 Qi-Wen Wang
Qi-Wen Wang Haorui Zheng†
Haorui Zheng† Yang Yang
Yang Yang Zhuan Liao
Zhuan Liao- Department of Gastroenterology, Shanghai Institute of Pancreatic Diseases, Changhai Hospital, Naval Medical University, Shanghai, China
Background: Recurrent acute pancreatitis (RAP) poses significant clinical challenges, with 32.3% developing to chronic pancreatitis within 5 years. The underlying microbial factors contributing to RAP remain poorly understood. This study aims to identify blood microbial signatures associated with RAP and explore the potential microbial predictors for RAP.
Methods: In this prospective cohort, 90 acute pancreatitis patients are classified into non-recurrent acute pancreatitis (NRAP, n=68) and RAP (n=22) groups based on the number of pancreatitis episodes. Microbial composition of blood samples is analyzed using 5-region (5R) 16S rRNA gene sequencing. Key microbial taxa and functional predictions are made. A random forest model is used to assess the predictive value of microbial features for RAP. The impact of Staphylococcus hominis (S. hominis) on RAP is further evaluated in an experimental mouse model.
Results: Linear discriminant analysis effect size (LEfSe) analysis highlights significant microbial differences, with Paracoccus aminovorans, Corynebacterium glucuronolyticum and S. hominis being prominent in RAP. Functional predictions indicate enrichment of metabolic pathways in the RAP group. Random forest analysis identifies key microbial taxa with an AUC value of 0.759 for predicting RAP. Experimental validation shows that S. hominis exacerbates pancreatic inflammation in mice.
Conclusions: This study identifies distinct clinical and microbial features associated with RAP, emphasizing the role of specific bacterial taxa in pancreatitis recurrence. The findings suggest that microbial profiling could enhance the diagnosis and management of RAP, paving the way for personalized therapeutic approaches.
Introduction
Acute pancreatitis (AP) is an acute inflammation of the pancreas caused by etiologic factors such as gallstones, alcohol use, and hypertriglyceridemia that can lead to significant morbidity and mortality (1). The incidence of AP is estimated at 13 to 49 cases per 100,000 persons per year (2). Despite advances in medical care, a substantial proportion of AP patients experience recurrent episodes, termed recurrent acute pancreatitis (RAP), with a reported recurrence rate of 11%–36% (3, 4). RAP represents a critical turning point in AP-RAP-chronic pancreatitis (CP) continuum, as it remarkably increases the risk of life-threatening complications, up to 32.3% progression to CP within 5 years and substantial socioeconomic burdens, underscoring an urgent need for novel predictive biomarkers and precision intervention paradigms (5–10).
Recent decades have witnessed the gut-pancreas axis dysbiosis in pancreatitis pathogenesis. AP patients exhibit decreased commensal Lactobacillus and increased pathogenic Enterobacteriaceae, correlating with systemic inflammation and pancreatic necrosis (11, 12). Crucially, researches have reported that there is an “authentic circulating microbiome”—the collection of microbial DNA, RNA, and other microbial components found in the bloodstream (13–15). Païssé et al. confirmed that there was a stable microbial community with low biomass (< 0.1pg/μL) but taxonomically diverse in the blood of healthy people, mainly from Proteobacteria phylum (80%) and Actinobacteria phylum (10%) (16). Unlike the gut microbiome, blood microbial communities may originate from intestinal translocation, or infection or bacteremia, acting as “stealth contributors” to inflammation (17–20). However, conventional single-region 16S sequencing (e.g., V3-V4) or shotgun metagenomics lack resolution to detect low-biomass blood microbiota as high abundance of eukaryotic DNA products complicate dramatically the sequencing of bacterial DNA. Besides, no studies have explored blood microbiome dynamics between AP and RAP; and the predictive capacity of blood microbial signatures for RAP remains uncharacterized.
In light of these challenges, we collect serum samples from patients within 24 hours of the onset of AP, and divide patients into NRAP and RAP groups based on medical history. By pioneering 5-region (5R) 16S rDNA sequencing—a high-resolution approach especially for the detection of microbial samples with low biomass (21), we aim to decipher blood microbiome dynamics across AP-RAP trajectories, identify RAP-specific microbial signatures via 5R-enhanced resolution, and develop machine learning models integrating microbial biomarkers with clinical variables for RAP prediction, and investigate the effects of specific pathogenic bacteria on RAP in animal experiments.
Methods
Human sample collection
This is a single-center, cross-sectional study. Patients are enrolled in the First Affiliated Hospital of Naval Medical University, Shanghai, China, between September 2022 and April 2023. Serum samples are collected from patients diagnosed with AP according to the diagnostic criteria within 24 hours of disease onset (1). All participants provide informed consent. According to the diagnostic criteria for RAP, which includes clinical symptoms, serological, and imaging tests confirming AP, with ≥ 2 episodes, no permanent histological changes such as exocrine or endocrine dysfunction, fibrosis, or calcification, and an interval of ≥ 3 months between the two AP episodes, AP patients are further classified into NRAP and RAP groups (9). The major exclusion criteria are chronic pancreatitis, inflammatory bowel disease, immunosuppressive disease, cancer, the use of antibiotics within two months of enrollment. Detailed demographic and clinical information, including age, gender, medical history, and laboratory findings, are recorded for each patient. Ethical approval is obtained from the Shanghai Changhai Hospital Ethics Committee (CHEC2021-198). The samples are stored at -80°C refrigerator before analysis.
DNA extraction and 5R 16S rRNA gene sequencing
DNA is extracted from serum samples using standardized protocols to ensure high-quality genetic material. 5R 16S rRNA sequencing is used in the study to mitigate host DNA interference and facilitate microbiota detection in samples with low microbial content but high host proportions. The experiment utilizes polymerase chain reaction (PCR) to amplify variable regions of the 16S rRNA gene (V2, V3, V5, V6, and V8). The purified PCR products are evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) and the Illumina 16S Metagenomic Sequencing Library (Kapa Biosciences, Woburn, MA, USA), ensuring that the qualified library concentration was above 0.3 ng/μL. Paired-end sequencing is performed using the Illumina NovaSeq 6000 sequencing platform in PE150 sequencing mode, generating 20 Gb/sample.
Bioinformatics analysis and visualization
The sequencing data is analyzed using the Short MUltiple Regions Framework (SMURF) analysis pipeline, employing the optimized Greengenes (May 2013 version, Second Genome) database. The purified amplicon sequence variants (ASVs) are generated to identify microbial profiles. Alpha diversity analysis is conducted based on the resulting species-level abundance tables, using indices such as observed_species, Shannon, Simpson, Chao1, and goods_coverage to evaluate intra-sample diversity. Beta diversity is assessed by calculating two distances (Bray-Curtis) and performing six analyses to evaluate inter-sample/group diversity. A Venn diagram showing overlapping ASVs between the two groups is created using the VennDiagram package in R (version 4.3.1). The ggplot2 package is used for visualization. Linear discriminant analysis effect size (LEfSe) is used to compare microbial compositions, with thresholds set at an LDA score >3 and P < 0.05.
Microbiota function prediction and clinical correlation analysis
The functional potential of the microbiota is predicted using PICRUSt2 software (Version 2.4.1). The predicted functional profiles are analyzed for differences using STatistical Analysis of Metagenomic Profiles (STAMP) (version 2.1.3, Beiko Lab). Correlation analysis between clinical information and microbiota is performed using Spearman’s rank correlation coefficient and then visualized using the pheatmap package (Version 1.0.12, CRAN) in R (version 4.3.1), which assesses the strength and direction of association between clinical variables and microbial abundance.
Disease diagnostic model construction and validation
Based on the sequencing data, a random forest regression model for RAP is constructed in R (version 4.3.1) from the randomForest package, with 70% of the samples (n=63) assigned to the training cohort and the remaining 30% (n=27) to the validation cohort. Ten-fold cross-validation is applied to the training cohort. The most important variables are used to build the predictive model, and ROC curves are calculated to distinguish between NRAP and RAP patients. The confidence intervals for the ROC curves are calculated using the pROC package in R (version 4.3.1).
