
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 April 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1561719
This article is part of the Research Topic Tuberculosis: Recent Updates in Basic Research, Drug Discovery and Treatment View all 6 articles
 Chunju Fang1†Xuanlu He2†Fei Tang1†
Chunju Fang1†Xuanlu He2†Fei Tang1† Zi Wang1
Zi Wang1 Cong Pan3,4Qi Zhang1Jing Wu1Qinglan Wang5*Daishun Liu6*
Cong Pan3,4Qi Zhang1Jing Wu1Qinglan Wang5*Daishun Liu6* Yu Zhang1,7*
Yu Zhang1,7*Lung cancer (LC) and tuberculosis (TB) represent two major global public health issues. Prior evidence has suggested a link between TB infection and an increased risk of LC. As advancements in LC treatment have led to extended survival rates for LC patients, the co-occurrence of TB and LC has grown more prevalent and poses novel clinical challenges. The intricate molecular mechanisms connecting TB and LC are closely intertwined and many issues remain to be addressed. This review focuses on resemblance between the immunosuppression in tumor and granuloma microenvironments, exploring immunometabolism, cell plasticity, inflammatory signaling pathways, microbiomics, and up-to-date information derived from spatial multi-omics between TB and LC. Furthermore, we outline immunization-related molecular mechanisms underlying these two diseases and propose future research directions. By discussing recent advances and potential targets, this review aims to establish a foundation for developing future therapeutic strategies targeting LC with concurrent TB infection.
Globally, about one in five people will develop cancer in their lifetime, with roughly one in nine men and one in twelve women dying from the disease, as per the 2022 data from the International Agency for Research on Cancer (IARC) (1). In 2022, lung cancer (LC) emerged as the most commonly diagnosed cancer, accounting for nearly 2.5 million new cases, which is about 12.4% of all cancers worldwide (1). LC was also the primary cause of cancer-related deaths in 23 countries, with an estimated 1.8 million deaths (18.7%) (1).
Tuberculosis (TB), a contagious disease triggered by the bacterium Mycobacterium tuberculosis (Mtb), significantly contributes to global health issues and ranks among the leading causes of death worldwide. The World Health Organization’s (WHO) Global Tuberculosis Report 2024 estimates that there were an estimated 10.8 million new TB cases worldwide in 2023, resulting in 1.25 million deaths (2). Yet in 2023, TB probably re-emerged as the primary global driver of mortality attributed to a singular infectious pathogen, following 3 years in which it was overtaken by coronavirus disease (COVID-19) (2). TB primarily affects the lungs (pulmonary TB, PTB) but can also impact other body parts.
Emerging evidence reveals a compounding tuberculosis risk profile in LC patients through both disease pathophysiology and therapeutic interventions. Population-level analyses consistently reported that patients with solid cancers face a two to three-fold higher risk of developing active TB compared to individuals without cancer (3–6), with LC patients exhibiting particularly pronounced vulnerability - a meta-analysis of 23 studies quantified this risk at 9-fold higher than the general population (4). This baseline predisposition is amplified by oncologic therapies through distinct immunological mechanisms. Conventional cytotoxic regimens and molecularly targeted agents have been epidemiologically linked to progressive TB risk escalation (7–10). Notably, modern immunotherapies introduce novel immunological susceptibilities: programmed cell death 1/programmed cell death ligand 1 (PD-1/PD-L1) blockade demonstrates dramatic TB reactivation rates (7–17), evidenced by a Singaporean cohort reporting 2.09% incidence post-ICI treatment (17) and a meta-analysis of 27 studies quantifying 2,000 TB cases per 100,000 PD-1/PD-L1 individuals - representing 35-fold population excess risk (18). Moreover, LC patients treated with ICIs had a 6-fold higher rate of developing active TB compared to the general population (19–21). LC itself may increase the risk of TB activation, and the treatment for LC may further elevate this risk. However, more stratified and detailed studies are needed to elucidate this correlation.
Several studies (22–26) increasingly suggest a strong correlation between prior pulmonary TB infection and the development of LC. Furthermore, several recent meta-analyses (27–29) reported a 2–5-fold higher risk of LC for PTB patients. A systematic meta-analysis and review of 29 cohorts and 44 case-control researches (27) indicated a heightened risk of LC for up to two years following a diagnosed TB (HR 5.01; two studies). Moreover, a study (24) discovered that patients with concurrent LC and TB exhibit an 8-fold higher mortality rate compared to those with LC alone. Another study (30) demonstrated that the coexistence of TB and LC increases mortality risk and worsens the prognosis of patients. However, global epidemiological data on their co-occurrence are currently unavailable (31). Therefore, the coexistence of these two diseases warrants significant attention.
The simultaneous occurrence of PTB and LC is becoming more frequent in clinical settings. This emerging comorbidity constitutes a major public health challenge (32), driven by their interrelated epidemiological connections that potentiate mutual disease progression. Such bidirectional intersection significantly complicates clinical diagnosis and treatment approaches. However, little is currently known about immunization-related molecular mechanisms underlying these two diseases. Thus, this review focuses on immunosuppression microenvironments, immunometabolism, cell plasticity, inflammatory signaling pathways, and up-to-date knowledge derived from spatial multi-omics. Such exploration aims to identify viable immunological intervention targets that can offer significant insights for the clinical management of patients suffering from both LC and TB.
Both cancer and TB involve processes that reflect a coordinated and highly evolved program of immune escape strategies that interfere with innate and adaptive immune response (33). Tumor cells in LC create an immunosuppressive microenvironment, including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and exhausted T cells (characterized by impaired immunity) (34–38). On the other hand, TB primarily affects the lungs, triggers a robust immune effect, and inhibits bacterial growth. However, in some cases, the immune system fails to control the infection, leading to granulomas formation, which is a complex process involving immune evasion of the mycobacterium through continued interaction between Mtb-infected macrophages and T cells in response to chronic inflammation (39).
Tregs (a heterogenous sub-group of CD4+ T cells) are typically characterized by their role in inhibiting effector cells through mechanisms dependent on direct cell contact or by releasing anti-inflammatory cytokines, including interleukin-10 (IL-10), interleukin-35 (IL-35), and transforming growth factor-beta (TGF-β) (40, 41). Although Tregs were known for their protective role in lessening pathological inflammation in tissues, they also exhibit a suppressive influence on particular anti-tumor and anti-Mtb effects. Various factors, such as increased production of TGF-β or IL-10, and immature/tolerogenic DCs (40), might stimulate the growth of Tregs, leading to negative effector T cell regulation in both the tumor microenvironments (TMEs) (36, 41) and the granuloma microenvironments (GMEs) (42–44).
MDSCs, a diverse group of myeloid cells, proliferate in response to persistent and low-grade inflammation (45). Along with Tregs, they play a role in creating an immunosuppressive setting and blocking T-cell activation in both TB (45) and LC (34, 37). MDSCs produce elevated amounts of molecules that suppress T-cell reactions, such as indoleamine 2,3-dioxygenase (IDO), arginase-1, TGF-β, inducible nitric oxide synthase (iNOS), IL-10, and cyclooxygenase-2 (COX2) (34, 37, 45). PD-L1, IDO, and arginase-1, are crucial in their suppressive functions by breaking down essential metabolites and producing harmful byproducts that accumulate in the TME, thereby hindering T cell proliferation (34, 35, 37, 46). Recent spatial investigations have revealed a consistent structural pattern in TB granulomas, where PD-L1 and IDO1 are spatially synchronized, with myeloid core-infiltrating Tregs and a notable absence of activated T cells (44). Elevated levels of MDSCs have been found in the lungs of patients with TB and LC, suppressing the growth and cytokine release by CD4+ and CD8+ T cells and regulating T-cell movement (47, 48).
At the core of both the GME and TME is the shared characteristic of exhausted T cells within their local regions. This exhaustion is linked to the decline of robust effector functions, the upregulation of various inhibitory receptors, including immune checkpoints like CTLA-4, TIM-3, LAG-3, and PD-1 (49), and an altered transcriptional program (50). These dysfunctional T cells exhibit various suppressive receptors and/or immune-modulating cytokines, triggering negative regulatory pathways linked to inadequate management of tumors (51, 52), and chronic Mtb infection (53, 54). Moreover, the reduced levels of perforin and granzymes in CD8+ T cells has been connected to both Mtb infection (55) and cancer (56), potentially leading to reduced target cell killing.
In summary, LC and TB weaken the immune system both locally and systemically (Figure 1) and highlight the clinical significance of their coexistence. However, the interplay between the similar immunosuppressive microenvironments of tumors and granulomas, particularly the immune subtypes and their differences, requires further investigation. New detection methods, such as spatial multi-omics, are needed to provide insights into these interactions. A recent study (57) demonstrated that the relationship between KRT80 and the progression of lung adenocarcinoma was confirmed through both bulk transcriptome and single-cell transcriptome analyses. However, as the study cohorts were sourced from public databases and the sample size was limited, large-scale cohort studies are needed to fully evaluate the model’s effectiveness. Ideally, sequencing of granuloma and tumor tissue samples from clinical patients with tuberculosis and lung cancer would provide valuable insights. The weakened immune response in TB and LC arises from interactions with specific cells in their surrounding microenvironment. In the following sections, we will systematically compare how these two diseases share common mechanisms in three key areas: persistent inflammatory signals, cellular adaptation mechanisms, and metabolic reprogramming of immune cells.

Figure 1. Similarity of molecular mechanisms in TB and LC microenvironment. ① Inflammation-driven metabolic and immune reprogramming, such as lactate, arginine, and glutamine. Expansion and activation of immunosuppressive subsets, including Tregs, MDSCs, and exhausted T cells, leading to immune escape and disease progression. ② Balanced inflammatory processes (Th1 and Th2) and cell plasticity, including immune cell polarization (e.g. M1 and M2 polarization) and spatiotemporal cellular plasticity. ③ This plasticity provides an opportunity for therapeutic reprogramming of the immune system to potentiate anti-Mtb and antitumor activity. For instance, JHU083, a glutamine metabolism antagonist, has been shown to enhance antimycobacterial activity. Abbreviations: Trp, Tryptophan; Kyn, kynurenine; quinolinic acid, a product of the tryptophan/kynurenine pathway; Mø, macrophage; Th, helper T cell; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; NETs, neutrophil extracellular traps; NO, nitric oxide; VEGF, vascular endothelial growth factor; IFN-γ, interferon-γ; iNOS, inducible nitric oxide synthase; CTL, cytotoxic T cell; IDO, indoleamine 2,3-dioxygenase; Arg1, L-arginase; IL, interleukin; TGF, transforming growth factor; PD-1, programmed cell death 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; LAG-3, lymphocyte activating 3; TIM-3, T cell immunoglobulin and mucin-domain containing-3; TCA cycle, tricarboxylic acid cycle. Created in https://BioRender.com.
Immune system activity is influenced by the proportion of different cell types within specific tissues, the stage of tumor progression or Mtb infection, and the interplay of signaling within the TME or GME, including pro-inflammatory and anti-inflammatory processes (44, 58–60) (Figure 1). The characteristics and roles of immune cells are dynamic, changing with their local environment (44, 58–60).
First, this plasticity is evident in diverse cell polarization types, each directed by unique transcriptional programs. For instance, macrophages associated with Mtb infection or tumors originate from tissue-resident cells as well as peripheral blood monocytes are drawn to disease sites as a reaction to inflammatory signals (61–63). Cytokines from the Th1 class, including IFN-γ, encourage the polarization of classically activated M1 macrophages (61, 62). These macrophages, in turn, release pro-inflammatory cytokines such as TNF-α and IL-1β, and exhibit robust antigen processing capabilities, presenting significant antimicrobial and antitumor activities (61, 62). Conversely, Th2 or anti-inflammatory cytokines, such as TGF-β, IL-10, and IL-4, trigger the activation of M2 macrophages, which express arginase-1. These macrophages exhibit weak cytotoxicity, and are associated with wound healing and extracellular matrix remodeling (62, 64, 65).
Macrophages balance pro-inflammatory and anti-inflammatory responses to fight against Mtb infection and tumor cells while controlling tissue pathology. High-dimensional profiling techniques have recently enabled more precise, marker-based definitions of the diverse macrophage populations within tumors and their plastic evolution throughout disease progression or therapy (63, 66). A recent study (67) revealed that patients with PTB exhibit numerous non-necrotizing leukocyte aggregates around necrotizing granulomas. These lesions were more compositionally diverse than the necrotizing type and can be classified into four categories based on the cellular composition and spatial distribution of B cells and macrophages (67). This inherent macrophage plasticity poses a challenge when understanding their complex biology and presents therapeutic targeting opportunities.
