 Xinqi Xu1
Xinqi Xu1 Tingyu Yang
Tingyu Yang Zhimin Dou
Zhimin Dou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 March 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1575554
This article is part of the Research TopicExploring Immunometabolism: Metabolic Pathway and Immune Response in SepsisView all articles
Sepsis is defined as a condition related to infection that manifests with multiorgan dysfunction, representing a life-threatening state. Consequently, severe complications frequently occur, with liver injury being one of the most prevalent serious complications of sepsis. Liver dysfunction during sepsis serves as an independent predictor of mortality. This review provides a comprehensive overview of current research on sepsis-induced liver injury (SILI), encompassing the clinical manifestations, diagnostic criteria, pathogenesis and therapeutic strategies associated with this condition. SILI may manifest as hypoxic hepatitis due to ischemia and shock, cholestasis resulting from abnormal bile metabolism, or bile duct sclerosis. The pathophysiology of sepsis involves intricate interactions among the inflammatory response, oxidative stress, and cell death. All of these factors complicate treatment and represent potential targets for therapeutic intervention. Furthermore, this review addresses the limitations inherent in conventional therapies currently employed for managing SILI and emphasizes the potential of novel targeted strategies aimed at addressing the fundamental mechanisms underlying this condition.
Sepsis is a potentially fatal syndrome that arises from multiorgan dysfunction stemming from the host’s dysregulated response to infection. Currently, it has emerged as one of the leading contributors to infection-related mortality worldwide (1). According to epidemiological data, the annual incidence of adult sepsis is approximately 189 cases/100,000 individuals, with a mortality rate as high as 26.7% (2). Notably, 24.4% of sepsis cases occur within the intensive care unit (ICU) (3). Sepsis represents a central focus of medical research because of its high incidence, complex pathogenesis, critical nature, and unfavorable prognosis.
As a crucial metabolic immune organ, the liver clears bacteria and synthesizes acute phase proteins and cytokines during sepsis. Thus, immune defense can be effectively activated through inflammatory responses (4). However, an excessive inflammatory response may cause significant hepatocyte injury. This condition is specifically known as SILI (5). Research has shown that in patients with sepsis complicated by liver dysfunction or failure, the mortality rates range from 54% to 68% (6). These rates are significantly higher than those in patients with respiratory dysfunction or failure (7). Consequently, minimizing liver damage and promoting the recovery of liver function are essential to reduce the mortality rate among patients with sepsis.
With respect to the pivotal role of the liver in the pathophysiology of sepsis, this paper discusses the various types and clinical features of hepatic injury associated with sepsis. It subsequently reviews the critical mechanisms underlying the pathogenesis of hepatic injury, including interactions among inflammatory responses, oxidative stress, mitochondrial dysfunction, pyroptosis, and autophagy. This analysis aims to provide new insights and strategies for addressing hepatic injury in sepsis. More specifically, this paper evaluates current clinical trials and emerging therapies that are grounded in these pathogenic mechanisms. This study provides a theoretical framework and practical reference for future treatment strategies targeting SILI.
In clinical practice, sepsis may cause various types of liver injury, including hypoxic hepatitis, septic cholestasis, and secondary sclerosing cholangitis (Figure 1). These diverse clinical features associated with SILI complicate the diagnostic process. A better understanding of the specific clinical features related to each type is essential for the early and accurate diagnosis of liver injury in sepsis patients.
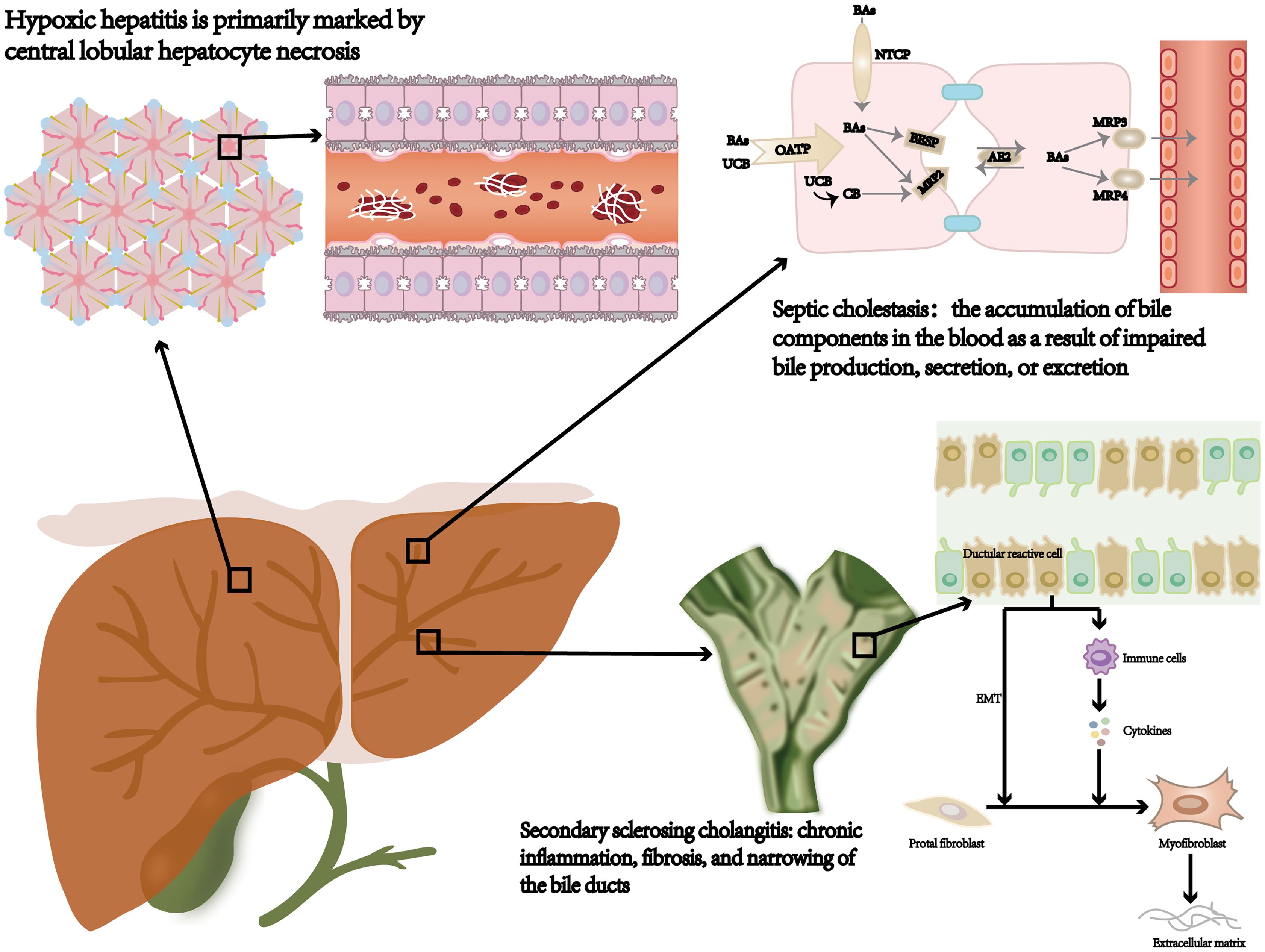
Figure 1. Septic liver injury type: hypoxic hepatitis, septic cholestasis, and secondary sclerosing cholangitis. NTCP, sodium‐taurocholate cotransporting polypeptide; OATP, organic anion transporting polypeptide; AE2, anion exchanger 2; BSEP, bile salt export pump; MDR3/4, multidrug resistance gene 3/4; Bas, bile acids; CB, conjugated bilirubin; UCB, unconjugated bilirubin; EMT, epithelial-mesenchymal transition.
HH typically develops within 48 hours after the occurrence of heart, circulatory, or respiratory failure. It is distinguished by a marked elevation in aminotransferase levels, frequently exceeding the standard upper limit by more than 10 times while ruling out alternative causes of hepatocellular necrosis (8, 9). Histologically, HH is predominantly characterized by central lobular necrosis of hepatocytes (10). Sepsis-induced systemic inflammation in HH adversely affects cardiac pump function, diminishing blood flow to the liver and consequently reducing the oxygen supply. The most common causes of HH include shock (48%), cardiac arrest (25%), and hypoxia (13%) (8). Laboratory features usually include elevated levels of lactate dehydrogenase (LDH), mildly increased total bilirubin (mainly unconjugated), and a prolonged international normalized ratio (INR), indicating coagulation dysfunction (8, 11, 12).
The SC is defined as the accumulation of bile components in the bloodstream due to impaired bile formation, secretion, or excretion during sepsis (13). Dysfunction is typically related to impaired cholangiocyte function without evidence of significant bile duct damage or hepatocyte loss (14). Laboratory tests revealed that the bilirubin level, mainly direct bilirubin, exceeded 2 μmol/L. Additionally, alkaline phosphatase (ALP) and γ-glutamyltransferase (GGT) levels are elevated to more than 2–3 times the normal range, whereas AST and ALT levels are generally within normal limits (9, 15).
Severe sepsis can lead to SSC, which encompasses chronic inflammation, fibrosis, and stenosis of the bile ducts (16, 17). These pathological changes can ultimately progress to cirrhosis and liver failure (18). More importantly, SSC is positively correlated with severe systemic hypotension, trauma, acute respiratory distress syndrome (ARDS), and systemic inflammatory response syndrome (SIRS) (19, 20). The major clinical features of SSC include chronic cholestasis, thickening of the bile duct walls, and either narrowing or dilation. These manifestations can be diagnosed through abdominal CT scans or magnetic resonance cholangiopancreatography (MRCP) (20–22). A distinction should be made from primary sclerosing cholangitis, which is a chronic autoimmune disease.
Considering the high mortality rate of patients with sepsis who experience complications of liver dysfunction, early identification and prompt intervention are crucial for improving patient prognosis.
The clinical manifestations of hepatic injury in sepsis, such as jaundice, elevated liver enzymes, and prolonged prothrombin time, are closely linked to the underlying pathogenic mechanisms. Understanding these mechanisms can provide insights into why and how liver dysfunction occurs in septic patients. The liver serves a dual function in sepsis: it is responsible for maintaining body homeostasis through the elimination of pathogens and toxins. They are also susceptible to injury due to excessive inflammatory responses and immune dysregulation. The systemic inflammatory cascade in sepsis damages liver cells and impairs their functionality. Moreover, advanced age, alcoholism, diabetes, mellitus and cardiac dysfunction are factors that markedly increase the risk of liver injury in septic patients (23). Therefore, understanding the pathogenesis of SILI is important for elucidating the various clinical manifestations of liver damage observed in sepsis.
Research has shown that the pathogenesis of SILI is intricately related to inflammation and its subsequent cascade reactions, oxidative stress, pyroptosis, autophagy, and the regulation of microRNAs (miRNAs) (Figure 2).
Figure 2. Pathogenesis of hepatic injury in sepsis.
