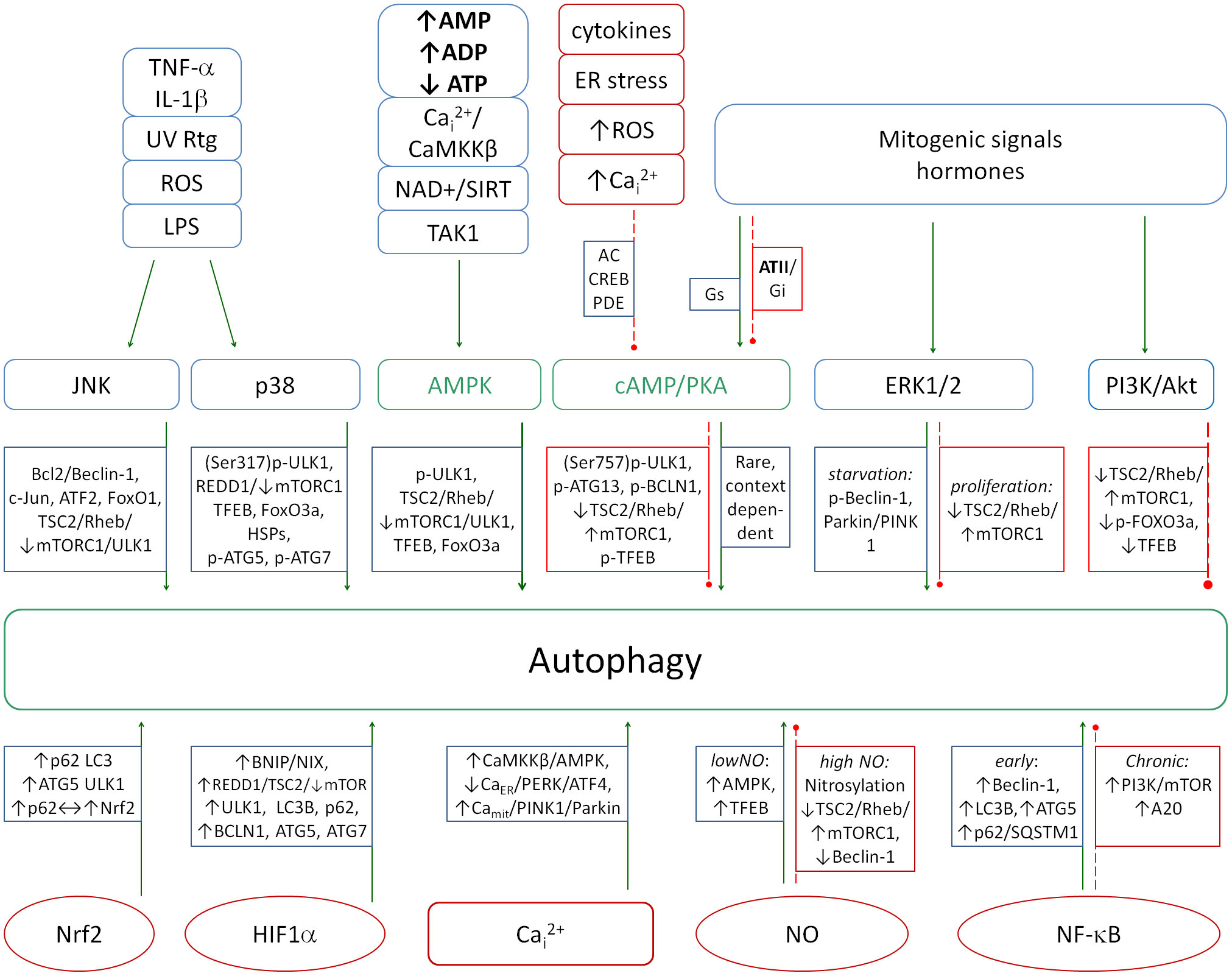
Figure 3. A diagram showing the action of major signaling pathways (top row) and selected molecules (bottom row) on the induction of autophagy. The proper balance between the pro- and anti-autophagic pathways is necessary to properly combat pathogens. Increased autophagy inhibition leads to the chronicity of infections and is a hallmark of several autoimmune diseases. Green solid arrows - activation, red dashed lines - inhibition.
The p38 MAPK pathway is known to be activated by various environmental stressors, including oxidative stress, hypoxia, UV or ionizing radiation, and osmotic disturbances (130), Additionally, the pathway is stimulated by inflammatory factors such as TNF-α (131, 132), IL-1β (133, 134) and TGF-β (135, 136)) as well as by pathogens including bacterial lipopolysaccharides (LPS) (137), which activate toll-like receptors (TLR). In response, p38 induces the expression of inflammatory genes (138–142) and promotes autophagy (143) as a protective mechanism.
ERK1/2 controls cell proliferation and the production of inflammatory cytokines (144–147). It is activated in response to inflammatory stimuli such as IL-17A, IL-1β (134, 148), bacterial LPS (149), or growth factors (e.g., VEGF, EGF) (150) Excessive activation of this pathway can lead to pathological inflammatory conditions, including psoriasis, rheumatoid arthritis, and Crohn’s disease (151, 152). ERK1/2 can induce autophagy by activating the Beclin-1 protein, although this effect is believed to occur at least partially as a consequence of PI3K/Akt pathway inhibition (153, 154). Conversely, under conditions that favor cell growth and proliferation, ERK1/2 may suppress autophagy by promoting mTOR activity via the TSC2/Rheb/mTORC1 axis, a mechanism that is particularly prominent in cancer (155) and is also implicated in neurodegenerative disorders (156).
The JNK pathway, by activating the transcription factor AP-1, increases the expression of pro-inflammatory mediators such as IL-6, IL-8, and TNF-α as well as chemokines that attract immune cells to the site of inflammation (157–159). In macrophages and neutrophils, JNK promotes the production of ROS, which contribute to the destruction of pathogens (160). The JNK pathway also plays a role in the resolution of inflammation by promoting apoptosis in dysfunctional cells, thereby limiting excessive inflammation (161), and by promoting autophagy which reduces the debris-mediated inflammation (161, 162). Its chronic activation has also been implicated in autoimmune diseases (163).
PI3K/Akt is a pathway that modulates pro- and anti-inflammatory processes, with the capacity to promote the production of pro-inflammatory cytokines (IL-6, IL-1β) (164–169) while concomitantly reducing excessive inflammation by inducing anti-inflammatory cytokines such as IL-10 (170–172) Inhibition of FOXO by Akt leads to the inhibition of autophagy, production of antioxidant enzyme and apoptosis, thereby promoting a state of chronic inflammation (173, 174). The hyperactivation of this pathway has been implicated in the development of various autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease and asthma (175, 176).
In contrast to the previously mentioned pathways, AMPK and cAMP/PKA function as a significant anti-inflammatory regulators (177–181) by inhibiting the activation of NF-κB (a potent pro-inflammatory transcription factor) (182, 183). PKA is also a potent inflammation resolving factor being reduced in acute phase of inflammation by increased PDE4 activity (breaking cAMP to AMP) (184) and being active when the inflammation finishes thus activating other resolving molecules (184, 185) and activating phagosomes (186). Additionally, both pathways enhance mitochondrial function and reduce levels of reactive oxygen species (ROS) levels (187–191) and reduce oxidative stress (187, 192).
AMPK promotes autophagy (193, 194) but influence of PKA depends on the metabolic content (195). A deficiency in AMPK has been associated with chronic inflammatory conditions including obesity and type 2 diabetes (196, 197) and deficiency of PKA is associated with multiple diseases including neurodegeneration, hypertension, type 2 diabetes, depression and anxiety (183, 198–201).
The equilibrium between the activities of the aforementioned pathways is pivotal in the modulation of inflammation. From the perspective of autophagy, certain pathways are stimulatory, while others are inhibitory, depending on the metabolic context. Consequently, an imbalance in the ratio of these pathways, resulting in excessive inhibition of autophagy, may serve as a significant catalyst for the transition of the regulatory system toward a state of chronic or hyperinflammation.
3.3 Concentration dependent roles of NO
Nitric oxide (NO) produced by iNOS (inducible nitric oxide synthase) exerts both pro- and anti-inflammatory effects, depending on its concentration and metabolic context. Pro-inflammatory cytokines such as IL-1, IL-6, IFN-γ, TNF-α and pro-inflammatory transcription factors: NF-κB (202), AP-1 (203), NFAT (204), STAT1 (205), and HIF-1α (206) induce the expression of iNOS and NO production (202). Subsequently, NO can then interact with O2-, to form toxic peroxynitrite (ONOO-). NO is known to have anti-inflammatory properties, mainly through the mechanisms of cyclic GMP (cGMP) (207–209) and S-nitrosylation of proteins (210). This effect is also partly related to the inhibition of NF-κB by cGMP (211). Another anti-inflammatory mechanism involves the induction of apoptosis in activated macrophages leading to a reduction in the inflammatory response (212, 213). Conversely, NO has been shown to activate inflammation by activating MAPK kinases (ERK, JNK, p38) through the induction of oxidative stress (e.g., by peroxynitrite) (214). In addition, excess NO has been demonstrated to exacerbate oxidative stress through nitrosylation and subsequent inhibition of antioxidant enzymes including superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase (CAT) (215). Furthermore, NO can also nitrosylate specific proteins within the endoplasmic reticulum, resulting in the accumulation of misfolded proteins, ER stress and subsequent NOX activation (216). The final NO regulatory effect appears to be concentration dependent and should be analyzed with caution in this context.
3.4 The regulatory role of autophagy and Nrf2
The main mechanisms that control inflammatory process and prevent self-destruction are autophagy and Nrf2. Autophagy removes abnormal proteins that induce inflammation through various mechanisms. These abnormal proteins include both viral proteins and abnormally folded cellular proteins resulting from ER stress. It is important to note that autophagy is a complex process involving more than 100 different proteins, and the block or inhibition of even one of these proteins by a pathogen protein can contribute to the loss of cellular regulatory capacity for autophagy. An imbalance in autophagy regulation can lead to its inhibition contributing to chronic and severe inflammation (217–219). This observation is significant when considered in the context of the fact that many intracellular pathogens chronically inhibit autophagy, thereby preventing their complete clearance from the cell (220–225).
Regulation of autophagy is closely linked to cytokines, ROS, NO, HIF-1α and Cai2+. Cytokines and ROS activate autophagy via pathways such as JNK (226), p38 (139, 227–230) and AMPK (231–233). However, in the context of inflammation, there is a delicate balance between the activatory and inhibitory influences of diverse signaling pathways on the autophagy process. The ERK1/2 pathway (234, 235) and NF-κB represent the primary inhibitory elements (236, 237). NF-κB functions in a positive feedback loop with the proautophagic p38 pathway (228–230, 238–240), thereby establishing a balance in intensity of autophagy.
Intracellular Ca2+ works as autophagy activator. Elevated calcium levels activate AMPK and inhibit mTOR, thereby promoting autophagy (241). In addition, Ca2+-activated calcineurin activates lysosomal biogenesis by phosphorylating TFEB (transcription factor EB).
HIF-1α also modulates autophagy in response to hypoxia by activating autophagy genes. In general, HIF-1α activates the autophagy (242, 243) and autophagy reduces the HIF-1α activity by degrading this molecule (244). NO exerts a dual regulatory effect on autophagy, functioning as both an activator and an inhibitor depending on its concentration and the specific cellular context. At physiological levels, NO promotes autophagy maintaining homeostasis. Moderate levels of NO stimulate autophagy mainly through the AMPK-mTOR pathway (245). In addition, NO increases ROS production by activating pathways such as JNK, which further enhances autophagic responses under conditions of oxidative stress. However, in pathological conditions such as chronic inflammation or excessive oxidative stress, excessive NO has been shown to inhibit autophagy via S-nitrosylation of key autophagic proteins such as ATG4, which impairs autophagosomal membrane elongation (246). NO has also been observed to nitrosylate JNK1 (blocking its autophagy-activating function) (247) and IKKβ, reducing AMPK phosphorylation while activating mTORC1, leading to autophagy suppression (248, 249). In summary, excess NO can significantly contribute to the maintenance of chronic inflammation and the persistence of chronic pathogens within the cell by both inhibiting autophagy and reducing Nrf2-induced antioxidant response. Chronic inflammation further exacerbates autophagy inhibition through sustained activation of NF-κB, which suppresses AMPK and Nrf2 and activates mTOR.
Nrf2 is a key transcription factor responsible for protecting cells from oxidative stress and regulating inflammatory processes. Its activity relies on complex feedbacks with NF-κB, ROS, NO, Ca²⁺ and HIF-1α, making it central to the antioxidant and anti-inflammatory system. Nrf2 is regulated by the protein Keap1, which under healthy conditions binds Nrf2 and targets it for degradation by proteasomes. Under conditions of oxidative stress, reactive oxygen species (ROS) and nitrogen (NO) modify cysteine residues in Keap1, leading to the release of Nrf2 and its translocation to the nucleus (250–253). Nrf2 then activates genes responsible for the production of antioxidant enzymes such as heme oxygenase 1 (HO-1), glutathione S-transferase (GST) and NAD(P)H:quinone oxidoreductase 1 (NQO1) which reduce ROS levels, thereby restoring redox balance. However, at excessive levels of NO and ONOO-, Nrf2-induced enzymes (e.g., HO-1, Mn-SOD, catalase, peroxiredoxin II E, glutathione peroxidase-thioredoxin reductase) are damaged or deactivated by nitrosylation, leading to a breakdown of the antioxidant barrier and promoting severe or chronic inflammation (250, 254–258).
Nrf2 also has an inhibitory effect on the cytokine system by blocking the activation of NF-κB and both ROS reduction and HO-1 production mediate this effect (259, 260). Conversely, NF-κB inhibits Nrf2 (261, 262), creating a specific double-negative regulatory system that enhances either the oxidative-inflammatory or antioxidant state, depending on which regulatory scale prevails. In chronic inflammation, decreased Nrf2 and increased NF-κB activity are often observed, leading to a loss of balance between pro- and anti-inflammatory mechanisms.
Intracellular calcium can activate various signaling pathways such as ERK1/2 and p38, which can increase Nrf2 activity by phosphorylating Nrf2 and increasing its stability (263, 264). Nrf2 also affects the regulation of Ca²⁺ in cells, reducing ER stress by increasing the expression of glutathione peroxidase GPx8 in the ER (265), reducing Cai2+ by modulating calcium channels (e.g. TRPC, RyR) (266) and increasing the activity of the calcium pump SERCA (266). In summary, Nrf2 and Cai2+ work in the classical negative coupling, where Cai2+ induces Nrf2 and Nrf2 reduces the calcium and ER stress.
In conclusion, Nrf2 is a key regulator of redox balance and inflammation whose function depends on dynamic interactions with ROS, NO, HIF-1α and Ca²⁺. Disruption of Nrf2 activation leads to an escalation of oxidative stress and inflammation. Interventions aimed at stabilizing Nrf2 are therefore an important therapeutic strategy in inflammatory and degenerative diseases.
4 Metabolic alterations generated by SARS-CoV-2
The SARS-CoV-2 genome encodes 29 proteins: 16 non-structural proteins (NSP1-16), 4 structural proteins: envelope protein [E], membrane protein [M], spike protein [S] and nucleocapsid protein [N], and 9 accessory proteins: ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10. A number of mechanisms have been described by which the virus as a whole or its individual proteins reprogram the cell’s metabolism to make it easier for itself to replicate and harder for the host to fight off. The mechanisms largely involve interference with and disruption of the feedback loops described above, leading to hyperinflammation. Key effects include mitochondrial dysfunction, inflammatory activation, inhibition of Nrf2, induction of calcium stress, inhibition of autophagy and induction of HIF-1α. A summary of all viral protein interactions with host cell metabolism discussed in the following subsections is shown in Figure 4. Assuming that a significant proportion of LCS patients are those with active chronic viral presence in cells, the following analysis which primarily considers the acute infection state may also be useful in determining treatment strategies for LCS patients.
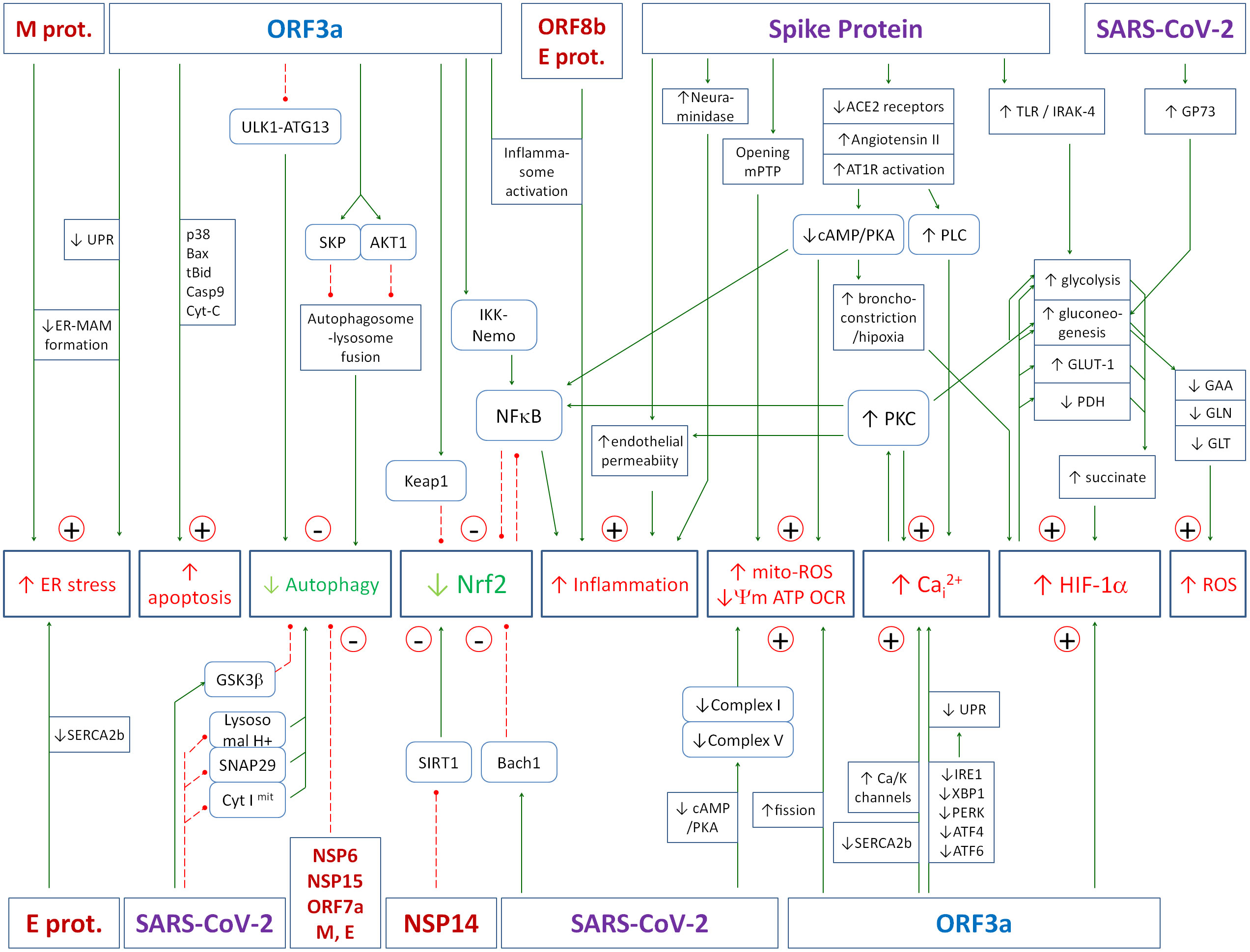
Figure 4. The figure summarizes the effects of the SARS-CoV-2 virus or its individual proteins on key elements of the cell’s antiviral defense. The numerous molecular mechanisms that contribute to the high pathogenicity of the virus, the generation of a hyperinflammatory state, and the propensity to progress to a state of active chronic infection are presented. Details of the various interactions are described in the text of the article. Green solid arrows - activation, red dashed lines - inhibition.