Animal experiment
Male C57BL/6 mice (6 - 8 weeks old) obtained from GemPharmatech Co., Ltd., (Nanjing, China), are treated with broad-spectrum antibiotics (ampicillin 1 g/l, neomycin 1 g/l, metronidazole 1 g/l, and vancomycin 0.5 g/l) in their drinking water for 4 weeks before experiment to deplete gut and systemic microbiota, as previously described (22). After antibiotic treatment, mice are randomly divided into PBS-gavaged group (n=8) and Staphylococcus hominis (S. hominis) - gavaged group (n=8). Mice are administered 200 ul PBS/mice only or at a dose of 107 colony-forming units (CFU) of S. hominis intragastrically once every two days for 2 weeks. This colonization protocols ensures dominance of S. hominis over potential residual microbes (23). After the first attack of AP with 8 hourly injections of 100 ug/kg caerulein, mice are allowed to recover for 7 days and receive the second attack to induce RAP (24). At 24 h after the first injection of caerulein in second period, mice are sacrificed and analyzed. All animal care and experimental protocols are approved by the Animal Ethics Committee of Shanghai Changhai Hospital Ethics Committee (CHEC (A.E) 2022-008).
Enzyme-linked immunosorbent assays
Mice serum is obtained by centrifuging whole blood samples. Serum levels of Amylase (Abcam, ab102523), IL-1β (Abcam, ab197742) and TNF-α (Abcam, ab102523) is measured using commercial ELISA kits according to manufacturer’s instructions.
Histological analysis
Formaldehyde-fixed pancreas is embedded in paraffin, sectioned into slices, and stained with hematoxylin-eosin (HE). Pancreatic histological score is assessed under a light microscope (Olympus, Tokyo, Japan) according to Rongione’s standard (25). Inflammatory cells in pancreas are evaluated by detection of F4/80 by immunohistochemistry. Briefly, slides are heated in a 70°C oven for 30 min and then deparaffinized in xylene, followed by rehydration through a graded ethanol series. After antigen repairing procedure, slides are incubated with F4/80 antibody (Abcam, ab300421, 1:500) at 4 °C overnight. After washing with PBS, slides are developed with DAB substrate and counterstained with hematoxylin. The IHC positive staining is analyzed by ImageJ (Version 1.8, NIH, USA).
Flow cytometry
Pancreatic tissue samples are harvested and digested in collagenase IV solution at 37°C for 30 min. Pancreatic cells are subsequently filtered through a 70 μm cell strainer. After washing and lysis of erythrocytes, Single-cell suspensions are incubated for 15 min at room temperature in stain buffer with the following antibodies for surface markers: mCD45 (#103108, BioLegend, FITC conjugate), mCD11b (#101230, Biolegend, PerCP conjugate) and mF4/80 (#123116, BioLegend, APC conjugate). Sample acquisition is carried out on Cytoflex flow cytometer (Beckman Cytoflex S, USA) and data are analyzed using FlowJo 10.8 (BD Biosciences, San Jose, CA).
Statistical analysis
Clinical characteristics of the two patient groups are analyzed using SPSS software (version 19.0, IBM Corp, USA). For normally distributed continuous variables, data are presented as mean ± standard deviation (SD); for non-normally distributed continuous variables, data are presented as median (interquartile range [IQR]). Categorical variables are presented as numbers (percentages). The P-value for categorical variables is calculated using the chi-square test or Fisher’s exact test. Continuous variables are analyzed using the t-test or the non-parametric Kruskal-Wallis test. A two-sided P-value of less than 0.05 is considered statistically significant.
Results
Clinical baseline information
90 patients are included in this study, classified into 68 NRAP cases and 22 RAP cases based on recurrence history. The demographic and clinical characteristics of the participants are summarized in Table 1. No significant differences are found among the key clinical variables, including age, gender, and etiology. The NRAP group shows higher levels of LDH compared to the RAP group (median 311 U/L, IQR [198–492] vs. median 219 U/L, IQR [155–281]; P = 0.021). Similarly, BUN levels are higher in the NRAP group than in the RAP group (median 5.1 mmol/L, IQR [4.0–6.6] vs. median 4.5 mmol/L, IQR [3.2–4.9]; P = 0.025). The proportion of severe acute pancreatitis patients is higher in the NRAP group compared to the RAP group (35.3% vs. 22.7%; P = 0.138), but this difference is not statistically significant. The duration of hospitalization shows no significant difference between the two groups (NRAP: median 10 days, IQR [5-21] vs. RAP: median 8 days, IQR [5-12]; P = 0.171).
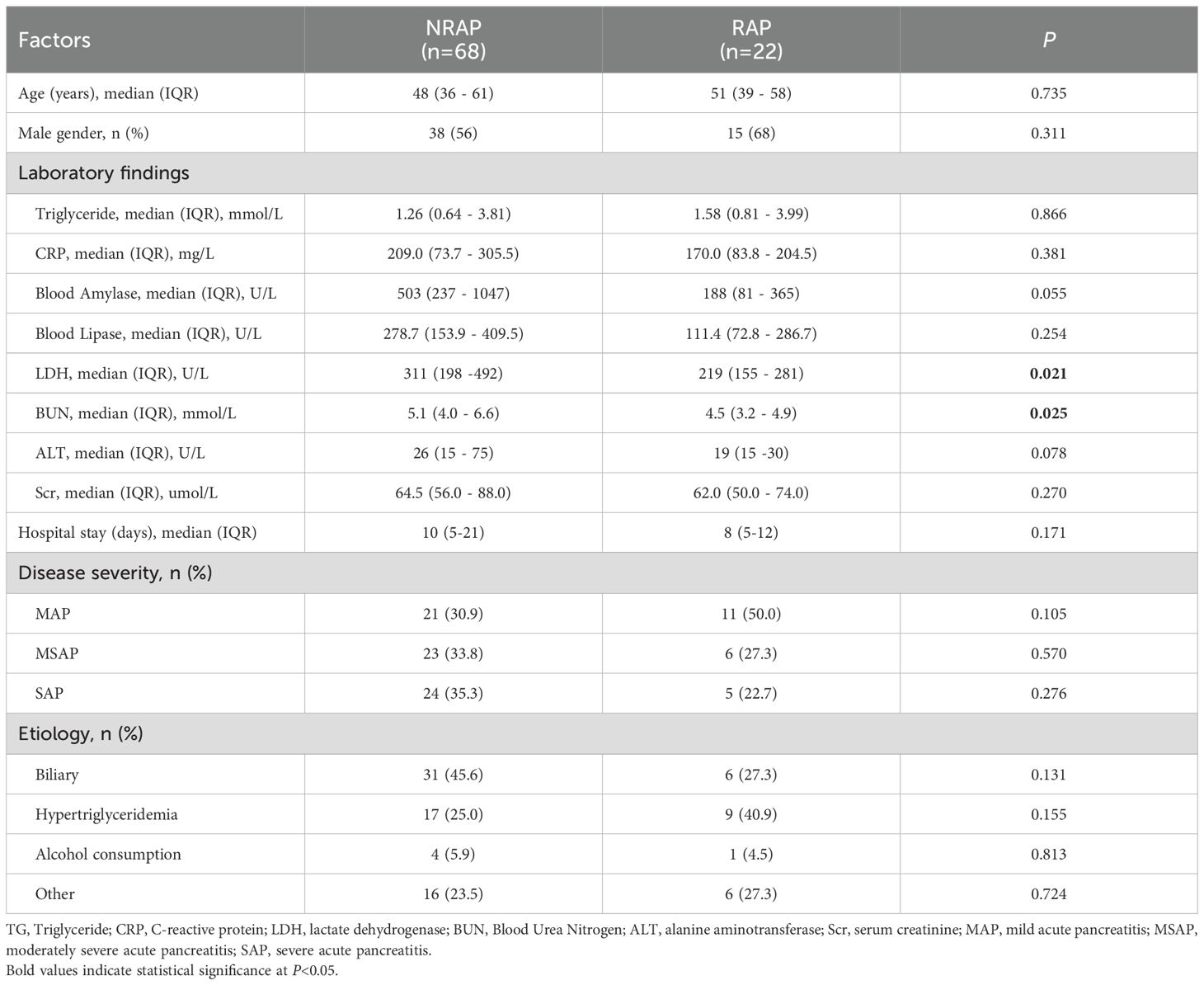
Table 1. Demographic and clinical characteristics of NRAP and RAP patients.
