 Xiaoying Lin
Xiaoying Lin Xiang Li
Xiang Li Zhifang Zhai
Zhifang Zhai Mingwang Zhang*
Mingwang Zhang*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 April 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1563286
Autoimmune Bullous Diseases (AIBDs), characterized by the formation of blisters due to autoantibodies targeting structural proteins, pose significant therapeutic challenges. Current treatments, often involving glucocorticoids or traditional immunosuppressants, are limited by their non-specificity and side effects. Cytokines play a pivotal role in AIBDs pathogenesis by driving inflammation and immune responses. The JAK-STAT pathway is central to the biological effects of various type I and II cytokines, making it an attractive therapeutic target. Preliminary reports suggest that JAK inhibitors may be a promising approach in PV and BP, but further clinical validation is required. In AIBDs, particularly bullous pemphigoid (BP) and pemphigus vulgaris (PV), JAK inhibitors have shown promise in modulating pathogenic cytokine signaling. However, the safety and selectivity of JAK inhibitors remain critical considerations, with the potential for adverse effects and the need for tailored treatment strategies. This review explores the role of cytokines and the JAK-STAT pathway in BP and PV, evaluating the therapeutic potential and challenges associated with JAK inhibitors in managing these complex disorders.
Autoimmune bullous diseases (AIBDs) encompass a spectrum of conditions characterized by the formation of vesicles, blisters, erosions, excoriations, and erythemas on the skin and/or mucosal membranes, which can lead to serious complications and even death due to superinfections, loss of body fluids and severely limited food intake (1). AIBDs can be broadly classified into two categories, pemphigus (intraepidermal blistering) and pemphigoid (subepidermal blistering), depending on the location of the blisters (2, 3). Pemphigus disorders are caused by autoantibodies directed against the desmosomal proteins desmoglein (Dsg) 1 and 3 and mainly include pemphigus vulgaris (PV), pemphigus foliaceus (PF), paraneoplastic pemphigus (PNP) and immunoglobulin A (IgA) pemphigus (4–6). In contrast, pemphigoid disorders are triggered by autoantibodies targeting various autoantigens within or underneath the basement membrane zone (BMZ), such as BP180, BP230, laminins and type VII collagen. This group mainly includes bullous pemphigoid (BP), linear IgA bullous dermatosis, dermatitis herpetiformis (DH), mucous membrane pemphigoid (MMP), anti-p200 pemphigoid (targeted laminin γ1) (7),epidermolysis bullosa acquisita (EBA), bullous systemic lupus erythematosus (BSLE) and herpes gestationis (8).
The treatment of AIBDs is predominantly involve glucocorticoids and immunosuppressive agents, which have serious adverse effects with prolonged use and whose efficacy is highly heterogeneous across patients (9, 10). In recent years, more precisely immunotherapy that targets cytokines or pivotal proteins extracellularly, such as rituximab against CD20, omalizumab against IgE, and dupilumab against IL-4Rα, are increasingly being utilized for the treatment of AIBDs that is unresponsive to glucocorticoids or immunosuppressants, and have demonstrated promising outcomes (11, 12). While these therapies offer benefits, some patients still exhibit unresponsive, which leads to mortality rates for patients with AIBDs remaining significantly higher as compared with the general population (13, 14). Hence, new strategies that target intracellular pathways activated by cytokines warrant consideration in the future.
Cytokines are a group of structurally distinct secreted proteins that bind to cellular receptors belonging to at least seven superfamilies which exert their biological effects through very different signaling pathways. Among them, Janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathways are required for the effective responses of innate and adaptive immune by regulating the signaling cascade of type I and type II cytokines (15). Dysfunction of the JAK-STAT signaling pathway has been linked to a range of inflammatory and autoimmune diseases, and JAK inhibitors have emerged as a promising therapeutic approach (16). Indeed, over the past decade, several small-molecule JAK inhibitors have been approved for the treatment of immune-mediated diseases such as rheumatoid arthritis (RA), psoriatic arthritis, ankylosing spondylitis, atopic dermatitis (AD), alopecia areata (AA) and others (17). In contrast to JAK inhibitors, until now, STAT inhibitors have not been marketed due to the structural peculiarities and functional complexities of STAT, which are still in the clinical research stage and are primarily concentrated in the field of cancer (18). This review will elucidate the present recognition of JAK-STAT signaling and the pathway-dependent type I/II cytokines in immune homeostasis. Furthermore, we describe the involvement of the cytokine network and JAK-STAT pathway in the pathogenesis of PV and BP, and discuss advancements in JAK-targeted therapeutics for these conditions. Finally, we explore the safety and selectivity in the clinical implementation of the JAK inhibitors.
The JAK family comprises highly conserved mammalian protein non-receptor tyrosine kinases, which include JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2). JAKs are ubiquitously expressed, with the exception of JAK3, which is predominantly found in immune cells (19). The canonical JAK/STAT signaling pathway is as follows (Figure 1): JAKs are noncovalently associated with the cytoplasmic domain of the cytokine receptors’ signaling chain. The binding of type I and II cytokines to the receptor induces dimerization or aggregation of the receptor’s signaling chains. This process brings JAKs into close proximity, leading to autophosphorylation or transphosphorylation. The activated JAKs then phosphorylate the associated receptor on specific tyrosine residues. The phosphorylated receptor, in turn, recruits a STAT protein (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, or STAT6), which is also phosphorylated by the activated JAK on its conserved C-terminal tyrosine residue. This event triggers the STAT protein’s homodimerization or heterodimerization and its subsequent translocation to the nucleus (20), where it functions as a transcription factor, regulating the expression of genes related to immune cell growth, proliferation, differentiation, and apoptosis (21–23). In addition to canonical signaling, studies have reported that JAK-STAT is also engaged in noncanonical signal transduction, which is relatively more complicated (24, 25). For example, unphosphorylated STAT3 could also bind to DNA and act as a transcriptional activator (26).
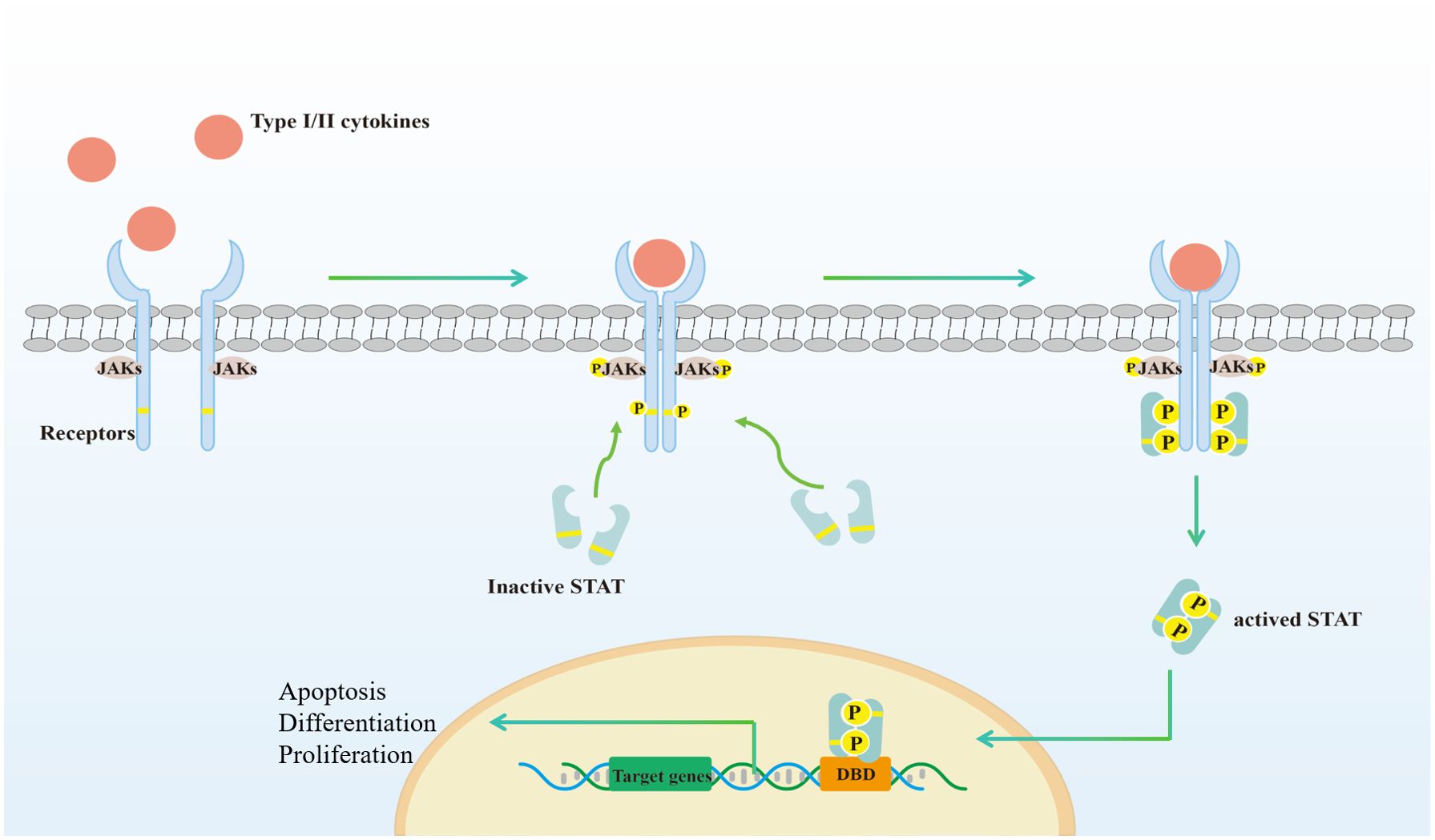
Figure 1. Activation of the canonical JAK-STAT signaling pathways. (1) Type I//II cytokines act through receptors associated with JAK. The receptors comprise at least two chains, each linked to a specific JAK; (2) Binding of ligand dimerizes the receptor, resulting in phosphorylation and activation of JAK for each other, which then phosphorylates the receptor. The STAT family has an N-terminal structural domain that allows STAT to form inactive dimers; (3) STAT bind to the phosphorylated receptor, which in turn phosphorylates by JAK; (4) STAT detach from the receptor to form activated dimers; (5) STAT dimers enter the nucleus, bind to DNA binding domain (DBD) and regulate transcription, which is involved in cell proliferation, differentiation and apoptosis. Created with Adobe Illustrator2023.
The JAK-STAT pathway is closely related to immune homeostasis and the development of autoimmune diseases and is thus subject to intricate regulation. This regulatory network includes three main negative regulators: a variety of protein tyrosine phosphatases (27–29), suppressors of cytokine signaling (SOCSs) (30–32), and protein inhibitors of activated STATs (PIASs) (33–35), as well as multiple positive regulators (e.g., cooperating transcription factors (36)). Mutations in genes encoding JAK or STAT proteins are associated with a variety of immune-related diseases. For example, inactivating mutations in JAK3 can cause severe combined immunodeficiency (SCID) (37, 38), while loss-of-function mutations in TYK2 result in milder immunodeficiency (39). Similarly, gain-of-function mutations in STAT1 are linked to a spectrum of infectious and autoimmune diseases (40), whereas loss-of-function in STAT3 leads to hyper-IgE syndrome, characterized by recurrent skin and pulmonary infections, elevated IgE levels, and chronic eczematous dermatitis (41). Thus, a substantial body of evidence indicates that abnormal activation of the JAK-STAT pathway is pivotal in the pathogenesis of human immune diseases.
Cytokines are classified into superfamilies based on the shared structural features of their cognate receptors. More than 50 cytokines, including interleukins (ILs), interferons (IFNs), hormones, and colony-stimulating factors (CSFs), are members of the type I/II cytokine families and signal via the JAK-STAT pathway. These cytokines can be further subdivided into groups based on their shared receptor subunits (Table 1).
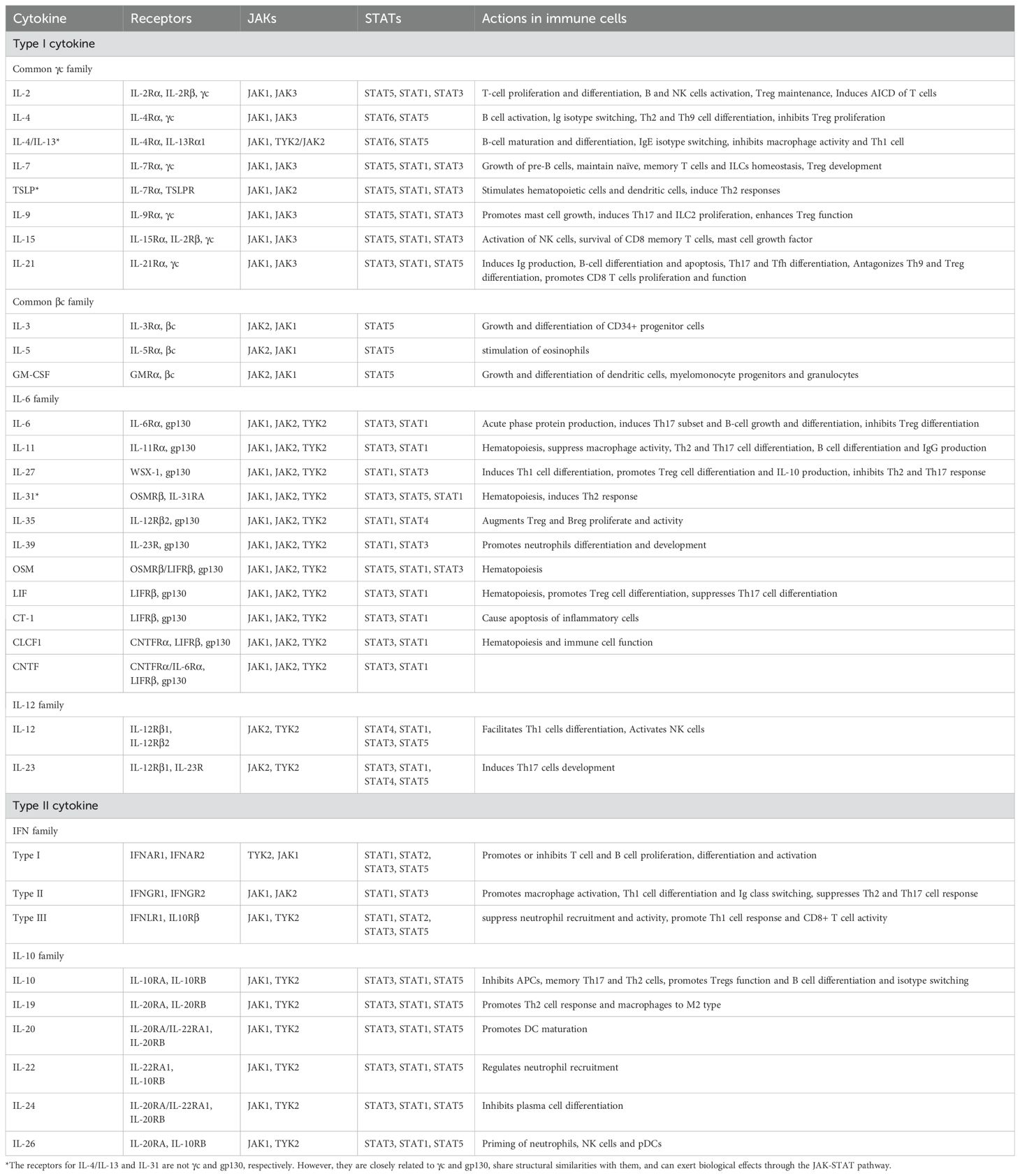
Table 1. Type I/type II cytokines, their receptors, JAK kinases, STAT proteins, and function.
The common γc family cytokines include IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. A mutation in the gene encoding the protein γc has been associated with SCID in humans, indicating that these cytokines play a fundamental role in the development of the immune system. The inaugural member of this family identified was IL-2, originally discovered as a T cell growth factor in 1976 (42). IL-2 transmits signals through JAK1 and JAK3, which covalently bind to the IL-2Rβ and γc, respectively (43), and predominantly recruit and activate STAT5A and STAT5B, as well as to a lesser extent, STAT3 and STAT1 (44). IL-2 has been shown to exhibit pleiotropic actions that are wide-ranging and significant (45). Specifically, IL-2 is known to promote the growth and differentiation of B cells (46), augment the proliferation of natural killer (NK) cells while enhancing their cytotoxicity (47). Additionally, IL-2 is essential for the development and expansion of T regulatory (Treg) cells (48), and for activation-induced cell death of T cells (49), which helps mediate tolerance and limit inappropriate immune responses. Furthermore, it aids in the differentiation of naïve CD4+ T cells into T helper 1 (Th1) (50), Th2 (51) and Th9 cells (52), while inhibiting the differentiation of Th17 (53) and T follicular helper (Tfh) cells (54).
IL-4 was originally discovered as a cytokine that stimulates B cell activation and increases the production of immunoglobulin class switch, resulting in elevated levels of IgG1 and IgE (55, 56). Further studies have shown that IL-4 plays a key role in driving Th2 responses (57) and promoting Th9 differentiation, while simultaneously suppressing the generation of transforming growth factor-β (TGF-β)-induced Foxp3+ Treg cells (58). IL-4 signals through two types of receptors: type I and type II. Type I IL-4R, expressed on hematopoietic cells, consists of IL-4Rα and γc subunits (59, 60) and is linked to JAK1 and JAK3, respectively (61). The type II IL-4R, composed of the IL-4Rα and IL-13Rα1, is primarily expressed on non-hematopoietic cells and is linked to JAK1 and TYK2/JAK2, which is also the functional receptor for IL-13 (62, 63). Both type I and type II IL-4 receptors mainly activate STAT6 and, to a lesser extent, STAT5 (64, 65).
IL-7 was first identified as a stromal-cell-derived factor that facilitates the growth of pre-B cells (66, 67). Apart from its role in pre-B cell development, IL-7 and its receptor are crucial for maintaining the development of T cells and innate lymphoid cells (ILCs) and protecting the survival of long-term memory T-cell (68, 69). Additionally, IL-7 contributes to the development of Treg cells (70). The IL-7 receptor is comprised of two subunits, IL-7Rα and γc (71, 72), which bind to JAK1 and JAK3, respectively, and initiate downstream signaling, most notably STAT5A and STAT5B (73). IL-7Rα is also a functional component of the receptor for thymic stromal lymphopoietin (TSLP), along with another component called TSLPR (74). TSLP mediates STAT5, STAT3 and STAT1 phosphorylation via kinases JAK1 and JAK2 (75), and is crucially important for stimulating hematopoietic cells and mediating type 2 immunity (76).
IL-9 was initially identified in mice as a cytokine that promotes the proliferation of T cells (77). Upon binding to its heterodimeric receptor composed of the IL-9Rα and γc, IL-9 triggers the cross-phosphorylation of JAK1 and JAK3, leading to the activation of STAT5, STAT3, and STAT1 (78, 79). Beyond its role in T cell growth, IL-9 acts as a growth factor for bone marrow mast cells and promotes the secretion of several cytokines, including IL-13 (80, 81). In addition, IL-9 facilitates the expansion of Th17 cell and ILC2 populations while enhancing the regulatory function of Treg cells (82, 83).
