 Filomena Napolitano
Filomena Napolitano Nunzia Montuori
Nunzia Montuori
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 March 2025
Sec. Molecular Innate Immunity
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1568629
This article is part of the Research Topic Pattern Recognition Receptors: Balancing Inflammation and Immune Homeostasis View all 3 articles
Pattern Recognition Receptors (PRRs) are a superfamily of receptors that detect molecular structures typical for pathogens and damaged cells and play a crucial role in the proper function of the innate immune system. A particular subgroup of membrane-bound PRRs is represented by the N-formyl peptide receptors (FPRs) that consist of transmembrane G-protein coupled receptors involved in inflammatory responses. FPRs were initially described in immune cells as transducers of chemotactic signals in phagocytes that react to tissue injury. Subsequently, FPRs were also identified in a wide variety of cell types, including cancer cells. Beyond broad cellular distribution, FPRs are also characterized by the ability to bind a variety of ligands with different chemical and biological properties, ranging from natural peptides to synthetic compounds. The binding of FPRs to specific agonists induces a cascade of functional biological events, such as cell proliferation, migration, angiogenesis, and oxidative stress. From all this evidence, it becomes clear that FPRs are multifaceted receptors involved in several pathophysiological processes associated with inflammation. In this review, we provide a comprehensive molecular description of structure-function relationship of FPRs and their pivotal role in the host defense, highlighting the regulatory functions in both the initiation and resolution of inflammation. In addition to their activity as PRRs during innate immune response, we focus on their involvement in pathological conditions, including chronic inflammatory disease, neurodegenerative disorders, and cancer, with special emphasis on FPR targeting as promising therapeutic strategies in the era of precision medicine.
The innate immune system serves as the first-line defense and provides rapid, non-specific protection against pathogens. It is now accepted that innate immune response is crucial for the development of adaptive immunity and the regulation of diverse physiological processes (1). Critical modulators of innate immune response are Pattern Recognition Receptors (PRRs), a superfamily of receptors that function as nonspecific sensors for pathogen-associated molecular patterns (PAMPs) and/or damage-related molecular patterns (DAMPs) (2). PAMPs are released from pathogens whereas DAMPs are endogenous molecules, including nucleic acids, proteins, ions, glycans, and metabolites, which are released following cell damage and/or stress. The ability of PRRs to recognize both PAMPs and DAMPs allows them to take part in non-sterile and sterile inflammation (3). After DAMP/PAMP recognition, PRRs induce transient pro-inflammatory gene expression and immune cell activation to remove pathogens or restore tissue homeostasis. However, chronic activation of PRR has been linked to chronic inflammation and autoimmune diseases.
Based on their localization, PRRs may be classified into membrane-bound PRRs implicated in the surveillance of extracellular milieu and intracellular PRRs activated by internalized inflammatory stimuli (4). Frequently, the description of PRR sub-families is incomplete as the most prevalent classification in literature includes only Toll-like receptors (TLRs), Nod-like receptors (NLRs), C-type lectin receptors (CLRs), and RIG-like receptors (RLRs) (4). Phagocytic leukocytes also express another class of innate immune receptors, named N-formyl peptide receptors (FPRs), which play a key role in host defense and inflammation. To date, three subtypes (FPR1, FPR2, and FPR3) have been identified in humans though the role of FPR2 in inflammation is much debated. They are members of the seven transmembrane receptors, also known as G protein-coupled receptors (GPCRs), which are responsible for transducing a wide range of signals across the plasma membrane (5).
The discovery of FPRs was made possible after the identification and characterization of formylated peptides from which they also take their name. Since prokaryotes, excluding Archaea, initiate protein synthesis with N-formyl methionine, chemically synthesized peptides starting with N-formyl methionine were used to evaluate neutrophil activation (6, 7). Moreover, formylated peptides are contained in mitochondria from which they are released following tissue and cellular trauma. The data that demonstrated leucocyte migration toward N-formyl peptides led to the discovery of FPR1. In early studies, N-formyl-methionyl-leucyl-phenylalanine (fMLF) resulted as the most potent agonist for neutrophil chemotaxis (7).
Apart from innate immune response, FPRs are an intriguing PRR class that may have more complex functions than are currently appreciated. This hypothesis is based on two important assumptions: i) FPRs are highly expressed in innate immune cells, but they are also detected on nonhematopoietic cells, including epithelial cells, endothelial cells, hepatocytes and neurons; ii) beyond formylated peptides, high number of FPR ligands eliciting different cellular responses has been reported in the literature. The broad tissue distribution of FPRs corresponds to their wide pathophysiological relevance as demonstrated by several studies ranging from chronic inflammation to cancer. However, the development of FPR-targeting drugs encounters many difficulties due to ligand promiscuity and marked signaling bias downstream of FPRs.
In this review, we discussed the most recent advancements on the biological functions of FPRs, the role of FPRs in the development and progression of inflammatory chronic diseases and how do FPRs function in the tumor microenvironment. This thorough analysis aims to offer a fresh insight into the pathophysiological role of FPRs and establish a sound scientific foundation for future therapeutic approaches.
In humans, the N-formyl peptide receptor family consists of three isoforms, named FPR1, FPR2, and FPR3, each encoded by a separate gene located on chromosome 19q13 (8). Given that early studies demonstrated that these receptors recognize formylated peptides deriving from bacterial and mitochondrial protein synthesis, FPRs were so named following the criteria of the International Union of Basic and Clinical Pharmacology (IUPHAR) nomenclature of a receptor based on its agonist. Traditionally, FPRs are considered as important mediators of inflammatory and immune responses in pathogen infection and cellular damage though a new scenario is emerging from several studies that reveal new insights on the heterogeneity and the promiscuity of these receptors. In fact, FPRs play a crucial role in pathophysiological processes such as tissue repair and chronic inflammation independently of binding to formylated peptides (9–11).
FPRs are G protein-coupled receptors (GPCRs), also known as seven-(pass)-transmembrane domain receptors containing an extracellular amino terminus, seven transmembrane helices, and an intracellular carboxy terminus. In contrast to the conventional localization of FPRs to the plasma membrane, new paradigms indicate that FPRs also localize to the nucleus, thus participating in intra-nuclear signaling, as demonstrated for FPR2 in lung carcinoma and gastric adenocarcinoma human cell lines (12). Subcellular fractionation has been described also for FPR1 that is stored in primary granules and secretory vesicles of circulating neutrophils (13). Further studies on subcellular fractionation, FPR transport from intracellular compartments to plasma membrane, and receptor cycling are needed.
However, binding of a signaling molecule to FPRs results in G protein activation, which in turn triggers the activation of multiple downstream second messengers. Particularly, FPRs are coupled with inhibitory G subfamily (Gi) of G proteins, as demonstrated by the total loss of cell response to agonists after the exposition to pertussis toxin (PTX) (14). Upon activation, intracellular domain of FPRs mediate signaling to heterotrimeric G-proteins, which dissociate into α and βγ subunits and trigger distinct and divergent signaling pathways. βγ subunit activates phospholipase C (PLC) responsible for calcium mobilization and activation of protein kinases C (PKC), which are crucial for degranulation and superoxide production. βγ subunit also activates phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway that is necessary for cell chemotaxis and superoxide production. α subunit of G proteins regulates the Ras superfamily of small GTPases, which are critical for cytoskeletal reorganization and lead to the activation of mitogen-activated protein kinase (MAPK) pathways, extracellular-regulated protein kinase 1/2 (ERK1/2), p38, and JUN-N-terminal protein kinase (JNK) (15). Notably, α subunit of G proteins also regulates the assembly and activation of NADPH oxidase, crucial for innate immunity as during phagocytosis it produces superoxide, thus contributing to the elimination of invading microorganisms. Particularly, FPR activation induces reactive oxygen species (ROS) production through GTP bound-Rac1 and ERK 1/2 pathways and the direct binding of Rac1 to the NADPH oxidase cytosolic component p67phox is crucial to form the active oxidase (10).
Since membrane lipid rafts are crucial to organizing the complicated trafficking and signaling of GPCRs, it is conceivable that lipid rafts can serve as platforms for integrating signal transduction processes in FPR signaling as well, but further investigations should be undertaken to explore the exact molecular and biochemical mechanisms. To date, cholesterol-rich plasma membrane rafts have been demonstrated to play a crucial role in intracellular transduction processes in FPR2 signaling (16). Moreover, recent data suggest that the constitutive internalization of FPR3 occurs through a mechanism dependent on caveolae, a distinct subset of lipid rafts enriched in the structural scaffolding protein caveolin (17).
Following activation by ligand, the control and the termination of FPR activities occur through uncoupling from G proteins (desensitization) and receptor internalization, which removes receptors from the cell surface. These mechanisms are crucial to prevent damage to the host in the continued exposure to activating ligand. In general, GPCR desensitization and internalization are mediated by receptor phosphorylation and arrestin binding to the cytosolic domains of the agonist-occupied receptor (18). The internalization of FPRs could occur in the absence of arrestins, which is however required for the recycling of internalized receptor (19, 20). It has been demonstrated that the processes of FPR desensitization and internalization are differentially regulated by phosphorylation at distinct sites within the carboxyl terminus of FPRs (19). In addition to homologous desensitization, FPR activation desensitizes several chemokine receptors, including CXCL8, CXCR4, CCR1, CCR5 by a mechanism defined heterologous desensitization (21–24). The complexity of FPR-mediated signaling pathways is partly linked to their ability to recognize a large variety of pathogen-derived and endogenous ligands and different agonists of the same FPR isoform can trigger distinct signaling cascade. Emerging studies have introduced the concept of “biased agonism” to explain the extensive variation in FPR-mediated signaling pathways. Biased agonism indicates that ligands stabilize the receptors in a specific conformation that triggers one signaling pathway between the different signal transduction pathways associated with the receptor itself. In practice, different agonists specific to an FPR isoform cause distinct effects. Therefore, cellular response is biased towards a specific signaling pathway (25). Of note, the multiple signals transduced by FPRs are also dependent both on ligand concentration and exposure time to ligand (26). Biased agonism dependent on ligand concentration could be determined by the ability of FPRs to express both high and low affinity binding sites for ligands and each binding site could elicit a specific intracellular signaling pathway. Alternatively, different concentrations of ligand and/or exposure time could induce distinct conformational changes in FPRs.
In addition to biased agonism, FPRs can undergo oligomerization that could be responsible for different signaling pathways. It has been reported that FPRs form different higher order structures, including FPR1 homodimers, FPR2 homodimers, FPR2/FPR1 heterodimers, and FPR2/FPR3 heterodimers (27–29). Surprisingly, anti-inflammatory and pro-inflammatory ligands might preferentially act on different FPR oligomers (27). It has been shown that pro-resolving ligands induce the formation of FPR1/FPR2 heterodimers and FPR2 homodimers, whereas pro-inflammatory ligands and FPR antagonists are not able to induce the formation of these structures. Moreover, Cooray et al. demonstrated that FPR2 homodimers might be linked to the activation of the p38/MAPK-activated protein kinase/heat shock protein 27 signaling pathway, exerting immunomodulatory and anti-inflammatory effects, including the release of IL-10 (27). However, the specificity and functional significance of homo- and hetero-dimerization of FPRs has not yet been completely understood. Future studies are needed to provide the molecular mechanism underlying the dual nature of FPRs, especially FPR2, and how agonist binding contributes to receptor dimerization.
