 Maria Mavridou
Maria Mavridou Simon H. Pearce
Simon H. Pearce- 1Translational and Clinical Research Institute, Newcastle University, BioMedicine West, Newcastle-upon-Tyne, United Kingdom
- 2Endocrine Unit, Royal Victoria Infirmary, Newcastle-upon-Tyne, United Kingdom
Autoimmune disorders develop owing to a misdirected immune response against self-antigen. Genetic studies have revealed that numerous variants in genes encoding immune system proteins are associated with the development of autoimmunity. Indeed, many of these genetic variants in key immune receptors or transcription factors are common in the pathogenesis of several different autoimmune conditions. In contrast, the proclivity to develop autoimmunity to any specific target organ or tissue is under-researched. This has particular relevance to autoimmune endocrine conditions, where organ-specific involvement is the rule. Genetic polymorphisms in the genes encoding the targets of autoimmune responses have been shown to be associated with predisposition to several autoimmune diseases, including type 1 diabetes, autoimmune thyroid disease and Addison’s disease. Mechanistically, variations leading to decreased intrathymic expression, overexpression, different localisation, alternative splicing or post-translational modifications can interfere in the tolerance induction process. This review will summarise the different ways genetic variations in certain genes encoding endocrine-specific antigens (INS, TSHR, TPO, CYP21A2, PIT-1) may predispose to different autoimmune endocrine conditions.
Introduction
Autoimmune endocrinopathies are, almost by definition, organ-specific autoimmune diseases whereby one specific hormone-secreting gland or tissue becomes the target of the immune response leading to immune-mediated destruction and, over time, clinical hormonal deficiency (1–3). Only Graves’ disease (GD) deviates from this paradigm with stimulatory antibodies rather than a destructive immune response leading to organ hyperactivity and frequent clinical manifestations outside the primary organ of involvement (i.e. Graves’ orbitopathy). These disorders are mostly inherited as complex genetic traits with a multigenic basis, that is variations in numerous genes (typically 50 or more identified to date) each contribute a small degree towards the proclivity to clinical autoimmune disease (2, 4). Perhaps unsurprisingly, it has been more straightforward to identify genetic variations in immune-related genes that contribute to multiple autoimmune disorders, than to identify those disease or organ-specific variations. Many of these immune-related variants contribute to the susceptibility to more than one autoimmune condition, including both endocrine conditions and additional non-endocrine, organ-specific disorders (1–4). Some of the variants involved in several conditions are briefly reviewed in the next section.
Whilst the common autoimmune endocrinopathies are inherited as complex multigenic traits, there are two Mendelian conditions which provide relevant insights into mechanisms of immune tolerance. The first condition is the APECED (Autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy)/APS1 (autoimmune polyglandular type 1) syndrome, which is most frequently owing to loss of function mutations in both alleles of the AutoImmune REgulator (AIRE) gene (5). AIRE is expressed in thymic epithelial cells where it controls expression of transcripts relevant in thymic T cell selection. Defective AIRE function abrogates expression of certain self-peptides in antigen-presenting cells which is essential for educating the developing thymic T cell repertoire and leads to failure to delete T cells expressing potentially autoreactive T cell receptors. APS1 is characterised by high penetrance (70-90%) of autoimmune Addison’s disease (AAD, autoimmune primary adrenal insufficiency) and autoimmune hypoparathyroidism (5). Both of these conditions are rare outside the context of APS1, with a prevalence of one in 8,000 in the sporadic population for AAD, showing the critical role of central thymic tolerance in this condition.
More recent findings have also highlighted the contribution of common genetic variants in the AIRE gene to the susceptibility of AAD in a Swedish cohort. The minor alleles of four intronic strongly linked SNPs, rs9983695, rs2075875, rs2075876, and rs6220374, have been shown to have significantly lower frequency in Swedish AAD patients (approximately 4%) compared to healthy controls (ranging from 10% to 11%) (6). Although none of these variants overlap with known transcription factor binding sites, their strong linkage suggests they may play a regulatory role in AIRE’s function. This suggests that thymic antigen presentation is not just important in Addison’s disease associated with APS1, but perhaps unsurprisingly with sporadic AAD as well (6).
The role of AIRE gene variants in APS1 contrasts to the IPEX (Immune dysregulation, polyendocrinopathy, enteropathy, X- linked) syndrome, caused by defects in the FOXP3 gene, encoding the critical transcription factor for development of regulatory T cells (7). IPEX is X-linked, giving rise to autoimmune diabetes before the age of 1yr in around 50% of affected boys, and signalling failure of peripheral tolerance as the primary mechanism. Thus, these 2 monogenic disorders demonstrate that genetic variants can impact the establishment and maintenance of immune tolerance via different mechanisms including on central T cell selection or by influencing peripheral regulatory lymphocyte function (5, 7).
Brief review of immune system genetic variation in endocrine autoimmunity
Genome-wide association studies (GWAS) on patient cohorts with autoimmune endocrine disorders have revealed that most of the gene variants currently associated with autoimmunity encode proteins that have a role in the antigen recognition, and lymphocyte activation and inactivation pathways (3, 8). Whilst the focus of this review concerns gene variants that confer organ-specificity to the disease, that is the antigen or target-organ specific gene variants, we first make a brief review of the variants in immune system genes that have well-established roles in endocrine autoimmunity, with many of these variants contributing to disease risk in multiple different conditions (Table 1).
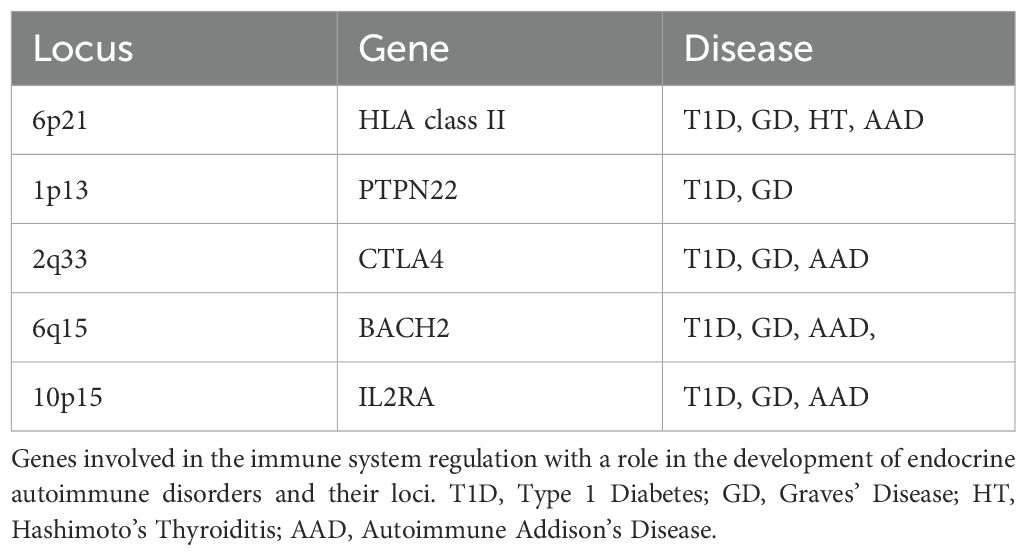
Table 1. Immune system genes linked to autoimmune endocrinopathies.
The locus that has the strongest effect in the predisposition to most autoimmune disorders is the MHC, encoding the human leukocyte antigen (HLA) molecules. This locus spans a large interval on the short arm of chromosome 6, with numerous different HLA genes, each with many different alleles which may either confer risk or protection from autoimmunity. Most studies have identified associations of HLA class II alleles and certain HLA-DRB1-DQA1-DQB1 haplotypes with autoimmune endocrine disorders, implying the essential role of binding and presentation of endocrine antigens to CD4+ T cells in the pathogenesis of particular diseases. It is thought that variants in DR and DQ genes encode proteins that might bind self-peptides with lower affinity, potentially inadequate for the central elimination of autoreactive T cells resulting in impaired tolerance (8, 9). An alternative scenario suggests that certain DR and DQ proteins communicate inefficiently with the TCRs on Tregs leading to the generation of a Treg population that is unable to control autoreactive T cells. Additionally, variations in HLA class I genes have been also linked with susceptibility to autoimmunity. However, the strong Linkage Disequilibrium (LD), a term used to describe the non-random association between alleles of two or more genetic loci, between HLA class I and class II loci and the abundance of alleles often poses a difficulty in the identification of linked alleles having independent association with a phenotype (9, 10).
The PTPN22 gene encodes the Lymphoid-tyrosine phosphatase (LYP) which is implicated not only in T and B cell signalling cascades, but also in natural killer (NK) cell and myeloid cell pathways. LYP acts by blocking T cell activation as it dephosphorylates CSK kinase and stops the downstream signal transduction. The rs2476601*T allele which leads to the Arg620*Trp substitution has been associated with autoimmune disorders including T1D and GD, with the variant having also been linked with relapse of GD after antithyroid treatment (11, 12). The Arg620*Trp variant is located in a highly conserved point in the P1 motif of the catalytic domain of LYP, suggesting the potential to directly affect phosphatase activity (13). The exact underlying mechanism is unclear as some studies have shown that this variant results in an LYP with loss of function that cannot inhibit T cell activation, whilst others suggest that the variant produces an LYP with gain of function and elevated inhibitory effect on T cell signalling pathway. The relevance of these investigations might be limited, though, as several of these functional studies have been conducted on the ortholog of LYP in mice, which shares 90% similarity in the catalytic and 60% similarity in the non-catalytic domains with the human homolog (1, 3, 8, 10, 14–17). More recent studies, however, suggest that the role of the substitution might be different depending on the cellular context in which the protein is expressed, or the location of the protein, implying that LYP might have a multifunctional role in the autoimmunity development (18). In particular, the variant might have a gain of function effect leading to an even stronger blocking of TCR signalling, and a subsequent diminished negative selection of autoreactive T cells in the thymus, and a reduced response to the BCR signal, and consequently hyper-responsive B cell populations. However, Zhang et al. have shown that in Jurkat T cells transduced with plasmids carrying the mutated LYP, and in PBMC obtained from rheumatoid arthritis (RA) patients homozygous for the risk allele, the Arg620*Trp substitution makes the protein more susceptible to intracellular degradation and therefore the variant leads to weaker inhibitory effects (19). Additionally, the variant leads to an LYP with loss of function in myeloid cells as it exhibits reduced interaction with TRAF3 and reduced amounts of TLR-induced type I IFNs (16–18, 20–22). These results suggest that there are pleiotropic effects with a gain of function phenotype centrally, along with loss of function effect in peripheral immune cells and, even more interestingly, different regulatory effects in lymphocytes than in myeloid cells (16, 17, 19, 23–25).
The Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4) is encoded by the CTLA4 gene and is an inhibitory co-stimulatory receptor which binds to B7 molecules on the surface of antigen presenting cells, and consequently, downregulates their activation. Amongst the polymorphisms which have been associated with autoimmune disorders, the variant rs231775*G on the exon 1, and an (AT)n repeat in the 3’-untranslated region (UTR) of the CTLA4 gene have been linked to T1D and AAD. Both variants are associated with decreased expression of CTLA4, reducing levels of inhibitory signalling and, therefore, unchecked T cell activation. In addition, the frequency of rs3087243*G allele on the 3’-UTR of CTLA4 gene is higher in GD and Hashimoto's thyroiditis (HT) patients than in healthy controls, with the allele also being associated with a reduction on the beneficial effects of anti-thyroid treatment in a Polish cohort with GD (26–29). The importance of the molecule can be understood by the fact that people with null CTLA4 alleles develop immunodeficiency as a form of an autosomal dominant immune dysregulation syndrome, characterised by hypogammaglobulinemia and recurrent infections with several tissues being affected, whilst CTLA4-Knockout (CTLA4-KO) mice develop fatal lymphoproliferative disease in the first weeks of life (30).
BACH2 transcription factor (BTB and CNC Homology 1, Basic Leucine Zipper Transcription factor 2) is expressed predominantly in B lymphocytes and plays a vital role in regulating CD4+ T-cell differentiation. Its functions include repressing effector CD T-cell lineages (Th1, Th2, and Th17), and it is crucial for the formation of regulatory T cells. The intronic BACH2 variant rs3757247*T has been associated with susceptibility to AAD as the frequency of the risk T allele is significantly higher in UK AAD patients compared to controls (31). The frequency of the risk allele has been also found higher in Polish patients with autoimmune polyendocrine syndromes (APS) (and in 6 Japanese T1D patients (32)) compared to healthy individuals (33). Additionally, a recent study revealed a correlation between the risk T allele and the existence of circulating autoantibodies against thyroid peroxidase (TPO) in first-degree relatives of (Polish) AAD patients (34).
rs3757247 is in complete LD with the intronic rs11755527, rs72928017 and rs72928038 variants, with rs11755527 having been linked to susceptibility to T1D in northern European (35) and Pakistani populations (36), however no association was found in a Brazilian population (37). rs3757247 and rs11755527, along with another intronic SNP, rs2474619, have been associated with GD in a Chinese Han population with rs2474619 determined as the most disease-associated variant (11, 38). rs72928038*A allele has been associated with risk of development of T1D as the variant leads to lower expression of BACH2 in multiple cell types, but mainly in T cells (39). The risk allele has been shown to impede the binding of the ETS1 transcription factor leading to decreased enhancer activity (39, 40). rs72928038*A has been also correlated with GD and HT in a UK cohort (11, 41). Thus, the exact molecular mechanisms by which the genetic variants affect gene regulation and the unravelling of the causative variant remain unidentified and require further investigation.
Interleukin-2 (IL-2) is a key driver of T cell maintenance, proliferation, and development. Polymorphisms on the gene encoding the alpha subunit of the IL-2 receptor (CD25) have been associated with T1D, GD (and AAD although with contradictory results in different populations: UK-Norway). Amongst those, T1D patients carrying the intronic rs2104286*C allele have lower CD25 expression on their naïve CD4+ T cells, but higher levels of soluble CD25. Thus, the variant could lead to an impaired IL-2 signalling transduction and lower responsiveness of T cells to IL-2 effects. Such a decrease in IL-2 signalling might have a sequential impact on FOXP3 expression and Tregs activity, with the protective minor allele of the variant rs12722495 probably acting in an opposite way (10, 14, 42–44).
Identification of susceptibility loci is a complex process as the genes involved in disease pathogenesis are often in LD. Furthermore, power is frequently limited by cohort size, meaning that reproducibility is a key issue. Furthermore, genetic heterogeneity between affected patients of different ethnicities is not unexpected, complicating the interpretation of some studies (3, 8, 10, 14, 15). A deep understanding of the genetic – epigenetic interactions in combination with environmental triggering events (viral or bacterial infections, alteration of gut and oral microbiome) that might have additional effects is required for an accurate unravelling of the multiple pathogenesis mechanisms of autoimmunity (45).
Role of target-organ genes in endocrine autoimmunity
Insulin gene variation in Type 1 Diabetes
Type 1 Diabetes Mellitus (T1D) is a chronic disease which is characterised by the autoimmune destruction of the insulin secreting β cells in the pancreatic islets by T lymphocytes (CTLs), leading to insulin deficiency. Similar to other autoimmune disorders, it is believed that a combination of numerous genetic variants, each with a small effect, along with environmental factors are implicated in the pathogenesis (46). These various susceptibility loci have been labelled as IDDM1, IDDM2 etc. in chronological order of discovery, with IDDM1 residing in the MHC on chromosome 6p21. As well as being the hormone that is deficient in T1D, insulin autoantibodies and T cell responses against insulin peptides are also central to its pathogenesis (46, 47). However, autoantibodies to other islet cell protein targets including glutamic acid decarboxylase (GAD) and insulinoma-associated 2 (IA-2) are highly prevalent and may predate the onset of T1D by many years.
One of the non-HLA loci involved in the disease susceptibility is a variable number of tandem repeats (VNTR) region upstream of the start codon of the insulin gene, INS, on chromosome 11. The region, which has also been known as the IDDM2 locus, is a minisatellite as it consists of repeats of a 14bp sequence with variable length; alleles have been distinguished by repeat number with class I alleles having a shorter length, whilst classes II and III have more repeats of the sequence (47–54). Studies in T1D patients and healthy controls have shown that IDDM2 class I alleles are associated with the disease susceptibility, with class I homozygosity increasing the risk for the disease development by 2-5 times (53). On the contrary, class III alleles, even in heterozygosity, have been shown to be dominantly protective.
Studies on the mRNA levels of insulin have shown that class I alleles are associated with increased INS mRNA levels in the pancreas compared to class III alleles (47, 50, 52). However, the opposite effect has been observed in human thymus, where class I alleles have been linked to decreased INS expression compared to class III alleles. Therefore, it is believed that the mechanism by which IDDM2 class I and III alleles can regulate the predisposition to T1D is by controlling the thymic expression levels of the insulin gene and its subsequent epitope presentation by HLA molecules to maturing T cells (50). With lower thymic expression of insulin peptides in VNTR class I carriers, some INS-reactive TCR-carrying T cells escape clonal deletion leading to autoreactivity and ultimately T1D. Experiments in which insulin gene copy numbers were manipulated in knockout (KO) mice, confirm that thymic INS expression is strongly genetically determined and that animals with low or absent thymic insulin mRNA had autoreactive peripheral T cell clones against insulin. Conversely, mice with high thymic insulin expression were protected from insulin autoreactivity (55).
A genetic study involving detailed phenotyping of German families with children at risk of developing T1D confirmed that the short, class I VNTR was associated with early onset diabetes. Interestingly, individuals with the dominantly protective class III VNTR had a lower risk of developing insulin autoantibodies, but had similar rates of GAD and IA2 antibodies, supporting that this variation at the INS locus was acting directly to determine the prevalence of insulin autoreactivity (56).
Although it might not be intuitively obvious that variations leading to high expression of an autoantigen might protect against autoimmune disease, these experiments in both human and rodent demonstrated for the first time that thymic gene expression forms a critical tolerogenic mechanism that may be affected by naturally occurring human genetic variation.
Graves’ disease
Graves’ disease (GD) is a common autoimmune disorder of the thyroid gland characterised by the continuous activation of the thyroid stimulating hormone receptor (TSHR) by autoantibodies, leading to hyperthyroidism. Although the reason why these TSH receptor stimulating antibodies, known as TRAb, occur is unknown, it is believed that, again, an interaction of environmental and genetic factors might trigger their appearance (57–60). TRAb are central to disease pathogenesis and circulating TRAb concentrations correlate tightly with disease severity and clinical outcome (61). Unlike T1D which has an approximately equal gender distribution, GD affects 5-8 times more women than men. The TSHR is a G protein-coupled receptor, consisting of an extracellular A subunit, bound to a transmembrane domain (B) subunit by a flexible hinge region. The immunodominant T cell and B cell epitopes reside within the extracellular A-subunit. Soluble, circulating A subunits are shed from the cell membrane and found in the circulation, where they may consist of several homo-oligomeric forms (62).
Amongst the genetic loci associated with GD susceptibility, HLA-DRB1, PTPN22, CTLA-4, there is also the TSHR gene which encodes the main autoantigen. Studies on the non-coding regions of the TSHR, showed that the A allele in the intronic rs179247 SNP is strongly associated with disease susceptibility, whilst it is also linked to the earlier onset of the disease (57, 59, 60, 63). Analysis of the TSHR expression revealed that although the polymorphism does not have any effect on the expression levels in the thyroid gland, it can influence the mRNA levels in the thymus. More specifically, individuals homozygous for the high-risk A allele have been shown to have lower TSHR expression levels in their thymus compared to people carrying the protective G allele, with G/G homozygous showing the higher TSHR expression (57, 59). Thus, it has been suggested that, with similarities to the INS situation for T1D, a gene variant that regulates (mRNA or protein) expression quantitatively in the thymus, can lead to the disease development through inadequate recognition of ‘autoreactive’ T cells and breakdown of negative T cell selection.
An alternative mechanism which has also been suggested, though, links the high-risk rs179247*A allele with alternative splicing of the TSHR gene. Studies have shown that people carrying the rs179247*A allele have lower levels of the full-length TSHR mRNA, and increased levels of two alternative shorter truncated transcripts in the thyroid gland. These alternative forms lack the transmembrane and the hinge region and therefore lead to the production of soluble TSHR isoforms, similar to the A-subunit part of the receptor (59, 63–72). These secreted TSHR forms can be presented and contribute to the loss of peripheral tolerance (57, 59). Additionally, elevated levels of these soluble isoforms compared to the full-length receptor might result in increased production of autoantibodies against TSHR, as it has been shown that the A-subunit of the receptor is the main target of the autoantibodies and also promotes the affinity maturation of the B cells producing thyroid-stimulating antibodies (68, 72).
However, a more recent study measuring the expression levels of the different isoforms has shown that the disease-associated genotype was associated with higher levels of the alternative splice variants in the thymus, and not in the thyroid, implying that these might have a role in the induction of the central tolerance in the thymus instead (57). In contrast to the case of INS, it has been proposed that expression of non-immunodominant TSHR epitopes in thymus could have a role in inducing tolerance (57).
rs12101255, another SNP linked to the disease susceptibility, in intron 1 of the TSHR gene, is in complete LD with its adjacent rs12101261. The two polymorphisms were found to overlap with a region of monomethylated histone 3 lysine 4 (H3K4me1), a chromatin modification induced by IFN-α, during a viral infection (59, 60, 63). The region has been shown to be critical for the TSHR expression as it is the binding site of the promyelocytic leukaemia zinc finger (PLZF) transcription factor. PLZF binds with a stronger affinity in individuals homozygous for the high-risk rs12101261*T allele, and is correlated with lower intrathymic expression of TSHR, compared to people homozygous for the protective allele C (Figure 1) (59, 60, 63). Thus, an environmental event, such as a viral infection, might influence the expression of an autoantigen gene in individuals carrying the high-risk allele of this variant. Decreased expression of the TSHR in the thymus, induced by IFN-α production, in a susceptible individual could trigger autoimmunity development through failure to delete autoreactive T cells (59, 60, 63, 73–76).
