
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1561174
This article is part of the Research Topic Immunotherapy Resistance and Advancing Adaptive Cell Therapeutics View all 10 articles
 Karolina Rassek1†
Karolina Rassek1† Jan Misiak2†
Jan Misiak2† Tomasz Ołdak2,3†
Tomasz Ołdak2,3† Natalia Rozwadowska1,2*†
Natalia Rozwadowska1,2*† Grzegorz Basak4†
Grzegorz Basak4† Tomasz Kolanowski1,2*†
Tomasz Kolanowski1,2*†One of the most successful treatments in hematologic cancer is chimeric antigen receptor (CAR)-T cell-based immunotherapy. However, CAR-T therapy is not without challenges like the costly manufacturing process required to personalize each treatment for individual patients or graft-versus-host disease. Umbilical cord blood (UCB) has been most commonly used for hematopoietic cell transplant as it offers several advantages, including its rich source of hematopoietic stem cells, lower risk of graft-versus-host disease, and easier matching for recipients due to less stringent HLA requirements compared to bone marrow or peripheral blood stem cells. In this review, we have discussed the advantages and disadvantages of different CAR-T cell manufacturing strategies with the use of allogeneic and autologous peripheral blood cells. We compare them to the UCB approach and discuss ongoing pre-clinical and clinical trials in the field. Finally, we propose a cord blood bank as a readily available source of CAR-T cells.
One of the most widespread approaches in cancer immunotherapies is chimeric antigen receptor (CAR)-T cell therapy, in which genetically modified allogeneic or autologous T cells or natural killer (NK) cells are redirected against the tumor cells armored with CAR construct (1). The classic CAR combines antigen-binding domains, most commonly a single-chain variable fragment derived from antibody variable domains, with the signaling domains of the T cell receptor 3ζ chain and additional costimulatory domains from receptors such as 4‐1BB, OX40, or CD28 (2). In recent years, CAR-T cell therapy has revolutionized the treatment of relapsed or refractory hematological malignancies, resulting in Food and Drug Agency (FDA) approval of six products targeting non-Hodgkin lymphomas (NHLs), acute lymphoblastic leukemia (ALL) and multiple myeloma (MM).
Despite the enormous progress in the treatment, the therapies are associated with several limitations, such as manufacturing difficulties, risk of relapse, or the impossibility of generating clinically applicable doses of CAR-T cells from previously treated patients. Moreover, there are two main risks associated with the use of autologous CAR T cell therapy: cytokine release syndrome (CRS) and neurotoxicity (3). On the other hand, allogeneic CAR-T cells raise safety concerns, as the infusion of donor-derived cells may cause graft-versus-host disease (GvHD) or trigger graft rejection (4).
An appealing strategy to generate “off-the-shelf” allogeneic CAR-T cells with limited potential of triggering GvHD is to utilize umbilical cord blood (UCB) as a source of T/NK cells. To date, UCB has been successfully used as a hematopoietic stem cell (HSC) source in allogeneic HSC transplantations (alloHSCT) with lower rates of GvHD in transplant recipients compared to conventional sources (bone marrow or mobilized stem cells) (5). Crucially, the UCB alloHSCT procedure permits higher human leukocyte antigen (HLA) disparity between a donor and a recipient (6). These qualities are attributed to the naïve phenotype of cord blood T/NK cells (5, 7).
In this review, we discuss the pros and cons of different cell sources for adoptive CAR-T cell immunotherapy. We focus on the therapeutic application and summarize the results from the ongoing clinical trials regarding the use of UCB to provide the perspective for future research teams and clinicians interested in this field. Finally, we propose a novel approach to utilizing CB banks for CAR-T cell manufacturing.
One of the key aspects determining CAR-T cell therapy’s effectiveness and long-term remission is the in vivo stability of transferred cells. It has been shown that the persistence of CAR-T cells is correlated with the phenotype of the modified T cells and that prolonged detection of CAR-T cells is associated with a better response even in patients with high-grade cancer (8). Moreover, the differentiation stage of adoptively transferred T cells affects their proliferation and survival which strongly correlates with their antitumor activity (9). Depending on the stage of cell differentiation, T cells can be divided into naïve T cells (TN), stem cell memory T cells (TSCM), central memory T cells (TCM), memory effector T cells (TEM), and effector T cells (TEF) (10).
Naïve T lymphocytes are characterized by high proliferative ability with the surface expression of CD62L, CCR7, and CD45RA isoform and a lack of activation markers like CD25, CD44, CD69, or CD45RO isoform (11). Lymphocytes are considered naïve until they are presented with an antigen that activates TN lymphocytes to proliferate and differentiate into (memory T cells) TCM and/or TEM lymphocytes (12). TSCM is a subpopulation of the least differentiated of the memory T cell subset. Surface markers expressed by TSCM include both naïve T cell markers, such as CD45RA, high levels of CD27, CD28, CD127, CD62L, and CCR7, as well as markers of memory T cells, such as CD122, CD95, or CXCR3 but do not include CD45RO (13, 14). They have stem cell-like self-renewal capacity and can regenerate all TCM and TEF cell populations (15, 16). Compared to other subpopulations, the T subset of TSCM develops a faster response to antigenic stimulation and may persist for a long time (12). TCM cells guard lymph nodes, providing central immunosurveillance against known pathogens. Compared to naïve T cells, memory T cells respond more quickly and deliver an early batch of cytokines when stimulated by a specific antigen (17). TCM secrete tumor necrosis factor α (TNF-α), interleukin 2 (IL-2), and co-express L-selectin and CCR7, as well as CD45RO, CD62L, CD27, but not CD45RA (18). Next population - TEMs produce many inflammatory cytokines such as TNF-α and interferon γ (IFN-γ) as their most important role is related to the activation of the immune response taking place in peripheral lymphoid organs (19). Phenotypically, TEM cells are positive for activation markers CD38, CD69, and CD25, they sometimes co-express CD45RA and CD45RO at high levels, however, are negative for CD62L, CCR7, and CD31 (20). Finally, the TEF subpopulation contains fully differentiated T cells, responsible for strong effector functions (12). TEF overexpress several homing receptors to migrate to inflammation sites like CCR5, LFA-1, as well as CD45RA, CD95, CD122, and KLGR1 but do not express CD45RO, CCR7, CD27, CD62L or CD28 (21, 22). Additionally, TEFs exhibit limited expansion, low self-renewal, and survival capacity as a consequence become exhausted quickly (23) (Figure 1).
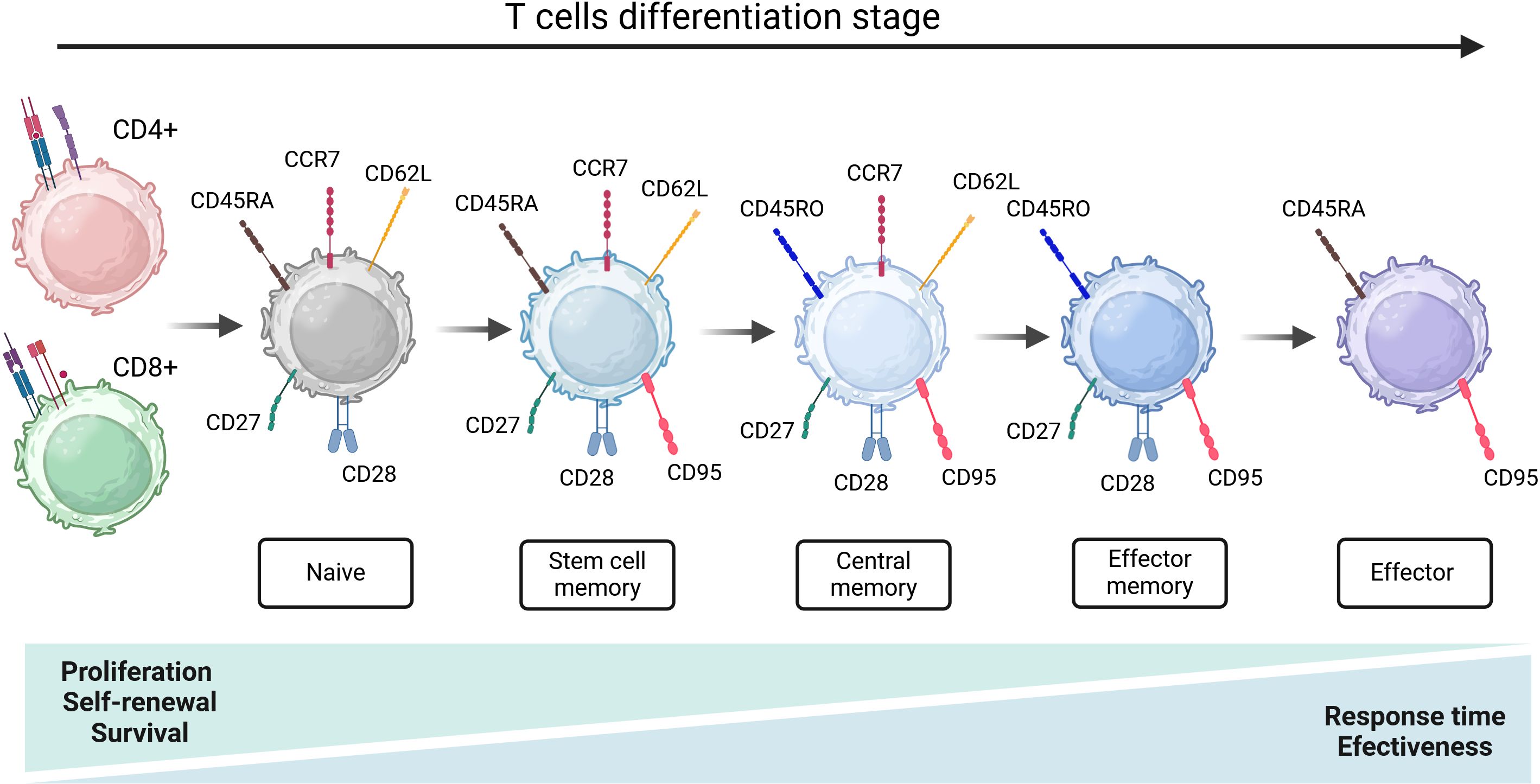
Figure 1. Linear model of stages and characteristics of T cells maturation. Following activation, naïve T cells differentiate into memory and effector cells. This differentiation is marked by the dynamic changes in the expression of CD45RA/RO, CCR7, CD28, CD62L, and CD95 antigens. Created in BioRender.
Naïve CD4+ T cells differentiate into several effector T cell subsets characterized by the ability to produce specific cytokines, namely helper T cells (Th) Th1, Th2, Th9, Th17, Th22, follicular helper T cells (Tfh) and regulatory T cells (Treg). Cell differentiation is regulated by the cytokines present in the environment and induced upon T cell receptor (TCR) signaling (24). Foreign antigens from infected or unknown cells are presented to T cells in the context of specific major histocompatibility complex I (MHC class I) called HLAs. Each cell subset has particular characteristics and releases cytokines that determine its function, such as Th1 and Th2 cells which produce IFN-γ and IL-4, respectively (25). CD4+ T cells are differentiated through the cytokines upon induction by master transcription factors such as T-bet, GATA-3, RORγt, Bcl6, or Foxp3 (26).
Studies showed that both CD4+ and CD8+ CAR-T cells derived from TCM and TM populations can be involved in killing malignant cells and achieve better results than those from TEM (27). Results from the clinical trials indicate that CAR-T cell products rich in TN, TSCM, or TCM are required for the sustained in vivo persistence of adoptively transferred CAR-T cells due to their ability to proliferate (28, 29). Moreover, the combination of the most potent CD4+ and CD8+ CAR-T cell subsets has a synergistic antitumor effect in vivo (30). These results demonstrate that naïve and TSCM/TCM cells are the most important players in CAR-T cell therapy due to their sustained proliferation and persistence in vivo.
Comprehending the phenotype of manufactured CAR-T products is critical to mitigate adverse events, as well as to improve antitumor effectiveness, such as CAR-T dosing or activation. The phenotypic profile of infused CAR-T cells has not only been linked to the cells’ persistence but also to the long-term outcomes of the treatment (18, 31).
CARs are created using the main signaling components of the T cell receptor and costimulatory molecules. In general, CAR consists of the extracellular single-chain variable fragment (scFv) specific for a cancer marker, and three main domains: an extracellular antigen-binding domain derived from a tumor-specific monoclonal antibody, a transmembrane domain that anchors the CAR to the T cell (derived from CD3, CD4, CD8 or CD28 proteins) and an intracellular T cell activation domain of CD3ζ with one or more costimulatory domains (usually from 4–1BB or CD28) that is required for full T cell activation (32–35).
