 Shoulian Huang1,2
Shoulian Huang1,2 Yu Kang
Yu Kang Qing Zhang
Qing Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 April 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1561968
This article is part of the Research Topic Harnessing Big Data for Precision Medicine: Revolutionizing Diagnosis and Treatment Strategies View all 36 articles
Immune checkpoints, such as PD-1 and CTLA-4, are crucial regulators of immune responses, acting as gatekeepers to balance immunity against foreign antigens and self-tolerance. These checkpoints play a key role in maintaining cardiac homeostasis by preventing immune-mediated damage to critical organs like the heart. In this study, we explored the involvement of PD-1 and CTLA-4 in cardiovascular complications, particularly atherosclerosis and myocarditis, which can lead to heart failure. We conducted a comprehensive analysis using animal models and clinical data to assess the effects of immune checkpoint inhibition on cardiac function. Our findings indicate that disruption of PD-1 and CTLA-4 pathways exacerbates myocardial inflammation, accelerates atherosclerotic plaque formation, and promotes the development of heart failure. Additionally, we observed that immune checkpoint inhibition in these models led to increased infiltration of T lymphocytes, higher levels of pro-inflammatory cytokines, and enhanced tissue damage. These results suggest that PD-1 and CTLA-4 are critical in preserving cardiac health, and their inhibition can result in severe cardiovascular toxicity. Our study emphasizes the need for careful monitoring of cardiovascular health in patients undergoing immune checkpoint inhibitor therapies.
The inflammation in the human body is modulated via immune checkpoints that facilitate the communication between immune and non-immune cells and can either activate or attenuate the immune response (1). Thus, immune checkpoints are the key drivers of adaptive immunity that act as gatekeepers to maintain the immune homeostasis in the body and keep autoimmunity at bay (2, 3). Immune checkpoints are an assembly of proteins expressed on the cellular facet of Thymocytes and antigen-presenting cells that act as corresponding receptors and ligands for each other (4). Immune checkpoints regulate the process of self-tolerance by governing the intensity and efficacy of Thymocytes, thus shielding tissues and organs from inflammatory immune action (5). Immune checkpoints comprise of both stimulatory as well as inhibitory signaling pathways and a balance in these pathways helps to maintain the immune homeostasis required for self-tolerance (2). The stimulatory signals activate the immune system to act while the inhibitory signals are required to halt the immune response and thus preventing self-tissue destruction (6).
The antigen-specific immune response is referred to as adaptive immunity. In this type of immune response, the antigen is presented, recognized and processed resulting in the production of an excessive number of immune cells that invade the antigen. Simultaneously, ‘memory’ cells are generated that help in efficient response against antigens in future. CD8+ cytotoxic T cells and CD4+ helper T cells are the major cells involved in acquired immunity (7). Activation of T cells occurs in three steps (1, 7–10)-
● Binding of T cells to Major Histocompatibility Complex (MHC)-bound peptides on Antigen-Presenting Cells (APCs): T-cell activation begins when the T-cell receptor (TCR) on a naïve T cell binds to the peptide-MHC complex presented on the surface of an antigen-presenting cell (APC), such as a dendritic cell. This interaction is highly specific; the TCR recognizes a unique peptide presented on MHC molecules, which are either Class I (for CD8+ cytotoxic T cells) or Class II (for CD4+ helper T cells). This initial binding is crucial for the recognition of foreign antigens and for initiating the immune response.
● Co-stimulatory Signal to Prevent Anergy: In addition to the TCR-peptide-MHC binding, a second signal is required for full T-cell activation. This co-stimulatory signal typically involves the interaction of CD28 on the T cell with its ligands, CD80 or CD86, on the APC. This interaction prevents T-cell anergy, which is a state of unresponsiveness that occurs when T cells are exposed to antigen in the absence of appropriate co-stimulation. Without this co-stimulatory signal, the T cell may become tolerant to the antigen, thereby avoiding an autoimmune response.
● Cytokine Signaling to Modulate Immune Response: Upon successful engagement of both the TCR and co-stimulatory receptors, APCs release various cytokines that play a critical role in shaping the intensity and quality of the immune response. These cytokines, such as interleukin-2 (IL-2), promote T-cell proliferation, differentiation, and survival. They also help determine whether the activated T cells become effector cells (such as Th1, Th2, or Th17 cells) or regulatory T cells (Tregs), each of which plays a distinct role in the immune response. Cytokines influence the immune response by modulating the differentiation of T cells into specific subsets that either promote inflammation or suppress it to maintain immune homeostasis.
Multiple interactions between Thymocytes and APCs, stimulatory or inhibitory act as a checkpoint that moderates Thymocytes immune reaction and immuno-homeostasis. The co-stimulatory or co-inhibitory signals are proteins that can either activate (turn up) or inhibit (turn down) the immune cell response and are referred as immune checkpoint proteins (11).
The body maintains a balance between immune response against antigens and normal body tissues and organs. Certain body parts like the eyes, brain and heart, are safeguarded from the inflammatory immune response by specific regulatory mechanisms. Healthy human myocardial cells possess a small set of Thymocytes and have regulatory mechanisms that prevent the recruitment and activation of Thymocytes and memory Thymocytes. However, for maintaining and regulating tissue homeostasis and the repair mechanism of tissue, dendritic cells and macrophages are found in the myocardium wherein, these cells have potential of initiating effector naïve Thymocytes (12). Additionally, the richly supplied microvasculature of the heart not only provides oxygen and nutrients to the heart cells but also aid in supplying circulating Thymocytes to the heart. The present review will emphasize understanding these regulatory immune mechanisms in the heart that help to protect the cardiac from the immune response. The current review will focus on understanding various vascular and cardiac immune checkpoints involved in cardiac homeostasis and their role and mechanism involved in numerous cardiovascular complications that can incite heart failure like, atherosclerosis and myocarditis.
Immune checkpoints, such as PD-1 and CTLA-4, are pivotal in regulating these immune processes within the cardiovascular system. These checkpoints control the activation, proliferation, and exhaustion of immune cells, ensuring that inflammation does not become excessive and that self-tolerance is maintained. Dysregulation of immune checkpoint pathways can lead to aberrant immune responses, contributing to cardiovascular diseases such as atherosclerosis and myocarditis. In atherosclerosis, immune checkpoint inhibition can exacerbate inflammation and promote plaque instability, while in myocarditis, the unchecked activation of immune cells can lead to myocardial injury and fibrosis. This review focuses on understanding the mechanisms by which immune checkpoints influence cardiovascular homeostasis and their involvement in the development of heart failure through conditions like atherosclerosis and myocarditis.
The association amid inflammation and cardiovascular diseases has been recognized and adaptive immunity pertaining to Thymocytes and its activation have been found to be instrumental in cardiovascular diseases (13–16). Chronic inflammation in the arteries propels the progression of atherosclerosis. Atherosclerosis is the predominant driver of cardiovascular complications and is marked by the growth of plaques in the arterial walls. The inflammation activates immune cells that induce the secretion of cytokines as well as chemokines, which in turn promotes the accretion of immune cells at the inflammation site (17, 18). Endothelial dysfunction also promotes cardiovascular complications wherein factors like hypertension and elevated levels of cholesterol predispose arterial walls to the enhanced infiltration and accumulation of macrophages and monocytes. Immune cells i.e., macrophages promote the formation of foam cells by engulfing oxidized oxidized low-density lipoproteins. Foam cells are the initial drivers of plaque formation or atherosclerosis by inducing the development of lipidic streaks in the artery wall (19–22). Apart from macrophages, Thymocytes are the predominant player in cardiovascular complications. CD4+ T cells or helper Thymocytes modify into different subsets with specialized functionality. T-helper 1 or Th1 cells secrete pro-inflammatory cytokines like interferon (IF)-γ, which is a promoter of inflammation and destabilizer of plaque (23–25). While T-helper 2 or Th2 cells induce the production of anti-inflammatory cytokines viz., interleukin (IL)-4, 10, which hinders the progression of plaque. T-helper 17 or Th17 cells have been associated with prolonged inflammation and increased risk for plaque formation (26). Regulatory cells or Treg cells halt inflammation and stabilize the plaque (27). In the heart, natural killer cells (Nk cells) exhibit cytotoxic activity and induce pro-inflammatory cytokines production which promotes the progression and destabilization of plaques (28). Alternatively, B-cells, also play a key role in systemic inflammation and the advancement of atherosclerosis (29).
Chronic inflammation marked with distinctly high levels of pro-inflammatory myocardial and systemic cytokines alters the left ventricular function and induces its remodeling as well (30, 31). The lack of harmony between inflammation-promoting cytokines and anti-inflammatory cytokines contributes to heart failure (32). Amplified pro-inflammatory cytokines has been found to be linked with heart failure (33). Inflammatory cytokines that have been found to be associated with cardiac complications comprise IL-1, 6, 8, 18, 33 and IL-1RA (34). Inflammatory cytokines, primarily released by neutrophils, induce cardiac remodeling and alter cardiac function by assisting in inducing apoptosis of cardiomyocytes and activating the matrix metalloproteinase (35, 36). Activation of IL-18 has been found to be related to an elevated risk for cardiovascular diseases with hospitalization risk, especially in patients with congestive heart failure (37). Similarly, a higher amount of IL-6 has been correlated with an amplified rate of hospitalization in patients with heart failure and even mortality (38). Cytokine IL-8 is also positively correlated with heart failure risk and hospitalization as compared to normal patients (36, 39, 40). Higher levels of cytokines IL-1RA are found in heart failure with reduced ejection fraction, irrespective of the diabetic status of the patients (40). IL-1β endorses infiltration of leucocytes at the sites of injury and has been found to be correlated with cardiac events promoting and leading to heart failure (41). On the contrary, levels of cytokine IL-33 are reduced in patients with heart failure as against healthy subjects (41).
Galectin (Gal)-3, a lectin of galectin family, is implicated in the signaling of different intracellular pathways of inflammation, cellular migration, immune response and signaling, by binding with intra-cellular as well as surface ligands (42). Cardiac remodeling has been found to be associated with Gal-3 and is considered a bio-signature of fibrosis and heart failure. Gal-3 is associated with inducing a signaling pathway for transforming growth factor-β SMAD3 and thus resulting in induction of collagen production, proliferation of myofibroblast, cardiac hypertrophy and infiltration of macrophages (43–46). A clinical study findings suggest Gal-3 to be a positive indicator of heart failure-related mortality in the follow-through period of 6.5 years (47). Further, in this clinical evaluation, Gal-3 was also found to be positively associated with left ventricular end-diastolic volume enhancement (48).
The necrotic cells secrete injury-associated molecular signatures i.e., heat shock protein 60 or HSP 60 and high-mobility group box 1 (HMGB1) protein, in amplified amounts, in case of cardiac stress, hypertension, metabolic syndrome and ischemic injury. These proteins are identified by natural immune cells via pattern recognition receptors. Consequently, the secretion of pro-inflammatory cytokines by M1 macrophages and non-immune cells which initiates the deployment of phagocytic cells of the immune system, removal of apoptotic cells, and renewal of tissues (49). C-reactive protein (CRP), IL-6, 1β and TNF-α expressed by innate immune cells upon inflammation have been associated with myocardial infarction, coronary disease and stroke (50). Increased vascular permeability and reduced amount of nitric oxide is associated with endothelial dysfunction. Pro-inflammatory cytokines released by macrophages promote rapid growth of mural cells and aggregation of particles of oxycholesterol-containing low-density lipoprotein (LDL) in vessels (51). Macrophages M1 is found to be associated with atherosclerotic plaque leading to acute coronary syndrome. Cytokines IL-6, 12, reactive nitrogen species and reactive oxygen species are pro-atherosclerotic as they increase the oxidative stress in atherosclerosis. The carotid artery plaques and its progression have been found to be linked with amplified levels of Th1, Th17 and IL-17, 23, 21 (52). Senescent CD14+CD16+ monocytes and CD4+ effector memory T cells (CD3+CD4+CD45RA−CD45RO+CCR7−) are found in atherosclerotic plaques (49, 53). While, late-differentiated CD4+CD28− peripheral Thymocytes have been linked with acute coronary events (54).
Originating from the thymus, matured naïve T lymphocytes (both CD8+ and CD+4) reach the secondary lymphatic organs via systemic circulation. Systemic circulation also helps in the accumulation of proteins and antigens.
Activation of T-lymphocytes can occur in two ways, one of them being activation mediated via dendritic cells. These antigens are then presented to the T-lymphocytes as peptide-MHC complexes expressed on the facet of antigen-presenting cells. The dendritic cells in the secondary lymphatic organs are the antigen-presenting cells that are critical for the activation of naïve T lymphocytes, However, the presentation of antigens by the antigen-presenting cells must be accompanied by the presence of tissue injury or pathogen, i.e., danger signals, for dendritic cells to express the co-stimulatory surface ligands. Thus, activation of T-lymphocytes occurs only when both the steps are present i.e., antigen presentation by the antigen-presenting cell, in combination with expression of co-stimulatory ligands on the facet of dendritic cells as a response to antigens or injury. Few costimulatory proteins for naïve T cells are from the immune serum globulin superfamily which includes, B7-1 (CD80) and B7-2 (CD86). The CD80 and CD86 are found on dendritic cells having prior exposure to pathogens as well as innate immune stimuli. These two synchronized steps result in the activation of naïve T lymphocytes prompting modification of Thymocytes to cytotoxic CD8+ or effector CD+4 helper cells. Thus, costimulatory signals resulting in immunological synapse formation of Thymocytes are limited to conditions wherein there are specific risks involved. Further, for functional activation, effector Thymocytes identify the antigen at the place of inflammation (55).
The alternative method of activation of T-lymphocytes is not dependent upon dendritic cells wherein antigen MHC presentation is carried out by cells other than dendritic cells. Also, in this case, the co-stimulation step is not pertinent for the immunological synapse formation of Thymocytes. In this approach, CD275 is an inducible thymocyte co-stimulator which is a part of an immunoglobulin superfamily found on B- cells and binds with the corresponding CD278 expressed on CD+4 T helper cells. The alternative costimulatory pathway also includes the binding of tumor necrosis factor protein receptors found on Thymocytes with corresponding tumor necrosis factor protein found on APCs, for e.g., for the stimulation of CD8+thymocyte response, ligand found on APCs i.e., 4-1BB ligand or 4-1BBL binds with 4-1BB or CD137 found on thymocytes. Similarly, another ligand found on antigen-presenting cells is CD252 or OX40 ligand interacts with CD134 or OX40 found on thymocytes (56, 57).
Apart from the above mechanisms, there are an additional mechanism that limits the activity of thymocytes and imparts the capability for self-tolerance toward its own cells/tissues. The mechanism that suppresses the action of thymocytes via programmed cell protein death-1 (PD-1) or CD279 and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) or CD152. These proteins are homologous to CD28 structurally but functionally contradictory i.e., PD-1 and CTLA-4 inhibit the functioning of thymocytes. Immediately after activation of thymocytes, CTLA-4 is found on the cellular facet of thymocytes, including naïve Thymocytes, regulatory T cells (Treg) and memory Thymocytes. However, Treg and memory Thymocytes express CTLA-4 when exposed chronically to antigens which results in Thymocyte exhaustion (58). The CTLA-4 expression is antigen exposure dependent and is expressed highly on exhausted thymocytes. The CTLA-4 competitively contests with CD28 to bind with B7, thus hampering the B7-CD28-dependent activation of T-lymphocytes (58, 59).
PD-1 or CD274/PD-2 or CD273 expression on T lymphocytes is initiated by the exposure to antigen. PD-1 and PD-2 are part of the immune serum globulin superfamily and are structurally homologous to CD28. The expression of ligands for PD-1 i.e., PD-L1, dendritic cell, epithelial parenchymal, endothelial cells and macrophages, is induced by the cytokines interferon-gamma (IFN-γ) and type-1 IFNs, while PD-L2 is found only on bone-marrow derived antigen-presenting cells (60, 61). Consequently, the binding of CD28 and thymocyte receptors activates the protein tyrosine kinases pathway, however, the binding of PD-1 and PD-L1 results in the deployment of protein tyrosine kinases, which hinders the protein tyrosine kinases pathway and suppresses thymocyte activity. Thus, both PD-1 and CTLA-4 shuts off the antigen-activated co-stimulatory thymocyte activation, thus PD-1 and CTLA-4 are referred to as co-inhibitors or immune checkpoints (62).
Apart from PD-1 and CTLA-4, other immune checkpoints are, Lymphocyte activation gene-3 (LAG-3) or CD223. LAG-3 also belongs to the immune serum globulin superfamily and is found on thymocytes, B-cells, macrophages and dendritic cells. LAG-3 inhibits the antigen presentation by binding with the class II MHC molecules (63–65). Another member of the immune serum globulin superfamily, which is found on natural killer (NK) cell, Thymocyte and macrophages, is the T cell immunoglobulin-3 (TIM-3) and inhibits the thymocyte activation by binding with carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM-1), galectin-9, and phosphatidyl serine (66–69). Another immune checkpoint is the thymocyte immune receptor containing Ig and ITIM domains (TIGIT), found upregulated in thymocytes and NK cells that bind with CD112 and CD155. TIGIT, LAG-3 and TIM-3 are found in amplified levels on the exhausted T cells (70, 71).
Another important component for self-tolerance and non-susceptibility of commensal microbes are the Treg cells, of which CD4+CD25+FoxP3+ Thymocytes are most widely studied. Studies show that mutation in FoxP3 can incite autoimmune diseases (72). Treg cells express PD-1, LAG-3 and CTLA-4 immune checkpoints (73, 74).