IL-15 was initially identified as a growth factor for T-cells (84). IL-15 binds to a heterotrimeric receptor composed of IL-15Rα, IL-2Rβ, and γc (85). IL-15 binds to IL-15Rα on one cell and then trans-presents the cytokine to neighboring cells that express IL-2Rβ and γc, followed by activating the JAK1/JAK3 and STAT5 pathways (86), and plays a crucial role in the development of NK cells 86 and the maintenance of memory CD8+ T cells 87. Additionally, it has been found to act as a growth factor for mast cells (87).
IL-21 was first discovered as the ligand for an orphan type 1 cytokine receptor, which was found to bear a striking resemblance to IL-2Rβ (88, 89). Upon binding to its receptor, IL-21 stabilizes the complex between the IL-21Rα and the common γc (90). This event consequently activates JAK1 and JAK3, thereby facilitating the recruitment and activation of STAT proteins, with STAT3 being the predominant species, followed by STAT1 and STAT5 (91, 92). In the immune system, IL-21 has the ability to support the production of IgG1 and simultaneously repress the production of IgE (93). Additionally, IL-21 is capable of driving B cells toward plasma cell differentiation, while also promoting apoptosis of incompletely activated B cells (94, 95). Furthermore, IL-21 collaborates with IL-7 or IL-15 to expand CD8+ T cells (96) and contributes to the formation of CD8+ T cell memory (97). IL-21 also plays a role in the differentiation of Tfh (98) and Th17 cells (99), but inhibits the differentiation of Treg (100) and Th9 cells (52).
The βc family of cytokines, comprising IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF), was initially identified as CSFs in the hematopoietic system. However, they are now recognized as pleiotropic in the immune system (101, 102). The functional receptors for IL-3, IL-5, and GM-CSF are IL-3α paired with βc, IL-5α with βc, and GM-CSFα with βc, respectively (103). Upon binding to their specific receptor-alpha chains, the cytokine-alpha chain binary complexes dimerize with βc to form heterodimers. This process activates JAKs, primarily JAK2, which bind to the cytoplasmic tail of the βc chain, ultimately leading to the phosphorylation of STAT5A/B for downstream signaling (104).
βc cytokines possess the capability to influence various types of cells in the hematopoietic system. For instance, IL-3 is involved in the growth and differentiation of CD34+ progenitor cells into basophils and mast cells, DCs, eosinophils, and monocytes-macrophages (105, 106). Recent studies suggest that IL-3 modulates type 1 DC function to induce Th2 responses (107). IL-5, primarily responsible for stimulating eosinophils, is essential for eosinophilic inflammation (108, 109). GM-CSF acts on DCs, myelomonocyte progenitors, and granulocytes (110) while inhibiting CD34+ progenitor cell differentiation into lymphoid progenitors or type 2 DCs, as well as the terminal differentiation of mast cells (111, 112).
The IL-6 family of cytokines encompasses a diverse group, including IL-6, IL-11, IL-27, IL-31, IL-35, IL-39, oncostatin M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), cardiotrophin 1 (CT-1) and cardiotrophin-like cytokine factor 1 (CLCF1). The unifying feature of this family is their shared reliance on the gp130 receptor signaling subunit, which is pivotal for their classification (113).
The cytokines within the IL-6 family utilize several distinct receptor arrangements. IL-6 and IL-11 engage a specific non-signaling receptor subunit (IL-6Rα and IL-11Rα, respectively) in addition to the signal-transducing receptor gp130, forming either an IL-6–IL-6Rα–gp130 or IL-11–IL-11Rα–gp130 hexameric complex for signal transduction (114). Conversely, the receptor complexes for LIF, CT-1, OSM, IL-27, IL-35 and IL-39 comprise gp130 and a second signal transducing receptor subunit (LIFRβ, OSMRβ, WSX-1, IL-12Rβ2 and IL-23R, respectively), which share structural similarities with gp130 (115). The receptor for CNTF and CLCF1 comprises three individual receptor subunits, including a non-signaling receptor subunit CNTFRα and the heterodimer gp130 (LIFRβ and gp130) (116). The only cytokine exception to this “gp130 rule” is IL-31, which binds to a receptor complex containing OSMRβ and a unique gp130-like receptor chain, designated as IL-31RA (117). The cytokine receptor complexes within the IL-6 family transmit signals inside cells through the activation of JAK1, and to a lesser extent JAK2, or TYK2 (118, 119). These kinases are covalently bound to the cytoplasmic domains of their respective signal-transducing receptors. Upon activation, JAK proteins phosphorylate and thereby activate latent transcription factors, including STAT1, STAT3, and STAT5 (120, 121). The specific STAT proteins activated vary with the cytokine, correlating with the diverse biological functions of the IL-6 family cytokines. For example, IL-27 preferentially induces STAT1 activity, while IL-6 predominantly enhances STAT3 transcriptional activity (122). A summary of the specific immune functions of individual cytokines is presented in Table 1.
The IL-12 cytokine family, comprising IL-12 and IL-23, shares structural similarities with the IL-6 family. These cytokines leverage orthologs of the gp130 receptor subunit to activate the JAK-STAT signaling pathway (123). IL-12 was first identified as a protein released by a specific human lymphoblastoid cell line that activates NK cells and T cells to produce IFNγ (124). IL-12 forms a complex with two different subunits, namely IL-12Rβ1 and IL-12Rβ2 (125). Upon binding, this complex activates JAK2 and TYK2, which subsequently recruit and phosphorylate STATs, mainly STAT4 (126). The phosphorylated STATs form dimers, translocate to the nucleus, and regulate gene transcription, playing a pivotal role in immune homeostasis. IL-12, a pro-inflammatory cytokine, drives the differentiation of CD4+ T cells into Th1-like cells and stimulates IFNγ production (127). Additionally, IL-12 can enhance the release of IFNγ and TNF in CD8+ T cells, NK cells, and ILC1s (128).
IL-23 was discovered a decade after the identification of IL-12 and was first characterized for its effects on memory T-cells but not naïve T-cells (129). The IL-23 receptor is composed of the IL-12Rβ1 and IL-23R chains. Similar to IL-12, the binding of IL-23 leads to the activation of JAK2 and TYK2, culminating in the phosphorylation of STATs, primarily STAT3 (130, 131). Research indicates that IL-23 is instrumental in promoting the expansion and maintenance of Th17 cells, without influencing their differentiation (132).
Since the discovery of the first IFN in 1957, based on its antiviral activity, more than 20 signaling molecules belonging to the IFN family have been identified (133). IFNs can be classified into three distinct classes of cytokines: type I IFNs, type II IFNs, and type III IFNs. The type I IFN family encompasses IFN-α (which can be further divided into 13 subtypes), IFN-β, IFN-δ, IFN-ϵ, IFN-κ, IFN-τ, and IFN-ω. All type I IFNs bind to a common receptor, composed of two subunits, IFNAR1 and IFNAR2, that interact with JAK1 and TYK2, respectively. Activation of JAKs rapidly phosphorylates STAT1, STAT2, STAT3, or STAT5, which then form homodimers or heterodimers, translocate to the nucleus, and initiate transcription (134). Beyond their direct antiviral effects, type I IFNs exhibit a range of immunomodulatory mechanisms. These include the promotion or inhibition of T-cell proliferation, differentiation, and apoptosis, contingent upon the timing of T cell receptor stimulation (135) and the duration of IFN exposure (135, 136). Type I IFNs promote B-cell activation and Ig class switching (137) but also inhibit B-cell growth and proliferation (138, 139), depending on different environments and antigens. These diverse biological effects may be mediated by the recruitment of different STATs (140).
In contrast to type I IFNs, type II IFNs consist of a single cytokine, IFN-γ (141). An IFN-γ homodimer binds to two IFNGR1 receptor chains and subsequently recruits two IFNGR2 chains to form a complex. The aggregation of its receptor components by IFN-γ leads to the phosphorylation of JAK1 and JAK2, which constitutively bind to IFNGR1 and IFNGR2, respectively. The activated JAKs further phosphorylate the STAT1 docking site in IFNGR1, followed by the recruitment and phosphorylation of STAT1. The phosphorylated STAT1 then dimerizes and translocates to the nucleus, where it regulates gene transcription (142). In addition to its antiviral activity, IFN-γ possesses pleiotropic immunologic functions, including the promotion of macrophage activation, enhancement of antigen presentation, induction of Th1 cell differentiation while concomitant inhibition of Th2 and Th17 cell responses, and promotion of Ig isotype switching (143).
Type III IFNs were discovered almost 50 years after the identification of type I IFNs and comprise four subtypes, IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4 (144, 145). All type III IFN cytokines signal through a shared heterodimeric receptor, composed of IFNLR1 and IL10Rβ (146, 147). Like type I interferons, ligand binding activates JAK1 and TYK2, as well as downstream STATs transcription factors, primarily STAT1 and STAT2 (148). Although the downstream signaling pathways and transcriptional responses activated by type III IFNs are remarkably similar to those of type I IFNs, type III IFNs also display unique roles in the immune response. For example, type III IFNs protect epithelial cells from viral, bacterial, and fungal infections (149). In terms of immune cells, type III IFNs inhibit neutrophil recruitment and activity (150) and promote Th1 cell responses and CD8+ T cell activity (151).
The IL-10 family comprises IL-10 and five members of the IL-20 subfamily: IL-19, IL-20, IL-22, IL-24, and IL-26 (152). IL-10 is a cytokine that plays a crucial role in regulating the immune system. It signals through its heterodimeric receptor, which consists of two subunits: IL-10RA and IL-10RB (153, 154). The receptor complex associates with two JAKs, JAK1 and TYK2, respectively (155). Upon binding to the receptor, IL-10 primarily activates STAT3, although it can also activate STAT1 and STAT5 in specific cell types (156). This leads to suppressive effects on myeloid cells by inhibiting proinflammatory cytokines and antigen-presenting cells (APCs). Furthermore, IL-10 directly inhibits memory Th17 and Th2 cells, while promoting the survival and function of Tregs and the differentiation and isotype switching of B cell (157, 158).
The IL-20 subfamily of cytokines signals through various types of receptors. Specifically, IL-20RB, the common β chain, can form a functional heterodimeric receptor with either the IL-20RA, enabling binding of IL-19, IL-20, and IL-24, or with the IL-22RA1, which only allows signaling of IL-20 and IL-24. Conversely, IL-10RB, the other common β chain, heterodimerizes with either IL-22RA1 or IL-20RA to create a functional receptor for IL-22 or IL-26, respectively (159). All IL-20 subfamily cytokines transmit signals through the JAK-STAT pathway. Notably, all family members preferentially phosphorylate JAK1 and TYK2, which in turn primarily activate STAT3 (160). The IL-20 subfamily of cytokines possesses partially similar biological activities, such as promoting epidermal integrity and innate defense (161). However, each cytokine has distinctive functions. For instance, IL-19 is responsible for polarizing T-lymphocytes to Th2 type and macrophages to M2 type (162), whereas IL-20 is involved in the promotion of DC maturation (163). IL-20 has been found to regulate neutrophil recruitment (164) while IL-24 could stimulate peripheral blood mononuclear cell to produce pro-inflammatory cytokines and inhibit plasma cell differentiation (165, 166).
In addition to the aforementioned cytokines closely related to the JAK-STAT pathway, other cytokines, such as TNF-α, IL-1 or IL-17 family, etc., although not signaling directly through the JAK-STAT pathway, are influenced by JAK-STAT-dependent cytokines in terms of expression and biological function due to cytokine network effects (15). These further underscores the importance of the JAK-STAT pathway in immune homeostasis and immune-mediated diseases.
PV is a severe organ-specific AIBD. The underlying cause is the production of autoantibodies that attack the essential proteins Dsg1 and/or Dsg3. This attack results in the loss of cell adhesion of keratinocytes, a process known as acantholysis. This process ultimately leads to the formation of blisters and erosion of the skin and mucous membranes (4). Multiple factors, including drugs, environment, infections, and others, are known to initiate the formation of autoantibodies that expose Dsgs. Once exposed, these Dsgs are recognized by APCs such as DCs, macrophages, or B cells, which present them to T cells through the MHC II-antigen complex interaction. In susceptible individuals, T cells with autoreactive potential have evaded both central and peripheral clonal deletion. Upon recognition of the MHC II-self peptide ligand complex and in the presence of inflammatory mediators, these T cells are activated and differentiate into Th1, Th2, Th17, Tfh, or Treg. Ultimately, these cells secrete various cytokines that can drive B-cell proliferation, activation, pathogenic DSG-specific IgG antibody production, as well as differentiation into plasma cells and memory cells (Figure 2) (167).

Figure 2. Cytokine involvement in pemphigus vulgaris. Exposed desmogleins (Dsgs) identified by antigen-presenting cells such as dendritic cells and present the antigen to autoreactive naïve CD4+T cells. Subsequently, under the influence of various cytokines, these T cells differentiate into distinct subsets, secreting cytokines that predominantly exert their biological effects via the JAK-STAT pathway. Specifically, Th2 differentiation is enhanced, leading to the secretion of Th2 cytokines IL-4, IL-5, and IL-10, while simultaneously suppressing Th1 responses and downregulating Th1 cytokines such as IL-2 and IFN-γ. This shift may be associated with cytokines released by NK cells. Additionally, Th17 and Tfh cell differentiation is amplified, resulting in the secretion of IL-6, IL-17, IL-21, and IL-23. Notably, IL-6 and IL-17 promote inflammatory responses, IL-21 enhances B cell activation into plasma cells, and IL-4 and IL-10 facilitate antibody class switching, prompting plasma cells to secrete IgG4 antibodies against Dsgs, culminating in acantholysis. Furthermore, a reduction in Treg cell differentiation is crucial for the proliferation of autoreactive T cells and antibody production. Figure image created with Adobe Illustrator2023.
Th1 cells are primarily responsible for secreting IFN-γ and IL-2, which play a crucial role in cellular immunity against intracellular microorganisms. In contrast, Th2 cells secrete cytokines such as IL-4, IL-5, IL-10, and IL-13, which are associated with host immunity to extracellular pathogens and the development of allergic diseases (168). Early studies suggest that PV pathogenesis involves both Th1 and Th2 cells. During the active phase of PV, DSG3-reactive Th2 cells secreting IL-4 are predominant. Conversely, during the chronic phase, DSG3-reactive Th1 cells secreting IFN-γ become more prevalent (169). This pattern correlates with the production of IgG4 antibodies during active disease and IgG1 antibodies during clinical remission. Additionally, there is a significant correlation between the presence of IgG1 and IgG4 antibodies against Dsg3 and the ratio of Dsg3-reactive Th1/Th2 cells (170). However, recent evidence has highlighted the critical role of Th2 cells in the development of PV. By constructing mouse or human Dsg-3 reactive T cell lines, researchers have found that Dsg-3 reactive T cells predominantly secrete Th2 cytokines IL-4 and IL-10 both in vivo and in vitro (171, 172). Further studies on PV patients have shown that the number of DSG-reactive Th2 cells is positively correlated with Dsg-3 antibody titer and disease activity (173). Moreover, the expression of Th2 cytokines such as IL-4, IL-5, and IL-10 was significantly increased during the active phase of the disease but decreased after disease control (174, 175). Dupilumab, a fully human monoclonal antibody that simultaneously blocks IL-4 and IL-13 signals by targeting IL-4Rα, is anticipated to be a valuable addition to the therapeutic intervention for PV (176), as demonstrated in several case reports in recent years (177–179).
Upon differentiation of the naïve T cells into Th2, they release Th2 cytokines that impede the Th1 response and down-regulate the production of Th1 cytokines (180). This phenomenon has been substantiated in patients with PV. The study conducted by Satyam et al. has verified that PV patients exhibited elevated levels of serum Th2 cytokines (IL-4 and IL-10) and reduced levels of Th1 hallmark cytokines (IFN-γ and IL-2) (174). Analogously, other studies have also affirmed that patients with PV exhibit reduced levels of serum IFN-γ and heightened levels of IL-4. This suggests that during the active phase of the disease, the Th2 response inhibits the Th1 response (181, 182). The underlying reason might be attributed to the functional alterations in NK cells in PV patients. Studies have demonstrated that the peripheral blood of PV patients contains an increased number of NK cells, while the IL-12 signaling pathway is impaired, and the expression of IL-5 and IL-10 is elevated in NK cells, thereby promoting a predisposition toward a Th2 type immune response in PV patients (183).
Upon exposure to cytokines TGF-β, IL-23, and IL-6, T cells undergo differentiation into Th17 cells, characterized by the secretion of a range of cytokines including IL-17, IL-21, IL-22, IL-26, and GM-CSF. Several studies have reported a significant increase in the number of Th17 cells in the peripheral blood and skin lesions of PV patients compared to the general population (184–186). Yang et al. (187) found that in PV skin lesions, the predominant T cell population was composed of CD4+ T cells expressing IL-21 and IL-17A, rather than classical Tfh cells expressing CXCR5. They speculate that these IL-21+/IL-17+ CD4+ T cells may contribute to the activation and differentiation of B cells, ultimately leading to the production of pathogenic autoantibodies in PV lesions. Additionally, another study discovered a greater number of CD154+ CD4+ T cells in the peripheral blood of PV patients, which highly expressed IL-17 and IL-21 and positively correlated with the level of Dsg-3 titer (188). Analyses for Th17-related cytokines revealed significantly elevated serum levels of IL-17, IL-21, and IL-23, but decreased levels of IL-22 in patients with PV compared to normal controls (189–191), and similar results were obtained in PV lesions (186, 192). Andrés et al. (193) reported the treatment of a PV patient with ustekinumab, an inhibitor of the p40 subunit of IL-12 and IL-23, after one month, the patient showed significant improvement in clinical symptoms and reduced serum levels of IL-12, IL-17, IFN-γ, and IL-6 without any adverse effects. However, as the treatment continued, a complete relapse of clinical symptoms was observed.
Tfh cells are a specialized subset of CD4+ T cells that localize to lymphoid follicles. Under the influence of cytokines such as IL-6, IL-21, and IL-27, naïve T cells induce the expression of Bcl-6 through the activation of transcription factors STAT3 or STAT4, thereby promoting differentiation along the Tfh lineage. Tfh cells can secrete cytokines like IL-21, IL-6, and IL-10, which support the survival and proliferation of B cells in the germinal center. This process ultimately drives B cell differentiation into plasma cells, immunoglobulin class-switching, and affinity maturation of antibodies (194). Tina et al. (195) found that IL-27 levels in the plasma of PV patients were positively correlated with Dsg-specific autoantibodies, as well as a significant increase in the frequency of Tfh cells and IL-21 levels, a cytokine produced by both Th17 and Tfh cells. Two other recent studies have also confirmed that Tfh cell frequencies are increased in PV patients and are strongly correlated with Dsg antibody levels, and that serum levels of cytokines such as, IL-6, and IL-21 are also significantly elevated (196, 197). However, there have been no reports on the function of IL-21 and the effects of anti-IL-21 therapy in PV.