Microbial profile of NRAP and RAP patients
The microbial compositions of serum samples from NRAP and RAP patients are characterized by 5R 16S rRNA gene sequencing. The sequencing quality is eligible, with no samples discarded and average reads count of 33,312 per sample. As shown in Figure 1A, there is no significant difference in α-diversity between the two groups, indicating that the overall composition of blood microbiota is similar. The species richness rarefaction curve gradually levels off, indicating a reasonable number of individual samples (Figure 1B). NMDS and UPGMA cluster analysis based on the UniFrac algorithm shows no significant difference in microbial β-diversity between the two groups (Figures 1C, D). α/β diversity indices reflect the unique nature of blood microbiota, which are inherently similar in dominant phyla. Venn diagrams show the common and unique phyla and genera detected in the NRAP and RAP groups (Figures 1E, F).
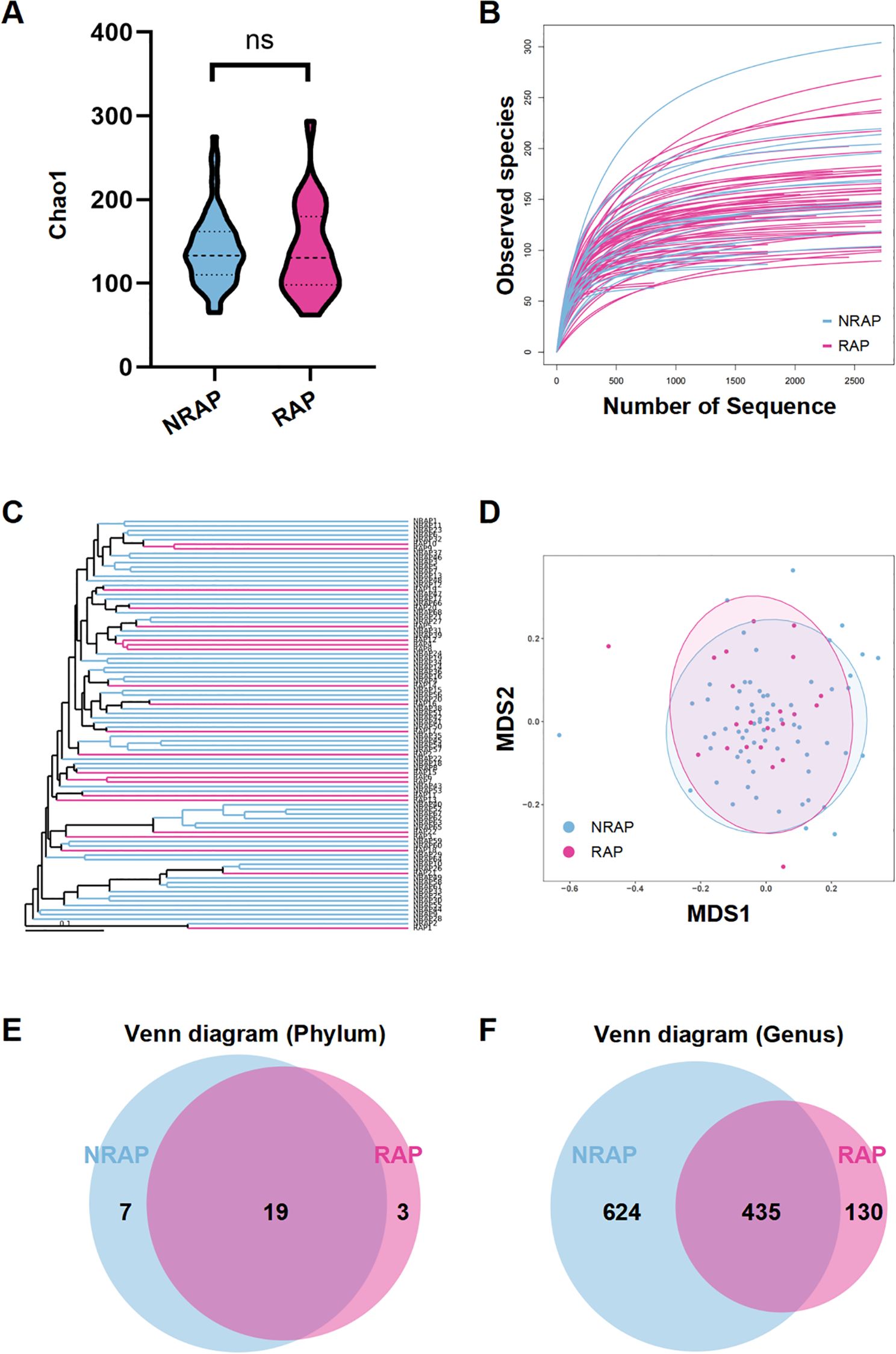
Figure 1. Comparison of microbial diversity between NRAP and RAP patients. (A) Alpha-diversity analysis; (B) Species richness rarefaction curve; (C) NMDS plot; (D) UPGMA clustering analysis; Venn diagram of shared and unique bacterial phyla (E) and genera (F) in NRAP and RAP groups. Top bars: ns, P > 0.05.
Figure 2A presents a stacked bar chart of the top 10 relative abundance bacterial species at the phylum level in NRAP and RAP groups, mainly composed of Proteobacteria (NRAP: 54.74% vs. RAP: 52.09%), Firmicutes (NRAP: 21.28% vs. RAP: 23.24%), and Actinobacteria (NRAP: 16.65% vs. RAP: 17.80%). Minor phyla (e.g., Bacteroidetes and Cyanobacteria) collectively account for < 8% of the microbiota. A chord diagram at the family level visualizes the microbial community composition and the associations between the two groups (Figure 2B). The top 10 genera profiles are shown in Figure 2C. Burkholderia is more frequent in the NRAP group than in the RAP group (NRAP: 8.67% vs. RAP: 5.25%), while the relative abundance of Corynebacterium is higher in the RAP group (NRAP: 7.11% vs. RAP: 8.95%). NRAP group shows a lower relative abundance of Lactobacillus when compared with the RAP group (NRAP: 1.69% vs. RAP: 3.64%).
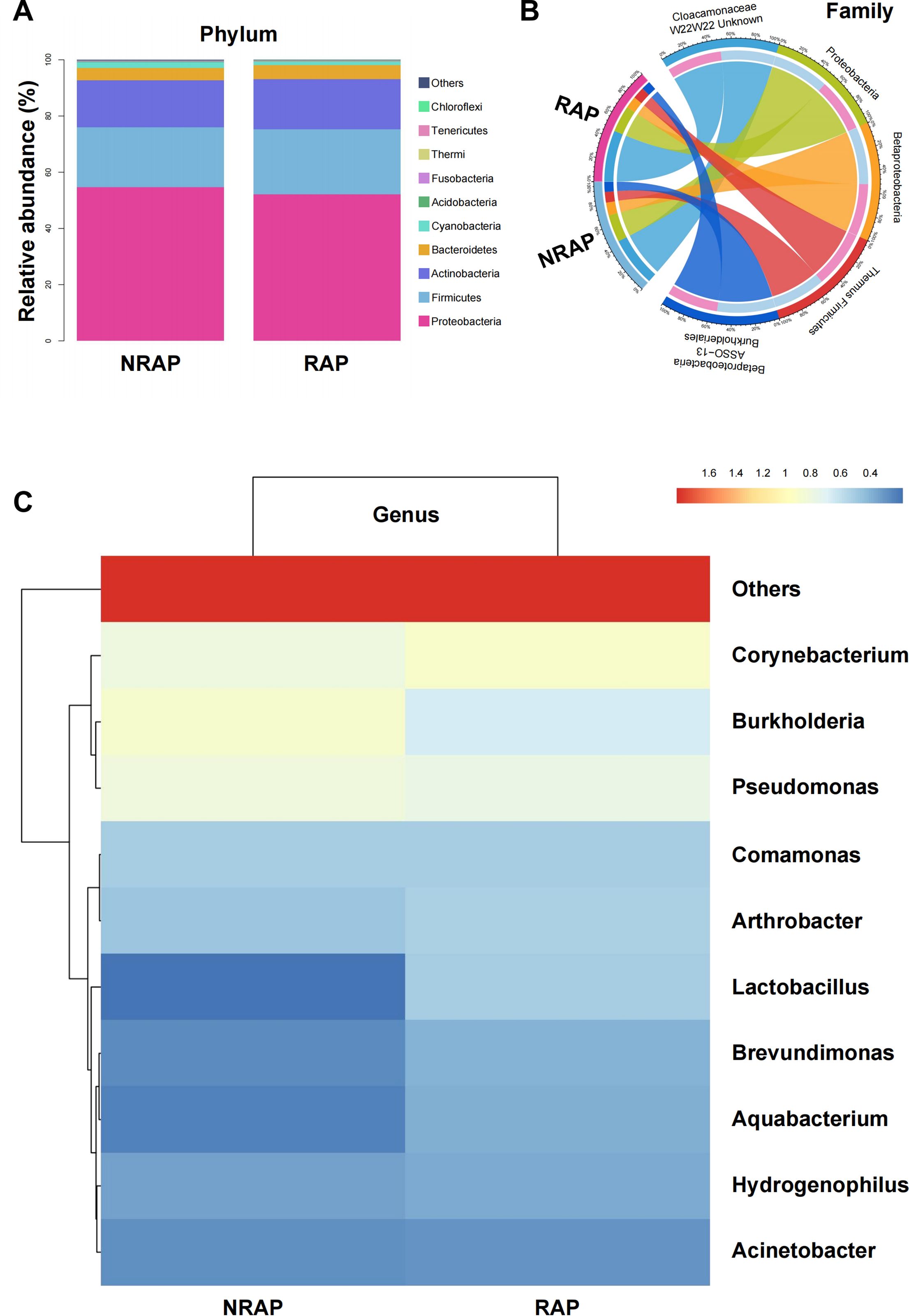
Figure 2. Relative Abundance of Major Bacterial Taxa in NRAP and RAP patients. (A) Stacked bar chart of top 10 bacterial phyla in NRAP and RAP groups; (B) Chord diagram showing associations of microbial families between NRAP and RAP groups; (C) Top 10 genera profiles comparing NRAP and RAP groups.