CTLA-4 is a surface receptor found on thymocytes (primarily on activated CD4+thymocytes, CD8+thymocytes and Treg cells), which is negatively linked with thymocyte activation and thus enforces preservation of cardiac tissues and maintains cardiac homeostasis (75). Primarily, CTLA-4 works as a regulator that controls the magnitude of activation of thymocytes (76, 77). CTLA-4 is a co-inhibitory receptor that functions in contradiction with the functioning of CD28. CD 28 is a co-stimulatory surface protein found on thymocytes which is a pre-requisite for the activation of thymocytes. The ligands for CD 28 are CD 80/86 or B-7 1/2 found on the cellular facet of antigen-presenting cells. The linkage between CD 28 and CD 80/86 incites the secretion of cytokines and cellular proliferation (78). CTLA-4 is structurally similar to CD28 and has a common affinity toward CD80/86, although CTLA-4 has a higher affinity to CD 80/86, thus CTLA-4 competitively and preferentially binds to CD80/86 which suppresses the multiplication of thymocytes and production of interleukin (79–83). The CTLA-4 acts as a ‘switch off’ indicator that controls the proliferation of thymocytes. The expression of CTLA-4 occurs post-activation of Thymocytes, however, it acts as a controller that confines the proliferation of thymocytes (84, 85). The immune response of CTLA-4 is manifested on CD4+thymocyte as, an amplification of Treg cells and halts activity of Th cell (86, 87). CTLA-4 promotes cardiac protection from the immune response (88). Animal studies have shown that CTLA-4 inhibitors resulted in enhanced myocardial inflammation and eventually myocarditis (89). Lymphoproliferative disease wherein infiltration of lymphocytes in multiple organs and tissue inflammation and necrosis like, myocarditis, is seen in animal knockout models devoid of CTLA-4 (90, 91). CTLA-4 deficit even in Treg cells is capable of myocarditis induction. The absence of expression of CTLA-4 in Treg cells results in lymphoproliferation wherein infiltration of myocardial cells and impairment of myocytes. Additionally, the absence of CTLA-4 expression further exacerbates the cardiac complications (86, 92). Study findings suggest that cardiac tissues are protected considerably from the action of cytotoxic Thymocytes due to the protective effect imparted by CTLA-4 (93). These findings indicate cardiotoxic effects pertaining to the suppression of CTLA-4 (94). The protective effect of CTLA-4 has been reported in several animal studies wherein increase in mRNA of CTLA-4 has been correlated with amplification of Treg cells and reduction in atherosclerotic plaque (95–97).
The function of preserving peripheral tolerance and fend off autoimmune response is carried out by PD-1/PD-L1 (98, 99). PD-L1 or CD274 or B7-H1, is the co-inhibitory ligand for PD-1 that controls the activity of thymocytes (100). PD-1 is found on the facet of activated thymocytes while its corresponding ligand, PD-L1 is found on antigen-presenting cells (101, 102). Latent and stimulated thymocytes, B-cells, dendritic cells, myeloid cells and immune-privileged tissues like, the placenta, brain, heart, endothelial cells, muscles and pancreas demonstrate PD-L1 while monocytes and dendritic cells solely demonstrate PD-L2 (103). PD-L1/L2 binding with PD-1 forms the basis of immune response for self-tolerance (104, 105). PD-1/L1 binding helps to restrain the excessive immune response which can induce cellular destruction and autoimmune effect. Activated PD-L1- PD-1 pathway results in suppression of effector thymocytes, which helps in preserving self-tolerance along with resolving inflammatory response (106). However, inhibition of PD-1/L1 interaction can induce an inflammatory immune response in protected tissues like cardiac tissues. Accordingly, the paucity of interaction between the PD-1/L1 pathway incites exacerbation of lesions in atherosclerosis (107). The development of cardiomyopathy in animals devoid of PD-1 also indicates the critical role of PD-1/PD-L1 in maintaining self-tolerance toward critical organs like the heart (99). Reduced levels of PD-1 and PD-L1 on thymocytes and antigen-presenting cells are observed in patients with coronary artery disease, which induces higher levels of CD4+ and CD8+ cells along with an amplified amount of pro-inflammatory cytokines (108).
In addition to PD-1 and CTLA-4, LAG-3 has recently emerged as a critical immune checkpoint involved in regulating immune responses in cardiac tissues. LAG-3 functions as an inhibitory receptor that downregulates T cell activation and contributes to immune tolerance. It is expressed on activated T cells and plays a role in preventing excessive immune responses that could lead to tissue damage. In the context of cardiovascular diseases, including atherosclerosis and myocarditis, LAG-3 may serve to modulate the inflammatory response, reducing the risk of autoimmune damage to the heart. Its expression in the myocardium during inflammatory conditions suggests that LAG-3 could be a potential target for therapeutic interventions aimed at controlling immune-mediated cardiovascular injuries. Future studies are needed to further elucidate its role in cardiac homeostasis and its potential as a therapeutic target in cardiovascular diseases.
The cardiotoxic events observed in patients treated with immune checkpoint inhibitors have highlighted the pivotal role of immune checkpoints in regulating cardiovascular health (109–116). For example, myocarditis, one of the most severe immune checkpoint inhibitor-related toxicities, has been documented in cases where patients receiving anti-PD-1 therapies, such as pembrolizumab and nivolumab, developed fatal myocarditis, resulting in cardiac arrest in some instances. The inhibition of immune checkpoints led to the activation of CD8+ T cells and macrophages, which infiltrated cardiac tissue and induced inflammation, compromising myocardial function. Additionally, left ventricular dysfunction, even in the absence of myocarditis, has been observed in patients treated with CTLA-4 inhibitors like ipilimumab. This dysfunction was linked to systemic inflammation and cytokine release, which impaired cardiomyocyte contractility and promoted tissue remodeling. Vasculitis has also been seen in patients treated with immune checkpoint inhibitors, as exemplified by a case where nivolumab-induced acute coronary vasculitis led to lymphocyte infiltration in the coronary vessels, causing endothelial dysfunction and plaque rupture. Lastly, pericarditis has been reported in patients, presenting with chest pain and fluid accumulation around the heart, which resulted from inflammation in the pericardium following immune checkpoint activation. These cases emphasize the complex mechanisms through which immune checkpoint inhibition can drive cardiovascular injury, underscoring the need for careful monitoring and management of cardiovascular health in patients receiving such therapies.
The unrestrained activity of the immune cells pertaining to association with the cardiac tissues, especially post-immune checkpoint inhibitor therapy can drive cardiac injury. Inhibition of immune checkpoint inhibitors drives an attack of lymphocytes on the normal, non-antigenic and non-cancerous tissues and cells of the body. Cardiac lymphocytic infiltration leading to myocarditis was observed in an animal study wherein mice were deficient of CTLA-4. A massive upsurge in levels IL-4, colony-stimulating factor and IFN-γ was observed in CTLA-4 deficient mice as against normal mice, which eventually resulted in the induction of lethal myocarditis in animals. Thus indicating the criticality of CTLA-4 in maintaining cardiac immune equilibrium. The findings of this study also depict the importance of estimating the levels of these cytokines as a prognostic indicator of myocarditis, especially in patients getting immune checkpoint inhibitor therapy (90).
Apart from CTLA-4, other immune checkpoints like myocardial PD-1 have also been found to affect cardiac homeostasis. The PD-1 immune checkpoint inhibit immune response mediated inflammatory response in cardiac tissues via increased expression of GADD153 (117, 118). PD-1 The animals that were devoid of PD-1 were found to develop autoimmune-based dilated cardiomyopathy wherein a significant amount of auto-immune antibodies targeted toward cardiomyocytes (119). The subjects treated with PD-1 inhibitors that developed myocarditis were found to have a significant amount of infiltrating thymocytes and macrophages similar to that found in the tumors as well as skeletal muscle cells (119). The role of PD-1 in cardiac injury was further confirmed by a study wherein IFN-γ mediated increase in PD-1 resulted in cardioprotective function. This also confirms that loss in PD-1 can drive cardiac injury (120). Loss or deficiency in the levels of PD-1 induces a rise in the levels of cardio-toxic cytokines resulting in cardiac injury as seen in the infarct area post reperfused acute myocardial infarction (117). In an animal study, wherein the animals were deficient in PD-1, exhibited the development of autoimmune myocarditis. The animals were found to have excessive levels of infiltrating myeloid cells, CD8+ and CD4+ thymocytes in the cardiac tissues along with elevated levels of auto-immune antibodies against cardiac myosin (121). A diagramatic representation of involvement of PD-1/PD-L1 in cardiac homeostasis and cardiac injury is shown in Figure 1.
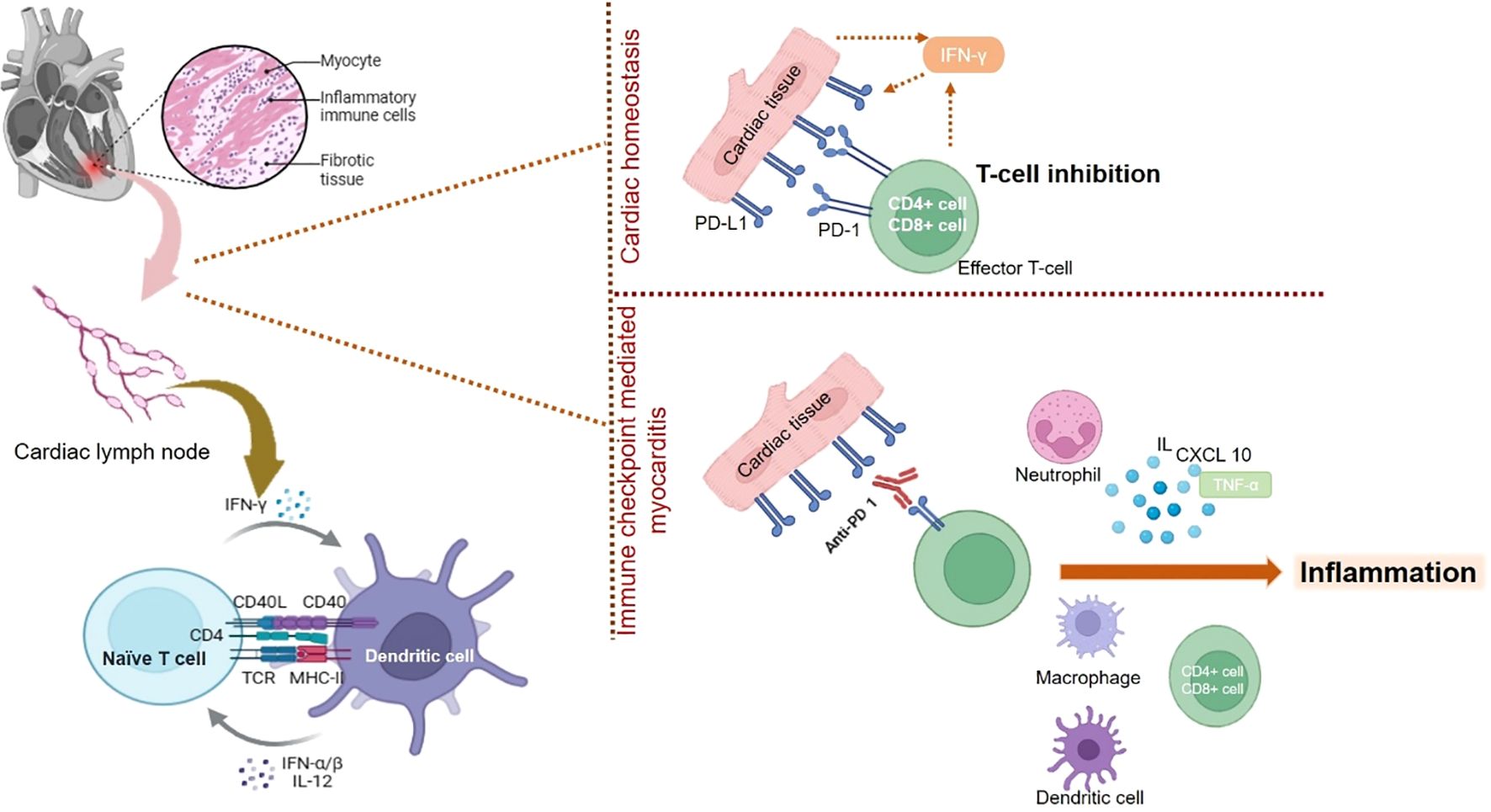
Figure 1. The mechanistic role played by PD-1/L1 in maintaining cardiac homeostasis and its inhibition results in immune checkpoint mediated myocarditis. Blockade of PD-1/L1 disrupts the cardiac homeostasis resulting activation of thymocytes or T-cells, which results in expression of pro-inflammatory condition leading to myocarditis.
Immune checkpoint-related cardiovascular events also involve vascular injury and subjects having a higher neutrophils to lymphocyte (NLR) ratio are at higher risk of developing vascular injury leading to cardiovascular events. However, levels of C-reactive protein were found to be unrelated to the incidence of vascular injury (122). The most prominent role of immune checkpoints in vascular injury is pertaining to the advancement of atherosclerosis. Deposition of lipidic cells beneath the endothelium of large arteries is referred to as Atherosclerosis (123). In atherosclerosis, under the influence of NF-κB and with the help of adhesion molecules like, vascular adhesion molecule-1 (VCAM-1), E-selectin and intracellular adhesion molecule-1 (ICAM-1), T-helper cells and endothelial macrophages are employed in the arterial endothelium. These macrophages phagocytose and oxidize the low-density lipoproteins to eventually result in the formation of foam cells via the induction of endothelial lesions. Post the foam cell creation, Thymocytes promote the formation and development of plagues via release of cytokines viz., IL-9, 11 and 12. Eventually, foam cells and plaque result in the formation of atheroma or atherosclerotic plaque (123, 124). The immune checkpoints, PD-1, PD-L1 and CTLA4 are inversely correlated with plaque formation and thus inhibition of the immune checkpoints results in enhanced recruitment of thymocytes which drive the development of plaque via induction of endothelial lesions (125, 126). This is also reflected in higher incidence (almost 4 to 5 fold) of cardiovascular adverse events viz., coronary revascularization, myocardial infarction and ischemic stroke, in cancer patients being administered immune checkpoint inhibitors (127). Experiments in animal models have shown that myeloid cells that are deficient in the expression of PD-1, promote the genes implicated in cholesterol synthesis and attenuate the genes involved metabolism of cholesterol, thus amplifying cholesterol levels, which is an established determinant of atherosclerosis (128). A diagrammatic representation of vascular injury leading to pro-atherogeneic activity due to inhibition of PD-1/L1 is shown in Figure 2.
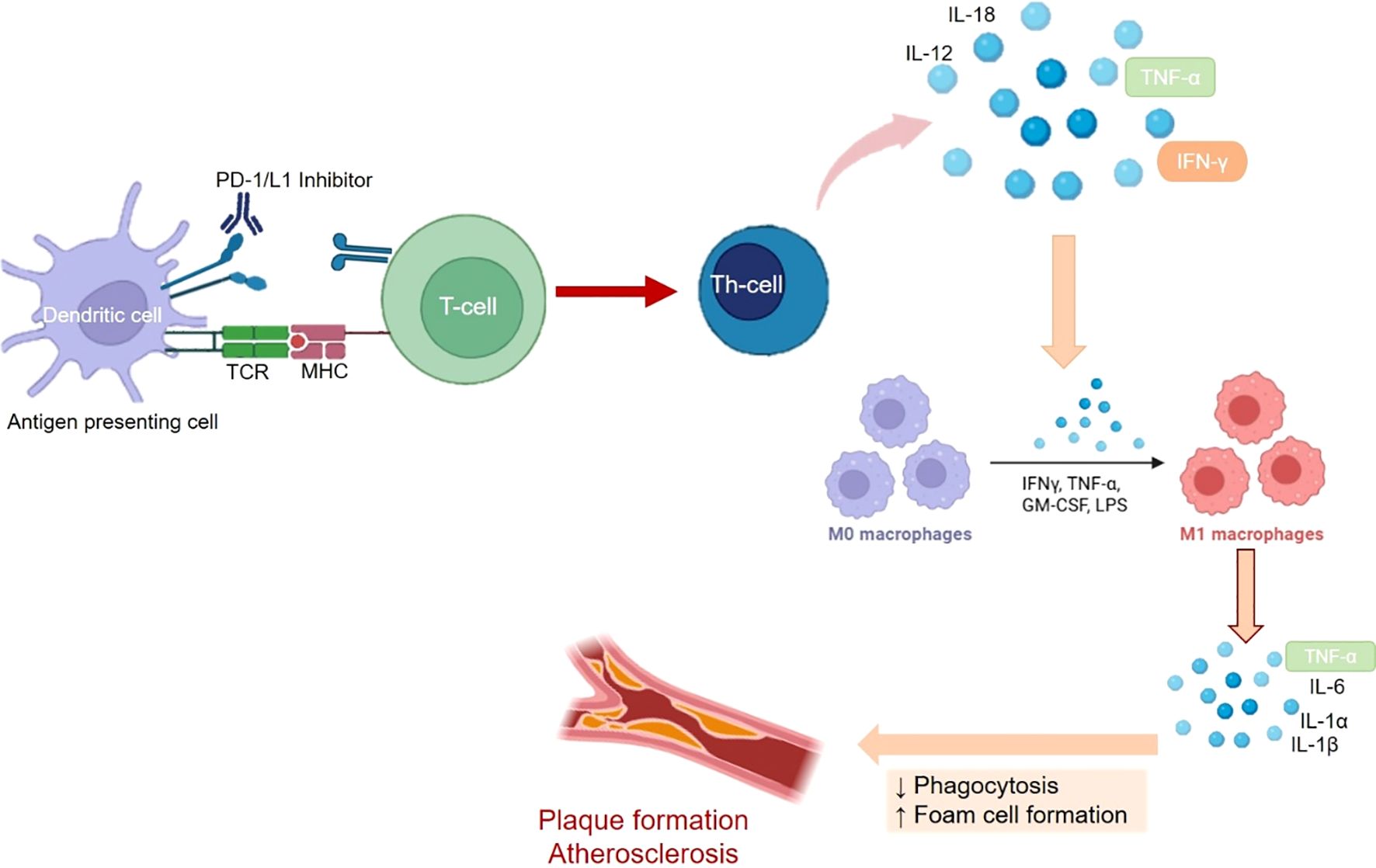
Figure 2. The mechanistic role of played by PD-1/L1 in immune system mediated development of Atheroscelrosis. Blocking of PD-1/L1 results in activation of thymoctes or T-cells resulting in expression of cytokines like IFN-γ, ILs and TNF-α. This further stimulates the differentiation of macrophages and subsequent release of cytokines like, TNF-α and Ils, which promote the development and proliferation of atherosclerotic plaque.