Since the introduction, several CAR-T cell generations have been developed. First-generation CARs had only one signaling domain, typically the CD3ζ that proved not to be sufficient due to the limited persistence and signaling ability of the cells (36). To improve that, the second generation of CARs included a co-stimulatory signaling domain (usually CD28 or 4-1BB) localized proximal to the membrane to enhance activation, and survival, and promote the expansion of the modified T cells (37). To achieve greater antitumor activity and increase CAR-T cell persistence, a second co-stimulatory signaling domain (e.g. CD28 or OX40) was incorporated into third-generation CARs (38). The fourth generation of CARs also known as “TRUCKs” (T cells redirected for universal cytokine killing) was based on the structure of the second-generation CARs. TRUCKs contain a transgenic expression cassette coding for a synthetic nuclear factor of activated T cell response elements to be produced and secreted upon antigen recognition, such as IL-12 (39). The secretion of cytokine enhances the immune response against cancer cells and recruits other immune cells to the tumor site (40). The next generations of CARs are often based on the second generation and contain several modifications including, truncated intracellular domains of cytokine receptors such as IL-2 with the addition of STAT3/STAT5 transcription factors binding motifs (41). These CAR constructs induce cytokine secretion through the activation of the JAK/STAT signaling pathway therefore driving CAR-T cells to remain active and generate TM cells (42).
Novel developments of CAR constructs are still being extensively studied and include logic gating with the example of tandem CAR-T cells targeting two antigens simultaneously or SynNotch-engineered CAR-T cells that use a synthetic Notch receptor to sense a specific antigen, which then triggers the expression of the CAR and many others CAR variants and augmentations (43, 44) (Figure 2). On the other hand, extensive clinical experience showed that overactivation of the peripheral T cells leads to extensive exhaustion and might not be the only possible way of CAR therapy development in the future. Genome editing is also being explored to improve next-generation CAR-T cells to overcome some of the current limitations of the therapy. To improve CAR-T cells’ persistence and function both knock-out/knock-in genes as well as epigenetic modifications are being extensively studied (45).
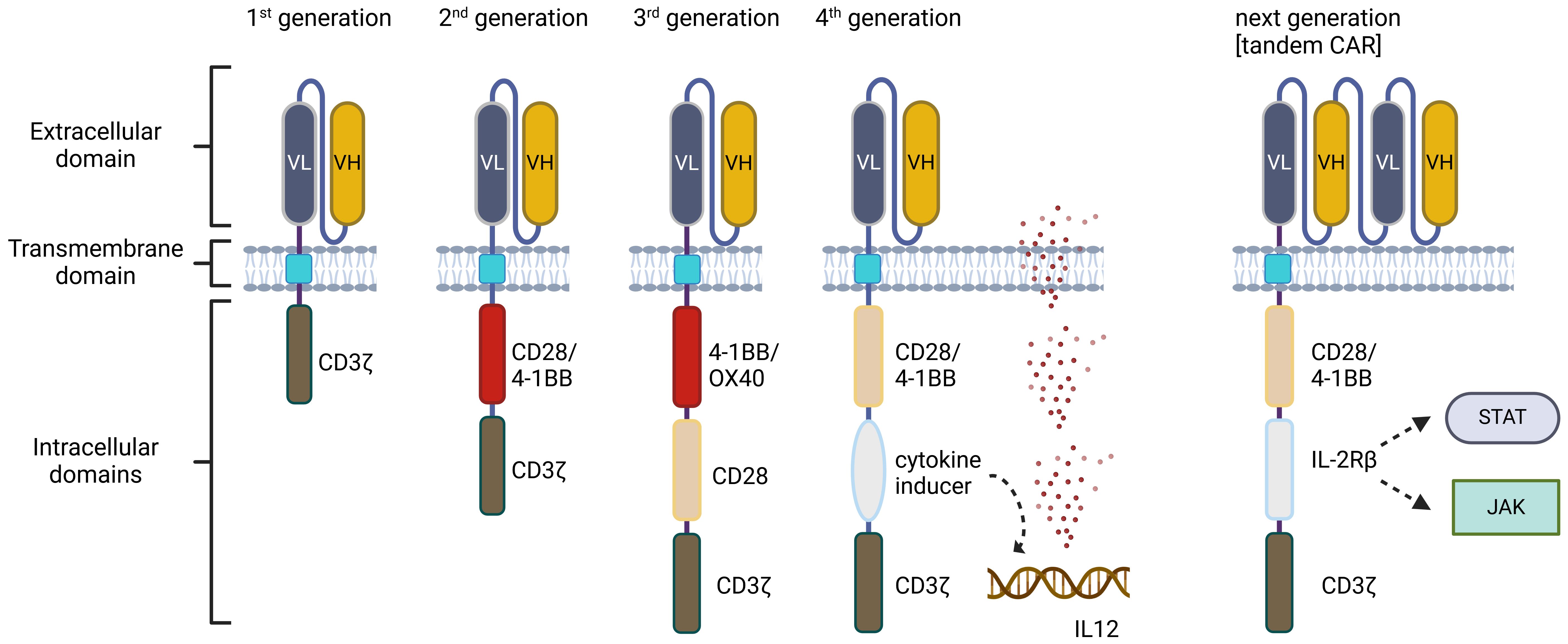
Figure 2. Generations of chimeric antigen receptors (CARs). The first generation of CARs is equipped with a single CD3ζ intracellular domain. The second generation includes additional costimulatory molecules, such as CD28 or 4-1BB. The third generation combines multiple co-stimulatory domains, like CD28-4-1BB or CD28-OX40. The fourth generation builds upon second-generation CARs, incorporating either constitutively or inducible expressed chemokines. The next generation of CARs retains the structure of the second generation but also incorporates cytokine receptors in the intracellular domain, such as the IL-2Rβ chain fragment. VL, variable region of light chain; VH, variable region of heavy chain. Created in BioRender.
While scientists and industry continue their work on novel generations of more persistent and capable CAR-T cells, the genetic modification of constructs involves certain risks and challenges, both in the laboratory during the genetic engineering process and in the patient’s body after the CAR-T cell transfer. One of the concerns relates to the safety of viral vectors. The use of viral vectors (lentiviruses or retroviruses) to deliver the CAR gene into T cells carries a risk of unintended genetic mutations or insertional oncogenesis, where genetic modification leads to the development of cancer (46). Although the FDA has called to label CAR-T cell products with a warning about secondary T cell mutagenesis, insertional mutagenesis was never proven to be a cause (47). Moreover, a recent study analyzed retrospective data from 61 centers worldwide including 3038 B-cell and non–B-cell malignancies treated with commercial or investigational CAR-T cell products in children, adolescents, and young adults. Remarkably, no cases of T-cell malignancies were reported following CAR-T cell treatment in this cohort (48).
Another risk applies to the off-target effect. Despite CAR-T cells being designed to target specific antigens on cancer cells, there is a risk of unintended attack of an antigen other than the intended one or activation of the CAR-T cells independently from their specificity (49).
Autologous CAR-T cell therapy, where the patient’s T cells are genetically engineered in vitro to express the CAR has proved its importance in clinical applications (50). Since the therapy uses the patient’s T cells, it is highly personalized and tailored to the person’s immune system. This reduces the risk of GvHD, a common complication of allogeneic stem cell therapy or CAR-T cell therapy that uses donor cells (51). Additionally, very high response rates, even in patients with limited treatment options followed by highly durable remissions have been observed after CD19-targeted CAR-T cell therapy (52–54).
However, this approach is not without drawbacks. Firstly, as it relies on autologous T cells, it depends on the nature and quality of T cells in the peripheral blood (PB) of the patient at the time of treatment. Frequently, patients have compromised immune systems and suffer from severe lymphopenia as a result of previous treatments which affects the quality and quantity of T cells harvested during apheresis (55). Besides, harvested T cells may have impaired functionality, reduced proliferative capacity, and altered phenotypes impacting their usefulness for CAR-T cell manufacturing (56).
A therapeutic dose of 107 – 109 cells is required for the treatment, hence too low T cell count or cell activation failure during the production process may be the cause of treatment failure. What is more, patient cells can be contaminated with malignant tumor cells (57). Autologous CAR-T cells may be also exposed to the patient’s tumor microenvironment before infusion. This leads to immune exhaustion, where CAR-T cells display decreased proliferative capacity, diminished anti-tumor activity, and attenuated persistence (58). Another issue is the high manufacturing cost of CARs due to the complex and personalized manufacturing process as autologous cell therapy is “tailor-made” meaning that that CAR-T cells must be generated de novo for each patient (59). Factors such as the time, materials, infrastructure, and skilled manpower required to generate CAR-T cells for each patient contribute to the astounding therapy cost. Additionally, as one batch is custom-made for one patient, the economy of scale cannot be applied (60). Time is another very important issue. In many cases, CAR-T cell therapy is the last treatment option for severely ill patients. In the case of autologous treatment, the production of personalized CARs requires a long manufacturing process. To successfully carry out the entire process of transporting a patient to the manufacturing facility and back to the treatment facility requires a thorough complex transport network, planning, and a skilled workforce that is highly prone to delays. This can result in an increase in treatment time of three to five weeks, which may be intolerable due to the progression of the patient’s disease (61). Recent pre-clinical studies developed a new platform called FasT CAR-T with a shortened next-day manufacturing time. The procedure was reported successfully with a median of 14 days from apheresis driven by 7 days of rapid sterility testing, which is a significant improvement from the median of 45 days. Although the first-in-human clinical study showed early promising efficacy in B-ALL patients, more data on additional patients and longer follow-up are needed to further evaluate the efficacy of this novel CAR-T cell therapy (62, 63). Lastly, while CAR-T cell therapy has demonstrated clinical efficacy in hematologic malignancies, severe toxicities such as CRS have also been reported (64). CRS is characterized by the release of cytokines (mainly IFN-γ, IL-6, IL-10, TNF-α, and IL-2) in response to the therapy. That causes systemic inflammatory responses resulting in flu-like symptoms and, in severe cases, organ dysfunction (65). The precise characterization and understanding of the composition of the infused T cells both in terms of T-cell memory differentiation and CD4/CD8 ratio are fundamental aspects of CAR-T cell therapy development and implementation. Collectively, directly impacts the antitumor activity, safety, and consistency, ultimately leading to better therapeutic outcomes (30, 66).
Donor-derived allogeneic CAR-T cell therapies offer many advantages compared with autologous treatments. One of the benefits of the allogeneic approach is the possibility of obtaining a higher quality of donor cells, which directly translates into the quality of the treatment (67). Healthy donors can be pre-screened for desirable characteristics of T cells like an appropriate number, phenotype, or optimal CD4:CD8 ratio that will minimize manufacturing errors. Selection of donors that have a high percentage of T cells is recommended, as contamination with non-T cell populations may affect downstream production (68). In contrast to autologous products, a large number of allogeneic CAR-T cells can be generated in a single manufacturing run making the production process cost-effective. Moreover, during production, standardized T-cell products from healthy donors are being created ahead of time, making it possible to generate many therapeutic doses from one manufacturing lot making it readily available for patients (69). Allogeneic CAR-T cells as “off-the-shelf” can reduce “vein-to-vein” time, decrease the risk of manufacturing failures, and alleviate logistical challenges (69). Eliminating treatment delays by reviving pre-frozen allogeneic CAR-T cells whenever the patient needs them may also improve the clinical outcomes of the therapy (70). Moreover, allogeneic CAR-T cell therapy does not carry the risk of contamination with cancer cells as T cells are manufactured from the healthy donor’s blood. Allogeneic CAR-T cells may also have a lower risk of immune exhaustion as they are not exposed to the patient’s tumor microenvironment or cytotoxic therapies prior to infusion (71).
Although allogeneic CAR-T cell therapy has numerous advantages, one of the most significant drawbacks is the risk of the development of GvHD (64). A condition where the T cell αβ receptor (TCRαβ) on the infused CAR-T cells recognizes cell surface HLA class I and class II molecules on the recipient’s cells and attacks them. Therefore, ensuring compatibility between the donor and recipient’s HLA types is essential to reduce the risk of GvHD, which may limit the availability of suitable donors (72). Until now, it has been established that for successful HLA-matching, the patient and potential donor should have high-resolution HLA typing using DNA-based methods for HLA-A, B, C, DRB1, and DQB1 (73).
Recently studies proposed a feasible alternative for HLA match assessment in terms of CAR-T cells derived from PB T cells. The higher efficacy of HLA-matched CAR-T cells (minimum of a 4/6 match to the patient) compared to HLA-haploidentical (sharing one haplotype) CAR-T cells has been observed (74). Additionally, results showed that haploidentical PB CAR-T cells induced only transient or no reduction in PB leukemia cell number without significant CAR-T cells expansion, which suggests rejection (74).
Achieving HLA compatibility is crucial not only to reduce the risk of GvHD but also to prevent CAR-T cell rejection. For that reason, patients receiving allogeneic CAR-T cells often require immunosuppressive drugs, which can weaken the anti-cancer immune response and increase susceptibility to infections (69). Hence, genome editing has been applied to prevent GvHD. Studies showed that the elimination of expression of the endogenous TCRαβ receptors using either zinc finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs) successfully prevented GvHD without compromising CAR-dependent effector functions (75, 76). In two phase 1 clinical trials in pediatric and adult patients with late-stage relapsed or refractory B-cell acute lymphoblastic leukemia (NCT02808442 and NCT02746952), TALENs were used to simultaneously disrupt cell surface expression of TCRαβ and CD52. Results proved that genetically modified CAR-T cells caused only minimal GvHD with no detrimental expansion of unedited TCRαβ-positive T cells (77). Another phase 1 study (NCT04227015) has utilized the alternative genome editing technique CRISPR/Cas9 to disrupt the TCRα subunit constant (TRAC) region previously shown to lead to loss of TCRαβ expression (78). Obtained data demonstrated that CD19/CD22-targeting CAR-T cells with a CRISPR/Cas9-disrupted TRAC region and CD52 gene infused in patients with relapsed/refractory acute lymphocytic leukemia showed manageable safety profile and notable antileukemia activity, with no GvHD observed (79).