The inflammatory response and cascade amplification are central aspects of the pathophysiology of SILI (Figure 3). Under sepsis conditions, bacteria and viruses produce pathogen-associated molecular patterns (PAMPs), which are recognized by pattern recognition receptors (PRRs) expressed on host cells. Among these receptors are Toll-like receptors (TLRs) (24). Gram-negative bacteria-derived lipopolysaccharides (LPSs) trigger immune cells, including Kupffer cells (KCs) and monocytes/macrophages, via the TLR4-mediated signaling pathway (25–27). This activation enhances the secretion of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). These cytokines not only exacerbate local inflammation but also propagate a systemic inflammatory response, commonly referred to as a “cytokine storm” (28, 29).
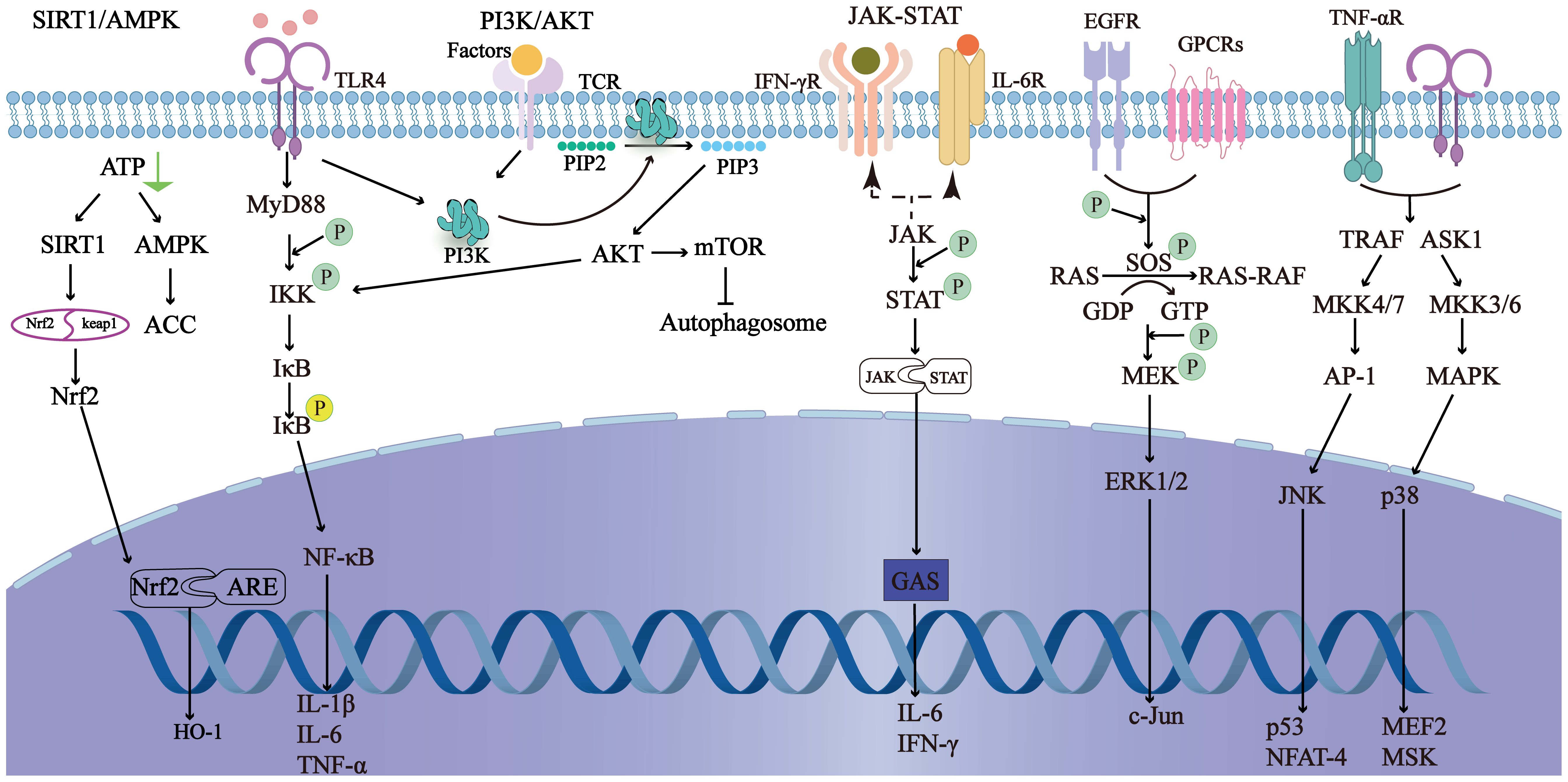
Figure 3. It shows the main related signaling pathways in sepsis-induced liver injury. By regulating the signaling pathways, the inflammatory response can be reduced, autophagy can be initiated, and oxidative stress can be prevented to improve sepsis-induced liver injury.
Additionally, NOD-like receptors (NLRs) play a crucial role in the inflammatory cascade. NLRs are intracellular PRRs that recognize damage-associated molecular patterns (DAMPs) and PAMPs. Activation of NLRs leads to the formation of inflammasomes, which in turn activate caspase-1 (30). Caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature forms, further amplifying the inflammatory response. The NF-κB signaling pathway is also a key component of this inflammatory cascade. Upon activation by PAMPs or DAMPs, NF-κB translocates to the nucleus and induces the transcription of proinflammatory cytokines, chemokines, and adhesion molecules, thereby promoting inflammation. Furthermore, infection with gram-positive bacteria, particularly Staphylococcus aureus, significantly increased the risk of acute liver injury among septic patients (23, 31).
Cytokines directly induce hepatocyte apoptosis or necrosis (32). Moreover, increased recruitment of immune cells to the inflamed site aggravates the inflammatory response, resulting in a cascade amplification effect that ultimately leads to multiple organ dysfunction syndrome (MODS). As the liver serves as a central metabolic organ, its dysfunction significantly impairs overall bodily function. Thus, targeting the disruption of this amplification cascade may be an effective therapy to alleviate liver injury.
Oxidative stress is a pivotal mediator of liver injury in sepsis, exacerbating pathophysiological processes through direct hepatocyte damage and compromising the antioxidant defense system.
In sepsis, the excessive generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is a critical factor. Studies have shown that in severe sepsis patients, the levels of ROS can increase by more than 50% compared with those in healthy individuals, while the activities of antioxidant enzymes such as superoxide dismutase (SOD) and catalase (CAT) are significantly reduced, indicating a state of antioxidant depletion (33, 34). ROS interact with proteins, carbohydrates, nucleic acids, and unsaturated lipids in hepatocytes, causing damage. For example, the interaction of ROS with lipids can lead to lipid peroxidation, which disrupts the integrity of cell membranes and organelles. Additionally, ROS can modify proteins, leading to loss of their function and the potential for aggregation, which can further impair cellular processes (35).
The interplay between oxidative stress and inflammation is intricate and forms a vicious cycle. ROS generation exacerbates inflammatory cascades in several ways (36). Firstly, ROS can activate NF-κB, a key transcription factor that induces the expression of pro-inflammatory cytokines such as tumor TNF-α and IL-6 (37). Studies have shown that in sepsis models, the activation of NF-κB can increase the production of TNF-α by more than 2-fold, further amplifying the inflammatory response (38). Secondly, ROS can also promote the activation of NOD-like receptors (NLRs), leading to the formation of inflammasomes and the activation of caspase-1, which in turn cleaves pro-IL-1β and pro-IL-18 into their mature forms, further amplifying inflammation (39).
Furthermore, oxidative stress contributes to endothelial dysfunction, microthrombosis, hepatic sinusoidal obstruction, and diminished hepatic perfusion, which worsen liver injury (40). For example, the increase in ROS can lead to the activation of endothelial cells, promoting the expression of adhesion molecules and the recruitment of leukocytes, which can cause microvascular inflammation and obstruction (41).
A promising strategy may be incorporating antioxidant therapy into the treatment of sepsis to mitigate oxidative stress-induced liver damage. For example, the use of N-acetylcysteine (NAC), a well-known antioxidant, has been shown to reduce ROS levels by more than 40% and increase antioxidant enzyme activities, thereby alleviating liver injury in sepsis models (42).
Mitochondrial dysfunction represents a critical factor contributing to disturbances in liver energy metabolism and organ damage during sepsis (43). Mitochondrial damage is frequently accompanied by increased oxidative stress, which promotes inflammatory responses and apoptosis. These processes are important mechanisms contributing to the deterioration of both the structure and function of the liver.
LPS, through the activation of hepatic cells, induces an overproduction of ROS, which causes damage to the mitochondrial membrane. This not only interferes with the binding of oxygen by cytochrome oxidase, blocking the respiratory chain and causing metabolic breakdown of liver energy but also triggers the release of cytochrome C, initiating hepatocyte apoptosis (33, 44). In septic mice, the release of cytochrome C from mitochondria can increase by up to 3-fold, significantly promoting apoptosis (44, 45). Liver mitochondria undergo structural injury, including mitochondrial swelling, disorganized crista proliferation, and decreased matrix electron density, in severe sepsis (46). Morphological alterations also include proton leakage, modifications in membrane permeability, and possible disruption of the mitochondrial membrane (33). Among these factors, changes in mitochondrial membrane permeability are critical at the beginning of apoptosis (47). Mitochondria induce changes in membrane permeability by releasing cytochrome C and apoptosis-inducing factors. These releases activate the caspase cascade, leading to the initiation of apoptosis or programmed cell death (48, 49). Regulating mitochondrial function represents a promising therapeutic target for mitigating SILI. This approach may offer a valuable strategy for clinical intervention.
In addition to the structural changes, mitochondrial dynamics, encompassing fusion and fission processes, also play a pivotal role in the progression of SILI. Mitochondrial fusion, which is mediated by proteins such as optic atrophy 1 (OPA1) and mitofusin 2 (MFN2), helps maintain mitochondrial integrity and function by facilitating the exchange of mitochondrial contents and the sharing of mitochondrial DNA (42). During sepsis, the balance between mitochondrial fusion and fission is disrupted, leading to impaired mitochondrial function and increased susceptibility to damage.
Conversely, mitochondrial fission, driven by proteins like dynamin-related protein 1 (DRP1), is essential for the segregation of damaged mitochondria and their subsequent degradation through mitophagy (50). However, excessive fission can result in the fragmentation of mitochondria, reducing their energy production efficiency and increasing the production of ROS. Studies have shown that in septic mice, the expression of DRP1 is upregulated, while the levels of OPA1 and MFN2 are decreased, indicating an imbalance in mitochondrial dynamics. This imbalance contributes to the exacerbation of liver injury by promoting mitochondrial dysfunction and oxidative stress.
Regulating mitochondrial dynamics could be a promising therapeutic strategy for mitigating SILI. For instance, promoting mitochondrial fusion by enhancing the expression of OPA1 or inhibiting excessive fission through the modulation of DRP1 activity may help restore mitochondrial function and reduce liver damage. Further research into the specific mechanisms and potential therapeutic targets related to mitochondrial dynamics in SILI is warranted to provide a more comprehensive understanding of this complex process (51).