Taxonomic characteristics of NRAP and RAP patients
LEfSe analysis depicts significant differences in bacterial taxa between the NRAP and RAP groups (Figure 3A). The key microbial taxa in the NRAP group include g_Prevotella, f_Xanthomonadaceae, and o_Xanthomonadales, whereas in the RAP group, g_Sphingopyxis is the dominant taxon. A boxplot visually displays the top 10 ranking genera with significant differences (Figure 3B). At the species level, compared to the NRAP group, the RAP group shows higher abundances of Paracoccus aminovorans (2.172% vs. 0.150%, q = 0.048), Staphylococcus hominis (0.503% vs. 0.210%, q = 0.01), and Corynebacterium glucuronolyticum (0.436% vs. 0.140%, q = 0.03) (Figure 3C).
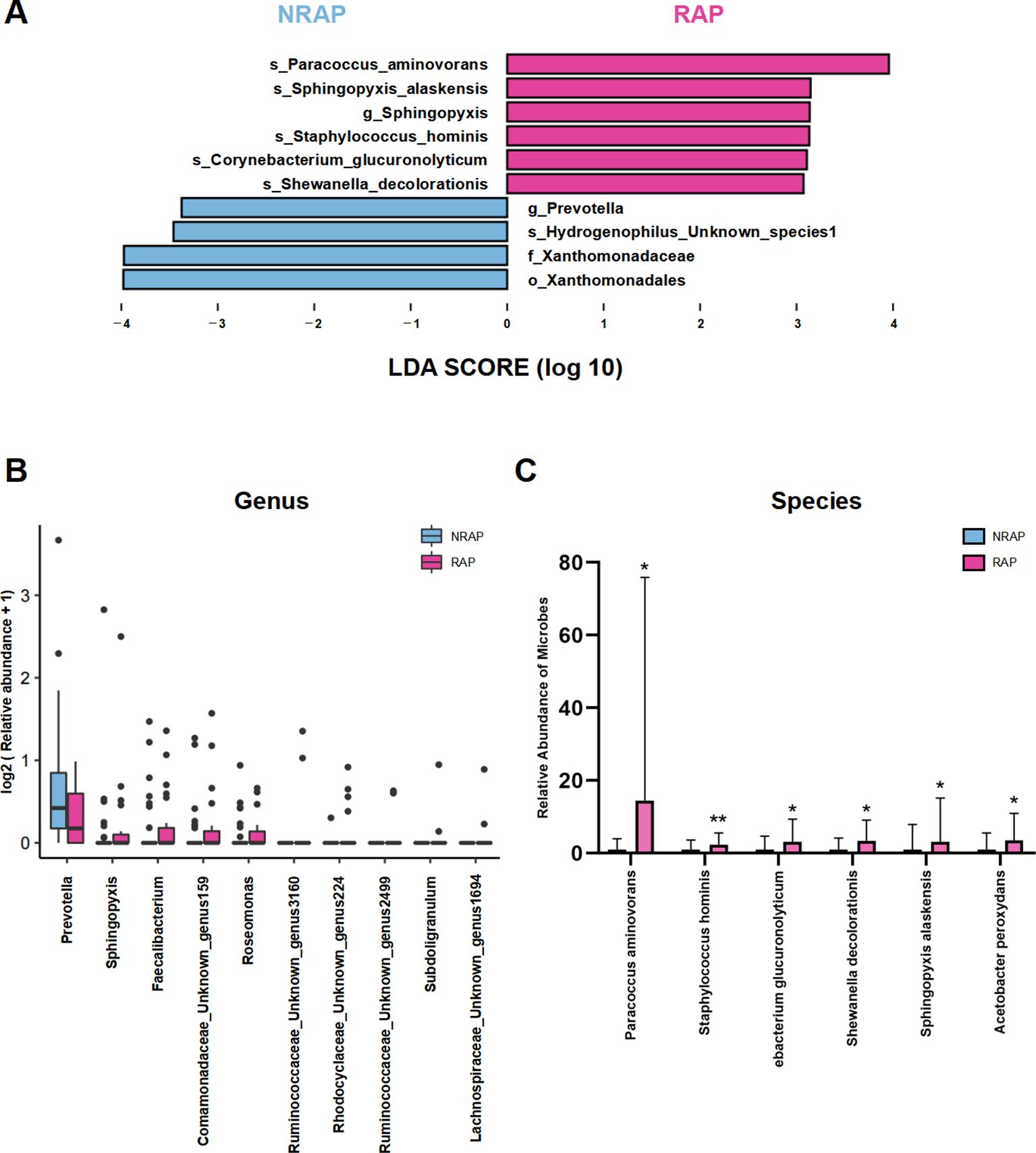
Figure 3. Differentially abundant taxa between NRAP and RAP patients. (A) LEfSe analysis; (B) Boxplot of top 10 genera with significant differences between NRAP and RAP groups; (C) Comparison of specific bacterial species abundances in NRAP and RAP groups. Top bars: *, wilcox-test P value < 0.05; **, wilcox-test P value < 0.01.
Microbial functions and correlation with clinical indicators in NRAP and RAP patients
Using PICRUSt analysis to predict gene functions of the microbiota, we find that the differential microbial community in the RAP group is enriched in KEGG pathways such as alanine, aspartate and glutamate metabolism, thiamine metabolism, and cell cycle-caulobacter; and COG pathways such as FoF1−type ATP synthase, membrane subunit b or b’ when compared to the NRAP group (Figures 4A, B).