4.1 Upregulation of the angiotensin 1 receptor
The key feature of the S1 protein is its ability to bind to the ACE2 (angiotensin converting enzyme) protein on the surface of cells, allowing it to enter the interior. ACE2 cleaves angiotensin II (ATII) into angiotensin (1-7), a hormone with opposite effects to angiotensin II (267, 268). During severe COVID-19 infection, ACE2 is blocked by the virus and the amount of this protein on the cell surface is reduced, leading to an increase in ATII and a decrease in its antagonist, angiotensin (1-7) (268). Excess ATII is an important factor in initiating a cascade of regulatory abnormalities in the cell (268–271). It acts through the angiotensin receptors AT1, AT2, AT3 and AT4, which are upregulated in COVID-19 (267).
AT receptors belong to the group of GPCRs (G-protein coupled receptors). Two main signaling pathways are involved in GPCR signaling: cAMP and phosphatidylinositol signaling (272). Depending on the type of cell, different types of GPCR receptors can be found on the surface of the cell, that trigger a specific response inside the cell. Most GPCR receptors are capable of activating more than one type of G protein, especially when the receptor is over-activated. This can happen, for example, when the concentration of ATII in the blood increases excessively. Activation of the AT1 receptor causes, among other things, activation of phospholipase C (PLC) and inhibition of adenylyl cyclase (AC) and cAMP/PKA pathway (273), which contribute to hyperinflammation in the course of COVID-19 (274). Activation of PLC leads to an increase in cytosolic Ca2+ concentration. Another effect of AT1 receptor overstimulation is the activation of NOX5, NF-κB, IL-6 and STAT3, which exacerbates oxidative stress and causes overproduction of pro-inflammatory cytokines (including IL-6, IL-1β and TNF-α) (268, 275–277).
Downregulation of PKA has several important effects on cellular metabolism. It causes deregulation of mitochondria by increasing electron leakage from cytochromes I and IV (188, 189). It also destabilizes the structure and decreases ATP production in cytochrome V (ATP synthase) (190, 191) and removes the regulatory inhibitory effect of high ATP levels on cytochrome IV (189), which is the natural negative feedback that decreases further ATP production and slows electron flow without its leakage. All of these effects contribute to the decrease in the mitochondrial membrane potential. It is also questionable whether the increased concentration of succinate, as an effect of HIF-1α induction, generates in such conditions the reverse flow of electrons from cytochrome II to I, generating the strong electron leakage in cytochrome I, as described in (188).
Next, the cAMP/PKA pathway causes relaxation of bronchial smooth muscle cells (278), thus, inhibition of AC by excess ATII promotes the contractile state of bronchial smooth muscle. Another effect of the downregulation of the cAMP/PKA pathway is the enhancement of inflammation and the increase of oxidative stress. The anti-inflammatory mechanism is that the CREB-CBP complex formed by CREB phosphorylation by PKA can result in the dissociation of the NF-κB-CBP complex, which blocks the action of NF-κB (183). PKA also inhibits the activation of the pro-inflammatory ERK, AKT, STAT3 and NF-κB pathways through phosphorylation and inhibition of the TNFR1 receptor (185), so down-regulation of PKA activity increases the strength of the coupling between inflammation and oxidative stress. As cAMP/PKA pathway is the important factor initiating the resolution of inflammation (185, 279), the slow recovery process after the disease may also depend on its downregulation. It should be noted that the inhibition of this pathway occurs in the early stage of the disease, so, it may be the important factor initiating and then sustaining the inflammatory-oxidative cascade.
4.2 Upregulation of calcium stress by SARS-CoV-2
There are several ways in which SARS-CoV-2 can increase cytosolic calcium. As mentioned above, the first is stimulation of the AT1 receptor by angiotensin II which further activates phospholipase C and its activation leads to an increase in cytosolic Ca2+ concentration. Increased Cai2+ activates the calmodulin pathway, which has a number of downstream effects, one of which is the activation of the proinflammatory transcription factor NFAT (274). The other important effect of Cai2+ is the activation of protein kinase C (PKC). Activation of PKC can lead to many metabolic changes that are involved in the severity of COVID-19. PKC contributes to the activation of NF-κB (280), which subsequently activates HIF-1α, inhibits Nrf2, and inhibits autophagy. PKC may also contribute to the activation of glycogenolysis and gluconeogenesis in the liver (281), which, in conjunction with HIF-1α overactivation, leads to increased glycolysis and increased succinate/fumarate concentration. Another role of PKC is to mediate the stimulation of proinflammatory cyclooxygenase-2 expression by viral spike protein through both calcium-dependent (PKCα) and calcium-independent mechanisms (PKCϵ, NF-κB, ERK, PI3K) (282).
4.3 Creating potassium/calcium channels by ORF3a
One of the important capabilities of the viral ORF3a protein is the ability to create ion channels that decrease the concentration of Ki+ and increase Cai2+ in the cell (3). This is particularly dangerous in the context of reduced energy production, as more energy amount is required to maintain the ion gradients. Decreased Ki+ concentration leads to decreased membrane potential, which can activate voltage-dependent calcium channels (VDCCc) (283, 284), which further increases Cai2+, and one of the possible effects is the activation of calcium-activated potassium channels (KCa), which further decreases Ki+ concentration (285). In the normal state, this process attempts to increase the membrane potential and reduce calcium influx, but in the pathological state, it drives cellular pathology. The above mechanism is postulated to induce apoptosis and necrosis (286, 287).
4.4 Reticuloendoplasmic stress caused by ORF3a and M proteins
The other pathway of calcium homeostasis disruption presented by Lee et al. (4) is the ability of SARS-CoV-2 ORF3a and M proteins to disrupt the ER membrane and mitochondrial-associated membrane (MAM) formation. This study demonstrated that ORF3a and M proteins affect the proteomic landscape of the ER. It was also shown that ORF3a-APEX2 constructs used for electron microscopy imaging significantly increased the formation of cubic membranes (CM), also known as convoluted membrane structures. Notably, CM structures were also observed in coronavirus-infected cells (288, 289). In the case of M protein, the ER was clearly disrupted in M-expressing cells and appeared to curl into whorl patterns (also referred to as aggresomes). Their results suggested that the ORF3a and M proteins of SARS-CoV-2 may be major contributors to the formation of ER-derived neoorganelles.
The interactome analysis for these two viral proteins also showed that a large proportion of plasma membrane and endoplasmic reticulum membrane proteins were present in both the ORF3a and M interactomes. This suggests that the function of the ER and especially the MAM is significantly disrupted by these proteins. The disruption of the ER and induction of the unfolded protein response (UPR) by SARS-CoV-2 has been described by several authors (5, 6). The detailed mechanisms include depletion of CaER2+ concentration and increase in Cai2+ (290, 291) and one of the mechanisms generating it is a direct interaction between E-protein and SERCA2b pump resulting in a decrease in SERCA-mediated ER Ca2+ reloading (7). It has also been postulated that ORF3a is capable of generating calcium channels that could reduce the Ca2+ gradient across the ER membrane (292).
4.5 Loss of endothelial monolayer integrity caused by Spike protein
Loss of the pulmonary endothelial barrier is one of the most dangerous effects caused by SARS-CoV-2 and several pathways are involved in this effect, most of which are related to S1 protein activity. The first pathway is increased PKC activity which can increase endothelial permeability (8). Even low levels of PKC activation can reverse cell chirality through PI3K/AKT signaling and alter the organization of junctional proteins between cells, leading to significant changes in endothelial permeability that may promote increased inflammation and pulmonary edema (8). The second pathway is the inhibition of adenylyl cyclase (AC) by ATII, one of the effects of which is bronchoconstriction contributing to hypoxia (278). In the experiment by Jana et al. (9), incubation of S1 protein with human pulmonary endothelial arterial cells resulted in the disruption of endothelial barrier function, an increase in the levels of numerous inflammatory molecules (VCAM-1, ICAM-1, IL-1β, CCL5, CXCL10), an increase in mitochondrial reactive oxygen species (ROS) and a slight increase in glycolytic reserve capacity. The effect of S1 was enhanced by the hypoxia that occurs during severe COVID-19 due to pulmonary hyperinflammation and reduced blood oxygen saturation, as hypoxia increases the ability of S1 to reduce ACE2 receptor levels.
S1 protein-mediated loss of endothelial monolayer integrity was also observed experimentally by Buzhdygan et al. using an advanced 3D microfluidic human blood-brain barrier model (10). They also found similar pro-inflammatory cytokine responses induced by spike protein, which are thought to contribute to the loss of membrane integrity (10). ICAM-1 and VCAM-1, key players in immune cell transendothelial migration following inflammatory challenge, were observed to be upregulated by S1 and associated with reduced barrier integrity (9).
4.6 Neuraminidase activity of the spike protein
One of the likely S1 activities is the enzymatic activity of neuraminidase (NEU) (293). Zhang et al. (293) used advanced 3D techniques to describe the similarity of S1-protein to the neuraminidase from influenza A and B viruses. Neuraminidase is the enzyme that cleaves sialic acid on the surface of leukocytes, which is the key stimulus for neutrophil activation to activate inflammation, cytokine storm, NOX-induced ROS production and neutrophil extracellular traps (NETs) formation (293).
In severe systemic inflammatory responses such as sepsis and COVID-19, neutrophils are central to organ damage. Hyperactivated neutrophils infiltrate vital organs and release cytotoxic molecules such as proteases, cytokines, ROS and NETs (294, 295). Although inflammatory mediators are essential for fighting infection, they can also damage the host cells (296).
Neuraminidase is one of the major activators of neutrophils, so neuraminidase inhibitors can be treated as important factors in inhibiting the inflammatory cascade. Formiga et al. (297) demonstrated the role of neuraminidase-1 (NEU1) and matrix metalloproteinase-9 (MMP-9) in triggering the hyperinflammatory state in COVID-19 patients. Lipopolysaccharide (LPS) has been shown to induce host membrane‐associated NEU activation in murine or human macrophages and dendritic cells (298). Upon LPS binding to toll-like receptor 4 (TLR4), matrix metalloproteinase 9 (MMP-9) induces NEU activity and contributes to the NF-κB-induced response of macrophages, suggesting a role for these enzymes in cell activation (298, 299). In another paper, Formiga et al. (300) showed that in vitro treatment of whole blood with the viral neuraminidase inhibitors oseltamivir or zanamivir, inhibited the activity of human neuraminidases as well as the exacerbated neutrophil response. These drugs also reduced neutrophil activation and increased survival in mice.
The neuraminidase inhibitor (NI) clinical trial presented by Wu et al. (301) showed that NI treatment reduced the mortality rate (5.7% vs. 10.3%) and the critical illness conversion rate (14.1% vs. 19.7%) compared with the non-NI group. They also showed that levels of N-acetylneuraminic acid and neuraminidase (predominantly the NEU3 isoform) were elevated in COVID-19 subjects and recovered 1 month after infection, suggesting increased desialylation in COVID-19 patients.
4.7 SARS-CoV-2 and mitochondria
Mitochondrial dysfunction, subsequent pathogenesis and multi-organ failure have been associated with COVID-19 infection (302). Functional mitochondrial analysis of COVID-19 peripheral blood mononuclear cells (PBMCs) revealed mitochondrial dysfunction, increased glycolysis and high mitokine levels (303). The presence of the oxidative stress in mitochondria in SARS-CoV-2 infected cells was confirmed by increased carbonyl content in the mitochondrial fractions in SARS-CoV-2-infected lung tissue lysates (304), suggesting that SARS-CoV-2 infection induces an oxidative stress environment in the lung cells in general, which also affects mitochondrial proteins. A reduction in mitochondrial respiration was also observed, as indicated by a loss of oxygen consumption rate (OCR) in isolated mitochondria from SARS-CoV-2-infected hamster lungs. Proteomic analysis also revealed specific deficits in the mitochondrial ATP synthase (Atp5a1) within complex V and in the ATP/ADP translocase (Slc25a4) (304). The other determinant of mitochondrial oxidative stress was the increased staining for 4-hydroxynonenal (4-HNE), a major end-product of lipid peroxidation (304).
SARS-CoV-2 infection also causes a reduction in the mitochondrial electron flow at several cytochrome complexes indicating a general virus-induced impairment of mitochondrial function. A lower basal OCR in the presence of pyruvate and malate was observed, indicating impaired complex I activity in the SARS-CoV-2-infected patients (304). In a complex V assay, a significant decrease in the ATP synthesis was observed in the SARS-CoV-2-infected lung mitochondria suggesting a possible impairment of the complex V ATP synthesis function (304).
The other important parameter of mitochondrial energy production is the mitochondrial membrane potential ΔΨm. The discussion of the energy production in mitochondria and the relationships between ΔΨm, Cai2+ and Ki+ concentrations, opening of mitochondrial permeability transition pores (mPTPs), and oxidative stress has been presented by Michalak et al. (305). In short, intracellular calcium and ROS cause the opening of mPTPs, which causes the efflux of protons from the intermembrane space and the decrease of ΔΨm. This leads to decreased energy production which may contribute to decreased Cai2+ removal from the cytoplasm and increased Cai2+, as this process is highly energy consuming.
Impaired complex I activity in the SARS-CoV-2 infected patients (304) may play a protective role to some extent, as it reduces the hydrogen entry into the ETC. However, electron leakage can vary depending on the detailed metabolic context. PKA inhibition caused by angiotensin II increases in electron leakage (188). On the other hand, metformin, a weak complex I inhibitor, reduces electron leakage in COVID-19 patients (306–311). However, other metabolic properties of metformin may also be involved in this protection (306, 307).
4.8 Disrupting the mitochondria by ORFs
ORF3a has an important disruptive effect on the mitochondrial function. ORF3a-induced ROS production and cell death appears to be a highly conserved activity (3, 312, 313), as these effects are observed in both fission yeast and human A549 and 293 T cells (3). However, the detailed molecular mechanisms of ORF3a-induced ROS production are not fully understood. ORF3a-mediated apoptosis has been described to be both mitochondria-dependent (287) and independent (286, 314). Exemples of mito-dependent proapoptotic effects include the activation of caspase-8, the increase in caspase-9 levels and release of cytochrome c from the mitochondrial membrane. Increased Bax oligomerization, Bid to tBid conversion, higher p53 levels and increased p38 activity mediate these effects (315–318).
Another study showed that ORF3a promotes mitochondrial fission, which can lead to the destabilization of normal physiological processes within the mitochondria. Increased fission is associated with impaired mitochondrial function and altered energy production (11). It has also been shown that ORF3a, ORF9b, ORF9c and ORF10 induce significant mitochondrial and metabolic reprogramming in A549 lung epithelial cells. While all four ORFs caused mitochondrial fragmentation and altered mitochondrial function, only ORF3a and ORF9c induced a marked structural change in mitochondrial cristae (11). ORF9b, ORF9c and ORF10 induced largely overlapping transcriptomes. In contrast, ORF3a induced a distinct transcriptome, including the downregulation of numerous genes for proteins with critical mitochondrial functions and morphology (11). Notably, reduced amino acid metabolism and increased metabolism of some lipids was induced by ORF3a (11).
4.9 Inhibition of Nrf2 by SARS-CoV-2
The Nrf2-induced cellular antioxidant response is an important element in the overall balance of the cellular antiviral response. In viral infections, oxidative cellular damage may be associated with inhibition of the Nrf2 pathway (319). The expression of Nrf2-related antioxidant genes has been shown to be suppressed in biopsies from COVID-19 patients (320). Rodrigues et al. (321) showed reduced nuclear accumulation of Nrf2 and expression of Nrf2 targeted genes in endothelial cells exposed to COVID-19 serum. In addition, these cells showed increased expression of Bach-1, a negative regulator of Nrf2 that competes with Nrf2 for DNA binding. All events were prevented by tocilizumab, the IL-6 receptor blocker, suggesting that IL-6 is the major cytokine involved in the inhibition of Nrf2-induced antioxidant defense (321). However, more complex regulatory mechanisms may have led to this effect as presented in Figure 1. Further research has shown that the inhibition of Nrf2 is due, at least in part, to the activity of the viral protein ORF3a. ORF3a promotes degradation of Nrf2 by recruiting Keap1, thereby attenuating cellular resistance to oxidative stress and facilitating ferroptotic cell death (322). Downregulation of Nrf2 and its dependent genes by SARS-CoV-2 infection exacerbates pulmonary inflammation and disease and therefore activation of Nrf2 appears to be an important element of the therapeutic approach during both chronic and acute SARS-CoV-2 infection (323, 324). In another study Shilei et al. showed that the SARS-CoV-2 NSP14 protein interacts with the catalytic domain of the NAD-dependent deacetylase sirtuin 1 (SIRT1) and inhibits its ability to activate the Nrf2/HO-1 pathway (325).
In conclusion, Nrf2 is the major down-regulator of most of the positive couplings that cause hyperinflammation and the inhibition of Nrf2 by viral proteins may be an important factor that amplifies the excessive oxidative stress and inflammatory couplings that cause the severe course of the disease or sustain the chronic state. In particular, reduced Nrf2 activity contributes to increased NF-κB activity and thus to the inhibition of autophagy. A reduction in Nrf2 activity is therefore an important pathway leading to an excessive positive feedback between cytokines, NOX, Cai2+ and HIF-1α, resulting in their excessive levels, leading to a cytokine storm and a state of hyperinflammation. In the case of chronic active SARS-CoV-2, it contributes to the chronic state of LCS.