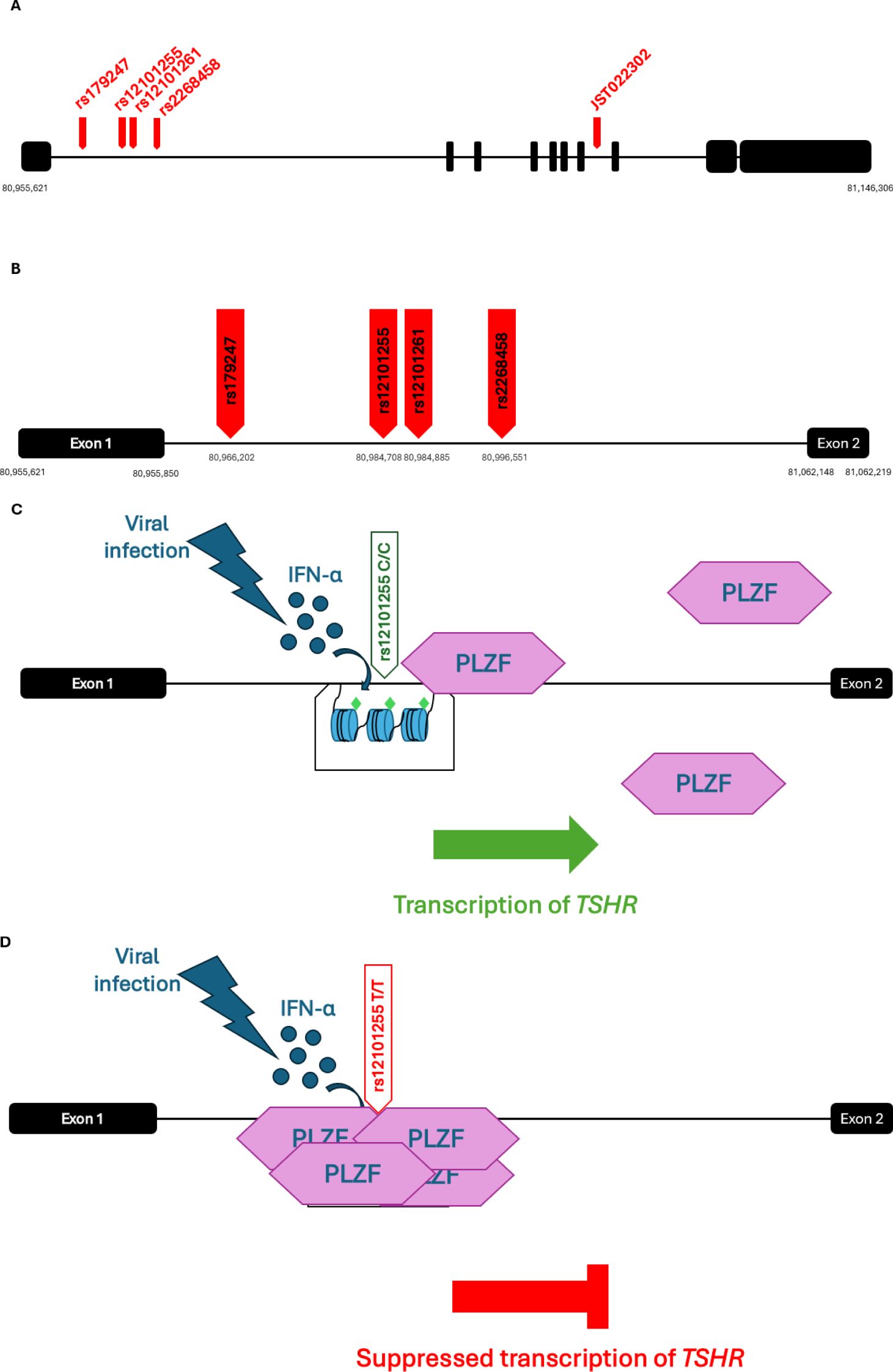
Figure 1. (A) Schematic representation of the GD-associated SNPs on the TSHR gene. The 10 exons are represented as black squares and SNPs as red arrows. The minor allele of the polymorphism JST022302 in intron 7 has been associated with the disease in a Japanese cohort (64, 79, 80) (B) Schematic representation of the GD-associated SNPs in intron 1 of the TSHR gene. (C, D) Effects of environmental events on individuals with genetic predisposition to GD; IFN-α is secreted in response to a viral infection event and induces monomethylation at the Lysine 4 of Histone 3 (H3K4me1) (represented as green diamond shapes) in the intronic region of TSHR containing the disease-associated variants. The transcription repressor PLZF binds with a stronger affinity in people homozygous for the high-risk rs12101261*T allele (D), than in individuals homozygous for the protective C allele (C). Thus, a viral infection can cause a reduction in the expression of TSHR in the thymus of people carrying the high-risk allele of the variant (D), and therefore lead to a decreased presentation of the protein epitopes to T cells. GD, Graves' Disease; TSHR, thyroid stimulating hormone receptor; IFN-α, Interferon alfa; PLZF, promyelocytic leukemia zinc finger transcription factor. [Image created in PowerPoint and adapted from (59, 73, 130)].
The first SNP which has been associated with the disease susceptibility in Caucasians (UK and Polish populations), rs2268458 (77), is located in the same 40-kb region of intron 1 of TSHR gene together with rs179247, rs12101255 and rs12101261, and all of them are in moderate LD posing additional difficulty in the dissection of the causative SNP. However, logistic regression analysis on the most highly associated SNPs, rs179247 and rs12101255, performed by a group studying the association of their minor alleles with elevated expression of the truncated isoforms of the receptor in UK and Polish Caucasians, revealed that the rs12101255 might drive the association of the nearby variants (64, 78, 79), although rs179247 had previously been shown as the variant with the strongest association with the disease in UK GD patients (64).
Meta-analysis of different studies showed that there is a level of consistency in the results amongst Caucasian and Asian populations. Although the high-risk alleles in intron 1 were confirmed in a Spanish GD cohort (63), interestingly, a study in Japanese GD patients in 2005 revealed the existence of SNPs in introns 7 and 8 of TSHR that were also associated with the disease, and this association was confirmed when these variants were tested in a UK GD cohort (64, 79, 80). More recent studies performed by the China Consortium for the Genetics of Autoimmune Thyroid Disease confirmed the association of the intron 1 variants (rs12101261) with the disease susceptibility in Chinese patients, but did not show significant association of the intron 7 variants that had been previously detected in a Japanese cohort (76). These results indicate that although there may be complex LD patterns that vary between ethnicities, variants on intron 1 of the TSHR play an important role on the disease susceptibility in Caucasian and Asian populations (64, 78–80).
Hashimoto’s thyroiditis
Hashimoto’s thyroiditis (HT), the most common autoimmune disorder, causes destruction of the thyroid gland by lymphocytic infiltration and leads to hypothyroidism. The presence of circulating autoantibodies against the main thyroid antigens, thyroid peroxidase (TPO) and thyroglobulin (Tg) is crucial for the diagnosis, but the mechanism behind their occurrence is not yet known. Twin studies have shown that genetic susceptibility plays a role in the disease development, but environmental events, such as stress and iodine consumption, also contribute to disease onset (81–83).
TPO is an enzyme involved in the biosynthesis of the thyroid hormones T4 and T3 and is encoded by the TPO gene located on chromosome 2. It is uniquely expressed in thyroid. Polymorphisms in the TPO gene have been associated with both the development of the disease and the levels of circulating autoantibodies. The rs2071400 SNP in the promoter of TPO has been shown to have a potential role in the disease pathogenesis. Studies have shown that the prevalence of the rs2071400*T allele is significantly higher in people with hypothyroidism than in healthy controls. In addition, homozygosity for the rs2071400*T variant is significantly more frequent in individuals with hypothyroidism compared to healthy people (81, 84–88). These findings suggest that the rs2071400*T allele is a risk factor for the development of hypothyroidism. Another variant which has been linked with disease predisposition (in an Indian population) is the missense variant, rs732609, in exon 12 of the TPO gene, with rs732609*C occurring more frequently in patients with hypothyroidism than in controls, and therefore conferring risk for the disease onset. Substitution of the A allele with C, leads to the change of Threonine725 to Proline, which is predicted to be a structurally tolerated substitution. However, this may still alter expression of TPO or its enzymatic activity (85, 88–90).
The large extracellular domain of TPO is structurally homologous to the enzyme myeloperoxidase (MPO) and the threonine 725 (Thr725) residue is located within this MPO-like domain of TPO (Figure 2). This domain is crucial for the activity of the enzyme, and therefore substitution of Threonine with Proline might have an impact on the enzyme’s peroxidase activity. The residue is also close to an immunodominant B cell epitope of TPO, consisting of nine amino acids, and is recognised by the well-characterised monoclonal TPO antibody, MAb-47. Studies have shown that the MAb-47 recognises the native structure of human TPO, and thus there is a possibility that the nearby residues play a crucial role in the maintenance of the conformational structure of the epitope (91–94).