Nevertheless, complications and toxicities may arise as a result of gene editing. This may include on- and off-target effects and issues such as CRS or neurotoxicity, especially when editing inadvertently causes the CAR-T cells to become more aggressive or activate faster than desired (80). Another challenge of allogeneic therapy combined with more extensive genetic modification is the risk of chromosomal aberrations. No currently available manufacturing process can provide 100% certainty that no aberrations are present in a therapy’s potentially hundreds of millions of cells. In 2021, the FDA put a 4-month hold on clinical trials of all allogeneic projects due to chromosomal abnormality found in a lymphoma patient in the Alpha-2 trial of the CD19-directed allogeneic CAR-T trial. Although the investigation has not shown any clinical consequences, such as aberrant proliferation or leukemogenesis of engineered cells, the issue of chromosomal aberrations risk is to be addressed (81).
CAR-NK cells are being investigated as an alternative cell source in allogeneic cell therapies as they are crucial in immune surveillance of invading viruses and killing cancer cells without the need for tumor-specific antigen presentation (82). Except for the CD3 signaling domain, as well as CD28 or 4-1BB as a co-stimulatory domain, CAR-NK cells enhance their cytotoxic capacity and cytokine production through additional co-stimulatory molecules, that is: NKG2D and CD244 (2B4) (83). The initial success of NK cell therapies has prompted further investigation into current allogeneic NK cell products, yielding encouraging outcomes in clinical trials. First, since NK cells do not recognize targets presented by the HLA system, therapeutic CAR-NK cells can be cultivated from donors with various genetic backgrounds and applied in diverse recipients without the concern of inducing GvHD (84). Second, due to a different spectrum of secreted cytokines, CAR-NK therapy does not cause CRS, therefore it is considered safer than CAR-T cells treatment (85). Third, NK cells are characterized by the abundance of CD16 (FcγRIIIA), which serves as a receptor for IgG1 and IgG3 and is essential for NK cell-mediated antibody-dependent cell-mediated cytotoxicity ADCC (86). Hence, they can also productively eliminate tumor cells in a CAR-independent manner through their stimulatory and inhibitory receptors which form the basis for the dual anti-tumor activity of CAR-NK cells.
In recent years several studies explored different approaches to genetically engineer CAR-NK cells. Especially in the treatment of T-ALL CAR-NK cell therapies targeting CD3, CD5, and CD7 have shown significant anti-tumor cytotoxicity both in vitro and in vivo (87–89). As of January 2025 currently ongoing clinical trials on CAR-NK focus not only on CD19 target but also on CD123 clinical trials.gov; ID (NCT05574608) and clinical trials.gov; ID CLL-1(NCT06307054) in acute myeloid leukemia (AML) or dual targeting of CD19 and CD70 clinical trials.gov; ID (NCT05842707) in B cell Non-Hodgkin lymphoma and CD33 and/or FLT3 in AML clinical trials.gov; ID (NCT06325748).
As pre-clinical and clinical data support the potential of CAR-NK cell therapies, still several challenges need to be addressed before clinical application. One of the main barriers to CAR-NK cell therapy is the lack of persistence in vivo in the cytokine support deficiency (90). Additionally, due to the inhibitory tumor microenvironment, CAR-NK ability of homing and infiltration is limited (91).
Over the past years, UCB transplantation has become an alternative therapeutic option for patients with cancer or genetic diseases for whom an HLA-matched family or unrelated adult PB stem cells or bone marrow donor has not been found (92). UCB therapy is currently used for the treatment of such hematological diseases as myelodysplastic syndrome (MDS), AML, aplastic anemia (AA), or ALL (93–96). It is the unique qualities of UCB compared to PB or bone marrow that make the therapy encouraging. In contrast to PB cells, UCB cells are characterized by higher proliferation, and expansion due to the higher percentage of hematopoietic stem cells, with the less mature phenotype of T cells and NK cells (97, 98). Crucially, the UCB T-cell population is characterized by a more naïve phenotype as it comprises approximately 85% naïve CD45RA+/RO- T cells and only 6% memory T cells in comparison to 39% and 50% in PB, respectively (99) (Figure 3). Remarkably, naïve T cells of UCB differ from PB naïve T cells (100, 101). UCB naïve T cells have higher proliferative and activation capacities, as well as delayed exhaustion compared to PB (100). They do display comparable activation levels with reduced secretion of cytokines such as IL-2, IFN-γ, and TNF-α upon CD3/CD28 targeted stimulation. Moreover, naïve UCB T cells are characterized by lower expression of CD40L and perforin thus, they exhibit lower cytotoxicity than PB cells. Furthermore, compared to PB, UCB contains a relatively high frequency of distinct CD4+CD25 bright subset of regulatory T cells. Thus, the likelihood of co-purification of activated or memory CD4 + 25+ T cells in UCB compared with PB is decreased. Last but not least, UCB cells display elevated self-renewal potential as well as multiple cell divisions attributed to longer telomeres and overexpression of transcription factors like NF- κB (102).
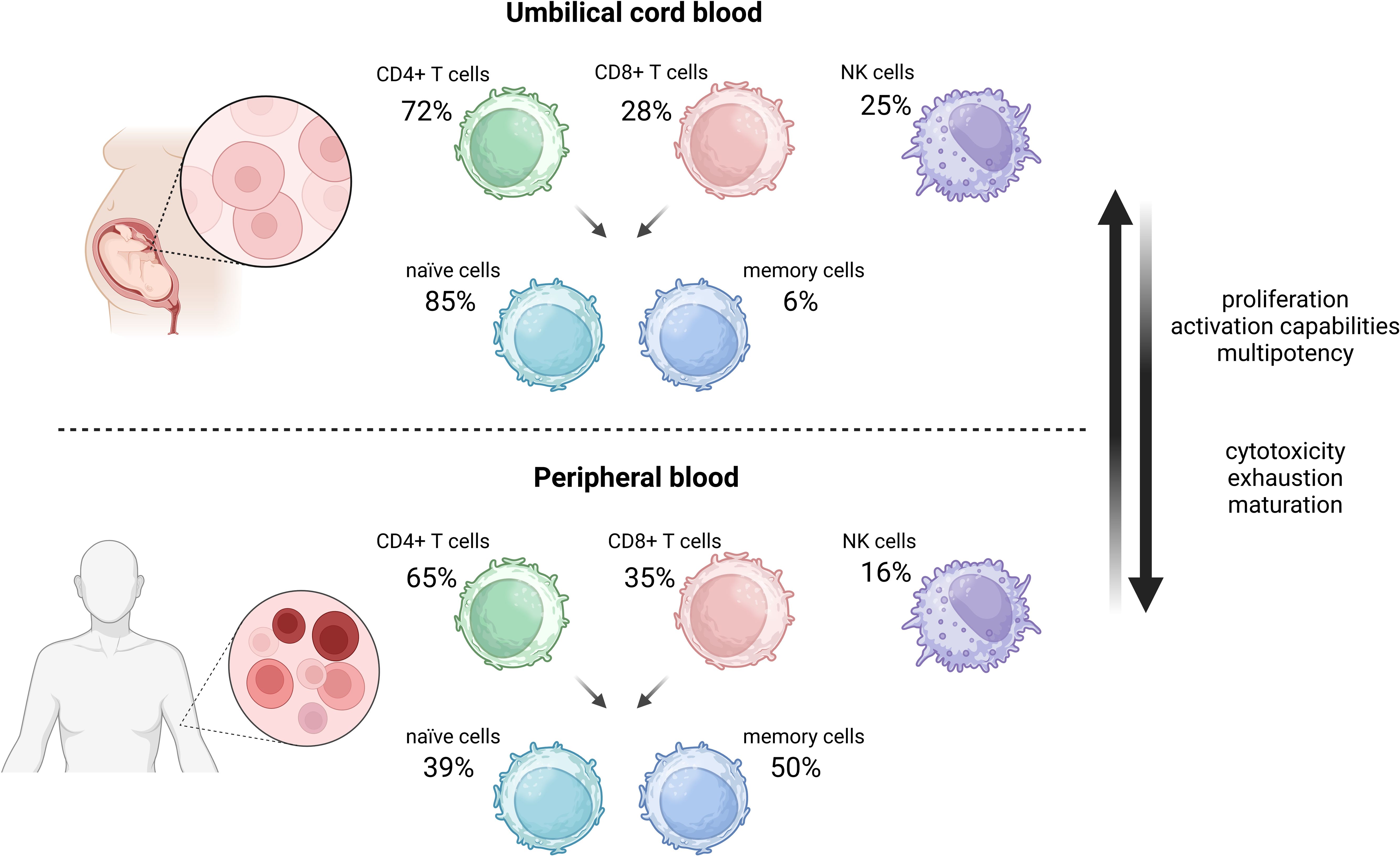
Figure 3. Comparison of umbilical cord blood and peripheral blood cells composition in terms of CAR-T cells application. Created in BioRender.
Therefore, one of the major advantages of UCB treatment is its safety. Compared to other donor cell resources, UCB collection not only painless but also carries a lower risk of viral infections and somatic mutations, which are the primary causes of morbidity following transplantation (103). Moreover, in the case of allogeneic transplantation, UCB allows for less stringent HLA matching criteria (4 of 6 HLA loci, considering a low resolution for HLA-A and HLA-B and high resolution for HLA-DR), which may be especially important for members of racial and ethnic minorities, increasing the inclusiveness of the therapies (104). UCB grafts could also reduce the mortality of transplant as due to its low immunogenicity decrease in both acute and chronic rates of GvHD are observed (105). Studies showed that PB consists higher concentration of CD8+ T cells comparing to UCB (97, 106). Given that GvHD is primarily driven by alloreactive donor CD8+ T cells, this difference in composition may help to explain the higher frequency of the chronic GvHD observed for example when the PB is used as a stem cell source for hematopoietic stem cell transplantation (107). Additionally, PB contains more mature monocytes, lower counts of plasmacytoid dendritic cells (pDCs), and reduced numbers of NK 56bright16− cells, further contributing to increased GvHD risk, when compared to UCB (108). At a molecular level, UCB T cells exhibit impaired nuclear factor of activated T cells (NFAT) signaling as well as lower activation of the NF-κB pathway that leads to reduced production of several pro-inflammatory cytokines, further decreasing GvHD frequency and intensity (109–111). On the other hand, there are several drawbacks to UCB cell usage. To begin with, in the case of autologous transplant and treatment of cancer or genetic disorders, stem cells derived from UCB may not be suitable as they might encompass the same abnormal cells or genetic variants that caused the onset of the disease at first (112). Furthermore, very often the amount of stem cells harvested from UCB is insufficient for the treatment of adults (113, 114). The last concern is the longer engraftment of CB stem cells (21-25 days) compared with adult donor cells (20 days) associated with higher hospital costs (115). To overcome the obstacles mainly related to the limited number of cells available in a single unit, several approaches have been investigated, including double UCB transplantation, in vitro UCB culture expansion, or direct intrabone transplantation of UCB cells (116–118).
Nowadays, haploidentical (haplo)-cord transplantation represents a promising approach to allogeneic transplantation. This technique involves combining two stem cell sources: UCB and CD34+ cells from haploidentical donors (119). Haplo-cord transplantation has shown several benefits such as rapid availability and less stringent HLA-matching requirements compared to unrelated traditional adult grafts as it combines the benefits of both haploidentical and UCB transplants (120). Since the haploidentical component is derived from a partially matched family member and the UCB component relies less on perfect HLA matches because of its naïve immune cells, the two stem cell sources complement each other and yield better outcomes than using either source alone. Furthermore, this approach is associated with accelerated neutrophil and platelet recovery, lower risks of acute and chronic GVHD, and excellent graft-versus-leukemia (GVL) effects (121). Additionally, when combined with reduced-intensity conditioning (RIC), haplo-cord transplantation has shown decreased susceptibility to delayed opportunistic infections, shortened hospital stays, and favorable long-term outcomes (122).