4.10 Activation of HIF-1α
HIF-1α plays a important role in the progression of SARS-CoV-2 infection by influencing metabolism, inflammatory responses, and viral replication. The viral protein capable of activating HIF-1α is ORF3a (313). Accumulation of HIF-1α results from both increased expression and inhibited proteasomal degradation (326). The primary cause of HIF-1α activation is hypoxia due to disruption of the alveolar-capillary barrier, leading to pulmonary edema, immune cell infiltration, and impaired gas exchange (9, 327). This damage can be induced by the S1 subunit of the viral spike protein, even in the absence of other viral components (9, 304). In addition, activation of AT1 receptors increases intracellular calcium, leading to bronchoconstriction and further hypoxia. The pathways involved are inhibition of adenylyl cyclase (328) and activation of phospholipase C, which increases Cai2+ and then activates PKC.
HIF-1α drives glucose metabolism by promoting glycolysis and gluconeogenesis and these processes are upregulated during SARS-CoV-2 infection (307, 329–337). Glycolysis is enhanced by upregulation of glucose transporters (GLUT-1) and activation of hexokinase, pyruvate kinase, lactate dehydrogenase and aldolase (304, 338–341). Gluconeogenesis is increased by hepatocyte infection, activation of enzymes such as PEPCK, and increased levels of GP73 (335, 336). These processes contribute to hyperglycemia and the accumulation of metabolites such as lactate and succinate, which further enhance HIF-1α activation and inflammation. It is worth noting that the natural inhibitor of glycolysis is citrate, so citrate can be considered as a natural support in the therapy of COVID-19 and other viruses that increase glycolysis.
Stabilization of HIF-1α also promotes viral replication. Proteins such as ORF3a enhance HIF-1α activity, leading to increased viral replication and cytokine production. Excess lactate from glycolysis inhibits mitochondrial antiviral signaling (MAVS), further facilitating viral replication (331). This creates a feedback loop in which HIF-1α enhances the effects of SARS-CoV-2 within the host. Targeting HIF-1α has therapeutic potential, but general inhibition should be avoided because HIF-1α has a the protective effect on mitochondria by reducing mitochondrial electron leakage and mito-stress (329). Instead, therapies should focus on specific effects, such as glucose metabolism and inflammatory responses, driven by HIF-1α activation.
4.11 Inhibition of autophagy by ORF3a, NSP6, ORF7a, NSP15, M and E proteins
The final metabolic effect of SARS-CoV-2 discussed here is the inhibition of autophagy (16). Recent studies have shown that different SARS-CoV-2 proteins inhibit autophagy through multiple pathways (16). NSP6 inhibits lysosomal acidification, the formation of acidic autolysosomes, and the formation of the hybrid pre-autophagosomal structure (HyPAS) - a precursor structure in the autophagy pathway that integrates various components from the ER, Golgi apparatus, and endosomes. Next, NSP6 supports autophagosome formation but inhibits autophagosome maturation (79–81). ORF3a inhibits the formation of acidic autolysosomes by sequestration of the HOPS component VPS39. When VPS39 is sequestered by ORF3a, the fusion process of the autophagosome with the lysosome is disrupted. Next, ORF3a inhibits PI3K complex II assembly by sequestrating its component UVRAG resulting in impaired endosome maturation. Finally, ORF3a inhibits lysosomal acidification and promotes lysosomal exocytosis, facilitating viral release (18, 82, 83). ORF7a inhibits lysosomal acidification and the formation of acidic autolysosomes (84). NSP15 inhibits autophagy initiation (84), M protein inhibits autolysosome formation (18, 84) and E protein inhibits autophagosome maturation and autolysosome formation (84).
The other viral proteins that interfere with antiviral defense through autophagy are ORF8 and NSP13. ORF8 promotes autophagic degradation of MHC-I (342), leading to a reduction in the amount of MHC-I on the surface of infected cells. As a result, infected cells are no longer recognized by CD8⁺ T cells, which impairs their elimination. NSP13 promotes autophagic degradation of TBK1 (343). TBK1 is a key activator of IRF3, which is required for the expression of the type I antiviral interferons IFN-α and IFN-β.
Interestingly, ORF3a of SARS-CoV-2, but not SARS-CoV has the ability to block autophagy (18, 344). This is an important piece of information that points to the critical role of autophagy inhibition in exacerbating the severity of SARS-CoV-2 compared to SARS-CoV infection.
4.12 Upregulating the inflammation
All of the discussed effects of SRAS-CoV-2 on host cell metabolism generally contribute to increased levels of inflammation. However, direct activating effects have also been observed. Nie et al. (345) investigated SARS-CoV-2 genes and found that ORF3a activates the proinflammatory NF-κB pathway by interacting with IKK-β (beta subunit of the IκB kinase complex) and NEMO (also known as IKK-γ) and by enhancing the interaction of IKK-β-NEMO, thereby positively regulating NF-κB activity. Other viral proteins are also capable of activating inflammation. ORF3a, ORF8b and E proteins have been reported to enhance activation of the inflammasome, leading to increased secretion of IL-1 and IL-18, and subsequent pathological changes associated with inflammation. Similarly, the NSP9 and NSP10 proteins of SARS-CoV-2 induce overproduction of IL-6 and IL-8, which are the major causes of the cytokine storm in COVID-19 patients (346).
4.13 Inhibition of Interferon production
Interferon I is one of the main lines of antiviral defense. Activation of IFN-I production is a complex process regulated by multiple signaling pathways that detect the presence of viral RNA or DNA in cells. Interferon regulatory factors 3 and 7 (IRF3, IRF7) are key transcription factors that regulate interferon production. The main signaling pathways are RIG-I-like receptors (RLRs), Toll-like receptors (TLRs) and stimulator of interferon gene (STING). Among others NEMO (347, 348), MAVS (349, 350), and NLRP6 (351) modulate type I IFN production and inflammasome activation (352–355), whereas TRIM18 and TRIM29 suppress macrophage activation and IFN production (354, 356).
SARS-CoV-2 has several proteins which act on the type I IFN pathway. The proteins NSP1 (357, 358), NSP3 (359), NSP5 (357), NSP6 (357), NSP10 (360), NSP12 (359), NSP15 (43, 357, 358), NSP16 (358, 361), ORF3b (362, 363), ORF6 (357), ORF7b (357), ORF9b (364), ORF10 (357, 365) M (366) and N proteins (367), act on the type I IFN pathway either by inhibiting transcription or by acting on effector mechanisms. The inhibition of interferon production and the delayed host response to the presence of the virus in cells leads to significant molecular perturbations, as in the case of NSP15, which causes the upregulation of over 2800 genes including networks associated with the activation the unfolded protein response (UPR) and the proinflammatory response associated with viral pathogenesis (43).
The above studies suggest a completely different role for viral proteins during the incubation period of the disease, when the immune response is inhibited, as opposed to the late phase of the disease, when there is hyperactivation of the immune system and a cytokine storm, as described in the earlier sections of this article. It also suggests the need for different therapeutic targets during the period of potential disease incubation and during the period of its late, severe course. With regard to LongCOVID-19, it is necessary to study to what extent in this phase it is necessary to stimulate the production of interferons and activate inflammatory processes, and to what extent it is necessary to inhibit them. It is not excluded that the optimal therapeutic strategy might be the simultaneous activation of interferon I production pathways, e.g. by blocking TRIM18 or TRIM29 (354, 356), to improve the detection of virus-infected cells and to inhibit the inflammatory response, e.g. by inhibiting NF-κB, which may help to restore proper Nrf2 and autophagy activity.
5 Summary
Analysis of the interaction of different viral proteins on cellular metabolism generally indicates a synergistic effect in increasing inflammation, oxidative stress, nitrosative stress, mitochondrial stress and calcium stress in the cell which explains the tendency of the virus to generate the hyperinflammatory state. Reciprocal positive feedbacks between these elements enhance the pro-inflammatory effect of the virus. In addition, inhibition of autophagy and Nrf2 disrupts two important regulatory mechanisms that prevent excessive inflammation and all of the above stresses. The number of couplings disrupted by viral proteins is so large that eradication of the infection is sometimes beyond the capabilities of the regulatory system. Understanding, how these couplings work, is essential for planning therapeutic strategies not only for COVID-19 but also for many other infections, both acute and chronic.
Analysis of the feedback loops shows that the regulatory system is extremely complex. The multitude of positive feedback loops suggests that treatment strategies with a single drug acting on a single therapeutic pathway or transcription factor have little chance of bringing the patient out of hyperinflammatory or chronic state, as the remaining active positive feedback loops continue to drive inflammation and oxidative stress, leading all the time, but only slightly weaker, to the destruction of the system. Analyzing this system, it can be expected that only the alleviation of at least some of the positive feedbacks can bring about a rapid improvement in the patient’s health. According to the author, a special attention should be paid to the activation of autophagy and Nrf2. If autophagy is blocked, the cell has no alternative way to get rid of viruses and misfolded proteins. Its activation seems to be essential for therapeutic success. Activation of Nrf2 also seems to be an important element as it can lead to inhibition of NF-κB and reduction of inflammation, which is particularly important in a state of hyperinflammation. Another underestimated therapeutic approach, according to the author, is the inhibition of glycolysis, e.g. with citric acid, a natural glycolysis inhibitor (368). This could be an important natural element to protect mitochondria from electron leakage, mito-stress, succinate accumulation and thus reduce HIF-1α activation. Research is also needed into the use of drugs or herbs that reduce calcium and reticuloendoplasmic stress, as these are significantly involved in the development of hyperinflammation or LCS. In particular, activation of the cAMP/PKA pathway, which is blocked by ATII, is worth considering as a therapeutic target, as it is likely to be an important element generating mitochondrial stress.
A more complex case is LCS caused by the development of autoimmune processes as a result of COVID-19 infection. The profile of metabolic abnormalities induced by different autoantibodies can vary considerably from patient to patient. In addition, conditions in which SARS-CoV-2 has induced chronic active infection with other pathogens (e.g. EBV) may require the use of appropriate diagnostic tests and individualized therapies depending on the type of pathogen. For example, EBV and cytomegalovirus are capable of causing coagulopathies, including disseminated intravascular coagulation (DIC) (65–67). However, it appears that the profile of metabolic disturbances in such cases will be similar to that of COVID-19, as the process of pathogen persistence is very often associated with the blockade of autophagy by these pathogens (220–225), which further leads to the induction of oxidative, nitrosative, Cai2+ and ER stress. The concomitant induction of NF-κB leads to the inhibition of Nrf2.
An important conclusion derived from the analysis of interdependent feedback mechanisms is that influencing a single component of the system inevitably triggers changes throughout the entire network. This highlights the need to differentiate between the direct effects of a particular influence and the indirect consequences that propagate through other interconnected elements.
Looking ahead, future research should aim to mathematically characterize each connection within the network graph by formulating appropriate differential equations or functional relationships. A representative dynamic model utilizing ordinary differential equations, which captures the response of Nrf2, Keap1, Srxn1, and GSH under oxidative stress, was proposed by Hiemstra et al. (369). Once each interaction is quantitatively described, it will become feasible to construct a comprehensive system of equations capable of simulating the behavior of the network, both under normal conditions and in response to disruptive agents such as viral proteins or pharmacological interventions. This, in turn, would provide a foundation for optimizing therapeutic strategies through mathematical modeling.
To achieve this level of system complexity, future collaboration with experts in computational modeling and systems biology will be essential. Their expertise in analyzing the self-regulatory dynamics of multidimensional systems will be critical in developing accurate models of metabolic regulation.
Author contributions
KM: Conceptualization, Formal Analysis, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. AM: Visualization, Writing – original draft. AB-K: Writing – review & editing, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
A20: Tumor necrosis factor alpha-induced protein 3
ACE2: Angiotensin-converting enzyme 2
AKT1: RAC-alpha serine/threonine-protein kinase
AMPK: AMP-activated protein kinase
AP-1: Activator protein 1
AT1 receptor: Angiotensin II type 1 receptor
ATII: Angiotensin II
ATF2: Activating Transcription Factor 2
Bach1: BTB and CNC Homology 1
Bcl2: B-cell lymphoma 2
Beclin-1: Bcl-2-interacting coiled-coil protein
Bid: BH3-interacting domain death agonist
BNIP: Bcl-2/adenovirus E1B 19-kDa-interacting protein
CAT: Catalase
CaMKKβ: Calcium/calmodulin-dependent protein kinase kinase beta
cAMP: Cyclic adenosine monophosphate
CCL5: C-C motif chemokine ligand 5 (RANTES)
cGMP: Cyclic guanosine monophosphate
CMV: Cytomegalovirus
CREB: cAMP Response Element-Binding Protein
CXCL10: C-X-C motif chemokine ligand 10
DAG: Diacylglycerol
DIC: Disseminated Intravascular Coagulation
EBV: Epstein-Barr Virus
EGF: Epidermal growth factor
ER: Endoplasmic reticulum
MAM: mitochondrial-associated membrane
ERK1/2: Extracellular signal-regulated kinase 1/2
ETC: Electron transport chain
FoxO1: Forkhead box O1
GAA: glucogenic amino acids
GLN: glutamine
GLT: glutamate
GP73: Golgi protein 73
GPCR: G protein-coupled receptor
GLUT-1: Glucose transporter 1
GPx: Glutathione peroxidase
GSH: Reduced glutathione
GSK3β: Glycogen Synthase Kinase 3 Beta
HIF-1α: Hypoxia-inducible factor 1-alpha
HMOX1: Heme oxygenase 1
HO-1: Heme oxygenase-1
HOPS: Homotypic fusion and protein sorting complex
HSP: heat shock protein
ICAM-1: Intercellular adhesion molecule 1
IFN: Interferon
IL: Interleukin
IRAK-4: Interleukin-1 Receptor-Associated Kinase 4
IRF3: Interferon Regulatory Factor 3
IKKβ: IκB kinase beta
IP3: Inositol 1,4,5-trisphosphate
IRE1: Inositol-Requiring Enzyme 1
IRF3: interferon regulatory factor 3
JAK/STAT: Janus kinase/signal transducer and activator of transcription pathway
JNK: c-Jun N-terminal kinase
Keap1: Kelch-like ECH-associated protein 1
LC3: icrotubule-associated protein 1 light chain 3
LCS: Long Covid-19 Syndrome
LPS: Lipopolysaccharide
MAM: mitochondrial-associated membrane
MAPK: Mitogen-activated protein kinase
MHC-I: Major histocompatibility complex class I
MMP-9: Matrix metalloproteinase-9
mPTP: Mitochondrial permeability transition pore
mTOR: Mechanistic target of rapamycin
NEMO: NF-κB essential modulator
NET: Neutrophil extracellular trap
NEU: Neuraminidase
NFAT: Nuclear Factor of Activated T-cells
NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells
NIX: BNIP3L, BCL2/adenovirus E1B 19kDa-interacting protein 3-like; NOX, NADPH oxidase
NO: Nitric oxide
Nrf2: Nuclear factor erythroid 2-related factor 2
OCR: Oxygen consumption rate
ONOO−: Peroxynitrite
p38: p38 mitogen-activated protein kinase
p62: Sequestosome 1
