 Yiyang Wen1,2†
Yiyang Wen1,2† Huabao Xiong
Huabao Xiong Dongmei Shi
Dongmei Shi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1564130
The cadherin family, which includes T-cadherin, plays a significant role in angiogenesis, a critical process involved in tumor growth, metastasis, and recurrence. T-cadherin is extensively expressed in both normal and tumor vascular tissues and has been shown to facilitate the proliferation and migration of vascular cells in some studies. However, T-cadherin also exerts inhibitory effects on angiogenesis in various tumor tissues. The functional role of T-cadherin may vary depending on the tumor type and the interaction between tumor cells and vascular cells, suggesting that it acts as a modulator rather than a primary driver of angiogenesis. Additionally, T-cadherin exhibits distinct characteristics depending on the tumor microenvironment. This review provides an overview of recent research on the role of T-cadherin in tumor angiogenesis and discusses its potential as a diagnostic or therapeutic marker in the field of tumor biology.
Tumor progress relies on the expansion of blood vessels to supply essential nutrients and oxygen to malignant cells, resulting in an aberrant tumor microenvironment. This process promotes tumor proliferation, invasion, and metastasis (1–3). Intratumor vessels commonly exhibit structural and functional abnormalities, including disorganized vascular structure, disruption of vascular endothelial cell (EC) junctions, loss of pericyte coverage and an irregular or deficient basal membrane (4, 5). These anomalies impact tumor growth and dissemination, leading to alterations in the tumor microenvironment conducive to tumor progression (6, 7).
T-cadherin, also known as H-cadherin or cadherin-13, was first identified in 1991 (8). T-cadherin has a distinct structure compared to classical cadherins, lacking transmembrane and cytoplasmic domains, but is membrane-bound due to a covalently attached glycosylphosphatidylinisitol (GPI) anchor (9). Unlike other cadherins, T-cadherin does not function primarily as an intercellular adhesion molecule due to the absence of an intracellular domain. Instead, it is believed to be involved in particular intracellular signal pathways (10). Increasing evidence shows that T-cadherin plays an essential role in regulating not only the proliferation, invasion and metastasis of the tumor cell but also in tumor angiogenesis across various cancers including lung, ovarian, esophageal, bladder, cervical and prostate carcinoma (11, 12). However, there is still controversy surrounding the specific functions and underlying mechanisms of T-cadherin in tumor angiogenesis. This review offers a historical overview of T-cadherin research and highlights its significance in tumor angiogenesis.
Human CDH13 (Truncated cadherin, T-cadherin) is located on chromosome 16q24, adjacent to CDH5 (Vascular endothelial cadherin, VE-cadherin), CDH1 (Epithelial cadherin, E-cadherin), CDH3 (Placental cadherin, P-cadherin), CDH8 (Cadherin-8) and CDH11 (Osteoblast cadherin, OB-cadherin), and it is highly conserved across other species in evolution (13). CDH13 gene contains 14 exons, encoding a cDNA sequence of 2142 bps that can be translated into a protein consisting of 713 amino acids (14).
Classical cadherins contain extracellular cadherin repeats, a single transmembrane domain, and a cytoplasmic domain with highly conserved binding sites for downstream catenins, such as p120−catenin and β−catenin, which in turn bind to α-catenin, polymerizing actin microfilaments and maintaining the stability of cytoskeleton. In contrast, T-cadherin lacks transmembrane and cytoplasmic domains and is inserted to the membrane through glycosyl phosphatidylinositol (GPI) attached to the apical aspect plasma membrane (Figure 1) (15), but lacks key amino acids for the adhesive functions. Notably, T-cadherin does not have the canonical strand-exchange dimer and lacks the conserved HisAlaVal motif responsible for homophilic adhesion. These differences indicate that the adhesive mechanisms of T-cadherin may be distinct from classical cadherins (16–18).
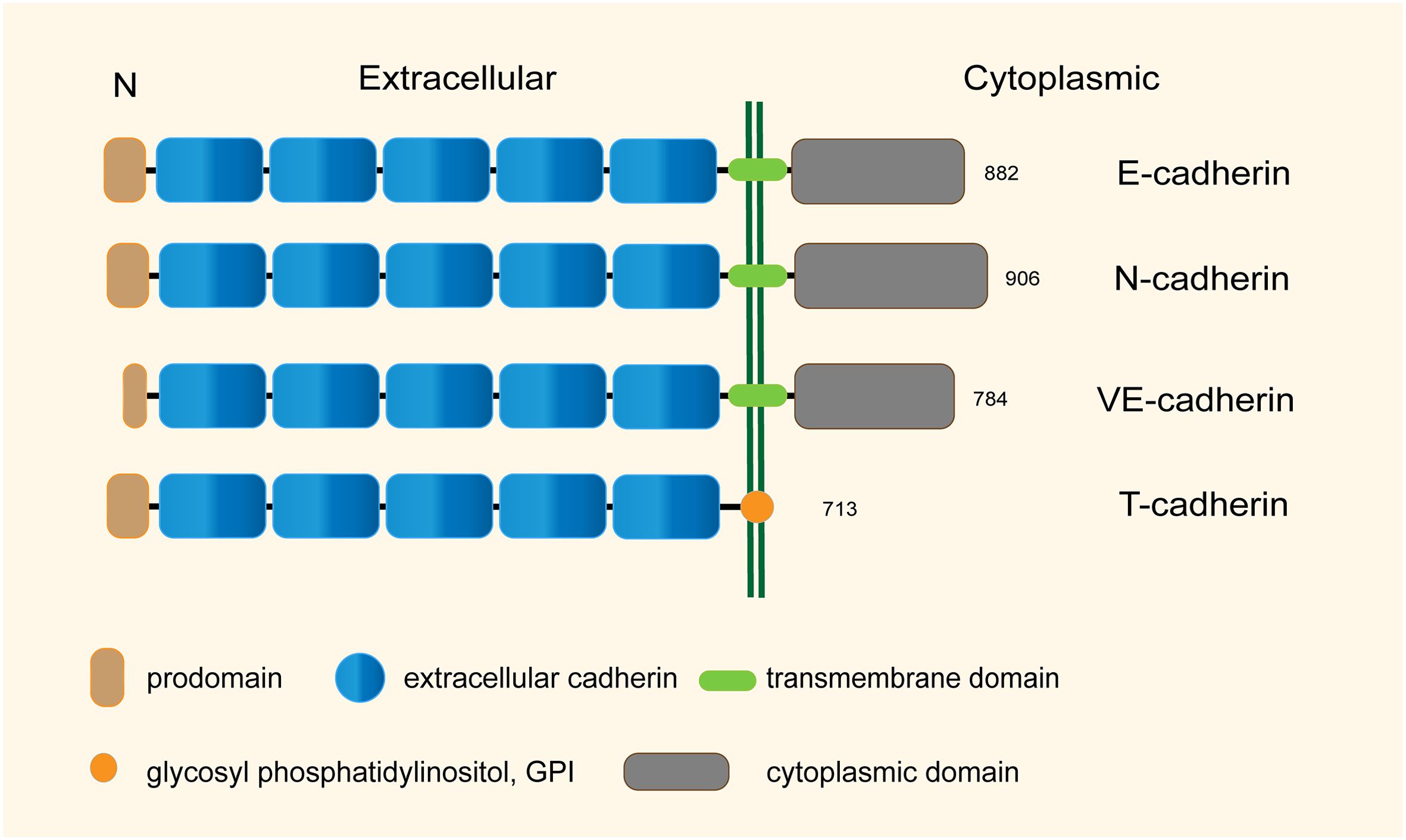
Figure 1. Structural characteristics of the representative human cadherins (total sizes in amino acid residues are shown).