Atherosclerosis is a major cardiovascular condition that contributes to the development of heart complications, leading to increased morbidity and mortality. It is primarily a chronic inflammatory disease of the arteries, marked by the accumulation of lipids in plaques (129, 130). The progression and destabilization of these plaques are significantly influenced by both innate and adaptive immunity. After endothelial injury, monocytes infiltrate the arterial walls and differentiate into macrophages. These macrophages uptake lipids and secrete pro-inflammatory cytokines, further aggravating the inflammation (131). Antigen-presenting cells, such as dendritic cells, are also recruited to the lesion site, where they activate Thymocytes, amplifying the inflammatory response. Effector Thymocytes, in turn, produce excessive pro-atherogenic cytokines, which increase plaque size and promote its destabilization (132).
Immune checkpoints, both co-stimulatory and inhibitory, regulate Thymocyte activation and play a crucial role in controlling the inflammatory response in atherosclerosis. The activation of these checkpoints can either stimulate or suppress Thymocyte function, influencing the development and growth of atherosclerotic plaques (133). Different immune checkpoints influence and drive the evolution and growth of plaque through their distinct pathway (Table 1). A digrammatic representation of involvement of different checkpoints in atherosclerosis is shown in Figure 3.
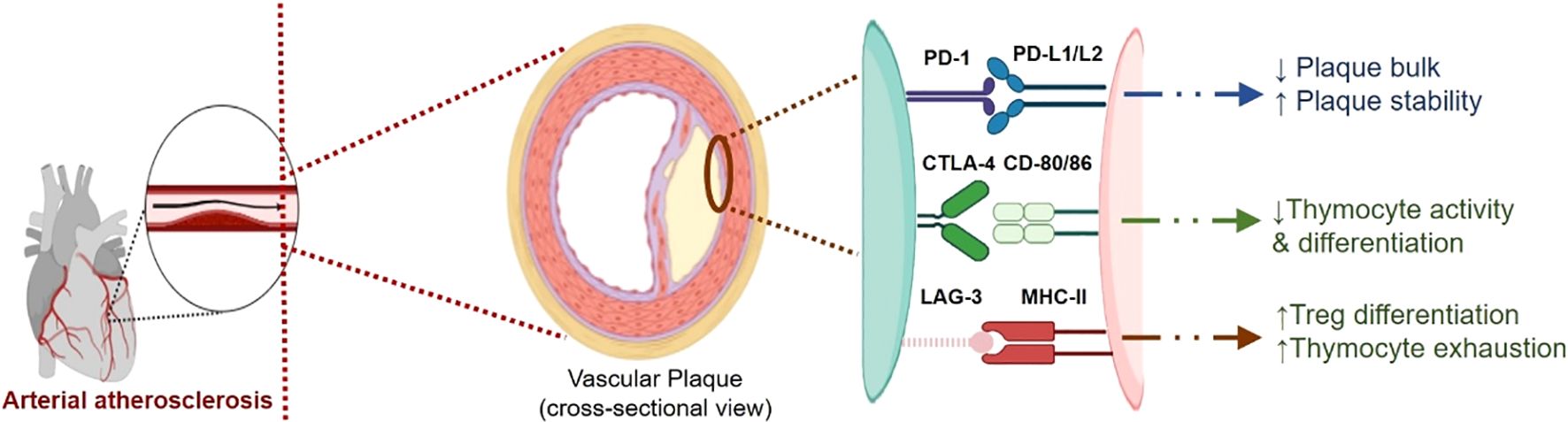
Figure 3. A digrammatic representation of involvement of different checkpoints viz., PD-1/L1, CTLA-4 and LAG-3, in atherosclerosis. The immune checkpoints imparts cardio-protective role and helps in maintaining cardio-homeostasis. These immune checkpoints imparts plaque stability and reduce the proliferation of plaque by controlling inflammation. Inhibition of these checkpoints disrupts this balance and thus promoting plaque development and proliferation.
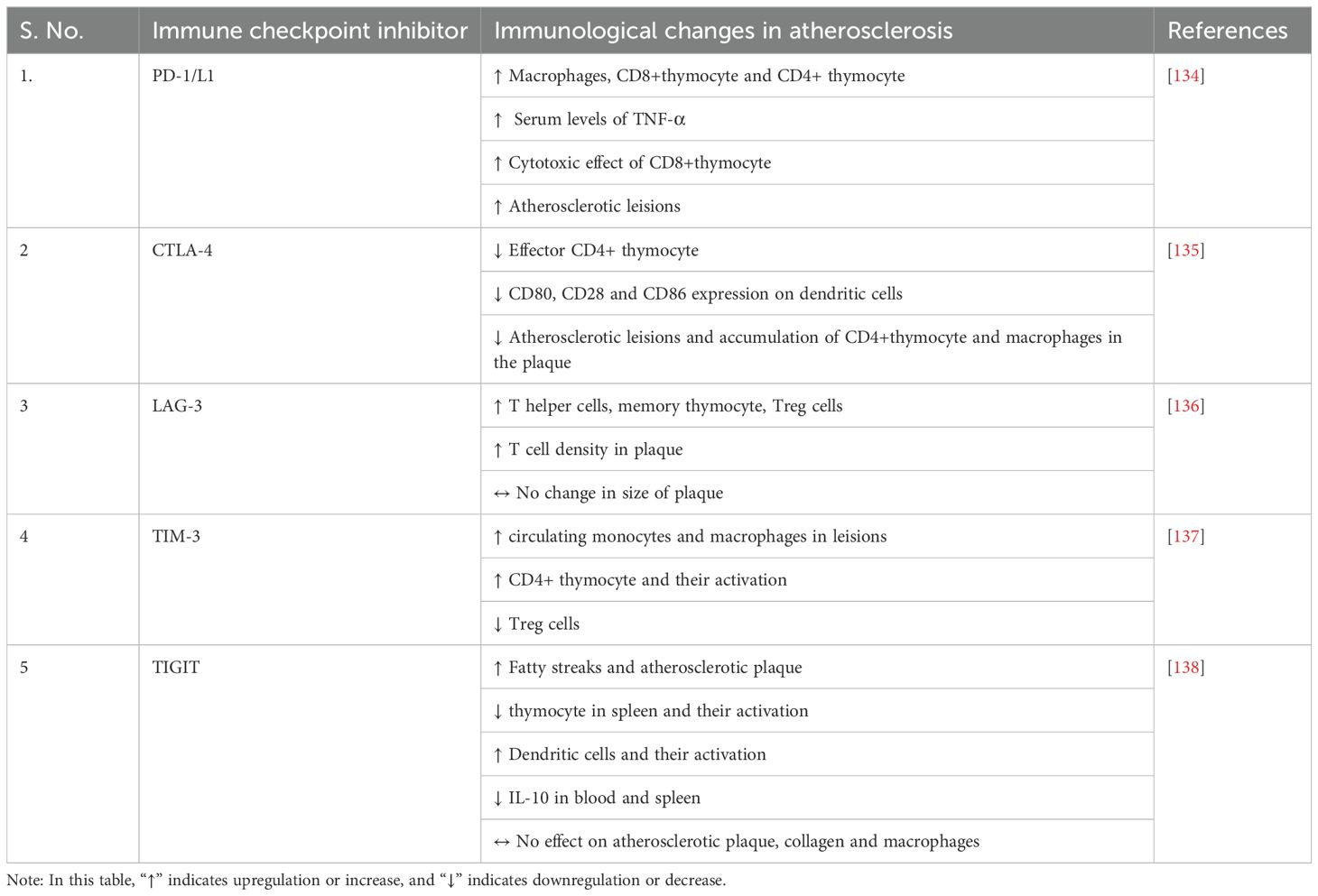
Table 1. Changes in immune checkpoints and other immune cells in atherosclerosis.
Several immune checkpoint pathways are involved in the progression of atherosclerosis, offering potential therapeutic targets for managing this disease. For instance, inhibition of immune checkpoints like PD-1 and CTLA-4 has shown promise in reducing inflammation and stabilizing plaques, though caution is needed due to the potential for immune-related adverse effects. Targeting these pathways may offer a dual benefit: reducing plaque progression and minimizing cardiovascular events, while avoiding excessive immune activation that could lead to other complications. Further research into these therapeutic strategies, particularly the use of immune checkpoint inhibitors in conjunction with traditional cardiovascular treatments, is needed to fully understand their potential in managing atherosclerosis.
Analysis of clinical investigation on immune checkpoint inhibitors has shown that there is a higher chance of developing cardiovascular complications viz., atherosclerosis, stroke and myocardial infarction (127, 139, 140). In fact, one of the studies has shown that almost 3-fold increase in the occurrence of atherosclerosis in human participants being administered immune checkpoint inhibitors (127). These findings lead to the assumption that thymocyte inhibition exerts a cardiovascular protective effect and thus reduces the chances of developing atherosclerosis. Thymocyte stimulation and growth is inhibited after interaction of PD-L1/PD-L2 with the corresponding PD-1 found on thymocytes. Activated Thymocytes exhibit inflammatory pro-atherogenic activity. Thus, PD-L1/PD-1 binding is inversely related to the pro-atherogenic activity.
This was confirmed in an animal study wherein knockout animals lacking PD-L1 or PD-L2 exhibited high atherosclerotic load along with elevated levels of CD4+ and CD8+ thymocytes. These animals also had amplified levels of Tumor Necrosis Factor (TNF)-α and amplified cytotoxicity of CD8+ thymocytes. Also, antigen-presenting cells from these knockout animals were found to have higher in-vitro efficiency in activating CD4+ thymocytes, in the presence as well as unavailability of cholesterol loading. Thus, the absence of PD-L1/PD-L2 was linked with a higher amount of plaque formation confirming the pivotal role played by PD-1/PD-L1 immune checkpoints in modulating plaque formation (141, 142).
Cytokine interferon (IFN)-γ, primarily produced by T-helper cells (Th cells) is a determinant in promoting atherosclerosis by deploying macrophages and thymocytes, promoting cytokine release and increase in endothelial antigen-presenting cells. Blocking or reduced expression of IFN-γ has been found to reduce atherosclerotic plaque size (143, 144). The linkage between PD-1 and PD-L1 results in differentiated Treg cells which interferes with the production of IFN-γ and TNF-α by the Th cells (145, 146). The stability of plaque is also influenced by IFN-γ. A stable atherosclerotic plaque reduces the risk of plaque breakdown and its subsequent dislocation, which may induce other cardiac complications like myocardial infarction and stroke. The cytokine IFN-γ can destabilize the plaque by hindering the propagation of vascular smooth muscles and the synthesis of cholesterol. Conversely, Treg cells express multiple immune checkpoints and promote the creation of anti-inflammatory cytokines viz., IL-10 and TGF-β, as they inhibit differentiation of inflammatory effector thymocytes. Thus, Treg cells exert and promote anti-atherogenic activity (147). This is confirmed in a study wherein attenuation in the population of Treg cells, deficit in IL-10 and interruption of TGF-β promote atherogenesis and can even worsen plaque formation (148, 149). Persistent exposure to inflammation and antigens can result in thymocytes exhaustion wherein these cells lose their function. This ensues reduction in the propagation of thymocytes, the release of cytokines and enhanced repressive expression of immune checkpoints like, LAG-3 and PD-1 (150). Exhausted thymocytes that express PD-1 are found in the atherosclerotic lesions. Consequently, inhibiting PD-1 by immune checkpoint inhibitors can reactivate the exhausted thymocytes, which can intensify and facilitate atherosclerosis (126).
Endothelial cells express PD-L1 under the influence of IFN-γ and TNF-α (151). This was confirmed in a study wherein PD-L1 was expressed by human umbilical cord vein endothelial cells (HUVECs) under the influence of IFN-γ (152). Constitutively HUVECs do not express PD-L1 (153), however, under the influence of IFN-α, β, γ and TNF-α HUVECs express PD-L1 (154). This indicates the critical role played by cytokines, IFN-α, β, γ and TNF-α, in promoting PD-L1 in the endothelial cells. Vascular endothelial growth factors (VEGF) also critically influence the appearance of PD-L1 in endothelial cells (155). It is well established that for cancer cells that exhibit PD-L1, protection from the cytotoxic and apoptotic effect of IFN-γ occurs by interfering with the JAK/STAT3/caspase7-dependent pathway (156, 157). Lymphatic endothelial cells expressing PD-L1 exhibit a similar pattern that helps them to prevent apoptosis (158). Thus, endothelial cells expressing PD-L1 exhibit the potential to counter the immune reaction by downregulating the CD8+ thymocytes and amplifying the activity of Treg cells, thus protecting endothelial cells from the pro-atherosclerotic effects (159).
CTLA-4 immune checkpoints have been found to facilitate the thymocyte-mediated inflammatory development of atherosclerosis. Thymocyte stimulation is negatively controlled by CTLA-4. This was confirmed in animal studies that were devoid of CTLA-4 wherein such animals exhibit enlarged lesions in the aorta. Also, the animal receiving antibodies blocking CTLA-4 exhibits an almost two-fold increase in plaque size and area with elevated Thymocyte and macrophage amounts in these plaques along with reduced collagen and smooth muscle cell content (160). Contrary to this study, animals that overexpress CTLA-4 or are administered an analogue of CTLA-4 decreased the population of CD4+thymocyte, reduction in cellular differentiation and generation of pro-inflammatory cytokines (IL-10 and IF-γ) and reduction (~58.5%) in thickness of the intima (135, 161). At the cellular level, induced pluripotent stem cell-derived cardiomyocytes (iPSC-CM), that were exposed to hypoxia which simulates the ischemic cardiac injury post-myocardial infarction, exhibit amplified amounts of CD80 and CD86 at the genetic level. The appearance of CD80 and CD86 at protein and genetic levels is increased post-cardiac injury (162). Amplification in the expression of CTLA-4 results in a drop in lesional CD4+ thymocytes. Also, a reduction in effector thymocytes in lymphatics due to disability of thymocytes to differentiate and grow under the conditions of reduced cytokine levels (135).
Intracellular dendritic cell binding of CTLA-4 and CD80/86 incites expression of Indeolamine 2,3-dioxygenase (IDO) (163). Augmentation in IDO results in halting thymocyte proliferation resulting in reduced activation of thymocytes and enhanced apoptosis of thymocytes. As a result, treatment with anti-CTLA-4 results in alleviating CD80/86 interaction which reduces the self-tolerance (164, 165).
The LAG-3, expressed on thymocytes, is the ligand that binds with MHC II (166). Clinical studies have shown that immune checkpoint LAG-3 has not been found to be correlated with cardiac complications like atherosclerosis, and coronary heart disease, because of which the FDA has approved them for anti-tumor therapy. Although long-term follow-up evaluation data is lacking. However, an independent relationship between LAG-3 and coronary heart disease has been reported and atherosclerotic plaque expresses LAG-3 in exhausted Thymocytes. LAG-3 is a potential predictive tool as a determinant of coronary heart disease and an indicator of plasma high-density lipoprotein-cholesterol (150, 167). The function of LAG-3 was evaluated in bone marrow chimeras hematopoietic cells devoid of LAG-3 and knockout mice devoid of LAG-3. The LAG-3 deficiency, as well as treatment with anti-LAG-3 antibodies, resulted in an enhanced population of memory cells and Thymocytes reducing IFN-γ, which was balanced with an amplification in Treg cells. Consequently, the plaque size remains unaffected, although there was an amplification in the stimulated thymocyte population in the plaque (136). LAG-3 and PD-L1 both aid in enhancing the stability of endothelial plaques (159).
Apart from the above immune checkpoint functionality in atherosclerosis, self-antigens also contribute to the development of plaque. A self-antigen, keratin-8 was found to increase the levels of PD-1 appearance in the human peripheral blood mononuclear cells (PBMCs) as against PBMCs obtained from coronary artery disease patients. This finding suggests that keratin-8 can potentially stimulate PD-1 expression thus restricting the thymocyte reaction and imparting protective function in anomalous patients of coronary artery disease (168, 169). Further, reports suggest that gastric adenocarcinoma cells with PD-L1, exhibit higher intracellular intake of lipids via fatty acid binding protein (Fabp4/5) elevation. Similar activity is exhibited by PD-L1-expressing cells. Consequently, interferance with PD-L1 results in reduced Fabp4/5 protein level and decreased intracellular intake of lipids. This results in higher availability of free lipids at the plaque site causing aggravation of atherosclerosis (170).
Myocarditis, an inflammation of the myocardium caused by infectious or non-infectious agents, can lead to severe complications, including heart failure and dilated cardiomyopathy (171). Although relatively rare, myocarditis is an important cause of heart failure and can manifest clinically through a range of symptoms such as palpitations, cardiogenic shock, arrhythmias, and heart failure (172–175). Chronic inflammation in myocarditis can lead to myocyte necrosis, fibrosis, and scarring, ultimately contributing to the development of dilated cardiomyopathy (174).