The possibility to selectively activate or inhibit specific pathways has attracted molecular pharmacology, thus leading to the development of biased agonists. In fact, a given FPR biased agonist might activate anti-inflammatory and pro-resolving pathways or inhibit Gi protein-mediated proinflammatory activities of the FPRs.
Remarkably, biased agonism-based regulation of FPR signaling and functions represents a valid therapeutic approach for inflammatory diseases. To shed light on the biased signaling mechanisms and favor the development of FPR-biased anti-inflammatory and pro-resolving therapies, conformational studies, crystallography, cryo-EM, and evidence for FPR higher-order structures are needed.
Several functions of FPRs occur through the interaction with the urokinase (uPA) receptor (uPAR). A specific region of uPAR, corresponding to amino acids 88-92 (SRSRY) can interact with FPRs, mediating uPA- or fMLF-dependent cell migration. uPA itself can promote uPAR interaction with FPRs, by determining the exposure of the uPAR88-92 region, upon binding to the receptor (30). fMLF-dependent cell migration of epithelial cells requires uPAR interaction with FPR1; on the other hand, uPAR interacts with FPR2 in monocytes and with FPR2 and FPR3 in basophils (9, 31, 32). Recently, it has been demonstrated that uPAR is able to induce cell proliferation in normal skin fibroblasts by interacting with FPR1, FPR2, and FPR3 and the inhibition of a single isoform with specific antibodies causes cell growth arrest (11). A cooperation between the three isoforms is conceivable, most likely through the formation of heterodimers. Moreover, FPRs/uPAR crosstalk induces a proliferative phenotype in skin fibroblasts, by Rac1 and ERK activation, c-Myc Ser62 phosphorylation and Cyclin D1 upregulation that drive cell cycle progression (11). To fulfill the wide range of functions, FPRs belong to a macromolecular complex on cell surface. Cell migration and ROS production require FPR interaction with uPAR and β1 integrin engagement, as demonstrated by experiments performed with P25 peptide, which disrupts uPAR interactions with β1 or β2 integrins (10, 33). A better understanding of the mechanisms by which FPRs interact with β1 integrin - with or without direct interaction - will provide new insights on the biological functions of these receptors.Taken together, these findings suggest that FPRs could modulate both pro- and anti-inflammatory response, depending on the nature and the concentration of the ligand and on the different binding sites of the receptor. The interplay of FPRs with different ligands and how different ligands interacting with the same FPR isoform can trigger opposing effects should be better examined to develop innovative approaches for the treatment of chronic diseases.Interestingly, FPRs are subjected to phenomena of dynamic evolution and neofunctionalization that could partly explain the complexity and promiscuity of these receptors. An early duplication induced FPR1 and FPR2/FPR3 splitting, whereas FPR3 originated from the latest duplication near the origin of primates, as shown by a phylogenetic analysis (34). High frequency of polymorphisms affects FPR expression or function and different responses of FPR haplotypes toward bacterial peptides have been detected (35, 36). Of note, the ligand specificity of FPRs seems to be conserved among species, suggesting a conserved function of FPRs in the immune system (36). Although evolutionary history indicates that FPRs are subjected to purifying selection, ligand-binding sites have experienced positive selection and significant sequence divergence has occurred among mammalian FPR1 genes (34). FPRs are also an example of genetic innovation; genomic events observed in rodents led to expansion and neofunctionalization of FPRs, which have evolved from pathogen sensors in immune cells to olfactory receptors in neurons (37).
Over the past 20 years, it has become increasingly clear that FPRs play a crucial role in host defense, as well as in immune and non-immune inflammatory disorders. Given that FPRs are a complex receptor system, the identity and the contribution of specific isoforms in health and disease remains largely unknown and, certainly, merit further research.
In the next chapter, we aim to report the main data relating to three members of FPRs to shed light on the single contribution of FPR1, FPR2 and FPR3 in pathophysiology through the description of the main ligands activating FPRs. Since the FPR family is known for the structural and biological diversity of their ligands, the multiple FPR ligands discovered over the past years are summarized in Table 1 and grouped into microbe-derived, endogenous, and synthetic ligands.
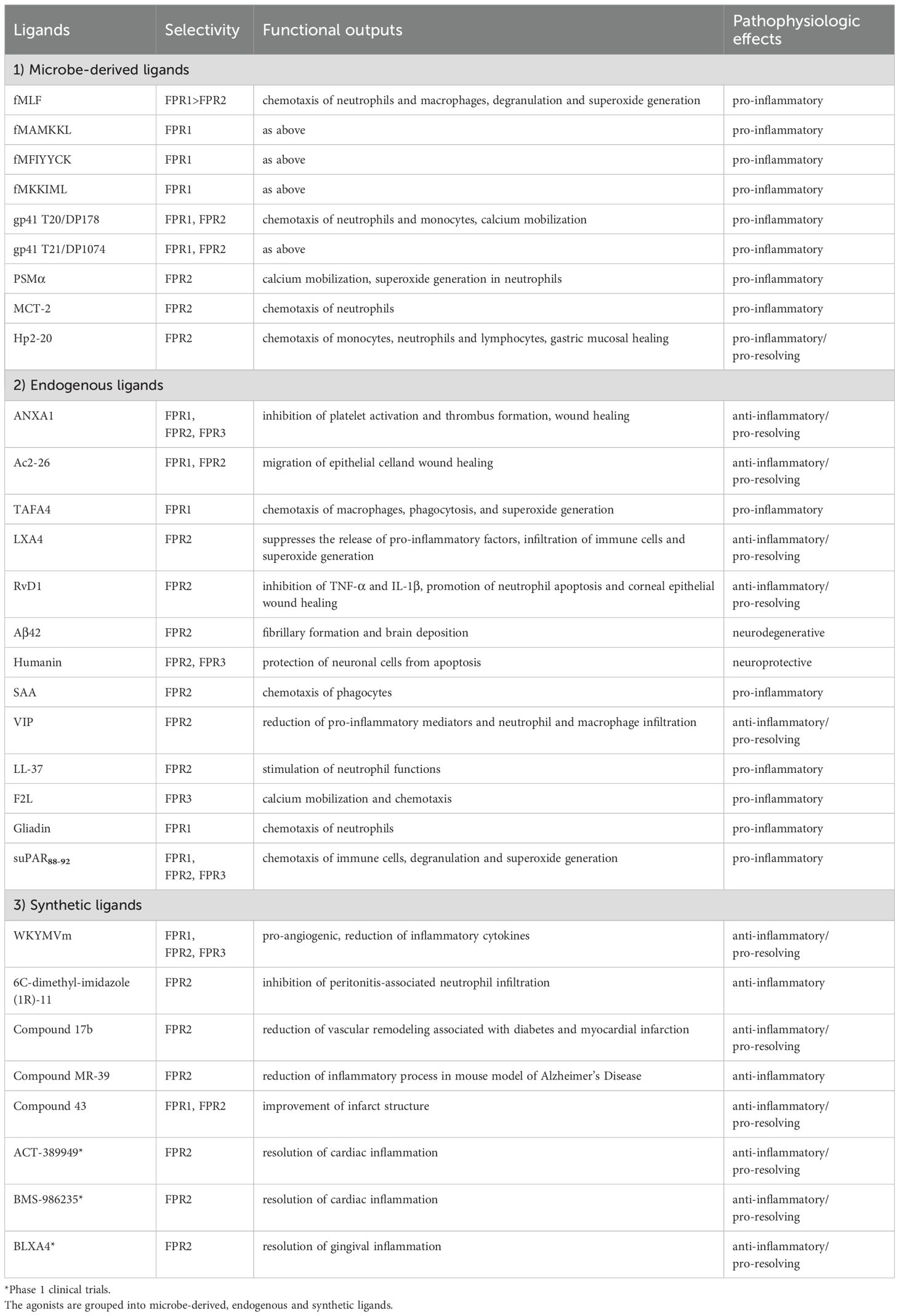
Table 1. Summary of the main ligands activating FPRs.
Among the three FPRs identified in humans, the first detected isoform was FPR1, which is a high-affinity receptor for various short formylpeptides such as the prototypical peptide N-formyl-Met-Leu-Phe (fMLF) (38).
The structure of FPR1 consists of 350 amino acids with two putative N-linked glycosylation residues at positions 4 and 10 and a disulfide bond at residues 98↔176 (39). Three putative phosphoserine sites (residues 328, 332, and 338) and four putative phosphothreonine sites (residues 329, 331, 334, 336, and 339) have also been identified (40). The secondary structure consists of seven transmembrane (TM) helixes (TM1 to TM7) characterized by six loops, among which three in the extracellular region and other three in the cytoplasm region (41). The restricted opening at the extracellular surface could explain the higher affinity of FPR1 for short peptides, unlike FPR2 (38).
FPR1 is mainly expressed in myeloid cells, including leukocytes, monocytes, macrophages, natural killer and dendritic cells. It has been also detected in nonhematopoietic cells, such as fibroblasts, epithelial cells, endothelial cells, smooth muscle cells, astrocytes, and hepatocytes (39).
The chemoattractant properties of FPR1 have been widely demonstrated; activated FPR1 plays a key role in the directional migration of phagocytes to the site of bacterial infections (42). During infections, FPR1 is also a crucial modulator of phagocyte degranulation and superoxide generation, by requiring Gi coupling (43).
Distinct FPR1-mediated phagocyte functions are regulated by different concentrations of chemoattractants. It has been demonstrated that FPR1 selectively triggers distinct signaling pathways in human peripheral blood neutrophils based on different concentrations of fMLF; subnanomolar concentrations of fMLF induced chemotaxis and conformational change of FPR1 whereas nanomolar and micromolar concentrations were responsible for degranulation and superoxide generation (44). Of note, the tight regulation of FPR1 by agonist concentration expands our knowledge of innate immunity: first, FPR1 responds with the chemotaxis of phagocytes to avoid damage to not affected tissues; second, it induces bactericidal activities of phagocytes in the site of infection, where the bacterial-derived peptide is at high concentrations.
FPR1 can recognize a variety of formylated and non-formylated, microbe- and host-derived ligands. Beyond the canonical E. coli-derived peptide fMLF, FPR1 interacts with other formylated bacterial peptides, including Salmonella-derived fMAMKKL peptide, Staphylococcus-derived fMFIYYCK peptide, and Listeria-derived fMKKIML peptide (45, 46). The microbe-derived non formylated peptides that interact with FPR1 are mainly viral, but further investigations are needed. Particularly, Human Immunodeficiency Virus (HIV) envelope proteins, such as gp41 T20/DP178 and gp41 T21/DP107, have been proposed as ligands of FPR1 though it has been shown that the chemotactic peptides released from HIV-1 gp41 act mainly through FPR2 (47). Both full-length Annexin-1 (ANXA1) and the N-terminal peptide of the protein, Ac2-26, have been proven to interact with FPR1 (48). FPR1 acts as a cytokine ligand, as demonstrated by TAFA4 or FAM19A that represents a novel class of chemokine like ligands involved in the regulation of immune responses within the central nervous system (40, 49). Furthermore, several synthetic low-molecular-weight ligands for FPR1 have been identified and some of these constitute novel promising compounds for the treatment of cancer and other inflammatory disorders (5).
Among the FPR family, FPR2 is the most versatile and promiscuous isoform both for the variety of structurally diverse ligands and for the ability to mediate also anti-inflammatory and pro-resolving actions. FPR2 is also indicated as ALX/FPR2 since it binds the endogenous ligand A4 lipoxin (LXA4).