Previous studies have detected Dsg3-specific T cells in healthy individuals carrying PV-associated HLA class II alleles (198, 199), However, these genetically susceptible individuals do not manifest PV phenotypes, which is attributed to the presence of IL-10- and TGF-β-secreting Dsg-3-specific Treg cells in this population—a cell type rarely found in PV patients (200). Naive T cells, upon stimulation by IL-2 and TGF-β, differentiate into Treg cells, which predominantly secrete TGF-β and IL-10 for immunosuppressive and immunomodulatory functions (201). Several studies have indicated that the proportion of Treg cells in the peripheral blood of PV patients is significantly reduced (202, 203) and negatively correlates with the number of Th17 cells (185, 191), which the latter is significantly increased in PV patients. Increasing Treg cell numbers through adoptive transfer or using the superagonistic anti-CD28 antibody D665 inhibits the production of Dsg-3 autoantibodies in a mouse model of PV, while Treg depletion enhances autoantibody production (204). A recent study found that Dsg3-specific CD4+ T cells could overcome peripheral tolerance in regulatory T cell depleted mice, inducing skin lesions, but not in wild-type mice (205). However, cytokine-level studies have found elevated levels of TGF-β and IL-10 in PV patients compared to normal subjects, suggesting the existence of other cellular sources of secretion besides Treg cells (174, 191, 206, 207). An explanation for this may relate to the pleiotropic nature of these cytokines; for instance, TGF-β contributes to the differentiation of Tregs while also promoting the development of Th17 cells. Similarly, IL-10 supports the differentiation of Tregs and regulatory B cells and acts as a key member of the Th2 cytokine family. Notably, PV patients have shown significant increases in both Th17 and Th2 responses.
IL-6, IL-8, TNF-α, and IL-1 are archetypal pro-inflammatory cytokines predominantly secreted by non-Th immune cells. Elevated levels of these cytokines have been observed in PV patients and are known to decrease post-treatment, aligning with disease activity (175, 206, 208–215). TNF-α and IL-1 are implicated in promoting keratinocyte acantholysis via C3 complement activation, plasminogen activator induction, or through independent mechanisms (211, 216). Notably, PV-susceptible mice lacking TNF-α and IL-1 receptors show attenuated disease upon passive transfer, underscoring the role of these cytokines (211). Emerging research highlights TNF-α’s capacity to compromise keratinocyte adhesion by upregulating ST18 in PV lesions (217). Encouragingly, TNF-α inhibitors, such as etanercept and infliximab, have been instrumental in managing refractory PV cases (218–221). Additionally, IL-6 receptor monoclonal antibodies have demonstrated therapeutic success in PV (222). However, the therapeutic potential of IL-8 and IL-1 monoclonal antibodies remains unexplored in PV.
In aggregate, cytokines including Th2 (IL-4, IL-5, IL-10), Th17 (IL-17, IL-21, IL-23, TGF-β), and other pro-inflammatory cytokines (IL-6, IL-8, TNF-α, IL-1) exhibit heightened expression in PV, with certain cytokines correlating positively with disease severity and autoantibody titers, and their diminished expression post-successful treatment. These cytokines have a significant role in promoting T-cell differentiation, B-cell maturation, and antibody production, indicating their crucial involvement in the pathogenesis of PV, rather than transcending a mere epiphenomenon during disease evolution. Despite the pronounced role of these cytokines, monoclonal antibodies targeting individual cytokines have not matched the anticipated therapeutic impact observed in conditions like psoriasis (223), AD (224), or RA (225). This divergence suggests a multi-cytokinetic interplay in PV, advocating for therapeutics targeting a constellation of cytokines. Notably, cytokines including IL-4, IL-5, IL-6, IL-10, IL-17, IL-21, and IL-23 exert their effects via the JAK-STAT pathway, with IL-8, TNF-α, IL-1, and TGF-β’s expression being modulated indirectly by this pathway. Consequently, JAK inhibitors emerge as a promising therapeutic contender for PV.
A research group scrutinized the expression levels of JAK3, STAT2, STAT4, and STAT6 proteins in skin and oral mucosal lesions of PV patients through immunohistochemistry. Their findings indicated a significant upregulation of these proteins in the lesions compared to those in healthy controls. The authors hypothesize that this upregulation correlates with the heightened levels of various cytokines in PV patients and propose that JAK/STAT proteins could emerge as novel therapeutic targets for PV (226, 227). Given that the activation of the JAK-STAT pathway is contingent upon the phosphorylation status of JAK and STAT proteins, and considering that cytokines implicated in PV primarily operate through STAT3 and STAT6, we assessed the levels of phosphorylated STAT3 and STAT6 in the skin lesions of PV patients and healthy controls. The results showed that pSTAT3 and pSTAT6 expression were significantly elevated in PV lesions (unpublished data).
Although clinical trials on JAK inhibitors for PV treatment are lacking, Tavakolpour et al. (228). regard tofacitinib, a JAK1/3-targeting small molecule inhibitor, as a potential therapeutic agent for refractory PV. Tofacitinib has been reported to inhibit a spectrum of cytokines from diverse cellular origins across a range of diseases, including IL-4, IL-5, IL-6, IL-10, IL-17, IL-21, IL-23, IL-8, TNF-α, and IL-1 (229–232), all of which are intricately linked to PV pathogenesis. Vander et al. (233). presented a case of PV with severe nail involvement, where a combination treatment of tofacitinib and rituximab led to a significant amelioration of skin and nail conditions within three weeks. The authors attribute the swift symptom improvement to tofacitinib, given rituximab’s protracted therapeutic onset. M Grace et al. recently reported the successful treatment of refractory oral mucosal lesions in a patient with PV using upadacitinib (234). Our team recently administered a treatment regimen of tofacitinib 5mg twice daily, in conjunction with prednisone 1mg/kg daily, to a patient with refractory PV. This treatment led to a noticeable improvement in the patient’s skin lesions and a decrease in corticosteroid reliance (unpublished data). These outcomes hint at tofacitinib’s potential as an efficacious drug for PV management. Nonetheless, further clinical trials are imperative to substantiate these findings. To date, no clinical applications of other JAK inhibitors in PV have been documented (Table 2).
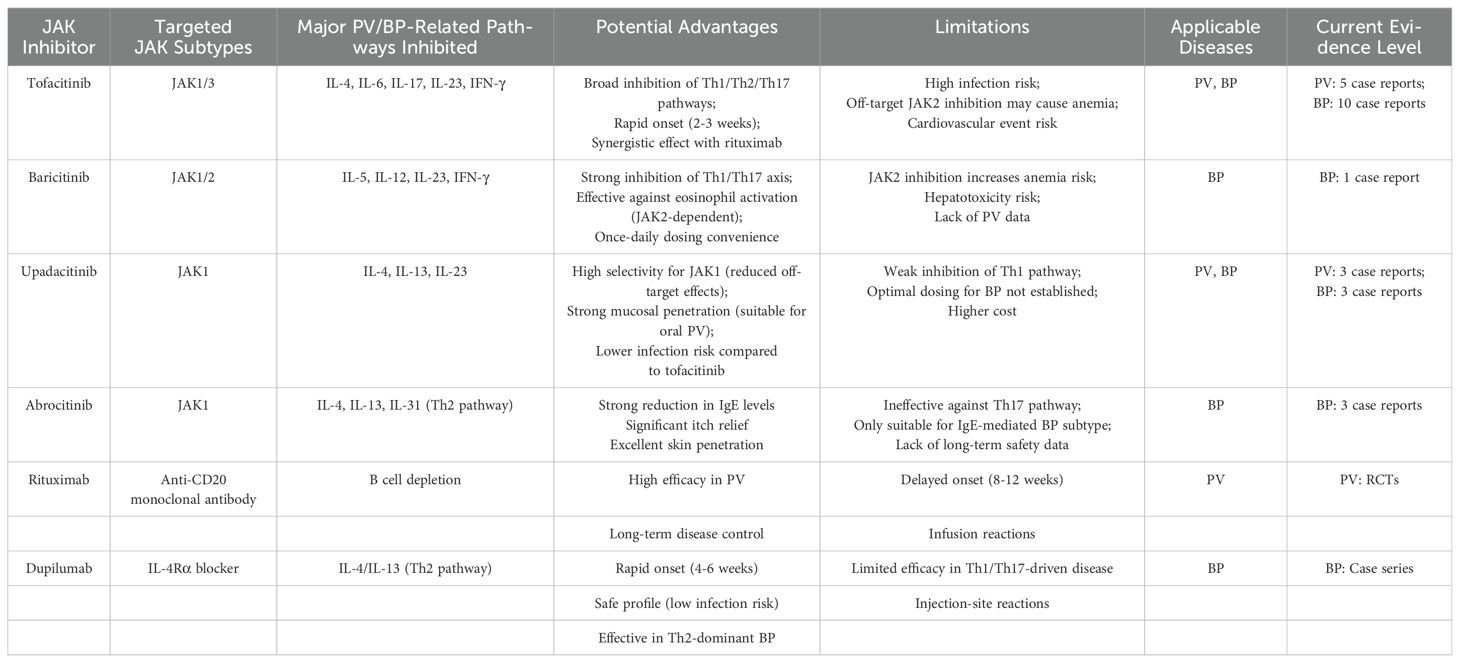
Table 2. Targeting characteristics of JAK inhibitors, rituximab, dupilumab and their applicability in PV/BP.
BP, akin to pemphigus, originates from a multifactorial interplay among patient-specific elements such as genetic susceptibility, regulatory Treg dysregulation, and aging, as well as external inciters like physical trauma, infections, malignancies, and medications. These elements may synergistically precipitate the collapse of immune tolerance to the BPAG protein, culminating in autoantibody production and disease manifestation (235). Specifically, DCs present a BPAG epitope via MHC class II, which subsequently activate BPAG-specific CD4+ T cells. These activated T cells further differentiate into distinct Th cell subsets, releasing a spectrum of cytokines encompassing IL-4, IL-13, IL-5, IL-6, IL-17, IL-21, and IL-31. This cytokine milieu fosters B cells differentiation and the production of IgG and IgE autoantibodies (236). The autoantibodies then bind to the basement membrane zone of the skin, instigating subepidermal blistering in BP via both complement-dependent and complement-independent mechanisms (Figure 3) (237, 238).
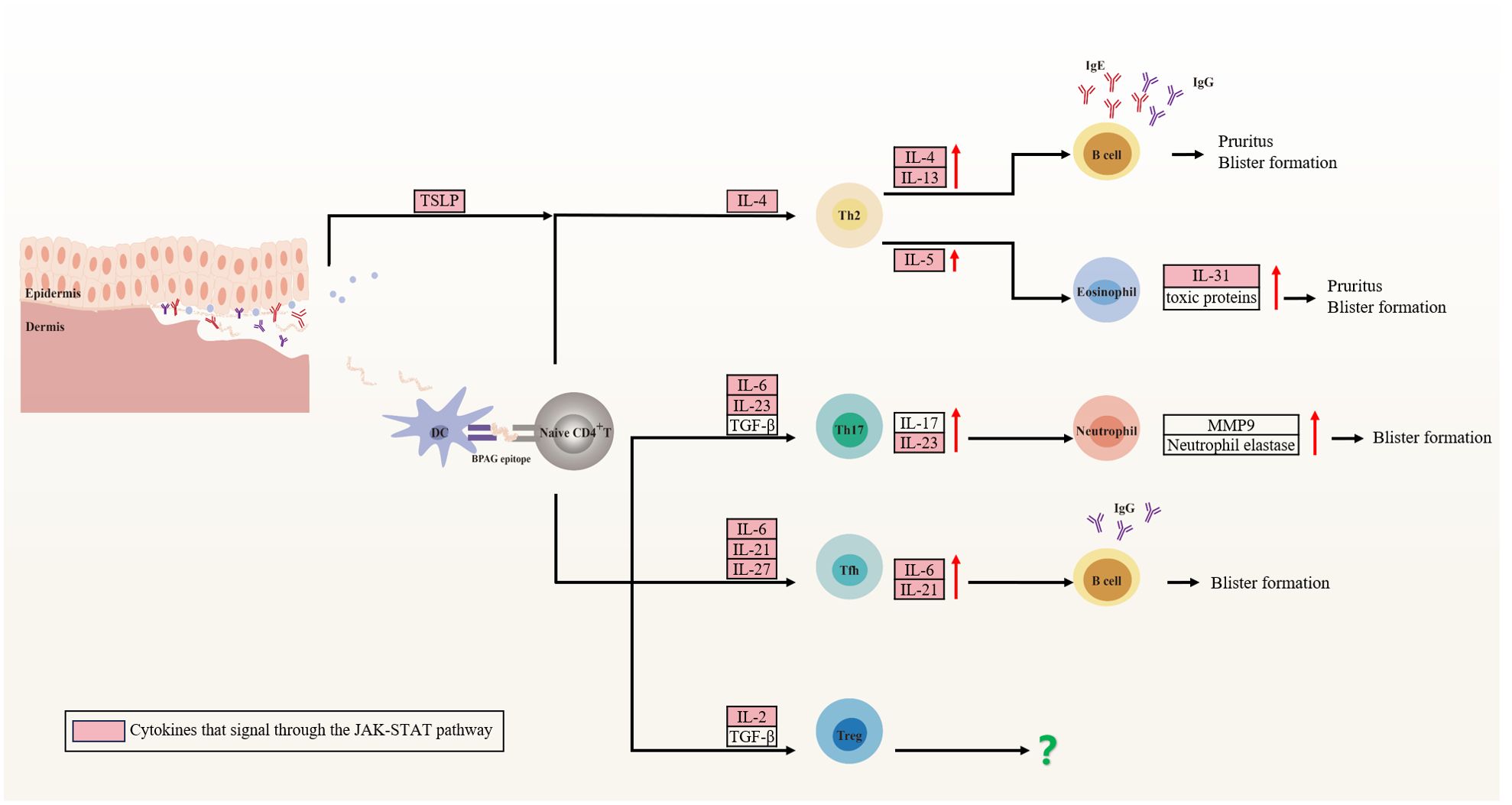
Figure 3. Cytokine involvement in bullous pemphigoid (BP). Patient-specific factors and external stimuli lead to the exposure of BPAG antigens, recognized and presented by dendritic cells to naïve CD4+ T cells. These cells differentiate into CD4+ T cell subsets that secrete cytokines inducing pruritus and vesicle formation, predominantly through the JAK-STAT pathway. Specifically, Th2 cells secrete IL-4 and IL-13, which regulates IgG and IgE isotype switching, while IL-5 enhances eosinophil accumulation and activation. Eosinophils secrete IL-31 and toxic proteins, contributing to local inflammation. Th17 cells produce IL-17 and IL-23, activating neutrophils that release neutrophil elastase and MMP-9, degrading the extracellular matrix and disrupting dermal-epidermal junctions. Tfh cells stimulate high-affinity autoantibody production by B cells via IL-21. However, the role of Treg cells in BP remains controversial. Figure image created with Adobe Illustrator2023.
In contrast to PV, BP is characterized by not only blister formation but also by the frequent presence of erythema, urticaria, eczematous-like rashes, and intense pruritus. This is due to the involvement of IgE and various innate immune cells, such as eosinophils and mast cells, in the pathogenesis of BP, indicating a close association between BP and type 2 inflammation. Indeed, Multiple studies have demonstrated elevated levels of type 2 inflammation-related cytokines, including IL-4, IL-5, IL-13, IL-31, and TSPL in BP patients (239–242).
IL-4 is particularly implicated in BP pathogenesis due to its role in Th2 cell differentiation and its facilitation of B cell immunoglobulin class switching to IgG1 and IgE (56). Pickford et al. reported that peripheral blood mononuclear cells from BP patients exhibit a significant increase in IL-4 secretion upon exposure to NC16A peptides (243). Furthermore, recent research has identified two major epitope peptides within BP180-NC16A that correlate with the induction of IL-4 production and autoantibody secretion in BP (244). IL-13, which shares functional similarities with IL-4 through its action on the IL-4Rα, is also implicated in BP. It synergizes with IL-4 to enhance B cell differentiation and IgE production (224). Researchers have reported the association between IL-13 gene polymorphism and the risk of BP (245, 246). In addition, IL-13 levels have been found to be positively correlated with the severity of pruritus in BP (247).
Due to their pivotal role in the pathogenesis of BP, several targeted therapeutic agents have been developed and are now in clinical use. Among these, dupilumab has garnered considerable attention. Since Alex et al. first reported the successful treatment of a refractory case of BP with dupilumab in 2018 (248), multiple case reports, case series, retrospective cohort studies, and systematic reviews have further substantiated its efficacy (249–252). These studies consistently demonstrate that dupilumab is a safe and effective therapeutic option, and its combined use with corticosteroids or immunosuppressants is recommended for the treatment of moderate to severe BP (253). Furthermore, a multicenter, randomized, double-blind, placebo-controlled clinical trial (NCT04206553) is underway, led by Dédée et al., to assess the efficacy and safety of dupilumab in adult patients with BP (254). Recently published data demonstrate that the dupilumab treatment group achieved all primary and secondary endpoints, including a significantly higher proportion of patients achieving complete remission, reduced disease severity, and alleviated pruritus compared to the control group, with no differential safety profile observed (255). Tralokinumab, a monoclonal antibody targeting IL-13, has also been documented in the successful treatment of a patient with BP complicated by end-stage kidney disease (256).
IL-5 serves as a pivotal cytokine for the maturation, survival, and functional activity of eosinophils, with its levels correlating with the severity of BP (257). In the presence of BP autoantibodies, IL-5 can activate eosinophils, which are implicated in the direct promotion of dermal-epidermal separation (258). Furthermore, another study has demonstrated that IL-5 is elevated in BP blister fluid, and is essential for the release of toxic proteins by eosinophils and the detachment of keratinocytes (259). Immunotherapies targeting the neutralization of IL-5 and its receptor have been documented in the treatment of, including mepolizumab, reslizumab, and benralizumab (260). A phase 2 double- blind trial (NCT01705795) of mepolizumab in BP demonstrated a significant reduction in peripheral blood eosinophil counts among treated patients. However, there were no significant differences in the proportion of patients remaining relapse-free or in the median time to relapse between the mepolizumab and placebo groups (261). For reslizumab, to date, two cases of refractory BP have been reported successfully managed with reslizumab, highlighting its efficacy in treating this condition (262, 263). In addition, recently, Arisa et al. reported a severe asthma patient developing BP during treatment with benralizumab. The authors hypothesized that BP may paradoxically arise through diverse mechanisms, even with therapeutic agents typically efficacious against the condition (264). A phase 3, double-blind clinical trial (NCT04612790) is currently evaluating benralizumab for BP treatment.