Early immunohistochemical studies have confirmed that T-cadherin is highly expressed in all cardiovascular tissues, including heart, aorta, arteries, post cava and capillaries (19). It is especially found in endothelial cells (ECs), smooth muscle cells (SMCs) and pericytes. In pathological conditions like atherosclerosis, restenosis after balloon angioplasty and tumor angiogenesis, T-cadherin is upregulated contributing to excessive migration, proliferation, and phenotypic modulation of vascular cells (20–22).
T-cadherin has been shown to promote proliferation in ECs and SMCs. It activates the PI3K/Akt/mTOR pathway and inhibits the p38MAPK pathway, protecting ECs from stress-induced apoptosis (23). T-cadherin also affects cell cycle progression, with an increased expression during early S-phase and promoting proliferation in both ECs and SMCs (24). Additionally, T-cadherin influences the expression of β-catenin and ILK, leading to cell proliferation and protection against apoptosis (25). However, T-cadherin knockdown strongly inhibited proliferation of pericytes (26).
Although the stable extracellular structural domain 1 (EC1) is not favorable for homologous binding, homophilic inhibition by recombinant T-cadherin protein against the T-cadherin EC1 domain significantly decreased the adhesion of SMCs and HUVECs (27). At the same time, adenovirus mediated overexpression of T-cadherin can increase detachment and migration of HUVECs, suggesting an anti-adhesive role of T-cadherin for vascular cells. Moreover, T-cadherin could promote SMCs to dedifferentiate upon GSK3β inactivation, which is characterized by acquisition of synthetic, migratory and proliferative properties in response to vascular injury or the presence of atherosclerosis (28). These observations were then confirmed in the subsequent study by using 2D-monolayer and 3D-spheroid migration models (29). The studies collectively demonstrate the function of T-cadherin in promoting vascular cell migration and inhibiting adhesion.
Another study reported that homophilic activation of T-cadherin in HUVECs induced morphological changes toward promigratory phenotype via RhoA/ROCK and Rac pathways and changed adhesion and polarization of the ECs (30). In the initial stage of angiogenesis, the contraction, stretching and remodeling of ECs has been shown to depend critically on RhoA/ROCK activation (31). The Rac activation is necessary for VEGF-induced migration, lamellipodia formation, and recruiting high-affinity integrins to lamellipodia, inducing formation of dynamic cellular protrusions and capillary structures at the leading edges of polarized cells during angiogenesis (32, 33). However, E. V. Semina et al. found that the expression of T-cadherin can lead to an activation of Rac1 and Cdc42, but have no effects on the RhoA signaling pathway (10). The difference in T-cadherin overexpression induced GTPases activation could possibly be related with different experimental conditions or the cell adhesion state.
T-cadherin overexpression leads to the degradation of VE-cadherin in lysosomes, disrupting endothelial barrier function and increasing permeability (34–36). T-cadherin’s involvement in GTPases-mediated signaling pathways affects actin stress fiber formation and microtubule polymerization, leading to decreased permeability of the endothelial monolayer (10, 37). T-cadherin also plays a significant role in regulating endothelial barrier function in response to serum and thrombin (38).
T-cadherin homophilic ligation induced a capillary-like structure consisting of ECs in a 2D model and stimulated a sprout outgrowth in an EC spheroid model whereas an overexpressed T-cadherin in ECs by adenoviral infection increased the sprouting from spheroids (39). In this study, the effects of vascular endothelial growth factor (VEGF) on neovascularization were enhanced by T-cadherin participation in mouse skeletal muscle in vivo (40). However, angiogenesis induced by T-cadherin was not eliminated by inhibition of the VEGF receptor. T-cadherin induced sprouting in both the absence and presence of VEGF, yet it did not trigger neovascularization in the absence of VEGF, suggesting a complex role of T-cadherin in capillary initiation that may be independent of VEGF (39). However, another study investigated the incubation of recombinant N-terminal EC1 domain of T-cadherin in stroma, which is crucial for intercellular recognition and adhesion (41), inhibited endothelial capillary growth in vitro and had no effects on endothelial cell proliferation, adhesion or apoptotic induction (42). The contrary results of T-cadherin in regulation of capillary initiation may be attributed to the different tumor cells used in the above models, or the variable roles of T-cadherin in different tumor angiogenesis mentioned above. It is worth noting that T-cadherin, located in the tumor microenvironment, seems to have a more significant effect on tumor capillary initiation than other mechanisms. This effect on the tumor microenvironment, in turn, participates in tumor angiogenesis (Figure 2).
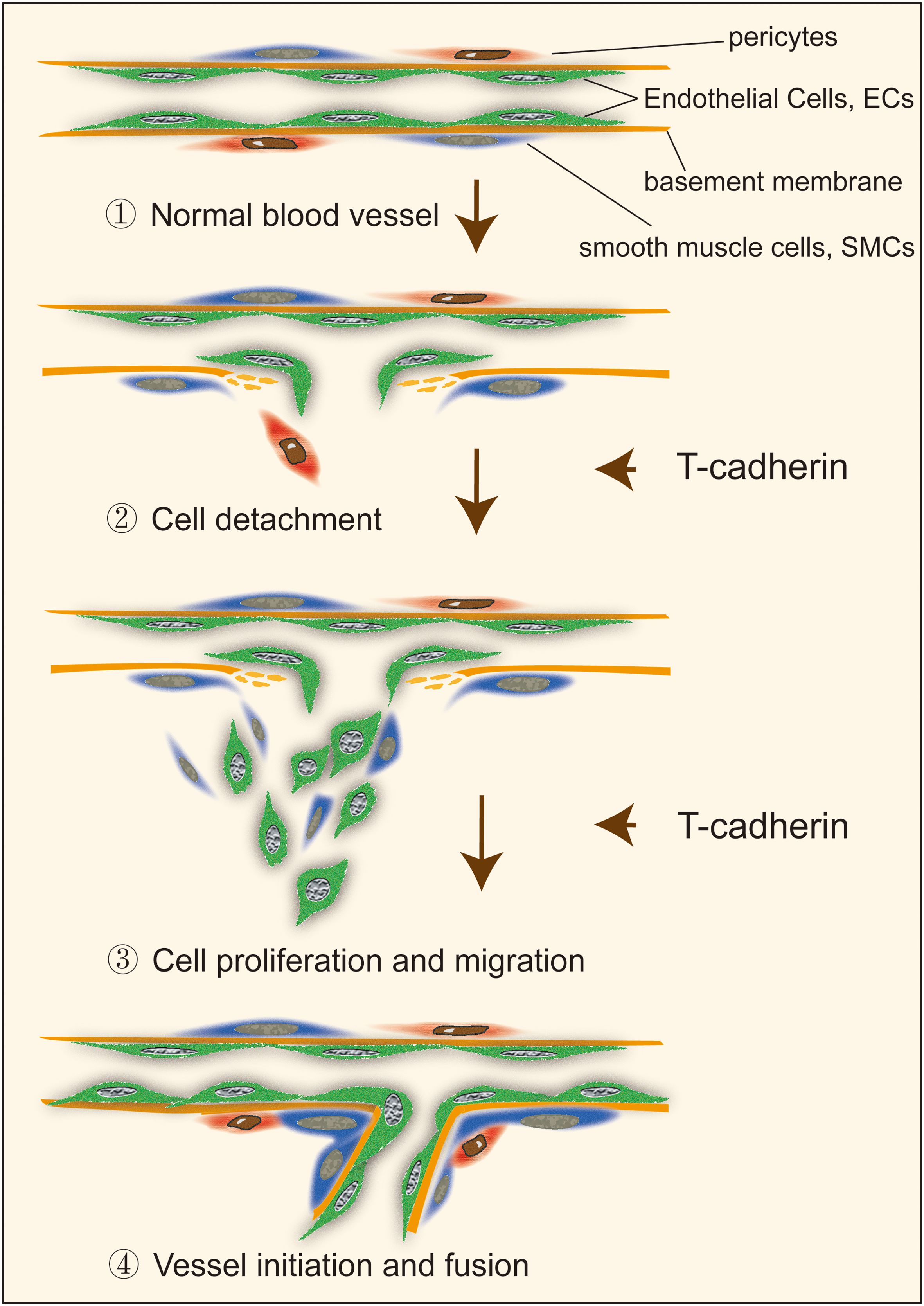
Figure 2. The role of T-cadherin in vascular cell detachment, migration and proliferation during capillary initiation.