Second, spatiotemporal cellular plasticity enables cells to transition between different states during cell migration and metastasis (58). Over the past decade, spatial multi-omics technologies have been employed to study the transcriptomes, proteomes, and metabolomes of the tumor immune microenvironment (TIME) in various cancers (68). For instance, Mark Sorin et al. (38) used highly multiplexed imaging mass cytometry (IMC) to analyze the cellular TIME landscape in lung adenocarcinoma. This approach revealed dynamic cellular behaviors and spatial patterns that were correlated with specific clinical outcomes, including variations in patient survival. Similarly, the polymorphic and elastic nature of TB granulomas is demonstrated by the coexistence of distinct GMEs within an infected individual, each with unique phenotypical and functional properties. Recent studies using MIBI-TOF technology (multiplexed ion beam imaging by time of flight) have identified eight representative microenvironments within TB granulomas (44). These microenvironments are characterized by local immunosuppression markers like PD-L1 and IDO-1 on expanding Tregs, myeloid cells, and elevated levels of TGF-β in the absence of IFN-γ (44). Moreover, the cellular composition of granulomas evolves with the infection stage; early high-burden granulomas (four weeks post-infection) show a type 2 wound healing effect, driven by IL-13 and IL-4, while late low-bacterial-burden granulomas (ten weeks post-infection) are dominated by a type 1 response, characterized by a greater presence of cytotoxic CD8+ T cells by pro-inflammatory signaling networks (69). These analyses illustrate that during various stages of tumor development and Mtb infection, cell types exhibit significant plasticity, which is shaped and controlled by factors such as cytokines, inflammation and growth signals, highlighting the importance of spatial relationships in understanding TIME and GME biology.
The tumor microenvironment and tuberculous infection are associated with three primary immunity-related metabolic pathways (glucose, fatty acids, and amino acids) and their products (70–72). The three major nutrients and their derivatives produce a broad range of metabolites, which are detected by specific sensors (metabolite sensing). This triggers a series of signal transduction pathways and epigenetic changes that influence gene expression (70). Metabolite sensing in cancer has emerged as a significant area of interest in recent years, but many questions remain unanswered. The regulation of metabolism, immunity, and inflammation via metabolite sensing allows the human body to coordinate its pathophysiology and maintain equilibrium with its external environment (70, 73, 74). Metabolic reprogramming in cancer cells causes them to exhibit different phenotypic characteristics from normal cells, including increased cell proliferation, migration, invasion, and angiogenesis (70, 73, 74). This metabolic disruption in cancer cells further fosters a microenvironment rich in various oncometabolites that promote cancer growth, thereby creating a vicious cycle. Meanwhile, baseline metabolic heterogeneity and inflammation-driven metabolic reprogramming during Mtb infection are associated with crucial immune functions, including mycobacterial growth control and the guidance of protective immunity (71).
The ways metabolites influence these immune cells and aid tumor and TB immune evasion (by impacting the function of immune cells) require further exploration. Given that both the pro- and anti-inflammatory immune cell phenotypes are connected to their cellular metabolism, it is crucial to define the molecular mechanisms that govern their metabolism (71). Arginine, lactate, and glutamine are among the metabolites that display commonality in LC and TB (Table 1). This section details how these metabolites and metabolic reprogramming influence immune cells in the tumor and granuloma microenvironments.
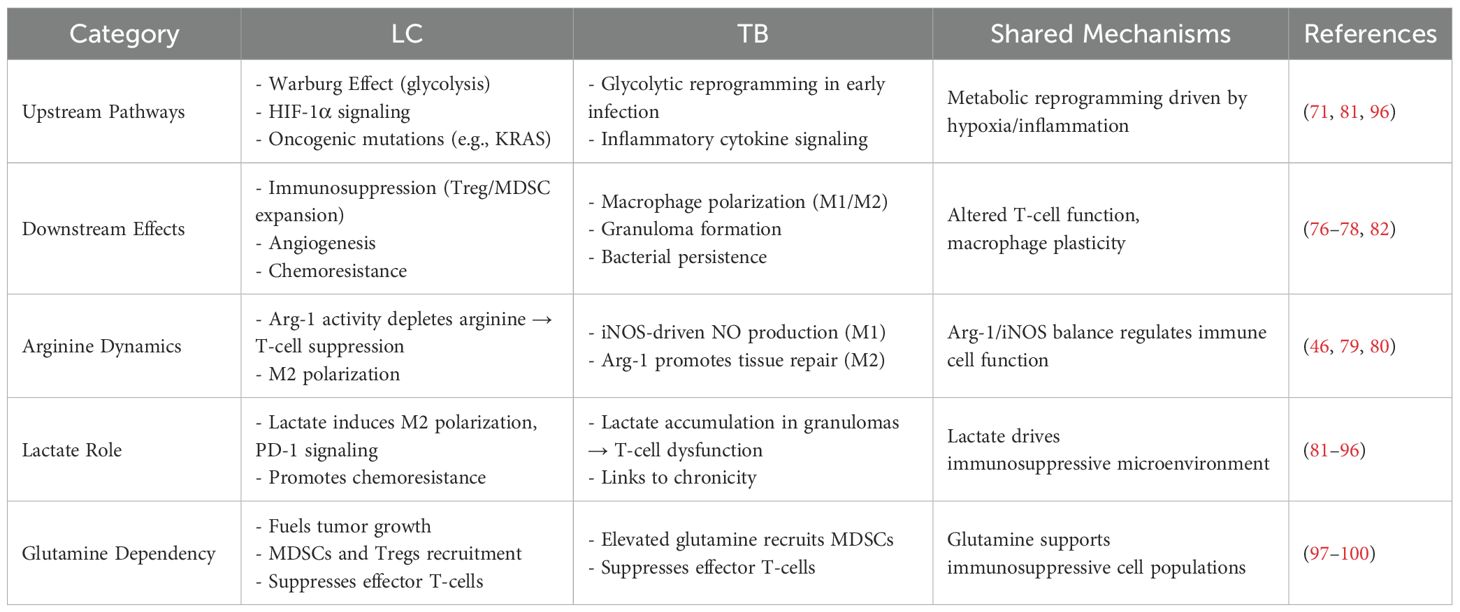
Table 1. Comparative analysis of immune-metabolic crosstalk in LC and TB microenvironments.
Classically activated M1 macrophages transform arginine into NO via iNOS activity, while M2 macrophages metabolize arginine to ornithine and urea by arginase-1 (64, 75). Pro-inflammatory glycolytic M1 macrophages, known for their antimicrobial activities, can transition to anti-inflammatory M2 macrophages, which depend on oxidative phosphorylation metabolism and lack antimicrobial effect (71, 76–78). This transition helps regulate inflammation and fosters tissue repair throughout the chronic stage of Mtb infection (71, 76–78). Increasing L-arginine levels have also been demonstrated to trigger metabolic alterations, including a transition from glycolysis to oxidative phosphorylation in activated T cells, promoting the formation of central memory-like cells with enhanced survival capacity and anti-tumor activity (46). Arginase-1 breaks down arginine into ornithine and contributes to the regulation of immune function in activated T-cells by depleting arginine in the local microenvironment (controlling arginine bioavailability results in nitric oxide production control), as demonstrated in cancer and tuberculosis experimental models (46, 79, 80). Overall, arginine influences macrophage polarization and affects T-cell function in both TB and LC. However, the specific impact of arginine on the immune microenvironment and systemic immune status in patients with concurrent TB-LC needs further investigation.
In solid tumors, lactate buildup occurs in the TME under hypoxic conditions and even in the presence of oxygen, driven by the Warburg Effect. This shift toward increased glycolytic flux is closely correlated with malignant transformation (81). Lactic acid has been linked to histone regulation and epigenetic lactylation in macrophage genomes (82–84). It promotes the transformation of macrophages from the pro-inflammatory, anti-cancer M1 to the anti-inflammatory, pro-cancer M2 type (82–84). Lactic acid can also enhance the immune suppressive function of Tregs (85), be converted to acetic acid by tumor-associated fibroblasts for cancer cell utilization (86), and subvert PD-1 inhibitor function to promote immune suppression (87). A recent study demonstrated that alanyl-tRNA synthetase (AARS1), which functions as a lactate sensor and lactyltransferase, can lactylate p53 and contribute to tumorigenesis (88). Additionally, lactate-induced lactylation of NBS1 was reported by Chen et al. (89) to promote homologous recombination (HR)-mediated DNA repair and lead to chemotherapy resistance.
Macrophages shift toward glycolytic metabolism in the early stages of Mtb infection, increasing lactate production (90, 91). Granuloma lesions can exhibit high lactate concentrations, reaching levels comparable to those observed in tumors (92, 93). Mtb expresses cytochrome bd oxidase to oxidize lactate into pyruvate, fueling bacterial respiration in human macrophages (94). This lactate oxidation capacity correlates with enhanced intracellular growth rates under hypoxia, suggesting lactate serves dual roles as both an immunosuppressive modulator and a pathogen nutrient source. The regulation of T-cell balance is crucial for either exacerbating or resolving TB pathology. The buildup of lactate within the granuloma lesions may significantly influence this balance, ultimately determining whether an anti-inflammatory or pro-inflammatory microenvironment develops (95). Therefore, it is promising to consider whether research approaches related to lactate in the tumor field can be applied to tuberculosis, particularly in exploring the mechanisms of drug-resistant tuberculosis.
High glutamine metabolism in tumors was discovered to have a strong correlation with the increased presence of immunosuppressive cells such as MDSCs and Tregs and concomitant suppression of effector T cells, which contributes to the spread of cancer and tumor development (70, 96, 97). Similarly, several studies have indicated that Mtb lung infection creates localized microenvironments characterized by increased elevated glutamine levels (98–100), subsequently leading to the accumulation of MDSCs, diminished effector T-cell activity, and reduced production of NO and citrulline (98). Treatment with JHU083, a glutamine metabolism antagonist, was linked with increased citrulline and NO production levels and potentiated antimycobacterial activity (98). Given the accumulation of glutamine in the tissue microenvironment and its association with immunosuppression, glutamine can be considered a potent target for immunometabolism in TB and LC.
To summarize, the function and persistence of immune cells in TB and cancer can be influenced by metabolic pathways through alterations in inflammatory processes within the tissue microenvironment (101) (Figure 1). Dynamic alterations in intracellular and extracellular metabolites, particularly variations in nutrient concentrations, can influence cell signaling pathways and epigenetic gene expression by acting as metabolite sensors (70). This creates a favorable microenvironment for tumor progression (70) or Mtb infection (90). It is important to emphasize that individual metabolic processes have been involved in eliciting effects during Mtb infection and LC progression. However, the inherent interconnectedness of metabolic reactions presents a significant challenge for researchers in identifying the specific contributions of these and other pathways to Mtb infection and LC progression, especially regarding the immune reprogramming related to metabolic pathways and host immune suppression. Additionally, metabolomics is closely intertwined with microbiomics. Notably, recent study (102) on the lung microbiome underscores its critical role in respiratory disease mechanisms, suggesting that spatial multi-omics analyses integrating metabolomics and microbiomics represent a promising direction for future research to further explore the molecular mechanisms linking TB and LC.
LC and TB show similarities in their shared immunosuppressive mechanisms, dysregulated inflammatory signaling, cellular plasticity, and immunometabolic reprogramming. These overlapping features may partially explain the pathophysiological basis for their clinical co-occurrence. However, it is important to note that distinct molecular mechanisms exist between the immune microenvironments of TB and LC. While a comprehensive comparative analysis of these nuanced differences is beyond the scope of this review due to space constraints, their exploration remains a critical frontier for future research—a primary motivation for this review. The following section will emphasis the specific molecular pathways, underlying their bidirectional pathological interactions.
Clinical case observations have shown that enhancing T-cell immunity in cancer patients by blocking the PD-1/PD-L1 axis may lead to latent tuberculosis reactivation (7–21). Studies in mice have demonstrated that PD-1-deficient mice experience intense inflammation and rapid mortality following Mtb infection (9, 103, 104). Additionally, in rhesus macaques, PD-1 inhibition results in larger TB granulomas (105). Several mechanisms may elucidate why ICIs are effective in treating lung cancer but also lead to TB re-activation.
The immune checkpoints balance pro-inflammatory and anti-inflammatory effects to control infections while avoiding immunopathology. In patients infected with Mtb, immune cells like CD4+ T cells, NK cells, neutrophils, and monocytes exhibit elevated PD-1 (106–108). In TB patients, PD-1 is present in granuloma areas where the interaction between host and pathogen is balanced but is missing in areas with caseating granulomas and marked immunopathology (107). Inhibiting PD-1 may disrupt the anti-Mtb-specific T-cell balance. This imbalance results in a pro-inflammatory environment rich in TNF-α, IL-6, and IL-12, which can promote Mtb growth, particularly TNF-α (107). It has been demonstrated that TNF-α can trigger necrosis in macrophages infected with Mtb. ROS production is essential for this effective immune response to TB (109–111). However, excessive TNF-α may impair macrophage function (109, 110). By enhancing the immune system’s response, immune checkpoint inhibition may cause inflammatory side activities, commonly known as immune-related adverse events (irAEs), which are autoimmune in nature (112). It has been found that autoreactive T-cells increase in TB patients, suggesting that autoimmunity maybe significant in TB pathology (113). Notably, irAEs caused by ICIs can be treated with anti-TNF-α antibodies (114), indicating that TNF-α may be a key factor in autoimmunity and TB pathology following PD-1 inhibition (107).