IL-31 serves as a principal inducer of pruritus and predominantly produced by eosinophils, is posited to exert immunomodulatory effects, specifically by promoting Th2-type immune responses (265). Elevated levels of IL-31 were detected in both lesional tissue and serum in BP patients (266, 267), and chemotactic and stimulated eosinophils to release reactive oxygen species and the chemokine CCL26 (268). While IL-31’s contribution to BP-associated itching is recognized, its role as the principal mediator of pruritus in BP remains to be definitively established. Nemolizumab, a monoclonal antibody directed against the IL-31RA, has shown efficacy in reducing pruritus in AD and prurigo nodularis (269). This suggests that nemolizumab may also be effective in the treatment of pruritus in BP, underscoring the need for further exploration of its therapeutic potential in this condition.
TSLP is a critical initiator of type 2 allergic responses, activating DCs, ILC2, naïve CD4+ T cells, and Th2 cells, and is implicated in the pathogenesis of itching (270). Elevated concentrations of TSLP have been reported in skin lesions, blister fluid, and sera of patients with BP (271–274). Dysfunction of BP180 has been shown to upregulate TSLP expression in keratinocytes, and this upregulation is strongly correlated with the severity of itch in a mouse strain. Moreover, the administration of an anti-TSLP neutralizing antibody could result in diminished scratching behavior, indicating the potential therapeutic efficacy of TSLP inhibition (271). Although no clinical trials have yet been conducted on TSLP inhibitors for BP treatment, Tezepelumab—a human monoclonal antibody targeting TSLP—is currently in trials for asthma and AD (275). Given its potential, Tezepelumab may emerge as an adjunctive therapy in BP treatment, offering a novel approach to alleviate pruritus and enhance the quality of life for patients.
The role of IL-17 and IL-23 in the pathogenesis of BP has been underscored by multiple studies demonstrating elevated levels of these cytokines in lesional skin, blister fluid, and serum of BP patients (276–280). Notably, persistently increased serum levels of IL-17 and IL-23 have been correlated with a higher risk of disease relapse post-treatment initiation (280). Genetic associations have also been identified, with two single-nucleotide polymorphisms (rs2201841 and rs7530511) in the IL-23R linked to BP (281). The absence of the NC14A domain of BP180 in mice triggers an autoimmune response against the cutaneous basement membrane, which is mitigated by anti-IL-17A treatment (282). IL17A-deficient mice exhibit resistance to autoantibody-induced BP, and pharmacological inhibition of IL-17A has been shown to reduce BP induction (283). These cytokines are implicated in BP development through the upregulation of MMP-9 and neutrophil elastase expression, processes that facilitate the separation of the dermis and epidermis (284, 285). Additionally, IL-17 and IL-23 have been shown to upregulate the expression of the glucocorticoid receptor-β, potentially contributing to glucocorticoid resistance in BP (286). Collectively, these findings suggest that the IL-17 axis plays a functional role in BP, and targeting this pathway with biologics presents a promising therapeutic strategy.
Indeed, targeting the IL-17/IL-23 axis has shown efficacy in patients with coexisting BP and psoriasis (287, 288). However, there are reports of new-onset BP in individuals treated with ustekinumab, secukinumab, or guselkumab for psoriasis, highlighting the complexity of cytokine-targeted therapies (289–291). A phase 2 open-label clinical trial (NCT03099538) investigating ixekizumab, a monoclonal antibody against IL-17A, did not meet its primary endpoint, as no cessation of blister formation within 12 weeks. Despite this setback, ongoing clinical trials are exploring the efficacy of biologics targeting IL-12/23 (Ustekinumab, NCT04117932) and IL-23 (Tildrakizumab, NCT04465292) in BP patients.
Tfh cells play a pivotal role in the generation of high-affinity autoantibodies by B cells within germinal centers through the secretion of IL-21 (292). The proportion of circulating Tfh cells and the plasma levels of IL-21 have been found to be markedly elevated and exhibit a positive correlation with both anti-BP180-NC16A autoantibody titers and disease severity in BP. In vitro studies have shown that the depletion of Tfh cells or the blockade of IL-21 can effectively suppress T cell-mediated B cell activation and the secretion of BP autoantibodies (293). Ohuchi et al. have reported an increase in both CXCL13, a chemokine critical for Tfh cell homing to germinal centers, and Tfh cells in the lesional skin and peripheral blood of BP patients, with a positive correlation to serum anti-BP180-NC16A titers (294). Furthermore, in STAT6-deficient scurfy mice, which are incapable of Tfh cell development, the Tfh cell population is markedly reduced, and these mice exhibit diminished production of autoantibodies against BP antigens (295). Collectively, these findings underscore the importance of the Tfh/IL-21 axis in the immunopathogenesis of BP and suggest that modulation of this pathway could represent a promising therapeutic approach for the disease.
The presence of Tregs and the cytokine IL-10 in peripheral blood and skin of BP patients is a subject of ongoing debate. Some studies have reported a decrease in the frequency of Tregs and IL-10 levels in BP, correlating with disease activity, while others have yielded conflicting results (240, 296–299). Nonetheless, it is established that Tregs are crucial for maintaining peripheral immune tolerance by suppressing the activation and proliferation of autoreactive T cells. In both human conditions such as immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, and in animal models like scurfy mice, Treg deficiency is associated with the spontaneous generation of autoantibodies against BP180 and BP230, suggests a significant role for Tregs in the regulation of autoantibody production in BP (295, 300). However, the precise role of Tregs in the pathogenesis of BP—whether they play a primary or ancillary role—remains to be elucidated through further investigation.
Elevated expression of multiple pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, has been observed in the serum of patients with BP and correlates positively with disease severity (207). IL-6 may be implicated in the development of BP by promoting the differentiation of Th17 and Tfh cells, as well as the production of antibodies by B cells (301). TNF-α can induce the release of inflammatory mediators such as IL-1, IL-6, IL-8, eotaxin-1, and MMP-9, and modulate the differentiation of T and B cells (302). Similarly, IL-1β can facilitate the release of TNF-α, IL-6, and MMP-9, leading to the recruitment of immune cells and the development of T-cell and B-cell-driven inflammatory responses (303). Despite the association of these cytokines with BP pathogenesis, their specificity in the disease is constrained. To date, no studies targeting IL-6/IL-6R or IL-1β for the treatment of BP have been reported. The application of TNF-α inhibitors in BP treatment has been documented in several case reports with inconsistent outcomes (304, 305), and in some instances, the use of TNF-α inhibitors has been associated with the onset of BP (306).
The presented data indicate that, similar to pemphigus vulgaris, the pathogenesis of BP is primarily linked to Th2 (IL-4, IL-5, IL-13, IL-31) and Th17 (IL-17, IL-23) cytokines. These cytokines are variably involved in the production of autoantibodies, the recruitment of eosinophils and neutrophils, the induction of pruritus, and the formation of bullae. Clinical trials targeting individual cytokines with biologic agents are currently in progress, with some showing favorable therapeutic outcomes, while others have demonstrated less satisfactory results. This variability suggests that BP is not a disease dominated by a single cytokine. Therefore, the concurrent inhibition of multiple cytokines, such as with JAK inhibitors, may offer a novel and potentially efficacious therapeutic alternative for this condition.
The involvement of the JAK/STAT signaling pathway in the pathogenesis of BP has been established. Juczynska et al. reported increased expression of all STAT proteins and JAK2 and JAK3 in BP skin lesions, and they proposed that JAK2 is implicated in the signaling pathways of IFN-γ and IL-5, whereas JAK3 predominantly influences the IL-4 and Th17 axes (307). Our recent findings indicate significantly elevated levels of phosphorylated STAT3 and STAT6 in BP lesions, reflecting overactivation of the JAK-STAT pathway at both the protein and transcriptional levels, as confirmed by immunohistochemistry (IHC) and transcriptome sequencing. Notably, these levels were substantially reduced following treatment with the JAK inhibitor tofacitinib (308). A review of the literature identified 17 cases of BP patients treated with JAK inhibitors since 2022 (309–317). The JAK inhibitors used included tofacitinib (JAK1/3, in 10 patients), baricitinib (JAK1/2, in 1 patient), upadacitinib (JAK1, in 3 patients), and abrocitinib (JAK1, in 3 patients). The treatment showed high efficacy and an acceptable safety profile (Table 2).
In summary, while JAK inhibitors present promising new therapeutic avenues for managing refractory PV/BP, the absence of large-scale randomized controlled trials (RCTs) and head-to-head comparison of JAK inhibitors and current first-line treatments— such as rituximab, corticosteroids, and dupilumab—limits our understanding of their relative efficacy, safety, and cost-effectiveness in PV/BP patients.
The safety profile of JAK inhibitors was initially regarded as comparable to biologic DMARDs (bDMARDs) based on early clinical trials in rheumatoid arthritis (RA) patients without severe comorbidities, which showed no significant increase in major adverse cardiovascular events (MACEs), venous thromboembolism (VTE), or malignancies, except for a higher risk of herpes zoster (318–327). However, this perspective shifted after the FDA’s 2021 black box warning, prompted by findings from the ORAL Surveillance trial (A3921133; NCT02092467).
The ORAL Surveillance was a 4-year, randomized, open-label, non-inferiority, post-authorization safety endpoint trial, compared tofacitinib (5 mg or 10 mg twice daily) with TNF inhibitors (etanercept or adalimumab) in RA patients (aged ≥50 years with at least one additional cardiovascular risk factors).The risks of MACE and cancers (excluding nonmelanoma skin cancer) were higher with the combined tofacitinib doses compared to TNF inhibitors, and the non-inferiority of tofacitinib was not demonstrated (328). Specifically, for tofacitinib at the approved dose of 5 mg twice daily, the ORAL Surveillance trial showed a numerically but not statistically higher risk of MACEs, serious infections, adjudicated hepatic events, VTE, deep vein thrombosis, pulmonary embolism, or adjudicated death from any cause compared to a TNF inhibitor. Additionally, there was a numerically and statistically significant increase in the risk of malignancies and adjudicated opportunistic infections, including herpes zoster and tuberculosis, as well as all cases of herpes zoster (both nonserious and serious) (328).
Following these results, FDA expanded safety warnings to all JAK inhibitors (baricitinib, upadacitinib, filgotinib), and the European Medicines Agency emphasizing caution in high-risk populations, including patients aged ≥65 years, smokers, and those with cardiovascular/metabolic comorbidities or prior histories of malignancies or VTE (329, 330). Subsequent post-hoc analyses of the ORAL Surveillance trial data have further elucidated which patients are at the highest risk for MACEs, malignancies, VTE, and death: (1) Patients with high risk or history of ASCVD have the highest risk of MACEs, and the appropriate use of statin therapy should be considered (331). (2) The increased incidence of malignancies is associated with a history of MACEs or ASCVD (332). (3) The risk of VTE is heightened in patients with a prior history of VTE, active RA, advanced age, obesity, or those undergoing hormone replacement therapy, when treated with either tofacitinib or TNF inhibitors. The causal role of JAK inhibitors in VTE remains inconclusive (333). (4) Elderly patients, smokers, and those with active disease are more susceptible to infections when treated with JAK inhibitors (334, 335). (5) Events including MACEs, malignancies, VTE, and serious infections nearly exclusively occur in “high-risk” patients (aged ≥ 65 years and/or former smokers), and are seldom seen in “low-risk” patients (aged < 65 years with no smoking history) (336). These studies provide valuable insights into better understanding of the true risks associated with JAK inhibitor use in clinical practice and how to mitigate such risks.
However, the ORAL Surveillance study has also raised several unresolved issues. Firstly, sustained systemic inflammation and disease activity have been identified as significant contributors to the development of MACEs, malignancies, VTE, and infections in patients with RA (337, 338). It remains contentious whether the adverse events observed in the ORAL Surveillance study are due to the disease itself or to the toxicity of tofacitinib. As several studies utilizing surrogate markers have revealed that JAK inhibitors can mitigate cardiovascular risk by reducing inflammation (339, 340). Secondly, the ORAL Surveillance study lacks a comparator group treated with placebo or conventional synthetic DMARDs (csDMARDs). This limitation precludes answering whether JAK inhibitors reduce the incidence of MACEs, malignancies, and VTE compared to inadequate treatment or csDMARD therapy in RA patients. The study only indicates that JAK inhibitors may not be as effective as TNF inhibitors in reducing the risk of these events, as TNF inhibitors have been shown to decrease the occurrence of MACEs (341).
Several clinical trials, meta-analyses, and integrated safety analyses for dermatological conditions such as AD, psoriasis, and AA have also confirmed the favorable safety profile of JAK inhibitors (342–344). Common adverse reactions include respiratory tract infections, nasopharyngitis, headache, and herpes zoster, all of which are self-limiting or can be alleviated with symptomatic treatment. The incidence of serious adverse events, such as MACEs, malignancies, and VTE, is not different from that of the control group. Similar to the ORAL Surveillance study, the incidence of these serious adverse events is also higher in dermatological patients aged 65 or older or those with a history of smoking (345). Due to the limited application of JAK inhibitors in AIBD, there have been no reports of serious adverse events to date. However, since pemphigus and pemphigoid diseases predominantly affect the elderly, and these patients have a higher risk of malignancies (346), VTE (347), and MACEs (348) compared to the general population, vigilance is warranted when using JAK inhibitors. Accordingly, prior to treatment initiation, a detailed medical history should be obtained to assess for VTE, malignancies, and associated risk factors. A comprehensive blood count, liver and kidney function tests, lipid panel, and screenings for hepatitis B, hepatitis C, HIV, and tuberculosis are recommended. Adult patients are advised to receive pneumococcal and herpes zoster vaccinations. Routine monitoring during treatment is essential, with particular attention to complete blood count, creatinine clearance, kidney and hepatic function. Dose adjustment or discontinuation is recommended in cases of significant hemoglobin decline (greater than 2 g/dL or levels below 8 g/dL), absolute neutrophil count between 500–1000/mm³, absolute lymphocyte count between 500–750/mm³, creatinine clearance between 30–60 mL/min or less than 30 mL/min, or in the presence of severe hepatic impairment (349).
JAKs are composed of approximately 1000 amino acids, with molecular weights ranging from 120 to 140 kDa, and consist of seven homology domains (JH1-7). The JH1 domain is the active kinase catalytic domain, containing conserved tyrosine residues that are the principal targets for JAK inhibitors. The high degree of homology among the JH1 domains of different JAK isoforms presents a challenge for the development of highly selective inhibitors. Selectivity of JAK inhibitors is evaluated in vitro assays using purified enzymes or cytokine stimulation with assessment of pSTAT activation. In vitro kinase assays and cellular assays have demonstrated that tofacitinib selectively inhibits cytokines signaling through JAK1 and JAK3 over JAK2, baricitinib shows specificity for JAK1 and JAK2 over JAK3, and both upadacitinib and abrocitinib are identified as selective JAK1 inhibitors (350–352). Table 3 illustrates the enzymatic and whole-cell activities of JAK inhibitors reported for the treatment of BP, where a lower IC50 value indicates greater potency. Selectivity for a specific JAK isoform is determined by the IC50 values and the ratios between them for the different JAK isoforms. The outcomes are contingent upon the assay substrates, cell lines, and cytokines being measured.
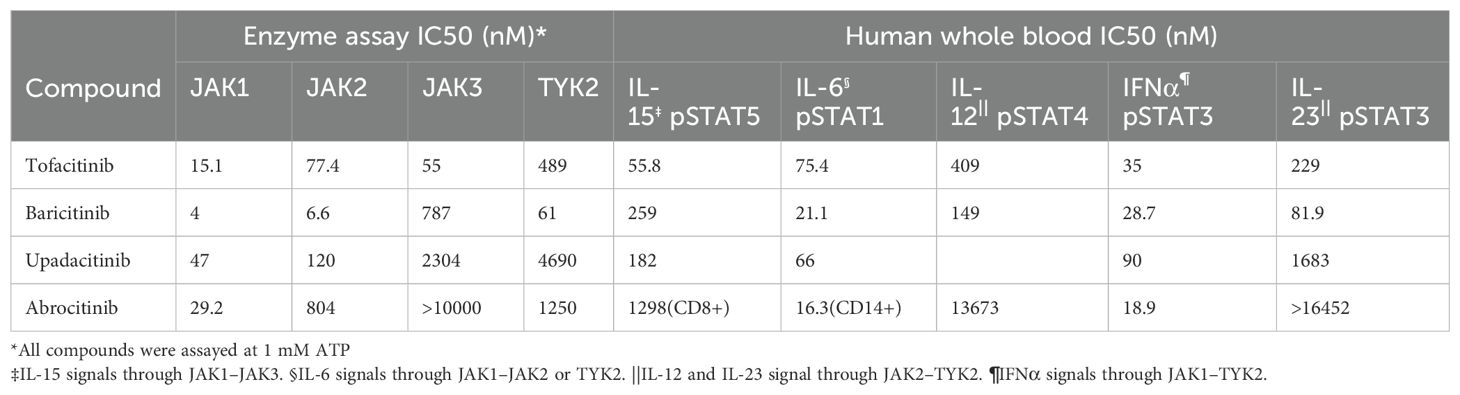
Table 3. The enzymatic and whole-cell activities of JAK inhibitors reported for the treatment of BP.
Although currently approved drugs have demonstrated varying degrees of selectivity for JAK isoform, the capacity of each JAK inhibitor to inhibit specific cytokine-signaling pathways is not readily predictable from preclinical selectivity data in direct cell-based assays. For instance, Dowty et al. compared the inhibitory effects of tofacitinib, baricitinib, upadacitinib, and filgotinib on cytokine-induced STAT phosphorylation patterns in whole blood cells at clinically efficacious doses, revealing generally similar cytokine receptor inhibition profiles with minor numerical differences (353). An additional in vitro pharmacological analysis examined the regulation of cytokine signaling by baricitinib, upadacitinib, and tofacitinib in human leukocyte subpopulations. While distinct cytokine pathways were modulated to varying degrees by different JAK inhibitors, no single agent potently or continuously inhibited an individual cytokine signaling pathway throughout the dosing interval (354). In an industry-sponsored study, filgotinib was found to inhibit JAK1-mediated signaling similarly to other JAK inhibitors but with reduced inhibition of JAK2-dependent and JAK3-dependent pathways (355). Moodley et al. assessed the in vivo impact of pan- and selective JAK inhibitors in mice through immunologic, genomic, and epigenomic profiling. While selective cell type-specific effects of JAK inhibitors were observable, there was a high overall overlap between these compounds (356). Clinically, no head-to-head trials have compared the efficacy of these agents; however, they have all demonstrated similar therapeutic effects in various autoimmune or immune-mediate diseases, such AR (357, 358), inflammatory bowel disease (351), AD (359) or AA (360). In summary, current experimental data do not conclusively indicate the potential advantages of higher selectivity in next-generation JAK inhibitors. It is only through rigorous clinical testing, including head-to-head studies and real-world application, that the clinical significance of differences between various JAK inhibitors will be determined.