As UCB T cells exhibit distinct naïve profiles, they recently became an attractive source for CAR-T cells as an alternative to autologous cell treatment, potentially broadening access to the therapy. UCB as a CAR-T cell source offers advantages such as the shift of CD4+25+ T cells toward a Treg suppressor cell phenotype that may contribute to a lower incidence of GvHD after CAR-T cell infusion (123). Furthermore, UCB cells express significantly lower markers of exhaustion such as programmed cell death-1 (PD-1), T-cell immunoglobulin, and mucin domain-containing protein-3 (TIM-3) and lymphocyte activation gene-3 (LAG-3) compared to PB T cells, allowing for long-term persistence and efficiency (124, 125). However, the use of UCB cells for CAR-T therapy raises several challenges. Firstly, due to their naïve state, UCB-derived CAR-T cells may require longer expansion time. While studies have reported successful generation of CAR-PB-derived CAR-T cell products in as little as 8 days, UCB-derived CAR-T cells may need more time to achieve comparable therapeutic dose (126, 127). It is unclear for now how the “fast-CAR” approaches would be working in UCB case as well. What is more, specific phenotype distribution plays a significant role in establishing the proliferative and expansive potential of UCB-derived cells (126). Therefore, the specific phenotype of naïve UCB-derived T cells must be considered when designing and interpreting UCB CAR-T studies. These include protocols for activation and cytokine stimulation in the manufacturing process, the ability to differentiate into effector cells, and cytotoxicity. Current manufacturing of CAR-T products involves induction of cell activation and expansion by targeting CD3/CD28 molecules and coincubation with interleukin (IL) 2 or IL-7 and IL-15. Notably, it is reported that IL-2 stimulation may cause cellular exhaustion, whereas IL-7 and IL-15 contribute to a higher percentage of memory cells. However, this evidence applies to conventional, PB-derived T cells so a more tailored and detailed study of UCB-derived T-cell activation pathways should be performed (128). Additionally, there is a question of whether the manufacturing process could deprive UCB T cells of their naivety features while providing them with sufficient anti-tumor cytotoxicity. According to studies, including our data, cord blood T cells retain a phenotype similar to T cells derived from peripheral blood upon CD3/CD28 stimulation and cytokine priming (129). Some differences could be attributed to the lower expression of exhaustion markers, nevertheless, the cytokine repertoire used in the manufacturing process significantly contributes to the phenotype of the resulting CAR-T cells (126).
Another concern is whether UCB-derived CAR-T cells could perform effector functions. Since T cells from UCB are more naive and less experienced in fighting infections or cancer compared to T cells from adult donors, it may take longer to adapt and respond effectively to cancer cells, potentially delaying the therapeutic effect. The seminal study completed by Serrano et al. proved that these cells are capable of CD19-specific cytotoxicity and was followed by results from other research groups (126, 130). However, in parallel with reports of satisfactory anti-tumor efficacy, concerns regarding adverse effects in allogeneic settings arise. Moreover, as UCB contains a relatively small number of T cells compared to PB, this limited T cell quantity may be insufficient for generating a therapeutic dose of CAR-T cells, especially for adult patients or those with high tumor burdens. Therefore, ex vivo expansion may be required to proceed with cellular transfer (131). The efficiency of the viral transduction, a crucial step in CAR gene introduction is yet another concern. Studies have shown variable transduction rates between units (15-85%), possibly due to T cell differentiation profiles (126, 132).
Besides the hurdles, several studies showed the feasibility and great therapeutic potential of UCB-derived CAR-T as such. Augmentation of attributes with targeted reduction of defective features should be in focus of new adoptive therapy approaches. Table 1 provides a comprehensive comparison of PB-derived and UCB-derived CAR-T therapies, highlighting key differences in aspects such as time, cost, cell quality, and risk profiles across autologous and allogeneic approaches.
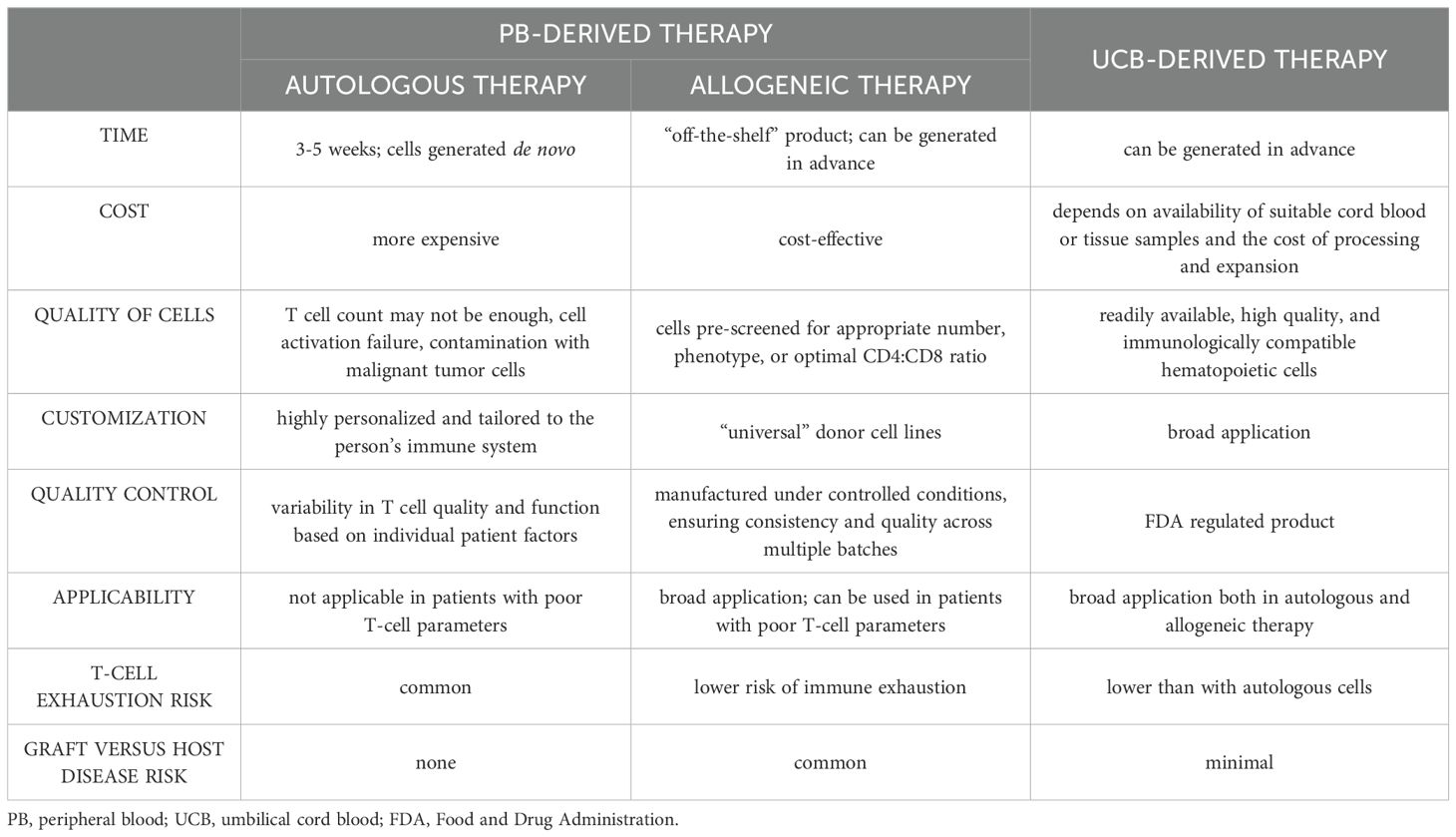
Table 1. Comparison of different approaches to generate CAR-T cells.
As the immunology of UCB-T cells supports their use as a source for CAR-T manufacturing, several both in vitro and in vivo strategies have been tested in preclinical settings worldwide.
First of all, it has been proven that UCB-T cells cells can be successfully engineered to express CAR receptors capable of recognizing specific markers (133). Serrano et al. first reported that naïve UCB-T cells cells could be engineered into CD19 CAR-T cells (130). Encouraging results showed a reduction in tumor bulk in the in vivo arm of the study, with 60% of mice achieving complete remission with no adverse reactions following CAR-T infusion observed. Later, Huang et al. engineered 2nd generation anti-CD19 product from UCB with what seems to be an exquisite transposon-based mechanism (134). In vitro assays on B-cell lymphoma and B-cell ALL cell lines showed the efficient killing of tumor cells by UCB-derived CAR-T cells. The researchers also showed that UCB-derived CAR-T cells were capable of short-term (10 days) anti-tumor efficacy in vivo regardless of IL-2 injections. Nevertheless, there was no mention of off-target toxicities (134). Notably, in a direct comparison between CAR-T cells manufactured from PB, UCB-derived CAR-T cells presented similar anti-tumor responses completely eradicating B-cell ALL blasts over 5 days of coculture in the in vitro assay (135).
Another study of UCB CAR-T cells targeting B-cell ALL was conducted by Liu et al. The researchers compared the efficacy of autologous CAR-T cells and donor-derived, either PB or UCB CAR-T cells (136). Unsurprisingly, both donor-derived PB and UCB CAR-T cells showed increased proportions of cells displaying an immature phenotype (including naïve T cells and central memory T cells) as well as induced better responses in vivo than ALL patient-derived CAR-T cells. The survival analysis showed that the mice in the CB CAR-T and PB CAR-T groups survived longer than those in the PB (patient) CAR-T group with the median survival times of the CB CAR-T, PB CAR-T, and PB (patient) CAR-T groups 51 (39–51) days, 51 (46–51) days, and 32 (25–33) days (p < 0.05), respectively. Moreover, CAR-T and tumor cell proportions in PB revealed that on days 14 and 28, mice in the UCB CAR-T and PB CAR-T groups had higher CAR-T cell expansion and lower tumor burden compared to those in the ALL CAR-T group, making it a promising source for allogeneic CAR-T cells.
Addressing the choice of the most suitable costimulatory domain for UCB-derived CAR, the study Yu et al. elucidated the anti-tumor capacity of UCB-derived CAR with 4-1BB co-stimulatory domain (137). Obtained results showed the target-specific killing of CD19+ T cell lymphoma cells in vitro (over 50% dead cells in 1:1 ratio up to 85% dead cells in 10:1 ratio) as well as inhibited tumor progression in vivo (<1000 mm3 vs >1500 mm3 tumor volume in controls). UCB CD19-CAR- T cells were also associated with minimal GvHD as no diarrhea, rash, or jaundice, which are common symptoms of GvHD, were observed during the observation period. Furthermore, Tammana et al. indicated that incorporating the 4-1BB domain yielded better responses in vivo than CD28. The results showed that the construction of 3rd generation CAR-T cells with both (4-1BB and CD28) domains was associated with even more potent anti-tumor response compared to CAR-T cells with single 4-1BB construct in mice CD19(+) leukemia and lymphoma tumor models, suggesting a synergistic role costimulation in engineering antileukemia UCB effector cells. A systemic NOD/SCID mouse model with established Raji tumors showed that UCB T cells expressing both CAR constructs exhibited significantly improved tumor control and reduced bioluminescence intensity compared with 4-1BB CAR and GFP controls, with bioluminescence intensity in the 4-1BB and CD28 accounting for one-third of the value in the 4-1BB group on day 8 (132). A unique approach was adopted by Pegram et al. The researchers constructed a novel anti-CD19 CAR construct (armored CAR), programmed to secrete IL-12 to interfere with the immunosuppressive cytokine profile within the tumor (138). Obtained results suggested that IL-12 is an important factor for phenotypic changes, such as increased CD62L, CD28, GzmB, and IFNγ. In accordance with previous studies, the UCB-derived CAR-T cells showed promising anti-tumor efficacy both in vitro and in vivo as they enhanced anti-tumor efficacy compared to the CD19 CAR alone. Notably, the transfer of UCB-T cells secreting IL-12 resulted in increased survival of CD19+ tumor-bearing mice (>40 days of survival vs <40 days of survival compared to controls; *P < 0.05) without a need for pretreatment (irradiation) or IL-2 support. Similar results were obtained while analyzing UCB-derived CAR-NK cells. The study by Herrera et al. highlighted the higher anticancer activity of UCB-derived CAR-NK cells in the treatment of primary chronic lymphocytic leukemia (CLL) compared to CAR-NK from adult cell sources. The study showed that UCB CAR-NK cells exhibited a more stable cell count per unit and demonstrated responsiveness to various interleukins to enhance their in vitro expansion, tumor cell killing activity, and promote prolonged cellular survival (139).