Pyroptosis is a form of programmed cell death that plays a vital role in the pathogenesis of SILI. The activation of immune cells initiates the pyroptotic pathway, which facilitates the release of inflammatory mediators. In turn, these inflammatory mediators aggravate inflammatory responses and further hepatocyte injury (52).
In the event of an attack by pathogens or inflammatory mediators, hepatocytes release PAMPs or damage-associated molecular patterns (DAMPs). Upon recognition by PRRs, these molecules initiate the activation of inflammasomes, especially the NOD-like receptor protein 3 (NLRP3) inflammasome, promoting the activation of Caspase-1 (53–55). This activation occurs through a two-step process (56). Initially, PRRs detect PAMPs or DAMPs, triggering the nuclear factor-κB (NF-κB) pathway and increasing NLRP3 and pro-IL-1β expression (57). This primes the cell for inflammation. A second signal, such as extracellular ATP or mitochondrial reactive oxygen species (mtROS), subsequently activates the NLRP3 inflammasome (58). Once activated, it recruits and activates Caspase-1 via the ASC adaptor protein, which then processes pro-IL-1β into its mature form for secretion (59). This secretion recruits immune cells to the liver, amplifying inflammation and exacerbating SILI. Activated Caspase-1 not only facilitates the maturation of proinflammatory cytokines such as IL-1β and IL-8 (60–62) but also cleaves gasdermin D (GSDMD), which forms pores in the cell membrane (63). The pores release cellular contents into the extracellular milieu, promoting inflammation and pyroptosis (64–67). The mature IL-1β cytokine recruits innate immune cells to the site of infection and enhances the activation of acquired immune cells. This process amplifies the inflammatory response and exacerbates liver injury. In addition to the classical Caspase-1-mediated pathway, pyroptosis can also be initiated through a nonclassical pathway involving the activation of Caspase-4, Caspase-5, or Caspase-11. Notably, Caspase-4/5 are especially important in humans (55, 68). These caspases can be directly activated by components derived from intracellular pathogens, such as LPS (69), leading to GSDMD cleavage and initiating pyroptosis (54).
Targeting the pyroptosis pathway and preventing its activation offers a promising strategy for reducing liver injury and may provide a novel therapeutic approach for the management of SILI.
Autophagy is crucial for cellular self-protection and the maintenance of homeostasis by degrading unnecessary proteins and damaged organelles (70). It also plays a vital role in regulating mitochondrial homeostasis by selectively removing damaged mitochondria through mitophagy, which helps maintain cellular energy production and prevent the accumulation of harmful reactive ROS (71, 72).
Mitophagy, the selective autophagy of damaged or dysfunctional mitochondria, is triggered by the loss of mitochondrial membrane potential. This process marks damaged mitochondria with ubiquitin, leading to their recognition and engulfment by autophagosomes. These autophagosomes then fuse with lysosomes, where lysosomal enzymes degrade the contents, ensuring only dysfunctional mitochondria are eliminated while preserving healthy ones for cellular energy needs (73).
Mitophagy is mediated by two main pathways: the ubiquitin-dependent PINK1/Parkin pathway and ubiquitin-independent pathways. In the PINK1/Parkin pathway, PINK1 stabilizes on the outer mitochondrial membrane upon membrane potential loss, recruiting Parkin (74). Parkin ubiquitinates mitochondrial proteins, marking them for degradation (74). Autophagy receptors like p62 recognize these ubiquitinated mitochondria and link them to autophagosomes for clearance (75). Ubiquitin-independent pathways involve mitochondrial proteins such as NIX and BNIP3, which can directly interact with LC3 to initiate autophagosome formation around mitochondria, offering alternative mechanisms for maintaining mitochondrial homeostasis (71, 76).
Research has revealed that SILI is closely associated with impaired autophagy activity in the liver (77). At an early stage of SILI, autophagic flux is increased, resulting in increased accumulation of autophagosomes. However, the number of autophagy-lysosomes decreases, leading to incomplete autophagy and further aggravating liver injury (77). Defective autophagy also results in the accumulation of damaged mitochondria, which further promotes cell death and worsens liver injury (78). When autophagy is overactivated, it may lead to the degradation of essential cellular components, resulting in cell dysfunction and death. In SILI, the excessive production of ROS and the activation of inflammatory signaling pathways can trigger excessive autophagy, which in turn exacerbates liver injury. For instance, the overactivation of autophagy can cause the loss of mitochondrial mass and impair cellular respiration, leading to energy deficiency and cell death (79). These findings suggest that autophagy dysfunction may be critical in the onset and progression of liver injury during sepsis.
Although most research on autophagy remains experimental, its critical role in maintaining cellular homeostasis and alleviating liver injury is undeniable. This pathway can modulate autophagy, particularly in the formation and degradation of autophagosomes, to restore the autophagy-lysosome system. This path offers a novel therapeutic approach for treating liver injury resulting from sepsis. This provides not only a fundamental theoretical basis but also new hope for addressing SILI.
Noncoding RNAs, including miRNAs and long noncoding RNAs (lncRNAs), modulate mRNA stability and translation efficiency post-transcriptionally by binding to target mRNAs. These RNAs play key regulatory roles in apoptosis, inflammation, and other cellular processes.
Epigenetic factors, such as DNA methylation and histone modifications, also significantly influence hepatic inflammatory responses by altering gene expression patterns without changing the DNA sequence (80). DNA methylation, primarily occurring at CpG islands, can lead to gene silencing by inhibiting the binding of transcription factors or recruiting methyl-binding proteins (81). Aberrant methylation patterns during sepsis affect genes involved in immunity and survival. For example, hypermethylation of the promoter region of the anti-inflammatory cytokine IL-10 reduces its expression, impairing the anti-inflammatory response (82).
Histone modifications, including acetylation, methylation, and phosphorylation, regulate gene transcription by altering chromatin structure (83). Acetylation of histone H3 at the promoter regions of proinflammatory cytokines such as TNF-α and IL-6 enhances their transcription, exacerbating liver injury (84). Conversely, deacetylation by histone deacetylases (HDACs) suppresses inflammatory responses and protects against liver damage (85). These epigenetic changes can create a vicious cycle where inflammation and oxidative stress synergistically worsen liver damage, highlighting the potential of targeting these mechanisms for therapeutic intervention.
Dysregulation of the expression of several miRNAs during sepsis is associated with liver injury. Studies have shown that miR-133a is highly expressed in both septic patients and cecal ligation and perforation (CLP) model mice and that its knockout attenuates liver injury, indicating that miR-133a might play a key role in SILI (86). Increased expression of miR-155 causes the inactivation of nuclear factor E2-related factor 2 (Nrf2), which elevates oxidative stress and exacerbates SILI (87). Additionally, miR-126-5p is highly upregulated in liver cells in both in vivo and in vitro models of sepsis. The overexpression of miR-126-5p induces cell apoptosis, thereby corroborating its involvement in SILI (88). In contrast, miR-122a has shown sensitivity and specificity in the diagnosis of SILI, suggesting its potential as a biomarker for SILI (89).
Previous studies have preliminarily verified the clinical value of lncRNAs in sepsis (90). For example, the lncRNA CRND is significantly downregulated in septic rats and LPS-treated hepatocytes. This downregulation of the lncRNA CRND might exacerbate the severity of SILI (88). In addition, the lncRNA NEAT1 can compete with Let-7a to regulate TLR4 to promote the development of SILI (91). The expression of the lncRNA MALAT is upregulated in SILI, where it binds to polypyrimidine tract-binding protein 1 (PTBP1). This interaction engages interferon-induced helicase C-domain protein 1 (IFIH1). The binding of these proteins promotes the polarization of M1 macrophages, thereby worsening SILI (92).
These findings illustrate that a complex mechanism underlies the regulatory effects of lncRNAs on SILI. LncRNAs function in SILI by modulating miRNAs through interactions as ceRNAs (also known as miRNA sponges), thereby inhibiting both their expression and function. The lncRNA CRNDE aggravates liver injury by regulating miR-126-5p and BCL2L2 (88). The lncRNA-220-miR-5101-ceRNA complex mediates LPS-induced liver injury through the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) signaling pathway. It influences the regulation of autophagy and apoptosis in Kupffer cells triggered by LPS (93). Similarly, the lncRNA CASC7 facilitates LPS-induced liver injury during sepsis through the miR-217/TLR4 axis (94). Moreover, the lncRNA SNGH11 mediates ferroptosis in SILI via the miR-324-3p/glutathione peroxidase 4 (GPX4) axis (95).
In addition, circRNAs have the capacity to adsorb and neutralize many miRNAs to influence gene expression regulation. For instance, circ-Katnal1 exacerbates inflammatory pyrogenesis in SILI through the miR-31-5p/GSDMD axis (96).
Therefore, dysregulation of the expression of miRNAs and lncRNAs, as well as their interaction, may disrupt critical physiological processes such as the inflammatory response and cell death, contributing to liver injury in sepsis. In this context, a thorough investigation into the roles and mechanisms of these noncoding RNAs is paramount for identifying new potential therapeutic targets.
Notably, all the aforementioned pathogenic mechanisms do not operate in isolation. Rather, they are interconnected and form a complex network of pathogenesis. The close relationship between inflammation and oxidative stress represents a critical element of this network. The inflammatory response triggers the release of numerous inflammatory mediators and cytokines that induce oxidative stress, leading to lipid peroxidation of the cell membrane, protein oxidation, and DNA damage. In turn, oxidative stress further activates inflammatory signaling pathways that enhance the infiltration of inflammatory cells and the secretion of mediators, thus aggravating the inflammatory response. In addition, both inflammation and oxidative stress influence pyroptosis, a form of programmed cell death that is precisely regulated by noncoding RNAs. These mechanisms interact synergistically. Each factor creates a vicious cycle that exacerbates liver damage and SILI. Consequently, a more in-depth investigation into the pathogenesis of SILI is warranted not only to elucidate its intricate pathogenic network but also to provide a scientific foundation for the development of new therapeutic strategies.
In addition to noncoding RNAs, epigenetic modifications such as histone modifications and DNA methylation also significantly influence gene expression and cellular functions during SILI.
Histone modifications, including acetylation, methylation, and phosphorylation, can alter the chromatin structure and accessibility, thereby regulating gene transcription. For example, acetylation of histone H3 at the promoter regions of proinflammatory cytokines such as TNF-α and IL-6 has been shown to enhance their transcription and exacerbate liver injury in sepsis models (97). Conversely, deacetylation of histones by HDACs can suppress inflammatory responses and protect against liver damage (98).
DNA methylation, which primarily occurs at CpG islands, can lead to gene silencing by inhibiting the binding of transcription factors or recruiting methyl-binding proteins. Aberrant DNA methylation patterns have been observed in the liver during sepsis, affecting the expression of genes involved in immune responses and cell survival. Hypermethylation of the promoter region of the anti-inflammatory cytokine IL-10 has been reported in septic patients, resulting in decreased IL-10 expression and impaired anti-inflammatory capacity (99). On the other hand, hypomethylation of the proapoptotic gene Bim has been associated with increased Bim expression and hepatocyte apoptosis in sepsis (100).