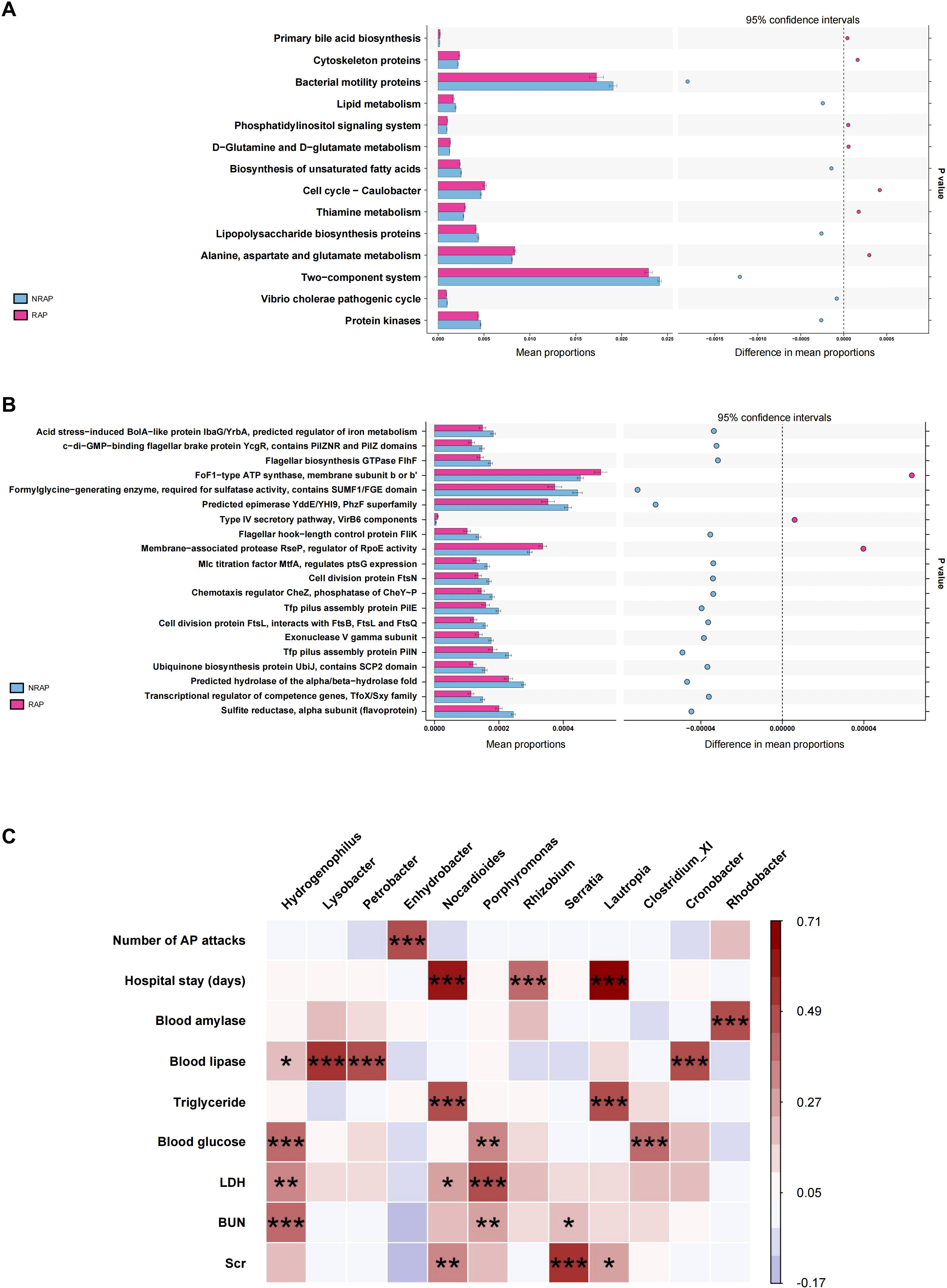
Figure 4. Functional predictions and clinical correlations of blood microbiota in NRAP and RAP patients. (A) KEGG pathway enrichment; (B) COG pathway enrichment; (C) Spearman correlation heatmap showing associations between microbiota and clinical indicators. Top bars: *, P value < 0.05; **, P value < 0.01, ***, P < 0.001.
Spearman correlation analysis is conducted to identify associations between blood microbiota and clinical indicators, including demographic characteristics, laboratory tests, duration of hospitalization, and number of pancreatitis episodes (Figure 4C). At the genus level, Enhydrobacter is positively correlated with the number of AP attacks (r = 0.45, P < 0.001). Lautropia shows a strong positive correlation with hospitalization duration (r = 0.71, P < 0.001) and a moderate positive correlation with triglyceride (TG) (r = 0.48, P < 0.001). Additionally, Nocardioides is moderately positively correlated with both hospitalization duration and TG, with statistical significance (r = 0.58, P < 0.001; r = 0.47, P < 0.001).
Potential microbial features predictive of RAP
To investigate the value of blood microbial features in predicting RAP, we use a random forest regression model to identify taxa associated with RAP. Increase in Mean Squared Error (%IncMSE) identifies Bacteroides (4.3%), Blautia (3.3%), and Rhodocyclaceae (2.4%) as top predictors (Figure 5A). Increase in Node Purity (IncNodePurity) reveals that Bacteroides (0.38), Prevotella (0.35), and Blautia (0.33) show strong classification power (Figure 5A). In the validation cohort, the diagnostic model has an AUC value of 0.759 (95% CI: 0.6346 - 0.8834, cutoff > 0.540, sensitivity: 100%, specificity: 62.8%), demonstrating moderate discriminative power (Figure 5B).
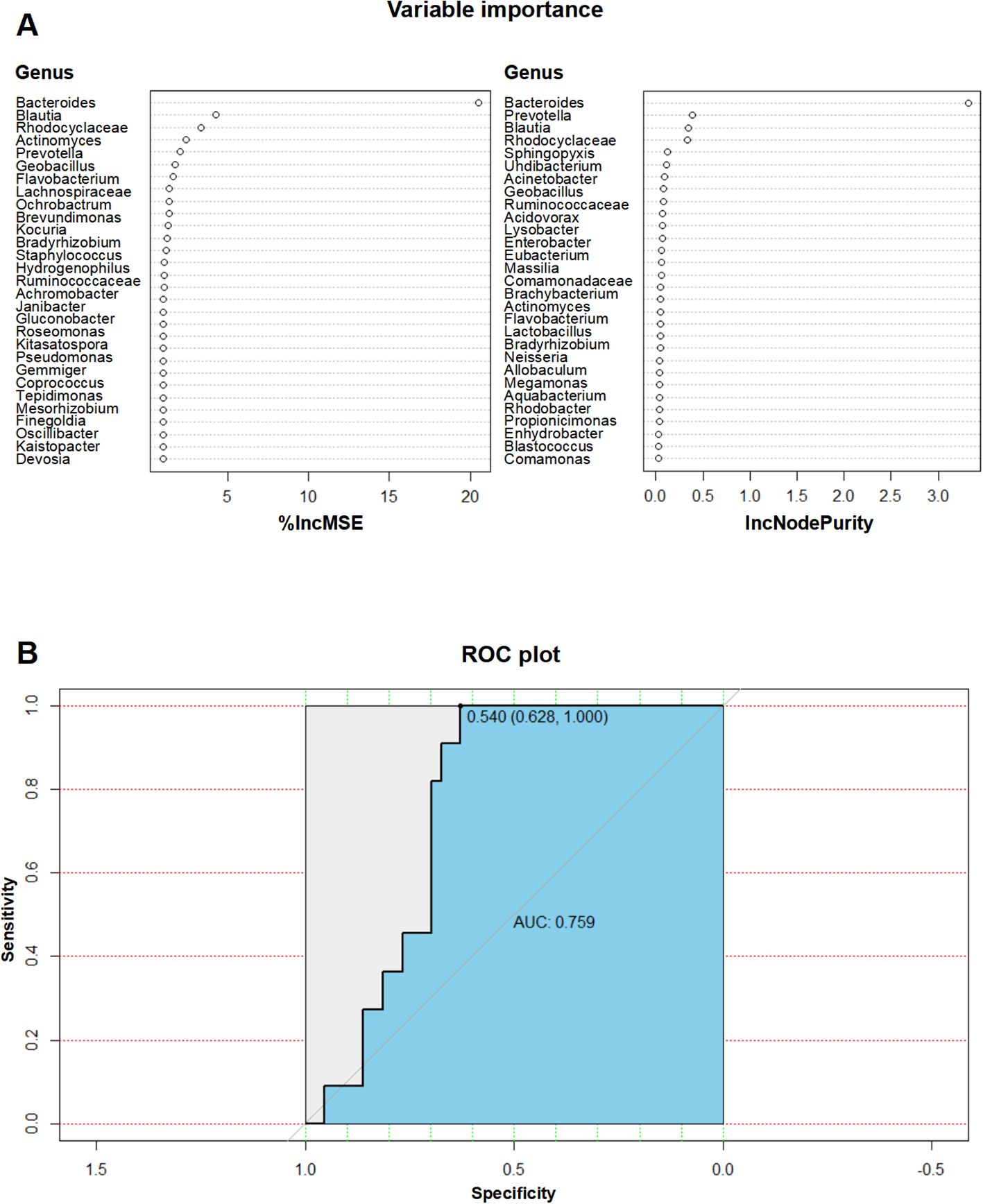
Figure 5. Predictive value of microbial features for RAP diagnosis. (A) IncMSE and IncNodePurity analysis from the random forest model identifying key taxa for RAP prediction; (B) ROC curve of the diagnostic model with an AUC value of 0.759 for RAP prediction.