In contrast to normal vascular structures, the vascular network of malignant tumors exhibits a disorganized and non-hierarchical arrangement, lacking the typical progression from arterioles to capillaries to venules. Malignant tumor vessels possess uneven distribution of the basal membrane, large differences in vessel caliber size, partially dissociated pericytes and SMCs from ECs and basal membrane (43, 44). Current studies have highlighted that a low or insufficient level of T-cadherin expression in a variety of tumor cells is often closely correlated with the malignant features, as observed in breast cancer, colorectal cancer, endometrial cancer, bladder cancer, melanoma, and squamous cell cancer (45–50). In contrast, high levels of T-cadherin expression have been detected in osteosarcoma, basal cell carcinoma and hepatocellular carcinoma (14, 51, 52). These contrasting findings strongly suggest a correlation between T-cadherin and tumor growth, indicating that the effects of T-cadherin on cell behavior can vary significantly across different cancer types (14, 53).
Overexpression of T-cadherin in tumor vessel endothelial cells was first observed in a Lewis lung carcinoma lung metastasis model (22). Increased expression of T-cadherin was observed in tumor penetrating vessels, while little or no T-cadherin was detected in tumor cells. Another study showed that T-cadherin was absent in 43% of 35 NSCLC tumors but present in all adjacent nonmalignant lung tissue (54). Aberrant promoter methylation may be an important mechanism underlying the low expression or inactivation of T-cadherin in lung cancer. However, there was no significant correlation between hypermethylation of T-cadherin promoter and clinicopathological features, smoking status, clinical stages, or EGFR (epidermal growth factor receptor) mutation status (55, 56). In addition, poorly differentiated NSCLC (non-small cell lung cancer) shows higher levels of T-cadherin promoter hypermethylation than moderately or highly differentiated NSCLC. furthermore, NSCLC patients without T-cadherin hypermethylation have longer overall survival than those with T-cadherin hypermethylation (56). Thus, T-cadherin may act as a tumor suppressor in lung cancer, and its inactivation could contribute to tumor progression and poor prognosis.
Early studies indicated that CDH13 gene is frequently methylated in breast cancer, leading to downregulation of T-cadherin expression (57, 58). Consistent with clinical findings in lung cancer, negative expression of T-cadherin was significantly associated with poor prognosis in patients with axillary lymph node-positive breast cancer or triple-negative breast cancer (59–61). In addition, DNA polymerase β (Pol β) upregulated T-cadherin expression by promoting T-cadherin promoter DNA demethylation, which in turn inhibited tumor migration and invasion, further validating the suppressor role of T-cadherin in breast cancer (62). However, T-cadherin may not be a monofunctional tumor suppressor. Using a transgenic mouse model, researchers found that deletion of T-cadherin limited tumor formation, restrains neovascularization, causes hypoxia, and increases metastases to the lungs (63). Importantly, T-cadherin-deficient tumors exhibit reduced vascular density, enhanced apoptosis, and enlarged hypoxic and necrotic regions. When compared with that in wild-type tumors, poorly differentiated tumors were more prone to be observed in the T-cadherin-deficient condition, suggesting a crucial role of T-cadherin in supporting tumor growth. Considering the significant role of T-cadherin in vascularization, it appears that the contradictory conclusions may be attributed to the complex mechanisms underlying tumor angiogenesis or to the possibility that T-cadherin plays different roles at different stages of tumor angiogenesis.
CDH13 gene methylation is also prevalent in melanoma and has been investigated more extensively. A prior investigation evaluated the expression levels of T-cadherin in 40 human melanoma cell lines using RT-PCR (49). The study revealed that T-cadherin expression was significantly reduced or absent in 28 (70%) melanoma cell lines. However, the hypermethylation of CpG islands in the promoter region of the CDH13 gene is not the only mechanism responsible for the loss of T-cadherin. DNA demethylation and inhibition of histone deacetylase do not result in the re-expression of T-cadherin in melanoma cell lines. Additionally, certain transcriptional repressors, such as BRN2 in melanoma cells and ZEB1 in gallbladder cancer cells, inhibit the transcriptional activity of the CDH13 promoter (64). Specifically, in melanoma cells, the transcriptional repression of CDH13 promoter activity by BRN2 enhances their migratory and invasive capabilities (65).
Multiple studies found that T-cadherin expression in B16F10 melanoma cells remarkably reduced cell proliferation and invasion and promoted apoptosis, which may be associated with antagonizing the AKT/CREB/AP-1/FoxO3a signaling pathway. In mouse tumor models, smaller tumor masses and significantly decreased vascularization were observed in T-cadherin overexpressed group (66–68). In addition, melanoma cells with positive expression of T-cadherin were more sensitive to garcinol (a chemically synthesized polyisoprenylated benzophenone), which also demonstrated the inhibitory effect of T-cadherin on melanoma (69). However, the inconsistent influence of T-cadherin on tumor angiogenesis has been noted in other studies. First, expression of T-cadherin leads to increased invasive potential due to the upregulation of pro-oncogenic integrins, chemokines, adhesion molecules and extracellular matrix components. Furthermore, the overexpression of T-cadherin in HMEC-1 cells leads to the formation of a vascular network within melanoma in a 3D multicellular tumor spheroid model. The authors propose an intriguing hypothesis that T-cadherin, expressed by endothelial cells, may facilitate tumor angiogenesis exclusively within a pro-angiogenic microenvironment (70). Therefore, the role of T-cadherin in melanoma functions beyond the regulation of tumor cells, being closely associated with the mechanisms underlying tumor angiogenesis.