In recent years, advancements in understanding cytokine biology and its regulatory interplay with autoimmunity have catalyzed a revolution in the treatment of autoimmune diseases. Despite this progress, the persistent burden of these conditions underscores the ongoing need for novel therapeutics. Small molecule targeted drugs, such as JAK inhibitors, capable of simultaneously blocking multiple cytokines signaling pathways, have demonstrated remarkable efficacy in a variety of autoimmune and immune-mediated inflammatory diseases, heralding a new era in treatment.
This review comprehensively summarizes the role of cytokines signaling through the JAK-STAT pathway in the pathogenesis of BP and PV, positing JAK inhibitors as a potential novel therapeutic approach for AIBD. However, the specific mechanisms of JAK/STAT in the pathogenesis and progression of AIBDs remain to be fully elucidated. The distinct roles of JAK1, JAK2, JAK3, and TYK2 in PV and BP remain unclear, necessitating further research to clarify their contributions to disease mechanisms. Additionally, the interplay between STAT-dependent and non-STAT-dependent pathways in cytokine signaling requires exploration, as they may differentially impact disease progression and treatment outcomes. The correlation between JAK-STAT activation and disease severity also remains poorly understood, highlighting the need for mechanistic studies to determine whether JAK inhibitors block autoantibody production or merely reduce inflammation.
In summary, despite the promising therapeutic prospects, challenges remain in optimizing the use of JAK inhibitors. Selectivity for different JAK isoforms, potential side effects, and long-term safety concerns are areas that require further investigation. Additionally, the identification of biomarkers to predict treatment response and the development of combination therapies to enhance efficacy and reduce toxicity are active areas of research. Currently, there is a lack of comprehensive data on these aspects, and future studies will be essential to fill these gaps.
XYL: Formal Analysis, Investigation, Software, Validation, Visualization, Writing – original draft. XL: Investigation, Validation, Writing – original draft. ZFZ: Conceptualization, Supervision, Validation, Writing – review & editing. MWZ: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article.This work was supported by National Natural Science Foundation of China (NO. 82373461).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Holtsche MM, Boch K, Schmidt E. Autoimmune bullous dermatoses. J Der Deutschen Dermatologischen Gesellschaft = J German Soc Dermatology: JDDG. (2023) 21:405–12. doi: 10.1111/ddg.15046
2. Hofmann SC, Juratli HA, Eming R. Bullous autoimmune dermatoses. J Der Deutschen Dermatologischen Gesellschaft = J German Soc Dermatology: JDDG. (2018) 16:1339–58. doi: 10.1111/ddg.2018.16.issue-11
3. Baum S, Sakka N, Artsi O, Trau H, Barzilai A. Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev. (2014) 13:482–9. doi: 10.1016/j.autrev.2014.01.047
4. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet (London England). (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
5. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. (2017) 3:17026. doi: 10.1038/nrdp.2017.26
6. Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a comprehensive review on pathogenesis, clinical presentation and novel therapeutic approaches. Clin Rev Allergy Immunol. (2018) 54:1–25. doi: 10.1007/s12016-017-8662-z
7. Goletz S PM, Lari TR, Hammers CM, Wang Y, Emtenani S, Aumailley M, et al. Laminin β4 is a constituent of the cutaneous basement membrane zone and additional autoantigen of anti-p200 pemphigoid. J Am Acad Dermatol. (2024) 90:790–7. doi: 10.1016/j.jaad.2023.11.014
8. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London England). (9863) 2013:320–32:381. doi: 10.1016/S0140-6736(12)61140-4
9. Buchman AL. Side effects of corticosteroid therapy. J Clin Gastroenterol. (2001) 33:289–94. doi: 10.1097/00004836-200110000-00006
10. Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in patients with pemphigus. Acta dermato-venereologica. (2017) 97:607–11. doi: 10.2340/00015555-2611
11. Amber KT, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
12. Olbrich H, Sadik CD, Schmidt E. Autoimmune blistering diseases: promising agents in clinical trials. Expert Opin On Investigational Drugs. (2023) 32:615–23. doi: 10.1080/13543784.2023.2242778
13. Kridin K, Schmidt E. Epidemiology of pemphigus. JID Innov. (2021) 1:100004. doi: 10.1016/j.xjidi.2021.100004
14. Li J, Zuo Y-G, Zheng H-Y. Mortality of bullous pemphigoid in China. JAMA Dermatol. (2013) 149:106–8. doi: 10.1001/archdermatol.2012.2994
15. Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
16. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discovery. (2017) 16:843–62. doi: 10.1038/nrd.2017.201
17. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
18. Li Y-J, Zhang C, Martincuks A, Herrmann A, Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer. (2023) 23:115–34. doi: 10.1038/s41568-022-00537-3
19. Yamaoka K, Saharinen P, Pesu M, Holt VET, Silvennoinen O, O’Shea JJ. The janus kinases (Jaks). Genome Biol. (2004) 5:253. doi: 10.1186/gb-2004-5-12-253
20. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3:651–62. doi: 10.1038/nrm909
21. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
22. Tanaka Y, Luo Y, O’Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol. (2022) 18:133–45. doi: 10.1038/s41584-021-00726-8
23. Villarino AV, Gadina M, O’Shea JJ, Kanno Y. SnapShot: jak-STAT signaling II. Cell. (2020) 181:1696–.e1. doi: 10.1016/j.cell.2020.04.052
24. Hixson KM, Cogswell M, Brooks-Kayal AR, Russek SJ. Evidence for a non-canonical JAK/STAT signaling pathway in the synthesis of the brain’s major ion channels and neurotransmitter receptors. BMC Genomics. (2019) 20:677. doi: 10.1186/s12864-019-6033-2
25. Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. (2008) 18:545–51. doi: 10.1016/j.tcb.2008.08.008
26. Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. (2012) 287:14192–200. doi: 10.1074/jbc.M111.323899
27. Lu D, Liu L, Ji X, Gao Y, Chen X, Liu Y, et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat Immunol. (2015) 16:1263–73. doi: 10.1038/ni.3278
28. Böhmer F-D, Friedrich K. Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT. (2014) 3:e28087. doi: 10.4161/jkst.28087
29. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. (2003) 3:900–11. doi: 10.1038/nri1226
30. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. (2007) 7:454–65. doi: 10.1038/nri2093
31. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
32. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. (2019) 40:298–308. doi: 10.1016/j.tips.2019.03.001
33. Niu GJ, Xu JD, Yuan WJ, Sun JJ, Wang JX. Protein inhibitor of activated STAT (PIAS) negatively regulates the JAK/STAT pathway by inhibiting STAT phosphorylation and translocation. Front Immunol. (2018) 9:2392. doi: 10.3389/fimmu.2018.02392
34. Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. (2006) 16:196–202. doi: 10.1038/sj.cr.7310027
35. Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. (2005) 5:3460. doi: 10.1038/nri1667
36. Stöcklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. (1996) 383:726–8. doi: 10.1038/383726a0
37. Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. (1995) 377:65–8. doi: 10.1038/377065a0
38. Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Sci (New York N.Y.). (1995) 270:797–800. doi: 10.1126/science.270.5237.797
39. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. (2006) 25:745–55. doi: 10.1016/j.immuni.2006.09.009
40. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
41. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. New Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
42. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Sci (New York N.Y.). (1976) 193:1007–8. doi: 10.1126/science.181845
43. Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Sci (New York N.Y.). (1994) 266:1045–7. doi: 10.1126/science.7973659
44. Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. (1995) 2:331–9. doi: 10.1016/1074-7613(95)90141-8
45. Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. (2013) 38:13–25. doi: 10.1016/j.immuni.2013.01.004
46. Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, et al. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature. (1984) 312:641–3. doi: 10.1038/312641a0
47. Siegel JP, Sharon M, Smith PL, Leonard WJ. The IL-2 receptor beta chain (p70): role in mediating signals for LAK, NK, and proliferative activities. Sci (New York N.Y.). (1987) 238:75–8. doi: 10.1126/science.3116668
48. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. (2002) 17:167–78. doi: 10.1016/S1074-7613(02)00367-9
49. Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. (1991) 353:858–61. doi: 10.1038/353858a0
50. Liao W, Lin J-X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. (2011) 12:551–9. doi: 10.1038/ni.2030
51. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh T-Y, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. (2008) 9:1288–96. doi: 10.1038/ni.1656
52. Liao W, Spolski R, Li P, Du N, West EE, Ren M, et al. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci United States America. (2014) 111:3508–13. doi: 10.1073/pnas.1301138111
53. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
54. Ballesteros-Tato A, León B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
55. Howard M, Farrar J, Hilfiker M, Johnson B, Takatsu K, Hamaoka T, et al. Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J Exp Med. (1982) 155:914–23. doi: 10.1084/jem.155.3.914
56. Isakson PC, Puré E, Vitetta ES, Krammer PH. T cell-derived B cell differentiation factor(s). Effect on the isotype switch of murine B cells. J Exp Med. (1982) 155:734–48. doi: 10.1084/jem.155.3.734
57. Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol (Baltimore Md.: 1950). (1990) 145:3796–806. doi: 10.4049/jimmunol.145.11.3796
58. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. (2008) 9:1347–55. doi: 10.1038/ni.1677
59. Kondo M, Takeshita T, Ishii N, Nakamura M, Watanabe S, Arai K, et al. Sharing of the interleukin-2 (IL-2) receptor gamma chain between receptors for IL-2 and IL-4. Sci (New York N.Y.). (1993) 262:1874–7. doi: 10.1126/science.8266076
60. Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-4 receptor. Sci (New York N.Y.). (1993) 262:1880–3. doi: 10.1126/science.8266078
61. McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine. (2015) 75:38–50. doi: 10.1016/j.cyto.2015.05.023
62. Hilton DJ, Zhang JG, Metcalf D, Alexander WS, Nicola NA, Willson TA. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci United States America. (1996) 93:497–501. doi: 10.1073/pnas.93.1.497
63. Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. (2018) 9:888. doi: 10.3389/fimmu.2018.00888
64. Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4-induced transcription factor: IL-4 Stat. Sci (New York N.Y.). (1994) 265:1701–6. doi: 10.1126/science.8085155
65. Rolling C, Treton D, Pellegrini S, Galanaud P, Richard Y. IL4 and IL13 receptors share the gamma c chain and activate STAT6, STAT3 and STAT5 proteins in normal human B cells. FEBS Lett. (1996) 393:53–6. doi: 10.1016/0014-5793(96)00835-6
66. Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, et al. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature. (1988) 333:571–3. doi: 10.1038/333571a0
67. Goodwin RG, Lupton S, Schmierer A, Hjerrild KJ, Jerzy R, Clevenger W, et al. Human interleukin 7: molecular cloning and growth factor activity on human and murine B-lineage cells. Proc Natl Acad Sci United States America. (1989) 86:302–6. doi: 10.1073/pnas.86.1.302
68. Cui G, Staron MM, Gray SM, Ho P-C, Amezquita RA, Wu J, et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell. (2015) 161:750–61. doi: 10.1016/j.cell.2015.03.021
69. Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol. (2019) 20:1584–93. doi: 10.1038/s41590-019-0479-x
70. Cosorich I, Filoni J, Di Dedda C, Ferrari A, Jofra T, Cesarano S, et al. Interleukin-7 improves the fitness of regulatory T cells for adoptive transfer. Immunology. (2023) 170:540–52. doi: 10.1111/imm.v170.4
71. Noguchi M, Nakamura Y, Russell SM, Ziegler SF, Tsang M, Cao X, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-7 receptor. Sci (New York N.Y.). (1993) 262:1877–80. doi: 10.1126/science.8266077
72. Kondo M, Takeshita T, Higuchi M, Nakamura M, Sudo T, Nishikawa S, et al. Functional participation of the IL-2 receptor gamma chain in IL-7 receptor complexes. Sci (New York N.Y.). (1994) 263:1453–4. doi: 10.1126/science.8128231
73. Kittipatarin C, Khaled AR. Interlinking interleukin-7. Cytokine. (2007) 39:75–83. doi: 10.1016/j.cyto.2007.07.183
74. Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma. Nat Commun. (2017) 8:14937. doi: 10.1038/ncomms14937
75. Wohlmann A, Sebastian K, Borowski A, Krause S, Friedrich K. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol Chem. (2010) 391:181–6. doi: 10.1515/bc.2010.029
76. Corren J, Ziegler SF. TSLP: from allergy to cancer. Nat Immunol. (2019) 20:1603–9. doi: 10.1038/s41590-019-0524-9
77. Uyttenhove C, Simpson RJ, Van Snick J. Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity. Proc Natl Acad Sci United States America. (1988) 85:6934–8. doi: 10.1073/pnas.85.18.6934
78. Demoulin JB, Van Roost E, Stevens M, Groner B, Renauld JC. Distinct roles for STAT1, STAT3, and STAT5 in differentiation gene induction and apoptosis inhibition by interleukin-9. J Biol Chem. (1999) 274:25855–61. doi: 10.1074/jbc.274.36.25855
79. Bauer JH, Liu KD, You Y, Lai SY, Goldsmith MA. Heteromerization of the gammac chain with the interleukin-9 receptor alpha subunit leads to STAT activation and prevention of apoptosis. J Biol Chem. (1998) 273:9255–60. doi: 10.1074/jbc.273.15.9255
80. Wiener Z, Falus A, Toth S. IL-9 increases the expression of several cytokines in activated mast cells, while the IL-9-induced IL-9 production is inhibited in mast cells of histamine-free transgenic mice. Cytokine. (2004) 26:122–30. doi: 10.1016/j.cyto.2004.01.006
81. Hültner L, Druez C, Moeller J, Uyttenhove C, Schmitt E, Rüde E, et al. Mast cell growth-enhancing activity (MEA) is structurally related and functionally identical to the novel mouse T cell growth factor P40/TCGFIII (interleukin 9). Eur J Immunol. (1990) 20:1413–6. doi: 10.1002/eji.1830200632
82. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci United States America. (2009) 106:12885–90. doi: 10.1073/pnas.0812530106
83. Turner J-E, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld J-C, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951–65. doi: 10.1084/jem.20130071
84. Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Sci (New York N.Y.). (1994) 264:965–8. doi: 10.1126/science.8178155
85. Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, et al. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J. (1994) 13:2822–30. doi: 10.1002/j.1460-2075.1994.tb06576.x
86. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity. (2002) 17:537–47. doi: 10.1016/S1074-7613(02)00429-6
87. Masuda A, Matsuguchi T, Yamaki K, Hayakawa T, Yoshikai Y. Interleukin-15 prevents mouse mast cell apoptosis through STAT6-mediated Bcl-xL expression. J Biol Chem. (2001) 276:26107–13. doi: 10.1074/jbc.M011475200
88. Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci United States America. (2000) 97:11439–44. doi: 10.1073/pnas.200360997
89. Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. (2000) 408:57–63. doi: 10.1038/35040504
90. Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, et al. Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol (Baltimore Md.: 1950). (2001) 167:1–5. doi: 10.4049/jimmunol.167.1.1
91. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood. (2007) 109:4135–42. doi: 10.1182/blood-2006-10-054973
92. Wan C-K, Andraski AB, Spolski R, Li P, Kazemian M, Oh J, et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc Natl Acad Sci United States America. (2015) 112:9394–9. doi: 10.1073/pnas.1511711112
93. Ozaki K, Spolski R, Feng CG, Qi C-F, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Sci (New York N.Y.). (2002) 298:1630–4. doi: 10.1126/science.1077002
94. Jin H, Carrio R, Yu A, Malek TR. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J Immunol (Baltimore Md.: 1950). (2004) 173:657–65. doi: 10.4049/jimmunol.173.1.657
95. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol (Baltimore Md.: 1950). (2003) 170:4111–8. doi: 10.4049/jimmunol.170.8.4111
96. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. (2005) 201:139–48. doi: 10.1084/jem.20041057
97. Novy P, Huang X, Leonard WJ, Yang Y. Intrinsic IL-21 signaling is critical for CD8 T cell survival and memory formation in response to vaccinia viral infection. J Immunol (Baltimore Md.: 1950). (2011) 186:2729–38. doi: 10.4049/jimmunol.1003009
98. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. (2008) 29:138–49. doi: 10.1016/j.immuni.2008.05.009
99. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. (2007) 448:480–3. doi: 10.1038/nature05969
100. Attridge K, Wang CJ, Wardzinski L, Kenefeck R, Chamberlain JL, Manzotti C, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood. (2012) 119:4656–64. doi: 10.1182/blood-2011-10-388546
101. Martinez-Moczygemba M, Huston DP. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol. (2003) 112:653–65; quiz 666. doi: 10.1016/S0091
102. Pant H, Hercus TR, Tumes DJ, Yip KH, Parker MW, Owczarek CM, et al. Translating the biology of β common receptor-engaging cytokines into clinical medicine. J Allergy Clin Immunol. (2023) 151:324–44. doi: 10.1016/j.jaci.2022.09.030
103. Broughton SE, Dhagat U, Hercus TR, Nero TL, Grimbaldeston MA, Bonder CS, et al. The GM-CSF/IL-3/IL-5 cytokine receptor family: from ligand recognition to initiation of signaling. Immunol Rev. (2012) 250:277–302. doi: 10.1111/j.1600-065X.2012.01164.x
104. Broughton SE, Nero TL, Dhagat U, Kan WL, Hercus TR, Tvorogov D, et al. The βc receptor family - Structural insights and their functional implications. Cytokine. (2015) 74:247–58. doi: 10.1016/j.cyto.2015.02.005
105. Varricchi G, Poto R, Marone G, Schroeder JT. IL-3 in the development and function of basophils. Semin Immunol. (2021) 54:101510. doi: 10.1016/j.smim.2021.101510
106. Strobl H. Molecular mechanisms of dendritic cell sublineage development from human hematopoietic progenitor/stem cells. Int Arch Allergy Immunol. (2003) 131:73–9. doi: 10.1159/000070921
107. Ebner S, Hofer S, Nguyen VA, Fürhapter C, Herold M, Fritsch P, et al. A novel role for IL-3: human monocytes cultured in the presence of IL-3 and IL-4 differentiate into dendritic cells that produce less IL-12 and shift Th cell responses toward a Th2 cytokine pattern. J Immunol (Baltimore Md.: 1950). (2002) 168:6199–207. doi: 10.4049/jimmunol.168.12.6199
108. Walsh GM, Hartnell A, Wardlaw AJ, Kurihara K, Sanderson CJ, Kay AB. IL-5 enhances the in vitro adhesion of human eosinophils, but not neutrophils, in a leucocyte integrin (CD11/18)-dependent manner. Immunology. (1990) 71:258–65.