FPR2 is primarily expressed in the membranes of myeloid cells, such as leukocytes, monocytes, macrophages, natural killer and dendritic cells. Non-immune FPR2-expressing cells are mainly epithelial cells, hepatocytes, astrocytoma and neuroblastoma cells, and microvascular endothelial cells (50).
Molecular details regarding the structure and ligand recognition of FPR2 are still under investigation though in the last 5 years numerous efforts have been made to reveal insights into ligand-binding modes of FPR2. To date, there is data on the structure of FPR2 associated only with agonists. It would be useful and interesting to perform studies on FPR2 in an inactive conformation, for example FPR2-bound to specific antagonists. The crystal structure of FPR2 complexed with synthetic peptide WKYMVm showed an active conformation and revealed a canonical seven-transmembrane helical bundle conformation. The extracellular region is composed of an N terminus and three extracellular loops, with β-hairpin conformation of second extracellular loop that is connected to helix III by a disulfide bridge (51). A recent study of the structure of FPR2-Gi complex revealed three important insights into ligand-recognition and signaling: i) a vast space in ligand-binding pocket could explain the ability to recognize long peptides and large proteins; ii) the high flexibility of extracellular region of FPR2 could be linked to the ability to recognize chemically diverse agonists; iii) the amphiphilic nature of ligand-binding pocket fit with the ability to bind pro-resolving lipid mediators (52). Notably, protein and lipid ligands bind FPR2 in distinct binding sites (28).
How FPR2 displays opposing activities has been studied by different approaches. Cooray et al. proposed an unusual molecular mechanism that governs the dual function of FPR2 by regulating pro- and anti-inflammatory activities. Agonist binding and dimerization state determine FPR2 functional response; particularly, anti-inflammatory and pro-resolving signals activate homodimers of FPR2 leading to the release of IL-10, as well as FPR2/FPR1 heterodimers (27). Instead, Zhang et al. proposed the allosteric modulation of FPR2 as molecular mechanism underlying signaling bias abilities. Of note, the preincubation with agonists with pro- and anti-inflammatory effects at very low concentrations elicited different conformational changes in FPR2 (28).
The main feature of FPR2 is represented by the wide variety of endogenous and exogenous ligands including proteins, lipids and various stimulatory mediators. Formylated peptides, including fMLF, bind to FPR1 with a higher binding affinity, as described above, though alpha-type phenol-soluble modulins (PSMα) produced by Staphylococcus aureus and mitochondria-derived mitocryptide-2 (MCT-2) prefer to bind to FPR2 (50). Helicobacter pylori-derived non-formylated peptide, abbreviated to Hp2-20, is a potent FPR2 agonist and this interaction plays a critical role in the pathological processes associated with Helicobacter pylori infection (46). On the other hand, Hp2-20 promotes gastric mucosal healing by interacting with FPR2, thus suggesting a pro-resolving action (53). Furthermore, FPR2 can recognize several host-derived peptides. Among the evidence that indicates the role of FPR2 in facilitating the resolution of inflammation, there is the binding to LXA4. In fact, LXA4, which derives from arachidonic acid, exerts strong anti-inflammatory and pro-resolving effects (54), but further studies are needed to determine the functional role of LXA4 as FPR2 agonist. FPR2 can also act as a pro-resolving mediator by binding to Resolvin D1 (RvD1), which has been proven to exert good regulatory effects on inflammation (55). Apart from the ability to mediate the pro-resolving signal of lipidic mediators, FPR2 binds diverse peptides involved in inflammatory disorders, especially neuroinflammatory disease. Of interest, FPR2 serves as a receptor for β-amyloid peptide (Aβ42), highlighting its involvement in the pathogenesis of Alzheimer’s disease (AD). In contrast to this data, FPR2 has been shown to mediate the neuroprotective effects of humanin (56). This evidence, once again, showed the multifaceted nature of FPR2.
The dual role of FPR2 also emerges from studies on the Serum Amyloid A (SAA) as an agonist for FPR2. SAA is considered as a major acute-phase protein and is involved in the chemotaxis of phagocytes in inflamed tissues. Further studies showed that SAA plays a crucial role in epithelial wound healing to be indicated as an epithelial pro-restitutive factor by Hinrichs BH et al. (57). Thus, SAA could mediate opposing effects via FPR2.
Vasoactive Intestinal Peptide (VIP) is another host-derived peptide that activates FPR2 and directs FPR2 signaling towards the resolution of inflammation (58), whereas LL-37 elicits the proinflammatory action of FPR2 (50).
It is important to underline that the ligands of FPR2 with anti-inflammatory and pro-resolutive action are under investigation to develop new therapeutic strategies for diverse inflammatory conditions. Furthermore, the two opposing roles of FPR2 are still under study and can be relevant in the context of chronic diseases.
New knowledge of the pro-resolving signaling pathways has been acquired. Pro-inflammatory ligands activate FPR2 by coupling to Gαi resulting in Ca2+ mobilization and ERK phosphorylation, whereas pro-resolving mediators induce β-arrestin 2 recruitment, cAMP production and p38 MAPK phosphorylation. β-arrestins probably mediates heterologous desensitization of FPR2. Subsequent FPR2 internalization is essential to pro-resolving ligands as the internalized receptor inhibits NF-κB (59). In neutrophils, the recruitment of β-arrestin is crucial for FPR2 reactivation and cell migration, as demonstrated by the cell stimulation with conventional FPR2 agonist WKYMVM (60).
On the note, recent studies indicated that FPR2 expression could be regulated by a combination of hormones, genetic, and epigenetic factors. Sex-specific differences of FPR2 expression have been observed in mice. Particularly, estrogen directly regulates FPR2 expression as a protective mechanism against nonalcoholic fatty liver disease (61). In pancreatic tumor, FPR2 exerts immunosuppressive function in females, suggesting a potential sex‐specific approach for immunotherapy (62). Accumulating evidence showed that FPR2 expression could be regulated by microRNAs, especially during inflammation resolution. Pierdomenico AM et al. demonstrated that miR-181b directly binds FPR2 3′-UTR and controls pro-resolution signals (63). Epigenetic regulation of FPR2 promoter and heritable genetic variants that impair promoter activity have been discovered (64). The promoter polymorphism rs11666254 downregulates FPR2/ALX expression and increases risk of sepsis (65). Overall, the regulatory mechanisms of FPR2 expression should be investigated in future studies to provide innovative approaches to inflammatory diseases.
FPR3 still remains poorly characterized. Unlike FPR1 and FPR2, FPR3 does not serve as receptor for fMLF, which represents the established peptide released at sites of bacterial infections and tissue injury. It shares 56% and 83% of its amino acid sequence with FPR1 and FPR2, respectively. FPR3-expressing cells are mainly monocytes/macrophages and endothelial cells (66).
The structure of FPR3 has been largely unknown, although some experimental techniques have been suggested for performing structural analysis. For example, T4-lysozyme (T4L) fusion in the intracellular loop of a G protein-coupled receptor (GPCR) of FPR3 were expressed in stable tetracycline-inducible HEK293 cells and guaranteed approximately 0.2 mg of highly purified monomeric human FPR3-T4L from 2 g of cells. Since T4L fusion did not disturb the proper folding and functionality of FPR3, this strategy could facilitate ongoing efforts in the structural analysis of FPR3 (67).
Of note, FPR3 binds non-formylated peptides. ANXA1, which mediates the anti-inflammatory effects of glucocorticoids, is the first endogenous ligand of FPR3 (68). However, the high affinity ligand for FPR3 is F2L peptide, an acetylated amino-terminal peptide of the human heme-binding protein. In FPR3-expressing leucocytes, F2L induces calcium mobilization and chemotaxis. F2L thereby may be considered as a pro-inflammatory stimulus for FPR3. It has been demonstrated that F2L is generated after tissue damage and induces the recruitment of macrophages and dendritic cells, which contribute to tissue repair and resolution of inflammation (69). This data suggests a pro-resolutive action of FPR3. Another agonist for FPR3 is humanin, a mitochondrial-derived peptide with cytoprotective effects in many cell types. Humanin does not bind exclusively FPR3, but also FPR2 (70).
Interestingly, FPR3 presents a significant basal level of phosphorylation in the absence of ligand, probably due to the presence of Pro329. The clathrin-pathway is required for its constitutive internalization (71). Corroborating data demonstrated that FPR3 is expressed in small intracellular vesicles, unlike FPR1 and FPR2 that are expressed on the cell surface. Besides, it has been recently found that FPR1 is expressed intracellularly, while FPR1 and FPR3 were expressed in the nuclei of naïve CD4 T cells. Intracellular and/or nuclear FPR1 but not FPR3 induced chemotactic migration of naïve CD4 T cells (72).
In 2011, Rabiet MJ proposed that the constitutive internalization of FPR3 may serve to regulate the function of FPR1 and FPR2, but these hypotheses have never been confirmed (71).
From data present in literature, it emerges that FPR3 prevalently exerts anti-inflammatory and pro-resolutive actions, as demonstrated by the binding to regulatory peptide against inflammatory processes. Moreover, the intracellular localization of FPR3 opens new questions about the role of FPR family and the identification of endogenous ligands for intracellular FPR members could clarify their functional roles in inflammation and tissue repair. It is conceivable that the lack of scientific data on FPR3 could be linked to the absence of known ligands for intracellular and extracellular FPR3. For example, a recent analysis of the effects of d−Peptide analogues of Boc−Phe−Leu−Phe−Leu−Phe−COOH (used extensively as a FPR1/FPR2 antagonist) on neovascularization showed that this peptide elicited the angiogenic activity via FPR3, which is expressed by endothelial cells (73).
Based on their expression on a wide variety of cell types and their ability to bind ligands with diverse structural and functional features, FPRs have been implicated in multiple human diseases. In particular, the contribution of FPRs to inflammation and cancer places these receptors in the list of molecular targets for new therapeutic approaches. In addition, the study of the functional roles of FPRs in the mucosal immune system has attracted great interest. Mucosal immune cells, including innate lymphoid cells, epithelial cells, and intraepithelial lymphocytes, express FPRs that contribute to maintaining tissue homeostasis at these sites. Recent research focuses on the controversial role of FPRs in the mucosal system depending on their ligands expressed under various pathophysiological conditions (74). Although many reports indicate that FPRs are crucial for maintaining epithelial barrier integrity, a link between FPRs and inflammatory conditions affecting mucosal system, especially gastrointestinal tract and respiratory system, have been found, as described below.
Given that FPR1 plays a crucial role in the inflammatory reactions implicated in disease pathogenesis, FPR1 antagonists represent a promising therapeutic approach in inflammation and cancer.
Zhou et al. were among the first to recognize the involvement of FPR1 in tumorigenesis, demonstrating that FPR1 is expressed by highly malignant human glioma cells and mediate motility, growth, and angiogenesis of human glioblastoma (74). The mechanism implicated in FPR1-mediated glioma growth has been elucidated. ANXA1 released from necrotic tumor cells induces the growth of glioblastoma via the activation of FPR1 (75). Later, Zheng et al. demonstrated that ANXA1 was mainly expressed in cancer cells from high-grade glioma (HGG), whereas FPR1 was predominantly expressed in macrophages and microglia, suggesting that cancer cells interact with macrophages and microglia through the ANXA1/FPR1 axis to inhibit the immune response against glioma (76). These data reveal a new perspective of immunotherapy for glioma, which is characterized by a highly immunosuppressive tumor microenvironment.