To date, four studies investigated the efficacy of UCB-derived CAR-T cells against cell lines other than B-cell lineage. Ma et al. investigated in vitro efficacy of uncommon HLA-A-targeting CAR T cells in AML (140). The group developed TCR-like monoclonal antibody (8F4) UCB-derived CAR-T cells that specifically recognized the PR1/HLA-A2 on the surface of AML cells and were capable of killing leukemia cell lines and primary AML blasts in an HLA-A2-dependent manner (>60% of killing and >40% of killing respectively in 4:1 effector: target ratio). A more advanced study was completed by Caël et al., who generated anti-CD123 UCB CAR-T cells, compared them with PB counterparts in vitro, and assessed the efficacy in vivo (126). Using blastic plasmacytoid dendritic cell neoplasm models they proved that UCB-derived CAR-T cell product retains the pool of less differentiated cells after nine days of expansion using IL-7 and IL-15. Notably, UCB CAR-T CD4+ T cells exhibited a less differentiated phenotype compared to PB CAR-T cells, with a higher proportion of TSCM and TCM (68.1% vs. 31.8%, p < 0.001) and a lower proportion of TEM and TEMRA (31.8% vs. 68.2%, p < 0.001). While UCB and PB CD8+ T cells showed no significant differences, UCB CAR-T cells trended toward a less differentiated profile, with 49.2% vs. 32.8% TSCM + TCM (p = 0.055) and 51.0% vs. 67.1% TEM + TEMRA (p = 0.064). Additionally, UCB-derived CAR-T cells presented comparable efficacy to PB product (94.3 ± 3.8% and 93.8 ± 3.9% cytotoxicity at E:T ratio 1:1 respectively) in vitro and contributed to significantly better overall survival (>120 days vs around 40 days in controls; P=0.004) in leukemia modelin vivo. Moreover, they demonstrated that thawed or fresh UCB as a source for CAR-T manufacturing does not affect the product’s functionality. In a study conducted by Pinz et al., 3rd generation UCB CAR-T cells were used to target peripheral T-cell lymphomas (PTCLs) in vitro (141). Although it was shown that UCB-derived CD4 CAR-T cells efficiently suppressed the growth of lymphoma cells in vitro (with the overnight elimination rate of 38, 62 and 85% at E:T ratios of 2:1, 5:1 and 10:1 respectively) while also significantly prolonging mouse survival (>30 days vs ~20 days in controls), unfortunately, the study provided little information concerning UCB efficacy. Finally, Olbrich et al. tested the effectiveness of UCB CAR-T cells against human cytomegalovirus (HCMV)-infected cells with promising results showing high on-target effect (~30- 40% of dead target cells) and cytotoxicity in vitro (142). In the in vivo model, one week after administration, response to CAR-T cell therapy was observed in five out of eight mice, defined by significant reduction of the bioluminescent signal in relation to untreated controls. More importantly, none of all the treated mice showed adverse clinical symptoms such as loss of body weight, observable change in behavior, eczema or GvHD.
In summary, all the above-mentioned studies provided evidence of the anti-tumor efficacy of allogeneic CAR-T cells generated from UCB. However, although the feasibility of the manufacturing process as well as the anti-tumor cytotoxicity of CAR-T cells has been proved, few efficacy comparison between PB and UCB cells has been made. Table 2 summarizes the clinical trials results, presenting information on CAR constructs as well as manufacturing details.
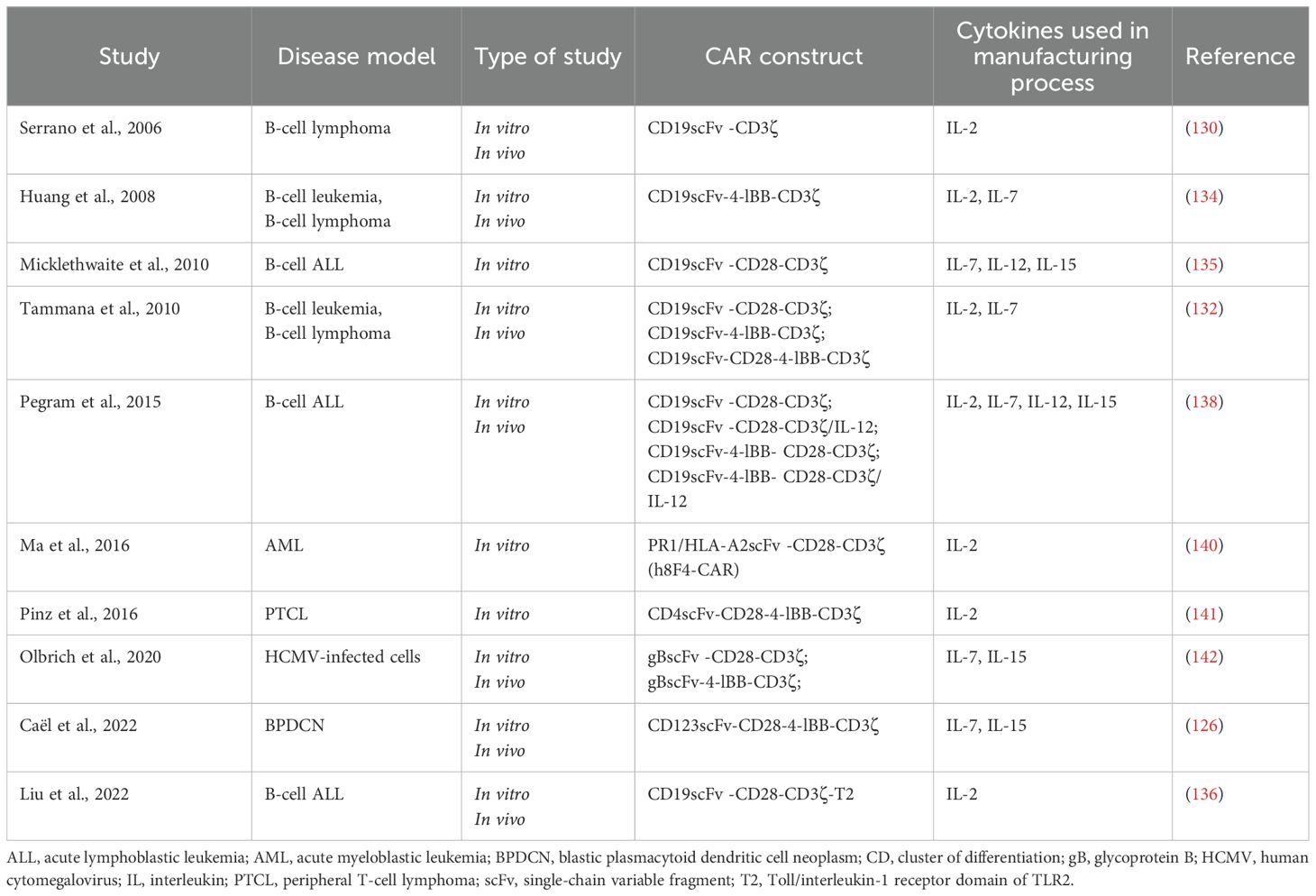
Table 2. Summary of preclinical studies investigating UCB-derived CAR T cells.
Following the limited but encouraging preclinical studies, there is a clear demand for additional advancements in CAR-T cell therapies utilizing UCB. We have searched the ClinicalTrials.gov database and found several registered clinical trials evaluating UCB CAR T cells (January 2025). A group from Henan Cancer Hospital is investigating the safety and efficacy of UCB CAR-T cells redirected against relapsed/refractory B-cell leukemia or lymphoma (clinical trials.gov; ID NCT03881774) (143). This phase 1 study involves patients with disease relapse following autologous CAR-T therapy and those unsuitable for autologous therapy due to the low quality of lymphocytes. Although the estimated completion date was January 2022, no results have been published yet. The group from the University College of London aims to investigate the manufacturability of allogeneic cord-blood derived T cells in a laboratory setting, evaluate their safety and efficacy of treatment in individuals with high-risk, relapsed/refractory B cell malignancies (clinical trials.gov; ID NCT05391490). Several studies are also evaluating the safety and efficacy of CB-derived CAR-NK therapies for treating various malignancies (clinical trials.gov; NCT05922930, NCT06066424, NCT05008536, NCT05703854, NCT05020015, NCT06358430, NCT05092451, and NCT05110742). For instance, the Xinqiao Hospital of Chongqing group is examining UCB-derived CAR-engineered NK cells in the treatment of patients with relapsed and refractory MM. The CAR incorporated in the NK cells facilitates the identification and elimination of MM cells by specifically targeting BCMA, a protein present on the surface of malignant plasma cells (clinicaltrials.gov; ID NCT05008536). Following the promising results of the phase 1 trial, Takeda Pharmaceuticals is currently conducting a phase 2 study of the safety and efficacy of UCB-derived NK CAR-T cells in adult patients with relapsed or refractory B-cell Non-Hodgkin Lymphoma (clinical trials.gov; ID NCT05020015). With several clinical trials ongoing, only a limited number of studies have demonstrated the safety and efficacy of CAR-NK therapy. Qian et al. evaluated the therapeutic potential and safety profile of CB-derived CAR-NK cells targeting CD19 in patients with relapsed or refractory B-cell cell lymphoma. The results demonstrated that CAR-NK cell therapy was well-tolerated, with no major adverse events such as CRS, neurotoxicity, or GvHD in none of the 9 treated patients. Moreover, the median progression-free survival was 9 months, and the complete responses were achieved in 55% of the cases, with a 58.33% overall response rate at the end of the study (clinicaltrials.gov; ID NCT05472558). Second study conducted by MD Anderson Cancer Centre has also been completed (clinical trials.gov; ID NCT03056339). The investigators enrolled 11 patients with CD-19-positive malignancies and treated them with next-generation anti-CD19 CAR NK cells engineered to express IL-15 and inducible caspase 9 safety switch (85). Of the 11 treated patients, eight (73%) had a response; of these patients, seven (four with lymphoma and three with CLL) had a complete remission, and one had remission of the Richter’s transformation component but had persistent CLL. Seven out of 11 patients achieved complete response, whereas none of the patients developed adverse events such as CRS or neurotoxicity despite HLA-mismatch (4/6 match in 9 patients). The study proved that UCB could be used as the allogeneic source for cellular therapies without requiring full HLA matching. Following the success of the Phase 1/2 results, the study progressed to an expansion phase (n = 26) and included 37 heavily pretreated patients with relapsed or refractory B cell malignancies demonstrating an ORR of 48.6% at both day 30 and day 100, with 1-year overall survival at 68% and progression-free survival at 32%. Rapid responses were observed at all dose levels: 100% of patients with low-grade NHL, 67% of patients with CLL without transformation and 41% of patients with diffuse large B cell lymphoma (DLBCL) achieved an OR. Most responses were complete responses (CRs), with 1-year cumulative CR rates of 83%, 50% and 29% for patients with NHL, CLL and DLBCL, respectively. Notably, CAR19/IL-15 cord blood units (CBU)-NK cells exhibited a comparable efficacy profile to autologous CAR19 T cells, while their safety profile was superior, showing no significant toxicities such as neurotoxicity, or GvHD and only one developed CRS (grade I). Although CAR-NK cells cannot be directly compared with CAR-T cells as NK cells possess distinguished immunogenic properties that decrease GvHD rates, CAR-NK cells present a promising, safe, and effective therapeutic option for patients with challenging B-cell malignancies (144, 145).
Stored in cord blood banks, UCB has established its role as an alternative source of hematopoietic stem cells for alloHSCT (146). Meanwhile, there is an increasing interest in using cord blood cells for new clinical and research applications. UCB has been regarded as an allogeneic and off-the-shelf source of NK cells as it contains relatively young and naïve NK cells, which may have greater potency and versatility in attacking target cells compared to those from adult donors (147) UCB is a rich source of progenitors and stem cells like mesenchymal stem cells (MSC), therefore UCB-derived MSCs have gained much interest for the use of potential therapeutic reasons (103, 148). Recently, it has been also proposed that a cord blood bank may function as a source for allogeneic CAR-T cell therapies (7, 126, 149).
The manufacturing of UCB CAR-T cells, as well as PB-derived CAR-T cells, consists of several key steps including T cell isolation, activation, gene modification and ex vivo CAR-T cell expansion (150). However, UCB-derived CAR-T cells face unique challenges due to the limited volume of UCB collections and their high nucleated red cell and mononuclear cell content which may complicate T cell isolation and processing (151). Additionally, because UCB is restricted by the small volume and low number of hematopoietic cells, ex vivo expansion is often required before adoptive cellular transfer (131). One remarkable advantage of UCB-derived CAR-T cells is their off-the-shelf availability, as UCB units are typically cryopreserved in UCB bank, easily accessible when needed. In contrast, autologous PB- derived T cells require patient-specific leukapheresis, leading to prolonged manufacturing and waiting time (110). What is more, allogeneic PB-derived CAR-T cells require additional genetic modifications such as elimination of the TCR receptor in order to prevent GvHD or immune rejection, a step that is not necessary in case of autologous CAR-T cells (152).
The quality of the starting material is crucial for successful CAR-T cell manufacturing, as demonstrated in autologous CAR-T therapies where baseline T cell characteristics, such as polyfunctionality, increased stemness, and reduced exhaustion, significantly impact clinical outcomes (153, 154). Similarly, for allogeneic CAR-NK cell production, donor-specific predictors of response and criteria for donor selection are vital. A study by Rezvani et al. investigated the safety and efficacy of CB-derived CAR19/IL-15 NK cells in a first-in-human phase 1/2 trial (155). The study reported day 30 overall response (OR) as the primary endpoint, with secondary objectives including day 100 response, progression-free survival, OS, and CAR19/IL-15 NK cell persistence. Among various UCB characteristics, multivariate analyses identified two key predictors of 1-year progression-free survival: a collection-to-cryopreservation time of ≤24 hours and a nucleated red blood cell (NRBC) content of ≤8 × 107 cells per CBU.
Cord blood banks offer three potential approaches for CAR-T cell manufacturing. Firstly, UCB could be used to generate genome-edited next-generation CAR-T cells, as gene editing in UCB-derived CAR-T cells offers significant advantages. Due to the naïve phenotype, UCB-derived T cells are highly sensitive to gene edits that enhance persistence and proliferation, such as upregulation of memory markers like CCR7 (156). Additionally, gene editing may improve their antitumor function through increasing of cytokine production (e.g., IL-7, IL-15) and elimination of exhaustion markers like PD-1, therefore prolonging their activity (157–159).