109. Takatsu K. Interleukin-5 and IL-5 receptor in health and diseases. Proc Jpn Acad Ser B Phys Biol Sci. (2011) 87:463–85. doi: 10.2183/pjab.87.463
110. Borish LC, Steinke JW. 2. Cytokines and chemokines. J Allergy Clin Immunol. (2003) 111:S460–75. doi: 10.1067/mai.2003.108
111. Iwasaki-Arai J, Iwasaki H, Miyamoto T, Watanabe S, Akashi K. Enforced granulocyte/macrophage colony-stimulating factor signals do not support lymphopoiesis, but instruct lymphoid to myelomonocytic lineage conversion. J Exp Med. (2003) 197:1311–22. doi: 10.1084/jem.20021843
112. Welker P, Grabbe J, Zuberbier T, Grützkau A, Henz BM. GM-CSF downmodulates c-kit, Fc(epsilon)RI(alpha) and GM-CSF receptor expression as well as histamine and tryptase levels in cultured human mast cells. Arch Dermatol Res. (2001) 293:249–58. doi: 10.1007/s004030100225
113. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. (2019) 50:812–31. doi: 10.1016/j.immuni.2019.03.027
114. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374:1–20. doi: 10.1042/bj20030407
115. Skiniotis G, Boulanger MJ, Garcia KC, Walz T. Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nat Struct Mol Biol. (2005) 12:545–51. doi: 10.1038/nsmb941
116. Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J Exp Med. (2020) 217:e20190347. doi: 10.1084/jem.20190347
117. Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth factor Rev. (2008) 19:347–56. doi: 10.1016/j.cytogfr.2008.08.003
118. Lütticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Sci (New York N.Y.). (1994) 263:89–92. doi: 10.1126/science.8272872
119. Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Sci (New York N.Y.). (1994) 263:92–5. doi: 10.1126/science.8272873
120. Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. (1998) 334:297–314. doi: 10.1042/bj3340297
121. Lai CF, Ripperger J, Morella KK, Wang Y, Gearing DP, Fey GH, et al. Separate signaling mechanisms are involved in the control of STAT protein activation and gene regulation via the interleukin 6 response element by the box 3 motif of gp130. J Biol Chem. (1995) 270:14847–50. doi: 10.1074/jbc.270.25.14847
122. Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciumè G, Laurence A, et al. Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity. (2015) 42:877–89. doi: 10.1016/j.immuni.2015.04.014
123. Chua AO, Chizzonite R, Desai BB, Truitt TP, Nunes P, Minetti LJ, et al. Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to gp130. J Immunol (Baltimore Md.: 1950). (1994) 153:128–36. doi: 10.4049/jimmunol.153.1.128
124. Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. (1989) 170:827–45. doi: 10.1084/jem.170.3.827
125. Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, et al. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci United States America. (1996) 93:14002–7. doi: 10.1073/pnas.93.24.14002
126. Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. (1996) 382:171–4. doi: 10.1038/382171a0
127. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Sci (New York N.Y.). (1993) 260:547–9. doi: 10.1126/science.8097338
128. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
129. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. (2000) 13:715–25. doi: 10.1016/S1074-7613(00)00070-4
130. Floss DM, Mrotzek S, Klöcker T, Schröder J, Grötzinger J, Rose-John S, et al. Identification of canonical tyrosine-dependent and non-canonical tyrosine-independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J Biol Chem. (2013) 288:19386–400. doi: 10.1074/jbc.M112.432153
131. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol (Baltimore Md.: 1950). (2002) 168:5699–708. doi: 10.4049/jimmunol.168.11.5699
132. Aggarwal S, Ghilardi N, Xie M-H, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
133. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discovery. (2007) 6:975–90. doi: 10.1038/nrd2422
134. Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Sci (New York N.Y.). (1994) 264:1415–21. doi: 10.1126/science.8197455
135. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. (2015) 15:231–42. doi: 10.1038/nri3806
136. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Sci (New York N.Y.). (2013) 340:207–11. doi: 10.1126/science.1235214
137. Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. (2001) 14:461–70. doi: 10.1016/S1074-7613(01)00126-1
138. Lin Q, Dong C, Cooper MD. Impairment of T and B cell development by treatment with a type I interferon. J Exp Med. (1998) 187:79–87. doi: 10.1084/jem.187.1.79
139. Fallet B, Narr K, Ertuna YI, Remy M, Sommerstein R, Cornille K, et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci Immunol. (2016) 1:eaah6817. doi: 10.1126/sciimmunol.aah6817
140. van-Boxel-Dezaire AHH, Rani MRS, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. (2006) 25:361–72. doi: 10.1016/j.immuni.2006.08.014
141. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
142. Tau G, Rothman P. Biologic functions of the IFN-gamma receptors. Allergy. (1999) 54:1233–51. doi: 10.1034/j.1398-9995.1999.00099.x
143. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
144. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
145. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. (2003) 4:69–77. doi: 10.1038/ni875
146. Hamming OJ, Terczyńska-Dyla E, Vieyres G, Dijkman R, Jørgensen SE, Akhtar H, et al. Interferon lambda 4 signals via the IFNλ receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. (2013) 32:3055–65. doi: 10.1038/emboj.2013.232
147. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. (2003) 4:63–8. doi: 10.1038/ni873
148. Wack A, Terczyńska-Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol. (2015) 16:802–9. doi: 10.1038/ni.3212
149. Kotenko SV, Rivera A, Parker D, Durbin JE. Type III IFNs: Beyond antiviral protection. Semin Immunol. (2019) 43:101303. doi: 10.1016/j.smim.2019.101303
150. Blazek K, Eames HL, Weiss M, Byrne AJ, Perocheau D, Pease JE, et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med. (2015) 212:845–53. doi: 10.1084/jem.20140995
151. Koltsida O, Hausding M, Stavropoulos A, Koch S, Tzelepis G, Ubel C, et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol Med. (2011) 3:348–61. doi: 10.1002/emmm.201100142
152. Sabat R. IL-10 family of cytokines. Cytokine Growth factor Rev. (2010) 21:315–24. doi: 10.1016/j.cytogfr.2010.11.001
153. Ho AS, Liu Y, Khan TA, Hsu DH, Bazan JF, Moore KW. A receptor for interleukin 10 is related to interferon receptors. Proc Natl Acad Sci United States America. (1993) 90:11267–71. doi: 10.1073/pnas.90.23.11267
154. Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. (1997) 16:5894–903. doi: 10.1093/emboj/16.19.5894
155. Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol (Baltimore Md.: 1950). (1995) 155:1079–90. doi: 10.4049/jimmunol.155.3.1079
156. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
157. Ouyang W, O’Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
158. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
159. Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines–from host defence to tissue homeostasis. Nat Rev Immunol. (2014) 14:783–95. doi: 10.1038/nri3766
160. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. (2004) 22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622
161. Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol (Baltimore Md.: 1950). (2007) 178:2229–40. doi: 10.4049/jimmunol.178.4.2229
162. Liao S-C, Cheng Y-C, Wang Y-C, Wang C-W, Yang S-M, Yu C-K, et al. IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol (Baltimore Md.: 1950). (2004) 173:6712–8. doi: 10.4049/jimmunol.173.11.6712
163. Bech R, Jalilian B, Agger R, Iversen L, Erlandsen M, Otkjaer K, et al. Interleukin 20 regulates dendritic cell migration and expression of co-stimulatory molecules. Mol Cell Ther. (2016) 4:1. doi: 10.1186/s40591-016-0046-x
164. Pavlidis P, Tsakmaki A, Pantazi E, Li K, Cozzetto D, Digby-Bell J, et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat Commun. (2022) 13:5820. doi: 10.1038/s41467-022-33331-8
165. Caudell EG, Mumm JB, Poindexter N, Ekmekcioglu S, Mhashilkar AM, Yang XH, et al. The protein product of the tumor suppressor gene, melanoma differentiation-associated gene 7, exhibits immunostimulatory activity and is designated IL-24. J Immunol (Baltimore Md.: 1950). (2002) 168:6041–6. doi: 10.4049/jimmunol.168.12.6041
166. Maarof G, Bouchet-Delbos L, Gary-Gouy H, Durand-Gasselin I, Krzysiek R, Dalloul A. Interleukin-24 inhibits the plasma cell differentiation program in human germinal center B cells. Blood. (2010) 115:1718–26. doi: 10.1182/blood-2009-05-220251
167. Das D, Akhtar S, Kurra S, Gupta S, Sharma A. Emerging role of immune cell network in autoimmune skin disorders: An update on pemphigus, vitiligo and psoriasis. Cytokine Growth Factor Rev. (2019) 45:35–44. doi: 10.1016/j.cytogfr.2019.01.001
168. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In vivo CD4+ T cell differentiation and function: revisiting the th1/th2 paradigm. Annu Rev Immunol. (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
169. Veldman C, Stauber A, Wassmuth R, Uter W, Schuler G, Hertl M. Dichotomy of autoreactive Th1 and Th2 cell responses to desmoglein 3 in patients with pemphigus vulgaris (PV) and healthy carriers of PV-associated HLA class II alleles. J Immunol (Baltimore Md.: 1950). (2003) 170:635–42. doi: 10.4049/jimmunol.170.1.635
170. Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR. Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci United States America. (1995) 92:5239–43. doi: 10.1073/pnas.92.11.5239
171. Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, Kuwana M. Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine pemphigus vulgaris. J Immunol (Baltimore Md.: 1950). (2008) 181:1526–35. doi: 10.4049/jimmunol.181.2.1526
172. Wucherpfennig KW, Yu B, Bhol K, Monos DS, Argyris E, Karr RW, et al. Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci United States America. (1995) 92:11935–9. doi: 10.1073/pnas.92.25.11935
173. Rizzo C, Fotino M, Zhang Y, Chow S, Spizuoco A, Sinha AA. Direct characterization of human T cells in pemphigus vulgaris reveals elevated autoantigen-specific Th2 activity in association with active disease. Clin Exp Dermatol. (2005) 30:535–40. doi: 10.1111/j.1365-2230.2005.01836.x
174. Satyam A, Khandpur S, Sharma VK, Sharma A. Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-Pemphigus vulgaris. Immunol investigations. (2009) 38:498–509. doi: 10.1080/08820130902943097
175. Keskin DB, Stern JNH, Fridkis-Hareli M, Razzaque Ahmed A. Cytokine profiles in pemphigus vulgaris patients treated with intravenous immunoglobulins as compared to conventional immunosuppressive therapy. Cytokine. (2008) 41:315–21. doi: 10.1016/j.cyto.2007.12.007
176. Tavakolpour S, Tavakolpour V. Interleukin 4 inhibition as a potential therapeutic in pemphigus. Cytokine. (2016) 77:189–95. doi: 10.1016/j.cyto.2015.09.017
177. Moore AY, Hurley K. Dupilumab monotherapy suppresses recalcitrant pemphigus vulgaris. JAAD Case Rep. (2023) 31:16–8. doi: 10.1016/j.jdcr.2022.10.035
178. Jiang C, Adjei S, Santiago S, Lu J, Duran M, Tyring S. Novel use of dupilumab in pemphigus vulgaris and pemphigus foliaceus. JAAD Case Rep. (2023) 42:12–5. doi: 10.1016/j.jdcr.2023.09.018
179. Chen S, Zhan S, Hua C, Tang Y, Cheng H. A novel combined use of dupilumab for treatment of aggressive refractory pemphigus vulgaris complicated with pulmonary tuberculosis: A case report and the RNA-seq analysis. Front Immunol. (2022) 13:825796. doi: 10.3389/fimmu.2022.825796
180. Charlton B, Lafferty KJ. The Th1/Th2 balance in autoimmunity. Curr Opin Immunol. (1995) 7:793–8. doi: 10.1016/0952-7915(95)80050-6
181. Lin MS, Swartz SJ, Lopez A, Ding X, Fernandez-Vina MA, Stastny P, et al. Development and characterization of desmoglein-3 specific T cells from patients with pemphigus vulgaris. J Clin Invest. (1997) 99:31–40. doi: 10.1172/JCI119130
182. Lee SH, Hong WJ, Kim S-C. Analysis of serum cytokine profile in pemphigus. Ann Dermatol. (2017) 29:438–45. doi: 10.5021/ad.2017.29.4.438
183. Takahashi H, Amagai M, Tanikawa A, Suzuki S, Ikeda Y, Nishikawa T, et al. T helper type 2-biased natural killer cell phenotype in patients with pemphigus vulgaris. J Invest Dermatol. (2007) 127:324–30. doi: 10.1038/sj.jid.5700527
184. Arakawa M, Dainichi T, Yasumoto S, Hashimoto T. Lesional Th17 cells in pemphigus vulgaris and pemphigus foliaceus. J Dermatol Sci. (2009) 53:228–31. doi: 10.1016/j.jdermsci.2008.09.008
185. Xu R-C, Zhu H-Q, Li W-P, Zhao X-Q, Yuan H-J, Zheng J, et al. The imbalance of Th17 and regulatory T cells in pemphigus patients. Eur J dermatology: EJD. (2013) 23:795–802. doi: 10.1684/ejd.2013.2177
186. Holstein J, Solimani F, Baum C, Meier K, Pollmann R, Didona D, et al. Immunophenotyping in pemphigus reveals a TH17/TFH17 cell-dominated immune response promoting desmoglein1/3-specific autoantibody production. J Allergy Clin Immunol. (2021) 147:2358–69. doi: 10.1016/j.jaci.2020.11.008
187. Yuan H, Zhou S, Liu Z, Cong W, Fei X, Zeng W, et al. Pivotal role of lesional and perilesional T/B lymphocytes in pemphigus pathogenesis. J Invest Dermatol. (2017) 137:2362–70. doi: 10.1016/j.jid.2017.05.032
188. Polakova A, Kauter L, Ismagambetova A, Didona D, Solimani F, Ghoreschi K, et al. Detection of rare autoreactive T cell subsets in patients with pemphigus vulgaris. Front In Immunol. (2022) 13:979277. doi: 10.3389/fimmu.2022.979277
189. Mortazavi H, Esmaili N, Khezri S, Khamesipour A, Vasheghani Farahani I, Daneshpazhooh M, et al. The effect of conventional immunosuppressive therapy on cytokine serum levels in pemphigus vulgaris patients. Iran J Allergy Asthma Immunol. (2014) 13:174–83.
190. Timoteo RP, da Silva MV, Miguel CB, Silva DAA, Catarino JDS, Rodrigues Junior V, et al. Th1/th17-related cytokines and chemokines and their implications in the pathogenesis of pemphigus vulgaris. Mediators Inflamm. (2017) 2017:7151285. doi: 10.1155/2017/7151285
191. Asothai R, Anand V, Das D, Antil PS, Khandpur S, Sharma VK, et al. Distinctive Treg associated CCR4-CCL22 expression profile with altered frequency of Th17/Treg cell in the immunopathogenesis of Pemphigus Vulgaris. Immunobiology. (2015) 220:1129–35. doi: 10.1016/j.imbio.2015.06.008
192. Xue J, Su W, Chen Z, Ke Y, Du X, Zhou Q. Overexpression of interleukin-23 and interleukin-17 in the lesion of pemphigus vulgaris: a preliminary study. Mediators Inflammation. (2014) 2014:463928. doi: 10.1155/2014/463928
193. Tirado-Sánchez A, Ponce-Olivera RM, Vazquez-González D, Bonifaz A. Th-17 and the lack of efficacy of ustekinumab in pemphigus vulgaris. Dermatol Online J. (2013) 19:15. doi: 10.5070/D39hw6p904
194. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. (2012) 8:337–47. doi: 10.1038/nrrheum.2012.58
195. Hennerici T, Pollmann R, Schmidt T, Seipelt M, Tackenberg B, Möbs C, et al. Increased frequency of T follicular helper cells and elevated interleukin-27 plasma levels in patients with pemphigus. PloS One. (2016) 11:e0148919. doi: 10.1371/journal.pone.0148919
196. Kim AR, Han D, Choi JY, Seok J, Kim S-E, Seo S-H, et al. Targeting inducible costimulator expressed on CXCR5+PD-1+ TH cells suppresses the progression of pemphigus vulgaris. J Allergy Clin Immunol. (2020) 146:1070–9.e8. doi: 10.1016/j.jaci.2020.03.036
197. Wang C, Zhang H, Liu T, Wang Z, Zhang Y, Yu G, et al. Circulating Tfr/Tfh cell imbalance may contribute to the immunopathogenesis of pemphigus. J Eur Acad Dermatol Venereology: JEADV. (2023) 37:e1399–402. doi: 10.1111/jdv.v37.12
198. Hertl M, Karr RW, Amagai M, Katz SI. Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol. (1998) 110:388–92. doi: 10.1046/j.1523-1747.1998.00156.x
199. Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grötzinger J, Schultz E, et al. T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol (Baltimore Md.: 1950). (2004) 172:3883–92. doi: 10.4049/jimmunol.172.6.3883
200. Veldman C, Höhne A, Dieckmann D, Schuler G, Hertl M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with pemphigus vulgaris. J Immunol (Baltimore Md.: 1950). (2004) 172:6468–75. doi: 10.4049/jimmunol.172.10.6468
201. Dikiy S, Rudensky AY. Principles of regulatory T cell function. Immunity. (2023) 56:240–55. doi: 10.1016/j.immuni.2023.01.004
202. Sugiyama H, Matsue H, Nagasaka A, Nakamura Y, Tsukamoto K, Shibagaki N, et al. CD4+CD25high regulatory T cells are markedly decreased in blood of patients with pemphigus vulgaris. Dermatol (Basel Switzerland). (2007) 214:210–20. doi: 10.1159/000099585
203. Alecu M, Ursaciuc C, Surcel M, Coman G, Ciotaru D, Dobre M. CD28 T-cell costimulatory molecule expression in pemphigus vulgaris. J Eur Acad Dermatol Venereology: JEADV. (2009) 23:288–91. doi: 10.1111/j.1468-3083.2008.03035.x
204. Yokoyama T, Matsuda S, Takae Y, Wada N, Nishikawa T, Amagai M, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. (2011) 23:365–73. doi: 10.1093/intimm/dxr020
205. Iriki H, Takahashi H, Wada N, Nomura H, Mukai M, Kamata A, et al. Peripheral tolerance by Treg via constraining OX40 signal in autoreactive T cells against desmoglein 3, a target antigen in pemphigus. Proc Natl Acad Sci United States America. (2021) 118:e2026763118. doi: 10.1073/pnas.2026763118
206. Khozeimeh F, Savabi O, Esnaashari M. Evaluation of interleukin-1α, interleukin-10, tumor necrosis factor-α and transforming growth factor-β in the serum of patients with pemphigus vulgaris. J Contemp Dent Pract. (2014) 15:746–9. doi: 10.5005/jp-journals-10024-1610
207. Kowalski EH, Kneibner D, Kridin K, Amber KT. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun Rev. (2019) 18:526–34. doi: 10.1016/j.autrev.2019.03.009
208. D’Auria L, Bonifati C, Mussi A, D’Agosto G, De Simone C, Giacalone B, et al. Cytokines in the sera of patients with pemphigus vulgaris: interleukin-6 and tumour necrosis factor-alpha levels are significantly increased as compared to healthy subjects and correlate with disease activity. Eur Cytokine Netw. (1997) 8:383–7.