Recent clinical studies have emphasized the growing concern of myocarditis associated with immune checkpoint inhibitors (ICIs). A retrospective analysis found that 50% of deaths related to immune checkpoint inhibitors were due to myocarditis (17). This condition can be fulminant, leading to cardiogenic shock, ventricular arrhythmias, and, in severe cases, cardiac arrest (176). The frequency of myocarditis is notably higher with PD-1/PD-L1 pathway inhibition compared to CTLA-4 blockade, with incidence rates of 0.41% and 0.07%, respectively (17). Recent studies have also shown that patients receiving PD-1/PD-L1 inhibitors have a higher risk of developing myocarditis (69.1% of cases), compared to those treated with CTLA-4 inhibitors (20%) (177, 178).
Furthermore, combination therapies involving PD-1/PD-L1 inhibitors and other immune checkpoint modulators, such as LAG-3 inhibitors, have led to an even higher incidence of myocarditis, with up to 1.7% of patients experiencing this adverse event, compared to 0.6% with single-agent PD-1/PD-L1 therapy (179). Moreover, there have been several reports of myocarditis occurring in patients receiving combination therapies of ICIs with radiotherapy or chemotherapy, exacerbating the risk of cardiotoxicity (180–183).
These findings highlight the critical need for careful monitoring and management of cardiac health in patients receiving immune checkpoint inhibitor therapies, particularly in combination with other treatments. A diagrammatic representation of the involvement of different immune checkpoints, including PD-1/PD-L1, CTLA-4, and LAG-3, in myocarditis is shown in Figures 4, 5.
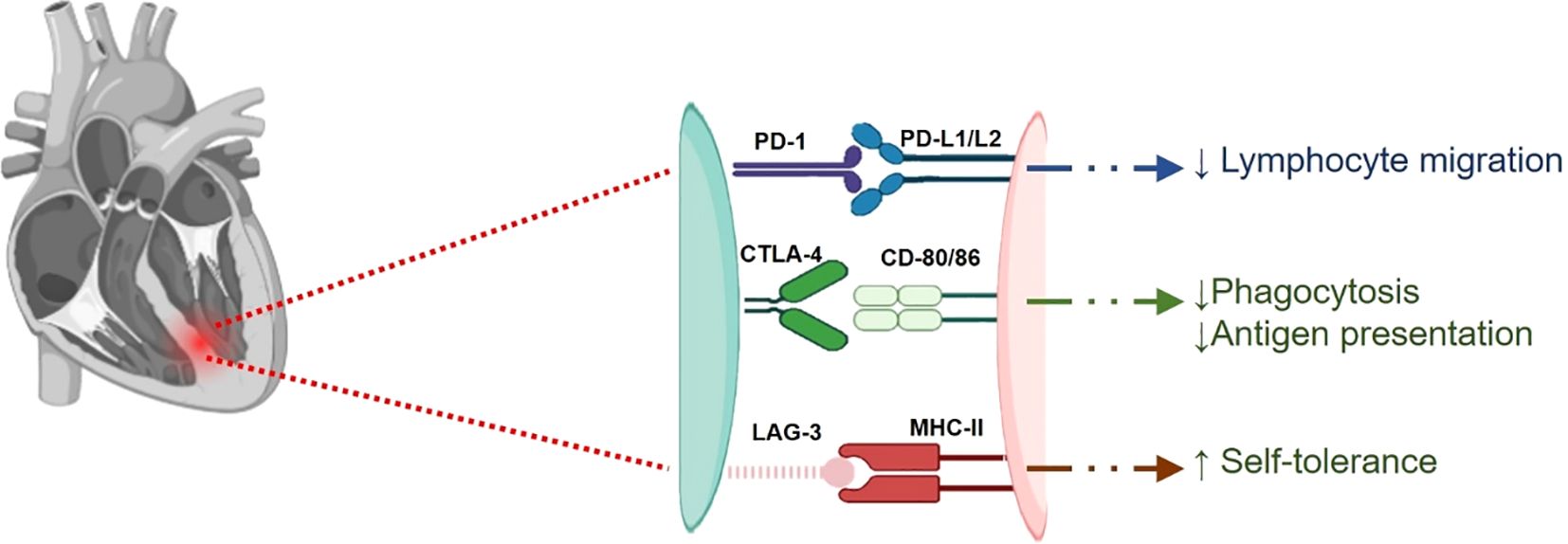
Figure 4. A digrammatic representation of involvement of different checkpoints viz., PD-1/L1, CTLA-4 and LAG-3, in myocarditis. PD-1/L1 and CTLA-4 are responsible for keeping autoimmunity in check and thus reducing cardiac inflammation. These immune checkpoints reduce lymphocyte infiltration and phagocytosis. Inhibition of these checkpoints disrupts this balance and thus resulting inflammatory conditions leading to myocarditis.
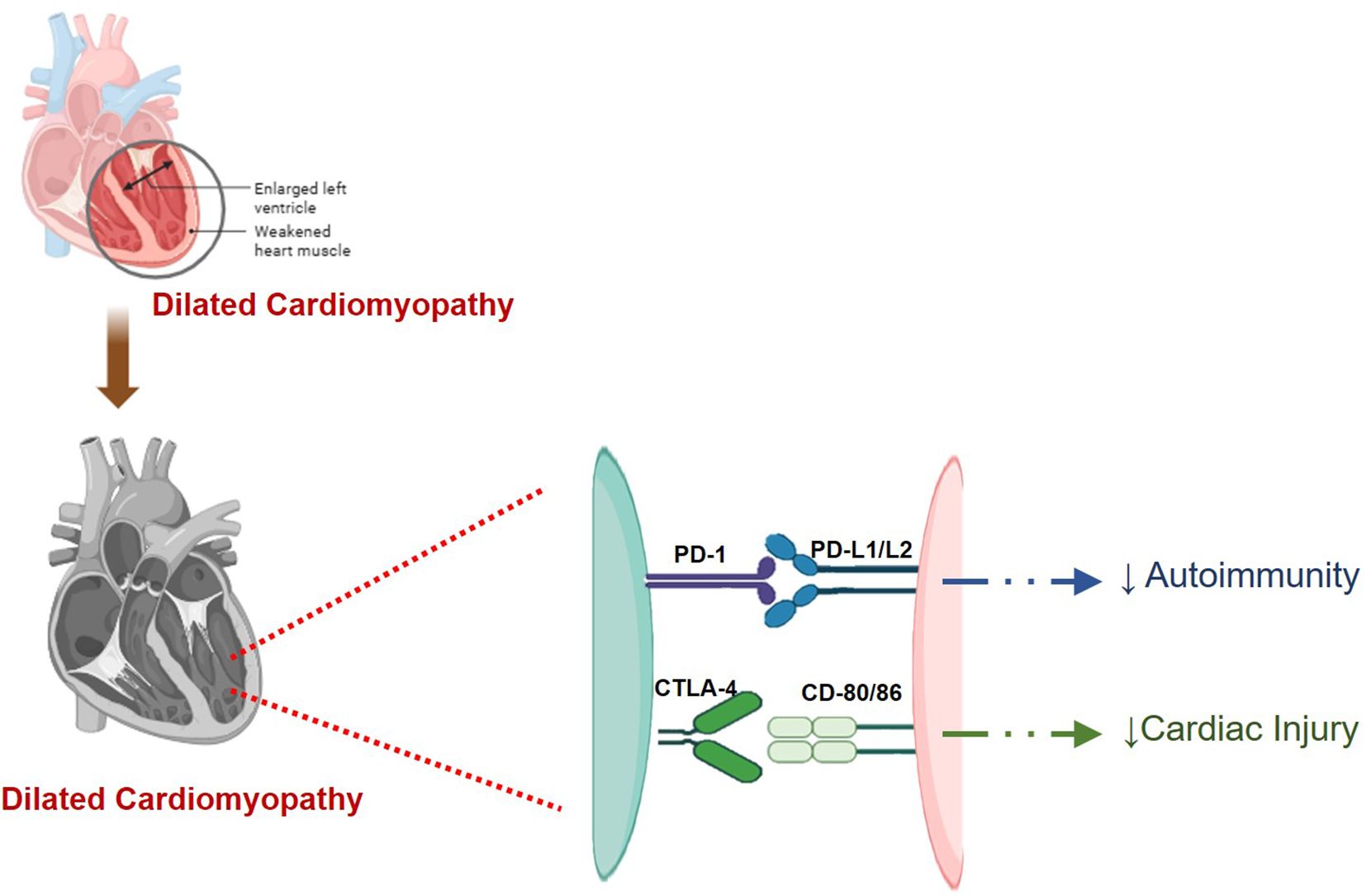
Figure 5. A digrammatic representation of involvement of different checkpoints viz., PD-1/L1, and CTLA-4 in cardiomyopathy. PD-1/L1 and CTLA-4 reduce cardiac injury and autoimmunity in check. Inhibition of these checkpoints disrupts this balance and thus resulting in cardiomyopathy.
A lack of harmony between self- tolerance and autoimmunity was the basic reason for the induction of immune checkpoint inhibition induced myocarditis (184). Although the exact relationship between myocarditis and immune checkpoints is not exactly established, however, certain studies suggest the influence of immune checkpoints on myocarditis. Clinical evaluation of patients of myocarditis, post-treatment with CTLA-4 and PD-1/PD-L1 inhibitors therapy, showed infiltration of thymocytes in the myocardial and skeletal cells (185). Enhanced activation and relocation of thymocytes in cardiac cells along with higher presentation of antigen and phagocytosis is observed when treated with inhibitors of PD-1/PD-L1 and CTLA-4 (186).
In a study, in mice with cardiomyopathy, cardiomyocytes expressed PD-L1 which interfered with and halted the thymocytes act by negatively controlling the production of IFN-γ (187). Immune checkpoint expression on the endothelial cells and cardiomyocytes seems to exert a protective function against induction of myocarditis and injury. This was confirmed in a study wherein PD-L1 and PD-L2 were found to be amplified under the influence of IFN-γ, in animals with CD8+thymocyte -moderated myocarditis. Interference with the production of IFN-γ as well as haltering or devoid of PD-L1/L2, thus exacerbated the disease condition (62, 188). Animal studies also show that the expression of PD-L1/PD-L2 was observed in endothelial cells that have been injured by the inflammation in myocarditis. Interestingly, endothelial cells of control animals did not exhibit PD-L1/L2 (154). These findings collectively indicate the protective function of PD-L1/PD-L2 in myocarditis (189, 190). Studies suggest that cardiac endothelial cell and cardiomyocyte apoptosis is inhibited by the intracellular PD-L1 as well as MHC-II signaling through MAPK/Erk and PI3K/Akt pathways (156, 157, 166).
A genetically modified animal model, wherein the animals were devoid of PD-L1/L2 completely while haploinsufficiency for CTAL-4 was present. The animals completely devoid of PD-1/L1 and haploid status for CTLA-4 were found to exhibit a high rate (~50%) of mortality due to myocarditis. Treatment of such animals with CTLA-4 inhibitor improved the survival of the animals along with the reduced infiltration of thymocytes at the cardiac injury site. Instead, animals devoid of PD-1/L1 but with homoallelic CTLA-4 did not develop myocarditis. This clearly indicates that PD-1/L1 and CTLA-4 interact together at the genetic level for the manifestation of myocarditis (191).
Giant cell myocarditis has been related to CTLA-4 inhibitors. Giant cell myocarditis is a deadly condition wherein acute dilated cardiomyopathy along with ventricular tachycardia and eventually cardiac block occurs (192). Histologically, in giant cell myocarditis Thymocyte mediated necrosis of myocytes and macrophages acquired multi-nucleated giant cells are formed (193). Giant cell myocarditis is chiefly a condition driven by CD4+Thymocytes and chemokines viz., C-X-C Motif Chemokine Receptor 3 (CXCR3). The CXCR3 is vital in the pathways, involved in the diversification, activation and deployment of CD4+ thymocytes, like, MAP kinase and PI3K/Akt pathways. CXACR3 is involved in the activation and deployment of CD4+ thymocytes and has not been found to be involved in the CD8+thymocytes. The upregulation of CD8+ thymocytes is primarily interrelated with immune checkpoint inhibitors and not CXACR3 expression. Inhibition of CTLA-4, however, has been found to upregulate the appearance of CXACR3, which in turn incites giant cell myocarditis (194).
Although LAG-3-associated myocarditis is rarely seen, few cases of myocarditis have been reported when administered with anti-LAG-3 agents. In the RELATIVITY-047 clinical trial carried out on more than 700 subjects, 1.7% of the subjects reported developing myocarditis when administered a blend of relatlimab (LAG-3–blocker) –nivolumab (PD-1 inhibitor) as against 0.6% of the subjects that received monotherapy of nivolumab only (179). Studies in knockout animal models that are devoid of LAG-3 also point out to lack of relation between LAG-3 and myocarditis (195). However, studies in the knockout model that was devoid of LAG-3 as well as PD-1, exhibited the development of myocarditis along with elevated levels of TNF-α and excessive infiltration of Thymocytes, though Treg cells exhibited a controlled functioning in such animals (196).
The alternative proposed mechanism to induce myocarditis encompasses the presence of shared cardiac and tumor antigens for cells expressing PD-L1 viz., endothelial cells and myocytes. Another proposed mechanism includes the existence of a pre-existent immune response that induces autoimmunity, like higher levels of anti-troponin T antibodies in patients developing myocarditis post PD-1 inhibitor therapy (197). Similarly, a PD-1 and CTLA-4 inhibitor treatment-induced rhabdomyolysis polymyositis patient exhibited higher levels of antibodies against striated muscles (198).
Animal models deficient in PD-1 have provided significant insights into the development of cardiomyopathy. These animals exhibited compromised cardiac contractile function, leading to untimely mortality. Notably, PD-1-deficient animals showed an accumulation of IgG on the surface of cardiomyocytes, along with circulating IgG antibodies targeting cardiac troponin I. However, no immune cell infiltration was observed, indicating that the mechanism behind cardiomyopathy in this model was primarily inflammatory rather than immune cell-driven (99, 119).
In human studies, PD-1 expression was found to be upregulated in the intercalated discs and myocardium of patients who had developed myocardial infarction and dilated cardiomyopathy. The expression of PD-1 in these patients was inversely correlated with left ventricular ejection fraction (LVEF) and directly associated with left ventricular end-diastolic volume, suggesting a role for PD-1 in regulating cardiac function during injury (199).
Furthermore, the upregulation of myocardial PD-L1 has been shown to mitigate cardiac injury by negatively regulating immune responses and reducing Thymocyte activity (200). A study on immune checkpoint inhibitor-induced dilated cardiomyopathy found that cardiomyocytes expressed PD-L1, which helped suppress the production of pro-inflammatory cytokines, including TNF-α and IFN-γ, ultimately reducing inflammation and tissue damage (187).
Associated cardiovascular complications, other immune checkpoints are alos being evaluated. These immune checkpoints include TIM-3 or Hepatitis A virus cellular receptor 2 (HAVCR2), V-set and transmembrane domain-containing protein 3 (Vstm3), TIGIT or Washington University cell adhesion molecule (WUCAM) and V-Set and immunoglobulin domain-containing protein 9 (VSIG9) (201). These new immune checkpoints can be utilized as novel targets for the treatment of tumors and are currently being clinically evaluated, alone or in conjunction with PD-1/L1 inhibitors or CTLA-4 inhibitors (202–217).
Immune checkpoint inhibitor, TIM-3 or HAVCR2, is part of TIM family and can bind to four corresponding ligands viz., Galectin-9 (LGALS9), high mobile protein B1 (HMGB1), phosphatidylserine (PtdSer) and surface-bound ligands carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) (67, 218–221). Under the influence of IFN-γ, CD4+thymocytes and CD+8 thymocytes amplify the expression of TIM-3 (204, 205). TIM-3, expresses on NK cells, Treg cells, Th17, dendritic cells and macrophages and inhibits cytokines IFN-γ and TNF, thereby suppressing type I immune response (222–232). Although some reports suggest that the TIM-3 pathway serves both stimulatory as well as inhibitory function (223). Structurally, TIM-3 possesses five tyrosine conserved residues, of which phosphorylation of Tyr 265 and Tyr272 is essential in humans. Interaction of TIM-3 with HLA-B-associated transcript 3 (Bat3) and Lymphocyte-Specific Protein Tyrosine Kinase (LCK) on the lipidic rafts to promote differentiation and survival of thymocytes (233–235). However, binding with CD148 and CD45 results in replacement of BAT-3 on TIM-3 and the deploying tyrosine phosphatase causing inactivation of LCK. This inhibits the proliferation of thymocytes by negatively regulating the signaling of Thymocyte receptors (235, 236). Thus, the inhibitory action of TIM-3 on Thymocytes is mediated by BAT-3. Clinical evaluation of the inhibitors of TIM-3 are underway and only few cases of cardiovascular complications has been reported. Infact, some report suggest that atherosclerosis is inversely related to TIM-3. The study findings show that TIM-3 inhibitors amplify the atherosclerotic lesions which is accompanied by enhanced levels of macrophages, CD+4 Tcells and monocytes, along with diminished levels of Treg cells and B cells (137). Another study evaluated the effect of co-treatment with PD-1 and TIM-3 inhibitor and it was found that cytokines which exert anti-atherogeneic effect were reduced but TNF-α and IFN-γ levels were amplified resulting in advancement of atherosclerosis (237).
Another immune checkpoint, TIGIT, or Vstm3/VSIG9/WUCAM, is categorically expressed on lymphocytes, specifically on NK cells, effector CD8+ thymocytes, regulatory CD4+ thymocytes cells and helper thymocytes (207, 238–241). TIGIT expression is amplified especially on Treg cells in the tumor microenvironment (242–244). TIGIT regulate the NK cells and thymocytes functioning via binding with CD155 or poliovirus receptor or Necl-5. TIGIT exhibit a lower affinity toward another nectin family receptor, CD112 or poliovirus receptor related-2 or Nectin-2 (245–248). TIGIT interferes with the proliferation and activation of CD8+thymocytes by exerting its effect on thymocyte receptor expression. This incites negative regulation of the thymocyte receptor α chain and other parts of the receptors (249, 250). TIGIT can suppress the cytotoxic activity and differentiation of CD8+ thymocyte cells via p-ERK signaling (251, 252). By competitively interacting with CD226 or DNAM-1, thymocyte stimulation can be reduced (253). Clinical evaluation of TIGIT inhibitors has so far not reported any cardiovascular complications and additional studies are desirable to establish the safety of TIGIT inhibitors (254–261).