However, due to the smaller number of CD3-positive cells compared to PB and the elimination of alloreactivity-inducing molecules in genome editing, UCB seems to be a second choice rather than the first option. The second strategy is to use UCB-banked cells as an autologous source for individuals who had their cells banked and need higher quality T cells for conventional CAR-T cell therapy. However, in this case, although T cells in the autologous cord blood unit could be expanded, overcoming technical limitations, the likelihood of an individual banking their cells (less than a few percent of the population), makes this proposal unlikely to materialize. Finally, another approach exploits the fact that in the setting of HSCT, UCB transplant requires a lower donor-recipient HLA match due to the naivety of T cells (160). In this situation, HLA-matching requires 4/6 allele complementarity (with at least one match at HLA-A, -B, and DRB1) (160). The fact that UCB units stored in the banks are HLA-typed supports the idea of generating “off-the-shelf” HLA-matched products (146). Accordingly, designing allogeneic CAR-T cells emerges as a possible application of this strategy. A population-wide bank of allogeneic CAR-T cells could be established provided that UCB-derived CAR-T cells demonstrated an acceptable safety profile in a clinical trial. Combining conventional CAR constructs with large-scale inventory would reduce costs significantly and enable product availability at request. However, such a proposal implies the calculation of a sustainable bank size. As UCB units are HLA-typed, we can estimate the required size of UCB CAR-T bank that would cover the proper population fraction at the 4/6 or higher HLA-match level. According to the analysis of the UK cord bank performed by Querol et al., a bank size of 50,000 units provides at least one donor for as much as 98% of patients (4/6 HLA match) (161). Nevertheless, decreasing the bank size has little impact on the probability of finding a 4/6 HLA-matched donor, with 10,000 unit banks still providing more than a 90% probability of donor finding (161). In the Finnish population, a bank size as small as approximately 200 units is associated with a 90% probability of finding a suitable 4/6 donor, whereas 1700 units are enough to provide 80% coverage in a 5/6 HLA setting (162). In the Korean population, a UCB donor pool required for a 95% probability of a 4/6 HLA match is estimated to be 2150 (163). For the 5/6 HLA match, the number is approximately 51,000 (163). It seems reasonable that the higher the homogeneity of the population, the lower the required size of the allogeneic CAR products. However, HLA allele and haplotype distribution within cord blood banks also play an important role, as uniformly distributed cord blood bank provides appropriate coverage while retaining a relatively small bank size (162).
Apart from biological arguments, more logistic and technical factors should be taken into account when considering cord blood banks for CAR-T production. The acquisition, processing, and depository of UCB are costly, thus relatively small percentage of the population decided on CB banking thus far, which may result in low utilization of UCB units from UCB banks (164). Additionally, the effect of long-term cryopreservation on UCB cell functionality remains unclear. Although transplantation results appear unchanged if UCB cells were cryopreserved for up to 10 years, it is yet to unravel whether longer preservation may impact UCB cell viability (165).
Moreover, both ethnic and economic challenges must be addressed to ensure fair access to UCB as a cell bank for CAR-T therapies. One of the main issues constitute lack of ethnic diversity as some of the groups are underrepresented. For instance, study by Akyurekli et al. showed that non-Caucasian ethnicity of the cord blood donor was associated with a higher risk of failing to meet banking criteria often due to lower collected volumes and reduced cell counts. This disparity can be attributed to a variety of factors, including socioeconomic barriers and lack of awareness of donation options (166). In addition, the high costs associated with collecting, processing, and storing UCB pose economic barriers, especially for lower-income families (167). Investing in cost-effective manufacturing techniques, subsidizing public UCB banking, and promoting nonprofit partnerships can help reduce the financial burden. Ethical considerations such as informed consent, donor rights, and equitable distribution of UCB-derived therapies also require clear rules to ensure transparency and trust. Addressing these issues through education, policy reform, and financial support programs will be essential to making UCB-derived CAR-T cell therapies accessible and beneficial to all patients, regardless of ethnic or economic background.
In recent years, the field of CAR-T cell therapies has attracted the attention of researchers worldwide. It is now well-known that conventional autologous regimens have several disadvantages attributed to high costs, long time of the manufacture leading to the extended length of the “vein-to-vein” time, and unavailability for patients with low-quality T cells. Thus, numerous studies have investigated the possibilities of harnessing donor-derived CAR-T cells to address these hurdles. The allogeneic CAR-T cells gained much interest and are perceived as a promising solution to the shortcomings of the current therapies. Still, the vast majority of studies utilize the concept of genome editing that provides potent CAR-T products deprived of TCR or MHC molecules.
UCB provides multiple advantages over PB allogeneic or autologous sources for CAR-T cells. The use of UCB as a source of CAR-T cells offers accessibility as they could be transduced with conventional CAR constructs and be available at request from the CAR-T bank. The immunology of UCB T cells favors them as they are mostly composed of TSCM and TCM and therefore retained less differentiated phenotype than PB CAR-T cells (126). Additionally, UCB CAR-T cells express exhaustion markers like PD1, LAG3, or TIM3 on significantly lower levels compared to allogeneic PB-derived CAR, which translates into the capability of longer persistence in vivo and decreased potential risk of GVHD (125). UCB-derived CAR-T cells could be transferred both autologously to reduce post-transplant recurrence and in an allogeneic setting with fewer HLA restrictions that enable more accessible donor-recipient matching. Finally, UCB-derived CAR-T cells provide the cost-effective and sustainable strategy for utilizing cord blood banks. On the other hand, UCB poses challenges related to T cell numbers, maturity, and antigen recognition.
Yet, as the field of CAR-T cell therapy continues to advance, ongoing research is aimed at addressing these challenges and optimizing the use of UCB as a source for CAR-T cells that will overcome the challenges associated with conventional autologous or allogeneic PB-derived therapies. Although current knowledge regarding the safety and efficacy of UCB CAR-T cells is limited to preclinical and few early clinical studies, reports from the ongoing research are optimistic, and further advancements are highly awaited.
KR: Writing – original draft, Writing – review & editing. JM: Writing – review & editing. TO: Writing – review & editing. NR: Supervision, Writing – review & editing. GB: Writing – review & editing. TK: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Medical Research Agency project no 2022/ABM/06/00006 to FamiCordTx S.A. and National Science Centre, decision no 2023/49/N/NZ6/00980 to Institute of Human Genetics Polish Academy of Sciences.
JM and TO are employees of FamiCordTx S.A. company developing CAR & TCR based gene therapies, including cord blood-derived cells; NR is an employee and a scientific advisory board of FamiCordTx S.A.; TK is a Chief Scientific Officer and Board Member of FamiCordTx S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
2. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
3. Xiao XY, Huang SK, Chen SF, Wang YZ, Sun QH, Xu XJ, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR-T cell therapy and associated prevention and management strategies. J Exp Clin Canc Res. (2021) 40:367. doi: 10.1186/s13046-021-02148-6
4. Sanber K, Savani B, Jain T. Graft–host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Brit J Haematol. (2021) 195:660–8. doi: 10.1111/bjh.17544
5. Brown JA, Boussiotis VA. Umbilical cord blood transplantation: basic biology and clinical challenges to immune reconstitution. Clin Immunol. (2008) 127:286–97. doi: 10.1016/j.clim.2008.02.008
6. Barker JN, Weisdorf DJ, DeFor TE, Blazar BR, McGlave PB, Miller JS, et al. Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic Malignancy. Blood. (2005) 105:1343–7. doi: 10.1182/blood-2004-07-2717
7. Tang TCY, Xu N, Nordon R, Haber M, Micklethwaite K, Dolnikov A. Donor T cells for CAR T cell therapy. biomark Res. (2022) 10:14. doi: 10.1186/s40364-022-00359-3
8. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
9. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
10. Zhang H, Zhao P, Huang H. Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol. (2020) 9:34. doi: 10.1186/s40164-020-00190-2
11. van den Broek T, Borghans JAM, van Wijk F. The full spectrum of human naive T cells. Nat Rev Immunol. (2018) 18:363–73. doi: 10.1038/s41577-018-0001-y
12. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. (2019) 97:664–74. doi: 10.1111/imcb.12254
13. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
14. Wang F, Cheng F, Zheng F. Stem cell like memory T cells: A new paradigm in cancer immunotherapy. Clin Immunol. (2022) 241:109078. doi: 10.1016/j.clim.2022.109078
15. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. (2012) 12:671–84. doi: 10.1038/nrc3322
16. Frumento G, Verma K, Croft W, White A, Zuo J, Nagy Z, et al. Homeostatic cytokines drive epigenetic reprogramming of activated T cells into a “Naive-memory” Phenotype. iScience. (2020) 23:100989. doi: 10.1016/j.isci.2020.100989
17. Samji T, Khanna KM. Understanding memory CD8(+) T cells. Immunol Lett. (2017) 185:32–9. doi: 10.1016/j.imlet.2017.02.012
18. Tantalo DG, Oliver AJ, von Scheidt B, Harrison AJ, Mueller SN, Kershaw MH, et al. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J Immunotherapy Cancer. (2021) 9(5):e002555. doi: 10.1136/jitc-2021-002555
19. Kwiecien I, Rutkowska E, Sokolowski R, Bednarek J, Raniszewska A, Jahnz-Rozyk K, et al. Effector memory T cells and CD45RO+ Regulatory T cells in metastatic vs. Non-metastatic lymph nodes in lung cancer patients. Front Immunol. (2022) 13:864497. doi: 10.3389/fimmu.2022.864497
20. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. (2013) 43:2797–809. doi: 10.1002/eji.201343751
21. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. (2018) 18:340–56. doi: 10.1038/nri.2017.146
22. Henson SM, Riddell NE, Akbar AN. Properties of end-stage human T cells defined by CD45RA re-expression. Curr Opin Immunol. (2012) 24:476–81. doi: 10.1016/j.coi.2012.04.001
23. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
24. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). (2016) 8(3):36. doi: 10.3390/cancers8030036
25. Takeuchi A, Saito T. CD4 CTL, a cytotoxic subset of CD4(+) T cells, their differentiation and function. Front Immunol. (2017) 8:194. doi: 10.3389/fimmu.2017.00194
26. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol. (2012) 12:136–48. doi: 10.1038/nri3152
27. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
28. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell Malignancies. Blood. (2016) 128:519–28. doi: 10.1182/blood-2015-11-683847
29. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
30. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. (2016) 30:492–500. doi: 10.1038/leu.2015.247
31. Rathod S. Phenotyping of CAR T cells. Method Cell Biol. (2022) 167:71–80. doi: 10.1016/bs.mcb.2021.10.001
32. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. (2014) 257:107–26. doi: 10.1111/imr.12131
33. Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T cells for cancer therapy. J Natl Cancer Inst. (2016) 108(7):djv439. doi: 10.1093/jnci/djv439
34. Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. (2017) 9:1183–97. doi: 10.15252/emmm.201607485
35. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. (2016) 3:16014. doi: 10.1038/mto.2016.14
36. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfus Med Hemother. (2019) 46:15–24. doi: 10.1159/000496870
37. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
38. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
39. Dragon AC, Zimmermann K, Nerreter T, Sandfort D, Lahrberg J, Kloss S, et al. CAR-T cells and TRUCKs that recognize an EBNA-3C-derived epitope presented on HLA-B*35 control Epstein-Barr virus-associated lymphoproliferation. J Immunother Cancer. (2020) 8(2):e000736. doi: 10.1136/jitc-2020-000736
40. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
41. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
42. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
43. Roex G, Feys T, Beguin Y, Kerre T, Poire X, Lewalle P, et al. Chimeric antigen receptor-T-cell therapy for B-cell hematological Malignancies: an update of the pivotal clinical trial data. Pharmaceutics. (2020) 12(2):194. doi: 10.3390/pharmaceutics12020194
44. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell. (2019) 35:489–503.e8. doi: 10.1016/j.ccell.2019.02.003
45. Mishra AK. Genome editing approaches for universal chimeric antigen receptor T cells. EJC Paediatric Oncol. (2024) 3:100149. doi: 10.1016j.ejcped.2024.100149
46. Lukjanov V, Koutna I, Simara P. CAR-T cell production using nonviral approaches. J Immunol Res. (2021) 2021:6644685. doi: 10.1155/2021/6644685
47. U.S. Food and Drug Administration (FDA). 18 April 2024. Available online at: https://www.fda.gov/ (Accessed: September 11, 2024).