209. Bhol KC, Desai A, Kumari S, Colon JE, Ahmed AR. Pemphigus vulgaris: the role of IL-1 and IL-1 receptor antagonist in pathogenesis and effects of intravenous immunoglobulin on their production. Clin Immunol (Orlando Fla.). (2001) 100:172–80. doi: 10.1006/clim.2001.5061
210. Ameglio F, D’Auria L, Cordiali-Fei P, Trento E, D’Agosto G, Mastroianni A, et al. Anti-intercellular substance antibody log titres are correlated with serum concentrations of interleukin-6, interleukin-15 and tumor necrosis factor-alpha in patients with Pemphigus vulgaris relationships with peripheral blood neutrophil counts, disease severity and duration and patients’ age. J Biol Regul Homeost Agents. (1999) 13:220–4.
211. Feliciani C, Toto P, Amerio P, Pour SM, Coscione G, Shivji G, et al. In vitro and in vivo expression of interleukin-1alpha and tumor necrosis factor-alpha mRNA in pemphigus vulgaris: interleukin-1alpha and tumor necrosis factor-alpha are involved in acantholysis. J Invest Dermatol. (2000) 114:71–7. doi: 10.1046/j.1523-1747.2000.00835.x
212. López-Robles E, Avalos-Díaz E, Vega-Memije E, Hojyo-Tomoka T, Villalobos R, Fraire S, et al. TNFalpha and IL-6 are mediators in the blistering process of pemphigus. Int J Dermatol. (2001) 40:185–8. doi: 10.1046/j.1365-4362.2001.01083.x
213. Alecu M, Alecu S, Coman G, Gălăţescu E, Ursaciuc C. ICAM-1, ELAM-1, TNF-alpha and IL-6 in serum and blister liquid of pemphigus vulgaris patients. Roum Arch Microbiol Immunol. (1999) 58:121–30.
214. Narbutt J, Lukamowicz J, Bogaczewicz J, Sysa-Jedrzejowska A, Torzecka JD, Lesiak A. Serum concentration of interleukin-6 is increased both in active and remission stages of pemphigus vulgaris. Mediators Inflamm 2008. (2008) p:875394. doi: 10.1155/2008/875394
215. Mortazavi H, Babaeijandaghi F, Akbarzadeh M, Rezaei N, Amirzargar AA, Daneshpazhooh M, et al. The influence of systemic therapy on the serum levels of IL-6 and IL-8 in pemphigus vulgaris. J Eur Acad Dermatol Venereology: JEADV. (2013) 27:387–90. doi: 10.1111/j.1468-3083.2011.04319.x
216. Feliciani C, Toto P, Wang B, Sauder DN, Amerio P, Tulli A. Urokinase plasminogen activator mRNA is induced by IL-1alpha and TNF-alpha in in vitro acantholysis. Exp Dermatol. (2003) 12:466–71. doi: 10.1034/j.1600-0625.2002.120415.x
217. Assaf S, Malki L, Mayer T, Mohamad J, Peled A, Pavlovsky M, et al. ST18 affects cell-cell adhesion in pemphigus vulgaris in a tumour necrosis factor-α-dependent fashion. Br J Dermatol. (2021) 184:1153–60. doi: 10.1111/bjd.v184.6
218. Fiorentino DF, Garcia MS, Rehmus W, Kimball AB. A pilot study of etanercept treatment for pemphigus vulgaris. Arch Dermatol. (2011) 147:117–8. doi: 10.1001/archdermatol.2010.409
219. Pardo J, Mercader P, Mahiques L, Sanchez-Carazo JL, Oliver V, Fortea JM. Infliximab in the management of severe pemphigus vulgaris. Br J Dermatol. (2005) 153:222–3. doi: 10.1111/j.1365-2133.2005.06672.x
220. Jacobi A, Shuler G, Hertl M. Rapid control of therapy-refractory pemphigus vulgaris by treatment with the tumour necrosis factor-alpha inhibitor infliximab. Br J Dermatol. (2005) 153:448–9. doi: 10.1111/j.1365-2133.2005.06744.x
221. Shetty A, Marcum CB, Glass LF, Carter JD. Successful treatment of pemphigus vulgaris with etanercept in four patients. J Drugs Dermatol. (2009) 8:940–3.
222. Andrew K, Dhariwal SK, Szczecinska W. Sarilumab (interleukin-6 receptor monoclonal antibody) for the treatment of pemphigus vulgaris. Clin Exp Dermatol. (2023) 48:1055–6. doi: 10.1093/ced/llad173
223. Ghoreschi K, Balato A, Enerbäck C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet (London England). (2021) 397:754–66. doi: 10.1016/S0140-6736(21)00184-7
224. Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy. (2020) 75:54–62. doi: 10.1111/all.13954
225. Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of Rheumatic diseases. Nat Rev Rheumatol. (2017) 13:217–33. doi: 10.1038/nrrheum.2017.22
226. Juczynska K, Wozniacka A, Waszczykowska E, Danilewicz M, Wągrowska-Danilewicz M, Zebrowska A. Expression of JAK3, STAT2, STAT4, and STAT6 in pemphigus vulgaris. Immunol Res. (2020) 68:97–103. doi: 10.1007/s12026-020-09122-y
227. Ociepa K, Danilewicz M, Wągrowska-Danilewicz M, Peterson-Jęckowska R, Wójcicka-Rubin A, Lewkowicz N, et al. Expression of the selected proteins of JAK/STAT signaling pathway in diseases with oral mucosa involvement. Int J Mol Sci. (2022) 24:323. doi: 10.3390/ijms24010323
228. Tavakolpour S. Tofacitinib as the potent treatment for refractory pemphigus: A possible alternative treatment for pemphigus. Dermatol Ther. (2018) 31:e12696. doi: 10.1111/dth.12696
229. Gao W, McGarry T, Orr C, McCormick J, Veale DJ, Fearon U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann Rheumatic Dis. (2016) 75:311–5. doi: 10.1136/annrheumdis-2014-207201
230. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheumatic Dis. (2014) 73:2192–8. doi: 10.1136/annrheumdis-2013-203756
231. Krueger J, Clark JD, Suárez-Fariñas M, Fuentes-Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: A randomized phase 2 study. J Allergy Clin Immunol. (2016) 137:1079–90. doi: 10.1016/j.jaci.2015.12.1318
232. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation. (2018) 137:1934–48. doi: 10.1161/CIRCULATIONAHA.117.030423
233. Vander Does A, Gamret AC, Yosipovitch G. Nail loss in mild to moderate pemphigus vulgaris. Skin Appendage Disord. (2022) 8:504–7. doi: 10.1159/000525462
234. Hren MG, Khattri S. Upadacitinib leading to resolution of refractory mucosal erosions in a case of pemphigus vulgaris. Int J Dermatol. (2025) 64(4):736–7. doi: 10.1111/ijd.17464
235. Ujiie H. What’s new in the pathogeneses and triggering factors of bullous pemphigoid. J Dermatol. (2023) 50:140–9. doi: 10.1111/1346-8138.16654
236. Lee AY, Kim T, Kim JH. Understanding CD4+ T cells in autoimmune bullous diseases. Front Immunol. (2023) 14:1161927. doi: 10.3389/fimmu.2023.1161927
237. Dainichi T, Chow Z, Kabashima K. IgG4, complement, and the mechanisms of blister formation in pemphigus and bullous pemphigoid. J Dermatol Sci. (2017) 88:265–70. doi: 10.1016/j.jdermsci.2017.07.012
238. Cole C, Vinay K, Borradori L, Amber KT. Insights into the pathogenesis of bullous pemphigoid: the role of complement-independent mechanisms. Front Immunol. (2022) 13:912876. doi: 10.3389/fimmu.2022.912876
239. Ameglio F, D’Auria L, Bonifati C, Ferraro C, Mastroianni A, Giacalone B. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. (1998) 138:611–4. doi: 10.1046/j.1365-2133.1998.02169.x
240. Schmidt E, Bastian B, Dummer R, Tony HP, Bröcker EB, Zillikens D. Detection of elevated levels of IL-4, IL-6, and IL-10 in blister fluid of bullous pemphigoid. Arch Dermatol Res. (1996) 288:353–7. doi: 10.1007/BF02507102
241. Teraki Y, Hotta T, Shiohara T. Skin-homing interleukin-4 and -13-producing cells contribute to bullous pemphigoid: remission of disease is associated with increased frequency of interleukin-10-producing cells. J Invest Dermatol. (2001) 117:1097–102. doi: 10.1046/j.0022-202x.2001.01505.x
242. Feliciani C, Toto P, Mohammad Pour S, Coscione G, Amerio P, Amerio P. A Th2-like cytokine response is involved in bullous pemphigoid. the role of IL-4 and IL-5 in the pathogenesis of the disease. Int J Immunopathol Pharmacol. (1999) 12:55–61. doi: 10.1177/205873929901200202
243. Pickford WJ, Gudi V, Haggart AM, Lewis BJ, Herriot R, Barker RN, et al. T cell participation in autoreactivity to NC16a epitopes in bullous pemphigoid. Clin Exp Immunol. (2015) 180:189–200. doi: 10.1111/cei.12566
244. Zhang J, Fang H, Shen S, Dang E, Li Q, Qiao P, et al. Identification of immunodominant th2-cell epitopes in chinese patients with bullous pemphigoid. J Invest Dermatol. (2018) 138:1917–24. doi: 10.1016/j.jid.2018.03.1515
245. Wang Y, Mao X, Liu Y, Yang Y, Jin H, Li L. IL-13 genetic susceptibility to bullous pemphigoid: A potential target for treatment and a prognostic marker. Front Immunol. (2022) 13:824110. doi: 10.3389/fimmu.2022.824110
246. Tabatabaei-Panah P-S, Moravvej H, Alirajab M, Etaaty A, Geranmayeh M, Hosseine F, et al. Association between TH2 cytokine gene polymorphisms and risk of bullous pemphigoid. Immunol investigations. (2022) 51:343–56. doi: 10.1080/08820139.2020.1832113
247. Hashimoto T, Kursewicz CD, Fayne RA, Nanda S, Shah SM, Nattkemper L, et al. Pathophysiologic mechanisms of itch in bullous pemphigoid. J Am Acad Dermatol. (2020) 83:53–62. doi: 10.1016/j.jaad.2019.07.060
248. Kaye A, Gordon SC, Deverapalli SC, Her MJ, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. (2018) 154:1225–6. doi: 10.1001/jamadermatol.2018.2526
249. Zhao L, Wang Q, Liang G, Zhou Y, Yiu N, Yang B, et al. Evaluation of dupilumab in patients with bullous pemphigoid. JAMA Dermatol. (2023) 159:953–60. doi: 10.1001/jamadermatol.2023.2428
250. Huang D, Zhang Y, Yu Y, Jiang Y, Kong L, Ding Y, et al. Long-term efficacy and safety of dupilumab for severe bullous pemphigoid: A prospective cohort study. Int Immunopharmacol. (2023) 125:111157. doi: 10.1016/j.intimp.2023.111157
251. Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: A systematic review. Front In Immunol. (2022) 13:928621. doi: 10.3389/fimmu.2022.928621
252. Abdat R, Waldman RA, de Bedout V, Czernik A, McLeod M, King B, et al. Dupilumab as a novel therapy for bullous pemphigoid: A multicenter case series. J Am Acad Dermatol. (2020) 83:46–52. doi: 10.1016/j.jaad.2020.01.089
253. Borradori L, Van Beek N, Feliciani C, Tedbirt B, Antiga E, Bergman R, et al. Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereology: JEADV. (2022) 36:1689–704. doi: 10.1111/jdv.v36.10
254. Murrell DF, Joly P, Werth VP, Ujiie H, Worm M, Mangold AR, et al. Study design of a phase 2/3 randomized controlled trial of dupilumab in adults with bullous pemphigoid: LIBERTY-BP ADEPT. Adv Ther. (2024) 41(7):3014–5. doi: 10.1007/s12325-024-02886-x
255. Press Release: Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. Available online at: https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237 (Accessed September 11, 2024).
256. Maglie R, Baffa ME, Senatore S, Pipitò C, Caproni M, Solimani F, et al. Rapid and sustained response to tralokinumab in a patient with severe bullous pemphigoid and end-stage kidney disease. Clin Exp Dermatol. (2024) 49:161–3. doi: 10.1093/ced/llad331
257. D’Auria L, Pietravalle M, Mastroianni A, Ferraro C, Mussi A, Bonifati C, et al. IL-5 levels in the serum and blister fluid of patients with bullous pemphigoid: correlations with eosinophil cationic protein, RANTES, IgE and disease severity. Arch Dermatol Res. (1998) 290:25–7. doi: 10.1007/s004030050272
258. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy. (2017) 72:1105–13. doi: 10.1111/all.2017.72.issue-7
259. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. (2000) 143:112–6. doi: 10.1046/j.1365-2133.2000.03599.x
260. Roufosse F. Targeting the interleukin-5 pathway for treatment of eosinophilic conditions other than asthma. Front Med (Lausanne). (2018) 5:49. doi: 10.3389/fmed.2018.00049
261. Simon D, Yousefi S, Cazzaniga S, Bürgler C, Radonjic S, Houriet C, et al. Mepolizumab failed to affect bullous pemphigoid: A randomized, placebo-controlled, double-blind phase 2 pilot study. Allergy. (2020) 75:669–72. doi: 10.1111/all.13950
262. Rhyou H-I, Han S-H, Nam Y-H. Successful induction treatment of bullous pemphigoid using reslizumab: a case report. Allergy Asthma Clin Immunol. (2021) 17:117. doi: 10.1186/s13223-021-00619-1
263. Kwon H-J, Kim K-H, Yoon J-H. Refractory bullous pemphigoid successfully treated with reslizumab: A possible novel therapeutic modality. Ann Dermatol. (2023) 35:S103–6. doi: 10.5021/ad.21.177
264. Tanaka A, Fujimura Y, Fuke S, Izumi K, Ujiie H. A case of bullous pemphigoid developing under treatment with benralizumab for bronchial asthma. J Dermatol. (2023) 50:1199–202. doi: 10.1111/1346-8138.16811
265. Gibbs BF, Patsinakidis N, Raap U. Role of the pruritic cytokine IL-31 in autoimmune skin diseases. Front In Immunol. (2019) 10:1383. doi: 10.3389/fimmu.2019.01383
266. Rüdrich U, Gehring M, Papakonstantinou E, Illerhaus A, Engmann J, Kapp A, et al. Eosinophils are a major source of interleukin-31 in bullous pemphigoid. Acta dermato-venereologica. (2018) 98:766–71. doi: 10.2340/00015555-2951
267. Salz M, Haeberle S, Hoffmann J, Enk AH, Hadaschik EN. Elevated IL-31 serum levels in bullous pemphigoid patients correlate with eosinophil numbers and are associated with BP180-IgE. J Dermatol Sci. (2017) 87:309–11. doi: 10.1016/j.jdermsci.2017.07.019
268. Kunsleben N, Rüdrich U, Gehring M, Novak N, Kapp A, Raap U. IL-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and CCL26 in eosinophils, which are capable to release IL-31. J Invest Dermatol. (2015) 135:1908–11. doi: 10.1038/jid.2015.106
269. Keam SJ. Nemolizumab: first approval. Drugs. (2022) 82:1143–50. doi: 10.1007/s40265-022-01741-z
270. Nakajima S, Kabata H, Kabashima K, Asano K. Anti-TSLP antibodies: Targeting a master regulator of type 2 immune responses. Allergol Int. (2020) 69:197–203. doi: 10.1016/j.alit.2020.01.001
271. Zhang Y, Hwang B-J, Liu Z, Li N, Lough K, Williams SE, et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc Natl Acad Sci United States America. (2018) 115:6434–9. doi: 10.1073/pnas.1721805115
272. Valdebran M, Kowalski EH, Kneiber D, Li J, Kim J, Doan L, et al. Epidermal expression of eotaxins and thymic stromal lymphopoietin in eosinophil rich dermatoses. Arch Dermatol Res. (2019) 311:705–10. doi: 10.1007/s00403-019-01954-5
273. Li S-Z, Jin X-X, Ge X-L, Zuo Y-G, Jin H-Z. Thymic stromal lymphopoietin is implicated in the pathogenesis of bullous pemphigoid by dendritic cells. J Immunol Res. (2020) 2020:4594630. doi: 10.1155/2020/4594630
274. Li S-Z, Jin X-X, Shan Y, Jin H-Z, Zuo Y-G. Expression of thymic stromal lymphopoietin in immune-related dermatoses. Mediators Inflammation. (2022) 2022:9242383. doi: 10.1155/2022/9242383
276. Arakawa M, Dainichi T, Ishii N, Hamada T, Karashima T, Nakama T, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. (2011) 20:1022–4. doi: 10.1111/j.1600-0625.2011.01378.x
277. Le Jan S, Plée J, Vallerand D, Dupont A, Delanez E, Durlach A, et al. Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol. (2014) 134:2908–17. doi: 10.1038/jid.2014.263
278. Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, Stasikowska-Kanicka O, Cynkier A, Sysa-Jedrzejowska A, et al. IL-17 expression in dermatitis herpetiformis and bullous pemphigoid. Mediators Inflammation. (2013) 2013:967987. doi: 10.1155/2013/967987
279. Żebrowska A, Woźniacka A, Juczyńska K, Ociepa K, Waszczykowska E, Szymczak I, et al. Correlation between IL36α and IL17 and activity of the disease in selected autoimmune blistering diseases. Mediators Inflammation. (2017) 2017:8980534. doi: 10.1155/2017/8980534
280. Plée J, Le Jan S, Giustiniani J, Barbe C, Joly P, Bedane C, et al. Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci Rep. (2015) 5:18001. doi: 10.1038/srep18001
281. Tabatabaei-Panah P-S, Moravvej H, Aghaei S, Akbari M, Rajabi S, Kia A, et al. TH17/IL23 cytokine gene polymorphisms in bullous pemphigoid. Mol Genet Genomic Med. (2020) 8:e1519. doi: 10.1002/mgg3.v8.12
282. Lindgren O, Le Menn G, Tuusa J, Chen ZJ, Tasanen K, Kokkonen N. Absence of NC14A domain of COLXVII/BP180 in mice results in IL-17–Associated skin inflammation. J Invest Dermatol. (2023) 143:48–56. doi: 10.1016/j.jid.2022.07.019
283. Chakievska L, Holtsche MM, Künstner A, Goletz S, Petersen B-S, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun. (2019) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
284. Riani M, Le Jan S, Plée J, Durlach A, Le Naour R, Haegeman G, et al. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. J Allergy Clin Immunol. (2017) 139:863–72.e3. doi: 10.1016/j.jaci.2016.08.012
285. Le Jan S, Muller C, Plee J, Durlach A, Bernard P, Antonicelli F. IL-23/IL-17 axis activates IL-1β-associated inflammasome in macrophages and generates an auto-inflammatory response in a subgroup of patients with bullous pemphigoid. Front Immunol. (2019) 10:1972. doi: 10.3389/fimmu.2019.01972
286. Vazquez-Tello A, Halwani R, Hamid Q, Al-Muhsen S. Glucocorticoid receptor-beta up-regulation and steroid resistance induction by IL-17 and IL-23 cytokine stimulation in peripheral mononuclear cells. J Clin Immunol. (2013) 33:466–78. doi: 10.1007/s10875-012-9828-3