These findings highlight the potential of targeting histone modifications and DNA methylation as therapeutic strategies for SILI. For instance, the use of histone deacetylase inhibitors (HDACis) has shown promise in reducing inflammation and improving liver function in sepsis models (101). Similarly, DNA methyltransferase inhibitors (DNMTis) may help restore the expression of beneficial genes and alleviate liver injury (102).
The conventional treatment strategy for SILI is systematic and multifaceted to improve outcomes. Central to this therapy is anti-infection treatment. The early initiation of antibiotic therapy is essential for the rapid elimination of pathogens and prevention of infection spread, thereby controlling the progression of SILI (103). Effective management of the infection source by surgical or interventional procedures, such as abscess drainage, can drastically reduce pathogen loads.
Initial fluid resuscitation and hemodynamic support are fundamental components for achieving circulatory stability in SILI patients (104). Nutritional support and metabolic control can improve the intestinal microbiota, enhance the immune response and improve patient recovery (105).
Liver function support constitutes a component of the treatment regimen. This may lead to the incorporation of artificial liver devices that help detoxify and regenerate hepatocytes. Blood purification techniques are employed to eliminate toxins from the body, alleviating the metabolic burden on the liver (106, 107).
However, conventional therapeutic options for SILI present several disadvantages in clinical treatment. With the continuous increase in antibiotic resistance, selecting valid antibiotics has become more complicated, complicating treatment strategies and possibly leading to persistent infections that exacerbate liver damage. Moreover, multiple drugs tend to produce drug interactions that can increase liver and kidney toxicity while limiting available therapeutic options. These factors might further worsen organ dysfunction and negatively impact treatment outcomes.
Moreover, septic liver injury is frequently accompanied by dysfunction in other organs, rendering comprehensive intervention based on single-organ support therapies significantly more challenging. Presently, there is a lack of effective therapeutic targets against the underlying pathophysiological processes involved in SILI. This limitation severely restricts the efficacy of existing treatments and fundamentally hinders improvements in patient prognosis.
As discussed above, SILI has a complex and multilevel pathogenesis. Effective treatment of SILI can be achieved only by addressing the interconnected pathogenic mechanisms in conjunction with managing liver injury itself. The optimal therapeutic intervention must target various stages of the disease, taking into account not only hepatic and systemic inflammatory responses but also organ dysfunction.
Nanodrug delivery systems utilize nanotechnology to achieve targeted and precise drug delivery to damaged liver tissues, which increases drug bioavailability while minimizing toxic side effects. Different delivery platforms, including liposomes, solid lipid nanoparticles, and polymer micelles, have been developed for the treatment of SILI and have shown promising therapeutic effects. Collectively, these findings provide robust support for the potential clinical application of nanotechnology-based therapies in the management of SILI (108–111).
Immunoregulatory therapies are designed to modulate the activities of immune cells to mitigate the inflammatory injury caused by sepsis. This modulation can be achieved through the administration of monoclonal antibodies that neutralize key inflammatory mediators, such as TNF-α and IL-6 (112). Alternatively, IL-1 receptor antagonists may be employed to inhibit the signaling pathways involved in inflammation (113, 114). Furthermore, the Decoy Receptor 3 (DcR3) analog DCR3-SUMO protein has emerged as both a biomarker and a therapeutic target in the inflammatory response. Research indicates that DCR3-SUMO has anti-inflammatory properties. It improves tissue morphology in sepsis models, increases survival rates, and significantly reduces serum levels as well as liver and lung concentrations of various inflammatory markers (115).
Antioxidative stress therapies aim to modulate critical signaling pathways that suppress oxidative stress and inflammation to protect the liver. These pathways include the NF-κB (116), Forkhead box O (FOXO) (117, 118), silent information regulator 1 (SIRT1)/NRF2/heme oxygenase-1(HO-1) pathways (119), and mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinase (JNKs), and p38 (120). NAC has been demonstrated to ameliorate liver dysfunction by reducing lipid peroxides and increasing antioxidant capacity (121, 122). Endocrine and metabolic regulation play crucial roles in modulating oxidative stress. Melatonin downregulates the expression of TLR4 and NF-κB, inhibits the production of ROS and RNS, and upregulates the antioxidant defense system (123, 124). In addition, it activates the SIRT3 pathway in mitochondria, protecting them from oxidative stress and alleviating liver dysfunction and glucose metabolism disorders (125, 126). Additionally, melatonin decreases the expression of iNOS mRNA in the livers of septic mice, decreasing nitric oxide (NO) production and protecting the liver (127, 128). Metformin further alleviates liver injury in sepsis models by reducing cytokine levels and high mobility group protein B1 (HMGB1) and MAPK activation while increasing adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) expression (129).
The activation of the NLRP3 inflammasome is a critical step in the pyroptotic process, and its upstream activators have been extensively studied. Mitochondrial ROS and lysosomal damage are two major upstream activators of the NLRP3 inflammasome. Mitochondrial dysfunction during sepsis leads to increased production of ROS, which can trigger the NLRP3 inflammasome (130). Additionally, lysosomal rupture and the release of cathepsins can also activate the NLRP3 inflammasome, contributing to pyroptosis (131).
Targeting pyroptosis is a promising therapeutic strategy for mitigating liver injury in sepsis. Several therapeutic inhibitors of pyroptosis are currently under investigation. Small-molecule inhibitors targeting GSDMD have shown potential in preclinical studies (132). For example, the compound Vx-765 has been reported to inhibit GSDMD cleavage and pyroptosis, thereby reducing inflammation and tissue injury (133–135). Moreover, inhibitors targeting the NLRP3 inflammasome itself are also in development (136). The compound MCC950 is a potent NLRP3 inflammasome inhibitor that has demonstrated efficacy in animal models of sepsis (135, 137). Drug-free tea polyphenols nanoparticles (TPNs) have intrinsic broad-spectrum RONS scavenging and pyroptosis blocking activity, and their ability to inhibit pyroptosis has been clearly demonstrated in a mouse model of sepsis and in human cells (138).
These therapeutic inhibitors provide new avenues for the treatment of sepsis-induced liver injury and warrant further investigation in clinical trials.
Traditional Chinese medicine (TCM) has already exhibited advantages in the treatment of SILI. The principal TCM active ingredients, curcumin and forskolin, exhibit anti-inflammatory, antioxidant, and hepatoprotective effects (36). Ginsenoside Rg3 alleviates SILI by modulating the lncRNA TUG1/miR-200c-3p/SIRT1 signaling axis (139). Moreover, Baweidu powder (BWBDS) ameliorates SILI through the modulation of the gut microbiota in murine models, emphasizing the therapeutic role of the microbiome in hepatic protection (140). Therefore, there is substantial research potential for the use of TCM in treating SILI.
Targeting autophagy pathways has emerged as a promising approach for alleviating SILI. Specific inhibitors of the pyroptosis pathway, such as those that activate peroxisome proliferator-activated receptor γ (PPARγ), can inhibit the thioredoxin-interacting protein (TXNIP)/NLRP3 signaling pathway (141). Moreover, immune-responsive gene 1 (IRG1)/4-octyl itaconate (OI) regulates the Nrf2 signaling pathway to suppress NLRP3 inflammasome activation and macrophage pyroptosis, reducing acute liver injury in septic mice (142). Furthermore, compounds such as anemonin-B4 and albiflorin increase the expression of autophagy-related proteins via the mTOR/p70S6K signaling pathway (143, 144), whereas dexmedetomidine regulates the SIRT1/AMPK axis (145). Acetaldehyde dehydrogenase-2 (ALDH2) also mediates vital signaling proteins, including AKT, AMPK, mTOR, STAT3, and Notch1, which cooperatively increase the flux of hepatic autophagy and prevent hepatocyte damage (146). These findings provide compelling evidence for the therapeutic potential of autophagy regulation in the treatment of SILI.
Epigenetic modulation of inflammatory pathways is a promising approach for SILI treatment. Strategies targeting specific miRNAs have been identified to modulate key signaling pathways, such as the NF-κB (87, 147), TLR4 (148), and MAPK (149)pathways. For example, several reports have demonstrated that paclitaxel ameliorates liver injury in septic mice by modulating the miR-27a/TAB3/NF-κB axis (150). The overexpression of miR-103a-3p targets HMGB1, leading to reduced inflammation and cellular damage, thereby improving SILI (151). Moreover, inhibiting miR-155 to promote the expression of suppressor of cytokine signaling (SOCS1) to inhibit the JAK/STAT pathway has been found to decrease SILI (152). The lncRNA CASC9 offers another approach for treating SILI by stabilizing SOCS1 through its interaction with fused in sarcoma (FUS) (153). Additionally, the inhibition of the lncRNA LINC00472 and its regulation of the miR-373-3p/TRIM8 axis has also presented potential benefits in alleviating SILI (154). Silencing the lncRNA XIST reduces BRD4 expression, which evades sepsis-induced acute liver injury, and could serve as a novel biomarker for diagnosing and treating sepsis (155). Although these findings are promising in preclinical models, there is an urgent need for future research to translate these strategies into clinical applications.
Mesenchymal stem cells (MSCs) have garnered significant attention as promising cell-based therapeutic strategies for the treatment of sepsis owing to their wide availability, high self-renewal capacity, multilineage differentiation potential, and low immunogenicity. Studies conducted in animal models have reported that MSCs can prevent liver inflammation during sepsis (156, 157). Human Wharton’s Jelly-derived mesenchymal stem cells (WJ-MSCs) significantly attenuate the expression of NF-κB and cytokines, thereby alleviating SILI in a murine model of sepsis (158). More specifically, MSCs mitigate SILI by suppressing the M1 polarization of KCs (159). In addition to whole MSCs, exosomes derived from MSCs have emerged as an applicable alternative. Exosomes are capable of encapsulating various bioactive molecules, such as RNA, cytokines, chemokines, proteins, and lipids (160). Through paracrine modulation, these exosomes mediate intracellular and intercellular signaling as well as metabolic processes (161). For example, exosomes alleviate sepsis-associated acute liver injury by downregulating MALAT1 via miRNA-26a-5p (162). Nevertheless, two major limitations affecting the clinical application of MSCs include their low efficiency in differentiation and minimal survival rates post transplantation. However, MSC-derived exosomes provide an alternative opportunity to increase therapeutic specificity and precision in treating sepsis while addressing these challenges.
Liver injury in sepsis is a complex pathophysiological process. The main factors are inflammatory mediators, oxidative stress, and apoptosis. Although these mechanisms have not been fully elucidated, further studies on the mechanism of SILI may lead to the identification of new therapeutic targets. These discoveries hold promise not only for advancing treatment strategies for septic liver injury but also for enhancing overall management approaches for sepsis.
Otherwise, clinical decision-making can be significantly enriched if further research identifies biomarkers that indicate disease status, predict disease progression, and monitor treatment efficacy. Such biomarkers would prove invaluable in selecting appropriate personalized treatments and optimizing patient outcomes. In parallel, it is essential to develop safe, effective, and accessible treatments that address the diverse needs of patients. The simplification of procedures, reduction in treatment duration, and decreased costs will also further optimize the existing treatment protocols and improve the overall efficacy of interventions for SILI.