Moreover, Mtb may possess pathogen-specific characteristics that enable it to thrive in a hyper-inflammatory environment (such as those induced by ICIs) (115). Enhanced T-helper (Th) type 1 (Th1) responses triggered by ICIs can lead to overproduction of cytokines and increased MMP expression, resulting in tissue damage and destabilizing the delicate host-pathogen balance in the lungs. It has been suggested that various inhibitory receptors play crucial roles in modulating TB immune control. For instance, mice deficient in the chemokine scavenger D6 or the negative regulator of the IL-1 system (Toll/IL-1 receptor 8; TIR8) succumbed quickly to low-dose Mtb challenges, experiencing considerable local and systemic inflammation (116, 117). Heightened inflammation can lead to several detrimental outcomes, including an over-recruitment of inflammatory cells and the breakdown of the extracellular matrix, which promote Mtb proliferation (118–120). In this context, blocking PD-1 can induce T-cell dysfunction marked by hyperactive inflammatory responses, leading to a loss of immune regulation (121, 122).
In summary, the emerging clinical findings, together with studies in animal models and advanced cell cultures, collectively indicate that PD-1/PD-L1 disruption may create hyperinflammatory environments that favor mycobacterial growth. However, immune checkpoint inhibition does not necessarily result in autoimmune disorders or excessive inflammation; it depends on whether the balance is disrupted. Immune checkpoint inhibitors are widely used in cancer therapy, but their potential application in the treatment of tuberculosis remains controversial and needs further investigation (123). The key is to identify which patient groups are at higher risk for tuberculosis reactivation when receiving immune checkpoint inhibitors, thereby providing guidance for clinical practice.
Epidemiological studies found that anti-tumor therapy (surgery, chemotherapy, or radiotherapy) further increased the risk of Mtb infection in cancer patients (20, 21). Recent case reports have indicated that triple cancer therapy -which combines immunotherapy and chemoradiotherapy- may be linked to an increased risk of TB reactivation (124, 125). Lung cancer itself may increase the risk of tuberculosis activation, and the treatment for lung cancer may further elevate this risk. However, more stratified and detailed studies are needed to elucidate this correlation.
The induction of lymphopenia in cancer patients treated with most chemotherapeutic drugs has been acknowledged since the 1980s (126). Radiotherapy can cause lymphopenia as a direct result of blood or bone marrow irradiation (127–129). Additionally, 30% of patients undergoing anti-EGFR therapy develop lymphopenia (130). Multi-tyrosine kinase inhibitors can also induce grade III/IV lymphopenia in 10–15% of patients (131). As the combination of immunotherapy with chemotherapy, chemoradiotherapy, or targeted therapy becomes increasingly common in lung cancer treatment (132–135), combined treatment-induced lymphopenia warrants greater attention (128, 129).
Lymphocytes are susceptible to radiotherapy, and a reduction in peripheral total lymphocyte count (TLC) following ionizing radiation indicates an impaired host immune response to cancer. Numerous studies have established a connection between radiation-induced lymphopenia and poor prognosis across various cancers, including lung cancer (127–129, 136). One study involving 268 patients with advanced NSCLC who received ICIs, with 146 of them also undergoing radiotherapy, found that patients with lymphopenia at the start of immunotherapy had markedly poorer progression-free survival (PFS) (2.2 vs. 5.9 months) and overall survival (OS) (5.7 vs. 12.1 months) (136). Another recent study (129) reported that the median TLC dropped from 1.52 × 10^9 cells/L to 0.72 × 10^9 cells/L after chemoradiation in patients with locally advanced NSCLC, with 23% developing severe lymphopenia (<0.5 × 10^9 cells/L). Severe lymphopenia presented at the start of immunotherapy continued to be an independent negative predictor of PFS, with an HR of 4.90 and p < 0.001. Additionally, higher TLCs have been associated with a higher response rate and more durable treatment effects in patients treated with ICIs (137–139). In recent years, stereotactic body radiation therapy (SBRT) for lung cancer has garnered significant attention. Compared to conventional fractionated radiotherapy, SBRT can reduce lymphocyte depletion, improve prognosis, alleviate immunosuppression, and synergistically enhance the efficacy of ICIs, thereby boosting antitumor effects (140).
To conclude, a primary commonality between TB and LC is immunosuppression. The immunosuppression resulting from LC itself could potentially heighten the risk of TB activation. Furthermore, cancer treatments, such as immunotherapy, surgery, chemotherapy, and radiotherapy, might cause myelosuppression and immune dysregulation, thereby elevating the risk of active tuberculosis (Figure 2). While this statement is a hypothesis, and the detailed immunization-related molecular mechanisms involved need to be further explored. In clinical practice, we need to be aware of the immunosuppression that occurs during anti-tumor treatment, particularly the reduction in lymphocyte counts, and disruptions in immune function. Additionally, high-risk populations for tuberculosis activation should be screened and monitored before and during lung cancer treatment.
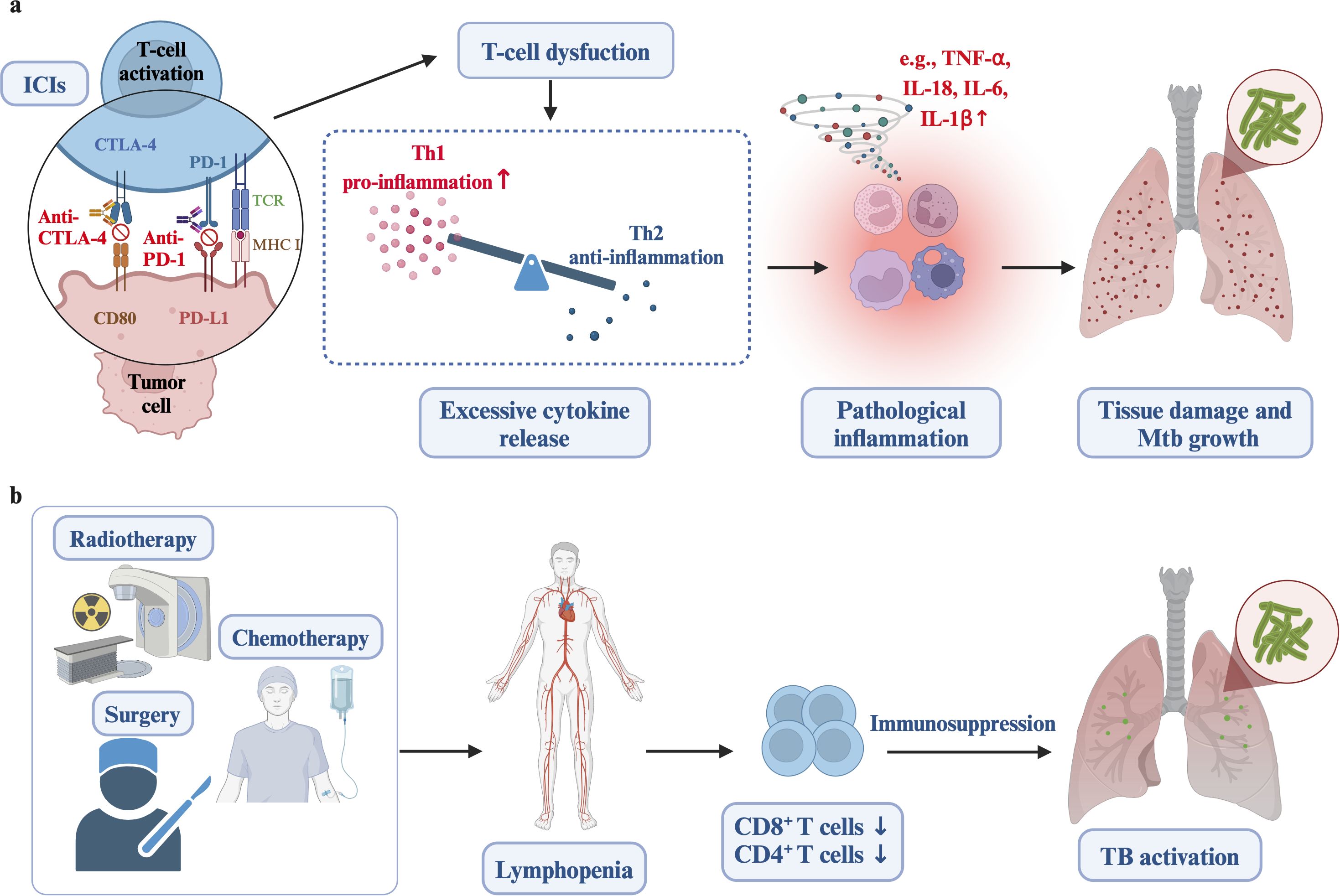
Figure 2. The relationship between cancer treatments and increased risk of active tuberculosis. (a) Immune checkpoint inhibitors (ICIs) can cause T-cell dysfunction and hyperinflammatory conditions, including excessive TNF-α, IL-6, and IL-12, resulting in tissue damage and mycobacterial growth. (b) Treatments such as radiotherapy, surgery, and chemotherapy lead to lymphopenia, decreased CD8+ T cells, systemic immunosuppression, and reduced infection barriers, ultimately increasing the susceptibility to active tuberculosis. Created in https://BioRender.com.
Successful tumor initiation requires two main interconnected events. 1) The buildup of mutations or epigenetic changes in genes and cellular signaling networks involved in tumor suppression (inactivation) and the activation of oncogenic signals is crucial (58). Inflammatory effects possess potent mechanisms that contribute to the accumulation of mutations and epigenetic changes in nearby epithelial cells, while these have traditionally been associated with environmental factors (such as variable radiation, carcinogens, and/or UV), and errors in DNA replication and repair. The initiation phase of tumorigenesis often involves irreversible DNA changes induced by reactive nitrogen species (RNS) and reactive oxygen species (ROS). These reactive species are mainly produced by macrophages and neutrophils, which is involved in inflammatory response derived from Mtb infection (58, 141–144). 2) For transformed or malignant clones to progress into a fully developed tumor, their emergence must be accompanied by substantial growth, a process significantly driven by inflammatory mechanisms (58).
The ‘initiation’ phase involves permanent DNA alterations that can remain in normal cells until the ‘promotion’ stage is triggered. The ‘promotion’ phase is crucial as it facilitates cell proliferation, ROS and RNS production, recruitment of inflammatory cells, and oxidative DNA damage (58, 59). Inflammatory cytokines such as IL-6, IL-17, and IL-11 play a vital role in shaping cell plasticity within the TME and enhancing tumor cell proliferation, particularly under sub-optimal in vivo conditions such as hypoxia, nutrient deficiency, and limited production of growth factors, while also counteracting anti-tumor immunity (58, 145). Moreover, inflammatory signals modulate the structural and metabolic properties of tumor and stromal cells by modulating the extracellular matrix, growth factor availability, and key metabolites linked to redox and amino acid metabolism (145). Prolonged pathogen-induced chronic inflammation significantly elevates the risk of cancer (58). Chronic inflammation triggered by TB infection induces DNA damage, DNA repair, heightened production of ROS and RNS, cell death, as well as cell proliferation (146). These processes remain in chronic inflammatory tissues, leading to uncontrolled DNA replication and cell proliferation, possibly contributing to LC initiation and promotion (Figure 3).
As previously discussed, the formation of granulomas is a complex process that involves Mtb immune evasion through ongoing interactions between infected macrophages and T cells in response to chronic inflammation (39). The TME and TB granulomas exhibit a central theme of exhausted T cell phenotypes (33). Numerous immune checkpoints, such as CTLA-4, TIM-3, LAG-3, and PD-1, are enriched in these exhausted T cells and are crucial in inhibiting T cell effects (49). The development of tumors may be influenced by the exhausted phenotype of T cells and the immunosuppressive effects triggered by Mtb infection.
Furthermore, neutrophils are important in the prolonged pulmonary inflammation associated with TB. As innate immune cells, neutrophils are involved in the early phases of Mtb infection and crucial for killing the bacteria (147). Beyond their phagocytic and lytic roles, neutrophils undergo necrosis and release neutrophil extracellular traps (NETs), which act to capture and sequester extracellular bacteria, thereby restricting their dissemination (148). However, during later phases of infection, uncontrolled neutrophil activity leads to neutrophil-driven inflammation, exacerbating lung tissue damage (147, 149–152). Studies show that NET formation can promote Mtb growth and contribute to disease severity and inflammation-mediated tissue damage (147, 149, 150). The inflammatory response is primarily driven by three neutrophil functions: oxidative burst, necrosis, and NETosis (153). The buildup of neutrophils at inflammation sites results in uncontrolled necrosis and inflammation in the surrounding tissues. Similarly, NETs are detected in necrotic lung lesions of TB patients who show poor responses to antibiotic therapy, highlighting their involvement in the pathogenesis of late-stage TB (141, 150, 154, 155).