48. Lamble AJ, Schultz LM, Nguyen K, Hsieh EM, McNerney K, Rouce RH, et al. Risk of T-cell Malignancy after CAR-T cell therapy in children, adolescents, and young adults. Blood Adv. (2024) 8:3544–8. doi: 10.1182/bloodadvances.2024013243
49. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR-modified T cells: toxicities and overcoming strategies. J Immunol Res. (2018) 2018:2386187. doi: 10.1155/2018/2386187
50. Mullard A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discovery. (2022) 21:249. doi: 10.1038/d41573-022-00048-8
51. Sanber K, Savani B, Jain T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Br J Haematol. (2021) 195:660–8. doi: 10.1111/bjh.17544
52. Tian L, Li C, Sun J, Zhai Y, Wang J, Liu S, et al. Efficacy of chimeric antigen receptor T cell therapy and autologous stem cell transplant in relapsed or refractory diffuse large B-cell lymphoma: A systematic review. Front Immunol. (2022) 13:1041177. doi: 10.3389/fimmu.2022.1041177
53. Elsallab M, Ellithi M, Hempel S, Abdel-Azim H, Abou-El-Enein M. Long-term response to autologous anti-CD19 chimeric antigen receptor T cells in relapsed or refractory B cell acute lymphoblastic leukemia: a systematic review and meta-analysis. Cancer Gene Ther. (2023) 30:845–54. doi: 10.1038/s41417-023-00593-3
54. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
55. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
56. Tuazon SA, Li A, Gooley T, Eunson TW, Maloney DG, Turtle CJ, et al. Factors affecting lymphocyte collection efficiency for the manufacture of chimeric antigen receptor T cells in adults with B-cell Malignancies. Transfusion. (2019) 59:1773–80. doi: 10.1111/trf.15178
57. Jenkins MJ, Farid SS. Cost-effective bioprocess design for the manufacture of allogeneic CAR-T cell therapies using a decisional tool with multi-attribute decision-making analysis. Biochem Eng J. (2018) 137:192–204. doi: 10.1016/j.bej.2018.05.014
58. Zhu X, Li Q, Zhu X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front Cell Dev Biol. (2022) 10:1034257. doi: 10.3389/fcell.2022.1034257
59. Cliff ERS, Kelkar AH, Russler-Germain DA, Tessema FA, Raymakers AJN, Feldman WB, et al. High cost of chimeric antigen receptor T cells: challenges and solutions. Am Soc Clin Oncol Educ Book. (2023) 43:e397912. doi: 10.1200/EDBK_397912
60. Harrison RP, Zylberberg E, Ellison S, Levine BL. Chimeric antigen receptor-T cell therapy manufacturing: modelling the effect of offshore production on aggregate cost of goods. Cytotherapy. (2019) 21:224–33. doi: 10.1016/j.jcyt.2019.01.003
61. Geethakumari PR, Ramasamy DP, Dholaria B, Berdeja J, Kansagra A. Balancing quality, cost, and access during delivery of newer cellular and immunotherapy treatments. Curr Hematol Malig Rep. (2021) 16:345–56. doi: 10.1007/s11899-021-00635-3
62. Yang JF, He JP, Zhang X, Li JJ, Wang ZG, Zhang YL, et al. Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J. (2022) 12:104. doi: 10.1038/s41408-022-00694-6
63. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat BioMed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
64. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR-T cell therapy. Mol Ther Oncolytics. (2016) 3:16011. doi: 10.1038/mto.2016.11
65. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
66. Bove C, Arcangeli S, Falcone L, Camisa B, El Khoury R, Greco B, et al. CD4 CAR-T cells targeting CD19 play a key role in exacerbating cytokine release syndrome, while maintaining long-term responses. J Immunotherapy Cancer. (2023) 11:e005878. doi: 10.1136/jitc-2022-005878
67. Bedoya DM, Dutoit V, Migliorini D. Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol. (2021) 12:640082. doi: 10.3389/fimmu.2021.640082
68. Li YH, Huo Y, Yu L, Wang JZ. Quality control and nonclinical research on CAR-T cell products: general principles and key issues. Engineering. (2019) 5:122–31. doi: 10.1016/j.eng.2018.12.003
69. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T cells: more than ease of access? Cells-Basel. (2018) 7:155. doi: 10.3390/cells7100155
70. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘sOff-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
71. Kim DW, Cho JY. Recent advances in allogeneic CAR-T cells. Biomolecules. (2020) 10(2):263. doi: 10.3390/biom10020263
72. Lv Z, Luo F, Chu Y. Strategies for overcoming bottlenecks in allogeneic CAR-T cell therapy. Front Immunol. (2023) 14:1199145. doi: 10.3389/fimmu.2023.1199145
73. Nunes E, Heslop H, Fernandez-Vina M, Taves C, Wagenknecht DR, Eisenbrey AB, et al. Definitions of histocompatibility typing terms. Blood. (2011) 118:e180–3. doi: 10.1182/blood-2011-05-353490
74. Jin X, Cao YQ, Wang LQ, Sun R, Cheng L, He XY, et al. HLA-matched and HLA-haploidentical allogeneic CD19-directed chimeric antigen receptor T-cell infusions are feasible in relapsed or refractory B-cell acute lymphoblastic leukemia before hematopoietic stem cell transplantation. Leukemia. (2020) 34:909–13. doi: 10.1038/s41375-019-0610-x
75. Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. (2014) 21:539–48. doi: 10.1038/gt.2014.26
76. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. (2012) 119:5697–705. doi: 10.1182/blood-2012-01-405365
77. Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet. (2020) 396:1885–94. doi: 10.1016/S0140-6736(20)32334-5
78. Morgan NV, Goddard S, Cardno TS, McDonald D, Rahman F, Barge D, et al. Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+ T cells. J Clin Invest. (2011) 121:695–702. doi: 10.1172/JCI41931
79. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.CCR-20-3863
80. Qasim W. Genome-edited allogeneic donor “universal” chimeric antigen receptor T cells. Blood. (2023) 141:835–45. doi: 10.1182/blood.2022016204
81. Sasu BJ, Opiteck GJ, Gopalakrishnan S, Kaimal V, Furmanak T, Huang D, et al. Detection of chromosomal alteration after infusion of gene-edited allogeneic CAR T cells. Mol Ther. (2023) 31:676–85. doi: 10.1016/j.ymthe.2022.12.004
82. Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, et al. Current perspectives on “Off-the-shelf” Allogeneic NK and CAR-NK cell therapies. Front Immunol. (2021) 12:732135. doi: 10.3389/fimmu.2021.732135
83. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
84. Berrien-Elliott MM, Jacobs MT, Fehniger TA. Allogeneic natural killer cell therapy. Blood. (2023) 141:856–68. doi: 10.1182/blood.2022016200
85. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
86. Terren I, Orrantia A, Vitalle J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
87. Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell Malignancies. Oncotarget. (2016) 7:56219–32. doi: 10.18632/oncotarget.11019
88. Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, et al. Preclinical targeting of aggressive T-cell Malignancies using anti-CD5 chimeric antigen receptor. Leukemia. (2017) 31:2151–60. doi: 10.1038/leu.2017.8
89. You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. (2019) 9:64–78.
90. Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discovery. (2021) 11:45–58. doi: 10.1158/2159-8290.Cd-20-0556
91. Li J, Hu H, Lian K, Zhang D, Hu P, He Z, et al. CAR-NK cells in combination therapy against cancer: A potential paradigm. Heliyon. (2024) 10:e27196. doi: 10.1016/j.heliyon.2024.e27196
92. Ballen KK, Verter F, Kurtzberg J. Umbilical cord blood donation: public or private? Bone Marrow Transplant. (2015) 50:1271–8. doi: 10.1038/bmt.2015.124
93. Madureira AB, Eapen M, Locatelli F, Teira P, Zhang MJ, Davies SM, et al. Analysis of risk factors influencing outcome in children with myelodysplastic syndrome after unrelated cord blood transplantation. Leukemia. (2011) 25:449–54. doi: 10.1038/leu.2010.285
94. Yanada M, Konuma T, Kuwatsuka Y, Kondo T, Kawata T, Takahashi S, et al. Unit selection for umbilical cord blood transplantation for adults with acute myeloid leukemia in complete remission: a Japanese experience. Bone Marrow Transplant. (2019) 54:1789–98. doi: 10.1038/s41409-019-0539-8
95. Peffault de Latour R, Chevret S, Jubert C, Sirvent A, Galambrun C, Ruggeri A, et al. Unrelated cord blood transplantation in patients with idiopathic refractory severe aplastic anemia: a nationwide phase 2 study. Blood. (2018) 132:750–4. doi: 10.1182/blood-2018-01-829630
96. Page KM, Labopin M, Ruggeri A, Michel G, Diaz de Heredia C, O'Brien T, et al. Factors associated with long-term risk of relapse after unrelated cord blood transplantation in children with acute lymphoblastic leukemia in remission. Biol Blood Marrow Transplant. (2017) 23:1350–8. doi: 10.1016/j.bbmt.2017.04.015
97. Szabolcs P, Park KD, Reese M, Marti L, Broadwater G, Kurtzberg J. Coexistent naive phenotype and higher cycling rate of cord blood T cells as compared to adult peripheral blood. Exp Hematol. (2003) 31:708–14. doi: 10.1016/S0301-472x(03)00160-7
98. Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. (2012) 73:248–57. doi: 10.1016/j.humimm.2011.12.015
99. Szabolcs P, Park KD, Reese M, Marti L, Broadwater G, Kurtzberg J. Coexistent naïve phenotype and higher cycling rate of cord blood T cells as compared to adult peripheral blood. Exp Hematol. (2003) 31:708–14. doi: 10.1016/s0301-472x(03)00160-7
100. Kloosterboer FM, van Luxemburg-Heijs SA, Willemze R, Falkenburg JH. Similar potential to become activated and proliferate but differential kinetics and profiles of cytokine production of umbilical cord blood T cells and adult blood naive and memory T cells. Hum Immunol. (2006) 67:874–83. doi: 10.1016/j.humimm.2006.02.040
101. Lin SJ, Yan DC, Lee YC, Hsiao HS, Lee PT, Liang YW, et al. Umbilical cord blood immunology: relevance to stem cell transplantation. Clin Rev Allergy Immunol. (2012) 42:45–57. doi: 10.1007/s12016-011-8289-4
102. Mayani H. Biological differences between neonatal and adult human hematopoietic stem/progenitor cells. Stem Cells Dev. (2010) 19:285–98. doi: 10.1089/scd.2009.0327
103. Liao Y, Geyer MB, Yang AJ, Cairo MS. Cord blood transplantation and stem cell regenerative potential. Exp Hematol. (2011) 39:393–412. doi: 10.1016/j.exphem.2011.01.002
104. Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. (2014) 371:339–48. doi: 10.1056/NEJMsa1311707
105. Chen YB, Wang T, Hemmer MT, Brady C, Couriel DR, Alousi A, et al. GvHD after umbilical cord blood transplantation for acute leukemia: an analysis of risk factors and effect on outcomes. Bone Marrow Transplant. (2017) 52:400–8. doi: 10.1038/bmt.2016.265
106. de Molla VC, Barbosa MCR, Junior AM, Goncalves MV, Guirao EKF, Yamamoto M, et al. Natural killer cells 56(bright)16(-) have higher counts in the umbilical cord blood than in the adult peripheral blood. Hematol Transfus Cell Ther. (2023) 45:419–27. doi: 10.1016/j.htct.2022.07.001
107. Busca A, Aversa F. In-vivo or ex-vivo T cell depletion or both to prevent graft-versus-host disease after hematopoietic stem cell transplantation. Expert Opin Biol Ther. (2017) 17:1401–15. doi: 10.1080/14712598.2017.1369949
108. Rodrigues CA, Rocha V, Dreger P, Brunstein C, Sengeloev H, Finke J, et al. Alternative donor hematopoietic stem cell transplantation for mature lymphoid Malignancies after reduced-intensity conditioning regimen: similar outcomes with umbilical cord blood and unrelated donor peripheral blood. Haematologica. (2014) 99:370–7. doi: 10.3324/haematol.2013.088997
109. Kadereit S, Mohammad SF, Miller RE, Woods KD, Listrom CD, McKinnon K, et al. Reduced NFAT1 protein expression in human umbilical cord blood T lymphocytes. Blood. (1999) 94:3101–7. doi: 10.1182/blood.V94.9.3101
110. Kaminski BA, Kadereit S, Miller RE, Leahy P, Stein KR, Topa DA, et al. Reduced expression of NFAT-associated genes in UCB versus adult CD4+ T lymphocytes during primary stimulation. Blood. (2003) 102:4608–17. doi: 10.1182/blood-2003-05-1732
111. Nitsche A, Zhang M, Clauss T, Siegert W, Brune K, Pahl A. Cytokine profiles of cord and adult blood leukocytes: differences in expression are due to differences in expression and activation of transcription factors. BMC Immunol. (2007) 8:18. doi: 10.1186/1471-2172-8-18
112. Devi S, Bongale AM, Tefera MA, Dixit P, Bhanap P. Fresh umbilical cord blood-A source of multipotent stem cells, collection, banking, cryopreservation, and ethical concerns. Life (Basel). (2023) 13(9):1794. doi: 10.3390/life13091794