A pro-metastatic role in ovarian cancer has been assigned to FPR1. uPAR triggers intra-abdominal dissemination of epithelial ovarian cancer cells through the interaction of its 84-95 sequence with the FPR1. The inhibition of this interaction by anti-uPAR84-95 Abs or RI-3 peptide significantly decreased the extent of cell adhesion (41). Elevated levels of FPR1 were associated with poor prognosis in cervical cancer patients (77). The over-expression of FPR1 has also been associated with drug-resistance in bladder cancer (78).
In contrast to the data described above, Prevete et al. demonstrated a protective role of FPR1 in tumor growth and progression. In gastric cancer, FPR1 suppresses tumor growth and angiogenesis due to a pro-resolving action (79). In the same way, the commensal bacterium Lactobacillus rhamnosus GG (LGG) exerts pro-resolving, antiangiogenic and homeostatic functions by activating FPR1 in colorectal carcinoma cells (80).
In other studies, the interaction between ANXA1 and FPR1 appears to be relevant in immune response against dying cancer cells. Of interest, anthracyclines-based chemotherapy didn’t exert therapeutic effects in FPR1 knockout tumor-bearing mice as Fpr1-deficient dendritic cells could not elicit T cell immunity against dying tumor cells (81).
The role of FPR1 in immunity and inflammation has been considered “ambiguous” (42). In infectious diseases, FPR1 plays an ambivalent role. In Escherichia coli and Listeria monocytogenes, FPR1 activation is crucial for host defense, as demonstrated in mouse strains deficient in Fpr1 (82, 83). Conversely, the absence of FPR1 provides protection against Yersinia pestis, which is the causative agent of plague (84). In COVID-19, FPR1 signaling is linked to pulmonary fibrosis and other long-term symptoms by inducing excessive alarmins S100A8/A9 (85).
FPR1 triggered the spread of inflammation, cardiomyocyte apoptosis and ventricular remodeling through a positive regulation of MAPK signaling pathway, as demonstrated in animal models of ischemia/reperfusion injury (86). In ischemic retinopathy, FPR1 contributed to inflammation and hindered reparative angiogenesis, thus implying its involvement in neuroretinal dysfunction (87).
In chronic inflammatory disease, there are contrasting data on the role of FPR1. In collagen‐induced arthritis, FPR1 induces beneficial effects by contributing to the inhibitory effects on TH1 and TH17 cell generation, which are crucial for disease progression (88). Conversely, other studies assign a pathogenetic role to FPR1 in diseases conditions associated with dysregulated inflammation, such as celiac disease, cigarette smoke-induced airway inflammation disease, and inflammatory bowel disease (89–91). For example, gliadin, a food antigen that binds to FPR1, decreases intestinal integrity and stimulates neutrophil migration (89). As demonstrated in the intestinal tract, FPR1 plays a crucial role also in inflammation-related diseases of respiratory system. FPR1 is overexpressed in neutrophils isolated from smokers, as compared to non-smokers. Since smoking cessation cannot revert lung inflammation, the FPR blockade prevents structural deterioration of the lungs in mice after smoking cessation (92).
These findings demonstrate the importance of FPR1-mediated neutrophilic chemotaxis that drives the persistence of inflammation and affects mucosal homeostasis in the gastrointestinal and respiratory tracts. As a result, FPR1-targeted therapy could be a novel direction for inflammatory diseases. Further studies could be extended to other mucosal surfaces, such as the oral cavity, the eye, and the reproductive tract.
Of interest, the gene encoding FPR1 is highly polymorphic and numerous single nucleotide polymorphisms (SNPs) have been positively or negatively linked to various diseases.
Variation affecting intracellular domain of FPR1 has been associated with failure of anti-cancer immunotherapies, the age at which the disease manifests, as well as negative prognosis. The loss-of-function provoked by rs867228 is associated with poor responses to anthracycline-based adjuvant chemotherapy in breast cancer and colorectal cancer patients (81). Epidemiological studies showed that both homo- and heterozygosity for rs867228 in FPR1 accelerates age at diagnosis of luminal B breast cancer by 4.9 years (93). rs867228 homozygosity is associated with anticipated diagnosis in other cancer types, such as esophageal carcinoma, head and neck and colorectal cancer (94).
Recently, it has been proposed that the most abundant SNPs in FPR1 may have been selected to enhance human survival against infectious diseases. The most fitting example is represented by rs5030880 that affects the extracellular loop of FPR1 and is considered as a human resistance allele that protects neutrophils from Yersinia pestis type III secretion system (T3SS), which selectively destroys human immune cells (84).
In the last decade, the complex network of mediators and pathways triggering the resolution of inflammation has been proposed as innovative therapeutic approaches for chronic diseases. In this perspective, FPR2 has been recently defined as a prototype to kick-start “resolution pharmacology” (95).
FPR2 pro-resolving agonists, including lipid mediators such as LXA4, RvD1, and ANXA1, have been shown to exert therapeutic potential in diverse experimental models of chronic diseases. For example, the outcome of Influenza A, caused by a virus from the Orthomixoviridae family, improved with LXA4 treatment in mice, by decreasing cell recruitment and pro-inflammatory cytokines release (96). In diabetic mice, RvD1 promoted corneal epithelial wound healing and the restoration of mechanical sensation (97). ANXA1 exerted a therapeutic potential in diabetes and microvascular disease, by protecting peripheral organs against the injury and dysfunction caused by hyperglycemia and hyperlipidemia (98).
To develop new FPR2-agonist biased compounds more resistant to degradation than natural peptides, different techniques and strategies have been adopted by drug design and discovery.
Among synthetic LXA4 mimetics, 6C-dimethyl-imidazole (1R)-11 was found to be the most potent and efficient anti-inflammatory agent by inhibiting peritonitis-associated neutrophil infiltration in vivo (99). Compound 17b, a small-molecule ANXA1 mimetic, mediated aorta vasodilation, reduced vascular remodeling associated with myocardial infarction and diabetes, and exerted anti-inflammatory action in pulmonary arterial hypertension (100, 101). Compound MR-39, an FPR2 ureidopropanamide agonist, alleviated the inflammatory process in mouse model of Alzheimer’s Disease (102).
An interesting field of investigation with ongoing clinical trials focused on cardiovascular disorders. Compound 43, a dual agonist of FPR1/2, preserved cardiac structure and function after myocardial infarction by stimulating pro-resolution macrophages and improving left ventricle and infarct structure (103). Two small molecule FPR2 agonists, ACT-389949 (NCT02099071, NCT02099201) and BMS-986235 (NCT03335553), have been investigated in phase 1 clinical trials, though the results on therapeutic efficacy have not been still published (104). ACT-389949 was shown to be safe and well tolerated in healthy subjects. BM986235 was shown to contribute to the resolution of cardiac inflammation and improve both cardiac structure and function post myocardial infarction in animal models. Interestingly, ACT-389949 induces a potent desensitization of FPR2, placing limits on the chronic administration of this compound in cardiovascular disorders (105).
Additionally, a phase 1 clinical trial to analyze the safety and preliminary efficacy of an LXA4 analog, methyl ester-benzo-lipoxin A4 (BLXA4), in patients with gingival inflammation was performed. BLXA4 treatment reduced local inflammation and increased the levels of pro-resolution molecules (106).
FPR2 agonists could be used as therapeutics in inflammatory diseases, but further studies on biased FPR2/ALX agonism towards pro-resolution are needed.
The involvement of FPR3 in human disease is still poorly understood. However, diverse groups of research have focused on the role of FPR3 in the regulation of tumor microenvironment. In breast cancer, FPR3 was found to be a prognostic marker in disease progression and individual survival, being significantly associated with FPR3 expression (107). In addition, a tumor suppressor role was assigned to over-expressed FPR3 in gastric cancer cells, by interfering with glycolytic metabolism, proliferation and stemness of tumor cells (108).
A different pathogenetic and clinical relevance of FPR3 was found in glioma, in which FPR3 expression levels were related to the infiltration of immune cells, affecting the glioma immune microenvironment. In fact, the author found that FPR3 correlated positively with immune checkpoint gene PD-1, which is an important immune checkpoint target in glioma immunotherapy (109). Additionally, FPR3 was found to be involved in the control of immune cells and immune response, as demonstrated by a greater abundance of PD-L1 in the gliomas with high-expression levels of FPR3. More interestingly, CD163+FPR+ macrophage subsets were identified by single-cell RNA sequencing in glioma patients and a prognostic model based on the risk score of these cell subsets were constructed using machine learning (110). In head and neck squamous cell carcinoma, a single-cell transcriptomic analysis of Treg demonstrated a significant decrease in ANXA1/FPR3 pair (111).
Whether FPR3 plays a negative or positive role in cancer progression is still debated. However, the emerging evidence described above indicates that FPR3 expression profile has clinical relevance in terms of tumor immunity assessment. All together, these findings indicate that FPR3 can be employed as a promising therapeutic target and/or prognostic indicator both for its ability to modulate glycolysis and stemness of cancer cells and its involvement in tumor immune escape. If future studies confirm that FPR3 is an immune-related biomarker predicting a poor prognosis for cancer, it could be useful for the management of immunotherapy protocols.
In inflammation-associated disease, the role of FPR3 is still uncertain. In a mouse sepsis model, the administration of a selective FPR3 pepducin agonist, which specifically targets the intracellular loops of G protein-coupled receptors, inhibited lung injury, splenocyte apoptosis, and inflammatory cytokine production, significantly increasing the survival rate (112). In systemic sclerosis, the overexpression of FPR3 was confirmed both at protein and mRNA levels in skin fibroblasts from 10 patients. These data agreed with the in vivo observations by histological analysis (9). Additionally, FPR3 emerged in the top 10 hub genes involved in cutaneous lupus erythematosus by bioinformatics analyses of gene expression profile (113). It has recently been proposed that FPR3 expressed in human dendritic cells might mediate allergic TH2 immune responses. Peptides derived from the allergenic lipocalins but not for peptides derived from the nonallergenic homologues might be binding partners of FPR3, suggesting an involvement of FPR3 in allergic responses to lipocalin allergens (114).
During the past decade, considerable progress has been made in understanding of the FPR role in inflammation and its resolution. Although our understanding of the downstream signaling events following FPR remains incomplete, we tried to put the pieces of intricate puzzle together. Therefore, we dissected the pro-inflammatory and pro-tumorigenic signaling from anti-inflammatory and pro-resolving signaling.
Pro-inflammatory and pro-tumorigenic signals by FPRs contribute to the activation and recruitment of immune cells and the assembly of NADPH oxidase, thereby promoting cell chemotaxis, calcium mobilization, and ROS production. Biochemical pathways involved in these activities are reported in Figure 1. Well-consolidated pro-inflammatory ligands are specific for FPR1, FPR2, and FPR3 or can stimulate all members of FPR family, such as WKYMVm synthetic peptide that is considered as a pan-agonist for FPRs. It is conceivable that FPR3, characterized by significant basal levels of phosphorylation, continuously recycles across the membrane when stimulated by pro-inflammatory ligands, having been found in early and late endosomes. FPRs can be involved in macromolecular complexes on cell surface, by the transactivation of EFGR and uPAR, thus enhancing cell proliferation, matrix deposition, epithelial-mesenchymal transition, ROS production, and pro-inflammatory cytokine release. Upon binding of agonists, FPRs activate heterotrimeric G proteins, which dissociates into α and βγ subunits. Intracellular effectors of α subunits include the mitogen-activated protein kinases ERK1/2 and p38, whereas βγ signaling activates PLC. ERK 1/2 could be involved in Myc protein stability by phosphorylation at serine 62 (Ser62) residue, which is crucial for cell cycle progression and cell proliferation. Hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2) by PLC generates IP3, which induces the release of calcium from endoplasmic reticulum stores, and diacylglycerol (DAG), which activates PKC isoforms. The phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway is also activated, thus inducing an increase in cyclin D1 expression and cell cycle progression from G0/G1 to S phase. Together, these findings support the existence of an NF-κB-mediated pathway and the activation of proangiogenic and inflammatory transcription factors including STAT, VEGF, and CXCL8. These events are accompanied by the release of pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, and IFN-γ.