Meanwhile, cancer cells can manipulate neutrophil recruitment and behavior by converting some into a pro-tumor phenotype (156, 157). Pro-tumor neutrophils can interact with cancer cells and other TME cells, including T cells, macrophages, and stromal cells, to promote tumor initiation, growth, and metastasis (156–159). This indicates that neutrophil-driven inflammation, characterized by the buildup of proinflammatory proteins linked to neutrophils and the formation of NETs, which work together to harm lung tissue in TB may contribute to LC progression and metastasis. However, the precise molecular mechanisms underlying this process require further investigation.
Repeated tissue damage and repair, caused by prolonged and intense TB pulmonary inflammation, ultimately lead to fibrotic scarring that is linked to a heightened risk of lung cancer (160–162). Recent studies in mouse models have documented that tuberculous fibrosis can elevate the tumorigenic potential of lung cells, potentially mediated by NOX4-associated autophagy (163). Inflammatory monocytes (IMs), identified as the “CCR2highCD14+ CD16low” phenotype in humans, are present in lung cancer and critical in forming scar tissue and promoting tumors (164–167). A recent study demonstrated that CCR2+ monocytes in the lungs of mice with cystic fibrosis drive pathogenic TGF-β signaling and maintain a pro-inflammatory environment by facilitating neutrophil recruitment (167). Lung squamous cell carcinomas (SCC) are marked by poor survival outcomes and significant infiltration of IMs, creating conditions that facilitate the distant metastasis of SCC (166). Tumors attract IMs by secreting monocyte chemoattractant protein-1 (MCP-1 or CCL2) (166), which can promote tumor cell proliferation, migration, and survival (168). High levels of CCL2 expression and increased macrophage infiltration have been linked to a poor prognosis and the development of metastatic disease in cancer (169). Additionally, elevated levels of CCL2 are also found in patients with pulmonary tuberculosis (170).
In summary, inflammatory signals can modify the TME to promote immunosuppression through the actions of Tregs, immature myeloid cells, and other suppressive elements. These signals also enhance the recruitment and proliferation of various pro-tumorigenic auxiliary cells within the TME while promoting their specialized functions (58). Chronic inflammation driven by Mtb may contribute to the progression and metastasis of lung cancer through mechanisms such as ROS and RNS production, DNA damage and repair, exhausted T cell phenotypes, induced immunosuppression, tuberculous fibrosis, and the infiltration of neutrophils and inflammatory monocytes (IMs) (Figure 3).
Advancements in sequencing technologies and analytical methods have enabled researchers to demonstrate that the gut microbiota performs various functions that impact the host’s overall health, such as nutrient metabolism and the regulation of the immune system by influencing immunological, inflammatory, neurological, metabolic, and endocrine functions (171–173). Studies have indicated that certain gut bacterial groups, like Bacteroides and Ruminococcus, in the microbiota of lung cancer patients are linked with early-stage lung cancer (174). Recent research has revealed that reduced gut (and lung) microbiota diversity, the abundance of specific microbiota, and enriched metabolic pathways may be related to lung adenocarcinoma development/progression (175, 176). A study analyzing lung cancer tissues found that bacterial genera such as Romboutsia, Novosphingobium, and Acinetobacter were more abundant than in adjacent normal tissues (177).
Broad-spectrum antibiotic treatment has been shown to heighten susceptibility to Mtb infection and modulate pulmonary inflammatory responses in a murine model (178). Analysis using random forest regression on microbiome-transcriptome-sputum data indicates that normalizing the TB inflammatory state is linked to the clearance of Mtb pathogens, a higher abundance of Clostridia Clusters IV and XIVa, a reduced presence of Bacilli and Proteobacteria (179). Mtb pathogen clearance is associated with the renormalization of the TB inflammatory state. This process is linked to an increase in Clostridia Clusters IV and XIVa and a decrease in Bacilli and Proteobacteria. Additionally, the alterations in inflammatory gene expression accompanying TB treatment are linked to the antimicrobial effects of the drugs, resulting in pathogen clearance and antibiotic-induced shifts in microbiome composition (179).
Moreover, another study demonstrated that gut microbiome dynamics and TB clearance are predictive cofactors in resolving TB-driven inflammation, thereby guiding therapeutic strategies for multi-drug-resistant tuberculosis (180). Antibiotic-induced reductions in pathogen load and microbiome alterations induced by antibiotics are independently linked to changes in inflammatory responses to the treatment of active TB (179). Microbial dysbiosis and decreased gut microbiome diversity (induced by long-term antibiotic treatment against TB) may be related to the occurrence and progression of lung cancer through microbiome-driven immunomodulation (Figure 3). However, more detailed research is needed to confirm this relationship. It is also important to explore which specific microbes are involved and play a key role. Additionally, investigating whether probiotics can help modulate the immune system, reduce drug-resistant tuberculosis, and lower the risk of cancer development is a promising area for future research.
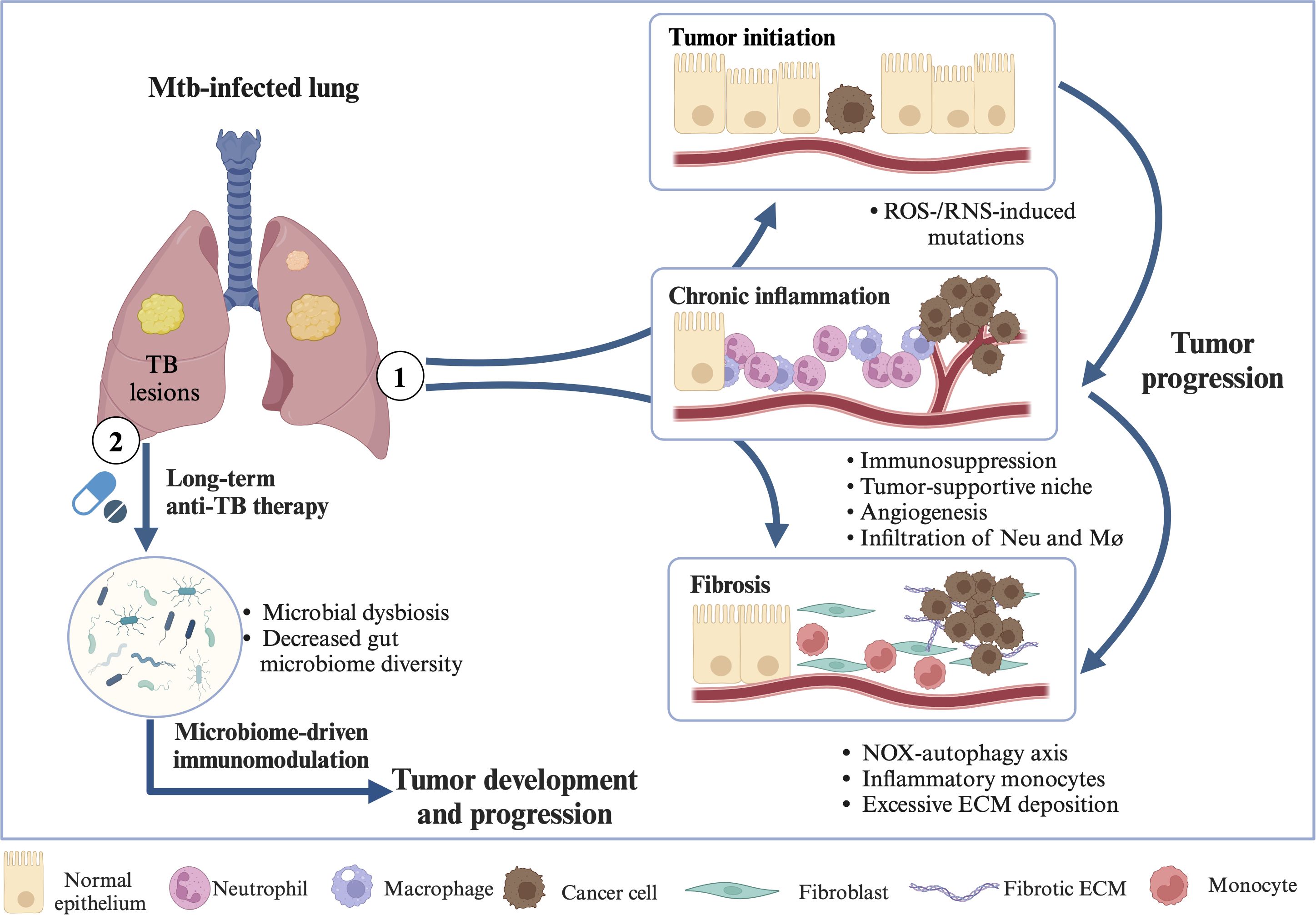
Figure 3. Molecular mechanisms in TB→LC. ① Chronic inflammation driven by Mtb may contribute to LC initiation, promotion, progression, and metastasis through mechanisms such as ROS and RNS production, DNA damage and repair, exhausted T cell phenotypes, induced immunosuppression, angiogenesis, tuberculous fibrosis, NOX-autophagy axis, excessive ECM deposition, and the infiltration of neutrophils, macrophages, and inflammatory monocytes. ② Microbial dysbiosis and reduced gut microbiome diversity, resulting from prolonged antibiotic treatment for tuberculosis, may be associated with the development and progression of LC through microbiome-driven immunomodulation. Abbreviations: ROS, reactive oxygen species; RNS, reactive nitrogen species; ECM, extracellular matrix; Neu, neutrophil; Mø, macrophage; NOX, non-phagocytic cell oxidase. Created in https://BioRender.com.
The concurrent LC and PTB pose substantial difficulties for differential diagnosis. The primary diagnosis methods rely on clinical symptoms, distinctive imaging findings, and pathogen detection. While clinical symptoms may provide indications, they often lack high specificity. Imaging signs reveal that patients with coexisting PTB and LC demonstrate a higher frequency of pleural indentations, satellite lesions, masses, spicule features, small vacuole features, nodular shadows, vacuole features, and lobulation features than those with PTB alone (181). Lung biopsy pathology remains the definitive standard for distinguishing LC from PTB, but acquiring biopsy samples from certain puncture-unfriendly locations can be problematic.
As an alternative, blood-based pathogen detection has emerged. The Hu laboratory (182) has pioneered an ultrasensitive CRISPR-mediated tuberculosis (CRISPR-TB) assay for detecting Mtb-cfDNA in serum, showing potential for improving the identification of pediatric tuberculosis and HIV-associated tuberculosis and allowing for early diagnosis and swift monitoring of tuberculosis treatment responses. Moreover, they have employed a nanoparticle-enhanced immunoassay for the multiplex detection of two interacting Mtb biomarkers on the surface of circulating extracellular vesicles, enhancing tuberculosis diagnosis in immunosuppressed children with HIV (183). Recently, circulating tumor DNA (ctDNA) has surfaced as a minimally invasive biomarker for tumor molecular profiling. The European Society of Medical Oncology (ESMO) has endorsed ctDNA testing in routine clinical practice for tumor genotyping, guiding molecularly targeted therapies in patients with metastatic cancer (184, 185).
With the advent of these non-invasive, highly sensitive technologies for detecting free DNA, the differentiation between PTB and LC is set to become more refined, paving the way for subsequent TB and LC treatments. In PTB-LC differential diagnosis, this CRISPR-TB technology provides two critical functions: 1) Active TB exclusion: A negative CRISPR-TB result (Mtb-cfDNA <5 copies/mL) in patients with lung masses strongly suggests LC-driven pathology, prompting immediate oncological intervention. 2) Treatment response monitoring: In confirmed PTB-LC cases, longitudinal Mtb-cfDNA quantitation (e.g., 80% reduction after 2-month HRZE therapy) helps distinguish TB recurrence from cancer progression when radiographic abnormalities persist. While CRISPR-TB and ctDNA were initially developed for standalone applications, their integration creates a synergistic diagnostic paradigm. The CRISPR-TB assay’s capacity to quantify Mtb-cfDNA load complements ctDNA’s ability to track tumor evolution, enabling real-time differentiation between TB-driven inflammation and LC progression. This dual-liquid biopsy approach may redefine clinical guidelines for TB-LC co-diagnosis in high-burden settings. However, its high cost currently limits widespread adoption.