Early studies found that T-cadherin was specifically localized at the basal layer of normal epidermis but was downregulated in cutaneous squamous cell carcinoma (71, 72). In squamous cell carcinoma HSC-1 cells, overexpression of T-cadherin increased surface β1 integrin expression, inhibited β1 integrin internalization, and promoted β1 integrin-mediated cell-matrix adhesion, which was possibly associated with reduced phosphorylation at Tyr845 of EGFR. This suggests that T-cadherin acts as a negative regulator of epidermal tumorigenesis (73). However, in another study, the overexpression of T-cadherin in cutaneous squamous cell carcinoma A431 cells inhibited the adhesion between tumor cells and vascular ECs, whereas T-cadherin deficiency induced adhesion between A431 cells and ECs (74). It is possible that the expression or functional alterations of other intercellular adhesion molecules are due to T-cadherin loss. Moreover, more blood vessels were observed in T-cadherin overexpressed tumors than those in T-cadherin silenced tumors in cutaneous squamous cell carcinoma xenografts. Interestingly, the promoting effects of T-cadherin on tumor angiogenesis in this study appeared to be a direct contribution of T-cadherin towards creation of a proangiogenic microenvironment (75, 76). Although overexpression of T-cadherin in A431 cells did not affect the tumor cell proliferation in vitro and in vivo, the culture supernatant of T-cadherin overexpressed A431 cells still promoted the sprout outgrowth (75). Therefore, T-cadherin expression in the tumor microenvironment promotes angiogenesis rather than that expressed in tumor cells, aligning with the concept that T-cadherin regulates cell proliferation and morphological changes in vascular cells.
Hepatocellular carcinoma (HCC) is generally believed to be a hypervascular tumor. Similar to the expression pattern of T-cadherin in other tumors, T-cadherin was underexpressed in hepatocellular carcinoma cells (26.5%, 13/49 cases) but was frequently (77.6%, 38/49 cases) overexpressed in tumor endothelial cells (77). Overexpression of T-cadherin induced G2/M cell cycle arrest, decreased cell proliferation, inhibited adherence-independent growth, and increased the sensitivity of HCC cells to TNFa-mediated apoptosis. They also found that T-cadherin significantly inhibited the activity of c-Jun, a key oncogene that is constitutively activated in hepatocellular carcinoma cells. T-cadherin was selectively expressed in intratumoral capillary endothelial cells, and the expression levels of T-cadherin was also positively correlated with tumor malignant progression (78). Specifically, the higher expression of T-cadherin was more likely detected in poorly differentiated tumor regions than in regions where the tumor cells were well differentiated, indicating that T-cadherin in sinusoidal vascular endothelial cells might be increasingly induced during the tumor progression (79).
Decreased expression of T-cadherin was associated with the larger tumor size, surrounding tissue infiltration, lymph node metastasis and poor differentiation in gastric cancer (80, 81). Moreover, PI3K/AKT/mTOR signaling pathway, an important role in regulating angiogenesis both in normal tissues and in cancers, was reported to be involved in T-cadherin related tumorigenesis of human gastric cancer and cervical cancer, since sustained activation of AKT1 in endothelial cells has been shown to induce the formation of abnormal blood vessels, which was similar to the aberrations of tumor vessels (82–84).
Another important role of T-cadherin is to function as the receptor of low-density lipoproteins (LDL) and adiponectin. In the early 1990s, T-cadherin was purified and identified from human and rat aorta, as well as cultured SMCs, as a receptor of LDL (85). However, the mechanism underlying the specificity interaction of T-cadherin and LDL in tumor angiogenesis has not been studied in detail.
Adiponectin is an adipose tissue-derived homeostatic factor that is mainly secreted by white adipocytes, but can also be produced by skeletal muscles, cardiac myocytes and ECs (86, 87). In plasma, Adiponectin exists in a variety of complexes, including trimers (LMW, low molecular weight), hexamers (MMW) and high molecular weight multimers (HMW), exerting protective functions in insulin−sensitizing, anti−inflammation, anti−proliferation, anti−atherosclerotic action and tumor suppression of various tissues (87). It is believed that the HMW adiponectin is the metabolically active form of adiponectin (88). However, the function of adiponectin in blood vessel remains controversial, both protective and promotive effects on blood vessel growth are reported (63, 89, 90).
Except AdipoRs (adiponectin receptors), T-cadherin seems to be another major receptor for native adiponectin in serum with still largely unknown role in intracellular signaling. T-cadherin was co- localized with adiponectin in vascular endothelium, pericytes, and skeletal muscles (91). Early studies have demonstrated that binding of eukaryotic-expressed adiponectin to cell-surface T-cadherin is dependent on the hexameric and HMW form (92). Subsequent research by Kita et al. demonstrated that native adiponectin selectively binds to cells expressing T-cadherin, while no binding was observed in cells expressing AdipoR1. Furthermore, the knockdown of T-cadherin led to a marked reduction in adiponectin binding to these cells (93). Additionally, cardiovascular tissues lacking T-cadherin appear to be insensitive to adiponectin, despite with the consistently expressed AdipoR1/AdipoR2 (94). Adiponectin induces EC differentiation into capillary-like structures and stimulates blood vessel growth by promoting cross-talk between AMPK and Akt signaling, which may be the same as the function of T-cadherin on ECs (95). Furthermore, T-cadherin expressed by tumor endothelial cells inhibits cell apoptosis during tumor angiogenesis by activating NF-κB upon binding to hexamer and/or HMW adiponectin (79). Contrarily, another study found that adiponectin can significantly inhibit proliferation and migration of ECs via the activation of the caspase-mediated ECs apoptosis, and recombinant adiponectin potently impaired primary tumor growth that is associated with decreased neovascularization in mice (89). Using an ischemia-induced revascularization model, the impaired revascularization phenotype could be rescued by overexpression of adiponectin in APN-KO (adiponectin knockout) mice but not in mice that were lacking T-cadherin, suggesting an essential role of T-cadherin in mediating the proangiogenic activity of adiponectin. Supportively, the study also found that knockdown of T-cadherin prevented adiponectin induced migration and proliferation of cultured ECs (96). Similarly, in MMTV-PyV-mT mice, T-cadherin was able to sequester adiponectin to the vasculature in a T-cadherin-dependent manner while the plasma adiponectin level was dramatically increased in T-cadherin-deficient mice (63). Moreover, in the absence of T-cadherin, adiponectin appears to lose its location in the vasculature, suggesting that T-cadherin plays a crucial role in the regulatory mechanisms of vascular function involving adiponectin in both normal tissues and malignant breast cancer. In the light of these findings, although the interaction of adiponectin and T-cadherin has been well established, the role of this binding on tumor angiogenesis is still not clear. In addition, T-cadherin seems to be a membrane binding protein for adiponectin rather than a receptor that needs to be confirmed by further studies in the future.
T-cadherin plays a crucial role in tumor angiogenesis by influencing blood vessel development in endothelial cells (ECs), smooth muscle cells (SMCs), and pericytes. Its function in angiogenesis relies on homophilic interactions or signal transductions as a membrane-binding protein (Figure 3). However, the specific effects of T-cadherin on vascular cell proliferation, detachment and migration have shown conflicting results in different studies. These contradictions may arise from the diverse nature of tumor types and limited expression of T-cadherin in tumor cells. While T-cadherin is not the primary initiator of angiogenesis, it acts as a modulator that requires initial vessel destabilization through angiogenic factors to facilitate phenotype conversion, proliferation, and survival of vascular cells (39, 97). T-cadherin’s presence in rapidly growing vessels suggests its role in guiding and directing tumor angiogenesis (98). Future research should focus on understanding the molecular mechanisms underlying T-cadherin’s effects on both normal and tumor blood vessels, particularly the intercellular signaling between tumor cells and vascular cells.