Parkin: Parkin RBR E3 ubiquitin-protein ligase
PDGF: Platelet-Derived Growth Factor
PDH: Pyruvate dehydrogenase
PEPCK: Phosphoenolpyruvate Carboxykinase
PERK: Protein kinase RNA-like ER kinase
PHD: Prolyl hydroxylase domain-containing protein
PI3K: Phosphoinositide 3-kinase
PINK1: PTEN-induced kinase 1; PKA, Protein kinase A
PKC: Protein kinase C
PLC: Phospholipase C
Rac1: Ras-related C3 botulinum toxin substrate 1
REDD1: Regulated in Development and DNA Damage responses 1 (DDIT4)
Rheb: Ras homolog enriched in brain
ROS: Reactive oxygen species
RyR: Ryanodine receptor
SERCA: Sarco/endoplasmic reticulum Ca2+-ATPase
SIRT: NAD-dependent deacetylase Sirtuin
SKP: S-phase kinase-associated protein 1
SNAP29: Synaptosomal-Associated Protein 29
Srxn1: Sulfiredoxin-1
SOD: Superoxide dismutase
STAT: Signal transducer and activator of transcription
TBK1: TANK-binding kinase 1
TFEB: Transcription factor EB
TGF-β: Transforming growth factor-beta
TLR: Toll-like receptor
TNF-α: Tumor necrosis factor-alpha
TRPC: Transient receptor potential canonical channel
TSC2: Tuberous Sclerosis Complex 2
VCAM-1: Vascular cell adhesion molecule-1
VEGF: Vascular endothelial growth factor
VPS39: Vacuolar Protein Sorting 39
ULK1: Unc-51 Like Autophagy Activating Kinase 1
UPR: Unfolded protein response
UVRAG: UV Radiation Resistance-Associated Gene
XBP1: X-box Binding Protein 1
References
1. Yan MZ, Yang M, Lai CL. Post-covid-19 syndrome comprehensive assessment: from clinical diagnosis to imaging and biochemical-guided diagnosis and management. Viruses. (2023) 15(2):533. doi: 10.3390/v15020533
2. Yan Z, Yang M, Lai C-L. Long covid-19 syndrome: A comprehensive review of its effect on various organ systems and recommendation on rehabilitation plans. Biomedicines. (2021) 9(8):966. doi: 10.3390/biomedicines9080966
3. Zhang J, Ejikemeuwa A, Gerzanich V, Nasr M, Tang Q, Simard JM, et al. Understanding the role of sars-cov-2 orf3a in viral pathogenesis and covid-19. Front Microbiol. (2022) 13:854567. doi: 10.3389/fmicb.2022.854567
4. Lee Y-B, Jung M, Kim J, Kang M-G, Kwak C, Kim J-S, et al. Endomembrane systems are reorganized by orf3a and membrane (M) of sars-cov-2. bioRxiv. (2021) bioRxiv 2021.06.01.446555. doi: 10.1101/2021.06.01.446555
5. Keramidas P, Pitou M, Papachristou E, Choli-Papadopoulou T. Insights into the activation of unfolded protein response mechanism during coronavirus infection. Curr Issues Mol Biol. (2024) 46:4286–308. doi: 10.3390/cimb46050261
6. Echavarría-Consuegra L, Cook GM, Busnadiego I, Lefèvre C, Keep S, Brown K, et al. Manipulation of the unfolded protein response: A pharmacological strategy against coronavirus infection. PloS Pathog. (2021) 17:e1009644. doi: 10.1371/journal.ppat.1009644
7. Berta B, Tordai H, Lukács GL, Papp B, Enyedi Aacute;, Padányi R, et al. Sars-cov-2 envelope protein alters calcium signaling via serca interactions. bioRxiv. (2023) bioRxiv 2023.06.13.544745. doi: 10.1101/2023.06.13.544745
8. Fan J, Ray P, Lu YW, Kaur G, Schwarz JJ, Wan LQ. Cell chirality regulates intercellular junctions and endothelial permeability. Sci Adv. (2018) 4:eaat2111. doi: 10.1126/sciadv.aat2111
9. Jana S, Heaven MR, Alayash AI. Cell-free hemoglobin does not attenuate the effects of sars-cov-2 spike protein S1 subunit in pulmonary endothelial cells. Int J Mol Sci. (2021) 22(16):9041. doi: 10.3390/ijms22169041
10. Buzhdygan TP, DeOre BJ, Baldwin-Leclair A, Bullock TA, McGary HM, Khan JA, et al. The sars-cov-2 spike protein alters barrier function in 2d static and 3d microfluidic in-vitro models of the human blood–brain barrier. Neurobiol Dis. (2020) 146:105131. doi: 10.1016/j.nbd.2020.105131
11. López-Ayllón BD, Marín S, Fernández MF, García-García T, Fernández-Rodríguez R, Lucas-Rius Ad, et al. Metabolic and mitochondria alterations induced by sars-cov-2 accessory proteins orf3a, orf9b, orf9c and orf10. bioRxiv. (2023) bioRxiv 2023.09.26.559506. doi: 10.1101/2023.09.26.559506
12. Stoian M, Procopiescu B, Seitan S, Scarlat G. Post-covid-19 syndrome: insights into a novel post-infectious systemic disorder. J Med Life. (2023) 16:195–202. doi: 10.25122/jml-2022-0329
13. Vojdani A, Vojdani E, Saidara E, Maes M. Persistent sars-cov-2 infection, ebv, hhv-6 and other factors may contribute to inflammation and autoimmunity in long covid. Viruses. (2023) 15(2):400. doi: 10.3390/v15020400
14. Naendrup JH, Garcia Borrega J, Eichenauer DA, Shimabukuro-Vornhagen A, Kochanek M, Boll B. Reactivation of ebv and cmv in severe covid-19-epiphenomena or trigger of hyperinflammation in need of treatment? A large case series of critically ill patients. J Intensive Care Med. (2022) 37:1152–8. doi: 10.1177/08850666211053990
15. Gassen NC, Papies J, Bajaj T, Emanuel J, Dethloff F, Chua RL, et al. Sars-cov-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat Commun. (2021) 12:3818. doi: 10.1038/s41467-021-24007-w
16. Chen D, Zhang H. Autophagy in severe acute respiratory syndrome coronavirus 2 infection. Curr Opin Physiol. (2022) 29:100596. doi: 10.1016/j.cophys.2022.100596
17. Singh K, Chen YC, Hassanzadeh S, Han K, Judy JT, Seifuddin F, et al. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in sars-cov-2 infection. Front Genet. (2021) 12:599261. doi: 10.3389/fgene.2021.599261
18. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, et al. Orf3a of the covid-19 virus sars-cov-2 blocks hops complex-mediated assembly of the snare complex required for autolysosome formation. Dev Cell. (2021) 56:427–42.e5. doi: 10.1016/j.devcel.2020.12.010
19. Lan L, Xu D, Ye G, Xia C, Wang S, Li Y, et al. Positive rt-pcr test results in patients recovered from covid-19. JAMA. (2020) 323:1502–3. doi: 10.1001/jama.2020.2783
20. Cheung CCL, Goh D, Lim X, Tien TZ, Lim JCT, Lee JN, et al. Residual sars-cov-2 viral antigens detected in gi and hepatic tissues from five recovered patients with covid-19. Gut. (2022) 71:226–9. doi: 10.1136/gutjnl-2021-324280
21. Liu L, Wang Y, Wang W, Ying W, Sun B, Wang X, et al. Increased expression of the tlr7/9 signaling pathways in chronic active ebv infection. Front Pediatr. (2022) 10:1091571. doi: 10.3389/fped.2022.1091571
22. Akazawa R, Otsuka S, Kato I, Imadome KI, Takita J. Transient remission of chronic active ebv infection after chemotherapy alone. Pediatr Int. (2022) 64:e14836. doi: 10.1111/ped.14836
23. Wei A, Ma H, Zhang L, Li Z, Guan Y, Zhang Q, et al. Clinical analysis of chronic active ebv infection with coronary artery dilatation and a matched case-control study. Orphanet J Rare Dis. (2021) 16:50. doi: 10.1186/s13023-021-01689-5
24. Aihara Y, Moriya K, Shimozato N, Nagamatsu S, Kobayashi S, Uejima M, et al. Chronic active ebv infection in refractory enteritis with longitudinal ulcers with a cobblestone appearance: an autopsied case report. BMC Gastroenterol. (2021) 21:6. doi: 10.1186/s12876-020-01589-1
25. Yonese I, Sakashita C, Imadome KI, Kobayashi T, Yamamoto M, Sawada A, et al. Nationwide survey of systemic chronic active ebv infection in Japan in accordance with the new who classification. Blood Adv. (2020) 4:2918–26. doi: 10.1182/bloodadvances.2020001451
26. Sakaki S, Imadome KI, Kawano F, Nakadate H, Ishiguro A. Shift in epstein-barr virus (Ebv)-infected cells in chronic active ebv disease. Pediatr Int. (2019) 61:825–6. doi: 10.1111/ped.13935
27. Schmaltz HN, Fried LP, Xue QL, Walston J, Leng SX, Semba RD. Chronic cytomegalovirus infection and inflammation are associated with prevalent frailty in community-dwelling older women. J Am Geriatr Soc. (2005) 53:747–54. doi: 10.1111/j.1532-5415.2005.53250.x
28. Dash S, Aydin Y, Widmer KE, Nayak L. Hepatocellular carcinoma mechanisms associated with chronic hcv infection and the impact of direct-acting antiviral treatment. J Hepatocell Carcinoma. (2020) 7:45–76. doi: 10.2147/JHC.S221187
29. Chandra PK, Gunduz F, Hazari S, Kurt R, Panigrahi R, Poat B, et al. Impaired expression of type I and type ii interferon receptors in hcv-associated chronic liver disease and liver cirrhosis. PloS One. (2014) 9:e108616. doi: 10.1371/journal.pone.0108616
30. Chang J, Yao Y, Sun X, Wang W, Qian H, Liu Y, et al. Jag1 mediates apoptosis in herpes simplex keratitis by suppressing autophagy via ros/jag1/notch1/pulk1 signaling pathway. Cell Biol Toxicol. (2024) 41:1. doi: 10.1007/s10565-024-09968-0
31. Ripa I, Andreu S, Josa-Prado F, Fernandez Gomez B, de Castro F, Arribas M, et al. Herpes simplex virus type 1 inhibits autophagy in glial cells but requires atg5 for the success of viral replication. Front Microbiol. (2024) 15:1411655. doi: 10.3389/fmicb.2024.1411655
32. Damoiseaux J, Dotan A, Fritzler MJ, Bogdanos DP, Meroni PL, Roggenbuck D, et al. Autoantibodies and sars-cov2 infection: the spectrum from association to clinical implication: report of the 15th dresden symposium on autoantibodies. Autoimmun Rev. (2022) 21:103012. doi: 10.1016/j.autrev.2021.103012
33. Zhou Y, Han T, Chen J, Hou C, Hua L, He S, et al. Clinical and autoimmune characteristics of severe and critical cases of covid-19. Clin Transl Sci. (2020) 13:1077–86. doi: 10.1111/cts.12805
34. Vojdani A, Vojdani E, Kharrazian D. Reaction of human monoclonal antibodies to sars-cov-2 proteins with tissue antigens: implications for autoimmune diseases. Front Immunol. (2021) 11:617089. doi: 10.3389/fimmu.2020.617089
35. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-onset igg autoantibodies in hospitalized patients with covid-19. Nat Commun. (2021) 12:5417. doi: 10.1038/s41467-021-25509-3
36. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse functional autoantibodies in patients with covid-19. Nature. (2021) 595:283–8. doi: 10.1038/s41586-021-03631-y
37. Angileri F, Legare S, Marino Gammazza A, Conway de Macario E, Jl Macario A, Cappello F. Molecular mimicry may explain multi-organ damage in covid-19. Autoimmun Rev. (2020) 19:102591. doi: 10.1016/j.autrev.2020.102591
38. Angileri F, Légaré S, Marino Gammazza A, Conway de Macario E, Macario AJL, Cappello F. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with covid-19? Br J Haematol. (2020) 190:e92–e3. doi: 10.1111/bjh.16883
39. Acosta-Ampudia Y, Monsalve DM, Rojas M, Rodríguez Y, Zapata E, Ramírez-Santana C, et al. Persistent autoimmune activation and proinflammatory state in post-coronavirus disease 2019 syndrome. J Infect Dis. (2022) 225:2155–62. doi: 10.1093/infdis/jiac017
40. Garcia-Martinez FJ, Moreno-Artero E, Jahnke S. Sars-cov-2 and ebv coinfection. Med Clin (Engl Ed). (2020) 155:319–20. doi: 10.1016/j.medcle.2020.06.010
41. Chen T, Song J, Liu H, Zheng H, Chen C. Positive epstein-barr virus detection in coronavirus disease 2019 (Covid-19) patients. Sci Rep. (2021) 11:10902. doi: 10.1038/s41598-021-90351-y
42. Drago F, Ciccarese G, Rebora A, Parodi A. Human herpesvirus-6, -7, and epstein-barr virus reactivation in pityriasis rosea during covid-19. J Med Virol. (2021) 93:1850–1. doi: 10.1002/jmv.26549
43. Volk A, Hackbart M, Deng X, Cruz-Pulido Y, O’Brien A, Baker SC. Coronavirus endoribonuclease and deubiquitinating interferon antagonists differentially modulate the host response during replication in macrophages. J Virol. (2020) 94:10.1128. doi: 10.1128/JVI.00178-20
44. Keita AK, Vidal N, Toure A, Diallo MSK, Magassouba Nf, Baize S, et al. A 40-month follow-up of ebola virus disease survivors in Guinea (Postebogui) reveals long-term detection of ebola viral ribonucleic acid in semen and breast milk. Open Forum Infect Dis. (2019) 6:ofz482. doi: 10.1093/ofid/ofz482
45. Paz-Bailey G, Rosenberg ES, Doyle K, Munoz-Jordan J, Santiago GA, Klein L, et al. Persistence of zika virus in body fluids - final report. N Engl J Med. (2018) 379:1234–43. doi: 10.1056/NEJMoa1613108
46. Chia JK, Chia AY. Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J Clin Pathol. (2008) 61:43–8. doi: 10.1136/jcp.2007.050054
47. Nash P, Chard M, Hazleman B. Chronic coxsackie B infection mimicking primary fibromyalgia. J Rheumatol. (1989) 16:1506–8.
48. Daba TM, Zhao Y, Pan Z. Advancement of mechanisms of coxsackie virus B3-induced myocarditis pathogenesis and the potential therapeutic targets. Curr Drug Targets. (2019) 20:1461–73. doi: 10.2174/1389450120666190618124722
49. Pretagostini R, Lai Q, Pettorini L, Garofalo M, Poli L, Melandro F, et al. Multiple organ failure associated with coxsackie virus in a kidney transplant patient: case report. Transplant Proc. (2016) 48:438–40. doi: 10.1016/j.transproceed.2016.02.005
50. Riddell MA, Moss WJ, Hauer D, Monze M, Griffin DE. Slow clearance of measles virus rna after acute infection. J Clin Virol. (2007) 39:312–7. doi: 10.1016/j.jcv.2007.05.006
51. Strzelczyk JK, Swietek A, Biernacki K, Golabek K, Gazdzicka J, Miskiewicz-Orczyk K, et al. Pcr detection of epstein-barr virus (Ebv) DNA in patients with head and neck squamous cell carcinoma, in patients with chronic tonsillitis, and in healthy individuals. BioMed Res Int. (2022) 2022:8506242. doi: 10.1155/2022/8506242
52. Acharya S, Ekalaksananan T, Vatanasapt P, Loyha K, Phusingha P, Promthet S, et al. Association of epstein-barr virus infection with oral squamous cell carcinoma in a case–control study. J Oral Pathol Med. (2015) 44:252–7. doi: 10.1111/jop.12231
53. Sixbey JW, Shirley P, Chesney PJ, Buntin DM, Resnick L. Detection of a second widespread strain of epstein-barr virus. Lancet. (1989) 2:761–5. doi: 10.1016/s0140-6736(89)90829-5
54. Scully C, Porter SR, Di Alberti L, Jalal M, Maitland N. Detection of epstein-barr virus in oral scrapes in hiv infection, in hairy leukoplakia, and in healthy non-hiv-infected people. J Oral Pathol Med. (1998) 27:480–2. doi: 10.1111/j.1600-0714.1998.tb01916.x
55. Gupta I, Nasrallah GK, Sharma A, Jabeen A, Smatti MK, Al-Thawadi HA, et al. Co-prevalence of human papillomaviruses (Hpv) and epstein-barr virus (Ebv) in healthy blood donors from diverse nationalities in Qatar. Cancer Cell Int. (2020) 20:107. doi: 10.1186/s12935-020-01190-2
56. Li H, Weng P, Najarro K, Xue QL, Semba RD, Margolick JB, et al. Chronic cmv infection in older women: longitudinal comparisons of cmv DNA in peripheral monocytes, anti-cmv igg titers, serum il-6 levels, and cmv pp65 (Nlv)-specific cd8(+) T-cell frequencies with twelve year follow-up. Exp Gerontol. (2014) 54:84–9. doi: 10.1016/j.exger.2014.01.010
57. Yang H, Zhou W, Lv H, Wu D, Feng Y, Shu H, et al. The association between cmv viremia or endoscopic features and histopathological characteristics of cmv colitis in patients with underlying ulcerative colitis. Inflammation Bowel Dis. (2017) 23:814–21. doi: 10.1097/MIB.0000000000001095
58. Kredel LI, Mundt P, van Riesen L, Johrens K, Hofmann J, Loddenkemper C, et al. Accuracy of diagnostic tests and a new algorithm for diagnosing cytomegalovirus colitis in inflammatory bowel diseases: A diagnostic study. Int J Colorectal Dis. (2019) 34:229–37. doi: 10.1007/s00384-018-3170-z
59. Justo D, Finn T, Atzmony L, Guy N, Steinvil A. Thrombosis associated with acute cytomegalovirus infection: A meta-analysis. Eur J Intern Med. (2011) 22:195–9. doi: 10.1016/j.ejim.2010.11.006
60. Atzmony L, Halutz O, Avidor B, Finn T, Zimmerman O, Steinvil A, et al. Incidence of cytomegalovirus-associated thrombosis and its risk factors: A case-control study. Thromb Res. (2010) 126:e439–43. doi: 10.1016/j.thromres.2010.09.006
61. van Hal S, Senanayake S, Hardiman R. Splenic infarction due to transient antiphospholipid antibodies induced by acute epstein-barr virus infection. J Clin Virol. (2005) 32:245–7. doi: 10.1016/j.jcv.2004.07.013
62. Mashav N, Saar N, Chundadze T, Steinvil A, Justo D. Epstein-barr virus-associated venous thromboembolism: A case report and review of the literature. Thromb Res. (2008) 122:570–1. doi: 10.1016/j.thromres.2008.03.005
63. Nawata A, Shirayama R, Oshida K, Sato T, Ito T, Shiba E, et al. Catastrophic antiphospholipid syndrome with epstein-barr virus-associated hemophagocytosis: A clinicopathological conference. Lupus. (2022) 31:1385–93. doi: 10.1177/09612033221118819
64. Suzuki Y, Shichishima T, Mukae M, Ohsaka M, Hayama M, Horie R, et al. Splenic infarction after epstein-barr virus infection in a patient with hereditary spherocytosis. Int J Hematol. (2007) 85:380–3. doi: 10.1532/IJH97.07208
65. van Steijn JH, van Tol KM, van Essen LH, Gans RO. Disseminated intravascular coagulation as an unusual presentation of an epstein-barr virus infection. Neth J Med. (2000) 57:169–71. doi: 10.1016/s0300-2977(00)00047-4