XX: Conceptualization, Writing – original draft, Writing – review & editing. TY: Writing – review & editing. JA: Writing – review & editing. BL: Conceptualization, Writing – review & editing. ZD: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Gansu Provincial Health Industry Research Program (GSWSKY2023-24), Gansu Provincial Youth Science and Technology Foundation (20JR10RA710), the Foundation of the First Hospital of Lanzhou University (ldyyyn2019-13).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AKT: Protein kinase B
ALDH: Acetaldehyde dehydrogenase-2
ALP: Alkaline phosphatase
AMPK: Adenosine 5’-monophosphate (AMP)-activated protein kinase
ARDS: Acute respiratory distress syndrome
BWBDS: Baweidu powder
CLP: Cecal ligation and perforation
DAMPs: Damage-associated molecular patterns
DcR3: Decoy Receptor 3
ERK: Extracellular signal-regulated kinases
FOXO: Forkhead box O
FUS: Fused in sarcoma
GGT: γ-glutamyltransferase
GPX4: Glutathione peroxidase 4
GSDMD: Gasdermin-D
HH: Hypoxic hepatitis
HMGB1: High mobility group protein B1
HO-1: Heme oxygenase-1
ICU: Intensive care unit
IFIH1: Interferon-induced helicase C-domain protein 1
IL-1β: Interleukin-1β
IL-6: Interleukin-6
IL-8: Interleukin-8;INR, International normalized ratio
IRG1: Immune-responsive gene 1
JNK: c-Jun N-terminal kinase
KCs: Kupffer cells
LDH: Lactate dehydrogenase
LPSs: Lipopolysaccharides
MAPKs: Mitogen-activated protein kinases
MODS: Multiple organ dysfunction syndrome
MRCP: Magnetic resonance cholangiopancreatography
MSCs: Mesenchymal stem cells
mTOR: Mechanistic target of rapamycin
NAC: N-acetylcysteine
NF-κB: Nuclear factor-κB
NLRP3: NOD-like receptor protein 3
NO: Nitric oxide
Nrf2: Nuclear factor E2-related factor 2
OI: 4-octyl itaconate
PAMPs: Pathogen-associated molecular patterns
PI3K: Phosphoinositide 3-kinase
PPARγ: Peroxisome proliferator-activated receptor γ
PRRs: Pattern recognition receptors
PTBP1: Polypyrimidine tract-binding protein 1
RNS: Reactive nitrogen species
ROS: Reactive oxygen species
SC: Septic cholestasis
SILI: Sepsis-induced liver injury
SIRS: Systemic inflammatory response syndrome
SIRT1: Silent information regulator 1
SOCS1: Suppressor of cytokine signaling
SSC: Secondary sclerosing cholangitis
TCM: Traditional Chinese medicine
TLRs: Toll-like receptors
TNF-α: Tumor necrosis factor-α
TXNIP: Thioredoxin-interactingprotein
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Fleischmann-Struzek C, Mellhammar L, Rose N, Cassini A, Rudd KE, Schlattmann P, et al. Incidence and mortality of hospital- and icu-treated sepsis: results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. (2020) 46:1552–62. doi: 10.1007/s00134-020-06151-x
3. Markwart R, Saito H, Harder T, Tomczyk S, Cassini A, Fleischmann-Struzek C, et al. Epidemiology and burden of sepsis acquired in hospitals and intensive care units: A systematic review and meta-analysis. Intensive Care Med. (2020) 46:1536–51. doi: 10.1007/s00134-020-06106-2
4. Sun J, Zhang J, Wang X, Ji F, Ronco C, Tian J, et al. Gut-liver crosstalk in sepsis-induced liver injury. Crit Care. (2020) 24:614. doi: 10.1186/s13054-020-03327-1
5. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
6. Yan J, Li S, Li S. The role of the liver in sepsis. Int Rev Immunol. (2014) 33:498–510. doi: 10.3109/08830185.2014.889129
7. Chen JW, Liu CY, Li S, Wu SW, Cai C, Lu MQ. Sepsis-associated liver injury: mechanisms and potential therapeutic targets. World J Gastroenterol. (2024) 30:4518–22. doi: 10.3748/wjg.v30.i42.4518
8. Jonsdottir S, Arnardottir MB, Andresson JA, Bjornsson HK, Lund SH, Bjornsson ES. Prevalence, clinical characteristics and outcomes of hypoxic hepatitis in critically ill patients. Scand J Gastroenterol. (2022) 57:311–8. doi: 10.1080/00365521.2021.2005136
9. Kasper P, Tacke F, Steffen HM, Michels G. Hepatic dysfunction in sepsis. Med Klin Intensivmed Notfmed. (2020) 115:609–19. doi: 10.1007/s00063-020-00707-x
10. Bynum TE, Boitnott JK, Maddrey WC. Ischemic hepatitis. Dig Dis Sci. (1979) 24:129–35. doi: 10.1007/bf01324740
11. Henrion J, Schapira M, Luwaert R, Colin L, Delannoy A, Heller FR. Hypoxic hepatitis: clinical and hemodynamic study in 142 consecutive cases. Medicine. (2003) 82:392–406. doi: 10.1097/01.md.0000101573.54295.bd
12. Aboelsoud MM, Javaid AI, Al-Qadi MO, Lewis JH. Hypoxic hepatitis - its biochemical profile, causes and risk factors of mortality in critically-ill patients: A cohort study of 565 patients. J Crit Care. (2017) 41:9–15. doi: 10.1016/j.jcrc.2017.04.040
13. Beyer D, Hoff J, Sommerfeld O, Zipprich A, Gaßler N, Press AT. The liver in sepsis: molecular mechanism of liver failure and their potential for clinical translation. Mol Med. (2022) 28:84. doi: 10.1186/s10020-022-00510-8
14. Ghenu MI, Dragoş D, Manea MM, Ionescu D, Negreanu L. Pathophysiology of sepsis-induced cholestasis: A review. JGH Open. (2022) 6:378–87. doi: 10.1002/jgh3.12771
15. Horvatits T, Drolz A, Trauner M, Fuhrmann V. Liver injury and failure in critical illness. Hepatology. (2019) 70:2204–15. doi: 10.1002/hep.30824
16. Kobashi H, Toshimori J, Yamamoto K. Sepsis-associated liver injury: incidence, classification and the clinical significance. Hepatol Res. (2013) 43:255–66. doi: 10.1111/j.1872-034X.2012.01069.x
17. Martins P, Verdelho MaChado M. Secondary sclerosing cholangitis in critically ill patients: an underdiagnosed entity. GE - Portuguese J Gastroenterol. (2019) 27:103–14. doi: 10.1159/000501405
18. Zhao J, Yue P, Mi N, Li M, Fu W, Zhang X, et al. Biliary fibrosis is an important but neglected pathological feature in hepatobiliary disorders: from molecular mechanisms to clinical implications. Med Rev (2021). (2024) 4:326–65. doi: 10.1515/mr-2024-0029
19. Leonhardt S, Veltzke-Schlieker W, Adler A, Schott E, Hetzer R, Schaffartzik W, et al. Trigger mechanisms of secondary sclerosing cholangitis in critically ill patients. Crit Care. (2015) 19:131. doi: 10.1186/s13054-015-0861-5
20. Möller K, Braden B, Culver EL, Jenssen C, Zadeh ES, Alhyari A, et al. Secondary sclerosing cholangitis and igg4-sclerosing cholangitis - a review of cholangiographic and ultrasound imaging. Endosc Ultrasound. (2023) 12:181–99. doi: 10.4103/eus-d-22-00208
21. Leonhardt S, Veltzke-Schlieker W, Adler A, Schott E, Eurich D, Faber W, et al. Secondary sclerosing cholangitis in critically ill patients: clinical presentation, cholangiographic features, natural history, and outcome: A series of 16 cases. Med (Baltimore). (2015) 94:e2188. doi: 10.1097/md.0000000000002188
22. Hentschel F, Bornscheuer T, Lüth S. Secondary cholangitis of the critically ill. Z Gastroenterol. (2019) 57:977–82. doi: 10.1055/a-0958-2843
23. Lin H, Liang Q, Cai Q, Huang M. Analysis of high-risk factors and clinical characteristics of sepsis-related liver injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. (2021) 33:186–91. doi: 10.3760/cma.j.cn121430-20201118-00627
24. Li P, Chang M. Roles of prr-mediated signaling pathways in the regulation of oxidative stress and inflammatory diseases. Int J Mol Sci. (2021) 22(14):7688. doi: 10.3390/ijms22147688
25. Wang H, Ma T, Bao Q, Zhu L, Ying T, Yu Y. Knockdown of protein interacting with C A Kinase 1 aggravates sepsis-induced acute liver injury by regulating the tlr4/nf-Kb pathway. Sci Rep. (2023) 13:11913. doi: 10.1038/s41598-023-38852-w
26. Gandhi CR. Pro- and anti-fibrogenic functions of gram-negative bacterial lipopolysaccharide in the liver. Front Med (Lausanne). (2020) 7:130. doi: 10.3389/fmed.2020.00130
27. Medzhitov R, Preston-Hurlburt P. Janeway CA, jr. A human homologue of the drosophila toll protein signals activation of adaptive immunity. Nature. (1997) 388:394–7. doi: 10.1038/41131
28. Kumar V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int Immunopharmacol. (2020) 89:107087. doi: 10.1016/j.intimp.2020.107087
29. Karki R, Kanneganti TD. The ‘Cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. (2021) 42:681–705. doi: 10.1016/j.it.2021.06.001
30. Sundaram B, Tweedell RE, Prasanth Kumar S, Kanneganti TD. The nlr family of innate immune and cell death sensors. Immunity. (2024) 57:674–99. doi: 10.1016/j.immuni.2024.03.012
31. Golden GJ, Toledo AG, Marki A, Sorrentino JT, Morris C, Riley RJ, et al. Endothelial heparan sulfate mediates hepatic neutrophil trafficking and injury during staphylococcus aureus sepsis. mBio. (2021) 12:e0118121. doi: 10.1128/mBio.01181-21
32. Alves GF, Aimaretti E, Einaudi G, Mastrocola R, de Oliveira JG, Collotta D, et al. Pharmacological inhibition of fak-pyk2 pathway protects against organ damage and prolongs the survival of septic mice. Front Immunol. (2022) 13:837180. doi: 10.3389/fimmu.2022.837180
33. Nagar H, Piao S, Kim CS. Role of mitochondrial oxidative stress in sepsis. Acute Crit Care. (2018) 33:65–72. doi: 10.4266/acc.2018.00157
34. Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, et al. Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. Int J Mol Sci. (2019) 20(18):4472. doi: 10.3390/ijms20184472
35. Jakubczyk K, Dec K, Kałduńska J, Kawczuga D, Kochman J, Janda K. Reactive oxygen species - sources, functions, oxidative damage. Pol Merkur Lekarski. (2020) 48:124–7. doi: 10.1155/2012/217037