287. Xiao Y, Gu Y, Xia D, Zhou X, Li W. Ixekizumab successfully treated refractory psoriasis concurrent bullous pemphigoid. J Dermatol. (2023) 50:e76–8. doi: 10.1111/1346-8138.16559
288. Loget J, Plée J, Antonicelli F, Bernard P. A successful treatment with ustekinumab in a case of relapsing bullous pemphigoid associated with psoriasis. J Eur Acad Dermatol Venereology: JEADV. (2017) 31:e228–30. doi: 10.1111/jdv.2017.31.issue-5
289. Kong L, Huang D, Lu J, Zhang Y, Li Y, Yi X, et al. Development of bullous pemphigoid during treatment of psoriasis with ustekinumab: a case report and literature review. Front Med (Lausanne). (2023) 10:1171802. doi: 10.3389/fmed.2023.1171802
290. Burlando M, Capurro N, Herzum A, Cozzani E, Parodi A. Guselkumab-associated bullous pemphigoid in a psoriasis patient: A case report and review of the literature. Dermatol Ther. (2022) 35:e15207. doi: 10.1111/dth.15207
291. Wang G, Hu X, Han S, Zhou M. A case report of bullous pemphigoid following secukinumab therapy for a patient with psoriasis. J Dermatolog Treat. (2024) 35:2366535. doi: 10.1080/09546634.2024.2366535
292. Olatunde AC, Hale JS, Lamb TJ. Cytokine-skewed Tfh cells: functional consequences for B cell help. Trends Immunol. (2021) 42:536–50. doi: 10.1016/j.it.2021.04.006
293. Li Q, Liu Z, Dang E, Jin L, He Z, Yang L, et al. Follicular helper T Cells (Tfh) and IL-21 involvement in the pathogenesis of bullous pemphigoid. PloS One. (2013) 8:e68145. doi: 10.1371/journal.pone.0068145
294. Ohuchi K, Fujimura T, Lyu C, Amagai R, Muto Y, Aiba S. Possible roles of CXCL13/CXCR5 axis in the development of bullous pemphigoid. J Dermatol. (2021) 48:353–9. doi: 10.1111/1346-8138.15713
295. Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. (2018) 142:1818–30.e6. doi: 10.1016/j.jaci.2018.03.014
296. Antiga E, Quaglino P, Volpi W, Pierini I, Del Bianco E, Bianchi B, et al. Regulatory T cells in skin lesions and blood of patients with bullous pemphigoid. J Eur Acad Dermatol Venereology: JEADV. (2014) 28:222–30. doi: 10.1111/jdv.2014.28.issue-2
297. Quaglino P, Antiga E, Comessatti A, Caproni M, Nardò T, Ponti R, et al. Circulating CD4+ CD25brightFOXP3+ regulatory T-cells are significantly reduced in bullous pemphigoid patients. Arch Dermatol Res. (2012) 304:639–45. doi: 10.1007/s00403-012-1213-9
298. Rensing-Ehl A, Gaus B, Bruckner-Tuderman L, Martin SF. Frequency, function and CLA expression of CD4+CD25+FOXP3+ regulatory T cells in bullous pemphigoid. Exp Dermatol. (2007) 16:13–21. doi: 10.1111/j.1600-0625.2006.00522.x
299. Bhol KC, Rojas AI, Khan IU, Ahmed AR. Presence of interleukin 10 in the serum and blister fluid of patients with pemphigus vulgaris and pemphigoid. Cytokine. (2000) 12:1076–83. doi: 10.1006/cyto.1999.0642
300. Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. (2018) 142:1831–42.e7. doi: 10.1016/j.jaci.2018.04.006
301. Jones BE, Maerz MD, Buckner JH. IL-6: a cytokine at the crossroads of autoimmunity. Curr Opin Immunol. (2018) 55:9–14. doi: 10.1016/j.coi.2018.09.002
302. Jang D-I, Lee AH, Shin H-Y, Song H-R, Park J-H, Kang T-B, et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α Inhibitors in therapeutics. Int J Mol Sci. (2021) 22:2719. doi: 10.3390/ijms22052719
303. Fetter T, de Graaf DM, Claus I, Wenzel J. Aberrant inflammasome activation as a driving force of human autoimmune skin disease. Front Immunol. (2023) 14:1190388. doi: 10.3389/fimmu.2023.1190388
304. Yamauchi PS, Lowe NJ, Gindi V. Treatment of coexisting bullous pemphigoid and psoriasis with the tumor necrosis factor antagonist etanercept. J Am Acad Dermatol. (2006) 54:S121–2. doi: 10.1016/j.jaad.2005.10.055
305. Nin M, Tokunaga D, Ishii N, Komai A, Hashimoto T, Katoh N. Case of coexisting psoriatic arthritis and bullous pemphigoid improved by etanercept. J Dermatol. (2013) 40:55–6. doi: 10.1111/j.1346-8138.2012.01659.x
306. Husein-ElAhmed H, Steinhoff M. Bullous pemphigoid induced by biologic drugs in psoriasis: a systematic review. J Dermatolog Treat. (2022) 33:2886–93. doi: 10.1080/09546634.2022.2089331
307. Juczynska K, Wozniacka A, Waszczykowska E, Danilewicz M, Wagrowska-Danilewicz M, Wieczfinska J, et al. Expression of the JAK/STAT signaling pathway in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflammation. (2017) 2017:6716419. doi: 10.1155/2017/6716419
308. Li X ZL, Gu H, He W, Zhai Z, Zhang M. Treatment and molecular analysis of bullous pemphigoid with tofacitinib: a case report and review of current literature. Front Immunol. (2024) 15:1464474. doi: 10.3389/fimmu.2024.1464474
309. Xiao Y, Xiang H, Li W. Concurrent bullous pemphigoid and plaque psoriasis successfully treated with Janus kinase inhibitor Baricitinib. Dermatol Ther. (2022) 35:e15754. doi: 10.1111/dth.v35.10
310. Youssef S, Gallitano S, Bordone LA. Two cases of bullous pemphigoid effectively treated with oral tofacitinib. JAAD Case Rep. (2023) 32:77–80. doi: 10.1016/j.jdcr.2022.10.028
311. Nash D, Kirchhof MG. Bullous pemphigoid treated with Janus kinase inhibitor upadacitinib. JAAD Case Rep. (2023) 32:81–3. doi: 10.1016/j.jdcr.2022.12.006
312. Fan B, Wang M. Tofacitinib in recalcitrant bullous pemphigoid: a report of seven cases. Br J Dermatol. (2023) 188:432–4. doi: 10.1093/bjd/ljac078
313. Gresham LM, Kirchhof MG. A case of drug-induced bullous pemphigoid secondary to immunotherapy treated with upadacitinib: A case report. SAGE Open Med Case Rep. (2023) 11:2050313X231160926. doi: 10.1177/2050313X231160926
314. Li H, Wang H, Qiao G, Liu Y, Zhang F, Pan F. Concurrent bullous pemphigoid and psoriasis vulgaris successfully treated with Janus kinase inhibitor tofacitinib: A case report and review of the literature. Int Immunopharmacol. (2023) 122:110591. doi: 10.1016/j.intimp.2023.110591
315. Sun L, Dong Z, Wang F. Highly selective JAK1 inhibitor for the treatment of refractory bullous pemphigoid: a case report. China J Leprosy Skin Dis. (2023) 39:906–8.
316. Jiang W, Ma X, Guo T, Song M, Zhang J. Abrocitinib-A promising option for patients with refractory bullous pemphigoid. J Eur Acad Dermatol Venereology: JEADV. (2024) 38:e119–21. doi: 10.1111/jdv.19475
317. Su F, Wang T, Qin Q, Xie Z. Upadacitinib for the management of bullous pemphigoid coexisting with psoriasis vulgaris: a case report and literature review. J Dermatolog Treat. (2024) 35:2302394. doi: 10.1080/09546634.2024.2302394
318. Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. (2019) 46:7–18. doi: 10.3899/jrheum.171361
319. Burmester GR, Cohen SB, Winthrop KL, Nash P, Irvine AD, Deodhar A, et al. Safety profile of upadacitinib over 15 000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open. (2023) 9:e002735. doi: 10.1136/rmdopen-2022-002735
320. Khosrow-Khavar F, Kim SC, Lee H, Lee SB, Desai RJ. Tofacitinib and risk of cardiovascular outcomes: results from the Safety of TofAcitinib in Routine care patients with Rheumatoid Arthritis (STAR-RA) study. Ann Rheumatic Dis. (2022) 81:798–804. doi: 10.1136/annrheumdis-2021-221915
321. Khosrow-Khavar F, Desai RJ, Lee H, Lee SB, Kim SC. Tofacitinib and risk of Malignancy: results from the safety of tofacitinib in routine care patients with rheumatoid arthritis (STAR-RA) study. Arthritis Rheumatol (Hoboken N.J.). (2022) 74:1648–59. doi: 10.1002/art.42250
322. Fleischmann R, Curtis JR, Charles-Schoeman C, Mysler E, Yamaoka K, Richez C, et al. Safety profile of upadacitinib in patients at risk of cardiovascular disease: integrated post hoc analysis of the SELECT phase III rheumatoid arthritis clinical programme. Ann Rheumatic Dis. (2023) 82:1130–41. doi: 10.1136/ard-2023-223916
323. Russell MD, Stovin C, Alveyn E, Adeyemi O, Chan CKD, Patel V, et al. JAK inhibitors and the risk of Malignancy: a meta-analysis across disease indications. Ann Rheumatic Dis. (2023) 82:1059–67. doi: 10.1136/ard-2023-224049
324. Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open. (2020) 6:e001395. doi: 10.1136/rmdopen-2020-001395
325. Taylor PC, Takeuchi T, Burmester GR, Durez P, Smolen JS, Deberdt W, et al. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: final results from long-term extension study and integrated database. Ann Rheumatic Dis. (2022) 81:335–43. doi: 10.1136/annrheumdis-2021-221276
326. Cohen SB, van Vollenhoven RF, Winthrop KL, Zerbini CAF, Tanaka Y, Bessette L, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheumatic Dis. (2021) 80:304–11. doi: 10.1136/annrheumdis-2020-218510
327. Winthrop KL, Tanaka Y, Takeuchi T, Kivitz A, Matzkies F, Genovese MC, et al. Integrated safety analysis of filgotinib in patients with moderately to severely active rheumatoid arthritis receiving treatment over a median of 1.6 years. Ann Rheumatic Dis. (2022) 81:184–92. doi: 10.1136/annrheumdis-2021-221051
328. Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. New Engl J Med. (2022) 386:316–26. doi: 10.1056/NEJMoa2109927
329. U.S.Food & Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions (2021). Available online at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death (Accessed September 01, 2021).
330. European Medicines Agency. EMA recommends measures to minimise risk of serious side effects with Janus kinase inhibitors for chronic inflammatory disorders (2022). Available online at: https://www.ema.europa.eu/en/news/ema-recommends-measures-minimise-risk-serious-side-effects-janus-kinase-inhibitors-chronic (Accessed October 28, 2022).
331. Charles-Schoeman C, Buch MH, Dougados M, Bhatt DL, Giles JT, Ytterberg SR, et al. Risk of major adverse cardiovascular events with tofacitinib versus tumour necrosis factor inhibitors in patients with rheumatoid arthritis with or without a history of atherosclerotic cardiovascular disease: a post hoc analysis from ORAL Surveillance. Ann Rheumatic Dis. (2023) 82:119–29. doi: 10.1136/ard-2022-222259
332. Curtis JR, Yamaoka K, Chen Y-H, Bhatt DL, Gunay LM, Sugiyama N, et al. Malignancy risk with tofacitinib versus TNF inhibitors in rheumatoid arthritis: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheumatic Dis. (2023) 82:331–43. doi: 10.1136/ard-2022-222543
333. Charles-Schoeman C, Fleischmann R, Mysler E, Greenwald M, Ytterberg SR, Koch GG, et al. Risk of venous thromboembolism with tofacitinib versus tumor necrosis factor inhibitors in cardiovascular risk-enriched rheumatoid arthritis patients. Arthritis Rheumatol. (2024) 76(8):1218–29. doi: 10.1002/art.42846
334. Balanescu A-R, Citera G, Pascual-Ramos V, Bhatt DL, Connell CA, Gold D, et al. Infections in patients with rheumatoid arthritis receiving tofacitinib versus tumour necrosis factor inhibitors: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheumatic Dis. (2022) 81:1491–503. doi: 10.1136/ard-2022-222405
335. Karpouzas GA, Szekanecz Z, Baecklund E, Mikuls TR, Bhatt DL, Wang C, et al. Rheumatoid arthritis disease activity and adverse events in patients receiving tofacitinib or tumor necrosis factor inhibitors: a post hoc analysis of ORAL Surveillance. Ther Adv Musculoskeletal Dis. (2023) 15:1759720X231201047. doi: 10.1177/1759720X231201047
336. Kristensen LE, Danese S, Yndestad A, Wang C, Nagy E, Modesto I, et al. Identification of two tofacitinib subpopulations with different relative risk versus TNF inhibitors: an analysis of the open label, randomised controlled study ORAL Surveillance. Ann Rheumatic Dis. (2023) 82:901–10. doi: 10.1136/ard-2022-223715
337. Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJL, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheumatic Dis. (2017) 76:17–28. doi: 10.1136/annrheumdis-2016-209775
338. Mellemkjaer L, Linet MS, Gridley G, Frisch M, Møller H, Olsen JH. Rheumatoid arthritis and cancer risk. Eur J Cancer. (1996) 32A:1753–7. doi: 10.1016/0959-8049(96)00210-9
339. Kume K, Amano K, Yamada S, Kanazawa T, Ohta H, Hatta K, et al. Tofacitinib improves atherosclerosis despite up-regulating serum cholesterol in patients with active rheumatoid arthritis: a cohort study. Rheumatol Int. (2017) 37:2079–85. doi: 10.1007/s00296-017-3844-9
340. Hamar A, Hascsi Z, Pusztai A, Czókolyová M, Végh E, Pethő Z, et al. Prospective, simultaneous assessment of joint and vascular inflammation by PET/CT in tofacitinib-treated patients with rheumatoid arthritis: associations with vascular and bone status. RMD Open. (2021) 7:e001804. doi: 10.1136/rmdopen-2021-001804
341. Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheumatic Dis. (2015) 74:480–9. doi: 10.1136/annrheumdis-2014-206624
342. King B, Mostaghimi A, Shimomura Y, Zlotogorski A, Choi G-S, Blume-Peytavi U, et al. Integrated safety analysis of baricitinib in adults with severe alopecia areata from two randomized clinical trials. Br J Dermatol. (2023) 188:218–27. doi: 10.1093/bjd/ljac059
343. Elmariah SB, Smith JS, Merola JF. JAK in the [Black] box: A dermatology perspective on systemic JAK inhibitor safety. Am J Clin Dermatol. (2022) 23:427–31. doi: 10.1007/s40257-022-00701-3
344. Zhang L, Guo L, Wang L, Jiang X. The efficacy and safety of tofacitinib, peficitinib, solcitinib, baricitinib, abrocitinib and deucravacitinib in plaque psoriasis - A network meta-analysis. J Eur Acad Dermatol Venereology: JEADV. (2022) 36:1937–46. doi: 10.1111/jdv.v36.11
345. Simpson EL, Silverberg JI, Nosbaum A, Winthrop K, Guttman-Yassky E, Hoffmeister KM, et al. Integrated safety update of abrocitinib in 3802 patients with moderate-to-severe atopic dermatitis: data from more than 5200 patient-years with up to 4 years of exposure. Am J Clin Dermatol. (2024) 25:639–54. doi: 10.1007/s40257-024-00869-w
346. Schulze F, Neumann K, Recke A, Zillikens D, Linder R, Schmidt E. Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol. (2015) 135:1445–7. doi: 10.1038/jid.2014.547
347. Chen T-L, Huang W-T, Loh C-H, Huang H-K, Chi C-C. Risk of incident venous thromboembolism among patients with bullous pemphigoid or pemphigus vulgaris: A nationwide cohort study with meta-analysis. J Am Heart Assoc. (2023) 12:e029740. doi: 10.1161/JAHA.123.029740
348. Bonnesen K, Poulsen CFB, Schmidt SAJ, Sørensen HT, Schmidt M. Autoimmune blistering disorders and cardiovascular risks: A population-based cohort study. J Am Acad Dermatol. (2024) 91:82–90. doi: 10.1016/j.jaad.2024.02.052
349. Nash P, Kerschbaumer A, Dörner T, Dougados M, Fleischmann RM, Geissler K, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann Rheumatic Dis. (2021) 80:71–87. doi: 10.1136/annrheumdis-2020-218398
350. Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (PF-04965842): A selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. (2018) 61:1130–52. doi: 10.1021/acs.jmedchem.7b01598
351. Danese S, Argollo M, Le Berre C, Peyrin-Biroulet L. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut. (2019) 68:1893–9. doi: 10.1136/gutjnl-2019-318448
352. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. (2018) 2:23. doi: 10.1186/s41927-018-0031-x
353. Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. (2019) 7:e00537. doi: 10.1002/prp2.v7.6
354. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. (2019) 21:183. doi: 10.1186/s13075-019-1964-1
355. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheumatic Dis. (2021) 80:865–75. doi: 10.1136/annrheumdis-2020-219012
356. Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci United States America. (2016) 113:9852–7. doi: 10.1073/pnas.1610253113
357. van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. New Engl J Med. (2012) 367:508–19. doi: 10.1056/NEJMoa1112072
358. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. New Engl J Med. (2017) 376:652–62. doi: 10.1056/NEJMoa1608345
359. Chovatiya R, Paller AS. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol. (2021) 148:927–40. doi: 10.1016/j.jaci.2021.08.009
Keywords: autoimmune bullous diseases, pemphigus vulgaris, bullous pemphigoid, JAK inhibitors, cytokines, JAK-STAT pathway
Citation: Lin X, Li X, Zhai Z and Zhang M (2025) JAK-STAT pathway, type I/II cytokines, and new potential therapeutic strategy for autoimmune bullous diseases: update on pemphigus vulgaris and bullous pemphigoid. Front. Immunol. 16:1563286. doi: 10.3389/fimmu.2025.1563286
Received: 19 January 2025; Accepted: 20 March 2025;
Published: 08 April 2025.
Edited by:
Sara Massironi, Vita-Salute San Raffaele University, ItalyReviewed by:
Marco Folci, University of Milan, ItalyCopyright © 2025 Lin, Li, Zhai and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhifang Zhai, emhhaXpmMTAwNEAxNjMuY29t; Mingwang Zhang, bWluZ3dhbmd6aGFuZzJAdG1tdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.