66. Muller NF, Schampera M, Jahn G, Malek NP, Berg CP, Hamprecht K. Case report: severe cytomegalovirus primary infection in an immunocompetent adult with disseminated intravascular coagulation treated with valganciclovir. BMC Infect Dis. (2016) 16:19. doi: 10.1186/s12879-016-1343-3
67. Niewold TB, Bundrick JB. Disseminated intravascular coagulation due to cytomegalovirus infection in an immunocompetent adult treated with plasma exchange. Am J Hematol. (2006) 81:454–7. doi: 10.1002/ajh.20602
68. Chen T, Tu S, Ding L, Jin M, Chen H, Zhou H. The role of autophagy in viral infections. J Biomed Sci. (2023) 30:5. doi: 10.1186/s12929-023-00899-2
69. Mouna L, Hernandez E, Bonte D, Brost R, Amazit L, Delgui LR, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus becn1/beclin 1-binding proteins. Autophagy. (2016) 12:327–42. doi: 10.1080/15548627.2015.1125071
70. Yao Q, Doyle Máire E, Liu Q-R, Appleton A, O’Connell Jennifer F, N-p W, et al. Long-term dysfunction of taste papillae in sars-cov-2. NEJM Evidence. (2023) 2:EVIDoa2300046. doi: 10.1056/EVIDoa2300046
71. André FR, Hiranmayi R, Junbum K, Alain CB, Olivier E, Robert ES. Persistent alveolar type 2 dysfunction and lung structural derangement in post-acute covid-19. medRxiv. (2022) medRxiv 2022.11.28.22282811. doi: 10.1101/2022.11.28.22282811
72. de Melo GD, Lazarini F, Levallois S, Hautefort C, Michel V, Larrous F, et al. Covid-19–related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci Trans Med. (2021) 13:eabf8396. doi: 10.1126/scitranslmed.abf8396
73. Hany M, Zidan A, Gaballa M, Ibrahim M, Agayby ASS, Abouelnasr AA, et al. Lingering sars-cov-2 in gastric and gallbladder tissues of patients with previous covid-19 infection undergoing bariatric surgery. Obes Surg. (2023) 33:139–48. doi: 10.1007/s11695-022-06338-9
74. Miura CS, Lima TM, Martins RB, Jorge DMM, Tamashiro E, Anselmo-Lima WT, et al. Asymptomatic sars-cov-2 infection in children’s tonsils. Braz J Otorhinolaryngol. (2022) 88:9. doi: 10.1016/j.bjorl.2022.10.016
75. Zuo T, Liu Q, Zhang F, Lui GC-Y, Tso EY, Yeoh YK, et al. Depicting sars-cov-2 faecal viral activity in association with gut microbiota composition in patients with covid-19. Gut. (2021) 70:276–84. doi: 10.1136/gutjnl-2020-322294
76. Tejerina F, Catalan P, Rodriguez-Grande C, Adan J, Rodriguez-Gonzalez C, Munoz P, et al. Post-covid-19 syndrome. Sars-cov-2 rna detection in plasma, stool, and urine in patients with persistent symptoms after covid-19. BMC Infect Dis. (2022) 22:211. doi: 10.1186/s12879-022-07153-4
77. Proal AD, VanElzakker MB, Aleman S, Bach K, Boribong BP, Buggert M, et al. Sars-cov-2 reservoir in post-acute sequelae of covid-19 (Pasc). Nat Immunol. (2023) 24:1616–27. doi: 10.1038/s41590-023-01601-2
78. Michalak KP, Michalak AZ. Understanding chronic inflammation: couplings between cytokines, ROS, NO, Ca i2+ , HIF-1a, Nrf2 and autophagy. Front Immunol. (2025) 16. doi: 10.3389/fimmu.2025.1558263
79. Kumar S, Javed R, Mudd M, Pallikkuth S, Lidke KA, Jain A, et al. Mammalian hybrid pre-autophagosomal structure hypas generates autophagosomes. Cell. (2021) 184:5950–69.e22. doi: 10.1016/j.cell.2021.10.017
80. Sun X, Liu YZ, Huang ZH, Xu WY, Hu W, Yi LN, et al. Sars-cov-2 non-structural protein 6 triggers nlrp3-dependent pyroptosis by targeting atp6ap1. Cell Death Differentiation. (2022) 29:1240–54. doi: 10.1038/s41418-021-00916-7
81. Cottam EM, Whelband MC, Wileman T. Coronavirus nsp6 restricts autophagosome expansion. Autophagy. (2014) 10:1426–41. doi: 10.4161/auto.29309
82. Chen D, Zheng Q, Sun L, Ji M, Li Y, Deng H, et al. Orf3a of sars-cov-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell. (2021) 56:3250–63.e5. doi: 10.1016/j.devcel.2021.10.006
83. Qu Y, Wang X, Zhu Y, Wang W, Wang Y, Hu G, et al. Orf3a-mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication. Front Cell Dev Biol. (2021) 9:716208. doi: 10.3389/fcell.2021.716208
84. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of sars-cov-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. (2021) 35(7):109126. doi: 10.1016/j.celrep.2021.109126
85. Quan Q, Ma X, Feng J, Li W, Li X. Ginsenoside rg1 improves autophagy dysfunction to ameliorate alzheimer’s disease via targeting fgr proto-oncogene. Neuropeptides. (2025) 111:102514. doi: 10.1016/j.npep.2025.102514
86. Dhapola R, Kumari S, Sharma P, Vellingiri B, HariKrishnaReddy D. Advancements in autophagy perturbations in alzheimer’s disease: molecular aspects and therapeutics. Brain Res. (2025) 1851:149494. doi: 10.1016/j.brainres.2025.149494
87. Keller CW, Adamopoulos IE, Lunemann JD. Autophagy pathways in autoimmune diseases. J Autoimmun. (2023) 136:103030. doi: 10.1016/j.jaut.2023.103030
88. Shen D, Liu K, Wang H, Wang H. Autophagy modulation in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. (2022) 209:140–50. doi: 10.1093/cei/uxac017
89. Yin H, Wu H, Chen Y, Zhang J, Zheng M, Chen G, et al. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front Immunol. (2018) 9:1512. doi: 10.3389/fimmu.2018.01512
90. Yuan M, Meng W, Liao W, Lian S. Andrographolide antagonizes tnf-alpha-induced il-8 via inhibition of nadph oxidase/ros/nf-kappab and src/mapks/ap-1 axis in human colorectal cancer hct116 cells. J Agric Food Chem. (2018) 66:5139–48. doi: 10.1021/acs.jafc.8b00810
91. Unger Benjamin L, Ganesan S, Comstock Adam T, Faris Andrea N, Hershenson Marc B, Sajjan Uma S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J Virol. (2014) 88:3705–18. doi: 10.1128/jvi.03039-13
92. Tattoli I, Carneiro LA, Jéhanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. Nlrx1 is a mitochondrial nod-like receptor that amplifies nf-Kb and jnk pathways by inducing reactive oxygen species production. EMBO Rep. (2008) 9:293–300. doi: 10.1038/sj.embor.7401161
93. Li J, Huang B, Shi X, Castranova V, Vallyathan V, Huang C. Involvement of hydrogen peroxide in asbestos-induced nfat activation. Mol Cell Biochem. (2002) 234-235:161–8. doi: 10.1023/A:1015962916195
94. Elbim C, Guichard C, Dang PMC, Fay M, Pedruzzi E, Demur H, et al. Interleukin-18 primes the oxidative burst of neutrophils in response to formyl-peptides: role of cytochrome B558 translocation and N-formyl peptide receptor endocytosis. Clin Diagn Lab Immunol. (2005) 12:436–46. doi: 10.1128/CDLI.12.3.436-446.2005
95. Vermot A, Petit-Härtlein I, Smith SME, Fieschi F. Nadph oxidases (Nox): an overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants. (2021) 10(6):890. doi: 10.3390/antiox10060890
96. Ladik M, Valenta H, Erard M, Vandenabeele P, Riquet FB. From tnf-induced signaling to nadph oxidase enzyme activity: methods to investigate protein complexes involved in regulated cell death modalities. Front Cell Death. (2023) 2. doi: 10.3389/fceld.2023.1127330
97. Tesoriere L, Attanzio A, Allegra M, Gentile C, Livrea MA. Indicaxanthin inhibits nadph oxidase (Nox)-1 activation and nf-Kb-dependent release of inflammatory mediators and prevents the increase of epithelial permeability in il-1β-exposed caco-2 cells. Br J Nutr. (2013) 111:415–23. doi: 10.1017/S0007114513002663
98. Li J, Lan T, Zhang C, Zeng C, Hou J, Yang Z, et al. Reciprocal activation between il-6/stat3 and nox4/akt signalings promotes proliferation and survival of non-small cell lung cancer cells. Oncotarget. (2015) 6:1031–48. doi: 10.18632/oncotarget.2671
99. Dwivedi G, Gran MA, Bagchi P, Kemp ML. Dynamic redox regulation of il-4 signaling. PloS Comput Biol. (2015) 11:e1004582. doi: 10.1371/journal.pcbi.1004582
100. Saha B, Jyothi Prasanna S, Chandrasekar B, Nandi D. Gene modulation and immunoregulatory roles of interferonγ. Cytokine. (2010) 50:1–14. doi: 10.1016/j.cyto.2009.11.021
101. Ha H, Debnath B, Neamati N. Role of the cxcl8-cxcr1/2 axis in cancer and inflammatory diseases. Theranostics. (2017) 7:1543–88. doi: 10.7150/thno.15625
102. Jendrysik MA, Vasilevsky S, Yi L, Wood A, Zhu N, Zhao Y, et al. Nadph oxidase-2 derived ros dictates murine dc cytokine-mediated cell fate decisions during cd4 T helper-cell commitment. PloS One. (2011) 6:e28198. doi: 10.1371/journal.pone.0028198
103. Kaur N, Naga OS, Norell H, Al-Khami AA, Scheffel MJ, Chakraborty NG, et al. T cells expanded in presence of il-15 exhibit increased antioxidant capacity and innate effector molecules. Cytokine. (2011) 55:307–17. doi: 10.1016/j.cyto.2011.04.014
104. Clemente TM, Augusto L, Angara RK, Gilk SD. Coxiella burnetii actively blocks il-17-induced oxidative stress in macrophages. bioRxiv. (2023) bioRxiv 2023.03.15.532774. doi: 10.1101/2023.03.15.532774
105. Meka RR, Venkatesha SH, Dudics S, Acharya B, Moudgil KD. Il-27-induced modulation of autoimmunity and its therapeutic potential. Autoimmun Rev. (2015) 14:1131–41. doi: 10.1016/j.autrev.2015.08.001
106. Rocic P, Lucchesi PA. Nad(P)H oxidases and tgf-B–induced cardiac fibroblast differentiation. Circ Res. (2005) 97:850–2. doi: 10.1161/01.RES.0000190403.87462.bf
107. Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and nadph oxidase in cultured rpe cells. Exp Eye Res. (2007) 85:462–72. doi: 10.1016/j.exer.2007.06.013
108. Didion SP. Cellular and oxidative mechanisms associated with interleukin-6 signaling in the vasculature. Int J Mol Sci. (2017) 18(12):2563. doi: 10.3390/ijms18122563
109. Ali MI, Chen X, Didion SP. Heterozygous enos deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol Rep. (2015) 3(12):e12630. doi: 10.14814/phy2.12630
110. Kuwano Y, Kawahara T, Yamamoto H, Teshima-Kondo S, Tominaga K, Masuda K, et al. Interferon-Γ Activates transcription of nadph oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. Am J Physiology-Cell Physiol. (2006) 290:C433–C43. doi: 10.1152/ajpcell.00135.2005
111. Werner LE, Wagner U. Calcium-sensing receptor-mediated nlrp3 inflammasome activation in rheumatoid arthritis and autoinflammation. Front Physiol. (2023) 13:1078569. doi: 10.3389/fphys.2022.1078569
112. Ruiz A, Matute C, Alberdi E. Endoplasmic reticulum ca(2+) release through ryanodine and ip(3) receptors contributes to neuronal excitotoxicity. Cell Calcium. (2009) 46:273–81. doi: 10.1016/j.ceca.2009.08.005
113. Bacsa B, Tiapko O, Stockner T, Groschner K. Mechanisms and significance of ca(2+) entry through trpc channels. Curr Opin Physiol. (2020) 17:25–33. doi: 10.1016/j.cophys.2020.06.005
114. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. (2016) 100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001
115. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links tca cycle dysfunction to oncogenesis by inhibiting hif-&X3b1; prolyl hydroxylase. Cancer Cell. (2005) 7:77–85. doi: 10.1016/j.ccr.2004.11.022
116. Korbecki J, Simińska D, Gąssowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, et al. Chronic and cycling hypoxia: drivers of cancer chronic inflammation through hif-1 and nf-Kb activation: A review of the molecular mechanisms. Int J Mol Sci. (2021) 22(19):10701. doi: 10.3390/ijms221910701
117. Minisini M, Cricchi E, Brancolini C. Acetylation and phosphorylation in the regulation of hypoxia-inducible factor activities: additional options to modulate adaptations to changes in oxygen levels. Life (Basel). (2023) 14(1):20. doi: 10.3390/life14010020
118. André-Lévigne D, Modarressi A, Pepper MS, Pittet-Cuénod B. Reactive oxygen species and nox enzymes are emerging as key players in cutaneous wound repair. Int J Mol Sci. (2017) 18(10):2149. doi: 10.3390/ijms18102149
119. Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces hif-1 activation in human monocytes via P44/42 mapk and nf-kappab. Biochem J. (2006) 396:517–27. doi: 10.1042/bj20051839
120. Rahman A, Tabassum T, Araf Y, Al Nahid A, Ullah MA, Hosen MJ. Silent hypoxia in covid-19: pathomechanism and possible management strategy. Mol Biol Rep. (2021) 48:3863–9. doi: 10.1007/s11033-021-06358-1
121. Malkov MI, Lee CT, Taylor CT. Regulation of the hypoxia-inducible factor (Hif) by pro-inflammatory cytokines. Cells. (2021) 10(9):2340. doi: 10.3390/cells10092340
122. Hellwig-B̈rgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1β and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. (1999) 94:1561–7. doi: 10.1182/blood.V94.5.1561
123. Zhou J, Schmid T, Brüne B. Tumor necrosis factor-A Causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-Kb-dependent pathway. Mol Biol Cell. (2003) 14:2216–25. doi: 10.1091/mbc.e02-09-0598
124. Kim KW, Lee SJ, Kim JC. Tnf-A Upregulates hif-1α Expression in pterygium fibroblasts and enhances their susceptibility to vegf independent of hypoxia. Exp Eye Res. (2017) 164:74–81. doi: 10.1016/j.exer.2017.08.008
125. Azimi I, Milevskiy MJG, Kaemmerer E, Turner D, Yapa KTDS, Brown MA, et al. Trpc1 is a differential regulator of hypoxia-mediated events and akt signalling in pten-deficient breast cancer cells. J Cell Sci. (2017) 130:2292–305. doi: 10.1242/jcs.196659
126. Hui AS, Bauer AL, Striet JB, Schnell PO, Czyzyk-Krzeska MF. Calcium signaling stimulates translation of hif-alpha during hypoxia. FASEB J. (2006) 20:466–75. doi: 10.1096/fj.05-5086com
127. Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of hif-1alpha expression by intermittent hypoxia: involvement of nadph oxidase, ca2+ Signaling, prolyl hydroxylases, and mtor. J Cell Physiol. (2008) 217:674–85. doi: 10.1002/jcp.21537
128. Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y, et al. Stim1 mediates hypoxia-driven hepatocarcinogenesis via interaction with hif-1. Cell Rep. (2015) 12:388–95. doi: 10.1016/j.celrep.2015.06.033
129. Westra J, Brouwer E, van Roosmalen IAM, Doornbos-van der Meer B, van Leeuwen MA, Posthumus MD, et al. Expression and regulation of hif-1alpha in macrophages under inflammatory conditions; significant reduction of vegf by camkii inhibitor. BMC Musculoskeletal Disord. (2010) 11:61. doi: 10.1186/1471-2474-11-61
130. Corre I, Paris F, Huot J. The P38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget. (2017) 8:55684–714. doi: 10.18632/oncotarget.18264
131. Winston BW, Chan ED, Johnson GL, Riches DW. Activation of P38mapk, mkk3, and mkk4 by tnf-alpha in mouse bone marrow-derived macrophages. J Immunol. (1997) 159:4491–7. doi: 10.4049/jimmunol.159.9.4491
132. Chen N-n, Wei F, Wang L, Cui S, Wan Y, Liu S. Tumor necrosis factor alpha induces neural stem cell apoptosis through activating P38 mapk pathway. Neurochemical Res. (2016) 41:3052–62. doi: 10.1007/s11064-016-2024-8
133. Wang X-j, Kong K-m, Qi W-l, Ye W-l, Song P-s. Interleukin-1 beta induction of neuron apoptosis depends on P38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacologica Sin. (2005) 26:934–42. doi: 10.1111/j.1745-7254.2005.00152.x
134. Liu X, Ye F, Xiong H, Hu D, Limb G, Xie T, et al. Il-1 beta upregulates il-8 production in human muller cells through activation of the P38 mapk and erk1/2 signaling pathways. Inflammation. (2014) 37:1486–95. doi: 10.1007/s10753-014-9874-5
135. Chen F, Wang Q, Li Y, Li F, Zhang L, Gu X. Tgf-B1-induced apoptosis in retinal endothelial cells is implicated in retinal vein occlusion. Exp Eye Res. (2025) 250:110168. doi: 10.1016/j.exer.2024.110168
136. Chen H, Wang M, Zhang Z, Lin F, Guo B, Lu Q, et al. Oxidative stress drives endometrial fibrosis via tgf-B1/mapk signaling pathway in breast cancer. FASEB J. (2024) 38:e70172. doi: 10.1096/fj.202401257RR
137. Kang YJ, Chen J, Otsuka M, Mols J, Ren S, Wang Y, et al. Macrophage deletion of P38α Partially impairs lipopolysaccharide-induced cellular activation1. J Immunol. (2008) 180:5075–82. doi: 10.4049/jimmunol.180.7.5075
138. Jung YD, Fan F, McConkey DJ, Jean ME, Liu W, Reinmuth N, et al. Role of P38 mapk, ap-1, and nf-Kb in interleukin-1β-induced il-8 expression in human vascular smooth muscle cells. Cytokine. (2002) 18:206–13. doi: 10.1006/cyto.2002.1034
139. Suzuki M, Tetsuka T, Yoshida S, Watanabe N, Kobayashi M, Matsui N, et al. The role of P38 mitogen-activated protein kinase in il-6 and il-8 production from the tnf-A- or il-1β-stimulated rheumatoid synovial fibroblasts. FEBS Lett. (2000) 465:23–7. doi: 10.1016/S0014-5793(99)01717-2
140. Westra J, Doornbos-van der Meer B, de Boer P, van Leeuwen MA, van Rijswijk MH, Limburg PC. Strong inhibition of tnf-A Production and inhibition of il-8 and cox-2 mrna expression in monocyte-derived macrophages by rwj 67657, a P38 mitogen-activated protein kinase (Mapk) inhibitor. Arthritis Res Ther. (2004) 6:R384. doi: 10.1186/ar1204
141. Chen K-H, Weng M-S, Lin J-K. Tangeretin suppresses il-1β-induced cyclooxygenase (Cox)-2 expression through inhibition of P38 mapk, jnk, and akt activation in human lung carcinoma cells. Biochem Pharmacol. (2007) 73:215–27. doi: 10.1016/j.bcp.2006.09.018