Cardiac implications are often overlooked in the clinical evaluation of immune checkpoint inhibitors (262–265). Inhibition of immune checkpoints has been found to induce myocarditis, vasculitis, pericardial disease and even heart failure (266). A brief overview of clinical implications of different immune checkpoints are enlisted in Table 2. Nevertheless, different studies have shown that animals devoid of PD-1 or administered with an inhibitor of CTLA-4 exhibit inflammation in the cardiac tissues (93, 99, 186, 275–278). Amplification of PD-L1 in animals with myocarditis has been reported (120, 279, 280). BALB/c mice devoid of PD-1 developed autoimmune cardiomyopathy (99). Inhibition or negatively regulating PD-1/PD-L1 has been found to amplify atherosclerosis in animals (142). Also, in such animals, troponin was found to be the cardiomyopathy-inducing antigen in animals devoid of PD-1 (119).
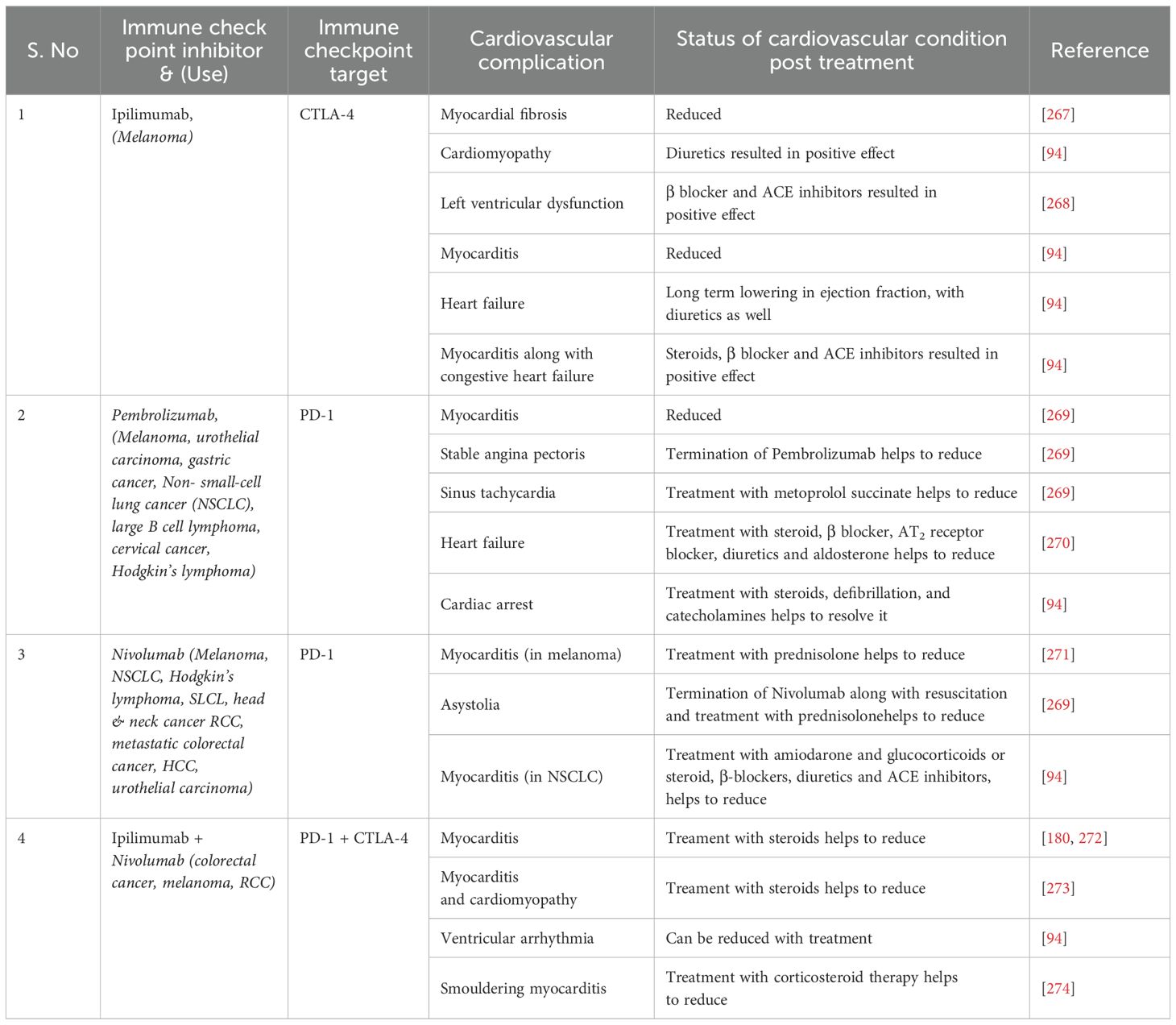
Table 2. Cardiovascular complications associated with different PD-1 and CTLA-4 inhibitors and status post- treatment.
Such cardiac complications have also been observed in human subjects undergoing clinical evaluation for immune checkpoint inhibitors (281). One such case of myocardial fibrosis was observed in a multi-center clinical evaluation of ipilimumab (267). Similarly, another case of autoimmune myocarditis that prompted heart failure was observed in subjects undergoing treatment for melanoma with pembrolizumab (270, 282). Ipilimumab instigated the late development of pericarditis in a patient undergoing treatment for melanoma (283). Several cases of cardiotoxicity were observed when administered ipilimumab and/or nivolumab/pembrolizumab in a multicenter clinical study. In fact, a couple of mortalities were also reported even though subjects were given treatment for the cardiac complications. Also, five cases develop myocarditis among eight cases of cardiac complications when administered inhibitors of PD-1 and/or CTLA-4 (94). In a small clinical evaluation, the administration of ipilimumab, in subjects having a history of cardiovascular disease or autoimmune condition, resulted in the exacerbation of autoimmune complications in almost 50% of subjects (284). Fatal fulminant myocarditis was reported in two subjects when administering a blend of ipilimumab and nivolumab in subjects suffering from melanoma. Both the subjects had a pre-existing condition of hypertension. Apart from these two cases, 18 cases out of 20594 subjects were reported to have developed myocarditis (94). In fact patients receiving a combination exhibit a severe and higher frequency of myocarditis (0.27%) as against those receiving nivolumab alone (0.06%) (185). In participants having coronary heart disease, levels of PD-L1 expressed in Treg cells were inversely related to the severity of coronary heart disease and has the potential to be utilized as a biomarker for it (285).
B-type natriuretic peptide (BNP) and troponin have been found to increase in case of immune checkpoint inhibitor-induced cardiotoxicity and can be utilized as biomarkers of cardiotoxicity (185, 270, 286, 287). Another potential biomarker for cardiotoxicity is anticonductive tissue autoantibodies (ACTA) (288, 289).
A retrospective analysis revealed that of the 424 subjects who received immune checkpoint inhibitors, 14.6% of them (62 subjects) reported cardiovascular condition. Further, of these 62 subjects, 5.6% had heart failure with monotherapy of immune checkpoint inhibitor while 6.1%, receiving combination or dual therapy of immune checkpoint inhibitor reported to have heart failure (290). Similar outcome was reported in meta-analysis of 13346 subjects, 3.1% of the subjects receiving either CTLA-4 inhibitor or PD-1/L1 inhibitor and 5.8% of the subjects receiving combination therapy reported to have heart failure. Interestingly, addition of chemotherapy along with immune checkpoint inhibitor did not significantly added to the incidence of heart failure (3.7%) (178). Further, administration of CTLA-4 inhibitors seems to induce higher frequency of cardiovascular complications viz., myocarditis, pericarditis etc. (94, 286, 291–296). Administration of PD-1/L1 inhibitors like, nivolumab and pembrolizumab etc. have been reported to cause cardiotoxicity like myocarditis (197, 270, 274, 297–310). These findings were further consolidated by the observation of a meta-analysis of data of 32518 subjects that immune checkpoint inhibitors can induce or amplify the risk of cardiotoxicity viz., myocardial infarction, myocarditis, pericarditis, cerebral arterial ischemia, dyslipidemia and heart failure. In fact heart failure is the most severe cardiotoxicity observed with treatment of immune checkpoint inhibitors (311). Thus, immune checkpoints are key determinants of cardiac homeostasis and alteration or blocking of the immune checkpoint can severely affect cardiac health (312–318).
Immune checkpoints are essential components of the immune system that help maintain self-tolerance and prevent autoimmune damage, particularly in critical organs such as the heart, eyes, brain, and kidneys. In the heart, immune checkpoints regulate cardiac homeostasis by controlling immune responses that can otherwise lead to cardiovascular complications, including atherosclerosis, myocarditis, and coronary heart disease, which can progress to heart failure. Clinical reports have highlighted the risks of cardiac injury and cardiotoxicity associated with immune checkpoint inhibition, particularly with the use of PD-1/L1 and CTLA-4 inhibitors. The development and progression of atherosclerosis have been found to be exacerbated by these inhibitors, while the role of LAG-3 in cardiotoxicity is still under investigation. Inhibition of the PD-1/PD-L1 pathway, in particular, has been shown to pose a higher risk for inducing myocarditis compared to CTLA-4 inhibition. The combination of PD-1/PD-L1 inhibitors with TIM-3 inhibition has also been linked to an increased incidence of atherosclerosis. These findings underscore the critical need for further understanding of immune checkpoint regulation in cardiac homeostasis.
On the other hand, modulating immune checkpoints presents a promising strategy for targeting specific stages or cell types involved in cardiovascular diseases, such as atherosclerosis and myocarditis. Given that immune checkpoints operate through both co-stimulatory and inhibitory pathways, selectively manipulating these pathways could lead to therapeutic benefits. Additionally, alterations in the levels of immune checkpoints and their ligands may serve as predictive biomarkers for cardiotoxicity. Future research should focus on identifying safe and effective strategies for regulating immune checkpoints in order to develop novel therapeutic and prognostic tools for managing cardiovascular health. Further clinical trials and preclinical studies are necessary to explore the long-term effects of immune checkpoint modulation on cardiovascular diseases and to refine therapeutic approaches that minimize the risk of cardiotoxicity.
SH: Data curation, Writing – original draft. YK: Data curation, Writing – review & editing. TL: Data curation, Writing – review & editing. YX: Data curation, Writing – review & editing. ZY: Data curation, Writing – review & editing. QZ: Conceptualization, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The article was funded by “0 to 1” Innovative research project of Sichuan University (Grant number 2023SCUH0063), and Science and Technology planning project of Yibin (Grant number 2023ZYY024).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
2. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
3. Borcherding N, Kolb R, Gullicksrud J, Vikas P, Zhu Y, Zhang W. Keeping tumors in check: A mechanistic review of clinical response and resistance to immune checkpoint blockade in cancer. J Mol Biol. (2018) 430:2014–29. doi: 10.1016/j.jmb.2018.05.030
4. Shao Y, Yang WY, Nanayakkara G, Saaoud F, Issa MB, Xu K, et al. Immune checkpoints are new therapeutic targets in regulating cardio-, and cerebro-vascular diseases and CD4+ Foxp3+ Regulatory T cell immunosuppression. Int J Drug Discovery Pharmacol. (2024) 3:100022. doi: 10.53941/ijddp.2024.100022
5. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
6. El Osta B, Hu F, Sadek R, Chintalapally R, Tang S-C. Not all immune-checkpoint inhibitors are created equal: meta-analysis and systematic review of immune-related adverse events in cancer trials. Crit Rev oncology/hematol. (2017) 119:1–12. doi: 10.1016/j.critrevonc.2017.09.002
7. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
8. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
9. Cope AP. Studies of T-cell activation in chronic inflammation. Arthritis Res Ther. (2002) 4:S197211. doi: 10.1186/ar557
10. Acuto O, Cantrell D. T cell activation and the cytoskeleton. Annu Rev Immunol. (2000) 18:165–84. doi: 10.1146/annurev.immunol.18.1.165
11. Lim S, Phillips JB, Madeira da Silva L, Zhou M, Fodstad O, Owen LB, et al. Interplay between immune checkpoint proteins and cellular metabolism. Cancer Res. (2017) 77:1245–9. doi: 10.1158/0008-5472.CAN-16-1647
12. Frodermann V, Nahrendorf M. Macrophages and cardiovascular health. Physiol Rev. (2018) 98:2523–69. doi: 10.1152/physrev.00068.2017
13. Simons KH, de Jong A, Jukema JW, de Vries MR, Arens R, Quax PH. T cell co-stimulation and co-inhibition in cardiovascular disease: a double-edged sword. Nat Rev Cardiol. (2019) 16:325–43. doi: 10.1038/s41569-019-0164-7
14. Mathieu P, Lemieux I, Després JP. Obesity, inflammation, and cardiovascular risk. Clin Pharmacol Ther. (2010) 87:407–16. doi: 10.1038/clpt.2009.311
15. Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. (2004) 109:II–2-II-10. doi: 10.1161/01.CIR.0000129535.04194.38
16. Henein MY, Vancheri S, Longo G, Vancheri F. The role of inflammation in cardiovascular disease. Int J Mol Sci. (2022) 23:12906. doi: 10.3390/ijms232112906
17. Salem J-E, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. (2018) 19:1579–89. doi: 10.1016/S1470-2045(18)30608-9
18. Martini E, Stirparo GG, Kallikourdis M. Immunotherapy for cardiovascular disease. J Leuko Biol. (2018) 103:493–500. doi: 10.1002/JLB.5MR0717-306R
19. Goldman A, Maor E, Bomze D, Liu JE, Herrmann J, Fein J, et al. Adverse cardiovascular and pulmonary events associated with chimeric antigen receptor T-cell therapy. J Am Coll Cardiol. (2021) 78:1800–13. doi: 10.1016/j.jacc.2021.08.044
20. Siegel-Axel D, Daub K, Seizer P, Lindemann S, Gawaz M. Platelet lipoprotein interplay: trigger of foam cell formation and driver of atherosclerosis. Cardiovasc Res. (2008) 78:8–17. doi: 10.1093/cvr/cvn015
21. Poznyak AV, Nikiforov NG, Starodubova AV, Popkova TV, Orekhov AN. Macrophages and foam cells: Brief overview of their role. linkage Targeting potential atherosclerosis Biomed. (2021) 9:1221. doi: 10.3390/biomedicines9091221
22. Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. (2005) 2:309–15. doi: 10.1038/ncpcardio0195
23. Behrouzi B, Udell JA. Moving the needle on atherosclerotic cardiovascular disease and heart failure with influenza vaccination. Curr Atheroscl Rep. (2021) 23:1–10. doi: 10.1007/s11883-021-00973-w
24. Gencer S, Evans BR, van der Vorst EP, Döring Y, Weber C. Inflammatory chemokines in atherosclerosis. Cells. (2021) 10:226. doi: 10.3390/cells10020226
25. Padgett LE, Araujo DJ, Hedrick CC, Olingy CE. Functional crosstalk between T cells and monocytes in cancer and atherosclerosis. J Leuco Biol. (2020) 108:297–308. doi: 10.1002/JLB.1MIR0420-076R
26. Ruzzenenti G, Maloberti A, Giani V, Biolcati M, Leidi F, Monticelli M, et al. Covid and cardiovascular diseases: direct and indirect damages and future perspective. High Blood Pressure Cardiovasc Prev. (2021) 28:439–45. doi: 10.1007/s40292-021-00464-8
27. Yeoh SG, Sum JS, Lai JY, Isa WHW, Lim TS. Potential of phage display antibody technology for cardiovascular disease immunotherapy. J Cardiovasc Trans Res. (2022) 15:360–80. doi: 10.1007/s12265-021-10169-x
28. Armenian SH, Chemaitilly W, Chen M, Chow EJ, Duncan CN, Jones LW, et al. National institutes of health hematopoietic cell transplantation late effects initiative: the cardiovascular disease and associated risk factors working group report. Biol Blood Marrow Transplant. (2017) 23:201–10. doi: 10.1016/j.bbmt.2016.08.019
29. Persaud L, De Jesus D, Brannigan O, Richiez-Paredes M, Huaman J, Alvarado G, et al. Mechanism of action and applications of interleukin 24 in immunotherapy. Int J Mol Sci. (2016) 17:869. doi: 10.3390/ijms17060869
30. Mann DL. Inflammatory mediators and the failing heart: past. present foreseeable future Circ Res. (2002) 91:988–98. doi: 10.1161/01.res.0000043825.01705.1b
31. Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. β-Adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines. Nitric oxide remodel Circ. (2000) 101:2103–9. doi: 10.1161/01.cir.101.17.2103
32. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. (2001) 103:2055–9. doi: 10.1161/01.CIR.103.16.2055
33. Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol. (1996) 27:1201–6. doi: 10.1016/0735-1097(95)00589-7
34. Boulet J, Sridhar VS, Bouabdallaoui N, Tardif J-C, White M. Inflammation in heart failure: pathophysiology and therapeutic strategies. Inflammation Res. (2024) 73:709–23. doi: 10.1007/s00011-023-01845-6
35. Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. (2000) 102:3060–7. doi: 10.1161/01.CIR.102.25.3060
36. Gullestad L, Ueland T, Brunsvig A, Kjekshus J, Simonsen S, Frøland SS, et al. Effect of metoprolol on cytokine levels in chronic heart failure—a substudy in the Metoprolol Controlled-Release Randomised Intervention Trial in Heart Failure (MERIT-HF). Am Heart J. (2001) 141:418–21. doi: 10.1067/mhj.2001.112785
37. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. (2020) 41:2153–63. doi: 10.1093/eurheartj/ehz542
38. Markousis-Mavrogenis G, Tromp J, Ouwerkerk W, Devalaraja M, Anker SD, Cleland JG, et al. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur J Heart fail. (2019) 21:965–73. doi: 10.1002/ejhf.1482
39. Nymo SH, Hulthe J, Ueland T, McMurray J, Wikstrand J, Askevold ET, et al. Inflammatory cytokines in chronic heart failure: interleukin-8 is associated with adverse outcome. Results CORONA Eur J Heart fail. (2014) 16:68–75. doi: 10.1093/eurjhf/hft125
40. Chaar D, Dumont B, Vulesevic B, Neagoe PE, Rakel A, Sirois MG, et al. Neutrophils pro-inflammatory and anti-inflammatory cytokine release in patients with heart failure and reduced ejection fraction. ESC Heart Fail. (2021) 8:3855–64. doi: 10.1002/ehf2.13539
41. Segiet OA, Piecuch A, Mielańczyk Ł, Michalski M, Nowalany-Kozielska E. Role of interleukins in heart failure with reduced ejection fraction. Anatolian J Cardiology/Anadolu Kardiyoloji Dergisi. (2019) 22:28799. doi: 10.14744/AnatolJCardiol.2019.32748
42. Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci. (2018) 131:jcs208884. doi: 10.1242/jcs.208884
43. Besler C, Lang D, Urban D, Rommel K-P, von Roeder M, Fengler K, et al. Plasma and cardiac galectin-3 in patients with heart failure reflects both inflammation and fibrosis: implications for its use as a biomarker. Circul: Heart fail. (2017) 10:e003804. doi: 10.1161/CIRCHEARTFAILURE.116.003804
44. Sharma UC, Pokharel S, Van Brakel TJ, Van Berlo JH, Cleutjens JP, Schroen B, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. (2004) 110:3121–8. doi: 10.1161/01.CIR.0000147181.65298.4D
45. Liu Y-H, D’Ambrosio M, Liao T-d, Peng H, Rhaleb N-E, Sharma U, et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiology-Heart Circulatory Physiol. (2009) 296:H404–12. doi: 10.1152/ajpheart.00747.2008
46. Lax A, Sanchez-Mas J, Asensio-Lopez MC, Fernandez-Del-Palacio MJ, Caballero L, Garrido IP, et al. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC: Heart Fail. (2015) 3:50–8. doi: 10.1016/j.jchf.2014.07.015