36. Srdić T, Đurašević S, Lakić I, Ružičić A, Vujović P, Jevđović T, et al. From molecular mechanisms to clinical therapy: understanding sepsis-induced multiple organ dysfunction. Int J Mol Sci. (2024) 25(14):7770. doi: 10.3390/ijms25147770
37. Chen W, Kim S, Kim SY, Beheshtian C, Kim N, Shin KH, et al. Gv1001, htert peptide fragment, prevents doxorubicin-induced endothelial-to-mesenchymal transition in human endothelial cells and atherosclerosis in mice. Cells. (2025) 14(2):98. doi: 10.3390/cells14020098
38. Souza ACP, Souza CM, Amaral CL, Lemes SF, Santucci LF, Milanski M, et al. Short-term high-fat diet consumption reduces hypothalamic expression of the nicotinic acetylcholine receptor A7 subunit (A7nachr) and affects the anti-inflammatory response in a mouse model of sepsis. Front Immunol. (2019) 10:565. doi: 10.3389/fimmu.2019.00565
39. Sundaram B, Pandian N, Kim HJ, Abdelaal HM, Mall R, Indari O, et al. Nlrc5 senses nad(+) depletion, forming a panoptosome and driving panoptosis and inflammation. Cell. (2024) 187:4061–77.e17. doi: 10.1016/j.cell.2024.05.034
40. Kantarjian H, Haddad FG, Jain N, Sasaki K, Short NJ, Loghavi S, et al. Results of salvage therapy with mini-hyper-cvd and inotuzumab ozogamicin with or without blinatumomab in pre-B acute lymphoblastic leukemia. J Hematol Oncol. (2023) 16:44. doi: 10.1186/s13045-023-01444-2
41. Melin N, Yarahmadov T, Sanchez-Taltavull D, Birrer FE, Brodie TM, Petit B, et al. A new mouse model of radiation-induced liver disease reveals mitochondrial dysfunction as an underlying fibrotic stimulus. JHEP Rep. (2022) 4:100508. doi: 10.1016/j.jhepr.2022.100508
42. Sun W, Liu J, Shi X, Bi Y, Liu H, Xu T. Emamectin benzoate and microplastics led to skeletal muscle atrophy in common carp via induced oxidative stress, mitochondrial dysfunction, and protein synthesis and degradation imbalance. J Agric Food Chem. (2025) 73(5):3106–16. doi: 10.1021/acs.jafc.4c10479
43. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
44. Prauchner CA. Oxidative stress in sepsis: pathophysiological implications justifying antioxidant co-therapy. Burns. (2017) 43:471–85. doi: 10.1016/j.burns.2016.09.023
45. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. (2011) 107:57–64. doi: 10.1093/bja/aer093
46. Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De-Wolf-Peeters C, Van den Berghe G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet. (2005) 365:53–9. doi: 10.1016/S0140-6736(04)17665-4
47. Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, et al. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. (1996) 184:1155–60. doi: 10.1084/jem.184.3.1155
48. Glover HL, Schreiner A, Dewson G, Tait SWG. Mitochondria and cell death. Nat Cell Biol. (2024) 26:1434–46. doi: 10.1038/s41556-024-01429-4
49. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome C by the mitochondrial channel vdac. Nature. (1999) 399:483–7. doi: 10.1038/20959
50. Zhou J, Zheng X, Xi C, Tang X, Jiang Y, Xie M, et al. Cr(Vi) induced hepatocyte apoptosis through the cth/H(2)S/drp1 signaling pathway. Sci Total Environ. (2024) 950:175332. doi: 10.1016/j.scitotenv.2024.175332
51. Lee H, Lee TJ, Galloway CA, Zhi W, Xiao W, de Mesy Bentley KL, et al. The mitochondrial fusion protein opa1 is dispensable in the liver and its absence induces mitohormesis to protect liver from drug-induced injury. Nat Commun. (2023) 14:6721. doi: 10.1038/s41467-023-42564-0
52. Wen R, Liu YP, Tong XX, Zhang TN, Yang N. Molecular mechanisms and functions of pyroptosis in sepsis and sepsis-associated organ dysfunction. Front Cell Infect Microbiol. (2022) 12:962139. doi: 10.3389/fcimb.2022.962139
53. Liu LL, Yan X, Xue KY, Wang XM, Li LY, Chen HY, et al. Prim-O-glucosycimifugin attenuates liver injury in septic mice by inhibiting nlrp3 inflammasome/caspase-1 signaling cascades in macrophages. Phytomedicine. (2022) 106:154427. doi: 10.1016/j.phymed.2022.154427
54. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
55. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. (2017) 29:201–10. doi: 10.1093/intimm/dxx018
56. Huang Y, Xu W, Zhou R. Nlrp3 inflammasome activation and cell death. Cell Mol Immunol. (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
57. Liu L, Lin L, Wang Y, Yan X, Li R, He M, et al. L-ap alleviates liver injury in septic mice by inhibiting macrophage activation via suppressing nf-Kb and nlrp3 inflammasome/caspase-1 signal pathways. J Agric Food Chem. (2024) 72:8460–75. doi: 10.1021/acs.jafc.3c02781
58. Wang Y, Zhao Y, Wang Z, Sun R, Zou B, Li R, et al. Peroxiredoxin 3 inhibits acetaminophen-induced liver pyroptosis through the regulation of mitochondrial ros. Front Immunol. (2021) 12:652782. doi: 10.3389/fimmu.2021.652782
59. Zhen H, Hu Y, Liu X, Fan G, Zhao S. The protease caspase-1: activation pathways and functions. Biochem Biophys Res Commun. (2024) 717:149978. doi: 10.1016/j.bbrc.2024.149978
60. Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature. (1994) 370:270–5. doi: 10.1038/370270a0
61. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. (1992) 356:768–74. doi: 10.1038/356768a0
62. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. (2020) 217(1):e20190314. doi: 10.1084/jem.20190314
63. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-em structure of the gasdermin A3 membrane pore. Nature. (2018) 557:62–7. doi: 10.1038/s41586-018-0058-6
64. Yu G, Choi YK, Lee S. Inflammasome diversity: exploring novel frontiers in the innate immune response. Trends Immunol. (2024) 45:248–58. doi: 10.1016/j.it.2024.02.004
65. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. (2021) 593:607–11. doi: 10.1038/s41586-021-03478-3
66. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
67. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
68. Wang G, Jin S, Ling X, Li Y, Hu Y, Zhang Y, et al. Proteomic profiling of lps-induced macrophage-derived exosomes indicates their involvement in acute liver injury. Proteomics. (2019) 19:e1800274. doi: 10.1002/pmic.201800274
69. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
70. Gómez-Virgilio L, Silva-Lucero MD, Flores-Morelos DS, Gallardo-Nieto J, Lopez-Toledo G, Abarca-Fernandez AM, et al. Autophagy: A key regulator of homeostasis and disease: an overview of molecular mechanisms and modulators. Cells. (2022) 11(15):2262. doi: 10.3390/cells11152262
71. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. (2021) 40:e104705. doi: 10.15252/embj.2020104705
72. Zhou J-C, Wang J-L, Ren H-Z, Shi X-L. Autophagy plays a double-edged sword role in liver diseases. J Physiol Biochem. (2022) 78:9–17. doi: 10.1007/s13105-021-00844-7
73. Killackey SA, Philpott DJ, Girardin SE. Mitophagy pathways in health and disease. J Cell Biol. (2020) 219(11):e202004029. doi: 10.1083/jcb.202004029
74. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. (2008) 183:795–803. doi: 10.1083/jcb.200809125
75. Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, et al. Mitochondrial stasis reveals P62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. (2018) 28:588–604.e5. doi: 10.1016/j.cmet.2018.06.014
76. Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, et al. The mitochondrial protein bnip3l is the substrate of park2 and mediates mitophagy in pink1/park2 pathway. Hum Mol Genet. (2015) 24:2528–38. doi: 10.1093/hmg/ddv017
77. Lin C-W, Lo S, Perng D-S, Wu DB-C, Lee P-H, Chang Y-F, et al. Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock. (2014) 41:241–9. doi: 10.1097/shk.0000000000000111
78. Iba T, Helms J, Maier CL, Ferrer R, Levy JH. Autophagy and autophagic cell death in sepsis: friend or foe? J Intensive Care. (2024) 12:41. doi: 10.1186/s40560-024-00754-y
79. Wang H, Zhu J, Wei L, Wu S, Shang L, Ye X, et al. Tslp protects against sepsis-induced liver injury by inducing autophagy via activation of the pi3k/akt/stat3 pathway. Pathol Res Pract. (2022) 236:153979. doi: 10.1016/j.prp.2022.153979
80. Binnie A, Tsang JLY, Hu P, Carrasqueiro G, Castelo-Branco P, dos Santos CC. Epigenetics of sepsis. Crit Care Med. (2020) 48:745–56. doi: 10.1097/ccm.0000000000004247
81. Isac T, Isac S, Rababoc R, Cotorogea M, Iliescu L. Epigenetics in inflammatory liver diseases: A clinical perspective (Review). Exp Ther Med. (2022) 23:366. doi: 10.3892/etm.2022.11293
82. Zheng Z, Huang G, Gao T, Huang T, Zou M, Zou Y, et al. Epigenetic changes associated with interleukin-10. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01105
83. Wu D, Shi Y, Zhang H, Miao C. Epigenetic mechanisms of immune remodeling in sepsis: targeting histone modification. Cell Death Dis. (2023) 14:112. doi: 10.1038/s41419-023-05656-9
84. Pan Y, Wang J, Xue Y, Zhao J, Li D, Zhang S, et al. Gskj4 Protects Mice against Early Sepsis Via Reducing Proinflammatory Factors and up-Regulating Mir-146a. Front Immunol. (2018) 9:2272. doi: 10.3389/fimmu.2018.02272
85. von Knethen A, Brüne B. Histone deacetylation inhibitors as therapy concept in sepsis. Int J Mol Sci. (2019) 20:346. doi: 10.3390/ijms20020346