S. hominis exacerbates experimental RAP in mice
To further validate the hypothesis that microbes associated with RAP can participate in disease progression, S. hominis, a Gram-positive bacterium from the Staphylococcus genus, is selected in this study based on the LEfSe analysis (Figure 3A), the top five differential species data between the two groups (Figure 3C), and previous literature reports. Antibiotic-treated, microbiota-depleted mice are gavaged with S. hominis or PBS as control for two weeks, and then induce with caerulein to create an RAP model (Figure 6A). The relative pancreatic weight ratio in the S. hominis group is higher than in the PBS group, indicating more severe pancreatic inflammation (Figures 6B, E). Histological evaluation of the pancreas shows more edema, significant inflammatory cell infiltration, and higher pancreatic tissue scores in the S. hominis group compared to the PBS group (Figures 6C, D). Serum biochemical analysis reveals elevated levels of serum amylase, pro-inflammatory cytokines TNF-α and IL-1β in the S. hominis group compared to the PBS group (Figures 6F–H). Immunohistochemical detection using F4/80 identifies increased macrophage infiltration in the pancreas of the S. hominis group compared to the PBS group (Figures 6I, J). Flow cytometry further confirms increased macrophage infiltration in the pancreas of the S. hominis group relative to the PBS group (Figures 6K, L).
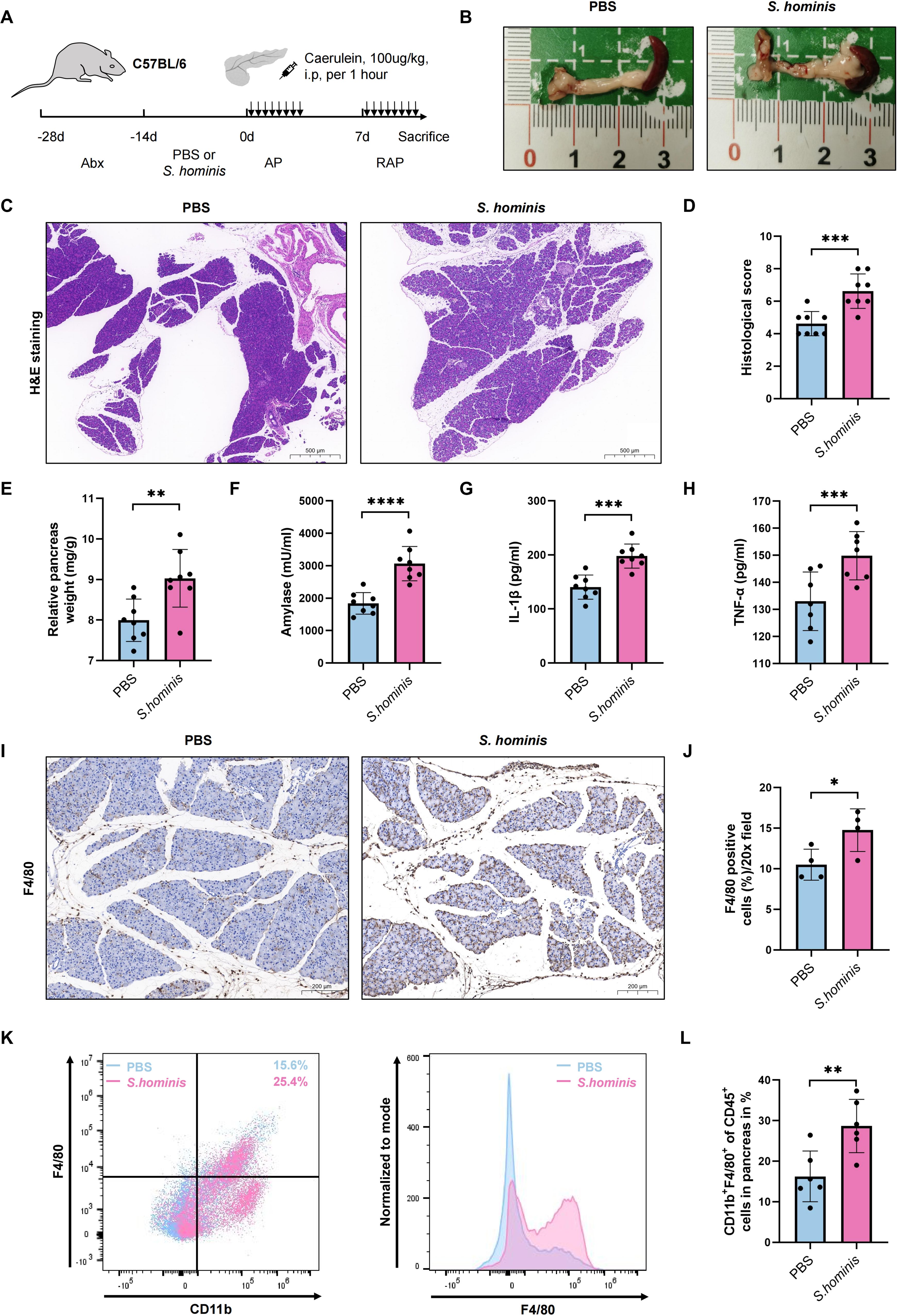
Figure 6. Impact of S. hominis on pancreatitis severity in a mouse RAP model. (A) Experimental design for PBS or S. hominis-gavaged mice in the RAP model; (B) Gross morphological appearance of pancreatitis; (C, D) H&E staining and histological scores of pancreatic tissues; (E) Relative pancreatic weight ratio; (F–H) Serum levels of amylase, TNF-α, and IL-1β; (I, J) Immunohistochemical detection of macrophage infiltration in the pancreas; (K, L) Quantification of CD11b+F4/80+cell infiltration in the pancreas via flow cytometry. Top bars: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Discussion
While the gut-pancreas axis has been extensively studied in pancreatitis pathogenesis—particularly focusing on gut dysbiosis-driven systemic inflammation, bacterial translocation, and entero-pancreatic metabolite crosstalk (26–28)—our work uncovers a previously unrecognized dimension of microbial influence: the blood microbiome as an independent mediator of RAP progression. Additionally, our findings suggest that real-time blood microbiome monitoring may offer clinical potential in acute care settings, underscoring its broader translational relevance compared to stool-based methods.
The clinical characteristics of the NRAP and RAP groups reveal subtle differences among microbial overall diversity, richness, and community structure. While the similar dominant phylum in blood microbiota, the nature of low-biomass rare taxa and technical ceiling may dilute subtle pathogen-specific shifts, LEfSe and functional analyses reveal that low-abundance taxa act in RAP through specific virulence and metabolic reprogramming.
The RAP group exhibited significant enrichment of Corynebacterium glucuronolyticum. Corynebacterium species, club-shaped gram-positive rods, are commonly found in animal hosts and are part of healthy human skin flora. Recently, they have recently been identified as causative agents of severe bloodstream infections, infective endocarditis, pneumonia and meningitis (29–32). Furthermore, growing evidence suggests that Corynebacterium species act as opportunistic pathogens in long-term hospitalized patients and have developed to drug resistance (33, 34). Its predominance in RAP suggests that they may be high-risk pathogens. Pseudomonas aeruginosa, identified by LEfSe as a top differential species, is well-known for causing acute or chronic infections in immunocompromised individuals and poses significant treatment challenges due to its rapid acquisition of drug resistance.
The enrichment of alanine, aspartate, and glutamate metabolis in the RAP group suggests a microbial role in modulating amino acid homeostasis, which is critical for pancreatic acinar cell survival and inflammation regulation. Elevated glutamate levels, for instance, are known to activate N-methyl-D-aspartate (NMDA) receptors in pancreatic neurons, exacerbating neurogenic inflammation and pain perception in chronic pancreatitis (35, 36). Similarly, the prominence of thiamine metabolism implies microbial involvement in energy production pathways, potentially compensating for oxidative stress-induced mitochondrial dysfunction in recurrent injury (37). Notably, the enrichment of FoF1-type ATP synthase highlights microbial adaptations to nutrient-depleted environments, possibly facilitating bacterial persistence in systemic circulation during recurrent attacks (38, 39). The positive correlation between Enhydrobacter abundance and AP attack frequency (r = 0.45, P < 0.001), which aligns with Szabó et al. observing Enhydrobacter abundance in septic patients (40). Lautropia’s strong association with prolonged hospitalization (r = 0.71) suggests its potential to cause invasive disease, which accords with another study obtaining five clinical strains of Lautropia from normally sterile sites (blood, peritoneal fluid) (41). Our Random Forest model achieves an AUC value of 0.759 with high sensitivity for predicting RAP. This underscores the potential for blood microbial profiling in clinical settings to predict and manage RAP effectively.