142. Beltrán AE, Briones AM, García-Redondo AB, Rodríguez C, Miguel M, Álvarez Y, et al. P38 mapk contributes to angiotensin ii-induced cox-2 expression in aortic fibroblasts from normotensive and hypertensive rats. J Hypertension. (2009) 27(1):142–54. doi: 10.1097/HJH.0b013e328317a730
143. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. P38 and jnk mapk pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. (2014) 344:174–9. doi: 10.1016/j.canlet.2013.11.019
144. Lucas RM, Luo L, Stow JL. Erk1/2 in immune signalling. Biochem Soc Trans. (2022) 50:1341–52. doi: 10.1042/BST20220271
145. Quément CL, Guénon I, Gillon J-Y, Lagente V, Boichot E. Mmp-12 induces il-8/cxcl8 secretion through egfr and erk1/2 activation in epithelial cells. Am J Physiology-Lung Cell Mol Physiol. (2008) 294:L1076–L84. doi: 10.1152/ajplung.00489.2007
146. Zhou J, Sun X, Zhang J, Yang Y, Chen D, Cao J. Il-34 regulates il-6 and il-8 production in human lung fibroblasts via mapk, pi3k-akt, jak and nf-Kb signaling pathways. Int Immunopharmacol. (2018) 61:119–25. doi: 10.1016/j.intimp.2018.05.023
147. Shan L, Redhu N, Saleh A, Halayko A, Chakir J, Gounni A. Thymic stromal lymphopoietin receptor-mediated il-6 and cc/cxc chemokines expression in human airway smooth muscle cells: role of mapks (Erk1/2, P38, and jnk) and stat3 pathways. J Immunol (Baltimore Md: 1950). (2010) 184:7134–43. doi: 10.4049/jimmunol.0902515
148. Chen J, Liao M-y, Gao X-l, Zhong Q, Tang T-t, Yu X, et al. Il-17a induces pro-inflammatory cytokines production in macrophages via mapkinases, nf-Kb and ap-1. Cell Physiol Biochem. (2013) 32:1265–74. doi: 10.1159/000354525
149. Wong E, Xu F, Joffre J, Nguyen N, Wilhelmsen K, Hellman J. Erk1/2 has divergent roles in lps-induced microvascular endothelial cell cytokine production and permeability. Shock. (2021) 55(3):349–56. doi: 10.1097/SHK.0000000000001639
150. Saryeddine L, Zibara K, Kassem N, Badran B, El-Zein N. Egf-induced vegf exerts a pi3k-dependent positive feedback on erk and akt through vegfr2 in hematological in vitro models. PloS One. (2016) 11:e0165876. doi: 10.1371/journal.pone.0165876
151. Chen J, Luo X, Liu M, Peng L, Zhao Z, He C, et al. Silencing long non-coding rna neat1 attenuates rheumatoid arthritis via the mapk/erk signalling pathway by downregulating microrna-129 and microrna-204. RNA Biol. (2021) 18:657–68. doi: 10.1080/15476286.2020.1857941
152. Huang Y, Xu L, Yang Q, Xiao X, Ye Z, Li R, et al. Nlrp12 C.1382dup promotes the development of crohn’s disease through the erk/nlrp3/il-1β Pathway. Gene. (2024) 931:148855. doi: 10.1016/j.gene.2024.148855
153. Menon MB, Dhamija S. Beclin 1 phosphorylation – at the center of autophagy regulation. Front Cell Dev Biol. (2018) 6:137. doi: 10.3389/fcell.2018.00137
154. Ferreira-Marques M, Carvalho A, Cavadas C, Aveleira CA. Pi3k/akt/mtor and erk1/2-mapk signaling pathways are involved in autophagy stimulation induced by caloric restriction or caloric restriction mimetics in cortical neurons. Aging (Albany NY). (2021) 13:7872–82. doi: 10.18632/aging.202805
156. Ahn EH, Park J-B. Molecular mechanisms of alzheimer’s disease induced by amyloid-B and tau phosphorylation along with rhoa activity: perspective of rhoa/rho-associated protein kinase inhibitors for neuronal therapy. Cells. (2025) 14:89. doi: 10.3390/cells14020089
157. Raivich G, Behrens A. Role of the ap-1 transcription factor C-jun in developing, adult and injured brain. Prog Neurobiol. (2006) 78:347–63. doi: 10.1016/j.pneurobio.2006.03.006
158. Namba S, Nakano R, Kitanaka T, Kitanaka N, Nakayama T, Sugiya H. Erk2 and jnk1 contribute to tnf-A-induced il-8 expression in synovial fibroblasts. PloS One. (2017) 12:e0182923. doi: 10.1371/journal.pone.0182923
159. Chen WC, Tseng CK, Chen YH, Lin CK, Hsu SH, Wang SN, et al. Hcv ns5a up-regulates cox-2 expression via il-8-mediated activation of the erk/jnk mapk pathway. PloS One. (2015) 10(7):e0133264. doi: 10.1371/journal.pone.0133264
160. Ventura JJ, Cogswell P, Flavell RA, Baldwin AS Jr., Davis RJ. Jnk potentiates tnf-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. (2004) 18:2905–15. doi: 10.1101/gad.1223004
161. Tang H-W, Liao H-M, Peng W-H, Lin H-R, Chen C-H, Chen G-C. Atg9 interacts with dtraf2/traf6 to regulate oxidative stress-induced jnk activation and autophagy induction. Dev Cell. (2013) 27:489–503. doi: 10.1016/j.devcel.2013.10.017
162. Zhou Y-Y, Li Y, Jiang W-Q, Zhou L-F. Mapk/jnk signalling: A potential autophagy regulation pathway. Bioscience Rep. (2015) 35:e00199. doi: 10.1042/BSR20140141
163. Denninger K, Rasmussen S, Larsen JM, Ørskov C, Seier Poulsen S, Sørensen P, et al. Jnk1, but not jnk2, is required in two mechanistically distinct models of inflammatory arthritis. Am J Pathol. (2011) 179:1884–93. doi: 10.1016/j.ajpath.2011.06.019
164. Hou C-H, Tang C-H, Hsu C-J, Hou S-M, Liu J-F. Ccn4 induces il-6 production through Avβ5 receptor, pi3k, akt, and nf-Kb singling pathway in human synovial fibroblasts. Arthritis Res Ther. (2013) 15:R19. doi: 10.1186/ar4151
165. Zhao M, Zhou A, Xu L, Zhang X. The role of tlr4-mediated pten/pi3k/akt/nf-Kb signaling pathway in neuroinflammation in hippocampal neurons. Neuroscience. (2014) 269:93–101. doi: 10.1016/j.neuroscience.2014.03.039
166. Tseng W-P, Su C-M, Tang C-H. Fak activation is required for tnf-A-induced il-6 production in myoblasts. J Cell Physiol. (2010) 223:389–96. doi: 10.1002/jcp.22047
167. Simão F, Matté A, Pagnussat AS, Netto CA, Salbego CG. Resveratrol Prevents Ca1 Neurons against Ischemic Injury by Parallel Modulation of Both Gsk-3β and Creb through Pi3-K/Akt Pathways. Eur J Neurosci. (2012) 36:2899–905. doi: 10.1111/j.1460-9568.2012.08229.x
168. Wan X, Zhou M, Huang F, Zhao N, Chen X, Wu Y, et al. Akt1-creb stimulation of pdgfrα Expression is pivotal for pten deficient tumor development. Cell Death Dis. (2021) 12:172. doi: 10.1038/s41419-021-03433-0
169. Callahan V, Hawks S, Crawford MA, Lehman CW, Morrison HA, Ivester HM, et al. The pro-inflammatory chemokines cxcl9, cxcl10 and cxcl11 are upregulated following sars-cov-2 infection in an akt-dependent manner. Viruses. (2021) 13(6):1062. doi: 10.3390/v13061062
170. Günzl P, Bauer K, Hainzl E, Matt U, Dillinger B, Mahr B, et al. Anti-inflammatory properties of the pi3k pathway are mediated by il-10/dusp regulation. J Leukocyte Biol. (2010) 88:1259–69. doi: 10.1189/jlb.0110001
171. Zhang X, Li N, Shao H, Meng Y, Wang L, Wu Q, et al. Methane limit lps-induced nf-Kb/mapks signal in macrophages and suppress immune response in mice by enhancing pi3k/akt/gsk-3β-mediated il-10 expression. Sci Rep-Uk. (2016) 6:29359. doi: 10.1038/srep29359
172. Lin L, Chen H, Zhang Y, Lin W, Liu Y, Li T, et al. Il-10 protects neurites in oxygen-glucose-deprived cortical neurons through the pi3k/akt pathway. PloS One. (2015) 10:e0136959. doi: 10.1371/journal.pone.0136959
173. Kim DH, Bang E, Ha S, Jung HJ, Choi YJ, Yu BP, et al. Organ-differential roles of akt/foxos axis as a key metabolic modulator during aging. Aging Dis. (2021) 12:1713–28. doi: 10.14336/ad.2021.0225
174. Cao G, Lin M, Gu W, Su Z, Duan Y, Song W, et al. The rules and regulatory mechanisms of foxo3 on inflammation, metabolism, cell death and aging in hosts. Life Sci. (2023) 328:121877. doi: 10.1016/j.lfs.2023.121877
175. Banham-Hall E, Clatworthy MR, Okkenhaug K. The therapeutic potential for pi3k inhibitors in autoimmune rheumatic diseases. Open Rheumatol J. (2012) 6:245–58. doi: 10.2174/1874312901206010245
176. Mercurio L, Albanesi C, Madonna S. Recent updates on the involvement of pi3k/akt/mtor molecular cascade in the pathogenesis of hyperproliferative skin disorders. Front Med-Lausanne. (2021) 8. doi: 10.3389/fmed.2021.665647
177. Park SY, Lee SW, Lee SY, Hong KW, Bae SS, Kim K, et al. Sirt1/adenosine monophosphate-activated protein kinase A Signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.01135
178. Gong H, Chen H, Xiao P, Huang N, Han X, Zhang J, et al. Mir-146a impedes the anti-aging effect of ampk via nampt suppression and nad(+)/sirt inactivation. Signal Transduct Target Ther. (2022) 7:66. doi: 10.1038/s41392-022-00886-3
179. Han X, Tai H, Wang X, Wang Z, Zhou J, Wei X, et al. Ampk activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular nad(+) elevation. Aging Cell. (2016) 15:416–27. doi: 10.1111/acel.12446
180. Kim J, Yang G, Kim Y, Kim J, Ha J. Ampk activators: mechanisms of action and physiological activities. Exp Mol Med. (2016) 48:e224. doi: 10.1038/emm.2016.16
181. Cui Y, Chen J, Zhang Z, Shi H, Sun W, Yi Q. The role of ampk in macrophage metabolism, function and polarisation. J Trans Med. (2023) 21:892. doi: 10.1186/s12967-023-04772-6
182. Chen X, Li X, Zhang W, He J, Xu B, Lei B, et al. Activation of ampk inhibits inflammatory response during hypoxia and reoxygenation through modulating jnk-mediated nf-Kb pathway. Metabolism. (2018) 83:256–70. doi: 10.1016/j.metabol.2018.03.004
183. Li C, Chen T, Zhou H, Feng Y, Hoi MPM, Ma D, et al. Bhdpc is a novel neuroprotectant that provides anti-neuroinflammatory and neuroprotective effects by inactivating nf-Kb and activating pka/creb. Front Pharmacol. (2018) 9:614. doi: 10.3389/fphar.2018.00614
184. Cherpokova D, Jouvene CC, Libreros S, DeRoo EP, Chu L, de la Rosa X, et al. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood. (2019) 134:1458–68. doi: 10.1182/blood.2018886317
185. Tavares LP, Negreiros-Lima GL, Lima KM, E Silva PMR, Pinho V, Teixeira MM, et al. Blame the signaling: role of camp for the resolution of inflammation. Pharmacol Res. (2020) 159:105030. doi: 10.1016/j.phrs.2020.105030
186. Lee H-J, Ko H-J, Song D-K, Jung Y-J. Lysophosphatidylcholine Promotes Phagosome Maturation and Regulates Inflammatory Mediator Production through the Protein Kinase a–Phosphatidylinositol 3 Kinase–P38 Mitogen-Activated Protein Kinase Signaling Pathway During Mycobacterium Tuberculosis Infection in Mouse Macrophages. Front Immunol. (2018) 9:920. doi: 10.3389/fimmu.2018.00920
187. Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel S-P, Andrzejewski S, et al. Ampk maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. (2017) 21:1–9. doi: 10.1016/j.celrep.2017.09.026
188. Signorile A, De Rasmo D. Mitochondrial complex I, a possible sensible site of camp pathway in aging. Antioxidants-Basel. (2023) 12:221. doi: 10.3390/antiox12020221
189. Kadenbach B. Regulation of cytochrome C oxidase contributes to health and optimal life. World J Biol Chem. (2020) 11:52–61. doi: 10.4331/wjbc.v11.i2.52
190. García-Bermúdez J, Sánchez-Aragó M, Soldevilla B, del Arco A, Nuevo-Tapioles C, Cuezva José M. Pka phosphorylates the atpase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H+-atp synthase. Cell Rep. (2015) 12:2143–55. doi: 10.1016/j.celrep.2015.08.052
191. Ould Amer Y, Hebert-Chatelain E. Mitochondrial camp-pka signaling: what do we really know? Biochim Biophys Acta (BBA) - Bioenergetics. (2018) 1859:868–77. doi: 10.1016/j.bbabio.2018.04.005
192. Wang Z, Zhang L, Liang Y, Zhang C, Xu Z, Zhang L, et al. Cyclic amp mimics the anti-ageing effects of calorie restriction by up-regulating sirtuin. Sci Rep-Uk. (2015) 5:12012. doi: 10.1038/srep12012
193. Kim J, Kundu M, Viollet B, Guan K-L. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
194. Sanduja S, Feng Y, Mathis RA, Sokol ES, Reinhardt F, Halaban R, et al. Ampk promotes tolerance to ras pathway inhibition by activating autophagy. Oncogene. (2016) 35:5295–303. doi: 10.1038/onc.2016.70
195. Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The tor and pka signaling pathways independently target the atg1/atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U.S.A. (2009) 106:17049–54. doi: 10.1073/pnas.0903316106
196. Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of ampk. Am J Physiology-Endocrinology Metab. (2016) 311:E730–E40. doi: 10.1152/ajpendo.00225.2016
197. Ruderman NB, Carling D, Prentki M, Cacicedo JM. Ampk, insulin resistance, and the metabolic syndrome. J Clin Invest. (2013) 123:2764–72. doi: 10.1172/JCI67227
198. Sepúlveda C, Hernández B, Burgos CF, Fuentes E, Palomo I, Alarcón M. The camp/pka pathway inhibits beta-amyloid peptide release from human platelets. Neuroscience. (2019) 397:159–71. doi: 10.1016/j.neuroscience.2018.11.025
199. Li L, Fan X, Zhang X-T, Yue S-Q, Sun Z-Y, Zhu J-Q, et al. The effects of chinese medicines on camp/pka signaling in central nervous system dysfunction. Brain Res Bull. (2017) 132:109–17. doi: 10.1016/j.brainresbull.2017.04.006
200. Gao F, Yang S, Wang J, Zhu G. Camp-pka cascade: an outdated topic for depression? Biomedicine Pharmacotherapy. (2022) 150:113030. doi: 10.1016/j.biopha.2022.113030
201. Chen D, Wang J, Cao J, Zhu G. Camp-pka signaling pathway and anxiety: where do we go next? Cell Signalling. (2024) 122:111311. doi: 10.1016/j.cellsig.2024.111311
202. Aktan F. Inos-mediated nitric oxide production and its regulation. Life Sci. (2004) 75:639–53. doi: 10.1016/j.lfs.2003.10.042
203. Mendes AF, Carvalho AP, Caramona MM, Lopes MC. Role of nitric oxide in the activation of nf-Kb, ap-1 and nos ii expression in articular chondrocytes. Inflammation Res. (2002) 51:369–75. doi: 10.1007/PL00000317
204. Obasanjo-Blackshire K, Mesquita R, Jabr RI, Molkentin JD, Hart SL, Marber MS, et al. Calcineurin regulates nfat-dependent inos expression and protection of cardiomyocytes: co-operation with src tyrosine kinase. Cardiovasc Res. (2006) 71:672–83. doi: 10.1016/j.cardiores.2006.05.026
205. Ganster RW, Taylor BS, Shao L, Geller DA. Complex regulation of human inducible nitric oxide synthase gene transcription by stat 1 and nf-Kb. Proc Natl Acad Sci. (2001) 98:8638–43. doi: 10.1073/pnas.151239498
206. Lu D-Y, Liou H-C, Tang C-H, Fu W-M. Hypoxia-induced inos expression in microglia is regulated by the pi3-kinase/akt/mtor signaling pathway and activation of hypoxia inducible factor-1α. Biochem Pharmacol. (2006) 72:992–1000. doi: 10.1016/j.bcp.2006.06.038
207. Li D-Y, Gao S-J, Sun J, Zhang L-Q, Wu J-Y, Song F-H, et al. Targeting the nitric oxide/cgmp signaling pathway to treat chronic pain. Neural Regeneration Res. (2023) 18(5):996–1003. doi: 10.4103/1673-5374.355748
208. Rossaneis AC, Longhi-Balbinot DT, Bertozzi MM, Fattori V, Segato-Vendrameto CZ, Badaro-Garcia S, et al. Ru(Bpy)2(No)So3](Pf6), a nitric oxide donating ruthenium complex, reduces gout arthritis in mice. Front Pharmacol. (2019) 10:229. doi: 10.3389/fphar.2019.00229
209. Manchope MF, Calixto-Campos C, Coelho-Silva L, Zarpelon AC, Pinho-Ribeiro FA, Georgetti SR, et al. Naringenin inhibits superoxide anion-induced inflammatory pain: role of oxidative stress, cytokines, nrf-2 and the no–cgmp–pkg–katpchannel signaling pathway. PloS One. (2016) 11:e0153015. doi: 10.1371/journal.pone.0153015
210. Corti A, Franzini M, Scataglini I, Pompella A. Mechanisms and targets of the modulatory action of S-nitrosoglutathione (Gsno) on inflammatory cytokines expression. Arch Biochem Biophysics. (2014) 562:80–91. doi: 10.1016/j.abb.2014.08.002
211. Nguyen TH, Axell A, Turek I, Wright B, Meehan-Andrews T, Irving HR. Modulation of inflammatory cytokine production in human monocytes by cgmp and irak3. Int J Mol Sci. (2022) 23(5):2552. doi: 10.3390/ijms23052552
212. Fürnrohr BG, Sheriff A, Munoz L, von Briesen H, Urbonaviciute V, Neubert K, et al. Signals, receptors, and cytokines involved in the immunomodulatory and anti-inflammatory properties of apoptotic cells. Signal Transduction. (2005) 5:356–65. doi: 10.1002/sita.200500071
213. Choi K-S, Song E-K, Yim C-Y. Cytokines secreted by il-2-activated lymphocytes induce endogenous nitric oxide synthesis and apoptosis in macrophages. J Leukocyte Biol. (2008) 83:1440–50. doi: 10.1189/jlb.1007701
214. Schieke SM, Briviba K, Klotz L-O, Sies H. Activation pattern of mitogen-activated protein kinases elicited by peroxynitrite: attenuation by selenite supplementation. FEBS Lett. (1999) 448:301–3. doi: 10.1016/S0014-5793(99)00372-5
215. Arora D, Jain P, Singh N, Kaur H, Bhatla SC. Mechanisms of nitric oxide crosstalk with reactive oxygen species scavenging enzymes during abiotic stress tolerance in plants. Free Radic Res. (2016) 50:291–303. doi: 10.3109/10715762.2015.1118473
216. Nakato R, Ohkubo Y, Konishi A, Shibata M, Kaneko Y, Iwawaki T, et al. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci Rep-Uk. (2015) 5:14812. doi: 10.1038/srep14812