87. Yang ZB, Chen WW, Chen HP, Cai SX, Lin JD, Qiu LZ. Mir-155 aggravated septic liver injury by oxidative stress-mediated er stress and mitochondrial dysfunction via targeting nrf-2. Exp Mol Pathol. (2018) 105:387–94. doi: 10.1016/j.yexmp.2018.09.003
88. Li Y, Song J, Xie Z, Liu M, Sun K. Long noncoding rna colorectal neoplasia differentially expressed alleviates sepsis-induced liver injury via regulating mir-126-5p. IUBMB Life. (2020) 72:440–51. doi: 10.1002/iub.2230
89. Rahmel T, Schäfer ST, Frey UH, Adamzik M, Peters J. Increased circulating microrna-122 is a biomarker for discrimination and risk stratification in patients defined by sepsis-3 criteria. PloS One. (2018) 13:e0197637. doi: 10.1371/journal.pone.0197637
90. Dai Y, Liang Z, Li Y, Li C, Chen L. Circulating long noncoding rnas as potential biomarkers of sepsis: A preliminary study. Genet Test Mol Biomarkers. (2017) 21:649–57. doi: 10.1089/gtmb.2017.0061
91. Zhang CC, Niu F. Lncrna neat1 promotes inflammatory response in sepsis-induced liver injury via the let-7a/tlr4 axis. Int Immunopharmacol. (2019) 75:105731. doi: 10.1016/j.intimp.2019.105731
92. Yang Z, Xia H, Lai J, Qiu L, Lin J. Artesunate alleviates sepsis-induced liver injury by regulating macrophage polarization via the lncrna malat1/ptbp1/ifih1 axis. Diagn Microbiol Infect Dis. (2024) 110:116383. doi: 10.1016/j.diagmicrobio.2024.116383
93. Yang Y, Tian T, Li S, Li N, Luo H, Jiang Y. Lncrna 220: A novel long non-coding rna regulates autophagy and apoptosis in kupffer cells via the mir-5101/pi3k/akt/mtor axis in lps-induced endotoxemic liver injury in mice. Int J Mol Sci. (2023) 24(13):11210. doi: 10.3390/ijms241311210
94. Sun C, Chen Y, Chen Z, Wang H, Yang W, Zhou X. Lncrna casc7 contributes to the progression of lps-induced liver injury by targeting mirna-217/tlr4 axis. Biomol BioMed. (2024). 25(2):493–504. doi: 10.17305/bb.2024.10543
95. Yang Y, Wang A, Zhou J, Yang Y, Wu H. Lncrna snhg11 induces ferroptosis in liver injury cells through mir-324-3p/gpx4 axis-mediated sepsis. Cell Mol Biol (Noisy-le-grand). (2023) 69:163–9. doi: 10.14715/cmb/2023.69.12.26
96. Kang K, Li N, Gao Y, Wang C, Chen P, Meng X, et al. Circ-katnal1 enhances inflammatory pyroptosis in sepsis-induced liver injury through the mir-31-5p/gsdmd axis. Mediators Inflammation. (2022) 2022:8950130. doi: 10.1155/2022/8950130
97. Li H, Chen X, Xu J, Zhu L, Li C, Sun X, et al. Grp/grpr enhances alcohol-associated liver injury through the irf1-mediated caspase-1 inflammasome and nox2-dependent ros pathway. Hepatology. (2024) 79:392–408. doi: 10.1097/hep.0000000000000531
98. Ghiboub M, Zhao J, Li Yim AYF, Schilderink R, Verseijden C, van Hamersveld PHP, et al. Hdac3 mediates the inflammatory response and lps tolerance in human monocytes and macrophages. Front Immunol. (2020) 11:550769. doi: 10.3389/fimmu.2020.550769
99. Hai L, Friedel D, Hinz F, Hoffmann DC, Doubrovinskaia S, Rohdjess H, et al. Distinct epigenetic and transcriptional profiles of epstein-barr virus (Ebv) positive and negative primary cns lymphomas. Neuro Oncol. (2024). doi: 10.1093/neuonc/noae251
100. Shi P, Du Y, Zhang Y, Yang B, Guan Q, Jing Y, et al. Ubiquitin-independent degradation of bim blocks macrophage pyroptosis in sepsis-related tissue injury. Cell Death Dis. (2024) 15:703. doi: 10.1038/s41419-024-07072-z
101. Truong N, Cottingham AL, Dharmaraj S, Shaw JR, Lasola JJM, Goodis CC, et al. Multimodal nanoparticle-containing modified suberoylanilide hydroxamic acid polymer conjugates to mitigate immune dysfunction in severe inflammation. Bioeng Transl Med. (2024) 9:e10611. doi: 10.1002/btm2.10611
102. Samanta S, Zhou Z, Rajasingh S, Panda A, Sampath V, Rajasingh J. Dnmt and hdac inhibitors together abrogate endotoxemia mediated macrophage death by stat3-jmjd3 signaling. Int J Biochem Cell Biol. (2018) 102:117–27. doi: 10.1016/j.biocel.2018.07.002
103. He XL, Chen JY, Feng YL, Song P, Wong YK, Xie LL, et al. Single-cell rna sequencing deciphers the mechanism of sepsis-induced liver injury and the therapeutic effects of artesunate. Acta Pharmacol Sin. (2023) 44:1801–14. doi: 10.1038/s41401-023-01065-y
104. Durand F, Kellum JA, Nadim MK. Fluid resuscitation in patients with cirrhosis and sepsis: A multidisciplinary perspective. J Hepatol. (2023) 79:240–6. doi: 10.1016/j.jhep.2023.02.024
105. Xu F, Lu G, Wang J. Enhancing sepsis therapy: the evolving role of enteral nutrition. Front Nutr. (2024) 11:1421632. doi: 10.3389/fnut.2024.1421632
106. Papamichalis P, Oikonomou KG, Xanthoudaki M, Valsamaki A, Skoura AL, Papathanasiou SK, et al. Extracorporeal organ support for critically ill patients: overcoming the past, achieving the maximum at present, and redefining the future. World J Crit Care Med. (2024) 13:92458. doi: 10.5492/wjccm.v13.i2.92458
107. Huber W, Ruiz de Garibay AP. Options in extracorporeal support of multiple organ failure. Med Klin Intensivmed Notfmed. (2020) 115:28–36. doi: 10.1007/s00063-020-00658-3
108. Lu Y, Shi Y, Wu Q, Sun X, Zhang WZ, Xu XL, et al. An overview of drug delivery nanosystems for sepsis-related liver injury treatment. Int J Nanomedicine. (2023) 18:765–79. doi: 10.2147/ijn.S394802
109. Marey AM, Dkhil MA, Abdel Moneim AA, Santourlidis S, Abdel-Gaber R, Alquraishi MI, et al. Fighting sepsis-induced liver damage with biosynthesized silver nanoparticles. Microsc Res Tech. (2024). 88(1):127–38. doi: 10.1002/jemt.24691
110. Xiao Y, Ren C, Chen G, Shang P, Song X, You G, et al. Neutrophil membrane-mimicking nanodecoys with intrinsic anti-inflammatory properties alleviate sepsis-induced acute liver injury and lethality in a mouse endotoxemia model. Mater Today Bio. (2022) 14:100244. doi: 10.1016/j.mtbio.2022.100244
111. Xu Y, Li Y, Liu X, Pan Y, Sun Z, Xue Y, et al. Spions enhances il-10-producing macrophages to relieve sepsis via cav1-notch1/hes1-mediated autophagy. Int J Nanomedicine. (2019) 14:6779–97. doi: 10.2147/ijn.S215055
112. Zaini A, Jawad HE, Hadi NR. Targeting vegf using bevacizumab attenuates sepsis-induced liver injury in a mouse model of cecal ligation and puncture. J Med Life. (2023) 16:1488–98. doi: 10.25122/jml-2023-0064
113. Wahab F, Tazinafo LF, Cárnio EC, Aguila FA, Batalhão ME, Rocha MJ. Interleukin-1 receptor antagonist decreases cerebrospinal fluid nitric oxide levels and increases vasopressin secretion in the late phase of sepsis in rats. Endocrine. (2015) 49:215–21. doi: 10.1007/s12020-014-0452-2
114. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase iii trial. Crit Care Med. (2016) 44:275–81. doi: 10.1097/ccm.0000000000001402
115. Su J, Tong Z, Feng Z, Wu S, Zhou F, Li R, et al. Protective effects of dcr3-sumo on lipopolysaccharide-induced inflammatory cells and septic mice. Int J Biol Macromol. (2024) 275:133703. doi: 10.1016/j.ijbiomac.2024.133703
116. Chen M, Zhang Y, Wang Y, Gulifeire T, Yu X. Role and mechanism of sirt1 in regulating nrf2/ho-1 signaling pathway in septic liver injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. (2023) 35:598–603. doi: 10.3760/cma.j.cn121430-20220815-00744
117. Xiao W, Oldham WM, Priolo C, Pandey AK, Loscalzo J. Immunometabolic endothelial phenotypes: integrating inflammation and glucose metabolism. Circ Res. (2021) 129:9–29. doi: 10.1161/circresaha.120.318805
118. Zhang M, Sui W, Xing Y, Cheng J, Cheng C, Xue F, et al. Angiotensin iv attenuates diabetic cardiomyopathy via suppressing foxo1-induced excessive autophagy, apoptosis and fibrosis. Theranostics. (2021) 11:8624–39. doi: 10.7150/thno.48561
119. Lee Y, Jeong GS, Kim KM, Lee W, Bae JS. Cudratricusxanthone a attenuates sepsis-induced liver injury via sirt1 signaling. J Cell Physiol. (2018) 233:5441–6. doi: 10.1002/jcp.26390
120. Xu D, Liao S, Lv Y, Wang J, Kong L. Nmr-based metabolomics approach reveals effects of antioxidant nutrients in sepsis-induced changes in rat liver injury. J Nutr Biochem. (2020) 85:108440. doi: 10.1016/j.jnutbio.2020.108440
121. Aisa-Alvarez A, Soto ME, Guarner-Lans V, Camarena-Alejo G, Franco-Granillo J, Martínez-Rodríguez EA, et al. Usefulness of antioxidants as adjuvant therapy for septic shock: A randomized clinical trial. Medicina (Kaunas). (2020) 56(11):619. doi: 10.3390/medicina56110619
122. Nabi T, Nabi S, Rafiq N, Shah A. Role of N-acetylcysteine treatment in non-acetaminophen-induced acute liver failure: A prospective study. Saudi J Gastroenterol. (2017) 23:169–75. doi: 10.4103/1319-3767.207711
123. Saha M, Manna K, Das Saha K. Melatonin suppresses nlrp3 inflammasome activation via tlr4/nf-Kb and P2x7r signaling in high-fat diet-induced murine nash model. J Inflammation Res. (2022) 15:3235–58. doi: 10.2147/jir.S343236
124. Zhang J-J, Meng X, Li Y, Zhou Y, Xu D-P, Li S, et al. Effects of melatonin on liver injuries and diseases. Int J Mol Sci. (2017) 18:673. doi: 10.3390/ijms18040673
125. Colunga Biancatelli RML, Berrill M, Mohammed YH, Marik PE. Melatonin for the treatment of sepsis: the scientific rationale. J Thorac Dis. (2020) 12:S54–s65. doi: 10.21037/jtd.2019.12.85
126. Chen J, Xia H, Zhang L, Zhang H, Wang D, Tao X. Protective effects of melatonin on sepsis-induced liver injury and dysregulation of gluconeogenesis in rats through activating sirt1/stat3 pathway. BioMed Pharmacother. (2019) 117:109150. doi: 10.1016/j.biopha.2019.109150