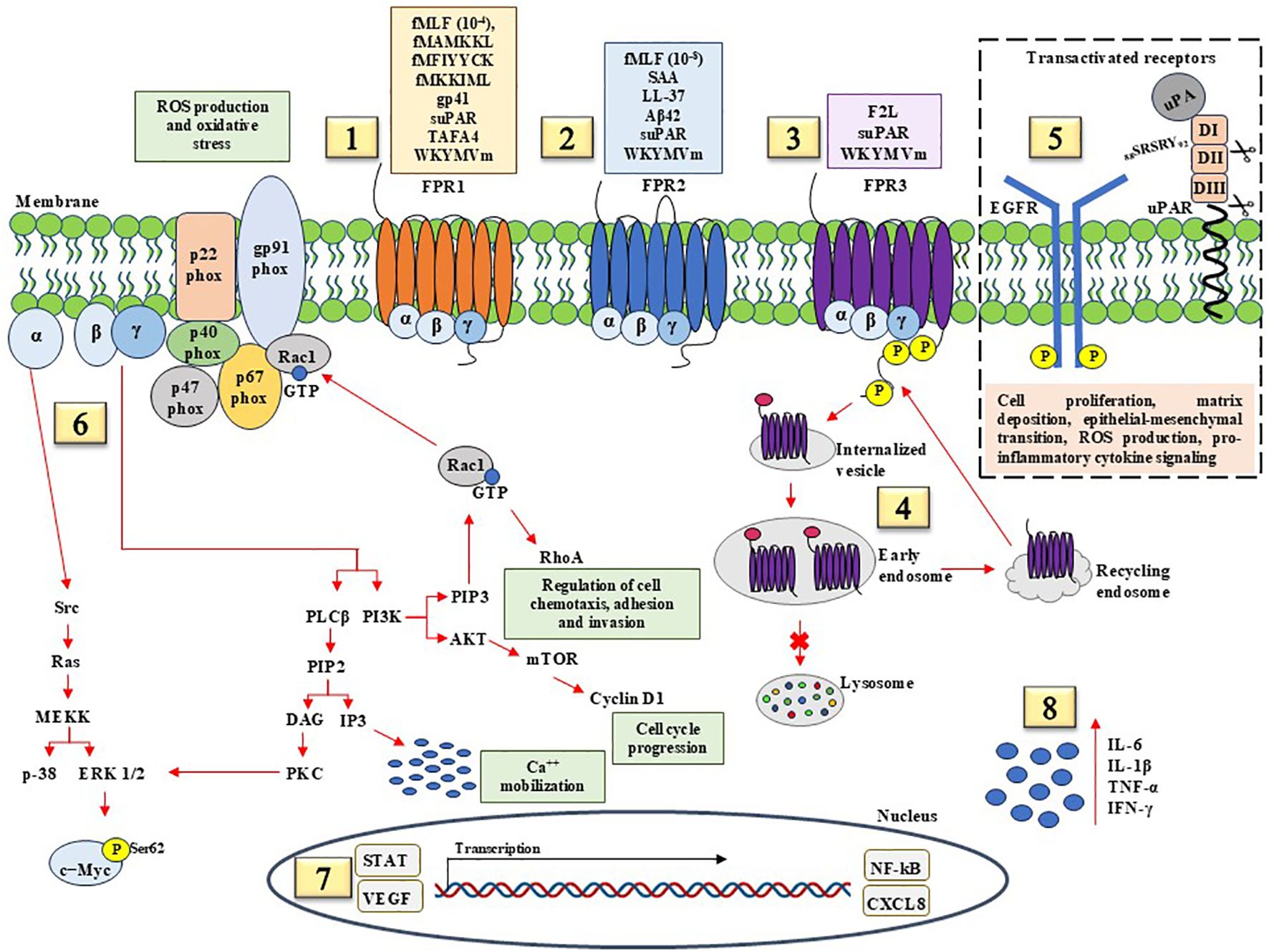
Figure 1. Pro-inflammatory and pro-tumorigenic signaling pathways mediated by FPRs. (1) FPR1 recognize microbe- derived formylated peptides, including E. Coli-derived fMLF, Salmonella-derived fMAMKKL, Staphylococcus-derived fMIYYCK, Listeria-derived fMKKIML; HIV-derived non formylated peptides gp41; soluble Urokinase Receptor (suPAR); TAFA chemokine like family member 4 (TAFA4); WKYMVm (Trp-Lys-Tyr-Met-Val-D-Met) synthetic hexapeptide. (2) FPR2 agonists with pro-inflammatory effects include fMLF, Serum Amyloid A (SAA), antimicrobial peptide LL-37, suPAR, and WKYMVm synthetic peptide. (3) FPR3, with significantly basal levels of phosphorylation, binds F2L, a peptide derived from heme-binding protein, suPAR, and WKYMVm synthetic peptide. (4) FPR3 is found mainly distributed in early and late endosome during acute inflammation, suggesting FPR3 continuous recycling following endocytosis in response to pro-inflammatory stimuli. (5) Pro-tumorigenic effects of FPRs are mediated in part by transactivation of epidermal growth factor (EGF) receptor (EGFR) and membrane-anchored uPAR. FPRs/EGFR and FPRs/uPAR crosstalk plays a central role in cell proliferation, matrix deposition, epithelial-mesenchymal transition, ROS production, and pro-inflammatory cytokine signaling. (6) The activation of FPRs from binding pro-inflammatory ligands results in the dissociation of the Gα from the Gβγ subunit. α subunit activates the Ras superfamily, which contribute to activation of the MAPK pathways, p38, and ERK1/2. FPR-mediated ERK activation results in c-Myc Ser62 phosphorylation that has been observed in numerous cancer cells. βγ subunits activate PLCβ, resulting in calcium release from intracellular stores, PKC and PI3K which further contribute to activation of Rac1 GTPase. Rac1 induces the assembly and activation of NADPH oxidase to produce reactive oxygen species (ROS), Rho GTPases to facilitate cancer cell metastasis by regulating actin and proteins associated with cell migration and invasion, and Akt/mTOR axis to enhance cyclin D1 and cell cycle progression. (7) The effects of pro-inflammatory and pro-tumorigenic ligands culminate in the activation of transcription factors including STAT, VEGF, NF-κB, and CXCL8. (8) Pro-inflammatory agonist peptides promote the production of pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, and IFN-γ.
On the other hand, anti-inflammatory and pro-resolving signals by FPRs contribute to activating and coordinating processes aimed at restoration of tissue integrity and function, as shown in Figure 2. FPR ligands with pro-resolving effects may cause receptor homodimerization or heterodimerization with other FPRs. FPR3 is constitutively internalized and can bind anti-inflammatory ligands. Pro-resolution and anti-inflammatory pathways start with β-arrestin 2 recruitment, which induces receptor desensitization and internalization and G-protein independent signaling. How this determinate the shift from pro-inflammatory to anti-inflammatory functions of FPRs and how FPR signaling pathways bifurcate into canonical and non-canonical transduction are still under investigation. Probably, biased allosteric modulators induce conformational changes in FPRs and generate pro- or anti-inflammatory signals, as demonstrated for FPR2. Specifically, FPR2 has two allosteric binding sites, each for one specific ligand, with pro- or anti-inflammatory activity. The recruitment of β-arrestin 2 to cytoplasmic portion of FPR2 is induced by anti-inflammatory ligands, whereas pro-inflammatory ligands elicit the release of calcium from endoplasmic reticulum stores and ERK phosphorylation (26). β-arrestin 2 promotes bcl-xL expression, leading to cell survival, cAMP-mediated signaling, and p38 MAPK phosphorylation. The internalization of FPR2 is crucial for the resolution of inflammation as the internalized receptor inhibits NF-κB activity. The activation of anti-inflammatory and pro-resolving pathways mediated by FPRs induces the transcription of cAMP response element-binding protein (CREB) that promotes anti-inflammatory responses through the inhibition of NF-κB activity and the induction of IL-10, peroxisome proliferator-activated receptor (PPARγ) that represses the NF-κB pathway, NRF2 that protects against oxidative damage. FPR pro-resolving agonists upregulate anti-inflammatory factors, including IL-10, and decrease pro-inflammatory cytokine production.