47. Lok DJ, van der Meer P, de la Porte P.W.B.-A., Lipsic E, Van Wijngaarden J, Hillege HL, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol. (2010) 99:323–8. doi: 10.1007/s00392-010-0125-y
48. Lok DJ, Lok SI, Bruggink-André-de-la-Porte PW, Badings E, Lipsic E, van Wijngaarden J, et al. Galectin-3 is an independent marker for ventricular remodeling and mortality in patients with chronic heart failure. Clin Res Cardiol. (2013) 102:103–10. doi: 10.1007/s00392-012-0500-y
49. Barbé-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME. The interplay between immunosenescence and age-related diseases. Semin immunopathol. (2020) 42:545–57. doi: 10.1007/s00281-020-00806-z
50. Biasucci LM, La Rosa G, Pedicino D, D’Aiello A, Galli M, Liuzzo G. Where does inflammation fit? Curr Cardiol Rep. (2017) 19:1–10. doi: 10.1007/s11886-017-0896-0
51. Sanz-González SM, Barquín L, García-Cao I, Roque M, González JM, Fuster JJ, et al. Increased p53 gene dosage reduces neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovasc Res. (2007) 75:803–12. doi: 10.1016/j.cardiores.2007.05.002
52. Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. (2020) 577:399–404. doi: 10.1038/s41586-019-1895-7
53. Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J. Senescent CD14+ CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. (2011) 186:1809–15. doi: 10.4049/jimmunol.1001866
54. Dumitriu IE, Araguás ET, Baboonian C, Kaski JC. CD4+ CD28null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. (2009) 81:11–9. doi: 10.1093/cvr/cvn248
55. Sharpe AH. Mechanisms of costimulation. Immunol Rev. (2009) 229:5. doi: 10.1111/j.1600-065X.2009.00784.x
56. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun. (2018) 95:77–99. doi: 10.1016/j.jaut.2018.08.007
57. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood J Am Soc Hematol. (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
58. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
59. Araki M, Chung D, Liu S, Rainbow DB, Chamberlain G, Garner V, et al. Genetic evidence that the differential expression of the ligand-independent isoform of CTLA-4 is the molecular basis of the Idd5. 1 type 1 diabetes region in nonobese diabetic mice. J Immunol. (2009) 183:5146–57. doi: 10.4049/jimmunol.0802610
60. Ishida M, Iwai Y, Tanaka Y, Okazaki T, Freeman GJ, Minato N, et al. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett. (2002) 84:57–62. doi: 10.1016/S0165-2478(02)00142-6
61. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
62. Grabie N, Lichtman AH, Padera R. T cell checkpoint regulators in the heart. Cardiovasc Res. (2019) 115:869–77. doi: 10.1093/cvr/cvz025
63. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol Rev. (2017) 276:80–96. doi: 10.1111/imr.2017.276.issue-1
64. Maruhashi T, I.-m.-Okazaki D, Takahashi S, Maeda TK, Shimizu K, Okazaki T. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol. (2018) 19:1415–26. doi: 10.1038/s41590-018-0217-9
65. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding LAG-3 signaling. Int J Mol Sci. (2021) 22:5282. doi: 10.3390/ijms22105282
66. Dankner M, Gray-Owen SD, Huang Y-H, Blumberg RS, Beauchemin N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology. (2017) 6:e1328336. doi: 10.1080/2162402X.2017.1328336
67. Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2015) 517:386–90. doi: 10.1038/nature13848
68. Yu J, Zhang H, Sun S, Sun S, Li L. The effects of Tim−3 activation on T−cells in gastric cancer progression. Oncol Lett. (2019) 17:1461–6. doi: 10.3892/ol.2018.9743
69. Farhad M, Rolig AS, Redmond WL. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology. (2018) 7:e1434467. doi: 10.1080/2162402X.2018.1434467
70. Joller N, Kuchroo VK. Tim-3, Lag-3, and TIGIT. In: Yoshimura A, ed. Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity. Curr Top Microbiol Immunol. (2017) 410:12756. doi: 10.1007/82_2017_62
71. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
72. Plitas G, Rudensky AY. Regulatory T cells: differentiation and function. Cancer Immunol Res. (2016) 4:721–5. doi: 10.1158/2326-6066.CIR-16-0193
73. Wing K, Yamaguchi T, Sakaguchi S. Cell-autonomous and-non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. (2011) 32:428–33. doi: 10.1016/j.it.2011.06.002
74. Zhang J-c, Chen W-d, Alvarez JB, Jia K, Shi L, Wang Q, et al. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol Sin. (2018) 39:1693–8. doi: 10.1038/s41401-018-0062-2
75. Van Coillie S, Wiernicki B, Xu J. Molecular and cellular functions of CTLA-4. Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy. (2020) 1248:7–32. doi: 10.1007/978-981-15-3266-5_2
76. Brunet J-F, Denizot F, Luciani M-F, Roux-Dosseto M, Suzan M, Mattei M-G, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature. (1987) 328:267–70. doi: 10.1038/328267a0
77. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. (1991) 174:561–9. doi: 10.1084/jem.174.3.561
78. Murphy K, Weaver C. Janeway’s immunobiology. New York London Garland Sci. (2016). doi: 10.1201/9781315533247
79. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
80. Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. (1993) 262:905–7. doi: 10.1126/science.7694361
81. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr., Lombard LA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. (1993) 262:909–11. doi: 10.1126/science.7694363
82. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. (1993) 366:76–9. doi: 10.1038/366076a0
83. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci. (1990) 87:5031–5. doi: 10.1073/pnas.87.13.5031
84. Van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti–cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. (1999) 190:355–66. doi: 10.1084/jem.190.3.355
85. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
86. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
87. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J Exp Med. (2009) 206:1717–25. doi: 10.1084/jem.20082492
88. Tajiri K, Aonuma K, Sekine I. Immune checkpoint inhibitor-related myocarditis. Japan J Clin Oncol. (2018) 48:7–12. doi: 10.1093/jjco/hyx154
89. Ying H, Yang L, Qiao G, Li Z, Zhang L, Yin F, et al. Cutting edge: CTLA-4–B7 interaction suppresses Th17 cell differentiation. J Immunol. (2010) 185:1375–8. doi: 10.4049/jimmunol.0903369
90. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
91. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8. doi: 10.1126/science.270.5238.985
92. Romo-Tena J, Gómez-Martín D, Alcocer-Varela J. CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev. (2013) 12:1171–6. doi: 10.1016/j.autrev.2013.07.002
93. Love VA, Grabie N, Duramad P, Stavrakis G, Sharpe A, Lichtman A. CTLA-4 ablation and interleukin-12–driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ Res. (2007) 101:248–57. doi: 10.1161/CIRCRESAHA.106.147124
94. Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, et al. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J immunother Cancer. (2016) 4:1–11. doi: 10.1186/s40425-016-0152-y
95. Van Puijvelde G, Hauer A, De Vos P, Van Den Heuvel R, Van Herwijnen M, van der Zee R, et al. Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation. (2006) 114:1968–76. doi: 10.1161/CIRCULATIONAHA.106.615609
96. van Puijvelde GH, van Es T, Van Wanrooij E, Habets K, De Vos P, van der Zee R, et al. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arteriosclerosis thrombosis Vasc Biol. (2007) 27:2677–83. doi: 10.1161/ATVBAHA.107.151274
97. Dietrich T, Hucko T, Schneemann C, Neumann M, Menrad A, Willuda J, et al. Local delivery of IL-2 reduces atherosclerosis via expansion of regulatory T cells. Atherosclerosis. (2012) 220:329–36. doi: 10.1016/j.atherosclerosis.2011.09.050
98. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
99. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
100. Sun Y, Li L, Wu Y, Yang K. PD-1/PD-L1 in cardiovascular disease. Clinica Chimica Acta. (2020) 505:26–30. doi: 10.1016/j.cca.2020.02.019
101. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. (2007) 19:813–24. doi: 10.1093/intimm/dxm057
102. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
103. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited, Annu. Rev Immunol. (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
104. Kythreotou A, Siddique A, Mauri FA, Bower M, Pinato DJ. PD-L1. J Clin Pathol. (2018) 71:189–94. doi: 10.1136/jclinpath-2017-204853
105. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. New Engl J Med. (2016) 375:1767–78. doi: 10.1056/NEJMra1514296
106. Varricchi G, Marone G, Mercurio V, Galdiero MR, Bonaduce D, Tocchetti CG. Immune checkpoint inhibitors and cardiac toxicity: an emerging issue. Curr med Chem. (2018) 25:1327–39. doi: 10.2174/0929867324666170407125017
107. Guan J, Lim KS, Mekhail T, Chang C-C. Programmed death ligand-1 (PD-L1) expression in the programmed death receptor-1 (PD-1)/PD-L1 blockade: a key player against various cancers. Arch Pathol Lab Med. (2017) 141:851–61. doi: 10.5858/arpa.2016-0361-RA
108. Lee J, Zhuang Y, Wei X, Shang F, Wang J, Zhang Y, et al. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J Mol Cell Cardiol. (2009) 46:169–76. doi: 10.1016/j.yjmcc.2008.10.028
109. Lyon AR, Yousaf N, Battisti NM, Moslehi J, Larkin J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. (2018) 19:e447–58. doi: 10.1016/S1470-2045(18)30457-1
110. Screever EM, Yousif LI, Moslehi JJ, Salem JE, Voors AA, Silljé HH, et al. Circulating immune checkpoints predict heart failure outcomes. ESC Heart Fail. (2023) 10:2330–7. doi: 10.1002/ehf2.14304
112. Jiménez-Alejandre R, Ruiz-Fernandez I, Martin P. Pathophysiology of immune checkpoint inhibitor-induced myocarditis. Cancers. (2022) 14:4494. doi: 10.3390/cancers14184494
113. Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiology-Heart Circulatory Physiol. (2017) 312:H1052–9. doi: 10.1152/ajpheart.00024.2017
114. Li X, Peng W, Wu J, Yeung S-CJ, Yang R. Advances in immune checkpoint inhibitors induced-cardiotoxicity. Front Immunol. (2023) 14:1130438. doi: 10.3389/fimmu.2023.1130438
115. Kalinoski H, Daoud A, Rusinkevich V, Jurčová I, Talor MV, Welsh RA, et al. Injury-induced myosin-specific tissue-resident memory T cells drive immune checkpoint inhibitor myocarditis. Proc Natl Acad Sci. (2024) 121:e2323052121. doi: 10.1073/pnas.2323052121
116. Inno A, Chiampan A, Lanzoni L, Verzè M, Molon G, Gori S. Immune checkpoint inhibitors and atherosclerotic vascular events in cancer patients. Front Cardiovasc Med. (2021) 8:652186. doi: 10.3389/fcvm.2021.652186
117. Michel L, Korste S, Spomer A, Hendgen-Cotta UB, Rassaf T, Totzeck M. PD1 deficiency modifies cardiac immunity during baseline conditions and in reperfused acute myocardial infarction. Int J Mol Sci. (2022) 23:7533. doi: 10.3390/ijms23147533
118. Baban B, Liu JY, Qin X, Weintraub NL, Mozaffari MS. Upregulation of programmed death-1 and its ligand in cardiac injury models: interaction with GADD153. PloS One. (2015) 10:e0124059. doi: 10.1371/journal.pone.0124059
119. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. (2003) 9:1477–83. doi: 10.1038/nm955
120. Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 Ligand 1 (PD-L1) regulates CD8+ T-cell–mediated injury in the heart. Circulation. (2007) 116:2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360
121. Wang J, Okazaki I-m, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. (2010) 22:443–52. doi: 10.1093/intimm/dxq026
122. Bar J, Markel G, Gottfried T, Percik R, Leibowitz-Amit R, Berger R, et al. Acute vascular events as a possibly related adverse event of immunotherapy: a single-institute retrospective study. Eur J Cancer. (2019) 120:122–31. doi: 10.1016/j.ejca.2019.06.021
123. Fan J, Watanabe T. Inflammatory reactions in the pathogenesis of atherosclerosis. J Atheroscl Thromb. (2003) 10:63–71. doi: 10.5551/jat.10.63
124. Hansson GK. Immune mechanisms in atherosclerosis. Arteriosclerosis thrombosis Vasc Biol. (2001) 21:1876–90. doi: 10.1161/hq1201.100220
125. Kondo S, Sato N, Aso K. The level of urinary epidermal growth factor is not influenced by the extent of psoriatic lesions. Arch Dermatol Res. (1991) 283:516–8. doi: 10.1007/BF00371925
126. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E-a, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
127. Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation. (2020) 142:2299–311. doi: 10.1161/CIRCULATIONAHA.120.049981
128. Strauss L, Mahmoud MA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. (2020) 5:eaay1863. doi: 10.1126/sciimmunol.aay1863
129. Gisterå A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. doi: 10.1038/nrneph.2017.51
130. Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular disease in Europe: epidemiological update 2016. Eur Heart J. (2016) 37:3232–45. doi: 10.1093/eurheartj/ehw334
131. Hansson GK, Robertson A-KL, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annu Rev Pathol Mech Dis. (2006) 1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100
132. Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol: Mech Dis. (2014) 9:73–102. doi: 10.1146/annurev-pathol-020712-163936
133. Foks A, Kuiper J. Immune checkpoint proteins: exploring their therapeutic potential to regulate atherosclerosis. Br J Pharmacol. (2017) 174:3940–55. doi: 10.1111/bph.v174.22
134. Bu DX, Tarrio M, Maganto-Garcia E, Stavrakis G, Tajima G, Lederer J, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1100–7. doi: 10.1161/ATVBAHA.111.224709
135. Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of cytotoxic T-lymphocyte–associated antigen-4 prevents atherosclerosis in mice. Arteriosclerosis thrombosis Vasc Biol. (2016) 36:1141–51. doi: 10.1161/ATVBAHA.115.306848
136. Mulholland M, Kritikou E, Katra P, Nilsson J, Björkbacka H, Lichtman AH, et al. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. Cardio Oncol. (2022) 4:635–45. doi: 10.1016/j.jaccao.2022.09.005
137. Foks AC, Ran IA, Wasserman L, Frodermann V, Ter Borg MN, de Jager SC, et al. T-cell immunoglobulin and mucin domain 3 acts as a negative regulator of atherosclerosis. Arteriosclerosis thrombosis Vasc Biol. (2013) 33:2558–65. doi: 10.1161/ATVBAHA.113.301879
138. Foks AC, Ran IA, Frodermann V, Bot I, van Santbrink PJ, Kuiper J, et al. Agonistic anti-TIGIT treatment inhibits T cell responses in LDLr deficient mice without affecting atherosclerotic lesion development. PloS One. (2013) 8:e83134. doi: 10.1371/journal.pone.0083134
139. Oren O, Yang EH, Molina JR, Bailey KR, Blumenthal RS, Kopecky SL. Cardiovascular health and outcomes in cancer patients receiving immune checkpoint inhibitors. Am J Cardiol. (2020) 125:1920–6. doi: 10.1016/j.amjcard.2020.02.016
140. Thuny F, Naidoo J, Neilan TG. Cardiovascular complications of immune checkpoint inhibitors for cancer. Eur Heart J. (2022) 43:4458–68. doi: 10.1093/eurheartj/ehac456
141. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. (2007) 117:2974–82. doi: 10.1172/JCI31344
142. D.-x.-Bu M, Maganto-Garcia E, Stavrakis G, Tajima G, Lederer J, Jarolim P, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arteriosclerosis thrombosis Vasc Biol. (2011) 31:1100–7. doi: 10.1161/ATVBAHA.111.224709
143. Gotsman I, Lichtman AH. Targeting interferon-γ to treat atherosclerosis. Am Heart Assoc. (2007) 101:333–4. doi: 10.1161/CIRCRESAHA.107.155838
144. Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler thrombosis: J Vasc Biol. (1991) 11:1223–30. doi: 10.1161/01.ATV.11.5.1223
145. Vuong JT, Stein-Merlob AF, Nayeri A, Sallam T, Neilan TG, Yang EH. Immune checkpoint therapies and atherosclerosis: mechanisms and clinical implications: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79:577–93. doi: 10.1016/j.jacc.2021.11.048
146. Frostegård J, Ulfgren A-K, Nyberg P, Hedin U, Swedenborg J, Andersson U, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. (1999) 145:33–43. doi: 10.1016/S0021-9150(99)00011-8
147. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