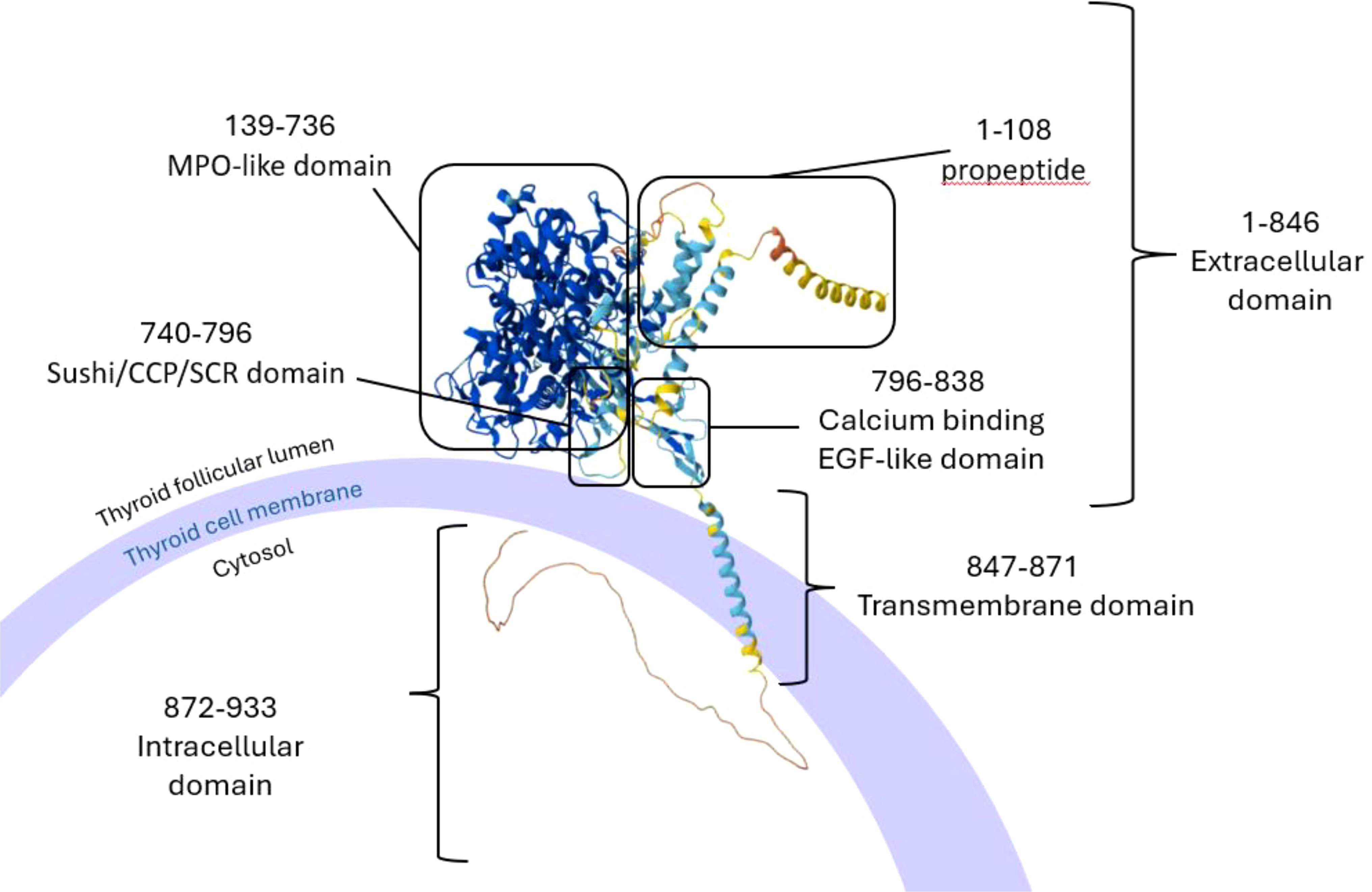
Figure 2. Schematic representation of the structure of TPO with its domains as it is bound on the membrane of thyroid cells. Image created in PowerPoint with the structure of the protein created by AlphaFold and the domains analysed by InterPro (result/InterProScan/iprscan5-R20241108-164444-0398-33458951-p1m/Overview). AlphaFold produces a per-residue model confidence score (pLDDT) between 0 and 100. Some regions below 50 pLDDT may be unstructured in isolation. Very high (pLDDT > 90) is shown in dark blue, high (90 > pLDDT > 70) in cyan blue, low (70 > pLDDT > 50) in yellow and very low (pLDDT < 50) in orange. TPO, thyroid peroxidase, MPO; myeloperoxidase; CCP, complement control protein; SCR, short consensus repeat; EGF, epidermal growth factor. Image adapted from (91, 92, 131).
Similar to sodium iodide symporter (NIS), and TSHR, expression of TPO has been detected in the Hassall’s corpuscles of human thymus tissue, although it was found to be expressed at lower levels compared to other thyroid antigens like Tg and TSHR by a Korean study. The same study showed a correlation between the expression levels of TPO and age, as TPO expression in the thymus was shown to be increased with increasing age (95). Another study by Misharin et al. on AIRE-KO mice, which investigated the correlation between AIRE and the expression of thyroid antigens in the thymus, revealed that intrathymic TPO expression was absent in AIRE deficient animals, whereas Tg and TSHR were still expressed, albeit at reduced levels (96, 97).
Thyroglobulin (Tg), the precursor molecule of T3 and T4 hormones, is encoded by the Tg gene which is located on chromosome 8. Although association studies have linked this locus with predisposition to autoimmune thyroid disorders in general, studies on HT patients have shown that the rs180195*G allele in the promoter region of the Tg gene is significantly more frequent in HT patients than in healthy individuals (75, 87, 98, 99). Substitution of the wild-type A allele with G, leads to an increase in the activity of the Tg promoter through the modification of the transcriptional activator interferon regulatory factor 1 (IRF-1) binding site. IRF-1 is activated in response to IFNAR signalling pathway and controls the expression of IFN-induced genes (75). It is possible, then, that an increase in IFN levels/activity as a protective mechanism against a potential infection could cause an increased expression of the Tg gene and therefore elevated presentation of the Tg epitopes. This finding can possibly explain the development of thyroid autoimmunity in people who have previously received IFN-α treatment and poses another example of genetic-epigenetic and environmental factors interaction for the induction of autoimmunity (75, 82, 87).
The rare splice site variant, c.1076-1G > C, which leads to the deletion of the whole of exon 9 of the Tg gene, has been found in all affected members of a family with early HT onset (100, 101). The shorter transcript, produced by the variant, might lead to the synthesis of a misfolded protein. Misfolding would either lead to an immunogenic isoform or expose different epitopes of the peptide sequence compared to the wild-type Tg (Figure 3). If the misfolded isoform is bound to MHC class II molecules, these alternative epitopes would not be recognised as ‘self’ by T cells, as they are not presented under normal conditions, and therefore activate an immune response; whilst if they manage to avoid intracellular degradation, they might also modify the level of post-translational modifications of the Tg, such as iodination (100–103). Although we cannot be sure about the mechanism causing this phenotype, the presence of autoantibodies against the Tg and the TPO in these patients implies that there is ongoing immunogenicity. Additionally, it is worth considering that any condition leading to increased thyroid cell turnover might predispose to autoimmunity. Co-existing autoimmune thyroiditis has been well-documented in conditions such as resistance to thyroid hormone beta (RTHβ) and Pendred’s syndrome, both of which are associated with goiter through distinct mechanisms. In RTHβ, mutations in the thyroid hormone receptor beta gene lead to impaired feedback regulation of the hypothalamic-pituitary-thyroid axis and persistent TSH stimulation of the thyroid with higher cell turnover and goiter, which may expose cryptic antigens and increase susceptibility to autoimmune thyroid diseases such as HT and GD (104–106). Similarly, Pendred’s syndrome is characterised by mutations in the SLC26A4 gene and causes thyroid dyshormonogenesis and deafness. Chronic TSH stimulation therefore leads to thyroid enlargement (goitre) and potentially in susceptible individuals increased thyroid antigen expression (106–109). Thus, loss of function Tg variants are known to impair thyroid hormone synthesis leading to congenital dyshormonogenetic goiter, and hypothyroidism. Genetic variants leading to less profoundly reduced Tg function (e.g. c.1076-1G > C), might be associated with inefficient rather than absent thyroid hormone synthesis and could lead to autoimmunity by means of increased TSH and thyrocyte turnover, goiter and higher thyroid antigen expression.
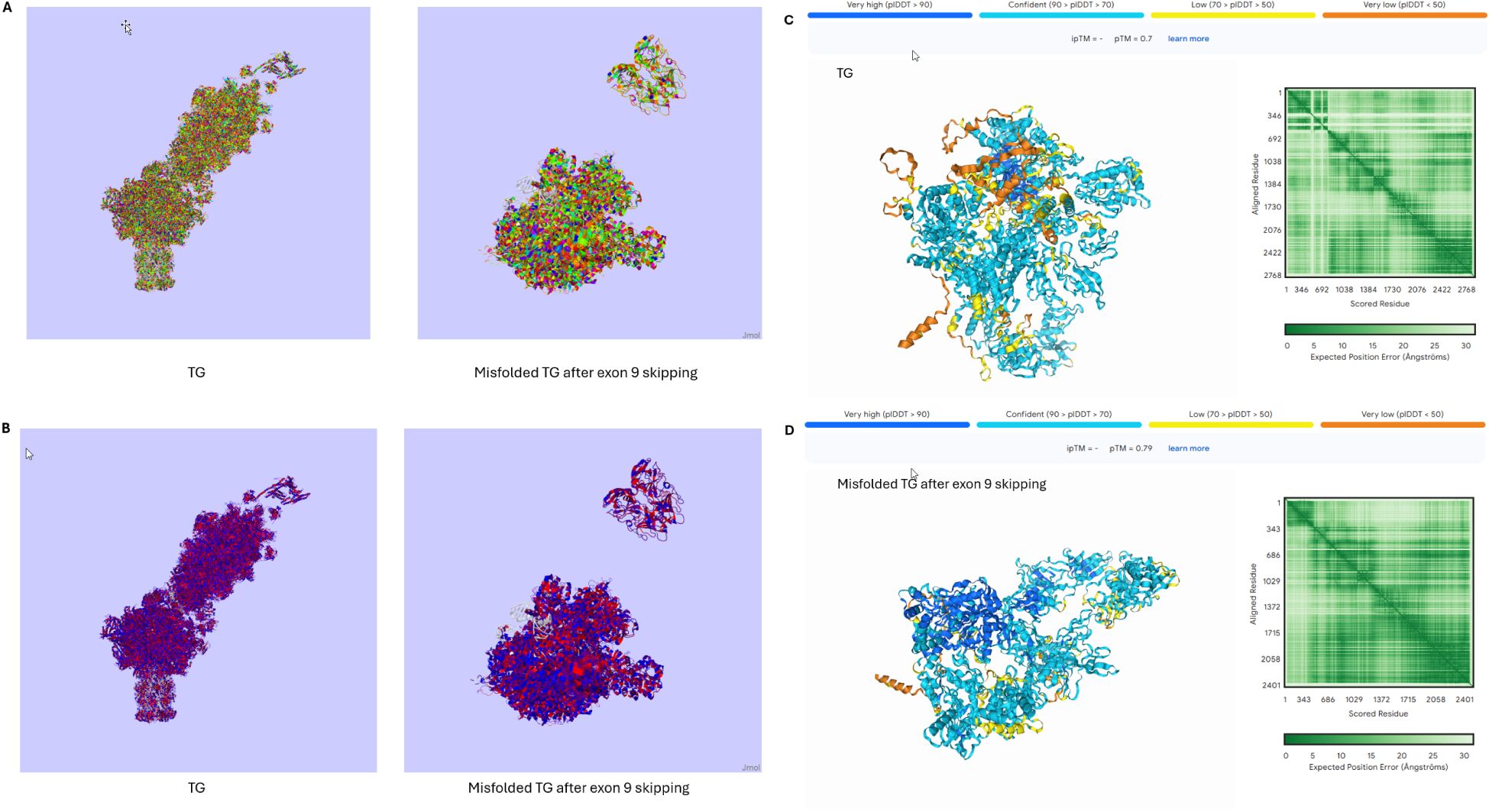
Figure 3. 3D structures of the normal TG and the misfolded TG after the skipping of exon 9 (introduced by the rare variant c.1076-1G > C). (A) Taylor colour schemes highlighting the amino acids based on their physicochemical properties. Figures created in Jalview 2.11.4.1. (B) Hydrophobicity colour schemes highlighting the amino acids based on their hydrophobicity. Figures created in Jalview 2.11.4.1. (C, D) Prediction of the 3D structures of the normal TG (C) and the misfolded TG protein (D) based on their amino acid sequences. Figures created in Alphafold server. AlphaFold produces a per-residue model confidence score (pLDDT) between 0 and 100. Some regions below 50 pLDDT may be unstructured in isolation. Very high (pLDDT > 90) is shown in dark blue, high (90 > pLDDT > 70) in cyan blue, low (70 > pLDDT > 50) in yellow and very low (pLDDT < 50) in orange. TG, Thyroglobulin.