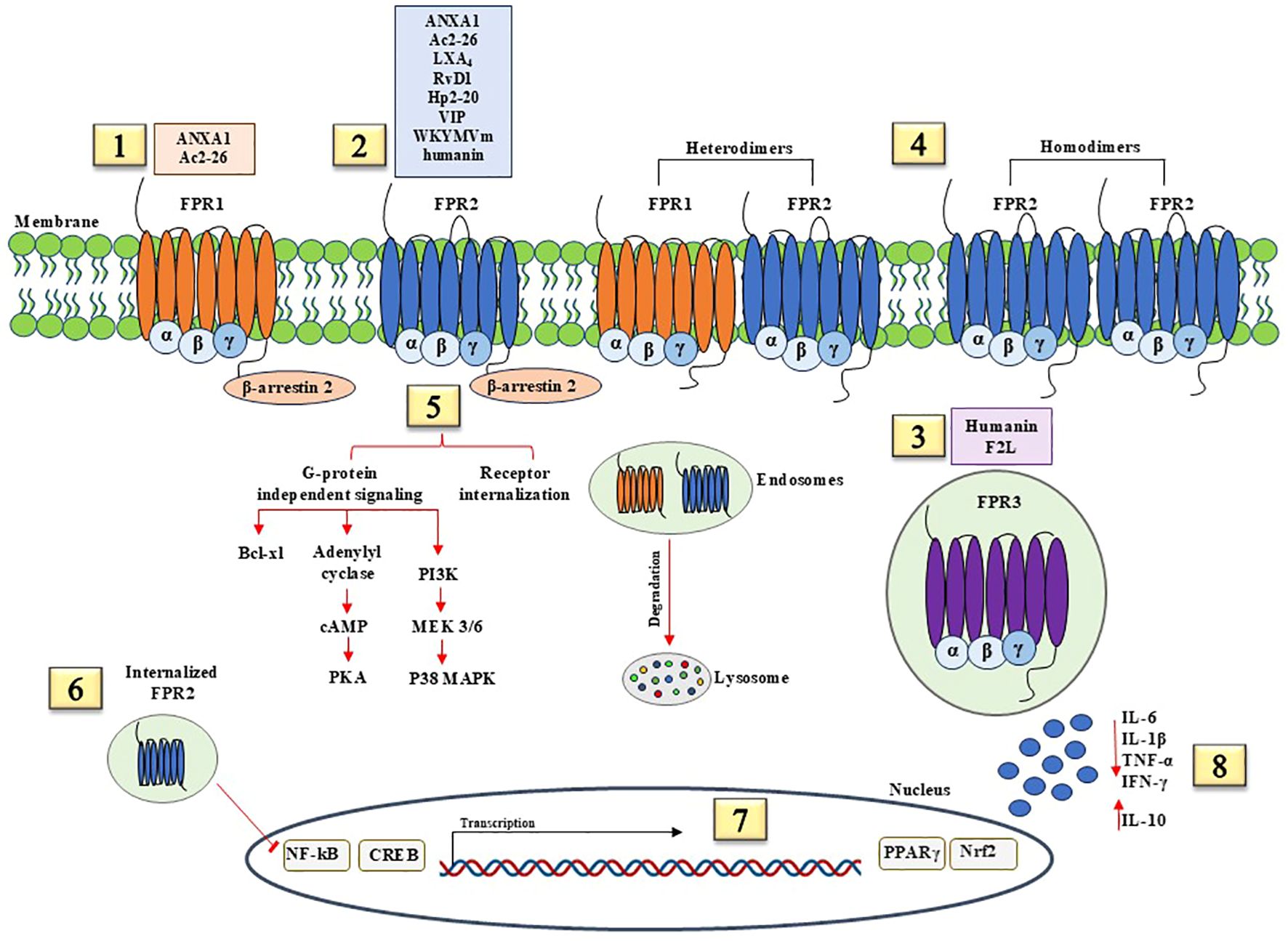
Figure 2. Anti-inflammatory and pro-resolving signaling pathways mediated by FPRs. (1) FPR1 agonists with anti-inflammatory effects include Annexin A1 (ANXA1) and N-terminal peptide of ANXA1, Ac2-26. (2) FPR1 agonists with anti-inflammatory effects include ANXA1, Ac2-26, lipid mediators Lipoxin A4 (LXA4) and Resolvin D1 (RvD1), Hp2-20 peptide of Helicobacter pylori, Vasoactive intestinal peptide (VIP), WKYMVm synthetic peptide, and humanin, a recently identified neuroprotective factor. (3) FPR3 is constitutively internalized and binds humanin and F2L. (4) FPR1/FPR2 heterodimers and FPR2 homodimers elicit pro-resolving and pro-inflammatory effects. (5) Pro-resolution and anti-inflammatory pathways start with β-arrestin 2 recruitment, which induces receptor desensitization and internalization and G-protein independent signaling. β-arrestin 2 promote bcl-xL expression, leading to cell survival, cAMP-mediated signaling, and p38 MAPK phosphorylation. (6) FPR2 internalization is crucial for the resolution of inflammation as the internalized receptor inhibits NF-κB activity. (7) The activation of anti-inflammatory and pro-resolving pathways mediated by FPRs induces the transcription of cAMP response element-binding protein (CREB), peroxisome proliferator-activated receptor (PPARγ), and nuclear factor erythroid 2–related factor 2 (NRF2). (8) FPR pro-resolving agonists upregulate anti-inflammatory factors, including IL-10, and block the release of pro-inflammatory cytokines, such as IL-6, IL-1β, TNF-α, and IFN-γ.
Finally, uncontrolled and unresolved inflammation may become chronic and lead to a pathological disease state; in this context, the modulation of FPR signaling could be instrumental in resolving pathologic inflammation. Particularly attractive are the immunomodulatory role of FPR2 by regulating proinflammatory and anti-inflammatory activities and the association of FPR3 with tumor immunity, indicating its availability as a prognostic biomarker in cancer. A better understanding of FPR signaling is necessary to aid the drug development process, in order to stimulate the desirable biological properties.
FN: Conceptualization, Data curation, Formal Analysis, Investigation, Software, Validation, Writing – original draft, Writing – review & editing, Supervision, Visualization. NM: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by Campania Bioscience PON03PE_00060_8 and by PRIN 2022 PNRR, Settore ERC: LS4 - Codice progetto MUR: P2022HB33K – CUP: E53D23019850001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wang R, Lan C, Benlagha K, Camara NOS, Miller H, Kubo M, et al. The interaction of innate immune and adaptive immune system. MedComm (2020). (2024) 5:e714. doi: 10.1002/mco2.714
2. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
3. Ma M, Jiang W, Zhou R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
4. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol. (2014) 258:5–16. doi: 10.1016/j.expneurol.2014.01.001
5. He HQ, Ye RD. The formyl peptide receptors: diversity of ligands and mechanism for recognition. Molecules. (2017) 22:455. doi: 10.3390/molecules22030455
6. Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci U S A. (1975) 72:1059–62. doi: 10.1073/pnas.72.3.1059
7. Showell HJ, Freer RJ, Zigmond SH, Schiffmann E, Aswanikumar S, Corcoran B, et al. The structure-activity relations of synthetic peptides as chemotactic factors and inducers of lysosomal secretion for neutrophils. J Exp Med. (1976) 143:1154–69. doi: 10.1084/jem.143.5.1154
8. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. (2009) 61:119–61. doi: 10.1124/pr.109.001578
9. Rossi FW, Napolitano F, Pesapane A, Mascolo M, Staibano S, Matucci-Cerinic M, et al. Upregulation of the N-formyl Peptide receptors in scleroderma fibroblasts fosters the switch to myofibroblasts. J Immunol. (2015) 194:5161–73. doi: 10.4049/jimmunol.1402819
10. Napolitano F, Rossi FW, Pesapane A, Varricchio S, Ilardi G, Mascolo M, et al. N-formyl peptide receptors induce radical oxygen production in fibroblasts derived from systemic sclerosis by interacting with a cleaved form of urokinase receptor. Front Immunol. (2018) 9:574. doi: 10.3389/fimmu.2018.00574
11. Napolitano F, Rossi FW, de Paulis A, Lavecchia A, Montuori N. The crosstalk between N-formyl peptide receptors and uPAR in systemic sclerosis: molecular mechanisms, pathogenetic role and therapeutic opportunities. Int J Mol Sci. (2024) 25:3156. doi: 10.3390/ijms25063156
12. Cattaneo F, Parisi M, Fioretti T, Sarnataro D, Esposito G, Ammendola R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch Biochem Biophys. (2016) 603:10–9. doi: 10.1016/j.abb.2016.05.006
13. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. (2015) 185:1172–84. doi: 10.1016/j.ajpath.2015.01.020
14. Gierschik P, Sidiropoulos D, Jakobs KH. Two distinct Gi-proteins mediate formyl peptide receptor signal transduction in human leukemia (HL-60) cells. J Biol Chem. (1989) 264:21470–3. doi: 10.1016/S0021-9258(20)88206-4
15. Weiß E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. (2018) 39:815–29. doi: 10.1016/j.it.2018.08.005
16. Kam AY, Liu AM, Wong YH. Formyl peptide-receptor like-1 requires lipid raft and extracellular signal-regulated protein kinase to activate inhibitor-kappa B kinase in human U87 astrocytoma cells. J Neurochem. (2007) 103:1553–66. doi: 10.1111/j.1471-4159.2007.04876.x
17. Rabiet MJ, Macari L, Dahlgren C, Boulay F. N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J Biol Chem. (2011) 286:26718–31. doi: 10.1074/jbc.M111.244590
18. Forsman H, Önnheim K, Andréasson E, Christenson K, Karlsson A, Bylund J, et al. Reactivation of desensitized formyl peptide receptors by platelet activating factor: a novel receptor cross talk mechanism regulating neutrophil superoxide anion production. PloS One. (2013) 8:e60169. doi: 10.1371/journal.pone.0060169
19. Maestes DC, Potter RM, Prossnitz ER. Differential phosphorylation paradigms dictate desensitization and internalization of the N-formyl peptide receptor. J Biol Chem. (1999) 274:29791–5. doi: 10.1074/jbc.274.42.29791
20. Vines CM, Revankar CM, Maestas DC, LaRusch LL, Cimino DF, Kohout TA, et al. N-formyl peptide receptors internalize but do not recycle in the absence of arrestins. J Biol Chem. (2003) 278:41581–4. doi: 10.1074/jbc.C300291200
21. Montuori N, Bifulco K, Carriero MV, La Penna C, Visconte V, Alfano D, et al. The cross-talk between the urokinase receptor and fMLP receptors regulates the activity of the CXCR4 chemokine receptor. Cell Mol Life Sci. (2011) 68:2453–67. doi: 10.1007/s00018-010-0564-7
22. Blackwood RA, Hartiala KT, Kwoh EE, Transue AT, Brower RC. Unidirectional heterologous receptor desensitization between both the fMLP and C5a receptor and the IL-8 receptor. J Leukoc Biol. (1996) 60:88–93. doi: 10.1002/jlb.60.1.88
23. Bednar F, Song C, Bardi G, Cornwell W, Rogers TJ. Cross-desensitization of CCR1, but not CCR2, following activation of the formyl peptide receptor FPR1. J Immunol. (2014) 192:5305–13. doi: 10.4049/jimmunol.1302983
24. Shen W, Li B, Wetzel MA, Rogers TJ, Henderson EE, Su SB, et al. Down-regulation of the chemokine receptor CCR5 by activation of chemotactic formyl peptide receptor in human monocytes. Blood. (2000) 96:2887–94. doi: 10.1182/blood.V96.8.2887
25. Gröper J, König GM, Kostenis E, Gerke V, Raabe CA, Rescher U. Exploring biased agonism at FPR1 as a means to encode danger sensing. Cells. (2020) 9:1054. doi: 10.3390/cells9041054
26. Zhang S, Gong H, Ge Y, Ye RD. Biased allosteric modulation of formyl peptide receptor 2 leads to distinct receptor conformational states for pro- and anti-inflammatory signaling. Pharmacol Res. (2020) 161:105117. doi: 10.1016/j.phrs.2020.105117
27. Cooray SN, Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJ, et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A. (2013) 110:18232–7. doi: 10.1073/pnas.1308253110
28. Tylek K, Trojan E, Regulska M, Lacivita E, Leopoldo M, Basta-Kaim A. Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: a link to brain pathology. Pharmacol Rep. (2021) 73:1004–19. doi: 10.1007/s43440-021-00271-x
29. Busch L, Vieten S, Brödel S, Endres K, Bufe B. Emerging contributions of formyl peptide receptors to neurodegenerative diseases. Biol Chem. (2021) 403:27–41. doi: 10.1515/hsz-2021-0258
30. Rossi FW, Prevete N, Rivellese F, Napolitano F, Montuori N, Postiglione L, et al. The Urokinase/Urokinase Receptor System in Mast Cells: Effects of its Functional Interaction with fMLF Receptors. Transl Med UniSa. (2016) 15:34–41.