148. Robertson A-KL, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-β signaling in T cells accelerates atherosclerosis. J Clin Invest. (2003) 112:1342–50. doi: 10.1172/JCI18607
149. Ait-Oufella H, Salomon BL, Potteaux S, Robertson A-KL, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. (2006) 12:178–80. doi: 10.1038/nm1343
150. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
151. Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol. (2002) 169:3581–8. doi: 10.4049/jimmunol.169.7.3581
152. Chen W-J, Hu X-F, Yan M, Zhang W-Y, Mao X-B, Shu Y-W. Human umbilical vein endothelial cells promote the inhibitory activation of CD4+ CD25+ Foxp3+ regulatory T cells via PD-L1. Atherosclerosis. (2016) 244:108–12. doi: 10.1016/j.atherosclerosis.2015.11.002
153. Wan B, Nie H, Liu A, Feng G, He D, Xu R, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. (2006) 177:8844–50. doi: 10.4049/jimmunol.177.12.8844
154. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. (2003) 33:3117–26. doi: 10.1002/eji.200324270
155. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
156. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood J Am Soc Hematol. (2008) 111:3635–43. doi: 10.1182/blood-2007-11-123141
157. Gato-Cañas M, Zuazo M, Arasanz H, Ibañez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. (2017) 20:1818–29. doi: 10.1016/j.celrep.2017.07.075
158. Lucas ED, Finlon JM, Burchill MA, McCarthy MK, Morrison TE, Colpitts TM, et al. Type 1 IFN and PD-L1 coordinate lymphatic endothelial cell expansion and contraction during an inflammatory immune response. J Immunol. (2018) 201:1735–47. doi: 10.4049/jimmunol.1800271
159. Yousif LI, Tanja AA, de Boer RA, Teske AJ, Meijers WC. The role of immune checkpoints in cardiovascular disease. Front Pharmacol. (2022) 13:989431. doi: 10.3389/fphar.2022.989431
160. Poels K, van Leent MM, Reiche ME, Kusters PJ, Huveneers S, de Winther MP, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. (2020) 9:1987. doi: 10.3390/cells9091987
161. Ewing M, Karper J, Abdul S, De Jong R, Peters H, De Vries M, et al. T-cell co-stimulation by CD28–CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int J Cardiol. (2013) 168:1965–74. doi: 10.1016/j.ijcard.2012.12.085
162. Screever E, Axelrod M, Blair M, Trykall D, Barnett J, Roden D, et al. Novel mechanism in cardiac injury: Immune checkpoints. Eur Heart J. (2020) 41:ehaa946. doi: 10.1093/ehjci/ehaa946.3625
163. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4–Ig regulates tryptophan catabolism in vivo. Nat Immunol. (2002) 3:1097–101. doi: 10.1038/ni846
164. Lohr J, Knoechel B, Jiang S, Sharpe AH, Abbas AK. The inhibitory function of B7 costimulators in T cell responses to foreign and self-antigens. Nat Immunol. (2003) 4:664–9. doi: 10.1038/ni939
165. Orabona C, Grohmann U, Belladonna ML, Fallarino F, Vacca C, Bianchi R, et al. CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol. (2004) 5:1134–42. doi: 10.1038/ni1124
166. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. (2011) 186:5173–83. doi: 10.4049/jimmunol.1002050
167. Golden D, Kolmakova A, Sura S, Vella AT, Manichaikul A, Wang X-Q, et al. Lymphocyte activation gene 3 and coronary artery disease. JCI Insight. (2016) 1:e88628. doi: 10.1172/jci.insight.88628
168. Mihailovic PM, Lio WM, Herscovici R, Chyu K-Y, Yano J, Zhao X, et al. Keratin 8 is a potential self-antigen in the coronary artery disease immunopeptidome: A translational approach. PloS One. (2019) 14:e0213025. doi: 10.1371/journal.pone.0213025
169. Veluswamy P, Wacker M, Scherner M, Wippermann J. Delicate role of PD-L1/PD-1 axis in blood vessel inflammatory diseases: current insight and future significance. Int J Mol Sci. (2020) 21:8159. doi: 10.3390/ijms21218159
170. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty acid oxidation controls CD8+ tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res. (2020) 8:479–92. doi: 10.1158/2326-6066.CIR-19-0702
171. Leone O, Pieroni M, Rapezzi C, Olivotto I. The spectrum of myocarditis: from pathology to the clinics. Virchows Archiv. (2019) 475:279–301. doi: 10.1007/s00428-019-02615-8
172. Brociek E, Tymińska A, Giordani AS, Caforio ALP, Wojnicz R, Grabowski M, et al. Myocarditis: etiology, pathogenesis, and their implications in clinical practice. Biol (Basel). (2023) 12:874. doi: 10.3390/biology12060874
173. Kociol RD, Cooper LT, Fang JC, Moslehi JJ, Pang PS, Sabe MA, et al. Recognition and initial management of fulminant myocarditis: a scientific statement from the American Heart Association. Circulation. (2020) 141:e69–92. doi: 10.1161/CIR.0000000000000745
174. Ammirati E, Frigerio M, Adler ED, Basso C, Birnie DH, Brambatti M, et al. Management of acute myocarditis and chronic inflammatory cardiomyopathy: an expert consensus document. Circul: Heart Fail. (2020) 13:e007405. doi: 10.1161/CIRCHEARTFAILURE.120.007405
175. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. (2013) 34:2636–48. doi: 10.1093/eurheartj/eht210
176. Wang DY, Salem J-E, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. (2018) 4:1721–8. doi: 10.1001/jamaoncol.2018.3923
177. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. (2018) 71:1755–64. doi: 10.1016/j.jacc.2018.02.037
178. Rubio-Infante N, Ramírez-Flores YA, Castillo EC, Lozano O, García-Rivas G, Torre-Amione G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: a meta-analysis. Eur J Heart fail. (2021) 23:173947. doi: 10.1002/ejhf.2289
179. Tawbi HA, SChadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. New Engl J Med. (2022) 386:24–34. doi: 10.1056/NEJMoa2109970
180. Semper H, Muehlberg F, Schulz-Menger J, Allewelt M, Grohé C. Drug-induced myocarditis after nivolumab treatment in a patient with PDL1-negative squamous cell carcinoma of the lung. Lung Cancer. (2016) 99:117–9. doi: 10.1016/j.lungcan.2016.06.025
181. Chang A, Nasti TH, Khan MK, Parashar S, Kaufman JL, Boise LH, et al. Myocarditis with radiotherapy and immunotherapy in multiple myeloma. J Oncol Pract. (2018) 14:561–4. doi: 10.1200/JOP.18.00208
182. Banfill K, Giuliani M, Aznar M, Franks K, McWilliam A, Schmitt M, et al. Cardiac toxicity of thoracic radiotherapy: existing evidence and future directions. J Thorac Oncol. (2021) 16:216–27. doi: 10.1016/j.jtho.2020.11.002
183. Liang S, Yang J, Lin Y, Li T, Zhao W, Zhao J, et al. Immune myocarditis overlapping with myasthenia gravis due to anti-PD-1 treatment for a chordoma patient: A case report and literature review. Front Immunol. (2021) 12:682262. doi: 10.3389/fimmu.2021.682262
184. Xavier CB, Lopes CDH, Harada G, Peres EDB, Katz A, Jardim DL. Cardiovascular toxicity following immune checkpoint inhibitors: A systematic review and meta-analysis. Trans Oncol. (2022) 19:101383. doi: 10.1016/j.tranon.2022.101383
185. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. New Engl J Med. (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
186. Ji C, Roy MD, Golas J, Vitsky A, Ram S, Kumpf SW, et al. Myocarditis in cynomolgus monkeys following treatment with immune checkpoint inhibitors. Clin Cancer Res. (2019) 25:4735–48. doi: 10.1158/1078-0432.CCR-18-4083
187. Tay WT, Fang Y-H, Beh ST, Liu Y-W, Hsu L-W, Yen C-J, et al. Programmed cell death-1: programmed cell death-ligand 1 interaction protects human cardiomyocytes against T-cell mediated inflammation and apoptosis response in vitro. Int J Mol Sci. (2020) 21:2399. doi: 10.3390/ijms21072399
188. Liu Y-x, Song Y-j, Liu X-h, Xu S-c, Kong C, Chen L-f, et al. PD-1 inhibitor induces myocarditis by reducing regulatory T cells, activating inflammatory responses, promoting myocardial apoptosis and autophagy. Cytokine. (2022) 157:155932. doi: 10.1016/j.cyto.2022.155932
189. Munir AZ, Gutierrez A, Qin J, Lichtman AH, Moslehi JJ. Immune-checkpoint inhibitor-mediated myocarditis: CTLA4, PD1 and LAG3 in the heart. Nat Rev Cancer. (2024) 24:540–53. doi: 10.1038/s41568-024-00715-5
190. Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun. (2013) 45:90–6. doi: 10.1016/j.jaut.2013.05.004
191. Wei SC, Meijers WC, Axelrod ML, Anang N-AA, Screever EM, Wescott EC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor–associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discovery. (2021) 11:614–25. doi: 10.1158/2159-8290.CD-20-0856
192. Cooper LT Jr., ElAmm C. Giant cell myocarditis. Diagn treatment Herz. (2012) 37:632–6. doi: 10.1007/s00059-012-3658-1
193. Xu J, Brooks EG. Giant cell myocarditis: a brief review. Arch Pathol Lab Med. (2016) 140:1429–34. doi: 10.5858/arpa.2016-0068-RS
194. Rikhi R, Karnuta J, Hussain M, Collier P, Funchain P, Tang WHW, et al. Immune checkpoint inhibitors mediated lymphocytic and giant cell myocarditis: uncovering etiological mechanisms. Front Cardiovasc Med. (2021) 8:721333. doi: 10.3389/fcvm.2021.721333
195. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
196. Okazaki T, I.-m.-Okazaki J, Sugiura D, Nakaki F, Yoshida T, Kato Y, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. (2011) 208:395–407. doi: 10.1084/jem.20100466
197. Martinez-Calle N, Rodriguez-Otero P, Villar S, Mejías L, Melero I, Prosper F, et al. Anti-PD1 associated fulminant myocarditis after a single pembrolizumab dose: the role of occult pre-existing autoimmunity. Haematologica. (2018) 103:e318. doi: 10.3324/haematol.2017.185777
198. Bilen MA, Subudhi SK, Gao J, Tannir NM, Tu S-M, Sharma P. Acute rhabdomyolysis with severe polymyositis following ipilimumab-nivolumab treatment in a cancer patient with elevated anti-striated muscle antibody. J immunother Cancer. (2016) 4:1–5. doi: 10.1186/s40425-016-0139-8
199. Kushnareva E, Kushnarev V, Artemyeva A, Mitrofanova L, Moiseeva O. Myocardial PD-L1 expression in patients with ischemic and non-ischemic heart failure. Front Cardiovasc Med. (2022) 8:759972. doi: 10.3389/fcvm.2021.759972
200. Hayashi T, Tiwary SK, Lavine KJ, Acharya S, Brent M, Adamo L, et al. The programmed death-1 signaling axis modulates inflammation and LV structure/function in a stress-induced cardiomyopathy model. Basic to Trans Sci. (2022) 7:1120–39. doi: 10.1016/j.jacbts.2022.05.006
201. Jo W, Won T, Daoud A, Čiháková D. Immune checkpoint inhibitors associated cardiovascular immune-related adverse events. Front Immunol. (2024) 15:1340373. doi: 10.3389/fimmu.2024.1340373
202. Lee JB, Ha S-J, Kim HR. Clinical insights into novel immune checkpoint inhibitors. Front Pharmacol. (2021) 12:681320. doi: 10.3389/fphar.2021.681320
203. Chauvin J-M, Zarour HM. TIGIT in cancer immunotherapy. J immunother Cancer. (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
204. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
205. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One. (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
206. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
207. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
208. Ge Z, Zhou G, Carrascosa LC, Gausvik E, Boor PP, Noordam L, et al. TIGIT and PD1 co-blockade restores ex vivo functions of human tumor-infiltrating CD8+ T cells in hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. (2021) 12:443–64. doi: 10.1016/j.jcmgh.2021.03.003
209. Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
210. Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome profiling identifies TIGIT as a marker of T-cell exhaustion in liver cancer. Hepatology. (2021) 73:1399–418. doi: 10.1002/hep.31466
211. Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ–mediated antitumor immunity and suppresses established tumors. Cancer Res. (2011) 71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096
212. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
213. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. (2017) 6:e1261779. doi: 10.1080/2162402X.2016.1261779
214. Cai C, Xu Y-F, Wu Z-J, Dong Q, Li M-Y, Olson JC, et al. Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J Urol. (2016) 34:561–7. doi: 10.1007/s00345-015-1656-7
215. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
216. Li X-Y, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression Via combined host and tumor-intrinsic mechanisms. J Clin Invest. (2022) 132:e159825. doi: 10.1172/JCI159825
217. Braun M, Aguilera AR, Sundarrajan A, Corvino D, Stannard K, Krumeich S, et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8+ T cells. Immunity. (2020) 53:805–823.e815. doi: 10.1016/j.immuni.2020.09.010
218. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
219. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood J Am Soc Hematol. (2009) 113:3821–30. doi: 10.1182/blood-2008-10-185884
220. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
221. Kang C-W, Dutta A, Chang L-Y, Mahalingam J, Lin Y-C, Chiang J-M, et al. Apoptosis of tumor infiltrating effector TIM-3+ CD8+ T cells in colon cancer. Sci Rep. (2015) 5:15659. doi: 10.1038/srep15659
222. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
223. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. (2017) 276:97–111. doi: 10.1111/imr.2017.276.issue-1
224. Tian T, Li Z. Targeting Tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front Oncol. (2021) 11:731175. doi: 10.3389/fonc.2021.731175
225. Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med. (2010) 207:505–20. doi: 10.1084/jem.20090397
226. Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci. (2011) 108:5009–14. doi: 10.1073/pnas.1100551108
227. Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol. (2012) 24:213–6. doi: 10.1016/j.coi.2011.12.005
228. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non–small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
229. So EC, Khaladj-Ghom A, Ji Y, Amin J, Song Y, Burch E, et al. NK cell expression of Tim-3: First impressions matter. Immunobiology. (2019) 224:362–70. doi: 10.1016/j.imbio.2019.03.001
230. Yong YK, Saeidi A, Tan HY, Rosmawati M, Enström PF, Batran RA, et al. Hyper-expression of PD-1 is associated with the levels of exhausted and dysfunctional phenotypes of circulating CD161++ TCR iVα7. 2+ mucosal-associated invariant T cells in chronic hepatitis B virus infection. Front Immunol. (2018) 9:472. doi: 10.3389/fimmu.2018.00472
231. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood J Am Soc Hematol. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
232. Li Y, Zhang J, Zhang D, Hong X, Tao Y, Wang S, et al. Tim-3 signaling in peripheral NK cells promotes maternal-fetal immune tolerance and alleviates pregnancy loss. Sci Signaling. 10:eaah4323. doi: 10.1126/scisignal.aah4323
233. Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, et al. T cell Ig and mucin Domain–Containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol. (2014) 192:782–91. doi: 10.4049/jimmunol.1302663
234. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med. (2012) 18:1394–400. doi: 10.1038/nm.2871
235. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J immunother Cancer. (2020) 8:e000911. doi: 10.1136/jitc-2020-000911
236. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. (2006) 351:571–6. doi: 10.1016/j.bbrc.2006.10.079
237. Qiu M-K, Wang S-C, Dai Y-X, Wang S-Q, Ou J-M, Quan Z-W. PD-1 and Tim-3 pathways regulate CD8+ T cells function in atherosclerosis. PloS One. (2015) 10:e0128523. doi: 10.1371/journal.pone.0128523
238. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. (2011) 41:902–15. doi: 10.1002/eji.201041136
239. Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol. (2021) 12:699895. doi: 10.3389/fimmu.2021.699895
240. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. (2009) 39:695–703. doi: 10.1002/eji.200839116
241. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell–cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci. (2012) 109:5399–404. doi: 10.1073/pnas.1120606109
242. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
243. Fourcade J, Sun Z, Chauvin J-M, Ka M, Davar D, Pagliano O, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. (2018) 3:e121157. doi: 10.1172/jci.insight.121157