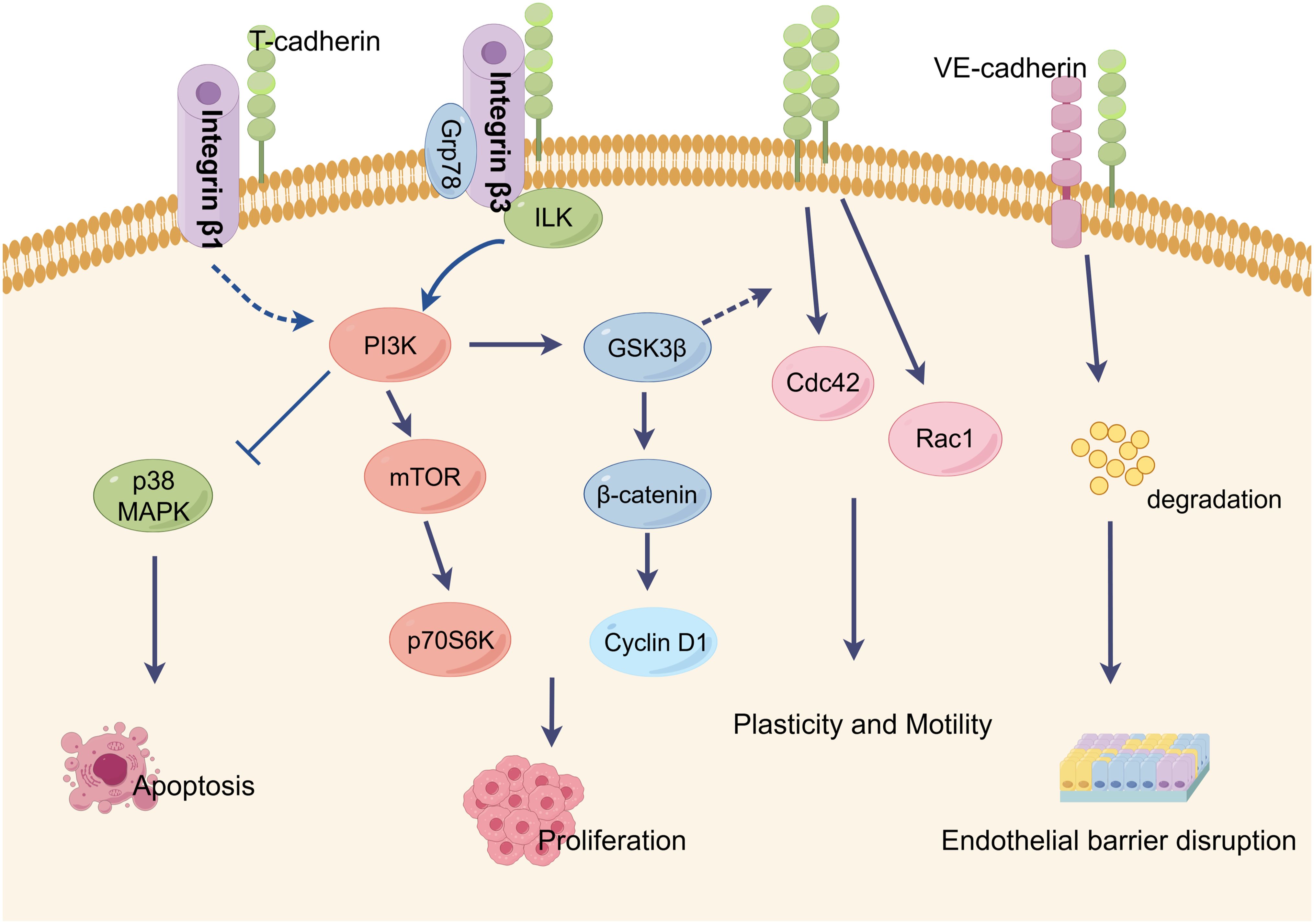
Figure 3. Characteristic T-cadherin-mediated intercellular signaling events and biological activities. T-cadherin lacks transmembrane and cytoplasmic domains and localizes in membrane lipid rafts through a glycosyl phosphatidylinositol (GPI). T-cadherin is thought to be a regulator of endothelial cell survival, proliferation, plasticity and motility, all of which are key processes in angiogenesis. By interactions with GRP78 or integrin-β3, T-cadherin stimulates cell proliferation and protects endothelial cells from apoptosis by activation of the PI3K/Akt pathway. T-cadherin also inhibits integrin-β1 internalization in squamous cell carcinoma cells. Homophilic activation of T-cadherin induced morphological changes toward promigratory phenotype via RhoA/ROCK and Rac pathways. T-cadherin is able to induce the degradation of VE-cadherin in lysosomes, resulting in the disruption of endothelial barrier function. This figure is drawn by Figdraw.
YW: Investigation, Validation, Writing – original draft. LM: Data curation, Software, Writing – original draft. YL: Data curation, Methodology, Writing – original draft. HX: Software, Writing – review & editing. DS: Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grants from the National Nature Science Foundation of China (NM 82272358), Key R&D Program of Jining (2023YXNS001, 2023YXNS055 and 2023YXNS247).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. (2005) 15:102–11. doi: 10.1016/j.gde.2004.12.005
2. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. (2000) 407:249–57. doi: 10.1038/35025220
3. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
4. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
5. McDonald DM, Baluk P. Imaging of angiogenesis in inflamed airways and tumors: newly formed blood vessels are not alike and may be wildly abnormal: Parker B. Francis lecture. Chest. (2005) 128:602S–8S. doi: 10.1378/chest.128.6_suppl.602S-a
6. Zanotelli MR, Reinhart-King CA. Mechanical forces in tumor angiogenesis. Adv Exp Med Biol. (2018) 1092:91–112. doi: 10.1007/978-3-319-95294-9_6
7. Kretschmer M, Rudiger D, Zahler S. Mechanical aspects of angiogenesis. Cancers (Basel). (2021) 13(19):4987. doi: 10.3390/cancers13194987
8. Ranscht B, Dours-Zimmermann MT. T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron. (1991) 7:391–402. doi: 10.1016/0896-6273(91)90291-7
9. Vestal DJ, Ranscht B. Glycosyl phosphatidylinositol–anchored T-cadherin mediates calcium-dependent, homophilic cell adhesion. J Cell Biol. (1992) 119:451–61. doi: 10.1083/jcb.119.2.451
10. Semina EV, Rubina KA, Rutkevich PN, Voyno-Yasenetskaya TA, Parfyonova YV, Tkachuk VA. T-cadherin activates Rac1 and Cdc42 and changes endothelial permeability. Biochem (Mosc). (2009) 74:362–70. doi: 10.1134/s0006297909040026
11. Yu W, Yang L, Li T, Zhang Y. Cadherin signaling in cancer: its functions and role as a therapeutic target. Front Oncol. (2019) 9:989. doi: 10.3389/fonc.2019.00989
12. Andreeva AV, Kutuzov MA. Cadherin 13 in cancer. Genes Chromosomes Cancer. (2010) 49:775–90. doi: 10.1002/gcc.20787
13. Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. (2001) 114:629–41. doi: 10.1242/jcs.114.4.629
14. Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, Resink TJ. A guide and guard: the many faces of T-cadherin. Cell Signal. (2009) 21:1035–44. doi: 10.1016/j.cellsig.2009.01.035
15. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. (2014) 14:121–34. doi: 10.1038/nrc3647
16. Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol. (2009) 41:349–69. doi: 10.1016/j.biocel.2008.09.027
17. Dames SA, Bang E, Haussinger D, Ahrens T, Engel J, Grzesiek S. Insights into the low adhesive capacity of human T-cadherin from the NMR structure of Its N-terminal extracellular domain. J Biol Chem. (2008) 283:23485–95. doi: 10.1074/jbc.M708335200
18. Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. (2005) 6:622–34. doi: 10.1038/nrm1699
19. Wang H, Tao L, Ambrosio A, Yan W, Summer R, Lau WB, et al. T-cadherin deficiency increases vascular vulnerability in T2DM through impaired NO bioactivity. Cardiovasc Diabetol. (2017) 16:12. doi: 10.1186/s12933-016-0488-0
20. Ivanov D, Philippova M, Antropova J, Gubaeva F, Iljinskaya O, Tararak E, et al. Expression of cell adhesion molecule T-cadherin in the human vasculature. Histochem Cell Biol. (2001) 115:231–42. doi: 10.1007/s004180100252
21. Kudrjashova E, Bashtrikov P, Bochkov V, Parfyonova Y, Tkachuk V, Antropova J, et al. Expression of adhesion molecule T-cadherin is increased during neointima formation in experimental restenosis. Histochem Cell Biol. (2002) 118:281–90. doi: 10.1007/s00418-002-0463-6
22. Wyder L, Vitaliti A, Schneider H, Hebbard LW, Moritz DR, Wittmer M, et al. Increased expression of H/T-cadherin in tumor-penetrating blood vessels. Cancer Res. (2000) 60:4682–88.