Studies on mice with experimental autoimmune thyroiditis (EAT), a murine model of HT, have proposed the co-existence of two T cell populations; the Tg-reactive T cells and the natural CD4+CD25+ nTregs that are specific for the Tg (110). These studies have shown that continued Tg epitope presentation is essential in order to maintain a population of nTregs which are critical for the control of Tg-reactive T cells in the periphery. Decreased levels of Tg in the circulation or presentation of alternative epitopes might disturb the balance required for the nTregs maintenance, and therefore lead to ‘escaped’ Tg-reactive T cells (81, 110, 111). This mechanism of peripheral tolerance contrasts to that demonstrated for insulin gene expression in the thymus.
Autoimmune Addison’s disease
Autoimmune Addison’s disease (AAD) is a rare autoimmune endocrine condition characterised by the destruction of the adrenal cortex. Autoantibodies against the steroid 21-hydroxylase (21-OHase) enzyme are detected in most patients, with the underlying mechanism for their production, though, remaining unclear (1, 8, 112). Given the fact that 21-OHase is an intracellular enzyme and unlikely to be accessible to circulating autoantibodies, their direct pathogenic role has never been demonstrated. However, the existence of antigen-specific T cells in the blood of AAD patients has been demonstrated and the presence of CD4+ and CD8+ T cells bearing 21-OHase-specific TCRs against two immunodominant 21-OHase epitopes has been confirmed, demonstrating the central contribution of Cytotoxic T cells to disease progression (8, 113, 114). Like in other autoimmune endocrine disorders, studies on twins and family members of AAD patients have shown that genetics play an essential role in the disease development. However, the rise in cases the recent years implies that environmental events, such as viral infections and stress, might also be involved (1, 5, 8, 115).
21-OHase is encoded by the CYP21A2 gene, which is located in the MHC class III region on chromosome 6, and mutations on this gene are the commonest cause for Congenital Adrenal Hyperplasia (CAH) (1, 116, 117). The MHC class III region is located in the middle of the MHC gene cluster, flanked by the MHC class II (HLA-DR, DQ) genes on one side and MHC class I (HLA-A -C) on the other. Very near the CYP21A2 locus, there is an adjacent highly homologous pseudogene, CYP21A1P, which cannot produce a functional enzyme due to an 8-bp deletion in the exon 3 which results in a truncated protein (1, 118). Both the location and the high sequence identity of the two genes contribute to recombination events which are considered the main reason for the deleterious mutations accumulating in the CYP21A2 gene that lead to CAH (118). The two genes, CYP21A2 and CYP21A1P, belong to a repeated genomic cassette, called the RCCX module. Apart from the two CYP21 genes, in its most frequent form in Caucasians, the locus contains the serine/threonine kinase 19 (STK19) gene, the tenascin-X (TNX) gene, the complement 4 A (C4A) and B(C4B) genes, and the pseudogenes STK19B and TNXA. Non-allelic homologous recombination (NAHR) events in the locus alter the module structure and cause gene conversions and deletions increasing the complexity of the region (118) (Figure 4). Sequencing of the CYP21A2 gene from AAD patients and healthy individuals showed five SNPs that occur more often in heterozygosity in AAD patients compared to controls (119). Three of them, rs61338903, rs6474 and rs6473, are located in the exonic sequence, whilst the other two, rs6467 and rs76565726, are intronic variants (119). The minor allele of variant rs6473 has been shown to regulate the transcriptional repressor CTCF binding site and has been associated with downregulation of HLA-C and HLA-DRB1 expression (120). In addition, AAD patients with 21-OHase antibodies were more likely to carry these variants in a heterozygous state. However, regression analysis revealed that all variants were in LD with HLA-DRB1*03:01, *04:01 and *04:04 alleles which are associated with high-risk for AAD (119, 121). In contrast, the rs6472*C allele was found to be protective against the disease, and independent from the HLA-DRB1 locus, with a significantly lower frequency in AAD patients compared to controls (119). Thus, it remains unclear whether the primary association at this locus is with HLA-DR alleles or whether variants in CYP21A2 itself could contribute to AAD susceptibility.
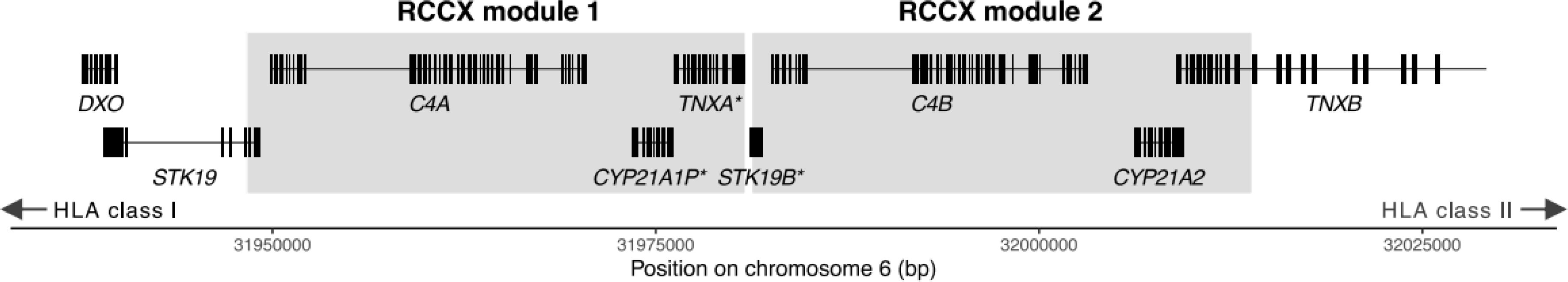
Figure 4. Schematic representation of the most common form of the RCCX cassette in healthy individuals, with pseudogenes noted with an asterisk. Figure adapted by Figure 1A of Lundtoft et al. (122).
Analysis of the copy number variation of the CYP21 genes showed different patterns for the functional CYP21A2 and the pseudogene CYP21A1P between healthy controls and AAD patients. Copy numbers of CYP21A2 were similar between AAD patients and healthy people, with 95% of the individuals carrying 2 copies of the gene. Interestingly, the CYP21A1P showed larger variation ranging from 0 to 5 copies, with AAD patients revealed as more likely to have lower copy numbers than the healthy controls, with each ‘lost’ copy being associated with a 3.4-fold odds ratio for disease (122). Examination of the copy number variation of the Complement-4 A (C4A) and B (C4B) genes, which are located in the same RCCX cassette, showed that there is a stronger correlation of CYP21A1P variation with C4A than with C4B, however lower copy numbers of the C4A gene were slightly more associated with AAD compared to those of CYP21A1P (122). Nevertheless, lower copies of both CYP21A1P and C4A are in close LD with the high risk HLA-DRB1*03:01 allele, as the SNPs described above, and therefore it remains unclear whether they might have an independent effect (119, 122, 123). It is not immediately obvious why copy number or expression of CYP21A1P, a pseudogene, should be relevant for disease pathogenesis. However, we speculate that AAD patients have reduced copy number or absence of CYP21A1P, because thymic expression of this pseudogene could underly central tolerance, in a similar way to that found for insulin gene variation.
Anti–pituitary-specific transcriptional factor-1 syndrome (Anti-PIT-1 hypophysitis)
Immune-mediated pituitary dysfunction is a rare condition, either involving the cells of the anterior pituitary alone, or involving the posterior pituitary to produce an associated vasopressin deficiency, known as infundibulohypophysitis. Anti-PIT-1 hypophysitis is a rare but recently recognised autoimmune pituitary disease, which is characterised by growth hormone (GH), prolactin (PRL), and thyroid stimulating hormone (TSH) deficiency (124–127). PIT-1 is an important transcription factor for the control of GH, PRL and TSH expression in the somatotroph, lactotroph and thyrotroph cells of the anterior pituitary gland respectively, and mutations on the PIT-1 gene cause congenital GH, PRL, and TSH deficiency. The presence of not only circulating anti-PIT-1 antibodies, but also CD8+ T cells in the pituitary gland of patients with anti-PIT-1 hypophysitis, indicate another example of a tissue-specific autoimmune disorder where CTLs are implicated in organ-specific cell damage (124, 125, 127–129). Thus far, the disease has only been found in the presence of thymoma or other types of neoplasms that have ectopic expression of the PIT-1 gene (124). Although it is currently unclear what types of malignancies coexist with the disease, and in which proportion, the underlying mechanism involves ectopic expression and presentation of PIT-1. Thus, somatic mutations leading to ectopic expression of PIT-1 in thymoma(s), or other malignancies, represents another mechanism by which dysregulation of antigenic gene expression can precipitate an organ-specific autoimmune endocrine disease.
Perspective for other autoimmune conditions
Autoimmune endocrine disorders may be considered relatively unique amongst autoimmune conditions in that the clinical manifestations occur largely after a hormone-secreting cell-type is irreversibly damaged leading to hormonal deficiency. This contrasts to a disease such as systemic lupus erythematosus (SLE), where the nuclear antigens are ubiquitous in all cell types and damage causing some of the clinical presentations is mediated by immune-complex deposition. Thus, there are some striking differences in antigen distribution and it is easy to envisage how peripheral immune tolerance may have a more important role in systemic autoimmune diseases with less restricted antigen distribution/expression than for organ-specific ones. Even within the spectrum of autoimmune thyroid disease, it is notable that patients with APS1, a ‘pure thymic defect’ in tolerance (5), very rarely develop GD, but quite frequently develop HT. This may reflect that TSH receptor A-subunit is shed from the thyrocyte membrane (68) leading to degree of peripheral tolerance, whereas thyroid peroxidase has an apical (internal) membrane localisation and perhaps restricted access from ongoing immune surveillance. As mentioned above, role of antigen itself in autoimmunity is relatively neglected and further studies could cast important light on this subject.
Conclusion
Genetic variation in organ-specific antigens remain understudied in autoimmunity in general. However, because of the discrete involvement of endocrine glands/tissues in autoimmune endocrinopathy, we are starting to glimpse potential antigen-specific mechanisms that underly disease proclivity for these conditions. Ectopic expression of many antigens in thymus may be a critical stage in T lymphocyte selection and under-expression may lead to failure to delete auto-reactive TCR-bearing T cell populations. Importantly, thymic gene/antigen expression may be regulated differently to that of the native tissue, leading to complexity in understanding the pathophysiological link. In contrast, continuous exposure of the peripheral immune system to endogenous antigen, such as Tg, may also provide an important mechanism for tolerance. We have much to learn in this area, but a fuller understanding of the antigenic stimulus and the detail of how this is involved in pathogenesis holds the promise of a tissue-specific solution rather than a general ‘immunosuppressive’ approach to therapy for autoimmunity.