244. Kurtulus S, Sakuishi K, Ngiow S-F, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
245. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
246. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med. (2016) 213:167–76. doi: 10.1084/jem.20150785
247. Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R, et al. Mouse PVRIG has CD8+ T cell–specific coinhibitory functions and dampens antitumor immunity. Cancer Immunol Res. (2019) 7:244–56. doi: 10.1158/2326-6066.CIR-18-0460
248. Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, Leung L, et al. PVRIG and PVRL2 are induced in cancer and inhibit CD8+ T-cell function. Cancer Immunol Res. (2019) 7:257–68. doi: 10.1158/2326-6066.CIR-18-0442
249. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
250. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. (2017) 38:20–8. doi: 10.1016/j.it.2016.10.002
251. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB, et al. T cells expressing checkpoint receptor TIGIT are enriched in follicular lymphoma tumors and characterized by reversible suppression of T-cell receptor signaling. Clin Cancer Res. (2018) 24:870–81. doi: 10.1158/1078-0432.CCR-17-2337
252. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
253. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
254. Chu X, Tian W, Wang Z, Zhang J, Zhou R. Co-inhibition of TIGIT and PD-1/PD-L1 in cancer immunotherapy: mechanisms and clinical trials. Mol Cancer. (2023) 22:93. doi: 10.1186/s12943-023-01800-3
255. Rotte A, Sahasranaman S, Budha N. Targeting TIGIT for immunotherapy of cancer: update on clinical development. Biomedicines. (2021) 9:1277. doi: 10.3390/biomedicines9091277
256. Van den Mooter T, Migeotte A, Jungels C, Delafontaine B, Nguyen T, Warot S, et al. Preliminary data from phase I first-in-human study of EOS884448, a novel potent anti-TIGIT antibody, monotherapy shows favorable tolerability profile and early signs of clinical activity in immune-resistant advanced cancers. Head Neck. (2021) 3:14. doi: 10.1158/1538-7445.AM2021-CT118
257. Niu J, Maurice-Dror C, Lee D, Kim D-W, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer☆. Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
258. Kim TW, Bedard PL, LoRusso P, Gordon MS, Bendell J, Oh D-Y, et al. Anti-TIGIT antibody tiragolumab alone or with atezolizumab in patients with advanced solid tumors: a phase 1a/1b nonrandomized controlled trial. JAMA Oncol. (2023) 9:1574–82. doi: 10.1001/jamaoncol.2023.3867
259. Zhang P, Liu X, Gu Z, Jiang Z, Zhao S, Song Y, et al. Targeting TIGIT for cancer immunotherapy: recent advances and future directions. biomark Res. (2024) 12:7. doi: 10.1186/s40364-023-00543-z
260. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). Am Soc Clin Oncol. (2020) 38:9503. doi: 10.1200/JCO.2020.38.15_suppl.9503
261. Rui S, Kong X, Liu J, Wang L, Wang X, Zou X, et al. The landscape of TIGIT target and clinical application in diseases. MedComm–Oncology. (2022) 1:e18. doi: 10.1002/mog.v1.2
262. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. (2016) 13:473–86. doi: 10.1038/nrclinonc.2016.58
263. Costa R, Carneiro BA, Agulnik M, Rademaker AW, Pai SG, Villaflor VM, et al. Toxicity profile of approved anti-PD-1 monoclonal antibodies in solid tumors: a systematic review and meta-analysis of randomized clinical trials. Oncotarget. (2016) 8:8910. doi: 10.18632/oncotarget.13315
264. Eigentler TK, Hassel JC, Berking C, Aberle J, Bachmann O, Grünwald V, et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat Rev. (2016) 45:7–18. doi: 10.1016/j.ctrv.2016.02.003
265. Naidoo J, Page D, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. (2015) 26:2375–91. doi: 10.1093/annonc/mdv383
266. Hu J-R, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. (2019) 115:854–68. doi: 10.1093/cvr/cvz026
267. Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PloS One. (2013) 8:e53745. doi: 10.1371/journal.pone.0053745
268. Roth ME, Muluneh B, Jensen BC, Madamanchi C, Lee CB. Left ventricular dysfunction after treatment with ipilimumab for metastatic melanoma. Am J Ther. (2016) 23:e1925–8. doi: 10.1097/MJT.0000000000000430
269. Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer. (2016) 60:210–25. doi: 10.1016/j.ejca.2016.02.024
270. Läubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J immunother Cancer. (2015) 3:11. doi: 10.1186/s40425-015-0057-1
271. Tadokoro T, Keshino E, Makiyama A, Sasaguri T, Ohshima K, Katano H, et al. Acute lymphocytic myocarditis with anti-PD-1 antibody nivolumab, circulation. Heart fail. (2016) 9:e003514. doi: 10.1161/CIRCHEARTFAILURE.116.003514
272. Gibson R, Delaune J, Szady A, Markham M. Suspected autoimmune myocarditis and cardiac conduction abnormalities with nivolumab therapy for non-small cell lung cancer. Case Rep. (2016) 2016):bcr2016216228. doi: 10.1136/bcr-2016-216228
273. Mehta A, Gupta A, Hannallah F, Koshy T, Reimold S. Myocarditis as an immune-related adverse event with ipilimumab/nivolumab combination therapy for metastatic melanoma. Melanoma Res. (2016) 26:319–20. doi: 10.1097/CMR.0000000000000251
274. Norwood TG, Westbrook BC, Johnson DB, Litovsky SH, Terry NL, McKee SB, et al. Smoldering myocarditis following immune checkpoint blockade. J immunother Cancer. (2017) 5:1–6. doi: 10.1186/s40425-017-0296-4
275. Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J Immunol. (2012) 188:487684. doi: 10.4049/jimmunol.1200389
276. Bockstahler M, Fischer A, Goetzke CC, Neumaier HL, Sauter M, Kespohl M, et al. Heart-specific immune responses in an animal model of autoimmune-related myocarditis mitigated by an immunoproteasome inhibitor and genetic ablation. Circulation. (2020) 141:1885–902. doi: 10.1161/CIRCULATIONAHA.119.043171
277. Ronen D, Bsoul A, Lotem M, Abedat S, Yarkoni M, Amir O, et al. Exploring the mechanisms underlying the cardiotoxic effects of immune checkpoint inhibitor therapies. Vaccines (Basel). (2022) 10:540. doi: 10.3390/vaccines10040540
278. Wu MM, Yang YC, Cai YX, Jiang S, Xiao H, Miao C, et al. Anti-CTLA-4 m2a antibody exacerbates cardiac injury in experimental autoimmune myocarditis mice by promoting ccl5-neutrophil infiltration. Adv Sci. (2024) 11:2400486. doi: 10.1002/advs.202400486
279. Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol (Baltimore Md.: 1950). (2008) 181:2513–21. doi: 10.4049/jimmunol.181.4.2513
280. Dong H, Qi Y, Kong X, Wang Z, Fang Y, Wang J. PD-1/PD-L1 inhibitor-associated myocarditis: epidemiology, characteristics, diagnosis, treatment, and potential mechanism. Front Pharmacol. (2022) 13:835510. doi: 10.3389/fphar.2022.835510
281. Zarifa A, Kim JW, Lopez-Mattei J, Palaskas N, Iliescu C, Kim PY. Cardiac toxicities associated with immune checkpoints inhibitors: mechanisms, manifestations and management. Korean Circ J. (2021) 51:579–97. doi: 10.4070/kcj.2021.0089
282. Banerjee A, Narasimhulu CA, Singla DK. Immune interactions in pembrolizumab (PD-1 inhibitor) cancer therapy and cardiovascular complications. Am J Physiology-Heart Circulatory Physiol. (2023) 325:H751–67. doi: 10.1152/ajpheart.00378.2023
283. Yun S, Vincelette ND, Mansour I, Hariri D, Motamed S. Late onset ipilimumab-induced pericarditis and pericardial effusion: a rare but life threatening complication. Case Rep oncol Med. (2015) 2015:794842. doi: 10.1155/2015/794842
284. Johnson DB, Khushalani NI, Puzanov I, Mudigonda T, Kaufman H, Sosman JA, et al. Ipilimumab in metastatic melanoma patients with pre-existing autoimmune disorders. Am Soc Clin Oncol. (2015) 33(15_suppl):9019. doi: 10.1200/jco.2015.33.15_suppl.9019
285. Li S-H, Chen W-J, Yan M, Shu Y-W, Liao Y-H. Expression of coinhibitory PD-L1 on CD4+CD25+FOXP3+ regulatory T cells is elevated in patients with acute coronary syndrome. Coronary Artery Dis. (2015) 26:598–603. doi: 10.1097/MCA.0000000000000282
286. Geisler BP, Raad RA, Esaian D, Sharon E, Schwartz DR. Apical ballooning and cardiomyopathy in a melanoma patient treated with ipilimumab: a case of takotsubo-like syndrome. J immunother Cancer. (2015) 3:1–4. doi: 10.1186/s40425-015-0048-2
287. Novo G, Cadeddu C, Sucato V, Pagliaro P, Romano S, Tocchetti CG, et al. Role of biomarkers in monitoring antiblastic cardiotoxicity. J Cardiovasc Med. (2016) 17:e27–34. doi: 10.2459/JCM.0000000000000379
288. Caio G, Volta U, Cerrato E, Clavenzani P, Montali N, Cogliandro R, et al. Detection of anticonductive tissue autoantibodies in a patient with chronic intestinal pseudo-obstruction and sick sinus syndrome. Eur J Gastroenterol Hepatol. (2013) 25:1358–63. doi: 10.1097/MEG.0b013e3283632dbc
289. Caio G. Myocarditis with immune checkpoint blockade. New Engl J Of Med. (2017) 376:291–2. doi: 10.1056/NEJMc1615251
290. Waheed N, Fradley MG, DeRemer DL, Mahmoud A, Shah CP, Langaee TY, et al. Newly diagnosed cardiovascular disease in patients treated with immune checkpoint inhibitors: a retrospective analysis of patients at an academic tertiary care center. Cardio-oncology. (2021) 7:1–7. doi: 10.1186/s40959-021-00097-9
291. Shalata W, Abu-Salman A, Steckbeck R, Mathew Jacob B, Massalha I, Yakobson A. Cardiac toxicity associated with immune checkpoint inhibitors: a systematic review. Cancers. (2021) 13:5218. doi: 10.3390/cancers13205218
292. Quagliariello V, Passariello M, Rea D, Barbieri A, Iovine M, Bonelli A, et al. Evidences of CTLA-4 and PD-1 blocking agents-induced cardiotoxicity in cellular and preclinical models. J Personal Med. (2020) 10:179. doi: 10.3390/jpm10040179
293. Pirozzi F, Poto R, Aran L, Cuomo A, Galdiero MR, Spadaro G, et al. Cardiovascular toxicity of immune checkpoint inhibitors: clinical risk factors. Curr Oncol Rep. (2021) 23:1–8. doi: 10.1007/s11912-020-01002-w
294. Esposito R, Fedele T, Orefice S, Cuomo V, Prastaro M, Canonico ME, et al. An emergent form of cardiotoxicity: acute myocarditis induced by immune checkpoint inhibitors. Biomolecules. (2021) 11:785. doi: 10.3390/biom11060785
295. Behravesh S, Shomali N, Danbaran GR, Aslani S, Hemmatzadeh M, Hosseinzadeh R, et al. Cardiotoxicity of immune checkpoint inhibitors: An updated review. Biotechnol Appl Biochem. (2022) 69:61–9. doi: 10.1002/bab.v69.1
296. Hu J, Tian R, Ma Y, Zhen H, Ma X, Su Q, et al. Risk of cardiac adverse events in patients treated with immune checkpoint inhibitor regimens: a systematic review and meta-analysis. Front Oncol. (2021) 11:645245. doi: 10.3389/fonc.2021.645245
297. Honton B, Despas F, Dumonteil N, Rouvellat C, Roussel M, Carrie D, et al. Bortezomib and heart failure: case-report and review of the French P harmacovigilance database. Fundam Clin Pharmacol. (2014) 28:349–52. doi: 10.1111/fcp.2014.28.issue-3
298. Giovannini E, Bonasoni MP, D’Aleo M, Tamagnini I, Tudini M, Fais P, et al. Pembrolizumab-induced fatal myasthenia, myocarditis, and myositis in a patient with metastatic melanoma: autopsy, histological, and immunohistochemical findings—a case report and literature review. Int J Mol Sci. (2023) 24:10919. doi: 10.3390/ijms241310919
299. Su L, Liu C, Wu W, Cui Y, Wu M, Chen H. Successful therapy for myocarditis concomitant with complete heart block after pembrolizumab treatment for head and neck squamous cell carcinoma: A case report with literature review. Front Cardiovasc Med. (2022) 9:898756. doi: 10.3389/fcvm.2022.898756
300. Pathak R, Katel A, Massarelli E, Villaflor VM, Sun V, Salgia R. Immune checkpoint inhibitor–induced myocarditis with myositis/myasthenia gravis overlap syndrome: a systematic review of cases. Oncol. (2021) 26:1052–61. doi: 10.1002/onco.13931
301. Ganatra S, Neilan TG. Immune checkpoint inhibitor-associated myocarditis. oncol. (2018) 23:879–86. doi: 10.1634/theoncologist.2018-0130
302. Ndjana Lessomo FY, Wang Z, Mukuka C. Comparative cardiotoxicity risk of pembrolizumab versus nivolumab in cancer patients undergoing immune checkpoint inhibitor therapy: A meta-analysis. Front Oncol. (2023) 13:1080998. doi: 10.3389/fonc.2023.1080998
303. Sakai T, Sasada S, Jyo C, Ishioka K, Takahashi S, Nakamura M. Acute myocarditis and pericarditis after nivolumab treatment in patients with non-small cell lung cancer. Ann Oncol. (2017) 28:ix90. doi: 10.1093/annonc/mdx697.072
304. Zhang C, Wei F, Ma W, Zhang J. Immune-related cardiovascular toxicities of PD-1/PD-L1 inhibitors in solid tumors: an updated systematic review and meta-analysis. Front Immunol. (2024) 15:1255825. doi: 10.3389/fimmu.2024.1255825
305. Varricchi G, Galdiero MR, Tocchetti CG. Novel actors on the stage of cardiac dysfunction induced by anti-PD1 oncological treatments. Oxford Univ Press. (2022) pp:330–2. doi: 10.1093/eurheartj/ehab584
306. Abulizi A, Yan G, Xu Q, Muhetaer R, Wu S, Abudukelimu K, et al. Cardiovascular adverse events and immune-related adverse events associated with PD-1/PD-L1 inhibitors for head and neck squamous cell carcinoma (HNSCC). Sci Rep. (2024) 14:25919. doi: 10.1038/s41598-024-75099-5
307. Quagliariello V, Passariello M, Coppola C, Rea D.d., Barbieri A, Scherillo M, et al. Cardiotoxicity and pro-inflammatory effects of the immune checkpoint inhibitor Pembrolizumab associated to Trastuzumab. Int J Cardiol. (2019) 292:171–9. doi: 10.1016/j.ijcard.2019.05.028
308. Shah D, Young K. Exploring pembrolizumab-induced myocarditis, myositis, and myasthenia gravis: A comprehensive literature review and case presentation on bladder cancer. Cureus. (2023) 15:e49867. doi: 10.7759/cureus.49867
309. Milutinovic S, Jancic P, Jokic V, Petrovic M, Dumic I, Rodriguez AM, et al. Pembrolizumab-associated cardiotoxicity: A retrospective analysis of the FDA adverse events reporting system. Pharmaceuticals. (2024) 17:1372. doi: 10.3390/ph17101372
310. Song W, Zheng Y, Dong M, Zhong L, Bazoukis G, Perone F, et al. Electrocardiographic features of immune checkpoint inhibitor-associated myocarditis. Curr Problems Cardiol. (2023) 48:101478. doi: 10.1016/j.cpcardiol.2022.101478
311. Dolladille C, Akroun J, Morice P-M, Dompmartin A, Ezine E, Sassier M, et al. Cardiovascular immunotoxicities associated with immune checkpoint inhibitors: a safety meta-analysis. Eur Heart J. (2021) 42:4964–77. doi: 10.1093/eurheartj/ehab618
312. Gergely TG, Drobni ZD, Kallikourdis M, Zhu H, Meijers WC, Neilan TG, et al. Immune checkpoints in cardiac physiology and pathology: therapeutic targets for heart failure. Nat Rev Cardiol. (2024) 21:44362. doi: 10.1038/s41569-023-00986-9
313. Gergely TG, Drobni ZD, Sayour NV, Ferdinandy P, Varga ZV. Molecular fingerprints of cardiovascular toxicities of immune checkpoint inhibitors. Basic Res Cardiol. (2024) 120:187205. doi: 10.1007/s00395-024-01068-8
314. Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int. (2018) 12:223–36. doi: 10.1007/s12072-018-9867-9
315. Blum SM, Zlotoff DA, Smith NP, Kernin IJ, Ramesh S, Zubiri L, et al. Immune responses in checkpoint myocarditis across heart, blood and tumour. Nature. (2024) 636:21523. doi: 10.1038/s41586-024-08105-5
316. Michel L, Helfrich I, Hendgen-Cotta UB, Mincu R-I, Korste S, Mrotzek SM, et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur Heart J. (2022) 43:316–29. doi: 10.1093/eurheartj/ehab430
317. Zheng W, Tang WW. Keeping the Failing Heart in Check: Can Modulating Immune Checkpoints Promote Myocardial Recovery? JACC Basic Transl Sci. (2022) 7:11402. doi: 10.1016/j.jacbts.2022.07.009
Keywords: atherosclerosis, myocarditis, programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), cardiotoxicity
Citation: Huang S, Kang Y, Liu T, Xiong Y, Yang Z and Zhang Q (2025) The role of immune checkpoints PD-1 and CTLA-4 in cardiovascular complications leading to heart failure. Front. Immunol. 16:1561968. doi: 10.3389/fimmu.2025.1561968
Received: 16 January 2025; Accepted: 13 February 2025;
Published: 04 April 2025.
Edited by:
Dan Liu, Wuhan University, ChinaReviewed by:
Jimin Guo, Beijing University of Chemical Technology, ChinaCopyright © 2025 Huang, Kang, Liu, Xiong, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Zhang, cXpoYW5nMjAwMGNuQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.