Pulmonary complications such as fibrosis and cavitation, arise from persistent inflammation driven by host pro-inflammatory signals triggered by a substantial Mtb burden. The effort to shorten treatment durations and enhance clinical outcomes has prompted a two-fold strategy: developing new antimicrobials and host-directed therapies (HDT) that positively influence immune responses to Mtb (147). Targets for HDT in PTB include lipid mediators, matrix metalloproteinases, cytokines, and NETs (147). HDT strategies focus on modulating the immune system by boosting antimicrobial response while suppressing excessive or unhelpful inflammatory effects, thereby reducing lung damage and hastening symptom resolution (147).yole of inflammation as a link between LC and PTB was discussed in Section 2.2; this section will highlight the progress in targeting inflammatory signaling pathways with nonsteroidal anti-inflammatory drugs (NSAIDs).
NSAIDs, widely used to manage various chronic conditions, function as anti-inflammatory medications by inhibiting cyclooxygenase enzymes. Maintaining a balance between LXA-4 and PGE-2 plays a vital role in controlling inflammation derived by TB. The promotion of LXA-4 generation and the reduction of PGE-2 production by NSAIDs contribute to a decrease in TNF levels, ultimately resulting in reduced tissue inflammation (186, 187). A randomized phase II clinical trial revealed that aspirin has been employed as an adjuvant to standard anti-TB therapy for tuberculous meningitis, demonstrating a reduction in stroke risk and mortality rates (188). In a C3HeB/FeJ mouse model of Mtb infection, ibuprofen significantly decreased neutrophil infiltration and reduced pathological lung inflammation in mice (189). Recently, another phase IIb randomized control trial (NCT04575519) has been registered to evaluate the potential benefits of adding acetylsalicylic and acid ibuprofen to the standard TB therapy protocol, targeting both drug-sensitive and multidrug-resistant TB cases.
In line with inflammation’s role in promoting tumorigenesis, long-term NSAID use is linked to reduced cancer incidence (190), including a significant decrease in lung cancer incidence among chronic smokers (191). Furthermore, a phase III trial observed a notable reduction in LC incidence while investigating a blocking antibody aimed at the inflammatory mediator IL-1β for atherosclerosis (CANTOS; NCT01327846) (192). It is worth noting that not every chronic inflammatory disease is associated with an increased cancer risk. Rheumatoid arthritis and psoriasis, for instance, do not seem to contribute to cancer development. However, the use of anti-inflammatory drugs to manage these inflammatory conditions, including TB, might influence cancer risk. Therefore, targeting inflammatory pathways might reduce the severity of both TB and LC, offering a potentially effective approach to enhance long-term outcomes in patients affected by these conditions. Nevertheless, future challenges lie in validating the dose-response relationships of repurposed drugs (e.g., NSAIDs).
The previously discussed roles of immunometabolism in TB and LC highlight their similarities. As an emerging area of research, therapeutic approaches focusing on immunometabolism pathways have seen notable advancements. For example, the glutamine metabolism pathway could be a therapeutic target for tumors and TB. On one side, glutamate’s biological impacts can be lessened by blocking its receptors, including genetic alterations (193) or the use of metabolic glutamate receptor 5 antagonists (194). On the other side, direct inhibition of tumor growth can be achieved by blocking glutamine transport (195, 196), including using the recombinant solute carrier family 7, member 11 (SLC7A11 or xCT). Glutamic acid (a byproduct of glutamine hydrolysis) is able to be expelled from tumor cells via transporters like xCT. The disruption of the glutamate-cysteine anti-transporter xCT results in cellular glutamate retention, causing glutamate to lose its function outside the cells, leading to decreased NSCLC proliferation and invasion (197).
Similarly, several studies have indicated that Mtb lung infection creates localized microenvironments characterized by increased glutamine levels (98–100), subsequently leading to MDSC accumulation, diminished effector T-cell functionality, and a decrease in the synthesis of NO and citrulline (98). Parveen et al. (98) demonstrated that JHU083, a glutamine metabolism antagonist, could trigger earlier recruitment of T-cells, boost infiltration of proinflammatory myeloid cells, and decrease IL-10-producing MDSCs. Inhibiting glutamine metabolism with JHU083 provides both antibacterial and host-targeted actions against tuberculosis (98), offering new perspectives for treating drug-resistant TB. However, its penetration capability into human granuloma microenvironments remains to be validated.
Metabolic disturbances in the tumor microenvironment result in spatial gradients arising from varying lactate sensitivity among tumor cells and stromal cells, including tumor-associated macrophages. Limited perfusion plays a role in creating metabolic gradients, enabling cancer cells to endure high lactate and low oxygen (198). Comparable spatial gradients also exist within TB granulomas, which exhibit unique immune transcripts, proteomic profiles, and diverse cell subsets. These granulomas have locally coordinated immunoregulatory programs with broader systemic impacts that define active TB (44, 67, 69, 199).
Oxygen tension gradients and changes in vasculature linked to granulomas cause limited perfusion and central hypoxia (200, 201). Redox balance within granulomas also plays a critical role (202). Redox-modulating agents such as the thioredoxin reductase inhibitor auranofin (203) and cysteine-reactive inhibitors like ebselen (204, 205) may regulate the redox balance and immune effects in the granuloma microenvironment through distinct mechanisms, thereby exhibiting potential antimicrobial properties. In addition, it’s probable that spatial variations in lactate metabolism and varied interactions among different cell types occur within the context of TB granulomas, given the similarities between tumor and TB microenvironments. These factors might serve as potential HDT targets for TB treatment. Furthermore, Chen et al. (89) have shown that lactate dehydrogenase expression and NBS1 lactylation are associated with clinical resistance to neoadjuvant chemotherapy, resulting in reduced patient survival rates. They also demonstrated that stiripentol, an anti-epileptic drug, significantly reduced lactate production and NBS1 lactylation. In mouse models, stiripentol combined with chemotherapy or radiotherapy showed strong synergistic antitumor effects and prolonged survival time. Related clinical trials have been registered (registration number: ChicTR2400083649). Overall, lactate has demonstrated great potential in treating tuberculosis and lung cancer.
There has been substantial advancement in identifying therapeutic targets that exploit metabolite-sensing signaling pathways to halt cancer progression (70). Therefore, targeting shared metabolic changes in TB and LC, such as those involving arginine, lactate, and glutamine, might provide common therapeutic targets for TB and LC.
In summary, we have provided a comprehensive review of the complex relationship between LC and TB, covering topics from epidemiology to underlying molecular mechanisms. We focus on the latest advancements in spatial multi-omics, as well as developments in immune microenvironments, cellular elasticity, immunometabolism, and microbiomics. Based on these insights, we propose future research directions and potential treatment strategies for TB-LC.
YZ: Conceptualization, Formal Analysis, Funding acquisition, Supervision, Writing – review & editing. CF: Conceptualization, Data curation, Funding acquisition, Investigation, Software, Visualization, Writing – original draft. XH: Formal Analysis, Investigation, Software, Writing – original draft. FT: Data curation, Formal Analysis, Investigation, Writing – original draft. ZW: Data curation, Software, Writing – review & editing. CP: Investigation, Supervision, Writing – review & editing. QZ: Formal Analysis, Software, Writing – review & editing. JW: Formal Analysis, Visualization, Writing – review & editing. QW: Conceptualization, Investigation, Supervision, Writing – review & editing. DL: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Science and Technology Innovation Bureau of Zhuhai City (Grant Number: ZHKJ2024-01); Joint Fund Program of Guizhou Provincial Health Commission (Grant Number: 2024GZYXKYJJXM0031); Science and Technology Foundation of Guizhou Province (Grant Numbers: Qiankehejichu-ZK (2021) General 455, ZK (2023) General 212); National Natural Science Foundation of China (Grant Number: 82371851); Science and Technology Foundation of Guizhou Province (Outstanding Young Scientists of Guizhou Province (Grant Number: Qiankeherencai-YQK(2023)021); and Health Commission Science and Technology Foundation of Guizhou Province (Grant Number: gzwkj 2022-028).
Author CP was employed by eBond Pharmaceutical Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Adam T, Arinaminpathy N, Baddeley A, Bastard M, Boon SD, Falzon D, et al. Global tuberculosis report 2024. Geneva: World Health Organization (2024) Licence: CC BY-NC-SA 3.0 IGO. (Accessed March 25, 2025).
3. Dobler CC, Cheung K, Nguyen J, Martin A. Risk of tuberculosis in patients with solid cancers and haematological Malignancies: a systematic review and meta-analysis. Eur Respir J. (2017) 50:1700157. doi: 10.1183/13993003.00157-2017
4. Cheng MP, Abou Chakra CN, Yansouni CP, Cnossen S, Shrier I, Menzies D, et al. Risk of active tuberculosis in patients with cancer: A systematic review and meta-analysis. Clin Infect Dis. (2017) 64:635–44. doi: 10.1093/cid/ciw838
5. Seo GH, Kim MJ, Seo S, Hwang B, Lee E, Yun Y, et al. Cancer-specific incidence rates of tuberculosis: A 5-year nationwide population-based study in a country with an intermediate tuberculosis burden. Med (Baltimore). (2016) 95:e4919. doi: 10.1097/MD.0000000000004919
6. Simonsen DF, Farkas DK, Horsburgh CR, Thomsen RW, Sorensen HT. Increased risk of active tuberculosis after cancer diagnosis. J Infect. (2017) 74:590–8. doi: 10.1016/j.jinf.2017.03.012
7. Fujita K, Terashima T, Mio T. Anti-PD1 antibody treatment and the development of acute pulmonary tuberculosis. J Thorac Oncol. (2016) 11:2238–40. doi: 10.1016/j.jtho.2016.07.006
8. Bae S, Kim YJ, Kim MJ, Kim JH, Yun SC, Jung J, et al. Risk of tuberculosis in patients with cancer treated with immune checkpoint inhibitors: a nationwide observational study. J Immunother Cancer. (2021) 9:e002960. doi: 10.1136/jitc-2021-002960
9. Barber DL, Sakai S, KudChadkar RR, Fling SP, Day TA, Vergara JA, et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci Transl Med. (2019) 11:eaat2702. doi: 10.1126/scitranslmed.aat2702
10. Langan EA, Graetz V, Allerheiligen J, Zillikens D, Rupp J, Terheyden P. Immune checkpoint inhibitors and tuberculosis: an old disease in a new context. Lancet Oncol. (2020) 21:e55–65. doi: 10.1016/S1470-2045(19)30674-6
11. Chen HW, Kuo YW, Chen CY, Chang CH, Wang SM, Chien YC, et al. Increased tuberculosis reactivation risk in patients receiving immune checkpoint inhibitor-based therapy. Oncologist. (2024) 29:e498–506. doi: 10.1093/oncolo/oyad340
12. Zaemes J, Kim C. Immune checkpoint inhibitor use and tuberculosis: a systematic review of the literature. Eur J Cancer. (2020) 132:168–75. doi: 10.1016/j.ejca.2020.03.015
13. Lin C, Xu G, Gao S, Feng T, Li S. Tuberculosis infection following immune checkpoint inhibitor treatment for advanced cancer: a case report and literature review. Front Immunol. (2023) 14:1162190. doi: 10.3389/fimmu.2023.1162190
14. Fujita K, Yamamoto Y, Kanai O, Okamura M, Hashimoto M, Nakatani K, et al. Incidence of active tuberculosis in lung cancer patients receiving immune checkpoint inhibitors. Open Forum Infect Dis. (2020) 7:ofaa126. doi: 10.1093/ofid/ofaa126
15. Anand K, Sahu G, Burns E, Ensor A, Ensor J, Pingali SR, et al. Mycobacterial infections due to PD-1 and PD-L1 checkpoint inhibitors. ESMO Open. (2020) 5:e000866. doi: 10.1136/esmoopen-2020-000866
16. Im Y, Lee J, Kim SJ, Koh WJ, Jhun BW, Lee SH. Development of tuberculosis in cancer patients receiving immune checkpoint inhibitors. Respir Med. (2020) 161:105853. doi: 10.1016/j.rmed.2019.105853
17. Chan GH, Gwee YX, Low JL, Huang Y, Chan ZY, Choo JR, et al. Immune checkpoint inhibition for non-small cell lung cancer in patients with pulmonary tuberculosis or Hepatitis B: Experience from a single Asian centre. Lung Cancer. (2020) 146:145–53. doi: 10.1016/j.lungcan.2020.05.020
18. Liu K, Wang D, Yao C, Qiao M, Li Q, Ren W, et al. Increased tuberculosis incidence due to immunotherapy based on PD-1 and PD-L1 blockade: A systematic review and meta-analysis. Front Immunol. (2022) 13:727220. doi: 10.3389/fimmu.2022.727220
19. Alhashimi MM, Citron ML, Fossieck BE Jr., Steinberg SM, Johnston-Early A, Krasnow SH, et al. Lung cancer, tuberculin reactivity, and isoniazid. South Med J. (1988) 81:337–40. doi: 10.1097/00007611-198803000-00013
20. Kim HR, Hwang SS, Ro YK, Jeon CH, Ha DY, Park SJ, et al. Solid-organ Malignancy as a risk factor for tuberculosis. Respirology. (2008) 13:413–9. doi: 10.1111/j.1440-1843.2008.01282.x
21. Wu CY, Hu HY, Pu CY, Huang N, Shen HC, Li CP, et al. Aerodigestive tract, lung and haematological cancers are risk factors for tuberculosis: an 8-year population-based study. Int J Tuberc Lung Dis. (2011) 15:125–30.