Author contributions
MM: Investigation, Software, Writing – original draft, Writing – review & editing. SP: conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Work in this laboratory was funded by UKRI/MRC grants MR/S001611/1, MR/V005898/1 and a generous endowment from the Robotham family (Anthony Robotham PhD studentship).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mitchell AL, Pearce SH. Autoimmune Addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol. (2012) 8:306–16. doi: 10.1038/nrendo.2011.245
2. Todd JA. Etiology of type 1 diabetes. Immunity. (2010) 32:457–67. doi: 10.1016/j.immuni.2010.04.001
3. Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell. (2010) 140:791–7. doi: 10.1016/j.cell.2010.03.003
4. Saevarsdottir S, Olafsdottir TA, Ivarsdottir EV, Halldorsson GH, Gunnarsdottir K, Sigurdsson A, et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature. (2020) 584:619–23. doi: 10.1038/s41586-020-2436-0
5. Husebye ES, Pearce SH, Krone NP, Kämpe O. Adrenal insufficiency. Lancet. (2021) 397:613–29. doi: 10.1016/S0140-6736(21)00136-7
6. Eriksson D, Bianchi M, Landegren N, Dalin F, Skov J, Hultin-Rosenberg L, et al. Common genetic variation in the autoimmune regulator (AIRE) locus is associated with autoimmune Addison’s disease in Sweden. Sci Rep. (2018) 8:8395. doi: 10.1038/s41598-018-26842-2
7. Chatila Ta, Blaeser F, Ho N, Lederman Hm, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. (2000) 106:R75–81. doi: 10.1172/JCI11679
8. Pazderska A, Pearce SH, Mitchell AL. Adrenal Disorders: Physiology, Pathophysiology and Treatment. Levine AC, editor. Cham, Switzerland: S.I. Publishing (2018) p. 85–108. doi: 10.1007/978-3-319-62470-9
9. Gough SCL, Simmonds MJ. The HLA region and autoimmune disease: associations and mechanisms of action. Curr Genomics. (2007) 8:453–65. doi: 10.2174/138920207783591690
10. Hocking AM, Buckner JH. Genetic basis of defects in immune tolerance underlying the development of autoimmunity. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.972121
11. Grixti L, Lane LC, Pearce SH. The genetics of Graves’ disease. Rev Endocrine Metab Disord. (2023) 25:203–14. doi: 10.1007/s11154-023-09848-8
12. Vos XG, Endert E, Zwinderman AH, Tijssen JGP, Wiersinga WM. Predicting the risk of recurrence before the start of antithyroid drug therapy in patients with Graves’ Hyperthyroidism. J Clin Endocrinol Metab. (2016) 101:1381–9. doi: 10.1210/jc.2015-3644
13. Tautz L, Critton DA, Grotegut S. Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol Biol. (2013) 1053:179–221. doi: 10.1007/978-1-62703-562-0_13
14. Gootjes C, Zwaginga JJ, Roep BO, Nikolic T. Functional impact of risk gene variants on the autoimmune responses in type 1 diabetes. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.886736
15. Stephan K. Genetic modifiers of thymic selection and central tolerance in type 1 diabetes. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.889856
16. Ivashkiv LB. PTPN22 in autoimmunity: different cell and different way. Immunity. (2013) 39:91–3. doi: 10.1016/j.immuni.2013.07.007
17. Tizaoui K, Terrazzino S, Cargnin S, Lee KH, Gauckler P, Li H, et al. The role of PTPN22 in the pathogenesis of autoimmune diseases: A comprehensive review. Semin Arthritis Rheum. (2021) 51:513–22. doi: 10.1016/j.semarthrit.2021.03.004
18. Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol. (2014) 32:83–119. doi: 10.1146/annurev-immunol-032713-120249
19. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease–associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. (2011) 43:902–9. doi: 10.1038/ng.904
20. Hamerman JA, Pottle J, Ni M, He Y, Zhang Z-Y, Buckner JH. Negative regulation of TLRsignaling in myeloid cells—implications for autoimmunediseases. Immunol Rev. (2016) 269:212–27. doi: 10.1111/imr.2016.269.issue-1
21. Wallis AM, Wallace EC, Hostager BS, Yi Z, Houtman JCD, Bishop GA. TRAF3 enhances TCR signaling by regulating the inhibitors Csk and PTPN22. Sci Rep. (2017) 7:2081. doi: 10.1038/s41598-017-02280-4
22. Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. (2013) 39:111–22. doi: 10.1016/j.immuni.2013.06.013
23. Menard L, Saadoun D, Isnardi I, Ng Y-S, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. (2011) 121:3635–44. doi: 10.1172/JCI45790
24. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. (2007) 179:4704–10. doi: 10.4049/jimmunol.179.7.4704
25. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. (2005) 37:1317–9. doi: 10.1038/ng1673
26. Bufalo NatássiaE, Dos Santos RB, Rocha AngélicaG, Teodoro L, Romaldini JoãoH, Ward LS. Polymorphisms of the genes CTLA4, PTPN22, CD40, and PPARG and their roles in Graves’ disease: susceptibility and clinical features. Endocrine. (2020) 71:104–12. doi: 10.1007/s12020-020-02337-x
27. Kavvoura FK, Akamizu T, Awata T, Ban Y, Chistiakov DA, Frydecka I, et al. Cytotoxic T-lymphocyte associated antigen 4 gene polymorphisms and autoimmune thyroid disease: A meta-analysis. J Clin Endocrinol Metab. (2007) 92:3162–70. doi: 10.1210/jc.2007-0147
28. Ni J, Qiu L-J, Zhang M, Wen P-F, Ye X-R, Liang Y, et al. CTLA-4 CT60(rs3087243) polymorphism and autoimmune thyroid diseases susceptibility: a comprehensive meta-analysis. Endocrine Res. (2014) 39:180–8. doi: 10.3109/07435800.2013.879167
29. Pawlak-Adamska E, Frydecka I, Bolanowski M, Tomkiewicz A, Jonkisz A, Karabon L, et al. CD28/CTLA-4/ICOS haplotypes confers susceptibility to Graves’ disease and modulates clinical phenotype of disease. Endocrine. (2016) 55:186–99. doi: 10.1007/s12020-016-1096-1
30. Mitsuiki N, Schwab C, Grimbacher B. What did we learn from CTLA-4 insufficiency on the human immune system? Immunol Rev. (2019) 287:33–49. doi: 10.1111/imr.12721
31. Pazderska A, Oftedal BE, Napier CM, Ainsworth HF, Husebye ES, Cordell HJ, et al. A variant in the BACH2 gene is associated with susceptibility to autoimmune Addison’s disease in humans. J Clin Endocrinol Metab. (2016) 101:3865–9. doi: 10.1210/jc.2016-2368
32. Onuma H, Kawamura R, Tabara Y, Yamashita M, Ohashi J, Kawasaki E, et al. Variants in the BACH2 and CLEC16A gene might be associated with susceptibility to insulin-triggered type 1 diabetes. J Diabetes Investig. (2019) 10:1447–53. doi: 10.1111/jdi.13057
33. Fichna M, Żurawek M, Słomiński B, Sumińska M, Czarnywojtek A, Rozwadowska N, et al. Polymorphism in BACH2 gene is a marker of polyglandular autoimmunity. Endocrine. (2021) 74:72–9. doi: 10.1007/s12020-021-02743-9
34. Fichna M, Małecki PP, Żurawek M, Furman K, Gębarski B, Fichna P, et al. Genetic variants and risk of endocrine autoimmunity in relatives of patients with Addison’s disease. Endocr Connect. (2023) 12(6):e230008. doi: 10.1530/EC-23-0008
35. Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. (2008) 40:1399–401. doi: 10.1038/ng.249
36. Karim KA, John P, Bhatti A, Zia A, Shahid G, Akhtar P, et al. Association of 32 type 1 diabetes risk loci in Pakistani patients. Diabetes Res Clin Pract. (2015) 108:137–42. doi: 10.1016/j.diabres.2015.01.022
37. Dieter C, Lemos NatáliaE, Dorfman LE, Duarte GCK, Assmann TaísS, Crispim D. The rs11755527 polymorphism in the BACH2 gene and type 1 diabetes mellitus: case control study in a Brazilian population. Arch Endocrinol Metab. (2020) 64:138–43. doi: 10.20945/2359-3997000000214
38. Liu W, Wang Hai−Ning, Gu Zhao−Hui, Yang Shao−Ying, Ye Xiao−Ping, Pan Chun−Ming, et al. Identification of BACH2 as a susceptibility gene for Graves’ disease in the Chinese Han population based on a three-stage genome-wide association study. Hum Genet. (2014) 133:661–71. doi: 10.1007/s00439-013-1404-2
39. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen W-M, Santa Cruz DF, Yang H, et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet. (2021) 53:962–71. doi: 10.1038/s41588-021-00880-5
40. Marquez A, Martin J. Genetic overlap between type 1 diabetes and other autoimmune diseases. Semin Immunopathol. (2022) 44:81–97. doi: 10.1007/s00281-021-00885-6
41. Cooper JD, Simmonds MJ, Walker NM, Burren O, Brand OJ, Guo H, et al. Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet. (2012) 21:5202–8. doi: 10.1093/hmg/dds357
42. Dendrou CA, Plagnol V, Fung E, Yang JHM, Downes K, Cooper JD, et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet. (2009) 41:1011–5. doi: 10.1038/ng.434
43. Garg G, Tyler JR, Yang JHM, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ Regulatory T cell function. J Immunol. (2012) 188:4644–53. doi: 10.4049/jimmunol.1100272
44. Cerosaletti K, Schneider A, Schwedhelm K, Frank I, Tatum M, Wei S, et al. Multiple autoimmune-associated variants confer decreased IL-2R signaling in CD4+CD25hi T cells of type 1 diabetic and multiple sclerosis patients. PloS One. (2013) 8(12):e83811. doi: 10.1371/journal.pone.0083811
45. De Luca F, Shoenfeld Y. The microbiome in autoimmune diseases. Clin Exp Immunol. (2019) 195:74–85. doi: 10.1111/cei.13158
46. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
47. Pugliese A, Miceli D. The insulin gene in diabetes. Diabetes/Metabolism Res Rev. (2002) 18:13–25. doi: 10.1002/dmrr.v18:1
48. Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, et al. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes. (2004) 53:1884–9. doi: 10.2337/diabetes.53.7.1884