217. Liu Y, Deng S, Sun L, He H, Zhou Q, Fan H, et al. Compound sophorae decoction mitigates dss-induced ulcerative colitis by activating autophagy through pi3k-akt pathway: A integrative research combining network pharmacology and in vivo animal model validation. J Ethnopharmacology. (2025) 337:118885. doi: 10.1016/j.jep.2024.118885
218. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol. (2017) 176:55–62. doi: 10.1016/j.clim.2017.01.007
219. Herrington FD, Carmody RJ, Goodyear CS. Modulation of nf-Kb signaling as a therapeutic target in autoimmunity. J Biomolecular Screening. (2015) 21:223–42. doi: 10.1177/1087057115617456
220. Dutta S, Ganguly A, Ghosh Roy S. An overview of the unfolded protein response (Upr) and autophagy pathways in human viral oncogenesis. Int Rev Cell Mol Biol. (2024) 386:81–131. doi: 10.1016/bs.ircmb.2024.01.004
221. Liu L-K, Jian J-T, Jing S-S, Gao R-L, Chi X-D, Tian G, et al. The crustacean DNA virus tegument protein vp26 binds to snap29 to inhibit snare complex assembly and autophagic degradation. J Virol. (2024) 98:e01408–23. doi: 10.1128/jvi.01408-23
222. Wyatt S, Glover K, Dasanna S, Lewison M, González-García M, Colbert CL, et al. Epstein–barr virus encoded bcl2, bhrf1, downregulates autophagy by noncanonical binding of becn1. Biochemistry. (2023) 62:2934–51. doi: 10.1021/acs.biochem.3c00225
223. Nagdev PK, Agnivesh PK, Roy A, Sau S, Kalia NP. Exploring and exploiting the host cell autophagy during mycobacterium tuberculosis infection. Eur J Clin Microbiol Infect Dis. (2023) 42:1297–315. doi: 10.1007/s10096-023-04663-0
224. Shi Y, Wu Z, Zeng P, Song J, Guo J, Yang X, et al. Seneca valley virus 3c protease blocks epha2-mediated mtor activation to facilitate viral replication. Microbial Pathogenesis. (2024) 191:106673. doi: 10.1016/j.micpath.2024.106673
225. Chen B, Guo G, Wang G, Zhu Q, Wang L, Shi W, et al. Atg7/gaplinc/irf3 axis plays a critical role in regulating pathogenesis of influenza a virus. PloS Pathog. (2024) 20:e1011958. doi: 10.1371/journal.ppat.1011958
226. Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and tnf-A Regulate autophagy through C-jun N-terminal kinase and akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. (2006) 84:448–54. doi: 10.1111/j.1440-1711.2006.01454.x
227. Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. (2003) 206:1107–15. doi: 10.1242/jeb.00220
228. Ashwell JD. The many paths to P38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. (2006) 6:532–40. doi: 10.1038/nri1865
229. Cuenda A, Rousseau S. P38 map-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. (2007) 1773:1358–75. doi: 10.1016/j.bbamcr.2007.03.010
230. Wuyts WA, Vanaudenaerde BM, Dupont LJ, Demedts MG, Verleden GM. Involvement of P38 mapk, jnk, P42/P44 erk and nf-Kb in il-1β-induced chemokine release in human airway smooth muscle cells. Respir Med. (2003) 97:811–7. doi: 10.1016/S0954-6111(03)00036-2
231. Pu W, Chu X, Guo H, Huang G, Cui T, Huang B, et al. The activated atm/ampk/mtor axis promotes autophagy in response to oxidative stress-mediated DNA damage co-induced by molybdenum and cadmium in duck testes. Environ pollut. (2023) 316:120574. doi: 10.1016/j.envpol.2022.120574
232. Li R, Luo X, Zhu Y, Zhao L, Li L, Peng Q, et al. Atm signals to ampk to promote autophagy and positively regulate DNA damage in response to cadmium-induced ros in mouse spermatocytes. Environ pollut. (2017) 231:1560–8. doi: 10.1016/j.envpol.2017.09.044
233. Zhou K, Bellenguez C, Sutherland C, Hardie G, Palmer C, Donnelly P, et al. The role of atm in response to metformin treatment and activation of ampk. Nat Genet. (2012) 44:361–2. doi: 10.1038/ng.2234
234. Baregamian N, Song J, Bailey CE, Papaconstantinou J, Evers BM, Chung DH. Tumor necrosis factor-A and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy and C-jun N-terminal kinase/P38 phosphorylation during necrotizing enterocolitis. Oxid Med Cell Longevity. (2009) 2:893614. doi: 10.4161/oxim.2.5.9541
235. Sivaprasad U, Basu A. Inhibition of erk attenuates autophagy and potentiates tumour necrosis factor-A-induced cell death in mcf-7 cells. J Cell Mol Med. (2008) 12:1265–71. doi: 10.1111/j.1582-4934.2008.00282.x
236. Yi W, Wen Y, Tan F, Liu X, Lan H, Ye H, et al. Impact of nf-Kb pathway on the apoptosis-inflammation-autophagy crosstalk in human degenerative nucleus pulposus cells. Aging (Albany NY). (2019) 11:7294–306. doi: 10.18632/aging.102266
237. Zhu BS, Xing CG, Lin F, Fan XQ, Zhao K, Qin ZH. Blocking nf-Kb nuclear translocation leads to P53-related autophagy activation and cell apoptosis. World J Gastroenterol. (2011) 17:478–87. doi: 10.3748/wjg.v17.i4.478
238. Shang YY, Yao M, Zhou ZW, Jian C, Li X, Hu RY, et al. Alisertib promotes apoptosis and autophagy in melanoma through P38 mapk-mediated aurora a signaling. Oncotarget. (2017) 8:107076–88. doi: 10.18632/oncotarget.22328
239. Liu J, Chang F, Li F, Fu H, Wang J, Zhang S, et al. Palmitate promotes autophagy and apoptosis through ros-dependent jnk and P38 mapk. Biochem Biophys Res Commun. (2015) 463:262–7. doi: 10.1016/j.bbrc.2015.05.042
240. Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of nf-Kb activation by map kinase cascades. Immunobiology. (1997) 198:35–49. doi: 10.1016/S0171-2985(97)80025-3
241. Saikia R, Joseph J. Ampk: A key regulator of energy stress and calcium-induced autophagy. J Mol Med (Berl). (2021) 99:1539–51. doi: 10.1007/s00109-021-02125-8
242. Wu J, Lei Z, Yu J. Hypoxia induces autophagy in human vascular endothelial cells in a hypoxia-inducible factor 1−Dependent manner. Mol Med Rep. (2015) 11:2677–82. doi: 10.3892/mmr.2014.3093
243. Liang R, Liu N, Cao J, Liu T, Sun P, Cai X, et al. Hif-1α/foxo1 axis regulated autophagy is protective for B Cell survival under hypoxia in human islets. Biochim Biophys Acta (BBA) - Mol Basis Dis. (2022) 1868:166356. doi: 10.1016/j.bbadis.2022.166356
244. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (Hif-1α) for lysosomal degradation. J Biol Chem. (2013) 288:10703–14. doi: 10.1074/jbc.M112.414771
245. Yang JY, Park MY, Park SY, Yoo HI, Kim MS, Kim JH, et al. Nitric oxide-induced autophagy in mc3t3-E1 cells is associated with cytoprotection via ampk activation. kjpp. (2015) 19:507–14. doi: 10.4196/kjpp.2015.19.6.507
246. Li Y, Zhang Y, Wang L, Wang P, Xue Y, Li X, et al. Autophagy impairment mediated by S-nitrosation of atg4b leads to neurotoxicity in response to hyperglycemia. Autophagy. (2017) 13:1145–60. doi: 10.1080/15548627.2017.1320467
247. Sarkar S, Korolchuk Viktor I, Renna M, Imarisio S, Fleming A, Williams A, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. (2011) 43:19–32. doi: 10.1016/j.molcel.2011.04.029
248. Caviedes A, Maturana B, Corvalán K, Engler A, Gordillo F, Varas-Godoy M, et al. Enos-dependent S-nitrosylation of the nf-Kb subunit P65 has neuroprotective effects. Cell Death Dis. (2021) 12:4. doi: 10.1038/s41419-020-03338-4
249. Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EFM, et al. Nitric oxide represses inhibitory Kb kinase through S-nitrosylation. Proc Natl Acad Sci. (2004) 101:8945–50. doi: 10.1073/pnas.0400588101
250. Um H-C, Jang J-H, Kim D-H, Lee C, Surh Y-J. Nitric oxide activates nrf2 through S-nitrosylation of keap1 in pc12 cells. Nitric Oxide. (2011) 25:161–8. doi: 10.1016/j.niox.2011.06.001
251. Mattart L, Calay D, Simon D, Roebroeck L, Caesens-Koenig L, Van Steenbrugge M, et al. The peroxynitrite donor 3-morpholinosydnonimine activates nrf2 and the upr leading to a cytoprotective response in endothelial cells. Cell Signalling. (2012) 24:199–213. doi: 10.1016/j.cellsig.2011.09.002
252. Kim SG, Kim SO. Pkc downstream of pl3-kinase regulates peroxynitrite formation for nrf2-mediated gsta2 induction. Arch Pharm Res. (2004) 27:757–62. doi: 10.1007/bf02980145
253. Kang KW, Choi SH, Kim SG. Peroxynitrite activates nf-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. (2002) 7:244–53. doi: 10.1016/s1089-8603(02)00117-9
254. Kinobe R, Ji Y, Nakatsu K. Peroxynitrite-mediated inactivation of heme oxygenases. BMC Pharmacol. (2004) 4:26. doi: 10.1186/1471-2210-4-26
255. Sivrikaya A, Kolayli S, Kucuk M, Aliyazicioglu R. In vitro effects of peroxynitrite treatment on fish liver catalase activity. J Enzyme Inhibition Medicinal Chem. (2009) 24:432–6. doi: 10.1080/14756360802188313
256. MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34f) by peroxynitrite. Arch Biochem Biophysics. (1999) 366:82–8. doi: 10.1006/abbi.1999.1202
257. Romero-Puertas MC, Laxa M, Mattè A, Zaninotto F, Finkemeier I, Jones AME, et al. S-nitrosylation of peroxiredoxin ii E promotes peroxynitrite-mediated tyrosine nitration. Plant Cell. (2007) 19:4120–30. doi: 10.1105/tpc.107.055061
258. Benhar M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radical Bio Med. (2018) 127:160–4. doi: 10.1016/j.freeradbiomed.2018.01.028
259. Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the nrf2 and nf-Kb pathways in diabetic neuropathy. Redox Biol. (2013) 1:394–7. doi: 10.1016/j.redox.2013.07.005
260. Chen L-G, Zhang Y-Q, Wu Z-Z, Hsieh C-W, Chu C-S, Wung B-S. Peanut arachidin-1 enhances nrf2-mediated protective mechanisms against tnf-A-induced icam-1 expression and nf-Kb activation in endothelial cells. Int J Mol Med. (2018) 41:541–7. doi: 10.3892/ijmm.2017.3238
261. Liu GH, Qu J, Shen X. Nf-kappab/P65 antagonizes nrf2-are pathway by depriving cbp from nrf2 and facilitating recruitment of hdac3 to mafk. Biochim Biophys Acta. (2008) 1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002
262. Saha S, Buttari B, Panieri E, Profumo E, Saso L. An overview of nrf2 signaling pathway and its role in inflammation. Molecules. (2020) 25(22):5474. doi: 10.3390/molecules25225474
263. Zipper LM, Mulcahy RT. Inhibition of erk and P38 map kinases inhibits binding of nrf2 and induction of gcs genes. Biochem Biophys Res Commun. (2000) 278:484–92. doi: 10.1006/bbrc.2000.3830
264. Wang H, Liu K, Chi Z, Zhou X, Ren G, Zhou R, et al. Interplay of mkp-1 and nrf2 drives tumor growth and drug resistance in non-small cell lung cancer. Aging (Albany NY). (2019) 11:11329–46. doi: 10.18632/aging.102531
265. Granatiero V, Konrad C, Bredvik K, Manfredi G, Kawamata H. Nrf2 signaling links er oxidative protein folding and calcium homeostasis in health and disease. Life Sci Alliance. (2019) 2(5):e201900563. doi: 10.26508/lsa.201900563
266. Huang Y, Li F. Effects of nrf2 on sarcopplasmic retiulum calcium regulation in C2c12 cells and its mechanism. Acta Med Mediterr. (2019) 35:2917. doi: 10.19193/0393-6384_2019_6_455
267. Patra T, Meyer K, Geerling L, Isbell TS, Hoft DF, Brien J, et al. Sars-cov-2 spike protein promotes il-6 trans-signaling by activation of angiotensin ii receptor signaling in epithelial cells. PloS Pathog. (2020) 16:e1009128. doi: 10.1371/journal.ppat.1009128
268. El-Arif G, Khazaal S, Farhat A, Harb J, Annweiler C, Wu Y, et al. Angiotensin ii type I receptor (At1r): the gate towards covid-19-associated diseases. Molecules. (2022) 27:2048. doi: 10.3390/molecules27072048
269. Rurua M, Ratiani L, Sanikidze T, Machvariani K, Pachkoria E, Ormocadze G, et al. Impact of the angiotensin-converting enzyme (Ace) inhibitors on the course of the septic shock developed during covid-19 and other severe respiratory infections in presence of hyperferritinemia. Georgian Med News. (2023) 337):110–7.
270. Sarzani R, Giulietti F, Di Pentima C, Filipponi A, Spannella F. Antagonizing the renin-angiotensin-aldosterone system in the era of covid-19. Intern Emerg Med. (2020) 15:885–7. doi: 10.1007/s11739-020-02365-5
271. Sun Q, Li L, Jin F, Liu Y, Yang B, Meng W, et al. Sars-cov-2 spike protein S1 exposure increases susceptibility to angiotensin ii-induced hypertension in rats by promoting central neuroinflammation and oxidative stress. Neurochem Res. (2023) 48:3016–26. doi: 10.1007/s11064-023-03949-1
272. Engelhardt S, Rochais F. G proteins: more than transducers of receptor-generated signals? Circ Res. (2007) 100:1109–11. doi: 10.1161/01.RES.0000266971.15127.e8
273. Li Y, Anand-Srivastava MB. Role of gi proteins in the regulation of blood pressure and vascular remodeling. Biochem Pharmacol. (2023) 208:115384. doi: 10.1016/j.bcp.2022.115384
274. Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin Ii Signal Transduction through the At1 Receptor: Novel Insights into Mechanisms and Pathophysiology. Clin Sci (Lond). (2007) 112:417–28. doi: 10.1042/cs20060342
275. Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin ii and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res. (2010) 106:1363–73. doi: 10.1161/CIRCRESAHA.109.216036
276. Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrol (Carlton). (2005) 10 Suppl:S7–10. doi: 10.1111/j.1440-1797.2005.00448.x
277. Hordijk PL. Regulation of nadph oxidases: the role of rac proteins. Circ Res. (2006) 98:453–62. doi: 10.1161/01.RES.0000204727.46710.5e
278. Haynes J Jr., Robinson J, Saunders L, Taylor AE, Strada SJ. Role of camp-dependent protein kinase in camp-mediated vasodilation. Am J Physiol. (1992) 262:H511–6. doi: 10.1152/ajpheart.1992.262.2.H511
279. Lawrence T, Gilroy DW. Chronic inflammation: A failure of resolution? Int J Exp Pathol. (2007) 88:85–94. doi: 10.1111/j.1365-2613.2006.00507.x
280. Staal J, Driege Y, Haegman M, Kreike M, Iliaki S, Vanneste D, et al. Defining the combinatorial space of pkc::Card-cc signal transduction nodes. FEBS J. (2021) 288:1630–47. doi: 10.1111/febs.15522
282. Liu M, Yang Y, Gu C, Yue Y, Wu KK, Wu J, et al. Spike protein of sars-cov stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J. (2007) 21:1586–96. doi: 10.1096/fj.06-6589com
283. Hryciw DH. Special issue: “Recent advances in ion channels and ion channelopathies. Int J Mol Sci. (2024) 25(7):3598. doi: 10.3390/ijms25073598
284. Monat J, Altieri LG, Enrique N, Sedán D, Andrinolo D, Milesi V, et al. Direct inhibition of bk channels by cannabidiol, one of the principal therapeutic cannabinoids derived from cannabis sativa. J Natural Products. (2024) 87:1368–75. doi: 10.1021/acs.jnatprod.3c01274
285. Van NTH, Kim WK, Nam JH. Challenges in the therapeutic targeting of kca channels: from basic physiology to clinical applications. Int J Mol Sci. (2024) 25(5):2965. doi: 10.3390/ijms25052965
286. Freundt EC, Yu L, Goldsmith CS, Welsh S, Cheng A, Yount B, et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J Virol. (2010) 84:1097–109. doi: 10.1128/JVI.01662-09
287. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The orf3a protein of sars-cov-2 induces apoptosis in cells. Cell Mol Immunol. (2020) 17:881–3. doi: 10.1038/s41423-020-0485-9
288. Deng Y, Angelova A. Coronavirus-induced host cubic membranes and lipid-related antiviral therapies: A focus on bioactive plasmalogens. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.630242
289. Snijder EJ, Limpens RWAL, de Wilde AH, de Jong AWM, Zevenhoven-Dobbe JC, Maier HJ, et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down rna synthesis. PloS Biol. (2020) 18:e3000715. doi: 10.1371/journal.pbio.3000715
290. Preissler S, Rato C, Yan Y, Perera LA, Czako A, Ron D. Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising bip-substrate complexes. eLife. (2020) 9:e62601. doi: 10.7554/eLife.62601
291. Qu Y, Sun Y, Yang Z, Ding C. Calcium ions signaling: targets for attack and utilization by viruses. Front Microbiol. (2022) 13:889374. doi: 10.3389/fmicb.2022.889374
292. Zhang J, Hom K, Zhang C, Nasr M, Gerzanich V, Zhang Y, et al. Sars-cov-2 orf3a protein as a therapeutic target against covid-19 and long-term post-infection effects. Pathogens. (2024) 13:75. doi: 10.3390/pathogens13010075
293. Zhang XW, Yap YL. The 3d structure analysis of sars-cov S1 protein reveals a link to influenza virus neuraminidase and implications for drug and antibody discovery. Theochem. (2004) 681:137–41. doi: 10.1016/j.theochem.2004.04.065
294. Alves-Filho JC, Spiller F, Cunha FQ. Neutrophil paralysis in sepsis. Shock. (2010) 34(7):15–21. doi: 10.1097/SHK.0b013e3181e7e61b
295. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-cov-2–triggered neutrophil extracellular traps mediate covid-19 pathologysars-cov-2 directly triggers ace-dependent nets. J Exp Med. (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
296. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
297. de Oliveira Formiga R, Amaral FC, Souza CF, Mendes D, Wanderley CWS, Lorenzini CB, et al. Neuraminidase is a host-directed approach to regulate neutrophil responses in sepsis and covid-19. Br J Pharmacol. (2022) 180(11):1460–81. doi: 10.1111/bph.16013