Staphylococcus hominis, another dominant species in the RAP group, represents the second most prevalent Coagulase-Negative Staphylococcus species (CoNS) colonizing human skin microbiota (42). However, it has been reported to be an opportunistic pathogen implicated in bloodstream infections, endocarditis and peritonitis (43, 44). Although the precise mechanisms by which S. hominis exacerbates RAP in our research remain to be fully elucidated, we hypothesize multiple synergistic mechanisms involved. First, its ability to form biofilms could facilitate colonization in necrotic pancreatic foci or pancreatic ducts (45). Biofilm-associated persistence might shield the bacterium from immune clearance and antibiotic penetration, thereby sustaining localized inflammation and tissue damage. Second, S. hominis-derived enzymes, including proteases and lipases (46), could directly degrade pancreatic parenchymal cells or amplify trypsinogen activation, potentially accelerating autodigestion and necrosis. Third, metabolites produced by S. homini could modulate macrophage polarization, skewing the immune response toward a hyperinflammatory state. Finally, in the context of pancreatitis-associated gut barrier dysfunction (47), S. hominis overgrowth in the gut-pancreas axis might disrupt microbial homeostasis, promoting bacterial translocation and secondary infection.
While our study provides valuable insights, there are several limitations to consider. First, the sample size was relatively small, which may limit the generalizability of our findings. Larger cohort studies are needed to validate these results. Second, a critical consideration in microbiome studies is the potential influence of pharmacological interventions. In our cohort, though the enrollment of patients accomplishes within 24 hours of disease onset, and patients within two months antibiotics use are excluded, medications may still confound our findings. Besides, our study design is cross-sectional, making it challenging to establish causality between the identified microbial signatures and RAP. Longitudinal studies would be beneficial to assess the temporal relationship between microbial changes and pancreatitis recurrence. In addition, while we identify exacerbation of RAP in S. hominis-infected mice, the underlying mechanisms by which this bacterium influences pancreatitis remain unclear. Future studies should focus on elucidating these mechanisms through in-depth functional analyses and experimental models.
In conclusion, our study first elucidates distinct circulating bacterial taxa in RAP, uncovering bacteria taxa which may drive disease recurrence, and laying the groundwork for developing microbiota-directed diagnostics and targeted therapies to disrupt the AP-RAP-CP continuum.
Data availability statement
The datasets presented in this study have been deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1224315. These data will be accessible starting from the publication date via the following link:https://www.ncbi.nlm.nih.gov/sra/PRJNA1224315.
Ethics statement
All animal care and experimental protocols were approved by the Animal Ethics Committee of the First Affiliated Hospital of Naval Medical University, Shanghai, China. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. All animal care and experimental protocols were approved by the Animal Ethics Committee of the First Affiliated Hospital of Naval Medical University, Shanghai, China. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
QW: Project administration, Writing – original draft. HZ: Data curation, Methodology, Writing – review & editing. YY: Data curation, Methodology, Writing – review & editing. XC: Writing – review & editing, Resources. ZD: Resources, Writing – review & editing. ZH: Writing – review & editing, Data curation. Z-SL: Writing – review & editing, Funding acquisition, Supervision. ZL: Funding acquisition, Supervision, Writing – review & editing, Project administration.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication this article. This work was supported by the National Natural Science Foundation of China (82330016).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1558983/full#supplementary-material
References
1. Mederos MA, Reber HA, Girgis MD. Acute pancreatitis: A review. JAMA. (2021) 325:382–90. doi: 10.1001/jama.2020.20317
2. Trikudanathan G, Yazici C, Evans Phillips A, Forsmark CE. Diagnosis and management of acute pancreatitis. Gastroenterology. (2024) 167(4):673–88. doi: 10.1053/j.gastro.2024.02.052
3. Ahmed Ali U, Issa Y, Hagenaars JC, Bakker OJ, van Goor H, Nieuwenhuijs VB, et al. Risk of recurrent pancreatitis and progression to chronic pancreatitis after a first episode of acute pancreatitis. Clin Gastroenterol Hepatol. (2016) 14:738–46. doi: 10.1016/j.cgh.2015.12.040
4. Li S, Gao L, Gong H, Cao L, Zhou J, Ke L, et al. Recurrence rates and risk factors for recurrence after first episode of acute pancreatitis: A systematic review and meta-analysis. Eur J Intern Med. (2023) 116:72–81. doi: 10.1016/j.ejim.2023.06.006
5. Lankisch PG, Breuer N, Bruns A, Weber-Dany B, Lowenfels AB, Maisonneuve P. Natural history of acute pancreatitis: a long-term population-based study. Am J Gastroenterol. (2009) 104:2797–805; quiz 806. doi: 10.1038/ajg.2009.405
6. Nøjgaard C, Becker U, Matzen P, Andersen JR, Holst C, Bendtsen F. Progression from acute to chronic pancreatitis: prognostic factors, mortality, and natural course. Pancreas. (2011) 40:1195–200. doi: 10.1097/MPA.0b013e318221f569
7. Takeyama Y. Long-term prognosis of acute pancreatitis in Japan. Clin Gastroenterol Hepatol. (2009) 7:S15–7. doi: 10.1016/j.cgh.2009.08.022
8. Yadav D, O'Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. (2012) 107:1096–103. doi: 10.1038/ajg.2012.126
9. Testoni PA. Acute recurrent pancreatitis: Etiopathogenesis, diagnosis and treatment. World J Gastroenterol. (2014) 20:16891–901. doi: 10.3748/wjg.v20.i45.16891
10. Whitcomb DC, Frulloni L, Garg P, Greer JB, Schneider A, Yadav D, et al. Chronic pancreatitis: An international draft consensus proposal for a new mechanistic definition. Pancreatology. (2016) 16:218–24. doi: 10.1016/j.pan.2016.02.001
11. Tan C, Ling Z, Huang Y, Cao Y, Liu Q, Cai T, et al. Dysbiosis of intestinal microbiota associated with inflammation involved in the progression of acute pancreatitis. Pancreas. (2015) 44:868–75. doi: 10.1097/MPA.0000000000000355
12. Hu X, Gong L, Zhou R, Han Z, Ji L, Zhang Y, et al. Variations in gut microbiome are associated with prognosis of hypertriglyceridemia-associated acute pancreatitis. Biomolecules. (2021) 11(5):695. doi: 10.3390/biom11050695
13. Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. (2015) 39:567–91. doi: 10.1093/femsre/fuv013
14. McLaughlin RW, Vali H, Lau PC, Palfree RG, De Ciccio A, Sirois M, et al. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J Clin Microbiol. (2002) 40:4771–5. doi: 10.1128/JCM.40.12.4771-4775.2002
15. Nikkari S, McLaughlin IJ, Bi W, Dodge DE, Relman DA. Does blood of healthy subjects contain bacterial ribosomal DNA? J Clin Microbiol. (2001) 39:1956–9. doi: 10.1128/JCM.39.5.1956-1959.2001
16. Paisse S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion. (2016) 56:1138–47. doi: 10.1111/trf.13477
17. Amar J, Lange C, Payros G, Garret C, Chabo C, Lantieri O, et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the D.E.S.I.R. study. PloS One. (2013) 8:e54461. doi: 10.1371/journal.pone.0054461