49. Catignani KG, German MS, Rutter WJ. The minisatellite in the diabetes susceptibility locus IDDM2 regulates insulin transcription. Nat Genet. (1995) 9:293–8. doi: 10.1038/ng0395-293
50. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet. (1997) 15:289–92. doi: 10.1038/ng0397-289
51. Ahmed S, Bennett ST, Huxtable SJ, Todd JA, Matthews DR, Gough SCL. INS VNTR allelic variation and dynamic insulin secretion in healthy adult non-diabetic Caucasian subjects. Diabetic Med. (1999) 16:910–7. doi: 10.1046/j.1464-5491.1999.00169.x
52. Bennett ST, Lucassen AM, Gough SCL, Powell EE, Undlien DE, Pritchard LE, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet. (1995) 9:284–92. doi: 10.1038/ng0395-284
53. Bennett ST, Wilson AJ, Cucca F, Nerup Jørn, Pociot F, Mckinney PA, et al. IDDM2-VNTR-encoded susceptibility to type 1 diabetes: dominant protection and parental transmission of alleles of the insulin gene-linked minisatellite locus. J Autoimmun. (1996) 9:415–21. doi: 10.1006/jaut.1996.0057
54. Bennett ST, Wilson AJ, Esposito L, Bouzekri N, Undlien DE, Cucca F, et al. Insulin VNTR allele-specific effect in type 1 diabetes depends on identity of untransmitted paternal allele. Nat Genet. (1997) 17:350–2. doi: 10.1038/ng1197-350
55. Alami CA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes. (2002) 51:1383–90. doi: 10.2337/diabetes.51.5.1383
56. Walter M, Albert E, Conrad M, Keller E, Hummel M, Ferber K, et al. IDDM2/insulin VNTR modifies risk conferred by IDDM1/HLA for development of Type 1 diabetes and associated autoimmunity. Diabetologia. (2003) 46:712–20. doi: 10.1007/s00125-003-1082-z
57. Marín-Sánchez A, Álvarez-Sierra D, González O, Lucas-Martin A, Sellés-Sánchez A, Rudilla F, et al. Regulation of TSHR expression in the thyroid and thymus may contribute to TSHR tolerance failure in graves’ Disease patients via two distinct mechanisms. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01695
58. Prabhakar BS, Bahn RS, Smith TJ. Current perspective on the pathogenesis of graves’ Disease and ophthalmopathy. Endocrine Rev. (2003) 24:802–35. doi: 10.1210/er.2002-0020
59. Stefan M, Faustino LC. Genetics of thyroid-stimulating hormone receptor—Relevance for autoimmune thyroid disease. Front Endocrinol. (2017) 8. doi: 10.3389/fendo.2017.00057
60. Stefan M, Wei C, Lombardi A, Li CW, Concepcion ES, Inabnet WB, et al. Genetic–epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci. (2014) 111:12562–7. doi: 10.1073/pnas.1408821111
61. Struja T, Fehlberg H, Kutz A, Guebelin L, Degen C, Mueller B, et al. Can we predict relapse in Graves’ disease? Results from a systematic review and meta-analysis. Eur J Endocrinol. (2017) 176:87–97. doi: 10.1530/EJE-16-0725
62. Rapoport B, Aliesky HA, Chen C-R, Mclachlan SM. Evidence that TSH receptor A-subunit multimers, not monomers, drive antibody affinity maturation in Graves’ Disease. J Clin Endocrinol Metab. (2015) 100:E871–5. doi: 10.1210/jc.2015-1528
63. Colobran R, Armengol MDP, Faner R, Gärtner M, Tykocinski L-O, Lucas A, et al. Association of an SNP with intrathymic transcription of TSHR and Graves’ disease: a role for defective thymic tolerance. Hum Mol Genet. (2011) 20:3415–23. doi: 10.1093/hmg/ddr247
64. Brand OJ, Barrett JC, Simmonds MJ, Newby PR, Mccabe CJ, Bruce CK, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease. Hum Mol Genet. (2009) 18:1704–13. doi: 10.1093/hmg/ddp087
65. Chu Y-D, Yeh C-T. The molecular function and clinical role of thyroid stimulating hormone receptor in cancer cells. Cells. (2020) 9(7):1730. doi: 10.3390/cells9071730
66. Costagliola S, Khoo D, Vassart G. Production of bioactive amino-terminal domain of the thyrotropin receptor via insertion in the plasma membrane by a glycosylphosphatidylinositol anchor. FEBS Lett. (1998) 436:427–33. doi: 10.1016/S0014-5793(98)01177-6
67. Davies T, Marians R, Latif R. The TSH receptor reveals itself. J Clin Invest. (2002) 110:161–4. doi: 10.1172/JCI0216234
68. Rapoport B, Mclachlan SM. TSH receptor cleavage into subunits and shedding of the A-subunit; A molecular and clinical perspective. Endocr Rev. (2016) 37:114–34. doi: 10.1210/er.2015-1098
69. Tanaka K, Chazenbalk GD, Mclachlan SM, Rapoport B. Subunit structure of thyrotropin receptors expressed on the cell surface. J Biol Chem. (1999) 274:33979–84. doi: 10.1074/jbc.274.48.33979
70. Ploski R, Szymaski K, Bednarczuk T. The genetic basis of Graves’ Disease. Curr Genomics. (2011) 12:542–63. doi: 10.2174/138920211798120772
71. Kleinau G, Neumann S, Gruters A, Krude H, Biebermann H. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr Rev. (2013) 34:691–724. doi: 10.1210/er.2012-1072
72. Mizutori Y, Chen C-R, Latrofa F, Mclachlan SM, Rapoport B. Evidence that shed thyrotropin receptor A subunits drive affinity maturation of autoantibodies causing Graves’ disease. J Clin Endocrinol Metab. (2009) 94:927–35. doi: 10.1210/jc.2008-2134
73. Boguslawska J, Godlewska M, Gajda E, Piekielko-Witkowska A. Cellular and molecular basis of thyroid autoimmunity. Eur Thyroid J. (2022) 11(1):e210024. doi: 10.1530/ETJ-21-0024
74. Lee HJ, Stefan-Lifshitz M, Li CW, Tomer Y. Genetics and epigenetics of autoimmune thyroid diseases: Translational implications. Best Pract Res Clin Endocrinol Metab. (2023) 37:101661. doi: 10.1016/j.beem.2022.101661
75. Stefan M, Jacobson EM, Huber AK, Greenberg DA, Li CW, Skrabanek L, et al. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon α-modulated mechanism. J Biol Chem. (2011) 286:31168–79. doi: 10.1074/jbc.M111.247510
76. Chu X, Pan C-M, Zhao S-X, Liang J, Gao G-Q, Zhang X-M, et al. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat Genet. (2011) 43:897–901. doi: 10.1038/ng.898
77. Dechairo BM, Zabaneh D, Collins J, Brand O, Dawson GJ, Green AP, et al. Association of the TSHR gene with Graves’ disease: the first disease specific locus. Eur J Hum Genet. (2005) 13:1223–30. doi: 10.1038/sj.ejhg.5201485
78. Płoski Rafał, Brand OJ, Jurecka-Lubieniecka B, Franaszczyk M, Kula D, Krajewski Paweł, et al. Thyroid stimulating hormone receptor (TSHR) intron 1 variants are major risk factors for Graves’ disease in three European Caucasian cohorts. PloS One. (2010) 5:e15512. doi: 10.1371/journal.pone.0015512
79. Pujol-Borrell R, Gimenez-Barcons M, Marin-Sanchez A, Colobran R. Genetics of graves’ Disease: special focus on the role of TSHR gene. Horm Metab Res. (2015) 47:753–66. doi: 10.1055/s-0035-1559646
80. Hiratani H, Bowden DW, Ikegami S, Shirasawa S, Shimizu A, Iwatani Y, et al. Multiple SNPs in intron 7 of thyrotropin receptor are associated with Graves’ disease. J Clin Endocrinol Metab. (2005) 90:2898–903. doi: 10.1210/jc.2004-2148
81. Zaletel K, Gaberscek S. Hashimoto’s thyroiditis: from genes to the disease. Curr Genomics. (2011) 12:576–88. doi: 10.2174/138920211798120763
82. Ragusa F, Fallahi P, Elia G, Gonnella D, Paparo SR, Giusti C, et al. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract Res Clin Endocrinol Metab. (2019) 33(6):101367. doi: 10.1016/j.beem.2019.101367
83. Ris-Stalpers C, Bikker H. Genetics and phenomics of hypothyroidism and goiter due to TPO mutations. Mol Cell Endocrinol. (2010) 322:38–43. doi: 10.1016/j.mce.2010.02.008
84. Mohssen M, Radhi S, Fadel-Al-Jumaili E, Abdulhasan A-HM, Tariq A. TPO gene rs2071400 polymorphisms as an independent risk factor for hypothyroidism in Iraqi patients. Biomedicine. (2023) 43:775–8. doi: 10.51248/.v43i02.2701
85. Tomari S, Watanabe M, Inoue N, Mizuma T, Yamanaka C, Hidaka Y, et al. The polymorphisms in the thyroid peroxidase gene were associated with the development of autoimmune thyroid disease and the serum levels of anti-thyroid peroxidase antibody. Endocrine J. (2017) 64:1025–32. doi: 10.1507/endocrj.EJ17-0191
86. Al-Mofarji ST, Jasim HM, Mohammed SB. Association between TPO gene polymorphisms with susceptibility to hyperthyroidism. Biomedicine. (2024) 43:1744–9. doi: 10.51248/.v43i6.3499
87. Mizuma T, Watanabe M, Inoue N, Arakawa Y, Tomari S, Hidaka Y, et al. Association of the polymorphisms in the gene encoding thyroglobulin with the development and prognosis of autoimmune thyroid disease. Autoimmunity. (2017) 50:386–92. doi: 10.1080/08916934.2017.1344971
88. Al-Mofarji ST, Jasim HM, Mohammed SB, Al-Samerriae AY. TPO gene expression in relation with promoter SNPs in Iraqi patients with hyperthyroidism. Al-Rafidain J Med Sci. (2023) 5:S100–105. doi: 10.54133/ajms.v5i1S.313
89. Khoshi A, Sirghani A, Ghazisaeedi M, Zarei Mahmudabadi A, Azimian A. Association between TPO Asn698Thr and Thr725Pro gene polymorphisms and serum anti-TPO levels in Iranian patients with subclinical hypothyroidism. Hormones. (2017) 16:75–83. doi: 10.14310/horm.2002.1721
90. Balmiki N, Bankura B, Guria S, Das TK, Pattanayak AK, Sinha A, et al. Genetic analysis of thyroid peroxidase (TPO) gene in patients whose hypothyroidism was found in adulthood in West Bengal, India. Endocrine J. (2014) 61:289–96. doi: 10.1507/endocrj.EJ13-0237
91. Czarnocka B, Eschler DC, Godlewska M, Tomer Y. “Chapter 44 - Thyroid Autoantibodies: Thyroid Peroxidase and Thyroglobulin Antibodies”. In: Shoenfeld Y, Meroni PL, Gershwin ME, editors. Autoantibodies. (2014). p. 365–73. doi: 10.1016/B978-0-444-56378-1.00044-7