113. Waller-Wise R. Umbilical cord blood: information for childbirth educators. J Perinat Educ. (2011) 20:54–60. doi: 10.1891/1058-1243.20.1.54
114. Fuchs EJ, O'Donnell PV, Eapen M, Logan B, Antin JH, Dawson P, et al. Double unrelated umbilical cord blood vs HLA-haploidentical bone marrow transplantation: the BMT CTN 1101 trial. Blood. (2021) 137:420–8. doi: 10.1182/blood.2020007535
115. Webb S. Banking on cord blood stem cells. Nat Biotechnol. (2013) 31:585–8. doi: 10.1038/nbt.2629
116. Kalin B, Ter Borg M, Wijers R, Somers JAE, van der Holt B, van Bergen CAM, et al. Double umbilical cord blood transplantation in high-risk hematological patients: A phase II study focusing on the mechanism of graft predominance. Hemasphere. (2019) 3:e285. doi: 10.1097/HS9.0000000000000285
117. Bonifazi F, Dan E, Labopin M, Sessa M, Guadagnuolo V, Ferioli M, et al. Intrabone transplant provides full stemness of cord blood stem cells with fast hematopoietic recovery and low GVHD rate: results from a prospective study. Bone Marrow Transplant. (2019) 54:717–25. doi: 10.1038/s41409-018-0335-x
118. Bozhilov YK, Hsu I, Brown EJ, Wilkinson AC. In vitro human haematopoietic stem cell expansion and differentiation. Cells. (2023) 12(6):896. doi: 10.3390/cells12060896
119. Fernández MN, Regidor C, Cabrera R, García-Marco JA, Forés R, Sanjuán I, et al. Unrelated umbilical cord blood transplants in adults:: Early recovery of neutrophils by supportive co-transplantation of a low number of highly purified peripheral blood CD34 cells from an HLA-haploidentical donor. Exp Hematol. (2003) 31:535–44. doi: 10.1016/S0301-472x(03)00067-5
120. Kwon M, Balsalobre P, Serrano D, Perez Corral A, Buno I, Anguita J, et al. Single cord blood combined with HLA-mismatched third party donor cells: comparable results to matched unrelated donor transplantation in high-risk patients with hematologic disorders. Biol Blood Marrow Transplant. (2013) 19:143–9. doi: 10.1016/j.bbmt.2012.08.019
121. Liu H, van Besien K. Alternative donor transplantation–”mixing and matching”: the role of combined cord blood and haplo-identical donor transplantation (haplo-cord SCT) as a treatment strategy for patients lacking standard donors? Curr Hematol Malig Rep. (2015) 10:1–7. doi: 10.1007/s11899-014-0245-y
122. Liu HT, Rich ES, Godley L, Odenike O, Joseph L, Marino S, et al. Reduced-intensity conditioning with combined haploidentical and cord blood transplantation results in rapid engraftment, low GVHD, and durable remissions. Blood. (2011) 118:6438–45. doi: 10.1182/blood-2011-08-372508
123. Tolar J, Hippen KL, Blazar BR. Immune regulatory cells in umbilical cord blood: T regulatory cells and mesenchymal stromal cells. Br J Haematol. (2009) 147:200–6. doi: 10.1111/j.1365-2141.2009.07781.x
124. Kwoczek J, Riese SB, Tischer S, Bak S, Lahrberg J, Oelke M, et al. Cord blood–derived T cells allow the generation of a more naïve tumor-reactive cytotoxic T-cell phenotype. Transfusion. (2018) 58:88–99. doi: 10.1111/trf.14365
125. Lin Y, Lin J, Huang J, Chen Y, Tan J, Li Y, et al. Lower T cell inhibitory receptor level in mononuclear cells from cord blood compared with peripheral blood. Stem Cell Investig. (2019) 6:35. doi: 10.21037/sci.2019.09.01
126. Caël B, Galaine J, Bardey I, Marton C, Fredon M, Biichle S, et al. Umbilical cord blood as a source of less differentiated T cells to produce CD123 CAR-T cells. Cancers. (2022) 14:3168. doi: 10.3390/cancers14133168
127. Palen K, Zurko J, Johnson BD, Hari P, Shah NN. Manufacturing chimeric antigen receptor T cells from cryopreserved peripheral blood cells: time for a collect-and-freeze model? Cytotherapy. (2021) 23:985–90. doi: 10.1016/j.jcyt.2021.07.015
128. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
129. Parmar S, Robinson SN, Komanduri K, St John L, Decker W, Xing D, et al. Ex vivo expanded umbilical cord blood T cells maintain naive phenotype and TCR diversity. Cytotherapy. (2006) 8:149–57. doi: 10.1080/14653240600620812
130. Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Kim D, et al. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. (2006) 107:2643–52. doi: 10.1182/blood-2005-09-3904
131. Wang J, Metheny L. Umbilical cord blood derived cellular therapy: advances in clinical development. Front Oncol. (2023) 13:1167266. doi: 10.3389/fonc.2023.1167266
132. Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell Malignancies. Hum Gene Ther. (2010) 21:75–86. doi: 10.1089/hum.2009.122
133. Cany J, Dolstra H, Shah N. Umbilical cord blood&x2013;derived cellular products for cancer immunotherapy. Cytotherapy. (2015) 17:739–48. doi: 10.1016/j.jcyt.2015.03.005
134. Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid Malignancies. Mol Ther. (2008) 16:580–9. doi: 10.1038/sj.mt.6300404
135. Micklethwaite KP, Savoldo B, Hanley PJ, Leen AM, Demmler-Harrison GJ, Cooper LJ, et al. Derivation of human T lymphocytes from cord blood and peripheral blood with antiviral and antileukemic specificity from a single culture as protection against infection and relapse after stem cell transplantation. Blood. (2010) 115:2695–703. doi: 10.1182/blood-2009-09-242263
136. Liu D-D, Hong W-C, Qiu K-Y, Li X-Y, Liu Y, Zhu L-W, et al. Umbilical cord blood: A promising source for allogeneic CAR-T cells. Front Oncology Original Res. (2022) 12:944248. doi: 10.3389/fonc.2022.944248
137. Yu T, Luo C, Zhang H, Tan Y, Yu L. Cord blood-derived CD19-specific chimeric antigen receptor T cells: an off-the-shelf promising therapeutic option for treatment of diffuse large B-cell lymphoma. Front Immunol. (2023) 14:1139482. doi: 10.3389/fimmu.2023.1139482
138. Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, Barker JN, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia. (2015) 29:415–22. doi: 10.1038/leu.2014.215
139. Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. (2019) 9:18729. doi: 10.1038/s41598-019-55239-y
140. Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. (2016) 18:985–94. doi: 10.1016/j.jcyt.2016.05.001
141. Pinz K, Liu H, Golightly M, Jares A, Lan F, Zieve GW, et al. Preclinical targeting of human T-cell Malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia. (2016) 30:701–7. doi: 10.1038/leu.2015.311
142. Olbrich H, Theobald SJ, Slabik C, Gerasch L, Schneider A, Mach M, et al. Adult and cord blood-derived high-affinity gB-CAR-T cells effectively react against human cytomegalovirus infections. Hum Gene Ther. (2020) 31:423–39. doi: 10.1089/hum.2019.149
143. Cord blood derived CAR-T cells in refractory/relapsed B Cell malignancies. Available online at: https://ClinicalTrials.gov/show/NCT03881774 (Accessed October 9, 2024).
144. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
145. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
146. Querol S, Rubinstein P, Madrigal A. The wider perspective: cord blood banks and their future prospects. Br J Haematology. (2021) 195:507–17. doi: 10.1111/bjh.17468
147. Zhao X, Cai L, Hu Y, Wang H. Cord-blood natural killer cell-based immunotherapy for cancer. Front Immunol. (2020) 11:584099. doi: 10.3389/fimmu.2020.584099
148. Roura S, Bago JR, Soler-Botija C, Pujal JM, Galvez-Monton C, Prat-Vidal C, et al. Human umbilical cord blood-derived mesenchymal stem cells promote vascular growth in vivo. PloS One. (2012) 7:e49447. doi: 10.1371/journal.pone.0049447
149. Lee Y-H. The role of cord blood banks in the cell therapy era: future perspectives. br. (2017) 52:153–6. doi: 10.5045/br.2017.52.3.153
150. Aparicio C, Acebal C, Gonzalez-Vallinas M. Current approaches to develop “off-the-shelf” chimeric antigen receptor (CAR)-T cells for cancer treatment: a systematic review. Exp Hematol Oncol. (2023) 12:73. doi: 10.1186/s40164-023-00435-w
151. Georgiadis C, Nickolay L, Syed F, Zhan H, Gkazi SA, Etuk A, et al. Umbilical cord blood T cells can be isolated and enriched by CD62L selection for use in ‘off the shelf’ chimeric antigen receptor T-cell therapies to widen transplant options. Haematologica. (2024) 109:3941–51. doi: 10.3324/haematol.2024.285101
152. Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. (2020) 136:1407–18. doi: 10.1182/blood.2020005185
153. Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. (2020) 26:1878–87. doi: 10.1038/s41591-020-1061-7
154. Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. (2020) 4:4898–911. doi: 10.1182/bloodadvances.2020002394
155. Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat Med. (2024) 30:772–84. doi: 10.1038/s41591-023-02785-8
156. Kueberuwa G, Gornall H, Alcantar-Orozco EM, Bouvier D, Kapacee ZA, Hawkins RE, et al. CCR7(+) selected gene-modified T cells maintain a central memory phenotype and display enhanced persistence in peripheral blood in vivo. J Immunother Cancer. (2017) 5:14. doi: 10.1186/s40425-017-0216-7
157. Wang SY, Scurti GM, Dalheim AV, Quinn S, Stiff PJ, Nishimura MI. Nonactivated and IL-7 cultured CD19-specific CAR T cells are enriched in stem cell phenotypes and functionally superior. Blood Adv. (2024) 8:324–35. doi: 10.1182/bloodadvances.2023010607
158. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. (2019) 7:759–72. doi: 10.1158/2326-6066.CIR-18-0466
159. Abdoli Shadbad M, Hemmat N, Khaze Shahgoli V, Derakhshani A, Baradaran F, Brunetti O, et al. A systematic review on PD-1 blockade and PD-1 gene-editing of CAR-T cells for glioma therapy: from deciphering to personalized medicine. Front Immunol. (2021) 12:788211. doi: 10.3389/fimmu.2021.788211
160. Shi PA, Luchsinger LL, Greally JM, Delaney CS. Umbilical cord blood: an undervalued and underutilized resource in allogeneic hematopoietic stem cell transplant and novel cell therapy applications. Curr Opin Hematol. (2022) 29:317–26. doi: 10.1097/moh.0000000000000732
161. Querol S, Mufti GJ, Marsh SG, Pagliuca A, Little AM, Shaw BE, et al. Cord blood stem cells for hematopoietic stem cell transplantation in the UK: how big should the bank be? Haematologica. (2009) 94:536–41. doi: 10.3324/haematol.2008.002741
162. Haimila K, Penttilä A, Arvola A, Auvinen MK, Korhonen M. Analysis of the adequate size of a cord blood bank and comparison of HLA haplotype distributions between four populations. Hum Immunol. (2013) 74:189–95. doi: 10.1016/j.humimm.2012.10.018
163. Yoon JH, Oh S, Shin S, Park JS, Roh EY, Song EY, et al. The minimum number of cord blood units needed for Koreans is 51,000. Transfusion. (2014) 54:504–8. doi: 10.1111/trf.12284
164. Dessels C, Alessandrini M, Pepper MS. Factors influencing the umbilical cord blood stem cell industry: an evolving treatment landscape. Stem Cell Transl Med. (2018) 7:643–50. doi: 10.1002/sctm.17-0244
165. Mitchell R, Wagner JE, Brunstein CG, Cao Q, McKenna DH, Lund TC, et al. Impact of long-term cryopreservation on single umbilical cord blood transplantation outcomes. Biol Blood Marrow Tr. (2015) 21:50–4. doi: 10.1016/j.bbmt.2014.09.002
166. Akyurekli C, Chan JYS, Elmoazzen H, Tay J, Allan DS. Impact of ethnicity on human umbilical cord blood banking: a systematic review. Transfusion. (2014) 54:2122–7. doi: 10.1111/trf.12630
Keywords: CAR-T cells, umbilical cord blood, cord blood bank, allogeneic therapy, autologous therapy, off-the-shelf CAR-T cells
Citation: Rassek K, Misiak J, Ołdak T, Rozwadowska N, Basak G and Kolanowski T (2025) New player in CAR-T manufacture field: comparison of umbilical cord to peripheral blood strategies. Front. Immunol. 16:1561174. doi: 10.3389/fimmu.2025.1561174
Received: 15 January 2025; Accepted: 28 February 2025;
Published: 21 March 2025.
Edited by:
Luciana Rodrigues Carvalho Barros, University of São Paulo, BrazilReviewed by:
Fuwei Shang, German Cancer Research Center (DKFZ), GermanyCopyright © 2025 Rassek, Misiak, Ołdak, Rozwadowska, Basak and Kolanowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia Rozwadowska, bmF0YWxpYS5yb3p3YWRvd3NrYUBpZ2N6LnBvem5hbi5wbA==; Tomasz Kolanowski, dG9tYXN6LmtvbGFub3dza2lAZmFtaWNvcmR0eC5jb20=
†ORCID: Karolina Rassek, orcid.org/0000-0001-7758-7151
Jan Misiak, orcid.org/0000-0002-1834-4712
Tomasz Ołdak, orcid.org/0000-0002-9958-6755
Natalia Rozwadowska, orcid.org/0000-0002-5111-0870
Grzegorz Basak, orcid.org/0000-0003-3858-8180
Tomasz Kolanowski, orcid.org/0000-0003-3898-8272
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.