298. Amith SR, Jayanth P, Franchuk S, Siddiqui S, Seyrantepe V, Gee K, et al. Dependence of pathogen molecule-induced toll-like receptor activation and cell function on neu1 sialidase. Glycoconjugate J. (2009) 26:1197–212. doi: 10.1007/s10719-009-9239-8
299. Abdulkhalek S, Amith SR, Franchuk SL, Jayanth P, Guo M, Finlay T, et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for toll-like receptor activation and cellular signaling. J Biol Chem. (2011) 286:36532–49. doi: 10.1074/jbc.M111.237578
300. Formiga RO, Amaral FC, Souza CF, Mendes D, Wanderley CWS, Lorenzini CB, et al. Neuraminidase inhibitors rewire neutrophil function in vivo in murine sepsis and ex vivo in covid-19. bioRxiv. (2021) bioRxiv 2020.11.12.379115. doi: 10.1101/2020.11.12.379115
301. Wu J, Zhao M, Wei H, Li C, Hu D, Zheng L, et al. Neuraminidase inhibitor treatment is associated with decreased mortality in covid-19 patients: A retrospective analysis. Eur Heart J Cardiovasc Pharmacother. (2022) 8:392–401. doi: 10.1093/ehjcvp/pvac018
302. Shenoy S. Coronavirus (Covid-19) sepsis: revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflammation Res. (2020) 69:1077–85. doi: 10.1007/s00011-020-01389-z
303. Ajaz S, McPhail MJ, Singh KK, Mujib S, Trovato FM, Napoli S, et al. Mitochondrial metabolic manipulation by sars-cov-2 in peripheral blood mononuclear cells of patients with covid-19. Am J Physiology-Cell Physiol. (2020) 320:C57–65. doi: 10.1152/ajpcell.00426.2020
304. Jana S, Heaven MR, Stauft CB, Wang TT, Williams MC, D’Agnillo F, et al. Hif-1alpha-dependent metabolic reprogramming, oxidative stress, and bioenergetic dysfunction in sars-cov-2-infected hamsters. Int J Mol Sci. (2023) 24(1):558. doi: 10.3390/ijms24010558
305. Michalak K, Sobolewska-Wlodarczyk A, Wlodarczyk M, Sobolewska J, Wozniak P, Sobolewski B. Treatment of the fluoroquinolone-associated disability: the pathobiochemical implications. Oxid Med Cell Longev. (2017) 2017:8023935. doi: 10.1155/2017/8023935
306. Kow CS, Ramachandram DS, Hasan SS. Metformin therapy in covid-19: inhibition of netosis. J Thromb Thrombolysis. (2022) 54:217–8. doi: 10.1007/s11239-022-02667-9
307. Cory TJ, Emmons RS, Yarbro JR, Davis KL, Pence BD. Metformin suppresses monocyte immunometabolic activation by sars-cov-2 spike protein subunit 1. Front Immunol. (2021) 12:733921. doi: 10.3389/fimmu.2021.733921
308. Pujalte-Martin M, Belaid A, Bost S, Kahi M, Peraldi P, Rouleau M, et al. Targeting cancer and immune cell metabolism with the complex I inhibitors metformin and iacs-010759. Mol Oncol. (2024) 18(7):1719–38. doi: 10.1002/1878-0261.13583
309. Peralta S, Pinto M, Arguello T, Garcia S, Diaz F, Moraes CT. Metformin delays neurological symptom onset in a mouse model of neuronal complex I deficiency. JCI Insight. (2020) 5(21):e141183. doi: 10.1172/jci.insight.141183
310. Fontaine E. Metformin and respiratory chain complex I: the last piece of the puzzle? Biochem J. (2014) 463:e3–5. doi: 10.1042/BJ20141020
311. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. (2014) 3:e02242. doi: 10.7554/eLife.02242
312. Zhang J, Li Q, Cruz Cosme Ruth S, Gerzanich V, Tang Q, Simard JM, et al. Genome-wide characterization of sars-cov-2 cytopathogenic proteins in the search of antiviral targets. mBio. (2022) 13:e00169–22. doi: 10.1128/mbio.00169-22
313. Tian M, Liu W, Li X, Zhao P, Shereen MA, Zhu C, et al. Hif-1α Promotes sars-cov-2 infection and aggravates inflammatory responses to covid-19. Signal Transduction Targeted Ther. (2021) 6:308. doi: 10.1038/s41392-021-00726-w
314. Padhan K, Tanwar C, Hussain A, Hui PY, Lee MY, Cheung CY, et al. Severe acute respiratory syndrome coronavirus orf3a protein interacts with caveolin. J Gen Virol. (2007) 88:3067–77. doi: 10.1099/vir.0.82856-0
315. Law PTW, Wong CH, Au TCC, Chuck CP, Kong SK, Chan PKS, et al. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in vero E6 cells. J Gen Virol. (2005) 86:1921–30. doi: 10.1099/vir.0.80813-0
316. Padhan K, Minakshi R, Towheed MAB, Jameel S. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through P38 map kinase activation. J Gen Virol. (2008) 89:1960–9. doi: 10.1099/vir.0.83665-0
317. Ren L, Yang R, Guo L, Qu J, Wang J, Hung T. Apoptosis induced by the sars-associated coronavirus in vero cells is replication-dependent and involves caspase. DNA Cell Biol. (2005) 24:496–502. doi: 10.1089/dna.2005.24.496
318. Chan C-M, Tsoi H, Chan W-M, Zhai S, Wong C-O, Yao X, et al. The ion channel activity of the sars-coronavirus 3a protein is linked to its pro-apoptotic function. Int J Biochem Cell Biol. (2009) 41:2232–9. doi: 10.1016/j.biocel.2009.04.019
319. Komaravelli N, Casola A. Respiratory viral infections and subversion of cellular antioxidant defenses. J Pharmacogenomics Pharmacoproteomics. (2014) 5:141. doi: 10.4172/2153-0645.1000141
320. Olagnier D, Farahani E, Thyrsted J, Blay-Cadanet J, Herengt A, Idorn M, et al. Sars-cov2-mediated suppression of nrf2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat Commun. (2020) 11:4938. doi: 10.1038/s41467-020-18764-3
321. Rodrigues D, MaChado MR, Alves JV, Fraga-Silva TFC, Martins RB, Campos LCB, et al. Cytokine storm in individuals with severe covid-19 decreases endothelial cell antioxidant defense via downregulation of the nrf2 transcriptional factor. Am J Physiol Heart Circ Physiol. (2023) 325:H252–H63. doi: 10.1152/ajpheart.00096.2023
322. Liu L, Du J, Yang S, Zheng B, Shen J, Huang J, et al. Sars-cov-2 orf3a sensitizes cells to ferroptosis via keap1-nrf2 axis. Redox Biol. (2023) 63:102752. doi: 10.1016/j.redox.2023.102752
323. Cuadrado A, Pajares M, Benito C, Jiménez-Villegas J, Escoll M, Fernández-Ginés R, et al. Can activation of nrf2 be a strategy against covid-19? Trends Pharmacol Sci. (2020) 41:598–610. doi: 10.1016/j.tips.2020.07.003
324. Qu Y, Haas de Mello A, Morris DR, Jones-Hall YL, Ivanciuc T, Sattler RA, et al. Sars-cov-2 inhibits nrf2-mediated antioxidant responses in airway epithelial cells and in the lung of a murine model of infection. Microbiol Spectr. (2023) 11:e0037823. doi: 10.1128/spectrum.00378-23
325. Zhang S, Wang J, Wang L, Aliyari S, Cheng G. Sars-cov-2 virus nsp14 impairs nrf2/hmox1 activation by targeting sirtuin 1. Cell Mol Immunol. (2022) 19:872–82. doi: 10.1038/s41423-022-00887-w
326. Vassilaki N, Frakolaki E. Virus–host interactions under hypoxia. Microbes Infection. (2017) 19:193–203. doi: 10.1016/j.micinf.2016.10.004
327. D’Agnillo F, Walters K-A, Xiao Y, Sheng Z-M, Scherler K, Park J, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal covid-19. Sci Trans Med. 13(620):eabj7790. doi: 10.1126/scitranslmed.abj7790
328. Rang HP, Dale MM, Ritter JM, Moore PK. Pharmacology. 5th Ed. Churchill Livingstone, Edinburgh: Elsevier (2003).
329. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated glucose levels favor sars-cov-2 infection and monocyte response through a hif-1α/glycolysis-dependent axis. Cell Metab. (2020) 32:437–46.e5. doi: 10.1016/j.cmet.2020.07.007
330. Ozlem Zurnaci F, Guzel M. The effects of increased glucose level and glycolysis on sars cov-2 infection. Mini Rev Med Chem. (2022) 22:2344–9. doi: 10.2174/1389557522666220318115350
331. Ren Z, Yu Y, Chen C, Yang D, Ding T, Zhu L, et al. The triangle relationship between long noncoding rna, rig-I-like receptor signaling pathway, and glycolysis. Front Microbiol. (2021) 12:807737. doi: 10.3389/fmicb.2021.807737
332. Duan X, Tang X, Nair MS, Zhang T, Qiu Y, Zhang W, et al. An airway organoid-based screen identifies a role for the hif1alpha-glycolysis axis in sars-cov-2 infection. Cell Rep. (2021) 37:109920. doi: 10.1016/j.celrep.2021.109920
333. Medini H, Zirman A, Mishmar D. Immune system cells from covid-19 patients display compromised mitochondrial-nuclear expression co-regulation and rewiring toward glycolysis. iScience. (2021) 24:103471. doi: 10.1016/j.isci.2021.103471
334. Umar S, Palasiewicz K, Meyer A, Kumar P, Prabhakar BS, Volin MV, et al. Inhibition of irak4 dysregulates sars-cov-2 spike protein-induced macrophage inflammatory and glycolytic reprogramming. Cell Mol Life Sci. (2022) 79:301. doi: 10.1007/s00018-022-04329-8
335. Barreto EA, Cruz AS, Veras FP, Martins R, Bernardelli RS, Paiva IM, et al. Covid-19-related hyperglycemia is associated with infection of hepatocytes and stimulation of gluconeogenesis. Proc Natl Acad Sci U.S.A. (2023) 120:e2217119120. doi: 10.1073/pnas.2217119120
336. Wan L, Gao Q, Deng Y, Ke Y, Ma E, Yang H, et al. Gp73 is a glucogenic hormone contributing to sars-cov-2-induced hyperglycemia. Nat Metab. (2022) 4:29–43. doi: 10.1038/s42255-021-00508-2
337. Barberis E, Timo S, Amede E, Vanella VV, Puricelli C, Cappellano G, et al. Large-scale plasma analysis revealed new mechanisms and molecules associated with the host response to sars-cov-2. Int J Mol Sci. (2020) 21(22):8623. doi: 10.3390/ijms21228623
338. Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, et al. Transcription factor hif-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. (2001) 21:3436–44. doi: 10.1128/mcb.21.10.3436-3444.2001
339. Chen Z, Lu W, Garcia-Prieto C, Huang P. The warburg effect and its cancer therapeuthic implications. J Bioenerg Biomembr. (2007) 39:267–74. doi: 10.1007/s10863-007-9086-x
340. Faure H, Vigny A, Heddi S, Giraud S, Chautard D, Stepien G. Expression of oxydative phosphorylation genes in renal tumors and tumoral cell lines. Mol Carcinog. (1996) 16:165–72. doi: 10.1002/(SICI)1098-2744(199607)16:3<165::AID-MC7>3.0.CO;2-G
341. Unwin RD, Craven RA, Harnden P, Hanrahan S, Totty N, Knowles M, et al. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the warburg effect. Proteomics. (2003) 3:1620–32. doi: 10.1002/pmic.200300464
342. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, et al. The orf8 protein of sars-cov-2 mediates immune evasion through down-regulating mhc-I. Proc Natl Acad Sci. (2021) 118:e2024202118. doi: 10.1073/pnas.2024202118
343. Sui C, Xiao T, Zhang S, Zeng H, Zheng Y, Liu B, et al. Sars-cov-2 nsp13 inhibits type I ifn production by degradation of tbk1 via P62-dependent selective autophagy. J Immunol. (2022) 208:753–61. doi: 10.4049/jimmunol.2100684
344. Zhao Z, Lu K, Mao B, Liu S, Trilling M, Huang A, et al. The interplay between emerging human coronavirus infections and autophagy. Emerg Microbes Infect. (2021) 10:196–205. doi: 10.1080/22221751.2021.1872353
345. Nie Y, Mou L, Long Q, Deng D, Hu R, Cheng J, et al. Sars-cov-2 orf3a positively regulates nf-Kb activity by enhancing ikkβ-nemo interaction. Virus Res. (2023) 328:199086. doi: 10.1016/j.virusres.2023.199086
346. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of covid-19: current state of the science. Immunity. (2020) 52:910–41. doi: 10.1016/j.immuni.2020.05.002
347. Zhang H, Jiang P, Chen Z, Wang D, Zhou Y, Zhu X, et al. Norovirus 3c-like protease antagonizes interferon-beta production by cleaving nemo. Virology. (2022) 571:12–20. doi: 10.1016/j.virol.2022.04.004
348. Chen J, Wang D, Sun Z, Gao L, Zhu X, Guo J, et al. Arterivirus nsp4 antagonizes interferon beta production by proteolytically cleaving nemo at multiple sites. J Virol. (2019) 93(12). doi: 10.1128/JVI.00385-19
349. Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, et al. Correction: enterovirus 71 protease 2apro targets mavs to inhibit anti-viral type I interferon responses. PloS Pathog. (2024) 20:e1012209. doi: 10.1371/journal.ppat.1012209
350. Nelemans T, Tas A, Kikkert M, van Hemert MJ. Usutu virus ns4a suppresses the host interferon response by disrupting mavs signaling. Virus Res. (2024) 347:199431. doi: 10.1016/j.virusres.2024.199431
351. Xing J, Zhou X, Fang M, Zhang E, Minze LJ, Zhang Z. Dhx15 is required to control rna virus-induced intestinal inflammation. Cell Rep. (2021) 35(12):109205. doi: 10.1016/j.celrep.2021.109205
352. Xing J, Zhang A, Minze LJ, Li XC, Zhang Z. Trim29 negatively regulates the type I ifn production in response to rna virus. J Immunol. (2018) 201:183–92. doi: 10.4049/jimmunol.1701569
353. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. Trim29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
354. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for trim29 in the control of innate immunity in the respiratory tract. Nat Immunol. (2016) 17:1373–80. doi: 10.1038/ni.3580
355. Wang J, Wang L, Lu W, Farhataziz N, Gonzalez A, Xing J, et al. Trim29 controls enteric rna virus-induced intestinal inflammation by targeting nlrp6 and nlrp9b signaling pathways. Mucosal Immunol. (2025) 18:135–50. doi: 10.1016/j.mucimm.2024.10.004
356. Fang M, Zhang A, Du Y, Lu W, Wang J, Minze LJ, et al. Trim18 is a critical regulator of viral myocarditis and organ inflammation. J BioMed Sci. (2022) 29:55. doi: 10.1186/s12929-022-00840-z
357. Shemesh M, Aktepe TE, Deerain JM, McAuley JL, Audsley MD, David CT, et al. Sars-cov-2 suppresses ifnbeta production mediated by nsp1, 5, 6, 15, orf6 and orf7b but does not suppress the effects of added interferon. PloS Pathog. (2021) 17:e1009800. doi: 10.1371/journal.ppat.1009800
358. Deng X, Buckley AC, Pillatzki A, Lager KM, Faaberg KS, Baker SC. Inactivating three interferon antagonists attenuates pathogenesis of an enteric coronavirus. J Virol. (2020) 94(17). doi: 10.1128/JVI.00565-20
359. Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. Sars-cov-2 nsp12 attenuates type I interferon production by inhibiting irf3 nuclear translocation. Cell Mol Immunol. (2021) 18:945–53. doi: 10.1038/s41423-020-00619-y
360. Fang P, Hong Y, Xia S, Zhang J, Ren J, Zhou Y, et al. Porcine deltacoronavirus nsp10 antagonizes interferon-beta production independently of its zinc finger domains. Virology. (2021) 559:46–56. doi: 10.1016/j.virol.2021.03.015
361. Menachery VD, Gralinski LE, Mitchell HD, Dinnon KH 3rd, Leist SR, Yount BL Jr., et al. Middle east respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere. (2017) 2(6). doi: 10.1128/mSphere.00346-17
362. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. Sars-cov-2 orf3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. (2020) 32:108185. doi: 10.1016/j.celrep.2020.108185
363. Zhou P, Li H, Wang H, Wang LF, Shi Z. Bat severe acute respiratory syndrome-like coronavirus orf3b homologues display different interferon antagonist activities. J Gen Virol. (2012) 93:275–81. doi: 10.1099/vir.0.033589-0
364. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. Sars-cov-2 orf9b antagonizes type I and iii interferons by targeting multiple components of the rig-I/mda-5-mavs, tlr3-trif, and cgas-sting signaling pathways. J Med Virol. (2021) 93:5376–89. doi: 10.1002/jmv.27050
365. Han L, Zheng Y, Deng J, Nan ML, Xiao Y, Zhuang MW, et al. Sars-cov-2 orf10 antagonizes sting-dependent interferon activation and autophagy. J Med Virol. (2022) 94:5174–88. doi: 10.1002/jmv.27965
366. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (Sars-cov-2) membrane (M) protein inhibits type I and iii interferon production by targeting rig-I/mda-5 signaling. Signal Transduct Target Ther. (2020) 5:299. doi: 10.1038/s41392-020-00438-7
367. Yang Z, Li J, Li J, Zheng H, Li H, Lai Q, et al. Engagement of the G3bp2-trim25 interaction by nucleocapsid protein suppresses the type I interferon response in sars-cov-2-infected cells. Vaccines (Basel). (2022) 10(12):2042. doi: 10.3390/vaccines10122042
368. Ren JG, Seth P, Ye H, Guo K, Hanai JI, Husain Z, et al. Citrate suppresses tumor growth in multiple models through inhibition of glycolysis, the tricarboxylic acid cycle and the igf-1r pathway. Sci Rep. (2017) 7:4537. doi: 10.1038/s41598-017-04626-4
Keywords: SARS-CoV-2, autophagy, inflammation, NOX, Nrf2, calcium signaling, nitric oxide, HIF-1α
Citation: Michalak KP, Michalak AZ and Brenk-Krakowska A (2025) Acute COVID-19 and LongCOVID syndrome – molecular implications for therapeutic strategies - review. Front. Immunol. 16:1582783. doi: 10.3389/fimmu.2025.1582783
Received: 25 February 2025; Accepted: 28 March 2025;
Published: 17 April 2025.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Chandrima Gain, University of California, Los Angeles, United StatesCuncai Guo, Washington University in St. Louis, United States
Copyright © 2025 Michalak, Michalak and Brenk-Krakowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krzysztof Piotr Michalak, a21pY2hhbGFrQGFtdS5lZHUucGw=; Amelia Zofia Michalak, OTIyNjFAc3R1ZGVudC51bXAuZWR1LnBs; Alicja Brenk-Krakowska, YWxpY2phLmJyZW5rLWtyYWtvd3NrYUBhbXUuZWR1LnBs
†ORCID: Krzysztof Piotr Michalak, orcid.org/0000-0002-0267-5605
Amelia Zofia Michalak, orcid.org/0009-0007-5562-8478
Alicja Brenk-Krakowska, orcid.org/0000-0002-9346-9734