18. Dinakaran V, Rathinavel A, Pushpanathan M, Sivakumar R, Gunasekaran P, Rajendhran J. Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PloS One. (2014) 9:e105221. doi: 10.1371/journal.pone.0105221
19. Schierwagen R, Alvarez-Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, et al. Circulating microbiome in blood of different circulatory compartments. Gut. (2019) 68:578–80. doi: 10.1136/gutjnl-2018-316227
20. Lelouvier B, Servant F, Paisse S, Brunet AC, Benyahya S, Serino M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology. (2016) 64:2015–27. doi: 10.1002/hep.28829
21. Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. (2019) 10:5029. doi: 10.1038/s41467-019-13036-1
22. Kennedy EA, King KY, Baldridge MT. Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol. (2018) 9:1534. doi: 10.3389/fphys.2018.01534
23. Caballero S, Kim S, Carter RA, Leiner IM, Susac B, Miller L, et al. Cooperating commensals restore colonization resistance to vancomycin-resistant enterococcus faecium. Cell Host Microbe. (2017) 21:592–602 e4. doi: 10.1016/j.chom.2017.04.002
24. Geisz A, Sahin-Tóth M. Sentinel acute pancreatitis event increases severity of subsequent episodes in mice. Gastroenterology. (2021) 161:1692–4. doi: 10.1053/j.gastro.2021.06.013
25. Rongione AJ, Kusske AM, Kwan K, Ashley SW, Reber HA, McFadden DW. Interleukin 10 reduces the severity of acute pancreatitis in rats. Gastroenterology. (1997) 112:960–7. doi: 10.1053/gast.1997.v112.pm9041259
26. Li G, Liu L, Lu T, Sui Y, Zhang C, Wang Y, et al. Gut microbiota aggravates neutrophil extracellular traps-induced pancreatic injury in hypertriglyceridemic pancreatitis. Nat Commun. (2023) 14:6179. doi: 10.1038/s41467-023-41950-y
27. Qi-Xiang M, Yang F, Ze-Hua H, Nuo-Ming Y, Rui-Long W, Bin-Qiang X, et al. Intestinal TLR4 deletion exacerbates acute pancreatitis through gut microbiota dysbiosis and Paneth cells deficiency. Gut Microbes. (2022) 14:2112882. doi: 10.1080/19490976.2022.2112882
28. Li X, He C, Li N, Ding L, Chen H, Wan J, et al. The interplay between the gut microbiota and NLRP3 activation affects the severity of acute pancreatitis in mice. Gut Microbes. (2020) 11:1774–89. doi: 10.1080/19490976.2020.1770042
29. Díez-Aguilar M, Ruiz-Garbajosa P, Fernández-Olmos A, Guisado P, Del Campo R, Quereda C, et al. Non-diphtheriae Corynebacterium species: an emerging respiratory pathogen. Eur J Clin Microbiol Infect Dis. (2013) 32:769–72. doi: 10.1007/s10096-012-1805-5
30. Ishiwada N, Watanabe M, Murata S, Takeuchi N, Taniguchi T, Igari H. Clinical and bacteriological analyses of bacteremia due to Corynebacterium striatum. J Infect Chemother. (2016) 22:790–3. doi: 10.1016/j.jiac.2016.08.009
31. Otsuka Y, Ohkusu K, Kawamura Y, Baba S, Ezaki T, Kimura S. Emergence of multidrug-resistant Corynebacterium striatum as a nosocomial pathogen in long-term hospitalized patients with underlying diseases. Diagn Microbiol Infect Dis. (2006) 54:109–14. doi: 10.1016/j.diagmicrobio.2005.08.005
32. Tran TT, Jaijakul S, Lewis CT, Diaz L, Panesso D, Kaplan HB, et al. Native valve endocarditis caused by Corynebacterium striatum with heterogeneous high-level daptomycin resistance: collateral damage from daptomycin therapy? Antimicrob Agents Chemother. (2012) 56:3461–4. doi: 10.1128/AAC.00046-12
33. Navas J, Fernández-Martínez M, Salas C, Cano ME, Martínez-Martínez L. Susceptibility to Aminoglycosides and Distribution of aph and aac(3)-XI Genes among Corynebacterium striatum Clinical Isolates. PloS One. (2016) 11:e0167856. doi: 10.1371/journal.pone.0167856
34. Verroken A, Bauraing C, Deplano A, Bogaerts P, Huang D, Wauters G, et al. Epidemiological investigation of a nosocomial outbreak of multidrug-resistant Corynebacterium striatum at one Belgian university hospital. Clin Microbiol Infect. (2014) 20:44–50. doi: 10.1111/1469-0691.12197
35. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. (2000) 288:1765–9. doi: 10.1126/science.288.5472.1765
36. Bouwense SA, Buscher HC, van Goor H, Wilder-Smith OH. S-ketamine modulates hyperalgesia in patients with chronic pancreatitis pain. Reg Anesth Pain Med. (2011) 36:303–7. doi: 10.1097/AAP.0b013e3182177022
37. Palmieri F, Monne M, Fiermonte G, Palmieri L. Mitochondrial transport and metabolism of the vitamin B-derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD(+), and related diseases: A review. IUBMB Life. (2022) 74:592–617. doi: 10.1002/iub.v74.7
38. Escoll P, Platon L, Drame M, Sahr T, Schmidt S, Rusniok C, et al. Reverting the mode of action of the mitochondrial F(O)F(1)-ATPase by Legionella pneumophila preserves its replication niche. Elife. (2021) 10:e71978. doi: 10.7554/eLife.71978.sa2
39. Vlasov AV, Osipov SD, Bondarev NA, Uversky VN, Borshchevskiy VI, Yanyushin MF, et al. ATP synthase F(O)F(1) structure, function, and structure-based drug design. Cell Mol Life Sci. (2022) 79:179. doi: 10.1007/s00018-022-04153-0
40. Szabo BG, Kiss R, Makra N, Penzes K, Vad E, Kamotsay K, et al. Composition and changes of blood microbiota in adult patients with community-acquired sepsis: A pilot study from bench to bedside. Front Cell Infect Microbiol. (2022) 12:1067476. doi: 10.3389/fcimb.2022.1067476
41. Daneshvar MI, Douglas MP, Weyant RS. Cellular fatty acid composition of Lautropia mirabilis. J Clin Microbiol. (2001) 39:4160–2. doi: 10.1128/JCM.39.11.4160-4162.2001
42. Severn MM, Williams MR, Shahbandi A, Bunch ZL, Lyon LM, Nguyen A, et al. The Ubiquitous Human Skin Commensal Staphylococcus hominis Protects against Opportunistic Pathogens. mBio. (2022) 13:e0093022. doi: 10.1128/mbio.00930-22
43. Argemi X, Hansmann Y, Prola K, Prevost G. Coagulase-negative staphylococci pathogenomics. Int J Mol Sci. (2019) 20(5):1215. doi: 10.3390/ijms20051215
44. Szczuka E, Krzymińska S, Bogucka N, Kaznowski A. Multifactorial mechanisms of the pathogenesis of methicillin-resistant Staphylococcus hominis isolated from bloodstream infections. Antonie Van Leeuwenhoek. (2018) 111:1259–65. doi: 10.1007/s10482-017-1007-3
45. Paharik AE, Horswill AR. The staphylococcal biofilm: adhesins, regulation, and host response. Microbiol Spectr. (2016) 4(2). doi: 10.1128/microbiolspec.VMBF-0022-2015
46. Kolar SL, Ibarra JA, Rivera FE, Mootz JM, Davenport JE, Stevens SM, et al. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen. (2013) 2:18–34. doi: 10.1002/mbo3.2013.2.issue-1
Keywords: acute pancreatitis, recurrent acute pancreatitis, microbiome, 16S rRNA gene sequencing, Staphylococcus hominis
Citation: Wang Q-W, Zheng H, Yang Y, Chang X, Du Z, Hang Z-n, Li Z-S and Liao Z (2025) Distinct microbial signatures and their predictive value in recurrent acute pancreatitis: insights from 5-region 16S rRNA gene sequencing. Front. Immunol. 16:1558983. doi: 10.3389/fimmu.2025.1558983
Received: 11 January 2025; Accepted: 13 February 2025;
Published: 28 February 2025.
Edited by:
Minghua Ren, First Affiliated Hospital of Harbin Medical University, ChinaReviewed by:
Xinyi Jiang, Wuxi People’s Hospital, ChinaSher Zaman Safi, Mahsa University, Malaysia
Amita Joshi, Biobay Solutions, India
Copyright © 2025 Wang, Zheng, Yang, Chang, Du, Hang, Li and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuan Liao, bGlhb3podWFuQHNtbXUuZWR1LmNu; Zhao-Shen Li, emhhb3NoZW5saUBzbW11LmVkdS5jbg==
†These authors have contributed equally to this work