31. Resnati M, Pallavicini I, Wang JM, Oppenheim J, Serhan CN, Romano M, et al. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci U S A. (2002) 99:1359–64. doi: 10.1073/pnas.022652999
32. de Paulis A, Montuori N, Prevete N, Fiorentino I, Rossi FW, Visconte V, et al. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J Immunol. (2004) 173:5739–48. doi: 10.4049/jimmunol.173.9.5739
33. Gorrasi A, Petrone AM, Li Santi A, Alfieri M, Montuori N, Ragno P. New pieces in the puzzle of uPAR role in cell migration mechanisms. Cells. (2020) 9:2531. doi: 10.3390/cells9122531
34. Muto Y, Guindon S, Umemura T, Kőhidai L, Ueda H. Adaptive evolution of formyl peptide receptors in mammals. J Mol Evol. (2015) 80:130–41. doi: 10.1007/s00239-015-9666-z
35. Sahagun-Ruiz A, Colla JS, Juhn J, Gao JL, Murphy PM, McDermott DH. Contrasting evolution of the human leukocyte N-formylpeptide receptor subtypes FPR and FPRL1R. Genes Immun. (2001) 2:335–42. doi: 10.1038/sj.gene.6363787
36. Gripentrog JM, Mills JS, Saari GJ, Miettinen HM. Variable responses of formyl peptide receptor haplotypes toward bacterial peptides. Immunogenetics. (2008) 60:83–93. doi: 10.1007/s00251-008-0277-3
37. Boillat M, Carleton A, Rodriguez I. From immune to olfactory expression: neofunctionalization of formyl peptide receptors. Cell Tissue Res. (2021) 383:387–93. doi: 10.1007/s00441-020-03393-5
38. Zhuang Y, Wang L, Guo J, Sun D, Wang Y, Liu W, et al. Molecular recognition of formylpeptides and diverse agonists by the formylpeptide receptors FPR1 and FPR2. Nat Commun. (2022) 13:1054. doi: 10.1038/s41467-022-28586-0
39. Zhangsun Z, Dong Y, Tang J, Jin Z, Lei W, Wang C, et al. FPR1: A critical gatekeeper of the heart and brain. Pharmacol Res. (2024) 202:107125. doi: 10.1016/j.phrs.2024.107125
40. Zhu S, Hu X, Bennett S, Mai Y, Xu J. Molecular structure, expression and role of TAFA4 and its receptor FPR1 in the spinal cord. Front Cell Dev Biol. (2022) 10:911414. doi: 10.3389/fcell.2022.911414
41. Minopoli M, Polo A, Ragone C, Ingangi V, Ciliberto G, Pessi A, et al. Structure-function relationship of an Urokinase Receptor-derived peptide which inhibits the Formyl Peptide Receptor type 1 activity. Sci Rep. (2019) 9:12169. doi: 10.1038/s41598-019-47900-3
42. Vacchelli E, Le Naour J, Kroemer G. The ambiguous role of FPR1 in immunity and inflammation. Oncoimmunology. (2020) 9:1760061. doi: 10.1080/2162402X.2020.1760061
43. Bokoch GM, Gilman AG. Inhibition of receptor-mediated release of arachidonic acid by pertussis toxin. Cell. (1984) 39:301–8. doi: 10.1016/0092-8674(84)90008-4
44. Wang J, Ye RD. Agonist concentration-dependent changes in FPR1 conformation lead to biased signaling for selective activation of phagocyte functions. Proc Natl Acad Sci U S A. (2022) 119:e2201249119. doi: 10.1073/pnas.2201249119
45. Bufe B, Schumann T, Kappl R, Bogeski I, Kummerow C, Podgórska M, et al. Recognition of bacterial signal peptides by mammalian formyl peptide receptors: a new mechanism for sensing pathogens. J Biol Chem. (2015) 290:7369–87. doi: 10.1074/jbc.M114.626747
46. Cuomo P, Papaianni M, Capparelli R, Medaglia C. The role of formyl peptide receptors in permanent and low-grade inflammation: helicobacter pylori infection as a model. Int J Mol Sci. (2021) 22:3706. doi: 10.3390/ijms22073706
47. Wood MP, Cole AL, Eade CR, Chen LM, Chai KX, Cole AM. The HIV-1 gp41 ectodomain is cleaved by matriptase to produce a chemotactic peptide that acts through FPR2. Immunology. (2014) 142:474–83. doi: 10.1111/imm.12278
48. Kelly L, McGrath S, Rodgers L, McCall K, Tulunay Virlan A, Dempsey F, et al. Annexin-A1: The culprit or the solution? Immunology. (2022) 166:2–16. doi: 10.1111/imm.13455
49. Wang W, Li T, Wang X, Yuan W, Cheng Y, Zhang H, et al. FAM19A4 is a novel cytokine ligand of formyl peptide receptor 1 (FPR1) and is able to promote the migration and phagocytosis of macrophages. Cell Mol Immunol. (2015) 12:615–24. doi: 10.1038/cmi.2014.61
50. Lee C, Han J, Jung Y. Formyl peptide receptor 2 is an emerging modulator of inflammation in the liver. Exp Mol Med. (2023) 55:325–32. doi: 10.1038/s12276-023-00941-1
51. Chen T, Xiong M, Zong X, Ge Y, Zhang H, Wang M, et al. Structural basis of ligand binding modes at the human formyl peptide receptor 2. Nat Commun. (2020) 11:1208. doi: 10.1038/s41467-020-15009-1
52. Zhuang Y, Liu H, Edward Zhou X, Kumar Verma R, de Waal PW, Jang W, et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat Commun. (2020) 11:885. doi: 10.1038/s41467-020-14728-9
53. de Paulis A, Prevete N, Rossi FW, Rivellese F, Salerno F, Delfino G, et al. Helicobacter pylori Hp(2-20) promotes migration and proliferation of gastric epithelial cells by interacting with formyl peptide receptors in vitro and accelerates gastric mucosal healing in vivo. J Immunol. (2009) 183:3761–9. doi: 10.4049/jimmunol.0900863
54. Pan WH, Hu X, Chen B, Xu QC, Mei HX. The effect and mechanism of lipoxin A4 on neutrophil function in LPS-induced lung injury. Inflammation. (2022) 45:1950–67. doi: 10.1007/s10753-022-01666-5
55. Liu GJ, Tao T, Wang H, Zhou Y, Gao X, Gao YY, et al. Functions of resolvin D1-ALX/FPR2 receptor interaction in the hemoglobin-induced microglial inflammatory response and neuronal injury. J Neuroinflammation. (2020) 17:239. doi: 10.1186/s12974-020-01918-x
56. Zhu Y, Lin X, Zong X, Han S, Wang M, Su Y, et al. Structural basis of FPR2 in recognition of Aβ42 and neuroprotection by humanin. Nat Commun. (2022) 13:1775. doi: 10.1038/s41467-022-29361-x
57. Hinrichs BH, Matthews JD, Siuda D, O'Leary MN, Wolfarth AA, Saeedi BJ, et al. Serum amyloid A1 is an epithelial prorestitutive factor. Am J Pathol. (2018) 188:937–49. doi: 10.1016/j.ajpath.2017.12.013
58. Carion TW, Kracht D, Strand E, David E, McWhirter C, Ebrahim AS, et al. VIP modulates the ALX/FPR2 receptor axis toward inflammation resolution in a mouse model of bacterial keratitis. Prostaglandins Other Lipid Mediat. (2019) 140:18–25. doi: 10.1016/j.prostaglandins.2018.12.001
59. Yang WS, Wang JL, Wu W, Wang GF, Yan J, Liu Q, et al. Formyl peptide receptor 2 as a potential therapeutic target for inflammatory bowel disease. Acta Pharmacol Sin. (2023) 44:19–31. doi: 10.1038/s41401-022-00944-0
60. Gabl M, Holdfeldt A, Sundqvist M, Lomei J, Dahlgren C, Forsman H. FPR2 signaling without β-arrestin recruitment alters the functional repertoire of neutrophils. Biochem Pharmacol. (2017) 145:114–22. doi: 10.1016/j.bcp.2017.08.018
61. Lee C, Kim J, Han J, Oh D, Kim M, Jeong H, et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat Commun. (2022) 13:578. doi: 10.1038/s41467-022-28138-6
62. He F, Tay AHM, Calandigary A, Malki E, Suzuki S, Liu T, et al. FPR2 shapes an immune-excluded pancreatic tumor microenvironment and drives T-cell exhaustion in a sex-dependent manner. Cancer Res. (2023) 83:1628–45. doi: 10.1158/0008-5472
63. Pierdomenico AM, Recchiuti A, Simiele F, Codagnone M, Mari VC, Davì G, et al. MicroRNA-181b regulates ALX/FPR2 receptor expression and proresolution signaling in human macrophages. J Biol Chem. (2015) 290:3592–600. doi: 10.1074/jbc.M114.592352
64. Simiele F, Recchiuti A, Mattoscio D, De Luca A, Cianci E, Franchi S, et al. Transcriptional regulation of the human FPR2/ALX gene: evidence of a heritable genetic variant that impairs promoter activity. FASEB J. (2012) 26:1323–33. doi: 10.1096/fj.11-198069
65. Zhang H, Lu Y, Sun G, Teng F, Luo N, Jiang J, et al. The common promoter polymorphism rs11666254 downregulates FPR2/ALX expression and increases risk of sepsis in patients with severe trauma. Crit Care. (2017) 21:171. doi: 10.1186/s13054-017-1757-3
66. Devosse T, Guillabert A, D'Haene N, Berton A, De Nadai P, Noel S, et al. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. (2009) 182:4974–84. doi: 10.4049/jimmunol.0803128
67. Wang X, Cui Y, Wang J. T4-lysozyme fusion for the production of human formyl peptide receptors for structural determination. Appl Biochem Biotechnol. (2014) 172:2571–81. doi: 10.1007/s12010-013-0704-2
68. Ernst S, Lange C, Wilbers A, Goebeler V, Gerke V, Rescher U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J Immunol. (2004) 172:7669–76. doi: 10.4049/jimmunol.172.12.7669
69. Devosse T, Dutoit R, Migeotte I, De Nadai P, Imbault V, Communi D, et al. Processing of HEBP1 by cathepsin D gives rise to F2L, the agonist of formyl peptide receptor 3. J Immunol. (2011) 187:1475–85. doi: 10.4049/jimmunol.1003545
70. Harada M, Habata. Y, Hosoya M, Nishi K, Fujii R, Kobayashi M, et al. N-Formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem Biophys Res Commun. (2004) 324:255–61. doi: 10.1016/j.bbrc.2004.09.046
71. Lee HY, Jeong YS, Lee M, Kweon HS, Huh YH, Park JS, et al. Intracellular formyl peptide receptor regulates naïve CD4 T cell migration. Biochem Biophys Res Commun. (2018) 497:226–32. doi: 10.1016/j.bbrc.2018.02.060
72. Nawaz MI, Rezzola S, Tobia C, Coltrini D, Belleri M, Mitola S, et al. D-Peptide analogues of Boc-Phe-Leu-Phe-Leu-Phe-COOH induce neovascularization via endothelial N-formyl peptide receptor 3. Angiogenesis. (2020) 23:357–69. doi: 10.1007/s10456-020-09714-0
73. Jeong YS, Bae YS. Formyl peptide receptors in the mucosal immune system. Exp Mol Med. (2020) 52:1694–704. doi: 10.1038/s12276-020-00518-2
74. Zhou Y, Bian X, Le Y, Gong W, Hu J, Zhang X, et al. Formylpeptide receptor FPR and the rapid growth of Malignant human gliomas. J Natl Cancer Inst. (2005) 97:823–35. doi: 10.1093/jnci/dji142
75. Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol. (2011) 179:1504–12. doi: 10.1016/j.ajpath.2011.05.059
76. Zheng Y, Jiang H, Yang N, Shen S, Huang D, Jia L, et al. Glioma-derived ANXA1 suppresses the immune response to TLR3 ligands by promoting an anti-inflammatory tumor microenvironment. Cell Mol Immunol. (2024) 21:47–59. doi: 10.1038/s41423-023-01110-0