22. Moon SM, Choi H, Kim SH, Kang HK, Park DW, Jung JH, et al. Increased lung cancer risk and associated risk factors in tuberculosis survivors: A korean population-based study. Clin Infect Dis. (2023) 77:1329–39. doi: 10.1093/cid/ciad373
23. Ho LJ, Yang HY, Chung CH, Chang WC, Yang SS, Sun CA, et al. Increased risk of secondary lung cancer in patients with tuberculosis: A nationwide, population-based cohort study. PloS One. (2021) 16:e0250531. doi: 10.1371/journal.pone.0250531
24. Engels EA, Shen M, Chapman RS, Pfeiffer RM, Yu YY, He X, et al. Tuberculosis and subsequent risk of lung cancer in Xuanwei, China. Int J Cancer. (2009) 124:1183–7. doi: 10.1002/ijc.24042
25. Wu CY, Hu HY, Pu CY, Huang N, Shen HC, Li CP, et al. Pulmonary tuberculosis increases the risk of lung cancer: a population-based cohort study. Cancer. (2011) 117:618–24. doi: 10.1002/cncr.25616
26. Yu Y, Liao C, Hsu W, Chen H, Liao W, Muo C, et al. Increased lung cancer risk among patients with pulmonary tuberculosis: A population cohort study. J Thorac Oncol. (2011) 6:32–7. doi: 10.1097/JTO.0b013e3181fb4fcc
27. Cabrera-Sanchez J, Cuba V, Vega V, Stuyft Pvd, Otero L. Lung cancer occurrence after an episode of tuberculosis: a systematic review and meta-analysis. Eur Respir Rev. (2022) 31:220025. doi: 10.1183/16000617.0025-2022
28. Hwang SY, Kim JY, Lee HS, Lee S, Kim D, Kim S, et al. Pulmonary tuberculosis and risk of lung cancer: A systematic review and meta-analysis. J Clin Med. (2022) 11:765. doi: 10.3390/jcm11030765
29. Sodeifian F, Kian N, Atefi A, Naserghandi A, Zangiabadian M, Sadeghzade S, et al. Pulmonary tuberculosis and risk of lung cancer: A systematic review and meta-analysis. Respiration. (2024), 1–24. doi: 10.1159/000543319
30. Hong S, Mok Y, Jeon C, Jee SH, Samet JM. Tuberculosis, smoking and risk for lung cancer incidence and mortality. Int J Cancer. (2016) 139:2447–55. doi: 10.1002/ijc.30384
31. Yang L, Ye Z, Li L, Zhuang L, Guan J, Gong W. Visualization analysis of research progress and trends in coexistence of lung cancer and pulmonary tuberculosis using bibliometrics. Med Adv. (2024) 2:144–64. doi: 10.1002/med4.58
32. G. B. D. C. o. D. Collaborators. Global burden of 288 causes of death and life expectancy decomposition in 204 countries and territories and 811 subnational locations, 1990-2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. (2024) 403:2100–32. doi: 10.1016/S0140-6736(24)00367-2
33. Bickett TE, Karam SD. Tuberculosis-cancer parallels in immune response regulation. Int J Mol Sci. (2020) 21:6136. doi: 10.3390/ijms21176136
34. Lasser SA, Ozbay Kurt FG, Arkhypov I, Utikal J, Umansky V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. (2024) 21:147–64. doi: 10.1038/s41571-023-00846-y
35. de Souza N, Zhao S, Bodenmiller B. Multiplex protein imaging in tumour biology. Nat Rev Cancer. (2024) 24:171–91. doi: 10.1038/s41568-023-00657-4
36. Monkman J, Moradi A, Yunis J, Ivison G, Mayer A, Ladwa R, et al. Spatial insights into immunotherapy response in non-small cell lung cancer (NSCLC) by multiplexed tissue imaging. J Transl Med. (2024) 22:239. doi: 10.1186/s12967-024-05035-8
37. Zhao Z, Qin J, Qian Y, Huang C, Liu X, Wang N, et al. FFAR2 expressing myeloid-derived suppressor cells drive cancer immunoevasion. J Hematol Oncol. (2024) 17:9. doi: 10.1186/s13045-024-01529-6
38. Sorin M, Rezanejad M, Karimi E, Fiset B, Desharnais L, Perus LJM, et al. Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature. (2023) 614:548–54. doi: 10.1038/s41586-022-05672-3
39. Kaufmann SHE. How can immunology contribute to the control of tuberculosis? . Nat Rev Immunol. (2001) 1:20–30. doi: 10.1038/35095558
40. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
41. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. (2007) 13:4345–54. doi: 10.1158/1078-0432.CCR-07-0472
42. Green AM, Mattila JT, Bigbee CL, Bongers KS, Lin PL, Flynn JL. CD4(+) regulatory T cells in a cynomolgus macaque model of Mycobacterium tuberculosis infection. J Infect Dis. (2010) 202:533–41. doi: 10.1086/654896
43. Rahman S, Gudetta B, Fink J, Granath A, Ashenafi S, Aseffa A, et al. Compartmentalization of immune responses in human tuberculosis: few CD8+ effector T cells but elevated levels of FoxP3+ regulatory t cells in the granulomatous lesions. Am J Pathol. (2009) 174:2211–24. doi: 10.2353/ajpath.2009.080941
44. McCaffrey EF, Donato M, Keren L, Chen Z, Delmastro A, Fitzpatrick MB, et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat Immunol. (2022) 23:318–29. doi: 10.1038/s41590-021-01121-x
45. Dorhoi A, Kotze LA, Berzofsky JA, Sui Y, Gabrilovich DI, Garg A, et al. Therapies for tuberculosis and AIDS: myeloid-derived suppressor cells in focus. J Clin Invest. (2020) 130:2789–99. doi: 10.1172/JCI136288
46. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
47. du Plessis N, Loebenberg L, Kriel M, von-Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med. (2013) 188:724–32. doi: 10.1164/rccm.201302-0249OC
48. Magcwebeba T, Dorhoi A, du Plessis N. The emerging role of myeloid-derived suppressor cells in tuberculosis. Front Immunol. (2019) 10:917. doi: 10.3389/fimmu.2019.00917
49. Sun Q, Hong Z, Zhang C, Wang L, Han Z, Ma D. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. (2023) 8:320. doi: 10.1038/s41392-023-01522-4
50. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
51. Li W, Xu M, Li Y, Huang Z, Zhou J, Zhao Q, et al. Comprehensive analysis of the association between tumor glycolysis and immune/inflammation function in breast cancer. J Transl Med. (2020) 18:92. doi: 10.1186/s12967-020-02267-2
52. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e:1792. doi: 10.1038/cddis.2015.162
53. Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, et al. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PloS Pathog. (2016) 12:e:1005490. doi: 10.1371/journal.ppat.1005490
54. Khan N, Vidyarthi A, Amir M, Mushtaq K, Agrewala JN. T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol. (2017) 43:133–41. doi: 10.1080/1040841X.2016.1185603
55. Jiang H, Gong H, Zhang Q, Gu J, Liang L, Zhang J. Decreased expression of perforin in CD8(+) T lymphocytes in patients with Mycobacterium tuberculosis infection and its potential value as a marker for efficacy of treatment. J Thorac Dis. (2017) 9:1353–60. doi: 10.21037/jtd.2017.05.74
56. Hodge G, Barnawi J, Jurisevic C, Moffat D, Holmes M, Reynolds PN, et al. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-gamma by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin Exp Immunol. (2014) 178:79–85. doi: 10.1111/cei.12392
57. Feng F, Xu W, Lian C, Wang L, Wang Z, Chen H, et al. Tuberculosis to lung cancer: application of tuberculosis signatures in identification of lung adenocarcinoma subtypes and marker screening. J Cancer. (2024) 15:5329–50. doi: 10.7150/jca.97898
58. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
59. Maiorino L, Dassler-Plenker J, Sun L, Egeblad M. Innate immunity and cancer pathophysiology. Annu Rev Pathol. (2022) 17:425–57. doi: 10.1146/annurev-pathmechdis-032221-115501
60. Ashenafi S, Brighenti S. Reinventing the human tuberculosis (TB) granuloma: Learning from the cancer field. Front Immunol. (2022) 13:1059725. doi: 10.3389/fimmu.2022.1059725
61. Khan A, Zhang K, Singh VK, Mishra A, Kachroo P, Bing T, et al. Human M1 macrophages express unique innate immune response genes after mycobacterial infection to defend against tuberculosis. Commun Biol. (2022) 5:480. doi: 10.1038/s42003-022-03387-9
62. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci. (2019) 26:78. doi: 10.1186/s12929-019-0568-z
63. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
64. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
65. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
66. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. (2018) 175:1014–1030.e19. doi: 10.1016/j.cell.2018.09.030
67. Sawyer AJ, Patrick E, Edwards J, Wilmott JS, Fielder T, Yang Q, et al. Spatial mapping reveals granuloma diversity and histopathological superstructure in human tuberculosis. J Exp Med. (2023) 220:e20221392. doi: 10.1084/jem.20221392
68. Hsieh WC, Budiarto BR, Wang YF, Lin CY, Gwo MC, So DK, et al. Spatial multi-omics analyses of the tumor immune microenvironment. J BioMed Sci. (2022) 29:96. doi: 10.1186/s12929-022-00879-y
69. Gideon HP, Hughes TK, Tzouanas CN, Wadsworth MH, Tu 2AA, Gierahn TM, et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity. (2022) 55:827–846 e10. doi: 10.1016/j.immuni.2022.04.004
70. You M, Xie Z, Zhang N, Zhang Y, Xiao D, Liu S, et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduct Target Ther. (2023) 8:196. doi: 10.1038/s41392-023-01442-3
71. Sheedy FJ, Divangahi M. Targeting immunometabolism in host defence against Mycobacterium tuberculosis. Immunology. (2021) 162:145–59. doi: 10.1111/imm.13276
72. Howard NC, Khader SA. Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. (2020) 28:832–50. doi: 10.1016/j.tim.2020.04.010
73. O’Neill L. Immunometabolism and the land of milk and honey. Nat Rev Immunol. (2017) 17:217. doi: 10.1038/nri.2017.22
74. Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem. (2016) 291:1–10. doi: 10.1074/jbc.R115.693903
75. Lirk P, Hoffmann G, Rieder J. Inducible nitric oxide synthase–time for reappraisal. Curr Drug Targets Inflammation Allergy. (2002) 1:89–108. doi: 10.2174/1568010023344913
76. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
77. Hobson-Gutierrez SA, Carmona-Fontaine C. The metabolic axis of macrophage and immune cell polarization. Dis Model Mech. (2018) 11:dmm034462. doi: 10.1242/dmm.034462
78. Wang S, Liu R, Yu Q, Dong L, Bi Y, Liu G. Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. (2019) 452:14–22. doi: 10.1016/j.canlet.2019.03.015
79. Kim SH, Roszik J, Grimm EA, Ekmekcioglu S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front Oncol. (2018) , 8:67. doi: 10.3389/fonc.2018.00067
80. Duque-Correa MA, Kuhl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci U.S.A. (2014) 111:E4024–32. doi: 10.1073/pnas.1408839111
81. San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis. (2017) 38:119–33. doi: 10.1093/carcin/bgw127
82. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. (2018) 19:1319–29. doi: 10.1038/s41590-018-0226-8
83. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
84. Qian Y, Galan-Cobo A, Guijarro I, Dang M, Molkentine D, Poteete A, et al. Heymach: MCT4-dependent lactate secretion suppresses antitumor immunity in LKB1-deficient lung adenocarcinoma. Cancer Cell. (2023) 41:1363–1380.e7. doi: 10.1016/j.ccell.2023.05.015
85. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
86. Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol. (2018) 20:597–609. doi: 10.1038/s41556-018-0083-6
87. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–218.e9. doi: 10.1016/j.ccell.2022.01.001
88. Zong Z, Xie F, Wang S, Wu S, Zhang X, Yang B, et al. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell. (2024) 187:2375–92. doi: 10.1016/j.cell.2024.04.002
89. Chen H, Li Y, Li H, Chen X, Fu H, Mao D, et al. NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance. Nature. (2024) 631:663–9. doi: 10.1038/s41586-024-07620-9
90. Qualls JE, Murray PJ. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin Immunopathol. (2016) 38:139–52. doi: 10.1007/s00281-015-0534-0
91. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, et al. Cutting edge: mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol. (2016) 196:2444–9. doi: 10.4049/jimmunol.1501612
92. Somashekar BS, Amin AG, Rithner CD, Troudt J, Basaraba R, Izzo A, et al. Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected Guinea pigs: ex vivo 1H magic angle spinning NMR studies. J Proteome Res. (2011) 10:4186–95. doi: 10.1021/pr2003352
93. Somashekar BS, Amin AG, Tripathi P, MacKinnon N, Rithner CD, Shanley CA, et al. Metabolomic signatures in Guinea pigs infected with epidemic-associated W-Beijing strains of Mycobacterium tuberculosis. J Proteome Res. (2012) 11:4873–84. doi: 10.1021/pr300345x
94. Billig S, Schneefeld M, Huber C, Grassl GA, Eisenreich W, Bange FC. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci Rep. (2017) 7:6484. doi: 10.1038/s41598-017-05916-7
95. Kiran D, Basaraba RJ. Lactate metabolism and signaling in tuberculosis and cancer: A comparative review. Front Cell Infect Microbiol. (2021) 11:624607. doi: 10.3389/fcimb.2021.624607
96. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. (2020) 20:516–31. doi: 10.1038/s41568-020-0273-y
97. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. (2022) 20:114. doi: 10.1186/s12964-022-00909-0
98. Parveen S, Shen J, Lun S, Zhao L, Alt J, Koleske B, et al. Glutamine metabolism inhibition has dual immunomodulatory and antibacterial activities against Mycobacterium tuberculosis. Nat Commun. (2023) 14:7427. doi: 10.1038/s41467-023-43304-0
99. Harth G, Horwitz MA. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: demonstration of efficacy in vivo. Infect Immun. (2003) 71:456–64. doi: 10.1128/IAI.71.1.456-464.2003
100. Harth G, Maslesa-Galic S, Tullius MV, Horwitz MA. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Mol Microbiol. (2005) 58:1157–72. doi: 10.1111/j.1365-2958.2005.04899.x
101. Ramalho R, Rao M, Zhang C, Agrati C, Ippolito G, Wang FS, et al. Immunometabolism: new insights and lessons from antigen-directed cellular immune responses. Semin Immunopathol. (2020) 42:279–313. doi: 10.1007/s00281-020-00798-w
102. Li R, Li J, Zhou X. Lung microbiome: new insights into the pathogenesis of respiratory diseases. Signal Transduct Target Ther. (2024) 9:19. doi: 10.1038/s41392-023-01722-y
103. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. (2011) 186:1598–607. doi: 10.4049/jimmunol.1003304
104. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PloS Pathog. (2016) 12:e1005667. doi: 10.1371/journal.ppat.1005667
105. Kauffman KD, Sakai S, Lora NE, Namasivayam S, Baker PJ, Kamenyeva O, et al. PD-1 blockade exacerbates Mycobacterium tuberculosis infection in rhesus macaques. Sci Immunol. (2021) 6:eabf3861. doi: 10.1126/sciimmunol.abf3861
106. Day CL, Abrahams DA, Bunjun R, Stone L, de Kock M, Walzl G, et al. PD-1 expression on mycobacterium tuberculosis-specific CD4 T cells is associated with bacterial load in human tuberculosis. Front Immunol. (2018) 1995:1995. doi: 10.3389/fimmu.2018.01995
107. Tezera LB, Bielecka MK, Ogongo P, Walker NF, Ellis M, Garay-Baquero DJ, et al. Anti-PD-1 immunotherapy leads to tuberculosis reactivation via dysregulation of TNF-alpha. Elife. (2020) 9:e52668. doi: 10.7554/eLife.52668
108. Elkington PT, Friedland JS. Permutations of time and place in tuberculosis. Lancet Infect Dis. (2015) 15:1357–60. doi: 10.1016/S1473-3099(15)00135-8
109. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. (2013) 153:521–34. doi: 10.1016/j.cell.2013.03.022
110. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell. (2019) 178:1344–1361.e11. doi: 10.1016/j.cell.2019.08.004
111. Arias AA, Neehus AL, Ogishi M, Meynier V, Krebs A, Lazarov T, et al. Tuberculosis in otherwise healthy adults with inherited TNF deficiency. Nature. (2024) 633:417–25. doi: 10.1038/s41586-024-07866-3
112. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
113. Elkington P, Tebruegge M, Mansour S. Tuberculosis: an infection-initiated autoimmune disease? Trends Immunol. (2016) 37:815–8. doi: 10.1016/j.it.2016.09.007
114. Kim ST, Tayar J, Trinh VA, Suarez-Almazor M, Garcia S, Hwu P, et al. Successful treatment of arthritis induced by checkpoint inhibitors with tocilizumab: a case series. Ann Rheum Dis. (2017) 76:2061–4. doi: 10.1136/annrheumdis-2017-211560
115. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. (2018) 18:91–104. doi: 10.1038/nri.2017.112
116. Di Liberto D, Locati M, Caccamo N, Vecchi A, Meraviglia S, Salerno A, et al. Role of the chemokine decoy receptor D6 in balancing inflammation, immune activation, and antimicrobial resistance in Mycobacterium tuberculosis infection. J Exp Med. (2008) 205:2075–84. doi: 10.1084/jem.20070608
117. Garlanda C, Di Liberto D, Vecchi A, La Manna MP, Buracchi C, Caccamo N, et al. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol. (2007) 179:3119–25. doi: 10.4049/jimmunol.179.5.3119
118. Al Shammari B, Shiomi T, Tezera L, Bielecka MK, Workman V, Sathyamoorthy T, et al. The extracellular matrix regulates granuloma necrosis in tuberculosis. J Infect Dis. (2015) 212:463–73. doi: 10.1093/infdis/jiv076
119. Cambier CJ, O’Leary SM, O’Sullivan MP, Keane J, Ramakrishnan L. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity. (2017) 47:552–565.e4. doi: 10.1016/j.immuni.2017.08.003
120. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol. (2017) 2:17072. doi: 10.1038/nmicrobiol.2017.72
121. Ahmed M, Tezera LB, Elkington PT, Leslie AJ. The paradox of immune checkpoint inhibition re-activating tuberculosis. Eur Respir J. (2022) 60:2102512. doi: 10.1183/13993003.02512-2021
122. Morelli T, Fujita K, Redelman-Sidi G, Elkington PT. Infections due to dysregulated immunity: an emerging complication of cancer immunotherapy. Thorax. (2022) 77:304–11. doi: 10.1136/thoraxjnl-2021-217260
123. Yang L, Zhuang L, Ye Z, Li L, Guan J, Gong W. Immunotherapy and biomarkers in patients with lung cancer with tuberculosis: Recent advances and future Directions. iScience. (2023) 26:107881. doi: 10.1016/j.isci.2023.107881
124. Kato Y, Watanabe Y, Yamane Y, Mizutani H, Kurimoto F, Sakai H. Reactivation of TB during administration of durvalumab after chemoradiotherapy for non-small-cell lung cancer: a case report. Immunotherapy. (2020) 12:373–8. doi: 10.2217/imt-2020-0061
125. Crawley D, Breen RA, Elkington PT, Karapanagiotou E. Tuberculosis associated with Triplet therapy for lung cancer. Thorax. (2020) 75:609–10. doi: 10.1136/thoraxjnl-2019-213913
126. Blomgren H, Wasserman J, Rotstein S, Petrini B, von Stedingk LV. Impact of adjuvant chemotherapy on spontaneous and poke week mitogen triggered immunoglobulin secretion of blood lymphocytes in operable breast cancer. Anticancer Res. (1988) 8:839–44.
127. Upadhyay R, Venkatesulu BP, Giridhar P, Kim BK, Sharma A, Elghazawy H, et al. Risk and impact of radiation related lymphopenia in lung cancer: A systematic review and meta-analysis. Radiother Oncol. (2021) 157:225–33. doi: 10.1016/j.radonc.2021.01.034
128. Chen D, Verma V, Patel RR, Barsoumian HB, Cortez MA, Welsh JW. Absolute lymphocyte count predicts abscopal responses and outcomes in patients receiving combined immunotherapy and radiation therapy: analysis of 3 phase 1/2 trials. Int J Radiat Oncol Biol Phys. (2020) 108:196–203. doi: 10.1016/j.ijrobp.2020.01.032
129. Friedes C, Chakrabarti T, Olson S, Prichett L, Brahmer JR, Forde PM, et al. Association of severe lymphopenia and disease progression in unresectabl e locally advanced non-small cell lung cancer treated with definitive chemoradiation and immunotherapy. Lung Cancer. (2021) 154:36–43. doi: 10.1016/j.lungcan.2021.01.022
130. Ho C, Sangha R, Beckett L, Tanaka M, Lau DH, Eisen DB, et al. Escalating weekly doses of cetuximab and correlation with skin toxicity: a phase I study. Invest New Drugs. (2011) 29:680–7. doi: 10.1007/s10637-010-9396-4
131. Guevremont C, Alasker A, Karakiewicz PI. Management of sorafenib, sunitinib, and temsirolimus toxicity in metastatic renal cell carcinoma. Curr Opin Support Palliat Care. (2009) 3:170–9. doi: 10.1097/SPC.0b013e32832e4681
132. Zhang M, Zhang G, Niu Y, Zhang G, Ji Y, Yan X, et al. Sintilimab with two cycles of chemotherapy for the treatment of advanced squamous non-small cell lung cancer: a phase 2 clinical trial. Nat Commun. (2024) 15:1512. doi: 10.1038/s41467-024-45769-z
133. Banna GL, Hassan MA, Signori A, Giunta EF, Maniam A, Anpalakhan S, et al. Neoadjuvant chemo-immunotherapy for early-stage non-small cell lung cancer: A systematic review and meta-analysis. JAMA Netw Open. (2024) 7:e246837. doi: 10.1001/jamanetworkopen.2024.6837
134. Sorin M, Prosty C, Ghaleb L, Nie K, Katergi K, Shahzad MH, et al. Neoadjuvant chemoimmunotherapy for NSCLC: A systematic review and meta-analysis. JAMA Oncol. (2024) 10:621–33. doi: 10.1001/jamaoncol.2024.0057
135. Zhang Z, Liu X, Chen D, Yu J. Radiotherapy combined with immunotherapy: the dawn of cancer treatment. Signal Transduct Target Ther. (2022) 7:258. doi: 10.1038/s41392-022-01102-y
136. Cho Y, Park S, Byun HK, Lee CG, Cho J, Hong MH, et al. Impact of treatment-related lymphopenia on immunotherapy for advanced non-small cell lung cancer. Int J Radiat Oncol Biol Phys. (2019) 105:1065–73. doi: 10.1016/j.ijrobp.2019.08.047
137. Grossman SA, Ellsworth S, Campian J, Wild AT, Herman JM, Laheru D, et al. Survival in patients with severe lymphopenia following treatment with radiation and chemotherapy for newly diagnosed solid tumors. J Natl Compr Canc Netw. (2015) 13:1225–31. doi: 10.6004/jnccn.2015.0151
138. Campian JL, Ye X, Brock M, Grossman SA. Treatment-related lymphopenia in patients with stage III non-small-cell lung cancer. Cancer Invest. (2013) 31:183–8. doi: 10.3109/07357907.2013.767342
139. Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res. (2011) 17:5473–80. doi: 10.1158/1078-0432.CCR-11-0774
140. Zhao Q, Li T, Du S, He J, Zeng Z. Shortened radiation time promotes recovery from radiation-induced lymphopenia in early-stage non-small cell lung cancer patients treated with stereotactic body radiation therapy. Technol Cancer Res Treat. (2022) 21:15330338221112287. doi: 10.1177/15330338221112287
141. Nwongbouwoh Muefong C, Owolabi O, Donkor S, Charalambous S, Bakuli A, Rachow A, et al. Neutrophils contribute to severity of tuberculosis pathology and recovery from lung damage pre- and posttreatment. Clin Infect Dis. (2022) 74:1757–66. doi: 10.1093/cid/ciab729
142. Muefong CN, Sutherland JS. Neutrophils in tuberculosis-associated inflammation and lung pathology. Front Immunol. (2020) 11:962. doi: 10.3389/fimmu.2020.00962
143. Stanicka J, Russell EG, Woolley JF, Cotter TG. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J Biol Chem. (2015) 290:9348–61. doi: 10.1074/jbc.M113.510495
144. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
145. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
146. Xiong K, Sun W, He Y, Fan L. Advances in molecular mechanisms of interaction between Mycobacterium tuberculosis and lung cancer: a narrative review. Transl Lung Cancer Res. (2021) 10:4012–26. doi: 10.21037/tlcr-21-465
147. Tiwari D, Martineau AR. Inflammation-mediated tissue damage in pulmonary tuberculosis and host-directed therapeutic strategies. Semin Immunol. (2023) 65:101672. doi: 10.1016/j.smim.2022.101672
148. Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol Rev. (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
149. Borkute RR, Woelke S, Pei G, Dorhoi A. Neutrophils in tuberculosis: cell biology, cellular networking and multitasking in host defense. Int J Mol Sci. (2021) 22:4801. doi: 10.3390/ijms22094801
150. Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat Commun. (2020) 11:5566. doi: 10.1038/s41467-020-19412-6
151. Yeremeev V, Linge I, Kondratieva T, Apt A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb). (2015) 95:447–51. doi: 10.1016/j.tube.2015.03.007
152. Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919
153. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. (2017) 22:519–530.e3. doi: 10.1016/j.chom.2017.09.003
154. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J Innate Immun. (2013) 5:591–602. doi: 10.1159/000348676
155. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. (2014) 15:1017–25. doi: 10.1038/ni.2987
156. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. (2016) 16:431–46. doi: 10.1038/nrc.2016.52
157. Yu X, Li C, Wang Z, Xu Y, Shao S, Shao F, et al. Neutrophils in cancer: dual roles through intercellular interactions. Oncogene. (2024) 43:1163–77. doi: 10.1038/s41388-024-03004-5
158. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
159. Saini M, Szczerba BM, Aceto N. Circulating tumor cell-neutrophil tango along the metastatic process. Cancer Res. (2019) 79:6067–73. doi: 10.1158/0008-5472.CAN-19-1972
160. Shiels MS, Albanes D, Virtamo J, Engels EA. Increased risk of lung cancer in men with tuberculosis in the alpha-tocopherol, beta-carotene cancer prevention study. Cancer Epidemiol Biomarkers Prev. (2011) 20:672–8. doi: 10.1158/1055-9965.EPI-10-1166
161. Nalbandian A, Yan BS, Pichugin A, Bronson RT, Kramnik I. Lung carcinogenesis induced by chronic tuberculosis infection: the experimental model and genetic control. Oncogene. (2009) 28:1928–38. doi: 10.1038/onc.2009.32
162. Yu YY, Pinsky PF, Caporaso NE, Chatterjee N, Baumgarten M, Langenberg P, et al. Lung cancer risk following detection of pulmonary scarring by chest radiography in the prostate, lung, colorectal, and ovarian cancer screening trial. Arch Intern Med. (2008) 168:2326–32. doi: 10.1001/archinte.168.21.2326
163. Woo SJ, Kim Y, Jung H, Lee JJ, Hong JY. Tuberculous fibrosis enhances tumorigenic potential via the NOX4-autophagy axis. Cancers (Basel). (2021) 13:687. doi: 10.3390/cancers13040687
164. Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. (2014) , 5:127. doi: 10.3389/fimmu.2014.00127
165. Porrello A, Leslie PL, Harrison EB, Gorentla BK, Kattula S, Ghosh SK, et al. Factor XIIIA-expressing inflammatory monocytes promote lung squamous cancer through fibrin cross-linking. Nat Commun. (2018) 9:1988. doi: 10.1038/s41467-018-04355-w
166. Sapalidis K, Sardeli C, Pavlidis E, Koimtzis G, Koulouris C, Michalopoulos N, et al. Scar tissue to lung cancer; pathways and treatment. J Cancer. (2019) 10:810–8. doi: 10.7150/jca.30300
167. Oz HH, Cheng EC, Di Pietro C, Tebaldi T, Biancon G, Zeiss C, et al. Recruited monocytes/macrophages drive pulmonary neutrophilic inflammation and irreversible lung tissue remodeling in cystic fibrosis. Cell Rep. (2022) 41:111797. doi: 10.1016/j.celrep.2022.111797
168. Tan Z, Xue H, Sun Y, Zhang C, Song Y, Qi Y. The role of tumor inflammatory microenvironment in lung cancer. Front Pharmacol. (2021) 12:688625. doi: 10.3389/fphar.2021.688625
169. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
170. Hasan Z, Cliff JM, Dockrell HM, Jamil B, Irfan M, Ashraf M, et al. CCL2 responses to Mycobacterium tuberculosis are associated with disease severity in tuberculosis. PloS One. (2009) 4:e8459. doi: 10.1371/journal.pone.0008459
171. Mousavinasab F, Karimi R, Taheri S, Ahmadvand F, Sanaaee S, Najafi S, et al. Microbiome modulation in inflammatory diseases: Progress to microbiome genetic engineering. Cancer Cell Int. (2023) 23:271. doi: 10.1186/s12935-023-03095-2
172. Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat Rev Gastroenterol Hepatol. (2023) 20:323–37. doi: 10.1038/s41575-022-00698-4
173. Calder PC, Ortega EF, Meydani SN, Adkins Y, Stephensen CB, Thompson B, et al. Nutrition, immunosenescence, and infectious disease: an overview of the scientific evidence on micronutrients and on modulation of the gut microbiota. Adv Nutr. (2022) 13:S1–S26. doi: 10.1093/advances/nmac052
174. Zheng Y, Fang Z, Xue Y, Zhang J, Zhu J, Gao R, et al. Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microbes. (2020) 11:1030–42. doi: 10.1080/19490976.2020.1737487
175. Guo Y, Yuan W, Lyu N, Pan Y, Cao X, Wang Y, et al. Association studies on gut and lung microbiomes in patients with lung adenocarcinoma. Microorganisms. (2023) 11:546. doi: 10.3390/microorganisms11030546
176. Zhao F, An R, Wang L, Shan J, Wang X. Specific gut microbiome and serum metabolome changes in lung cancer patients. Front Cell Infect Microbiol. (2021) 11:725284. doi: 10.3389/fcimb.2021.725284
177. Kim OH, Choi BY, Kim DK, Kim NH, Rho JK, Sul WJ, et al. The microbiome of lung cancer tissue and its association with pathological and clinical parameters. Am J Cancer Res. (2022) 12:2350–62.
178. Yang F, Yang Y, Chen L, Zhang Z, Liu L, Zhang C, et al. The gut microbiota mediates protective immunity against tuberculosis via modulation of lncRNA. Gut Microbes. (2022) 14:2029997. doi: 10.1080/19490976.2022.2029997
179. Wipperman MF, Bhattarai SK, Vorkas CK, Maringati VS, Taur Y, Mathurin L, et al. Gastrointestinal microbiota composition predicts peripheral inflammatory state during treatment of human tuberculosis. Nat Commun. (2021) 12:1141. doi: 10.1038/s41467-021-21475-y
180. Bhattarai SK, Du M, Zeamer AL, M. MB, Kellogg TD, Firat K, et al. Commensal antimicrobial resistance mediates microbiome resilience to antibiotic disruption. Sci Transl Med. (2024) 16:eadi9711. doi: 10.1126/scitranslmed.adi9711
181. Sun W, Zhang L, Liang J, Li X, Yang Y, Sun W, et al. Comparison of clinical and imaging features between pulmonary tuberculosis complicated with lung cancer and simple pulmonary tuberculosis: a systematic review and meta-analysis. Epidemiol Infect. (2022) 150:e43. doi: 10.1017/S0950268822000176
182. Huang Z, LaCourse SM, Kay AW, Stern J, Escudero JN, Youngquist BM, et al. CRISPR detection of circulating cell-free Mycobacterium tuberculosis DNA in adults and children, including children with HIV: a molecular diagnostics study. Lancet Microbe. (2022) 3:e482–92. doi: 10.1016/S2666-5247(22)00087-8
183. Zheng W, LaCourse SM, Song B, Singh DK, Khanna M, Olivo J, et al. Diagnosis of paediatric tuberculosis by optically detecting two virulence factors on extracellular vesicles in blood samples. Nat BioMed Eng. (2022) 6:979–91. doi: 10.1038/s41551-022-00922-1
184. Amato O, Giannopoulou N, Ignatiadis M. Circulating tumor DNA validity and potential uses in metastatic breast cancer. NPJ Breast Cancer. (2024) 10:21. doi: 10.1038/s41523-024-00626-6
185. Fonseca NM, Maurice-Dror C, Herberts C, Tu W, Fan W, Murtha AJ, et al. Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer. Nat Commun. (2024) 15:1828. doi: 10.1038/s41467-024-45475-w
186. Pellegrini JM, Martin C, Morelli MP, Schander JA, Tateosian NL, Amiano NO, et al. PGE2 displays immunosuppressive effects during human active tuberculosis. Sci Rep. (2021) 11:13559. doi: 10.1038/s41598-021-92667-1
187. Bafica A, Scanga CA, Serhan C, MaChado F, White S, Sher A, et al. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest. (2005) 115:1601–6. doi: 10.1172/JCI23949
188. Rizvi I, Garg RK, Malhotra HS, Kumar N, Uniyal R. Role of aspirin in tuberculous meningitis: A systematic review and meta-analysis. Neurol India. (2019) 67:993–1002. doi: 10.4103/0028-3886.266232
189. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis. (2013) 208:199–202. doi: 10.1093/infdis/jit152
190. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. (2011) 377:31–41. doi: 10.1016/S0140-6736(10)62110-1
191. McCormack VA, Hung RJ, Brenner DR, Bickeboller H, Rosenberger A, Muscat JE, et al. Aspirin and NSAID use and lung cancer risk: a pooled analysis in the International Lung Cancer Consortium (ILCCO). Cancer Causes Control. (2011) 22:1709–20. doi: 10.1007/s10552-011-9847-z
192. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
193. Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiol (Bethesda). (2006) 21:362–9. doi: 10.1152/physiol.00024.2006
194. Herner A, Sauliunaite D, Michalski CW, Erkan M, De Oliveira T, Abiatari I, et al. Glutamate increases pancreatic cancer cell invasion and migration via AMPA receptor activation and Kras-MAPK signaling. Int J Cancer. (2011) 129:2349–59. doi: 10.1002/ijc.25898
195. Broer A, Gauthier-Coles G, Rahimi F, van Geldermalsen M, Dorsch D, Wegener A, et al. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J Biol Chem. (2019) 294:4012–26. doi: 10.1074/jbc.RA118.006378
196. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. (2018) 24:194–202. doi: 10.1038/nm.4464
197. Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene. (2018) 37:5007–19. doi: 10.1038/s41388-018-0307-z
198. Carmona-Fontaine C, Bucci V, Akkari L, Deforet M, Joyce JA, Xavier JB. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci U.S.A. (2013) 110:19402–7. doi: 10.1073/pnas.1311939110
199. Carow B, Hauling T, Qian X, Kramnik I, Nilsson M, Rottenberg ME. Spatial and temporal localization of immune transcripts defines hallmarks and diversity in the tuberculosis granuloma. Nat Commun. (2019) 10:1823. doi: 10.1038/s41467-019-09816-4
200. Datta M, Via LE, Kamoun WS, Liu C, Chen W, Seano G, et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U.S.A. (2015) 112:1827–32. doi: 10.1073/pnas.1424563112
201. Datta M, Kennedy M, Siri S, Via LE, Baish JW, Xu L, et al. Mathematical model of oxygen, nutrient, and drug transport in tuberculosis granulomas. PloS Comput Biol. (2024) 20:e1011847. doi: 10.1371/journal.pcbi.1011847
202. Phulera S, Akif M, Sardesai AA, Mande SC. Redox proteins of mycobacterium tuberculosis. J Indian Institute Sci. (2014) 94:1.
203. Yamashita M. Auranofin: past to present, and repurposing. Int Immunopharmacol. (2021) 101:108272. doi: 10.1016/j.intimp.2021.108272
204. Pathirage R, Favrot L, Petit C, Yamsek M, Singh S, Mallareddy JR, et al. Mycobacterium tuberculosis CitA activity is modulated by cysteine oxidation and pyruvate binding. RSC Med Chem. (2023) 14:921–33. doi: 10.1039/d3md00058c
Keywords: lung cancer, tuberculosis (TB), immunosuppressive microenvironments, immunometabolism, inflammation
Citation: Fang C, He X, Tang F, Wang Z, Pan C, Zhang Q, Wu J, Wang Q, Liu D and Zhang Y (2025) Where lung cancer and tuberculosis intersect: recent advances. Front. Immunol. 16:1561719. doi: 10.3389/fimmu.2025.1561719
Received: 16 January 2025; Accepted: 12 March 2025;
Published: 02 April 2025.
Edited by:
Ameya Bendre, Savitribai Phule Pune University, IndiaReviewed by:
Wenping Gong, The 8th Medical Center of PLA General Hospital, ChinaCopyright © 2025 Fang, He, Tang, Wang, Pan, Zhang, Wu, Wang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Zhang, c2t5bGluZV96eXVAMTYzLmNvbQ==; Daishun Liu, bGRzZG9jQDEyNi5jb20=; Qinglan Wang, d2FuZ3FpbmdsYW5Ac2N1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.