92. Le SN, Porebski BT, Mccoey J, Fodor J, Riley B, Godlewska M, et al. Modelling of thyroid peroxidase reveals insights into its enzyme function and autoantigenicity. PloS One. (2015) 10:e0142615. doi: 10.1371/journal.pone.0142615
93. Mclachlan SM, Rapoport B. Autoimmune response to the thyroid in humans: thyroid peroxidase–the common autoantigenic denominator. Int Rev Immunol. (2000) 19:587–618. doi: 10.3109/08830180009088514
94. Finke R, Seto P, Ruf J, Carayon P, Rapoport B. Determination at the molecular level of a B-cell epitope on thyroid peroxidase likely to be associated with autoimmune thyroid disease. J Clin Endocrinol Metab. (1991) 73:919–21. doi: 10.1210/jcem-73-4-919
95. Kim MiJ, Oh SoW, Youn H, Na J, Kang KW, Park DoJ, et al. Thyroid-related protein expression in the human thymus. Int J Endocrinol. (2017) 2017:1–10. doi: 10.1155/2017/8159892
96. Mclachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocrine Rev. (2014) 35:59–105. doi: 10.1210/er.2013-1055
97. Misharin AV, Nagayama Y, Aliesky HA, Rapoport B, Mclachlan SM. Studies in mice deficient for the autoimmune regulator (Aire) and transgenic for the thyrotropin receptor reveal a role for aire in tolerance for thyroid autoantigens. Endocrinology. (2009) 150:2948–56. doi: 10.1210/en.2008-1690
98. Lahooti H, Edirimanne S, Walsh JP, Delbridge L, Hibbert E, Wall J. Single nucleotide polymorphism 1623 A/G (rs180195) in the promoter of the Thyroglobulin gene is associated with autoimmune thyroid disease but not with thyroid ophthalmopathy. Clin Ophthalmol. (2017) 11:1337–45. doi: 10.2147/OPTH.S136070
99. Collins JE, Heward JM, Carr-Smith J, Daykin J, Franklyn JA, Gough SCL. Association of a rare thyroglobulin gene microsatellite variant with autoimmune thyroid disease. J Clin Endocrinol Metab. (2003) 88:5039–42. doi: 10.1210/jc.2003-030093
100. Lo MS, Towne M, Vannoy GE, Brownstein CA, Lane AA, Chatila TA, et al. Monogenic Hashimoto thyroiditis associated with a variant in the thyroglobulin (TG) gene. J Autoimmun. (2018) 86:116–9. doi: 10.1016/j.jaut.2017.09.003
101. Weetman AP. An update on the pathogenesis of Hashimoto’s thyroiditis. J Endocrinological Invest. (2020) 44:883–90. doi: 10.1007/s40618-020-01477-1
102. Latrofa F, Fiore E, Rago T, Antonangeli L, Montanelli L, Ricci D, et al. Iodine contributes to thyroid autoimmunity in humans by unmasking a cryptic epitope on thyroglobulin. J Clin Endocrinol Metab. (2013) 98:E1768–74. doi: 10.1210/jc.2013-2912
103. Arase N, Arase H. Cellular misfolded proteins rescued from degradation by MHC class II molecules are possible targets for autoimmune diseases. J Biochem. (2015) 158:367–72. doi: 10.1093/jb/mvv093
104. Barkoff MS, Kocherginsky M, Anselmo Joa˜O, Weiss RE, Refetoff S. Autoimmunity in patients with resistance to thyroid hormone. J Clin Endocrinol Metab. (2010) 95:3189–93. doi: 10.1210/jc.2009-2179
105. Ohba K, Sasaki S, Misawa NH, Matsushita A, Kuroda Go, Sakai Y, et al. Clinical outcomes of 34 patients with resistance to thyroid hormone beta: a twenty-year experience in Japan. Endocrine J. (2022) 69:179–88. doi: 10.1507/endocrj.EJ21-0390
106. Kühnen P, Turan S, Fröhler S, Güran Tülay, Abali S, Biebermann H, et al. Identification of PENDRIN (SLC26A4) mutations in patients with congenital hypothyroidism and “apparent” thyroid dysgenesis. J Clin Endocrinol Metab. (2014) 99:E169–76. doi: 10.1210/jc.2013-2619
107. Wemeau J-L, Kopp P. Pendred syndrome. Best Pract Res Clin Endocrinol Metab. (2017) 31:213–24. doi: 10.1016/j.beem.2017.04.011
108. Peter K. Mutations in the Pendred Syndrome (PDS/SLC26A) gene: an increasingly complex phenotypic spectrum from goiter to thyroid hypoplasia. J Clin Endocrinol Metab. (2014) 99:67–9. doi: 10.1210/jc.2013-4319
109. Vaidya B, Coffey R, Coyle B, Trembath R, San Lazaro C, Reardon W, et al. Concurrence of Pendred syndrome, autoimmune thyroiditis, and simple goiter in one family. J Clin Endocrinol Metab. (1999) 84:2736–8. doi: 10.1210/jcem.84.8.5903
110. Kong YM, Brown NK, Morris GP, Flynn JC. The essential role of circulating thyroglobulin in maintaining dominance of natural regulatory T cell function to prevent autoimmune thyroiditis. Hormone Metab Res. (2015) 47:711–20. doi: 10.1055/s-0035-1548872
111. Carayanniotis G. The cryptic self in thyroid autoimmunity: the paradigm of thyroglobulin. Autoimmunity. (2009) 36:423–8. doi: 10.1080/08916930310001602975
112. Napier C, Pearce SHS. Autoimmune Addison’s disease. Presse Med. (2012) 41:e626-35. doi: 10.1016/j.lpm.2012.09.010
113. Dawoodji A, Chen J-L, Shepherd D, Dalin F, Tarlton A, Alimohammadi M, et al. High frequency of cytolytic 21-hydroxylase-specific CD8+ T cells in autoimmune Addison’s disease patients. J Immunol. (2014) 193:2118–26. doi: 10.4049/jimmunol.1400056
114. Hellesen A, Aslaksen S, Breivik L, Røyrvik EC, Bruserud Øyvind, Edvardsen K, et al. 21-hydroxylase-specific CD8+ T cells in autoimmune Addison’s disease are restricted by HLA-A2 and HLA-C7 molecules. Front Immunol. (2021) 12:742848. doi: 10.3389/fimmu.2021.742848
115. Song H, Fang F, Tomasson G, Arnberg Fk, Mataix-Cols D, Fernández de la Cruz L, et al. Association of stress-related disorders with subsequent autoimmune disease. JAMA. (2018) 319:2388–400. doi: 10.1001/jama.2018.7028
116. Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Macut D. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol. (2019) 10. doi: 10.3389/fendo.2019.00432
117. Asghari Kollahi N, Rohani F, Baghbani-Arani F, Shojaei A. Complex alleles of CYP21A2 are the most frequent causes of congenital adrenal hyperplasia in Iranian population. Iranian J Pediatr. (2019) 29(6):e91994. doi: 10.5812/ijp.91994
118. Carrozza C, Foca L, De Paolis E, Concolino P. Genes and pseudogenes: complexity of the RCCX locus and disease. Front Endocrinol (Lausanne). (2021) 12:709758. doi: 10.3389/fendo.2021.709758
119. Brønstad I, Skinningsrud B, Bratland E, Løvas K, Undlien D, Husebye ES, et al. CYP21A2 polymorphisms in patients with autoimmune Addison’s disease, and linkage disequilibrium to HLA risk alleles. Eur J Endocrinol. (2014) 171:743–50. doi: 10.1530/EJE-14-0432
120. Strongest gene expression correlations in blood: HLA-C [Effect size: -0.319672, p-value(-log10): 3.3008076937936703], HLA-DRB1 [Effect size: -0.555033, p-value(-log10): 3.1042471621597683. Ensembl.org.
121. Mavridou M, Mitchell A, Allinson K, Lane L, Pearce S. HLA-DRB1*0404 is associated with the deletion of the 21-hydroxylase pseudogene in AAD patients. In: Society for Endocrinology BES 2022. Endocrine Abstracts, Harrogate, United Kingdom (2022).
122. Lundtoft C, Eriksson D, Bianchi M, Aranda-Guillén M, Landegren N, Rantapää-Dahlqvist S, et al. Relation between HLA and copy number variation of steroid 21-hydroxylase in a Swedish cohort of patients with autoimmune Addison’s disease. Eur J Endocrinol. (2023) 189:235–41. doi: 10.1093/ejendo/lvad102
123. Peterson P, Partanen J, Aavik E, Salmi H, Pelkonen R, Krohn KJE. Steroid 2 1 -hydroxylase gene polymorphsm in Addison’s disease patients. Tissue Antigens. (1995) 46:63–7. doi: 10.1111/j.1399-0039.1995.tb02478.x
124. Bando H, Iguchi G, Okimura Y, Odake Y, Yoshida K, Matsumoto R, et al. A novel thymoma-associated autoimmune disease: Anti-PIT-1 antibody syndrome. Sci Rep. (2017) 7:43060. doi: 10.1038/srep43060
125. Kanie K, Bando H, Iguchi G, Muguruma K, Matsumoto R, Hidaka-Takeno R, et al. Pathogenesis of anti–PIT-1 antibody syndrome: PIT-1 presentation by HLA class I on anterior pituitary cells. J Endocrine Soc. (2019) 3:1969–78. doi: 10.1210/js.2019-00243
126. Pfaffle R, Kim C, Otten B, Wit J-M, Eiholzer U, Heimann G, et al. Pit-1:Clinical aspects. Horm Res. (1996) 45:25–8. doi: 10.1159/000184824
127. Yamamoto M, Iguchi G, Bando H, Kanie K, Hidaka-Takeno R, Fukuoka H, et al. Autoimmune pituitary disease: new concepts with clinical implications. Endocrine Rev. (2020) 41:261–72. doi: 10.1210/endrev/bnz003
128. Yamamoto M, Bando H. A new insight into GH regulation and its disturbance from nutrition and autoimmune perspectives. Endocrine J. (2023) 70:867–74. doi: 10.1507/endocrj.EJ23-0264
129. Bando H, Iguchi G, Fukuoka H, Yamamoto M, Hidaka-Takeno R, Okimura Y, et al. Involvement of PIT-1-reactive cytotoxic T lymphocytes in anti-PIT-1 antibody syndrome. J Clin Endocrinol Metab. (2014) 99:E1744–9. doi: 10.1210/jc.2014-1769
130. Pujol-Borrell R, Álvarez-Sierra D, Jaraquemada D, Marín-Sánchez A, Colobran R. Central tolerance mechanisms to TSHR in Graves’ Disease: contributions to understand the genetic association. Horm Metab Res. (2018) 50:863–70. doi: 10.1055/a-0755-7927
Keywords: immunogenetics, autoimmune thyroid disease, thymus, central tolerance, Graves’ disease, Addison’s disease, type 1 diabetes
Citation: Mavridou M and Pearce SH (2025) Exploring antigenic variation in autoimmune endocrinopathy. Front. Immunol. 16:1561455. doi: 10.3389/fimmu.2025.1561455
Received: 15 January 2025; Accepted: 07 February 2025;
Published: 28 February 2025.
Edited by:
Anette S. B. Wolff, University of Bergen, NorwayReviewed by:
Kai Kisand, University of Tartu, EstoniaChristopher Pohlmeyer, Horizon Therapeutics, United States
Copyright © 2025 Mavridou and Pearce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon H. Pearce, c2ltb24ucGVhcmNlQG5jbC5hYy51aw==