127. Crespo E, Macías M, Pozo D, Escames G, Martín M, Vives F, et al. Melatonin inhibits expression of the inducible no synthase ii in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. (1999) 13:1537–46. doi: 10.1096/fasebj.13.12.1537
128. Tarocco A, Caroccia N, Morciano G, Wieckowski MR, Ancora G, Garani G, et al. Melatonin as a master regulator of cell death and inflammation: molecular mechanisms and clinical implications for newborn care. Cell Death Dis. (2019) 10:317. doi: 10.1038/s41419-019-1556-7
129. Song H, Zhang X, Zhai R, Liang H, Song G, Yuan Y, et al. Metformin attenuated sepsis-associated liver injury and inflammatory response in aged mice. Bioengineered. (2022) 13:4598–609. doi: 10.1080/21655979.2022.2036305
130. Guo NK, Si LN, Li PQ, Gan GF. Nano acacetin mitigates intestinal mucosal injury in sepsis rats by protecting mitochondrial function and regulating trx1 to inhibit the nlrp3 pyroptosis pathway. Int J Nanomedicine. (2024) 19:14125–41. doi: 10.2147/ijn.S497081
131. Niu Z, Xia X, Zhang Z, Liu J, Li X. (H)Ceo(2)@Ca-074me nanoparticles alleviate inflammation and improve osteogenic microenvironment by regulating the ctsb-nlrp3 signaling pathway. Int J Nanomedicine. (2025) 20:161–79. doi: 10.2147/ijn.S389156
132. Wright SS, Kumari P, Fraile-Ágreda V, Wang C, Shivcharan S, Kappelhoff S, et al. Transplantation of gasdermin pores by extracellular vesicles propagates pyroptosis to bystander cells. Cell. (2024) 188(2):280–291.e17. doi: 10.1016/j.cell.2024.11.018
133. Li M, Wang Y, Liu H, Huang X, Peng H, Yang Y, et al. Staphylococcus aureus membrane vesicles kill tumor cells through a caspase-1-dependent pyroptosis pathway. Int J Nanomedicine. (2024) 19:4007–19. doi: 10.2147/ijn.S455158
134. Jiao M, Wang J, Liu W, Zhao X, Qin Y, Zhang C, et al. Vx-765 inhibits pyroptosis and reduces inflammation to prevent acute liver failure by upregulating pparα Expression. Ann Hepatol. (2023) 28:101082. doi: 10.1016/j.aohep.2023.101082
135. Ye X, Lin Z-J, Hong G-H, Wang Z-M, Dou R-T, Lin J-Y, et al. Pyroptosis inhibitors mcc950 and vx-765 mitigate myocardial injury by alleviating oxidative stress, inflammation, and apoptosis in acute myocardial hypoxia. Exp Cell Res. (2024) 438:114061. doi: 10.1016/j.yexcr.2024.114061
136. Coll RC, Schroder K. Inflammasome components as new therapeutic targets in inflammatory disease. Nat Rev Immunol. (2025) 25:22–41. doi: 10.1038/s41577-024-01075-9
137. Lai Y, Zhuang L, Zhu J, Wang S, Guo C, Chen B, et al. Novel approach to alleviate lupus nephritis: targeting the nlrp3 inflammasome in cd8(+)Cd69(+)Cd103(+) T(Rm) cells. J Transl Med. (2024) 22:1139. doi: 10.1186/s12967-024-05951-9
138. Chen Y, Luo R, Li J, Wang S, Ding J, Zhao K, et al. Intrinsic radical species scavenging activities of tea polyphenols nanoparticles block pyroptosis in endotoxin-induced sepsis. ACS Nano. (2022) 16:2429–41. doi: 10.1021/acsnano.1c08913
139. Wu P, Yu X, Peng Y, Wang QL, Deng LT, Xing W. Ginsenoside rg3 alleviates septic liver injury by regulating the lncrna tug1/mir-200c-3p/sirt1 axis. J Inflammation (Lond). (2021) 18:31. doi: 10.1186/s12950-021-00296-2
140. Fan X, Mai C, Zuo L, Huang J, Xie C, Jiang Z, et al. Herbal formula baweibaidusan alleviates polymicrobial sepsis-induced liver injury via increasing the gut microbiota lactobacillus johnsonii and regulating macrophage anti-inflammatory activity in mice. Acta Pharm Sin B. (2023) 13:1164–79. doi: 10.1016/j.apsb.2022.10.016
141. Li Z, Liu T, Feng Y, Tong Y, Jia Y, Wang C, et al. Pparγ Alleviates sepsis-induced liver injury by inhibiting hepatocyte pyroptosis via inhibition of the ros/txnip/nlrp3 signaling pathway. Oxid Med Cell Longev. (2022) 2022:1269747. doi: 10.1155/2022/1269747
142. Zhou P, Yang L, Li R, Yin Y, Xie G, Liu X, et al. Irg1/itaconate alleviates acute liver injury in septic mice by suppressing nlrp3 expression and its mediated macrophage pyroptosis via regulation of the nrf2 pathway. Int Immunopharmacol. (2024) 135:112277. doi: 10.1016/j.intimp.2024.112277
143. Pei L, He L. Hepatoprotective Effect of Anemoside B4 against Sepsis-Induced Acute Liver Injury through Modulating the Mtor/P70s6k-Mediated Autophagy. Chem Biol Interact. (2021) 345:109534. doi: 10.1016/j.cbi.2021.109534
144. Liu Y, Feng L, Yao L. Albiflorin alleviates sepsis-induced acute liver injury through mtor/P70s6k pathway. Curr Mol Med. (2024) 24:344–54. doi: 10.2174/1566524023666230309124004
145. Yu Q, Zou L, Yuan X, Fang F, Xu F. Dexmedetomidine Protects against Septic Liver Injury by Enhancing Autophagy through Activation of the Ampk/Sirt1 Signaling Pathway. Front Pharmacol. (2021) 12:658677. doi: 10.3389/fphar.2021.658677
146. Liu Z, Ye S, Zhong X, Wang W, Lai CH, Yang W, et al. Pretreatment with the aldh2 activator alda−1 protects rat livers from ischemia/reperfusion injury by inducing autophagy. Mol Med Rep. (2020) 22:2373–85. doi: 10.3892/mmr.2020.11312
147. Funahashi Y, Kato N, Masuda T, Nishio F, Kitai H, Ishimoto T, et al. Mir-146a targeted to splenic macrophages prevents sepsis-induced multiple organ injury. Lab Invest. (2019) 99:1130–42. doi: 10.1038/s41374-019-0190-4
148. Xu X, Bu B, Tian H, Wu R, Yang J. Micrornas combined with the tlr4/tdag8 mrnas and proinflammatory cytokines are biomarkers for the rapid diagnosis of sepsis. Mol Med Rep. (2022) 26(5):334. doi: 10.3892/mmr.2022.12850
149. Cui S, Niu K, Xie Y, Li S, Zhu W, Yu L, et al. Screening of potential key ferroptosis-related genes in sepsis. PeerJ. (2022) 10:e13983. doi: 10.7717/peerj.13983
150. Yang Q, Zhang D, Li Y, Li Y, Li Y. Paclitaxel alleviated liver injury of septic mice by alleviating inflammatory response via microrna-27a/tab3/nf-Kb signaling pathway. BioMed Pharmacother. (2018) 97:1424–33. doi: 10.1016/j.biopha.2017.11.003
151. Chen L, Lu Q, Deng F, Peng S, Yuan J, Liu C, et al. Mir-103a-3p could attenuate sepsis-induced liver injury by targeting hmgb1. Inflammation. (2020) 43:2075–86. doi: 10.1007/s10753-020-01275-0
152. Lv X, Zhang Y, Cui Y, Ren Y, Li R, Rong Q. Inhibition of microrna−155 relieves sepsis−Induced liver injury through inactivating the jak/stat pathway. Mol Med Rep. (2015) 12:6013–8. doi: 10.3892/mmr.2015.4188
153. Bai J, Chen S. Lncrna casc9 enhances the stability of socs-1 by combining with fus to alleviate sepsis-induced liver injury. Cytokine. (2023) 171:156346. doi: 10.1016/j.cyto.2023.156346
154. Li L, He Y, He XJ, Bi MR, Qi YH, Zhu WW. Down-regulation of long noncoding rna linc00472 alleviates sepsis-induced acute hepatic injury by regulating mir-373-3p/trim8 axis. Exp Mol Pathol. (2020) 117:104562. doi: 10.1016/j.yexmp.2020.104562
155. Shen C, Li J. Lncrna xist silencing protects against sepsis-induced acute liver injury via inhibition of brd4 expression. Inflammation. (2021) 44:194–205. doi: 10.1007/s10753-020-01321-x
156. Yagi H, Soto-Gutierrez A, Kitagawa Y, Tilles AW, Tompkins RG, Yarmush ML. Bone marrow mesenchymal stromal cells attenuate organ injury induced by lps and burn. Cell Transplant. (2010) 19:823–30. doi: 10.3727/096368910x508942
157. Liu Y, Ren H, Wang J, Yang F, Li J, Zhou Y, et al. Prostaglandin E(2) secreted by mesenchymal stem cells protects against acute liver failure via enhancing hepatocyte proliferation. FASEB J. (2019) 33:2514–25. doi: 10.1096/fj.201801349RR
158. Cóndor JM, Rodrigues CE, Sousa Moreira R, Canale D, Volpini RA, Shimizu MH, et al. Treatment with human wharton’s jelly-derived mesenchymal stem cells attenuates sepsis-induced kidney injury, liver injury, and endothelial dysfunction. Stem Cells Transl Med. (2016) 5:1048–57. doi: 10.5966/sctm.2015-0138
159. Liang X, Li T, Zhou Q, Pi S, Li Y, Chen X, et al. Mesenchymal stem cells attenuate sepsis-induced liver injury via inhibiting M1 polarization of kupffer cells. Mol Cell Biochem. (2019) 452:187–97. doi: 10.1007/s11010-018-3424-7
160. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
161. Park EJ, Appiah MG, Myint PK, Gaowa A, Kawamoto E, Shimaoka M. Exosomes in sepsis and inflammatory tissue injury. Curr Pharm Des. (2019) 25:4486–95. doi: 10.2174/1381612825666191116125525
162. Cai J, Tang D, Hao X, Liu E, Li W, Shi J. Mesenchymal stem cell-derived exosome alleviates sepsis- associated acute liver injury by suppressing malat1 through microrna-26a-5p: an innovative immunopharmacological intervention and therapeutic approach for sepsis. Front Immunol. (2023) 14:1157793. doi: 10.3389/fimmu.2023.1157793
Keywords: sepsis, liver injury, inflammation, oxidative stress, pyroptosis, autophagy, noncoding regulation
Citation: Xu X, Yang T, An J, Li B and Dou Z (2025) Liver injury in sepsis: manifestations, mechanisms and emerging therapeutic strategies. Front. Immunol. 16:1575554. doi: 10.3389/fimmu.2025.1575554
Received: 12 February 2025; Accepted: 13 March 2025;
Published: 28 March 2025.
Edited by:
Lulong Bo, Navy Medical University, ChinaReviewed by:
Yukun Liu, Huazhong University of Science and Technology, ChinaCopyright © 2025 Xu, Yang, An, Li and Dou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Dou, bGR5eV9kb3V6aGltaW5AbHp1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.