23. Joshi MB, Philippova M, Ivanov D, Allenspach R, Erne P, Resink TJ. T-cadherin protects endothelial cells from oxidative stress-induced apoptosis. FASEB J. (2005) 19:1737–39. doi: 10.1096/fj.05-3834fje
24. Ivanov D, Philippova M, Allenspach R, Erne P, Resink T. T-cadherin upregulation correlates with cell-cycle progression and promotes proliferation of vascular cells. Cardiovasc Res. (2004) 64:132–43. doi: 10.1016/j.cardiores.2004.06.010
25. Joshi MB, Ivanov D, Philippova M, Erne P, Resink TJ. Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3beta in endothelial cells. FASEB J. (2007) 21:3083–95. doi: 10.1096/fj.06-7723com
26. Dasen B, Pigeot S, Born GM, Verrier S, Rivero O, Dittrich PS, et al. T-cadherin is a novel regulator of pericyte function during angiogenesis. Am J Physiol Cell Physiol. (2023) 324:C821–36. doi: 10.1152/ajpcell.00326.2022
27. Ivanov D, Philippova M, Tkachuk V, Erne P, Resink T. Cell adhesion molecule T-cadherin regulates vascular cell adhesion, phenotype and motility. Exp Cell Res. (2004) 293:207–18. doi: 10.1016/j.yexcr.2003.09.030
28. Frismantiene A, Dasen B, Pfaff D, Erne P, Resink TJ, Philippova M. T-cadherin promotes vascular smooth muscle cell dedifferentiation via a GSK3beta-inactivation dependent mechanism. Cell Signal. (2016) 28:516–30. doi: 10.1016/j.cellsig.2016.02.014
29. Frismantiene A, Kyriakakis E, Dasen B, Erne P, Resink TJ, Philippova M. Actin cytoskeleton regulates functional anchorage-migration switch during T-cadherin-induced phenotype modulation of vascular smooth muscle cells. Cell Adh Migr. (2018) 12:69–85. doi: 10.1080/19336918.2017.1319545
30. Philippova M, Ivanov D, Allenspach R, Takuwa Y, Erne P, Resink T. RhoA and Rac mediate endothelial cell polarization and detachment induced by T-cadherin. FASEB J. (2005) 19:588–90. doi: 10.1096/fj.04-2430fje
31. van Nieuw AG, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. (2003) 23:211–17. doi: 10.1161/01.atv.0000054198.68894.88
32. Hoang MV, Whelan MC, Senger DR. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc Natl Acad Sci U.S.A. (2004) 101:1874–79. doi: 10.1073/pnas.0308525100
33. Soga N, Connolly JO, Chellaiah M, Kawamura J, Hruska KA. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun Adhes. (2001) 8:1–13. doi: 10.3109/15419060109080703
34. Duong CN, Vestweber D. Mechanisms ensuring endothelial junction integrity beyond VE-cadherin. Front Physiol. (2020) 11:519. doi: 10.3389/fphys.2020.00519
35. Philippova M, Ivanov D, Tkachuk V, Erne P, Resink TJ. Polarisation of T-cadherin to the leading edge of migrating vascular cells in vitro: a function in vascular cell motility? Histochem Cell Biol. (2003) 120:353–60. doi: 10.1007/s00418-003-0584-6
36. Semina EV, Rubina KA, Sysoeva VY, Rutkevich PN, Kashirina NM, Tkachuk VA. Novel mechanism regulating endothelial permeability via T-cadherin-dependent VE-cadherin phosphorylation and clathrin-mediated endocytosis. Mol Cell Biochem. (2014) 387:39–53. doi: 10.1007/s11010-013-1867-4
37. Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, et al. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. (2004) 201:55–70. doi: 10.1002/jcp.20055
38. Andreeva AV, Han J, Kutuzov MA, Profirovic J, Tkachuk VA, Voyno-Yasenetskaya TA. T-cadherin modulates endothelial barrier function. J Cell Physiol. (2010) 223:94–102. doi: 10.1002/jcp.22014
39. Philippova M, Banfi A, Ivanov D, Gianni-Barrera R, Allenspach R, Erne P, et al. Atypical GPI-anchored T-cadherin stimulates angiogenesis in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2006) 26:2222–30. doi: 10.1161/01.ATV.0000238356.20565.92
40. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
41. Joshi MB, Kyriakakis E, Pfaff D, Rupp K, Philippova M, Erne P, et al. Extracellular cadherin repeat domains EC1 and EC5 of T-cadherin are essential for its ability to stimulate angiogenic behavior of endothelial cells. FASEB J. (2009) 23:4011–21. doi: 10.1096/fj.09-133611
42. Rubina K, Kalinina N, Potekhina A, Efimenko A, Semina E, Poliakov A, et al. T-cadherin suppresses angiogenesis in vivo by inhibiting migration of endothelial cells. Angiogenesis. (2007) 10:183–95. doi: 10.1007/s10456-007-9072-2
43. Sobierajska K, Ciszewski WM, Sacewicz-Hofman I, Niewiarowska J. Endothelial cells in the tumor microenvironment. Adv Exp Med Biol. (2020) 1234:71–86. doi: 10.1007/978-3-030-37184-5_6
44. Al-Ostoot FH, Salah S, Khamees HA, Khanum SA. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun. (2021) 28:100422. doi: 10.1016/j.ctarc.2021.100422
45. Riener MO, Nikolopoulos E, Herr A, Wild PJ, Hausmann M, Wiech T, et al. Microarray comparative genomic hybridization analysis of tubular breast carcinoma shows recurrent loss of the CDH13 locus on 16q. Hum Pathol. (2008) 39:1621–29. doi: 10.1016/j.humpath.2008.02.021
46. Polito R, Nigro E, Fei L, de Magistris L, Monaco ML, D’Amico R, et al. Adiponectin is inversely associated with tumour grade in colorectal cancer patients. Anticancer Res. (2020) 40:3751–57. doi: 10.21873/anticanres.14364
47. Suehiro Y, Okada T, Okada T, Anno K, Okayama N, Ueno K, et al. Aneuploidy predicts outcome in patients with endometrial carcinoma and is related to lack of CDH13 hypermethylation. Clin Cancer Res. (2008) 14:3354–61. doi: 10.1158/1078-0432.CCR-07-4609
48. Lin Y, Sun G, Liu X, Chen Y, Zhang C. Clinical significance of T-cadherin tissue expression in patients with bladder transitional cell carcinoma. Urol Int. (2011) 86:340–45. doi: 10.1159/000322962