77. Cao G, Zhang Z. FPR1 mediates the tumorigenicity of human cervical cancer cells. Cancer Manag Res. (2018) 10:5855–65. doi: 10.2147/CMAR.S182795
78. Jiang X, Lei T, Zhang M. Expression and functions of formyl peptide receptor 1 in drug-resistant bladder cancer. Technol Cancer Res Treat. (2018) 17:1533034618769413. doi: 10.1177/1533034618769413
79. Prevete N, Liotti F, Illiano A, Amoresano A, Pucci P, de Paulis A, et al. Formyl peptide receptor 1 suppresses gastric cancer angiogenesis and growth by exploiting inflammation resolution pathways. Oncoimmunology. (2017) 6:e1293213. doi: 10.1080/2162402X.2017.1293213
80. Liotti F, Marotta M, Sorriento D, Pagliuca C, Caturano V, Mantova G, et al. Probiotic Lactobacillus rhamnosus GG (LGG) restrains the angiogenic potential of colorectal carcinoma cells by activating a proresolving program via formyl peptide receptor 1. Mol Oncol. (2022) 16:2959–80. doi: 10.1002/1878-0261.13280
81. Vacchelli E, Enot DP, Pietrocola F, Zitvogel L, Kroemer G. Impact of pattern recognition receptors on the prognosis of breast cancer patients undergoing adjuvant chemotherapy. Cancer Res. (2016) 76:3122–6. doi: 10.1158/0008-5472.CAN-16-0294
82. Zhang M, Gao JL, Chen K, Yoshimura T, Liang W, Gong W, et al. A Critical Role of Formyl Peptide Receptors in Host Defense against Escherichia coli. J Immunol. (2020) 204:2464–73. doi: 10.4049/jimmunol.1900430
83. Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Wang A, et al. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. (2012) 2:786. doi: 10.1038/srep00786
84. Osei-Owusu P, Charlton TM, Kim HK, Missiakas D, Schneewind O. FPR1 is the plague receptor on host immune cells. Nature. (2019) 574:57–62. doi: 10.1038/s41586-019-1570-z
85. Wang Z, Wang Y, Yan Q, Cai C, Feng Y, Huang Q, et al. FPR1 signaling aberrantly regulates S100A8/A9 production by CD14+FCN1hi macrophages and aggravates pulmonary pathology in severe COVID-19. Commun Biol. (2024) 7:1321. doi: 10.1038/s42003-024-07025-4
86. Zhou QL, Teng F, Zhang YS, Sun Q, Cao YX, Meng GW. FPR1 gene silencing suppresses cardiomyocyte apoptosis and ventricular remodeling in rats with ischemia/reperfusion injury through the inhibition of MAPK signaling pathway. Exp Cell Res. (2018) 370:506–18. doi: 10.1016/j.yexcr.2018.07.016
87. Zheng F, Li W, Cheng C, Xiong D, Wei M, Wang T, et al. Formyl peptide receptor 1 inhibits reparative angiogenesis and aggravates neuroretinal dysfunction in ischemic retinopathy. Curr Eye Res. (2024) 49:1193–200. doi: 10.1080/02713683.2024.2363473
88. Park B, Lee M, Kim SD, Jeong YS, Kim JC, Yang S, et al. Activation of formyl peptide receptor 1 elicits therapeutic effects against collagen-induced arthritis. J Cell Mol Med. (2021) 25:8936–46. doi: 10.1111/jcmm.16854
89. Lammers KM, Chieppa M, Liu L, Liu S, Omatsu T, Janka-Junttila M, et al. Gliadin induces neutrophil migration via engagement of the formyl peptide receptor, FPR1. PloS One. (2015) 10:e0138338. doi: 10.1371/journal.pone.0138338
90. Gao L, Zeng N, Yuan Z, Wang T, Chen L, Yang D, et al. Knockout of formyl peptide receptor-1 attenuates cigarette smoke-induced airway inflammation in mice. Front Pharmacol. (2021) 12:632225. doi: 10.3389/fphar.2021.632225
91. McAllister MJ, Hall R, Whelan RJ, Fischer LJ, Chuah CS, Cartlidge PD, et al. Formylated peptide receptor-1-mediated gut inflammation as a therapeutic target in inflammatory bowel disease. Crohns Colitis 360. (2024) 6:otae003. doi: 10.1093/crocol/otae003
92. De Cunto G, Bartalesi B, Cavarra E, Balzano E, Lungarella G, Lucattelli M. Ongoing lung inflammation and disease progression in mice after smoking cessation: beneficial effects of formyl-peptide receptor blockade. Am J Pathol. (2018) 188:2195–206. doi: 10.1016/j.ajpath.2018.06.010
93. Carbonnier V, Le Naour J, Bachelot T, Vacchelli E, André F, Delaloge S, et al. Rs867228 in FPR1 accelerates the manifestation of luminal B breast cancer. Oncoimmunology. (2023) 12:2189823. doi: 10.1080/2162402X.2023.2189823
94. Sztupinszki Z, Le Naour J, Vacchelli E, Laurent-Puig P, Delaloge S, Szallasi Z, et al. A major genetic accelerator of cancer diagnosis: rs867228 in FPR1. Oncoimmunology. (2021) 10:1859064. doi: 10.1080/2162402X.2020.1859064
95. Perretti M, Godson C. Formyl peptide receptor type 2 agonists to kick-start resolution pharmacology. Br J Pharmacol. (2020) 177:4595–600. doi: 10.1111/bph.15212
96. Rago F, Melo EM, Miller LM, Duray AM, Batista Felix F, Vago JP, et al. Treatment with lipoxin A4 improves influenza A infection outcome, induces macrophage reprogramming, anti-inflammatory and pro-resolutive responses. Inflammation Res. (2024) 73:1903–18. doi: 10.1007/s00011-024-01939-9
97. Zhang Z, Hu X, Qi X, Di G, Zhang. Y, Wang. Q, et al. Resolvin D1 promotes corneal epithelial wound healing and restoration of mechanical sensation in diabetic mice. Mol Vis. (2018) 24:274–85.
98. Purvis GSD, Solito E, Thiemermann C. Annexin-A1: therapeutic potential in microvascular disease. Front Immunol. (2019) 10:938. doi: 10.3389/fimmu.2019.00938
99. de Gaetano M, Butler E, Gahan K, Zanetti A, Marai M, Chen J, et al. Asymmetric synthesis and biological evaluation of imidazole- and oxazole-containing synthetic lipoxin A4 mimetics (sLXms). Eur J Med Chem. (2019) 162:80–108. doi: 10.1016/j.ejmech.2018.10.049
100. Marshall SA, Qin CX, Jelinic M, O'Sullivan K, Deo M, Walsh J, et al. The novel small-molecule annexin-A1 mimetic, compound 17b, elicits vasoprotective actions in streptozotocin-induced diabetic mice. Int J Mol Sci. (2020) 21:1384. doi: 10.3390/ijms21041384
101. Studley WR, Lamanna E, Martin KA, Nold-Petry CA, Royce SG, Woodman OL, et al. The small-molecule formyl peptide receptor biased agonist, compound 17b, is a vasodilator and anti-inflammatory in mouse precision-cut lung slices. Br J Pharmacol. (2024) 181:2287–301. doi: 10.1111/bph.16231
102. Trojan E, Tylek K, Schröder N, Kahl I, Brandenburg LO, Mastromarino M, et al. The N-formyl peptide receptor 2 (FPR2) agonist MR-39 improves ex vivo and in vivo amyloid beta (1-42)-induced neuroinflammation in mouse models of alzheimer's disease. Mol Neurobiol. (2021) 58:6203–21. doi: 10.1007/s12035-021-02543-2
103. García RA, Ito BR, Lupisella JA, Carson NA, Hsu MY, Fernando G, et al. Preservation of post-infarction cardiac structure and function via long-term oral formyl peptide receptor agonist treatment. JACC Basic Transl Sci. (2019) 4:905–20. doi: 10.1016/j.jacbts.2019.07.005
104. Stalder AK, Lott D, Strasser DS, Cruz HG, Krause A, Groenen PM, et al. Biomarker-guided clinical development of the first-in-class anti-inflammatory FPR2/ALX agonist ACT-389949. Br J Clin Pharmacol. (2017) 83:476–86. doi: 10.1111/bcp.13149
105. Lupisella J, St-Onge S, Carrier M, Cook EM, Wang T, Sum C, et al. Molecular mechanisms of desensitization underlying the differential effects of formyl peptide receptor 2 agonists on cardiac structure-function post myocardial infarction. ACS Pharmacol Transl Sci. (2022) 5:892–906. doi: 10.1021/acsptsci.2c00042
106. Hasturk H, Schulte F, Martins M, Sherzai H, Floros C, Cugini M, et al. Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front Immunol. (2021) 12:704163. doi: 10.3389/fimmu.2021.704163
107. Qi J, Liu Y, Hu J, Lu L, Dou Z, Dai H, et al. Identification of FPR3 as a unique biomarker for targeted therapy in the immune microenvironment of breast cancer. Front Pharmacol. (2021) 11:593247. doi: 10.3389/fphar.2020.593247
108. Wang L, Mao X, Yu X, Su J, Li Z, Chen Z, et al. FPR3 reprograms glycolytic metabolism and stemness in gastric cancer via calcium-NFATc1 pathway. Cancer Lett. (2024) 593:216841. doi: 10.1016/j.canlet.2024.216841
109. Ye C, Li P, Chen B, Mo Y, Huang Q, Li Q, et al. Pan-cancer analysis and experimental validation of FPR3 as a prognostic and immune infiltration-related biomarker for glioma. Front Genet. (2024) 15:1466617. doi: 10.3389/fgene.2024.1466617
110. Zhou Q, Yan X, Guo Y, Jiang X, Cao T, Ke Y. Machine learning algorithms for predicting glioma patient prognosis based on CD163+FPR3+ macrophage signature. NPJ Precis Oncol. (2024) 8:201. doi: 10.1038/s41698-024-00692-w
111. Miraki Feriz A, Bahraini F, Khosrojerdi A, Azarkar S, Sajjadi SM, HosseiniGol E, et al. Deciphering the immune landscape of head and neck squamous cell carcinoma: A single-cell transcriptomic analysis of regulatory T cell responses to PD-1 blockade therapy. PloS One. (2023) 18:e0295863. doi: 10.1371/journal.pone.0295863
112. Lee HY, Kim HS, Jeong YS, Kim JC, Bae YS, Jo YH, et al. A membrane-tethering pepducin derived from formyl peptide receptor 3 shows strong therapeutic effects against sepsis. Biochem Biophys Res Commun. (2020) 524:156–62. doi: 10.1016/j.bbrc.2020.01.058
113. Gao ZY, Su LC, Wu QC, Sheng JE, Wang YL, Dai YF, et al. Bioinformatics analyses of gene expression profile identify key genes and functional pathways involved in cutaneous lupus erythematosus. Clin Rheumatol. (2022) 41:437–52. doi: 10.1007/s10067-021-05913-2
Keywords: chemoattractant receptors, G-protein coupled receptors, N-formyl peptide receptors, inflammation, receptor structure-function
Citation: Napolitano F and Montuori N (2025) The N-formyl peptide receptors: much more than chemoattractant receptors. Relevance in health and disease. Front. Immunol. 16:1568629. doi: 10.3389/fimmu.2025.1568629
Received: 30 January 2025; Accepted: 21 February 2025;
Published: 04 March 2025.
Edited by:
Liliana Oliveira, Universidade do Porto, PortugalReviewed by:
Xaria X Li, The University of Queensland, AustraliaCopyright © 2025 Napolitano and Montuori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nunzia Montuori, bm1vbnR1b3JAdW5pbmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.