49. Kuphal S, Martyn AC, Pedley J, Crowther LM, Bonazzi VF, Parsons PG, et al. H-Cadherin expression reduces invasion of Malignant melanoma. Pigment Cell Melanoma Res. (2009) 22:296–306. doi: 10.1111/j.1755-148X.2009.00568.x
50. Buechner SA, Resink TJ. T-cadherin expression in actinic keratosis transforming to invasive squamous cell carcinoma. Dermatopathology (Basel). (2019) 6:12–9. doi: 10.1159/000495609
51. Zucchini C, Bianchini M, Valvassori L, Perdichizzi S, Benini S, Manara MC, et al. Identification of candidate genes involved in the reversal of Malignant phenotype of osteosarcoma cells transfected with the liver/bone/kidney alkaline phosphatase gene. Bone. (2004) 34:672–79. doi: 10.1016/j.bone.2003.12.008
52. Riou P, Saffroy R, Chenailler C, Franc B, Gentile C, Rubinstein E, et al. Expression of T-cadherin in tumor cells influences invasive potential of human hepatocellular carcinoma. FASEB J. (2006) 20:2291–301. doi: 10.1096/fj.06-6085com
53. Rubina KA, Semina EV, Kalinina NI, Sysoeva VY, Balatskiy AV, Tkachuk VA. Revisiting the multiple roles of T-cadherin in health and disease. Eur J Cell Biol. (2021) 100:151183. doi: 10.1016/j.ejcb.2021.151183
54. Zhong Y, Delgado Y, Gomez J, Lee SW, Perez-Soler R. Loss of H-cadherin protein expression in human non-small cell lung cancer is associated with tumorigenicity. Clin Cancer Res. (2001) 7:1683–87.
55. Kim DS, Kim MJ, Lee JY, Kim YZ, Kim EJ, Park JY. Aberrant methylation of E-cadherin and H-cadherin genes in nonsmall cell lung cancer and its relation to clinicopathologic features. Cancer. (2007) 110:2785–92. doi: 10.1002/cncr.23113
56. Wang Z, Wang B, Guo H, Shi G, Hong X. Clinicopathological significance and potential drug target of T-cadherin in NSCLC. Drug Des Devel Ther. (2015) 9:207–16. doi: 10.2147/DDDT.S74259
57. Feng W, Orlandi R, Zhao N, Carcangiu ML, Tagliabue E, Xu J, et al. Tumor suppressor genes are frequently methylated in lymph node metastases of breast cancers. BMC Cancer. (2010) 10:378. doi: 10.1186/1471-2407-10-378
58. Lee SW. H-cadherin, a novel cadherin with growth inhibitory functions and diminished expression in human breast cancer. Nat Med. (1996) 2:776–82. doi: 10.1038/nm0796-776
59. Kong D, Wang MH, Yang J, Li L. Association of T-cadherin levels with the response to neoadjuvant chemotherapy in locally advanced breast cancer. Oncotarget. (2017) 8:13747–53. doi: 10.18632/oncotarget.14630
60. Kong DD, Yang J, Li L, Wang W, Chen YN, Wang SB, et al. T-cadherin association with clinicopathological features and prognosis in axillary lymph node-positive breast cancer. Breast Cancer Res Treat. (2015) 150:119–26. doi: 10.1007/s10549-015-3302-x
61. Kong DD, Wang MH, Yang J, Li L, Wang W, Wang SB, et al. T-cadherin is associated with prognosis in triple-negative breast cancer. Oncol Lett. (2017) 14:2975–81. doi: 10.3892/ol.2017.6505
62. Wang M, Long K, Li E, Li L, Li B, Ci S, et al. DNA polymerase beta modulates cancer progression via enhancing CDH13 expression by promoter demethylation. Oncogene. (2020) 39:5507–19. doi: 10.1038/s41388-020-1386-1
63. Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, Ranscht B. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res. (2008) 68:1407–16. doi: 10.1158/0008-5472.CAN-07-2953
64. Adachi Y, Takeuchi T, Nagayama T, Ohtsuki Y, Furihata M. Zeb1-mediated T-cadherin repression increases the invasive potential of gallbladder cancer. FEBS Lett. (2009) 583:430–36. doi: 10.1016/j.febslet.2008.12.042
65. Ellmann L, Joshi MB, Resink TJ, Bosserhoff AK, Kuphal S. BRN2 is a transcriptional repressor of CDH13 (T-cadherin) in melanoma cells. Lab Invest. (2012) 92:1788–800. doi: 10.1038/labinvest.2012.140
66. Iurlova EI, Rubina KA, Sysoeva V, Sharonov GV, Semina EV, Parfenova EV, et al. T-cadherin suppresses the cell proliferation of mouse melanoma B16F10 and tumor angiogenesis in the model of the chorioallantoic membrane. Ontogenez. (2010) 41:261–70. doi: 10.1134/S1062360410040028
67. Rubina KA, Surkova EI, Semina EV, Sysoeva VY, Kalinina NI, Poliakov AA, et al. T-cadherin expression in melanoma cells stimulates stromal cell recruitment and invasion by regulating the expression of chemokines, integrins and adhesion molecules. Cancers (Basel). (2015) 7:1349–70. doi: 10.3390/cancers7030840
68. Bosserhoff AK, Ellmann L, Quast AS, Eberle J, Boyle GM, Kuphal S. Loss of T-cadherin (CDH-13) regulates AKT signaling and desensitizes cells to apoptosis in melanoma. Mol Carcinog. (2014) 53:635–47. doi: 10.1002/mc.22018
69. Staebler S, Hoechst S, Thongmao A, Schneider N, Bosserhoff AK, Kuphal S. The role of T-cadherin (CDH13) in treatment options with garcinol in melanoma. Cancers (Basel). (2024) 16(10):1853. doi: 10.3390/cancers16101853
70. Ghosh S, Joshi MB, Ivanov D, Feder-Mengus C, Spagnoli GC, Martin I, et al. Use of multicellular tumor spheroids to dissect endothelial cell-tumor cell interactions: a role for T-cadherin in tumor angiogenesis. FEBS Lett. (2007) 581:4523–28. doi: 10.1016/j.febslet.2007.08.038
71. Takeuchi T, Liang SB, Matsuyoshi N, Zhou S, Miyachi Y, Sonobe H, et al. Loss of T-cadherin (CDH13, H-cadherin) expression in cutaneous squamous cell carcinoma. Lab Invest. (2002) 82:1023–29. doi: 10.1097/01.lab.0000025391.35798.f1
72. Zhou S, Matsuyoshi N, Liang SB, Takeuchi T, Ohtsuki Y, Miyachi Y. Expression of T-cadherin in Basal keratinocytes of skin. J Invest Dermatol. (2002) 118:1080–84. doi: 10.1046/j.1523-1747.2002.01795.x
73. Mukoyama Y, Utani A, Matsui S, Zhou S, Miyachi Y, Matsuyoshi N. T-cadherin enhances cell-matrix adhesiveness by regulating beta1 integrin trafficking in cutaneous squamous carcinoma cells. Genes Cells. (2007) 12:787–96. doi: 10.1111/j.1365-2443.2007.01092.x
74. Philippova M, Pfaff D, Kyriakakis E, Buechner SA, Iezzi G, Spagnoli GC, et al. T-cadherin loss promotes experimental metastasis of squamous cell carcinoma. Eur J Cancer. (2013) 49:2048–58. doi: 10.1016/j.ejca.2012.12.026
75. Pfaff D, Philippova M, Kyriakakis E, Maslova K, Rupp K, Buechner SA, et al. Paradoxical effects of T-cadherin on squamous cell carcinoma: up- and down-regulation increase xenograft growth by distinct mechanisms. J Pathol. (2011) 225:512–24. doi: 10.1002/path.2900
76. Lu Q, Huang Y, Wu J, Guan Y, Du M, Wang F, et al. T-cadherin inhibits invasion and migration of endometrial stromal cells in endometriosis. Hum Reprod. (2020) 35:145–56. doi: 10.1093/humrep/dez252
77. Chan DW, Lee JM, Chan PC, Ng IO. Genetic and epigenetic inactivation of T-cadherin in human hepatocellular carcinoma cells. Int J Cancer. (2008) 123:1043–52. doi: 10.1002/ijc.23634
78. Sun HC, Tang ZY. Angiogenesis in hepatocellular carcinoma: the retrospectives and perspectives. J Cancer Res Clin Oncol. (2004) 130:307–19. doi: 10.1007/s00432-003-0530-y
79. Adachi Y, Takeuchi T, Sonobe H, Ohtsuki Y. An adiponectin receptor, T-cadherin, was selectively expressed in intratumoral capillary endothelial cells in hepatocellular carcinoma: possible cross talk between T-cadherin and FGF-2 pathways. Virchows Arch. (2006) 448:311–18. doi: 10.1007/s00428-005-0098-9
80. Tang Y, Dai Y, Huo J. Decreased expression of T-cadherin is associated with gastric cancer prognosis. Hepatogastroenterology. (2012) 59:1294–98. doi: 10.5754/hge12016
81. Wei B, Shi H, Lu X, Shi A, Cheng Y, Dong L. Association between the expression of T-cadherin and vascular endothelial growth factor and the prognosis of patients with gastric cancer. Mol Med Rep. (2015) 12:2075–81. doi: 10.3892/mmr.2015.3592
82. Lin J, Chen Z, Huang Z, Chen F, Ye Z, Lin S, et al. Upregulation of T-cadherin suppresses cell proliferation, migration and invasion of gastric cancer in vitro. Exp Ther Med. (2017) 14:4194–200. doi: 10.3892/etm.2017.5090
83. Zhao J, Yang T, Ji J, Li C, Li Z, Li L. Garcinol exerts anti-cancer effect in human cervical cancer cells through upregulation of T-cadherin. BioMed Pharmacother. (2018) 107:957–66. doi: 10.1016/j.biopha.2018.08.060
84. Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. (2011) 4:51. doi: 10.3389/fnmol.2011.00051
85. Bochkov VN, Tkachuk VA, Philippova MP, Stambolsky DV, Buhler FR, Resink TJ. Ligand selectivity of 105 kDa and 130 kDa lipoprotein-binding proteins in vascular-smooth-muscle-cell membranes is unique. . Biochem J. (1996) 317:297–304. doi: 10.1042/bj3170297
86. Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin–a key adipokine in the metabolic syndrome. Diabetes Obes Metab. (2006) 8:264–80. doi: 10.1111/j.1463-1326.2005.00510.x
87. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. (2017) 18(6):1321. doi: 10.3390/ijms18061321
88. Nguyen T. Adiponectin: role in physiology and pathophysiology. Int J Prev Med. (2020) 11:136. doi: 10.4103/ijpvm.IJPVM_193_20
89. Brakenhielm E, Veitonmaki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, et al. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U.S.A. (2004) 101:2476–81. doi: 10.1073/pnas.0308671100
90. Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, et al. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res. (2009) 15:3265–76. doi: 10.1158/1078-0432.CCR-08-2649
91. Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. (2010) 120:4342–52. doi: 10.1172/JCI43464
92. Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U.S.A. (2004) 101:10308–13. doi: 10.1073/pnas.0403382101
93. Kita S, Fukuda S, Maeda N, Shimomura I. Native adiponectin in serum binds to mammalian cells expressing T-cadherin, but not AdipoRs or calreticulin. Elife. (2019) 8:e48675. doi: 10.7554/eLife.48675
94. Karnati HK, Panigrahi MK, Li Y, Tweedie D, Greig NH. Adiponectin as a potential therapeutic target for prostate cancer. Curr Pharm Des. (2017) 23:4170–79. doi: 10.2174/1381612823666170208123553
95. Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, et al. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. (2004) 279:1304–09. doi: 10.1074/jbc.M310389200
96. Parker-Duffen JL, Nakamura K, Silver M, Kikuchi R, Tigges U, Yoshida S, et al. T-cadherin is essential for adiponectin-mediated revascularization. J Biol Chem. (2013) 288:24886–97. doi: 10.1074/jbc.M113.454835
97. Philippova M, Ivanov D, Joshi MB, Kyriakakis E, Rupp K, Afonyushkin T, et al. Identification of proteins associating with glycosylphosphatidylinositol- anchored T-cadherin on the surface of vascular endothelial cells: role for Grp78/BiP in T-cadherin-dependent cell survival. Mol Cell Biol. (2008) 28:4004–17. doi: 10.1128/MCB.00157-08
Keywords: T-cadherin, endothelial cells, tumor angiogenesis, tumor, VEGF
Citation: Wen Y, Ma L, Liu Y, Xiong H and Shi D (2025) Decoding the enigmatic role of T-cadherin in tumor angiogenesis. Front. Immunol. 16:1564130. doi: 10.3389/fimmu.2025.1564130
Received: 21 January 2025; Accepted: 14 March 2025;
Published: 31 March 2025.
Edited by:
Constantinos Mikelis, University of Patras, GreeceReviewed by:
Florin Zugun-Eloae, Grigore T. Popa University of Medicine and Pharmacy, RomaniaCopyright © 2025 Wen, Ma, Liu, Xiong and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongmei Shi, c2hpZG9uZ21laTI4QDE2My5jb20=; Huabao Xiong, eGlvbmdoYmxAeWFob28uY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.