 Shunyu Xie
Shunyu Xie Xiaohui Wang
Xiaohui Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 April 2025
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1558386
This article is part of the Research Topic Deciphering Immune Responses in Infectious and Inflammatory Pathology View all 3 articles
The relationship between viral infection and autoimmune manifestations has been emerging as a significant focus of study, underscoring the intricate interplay between viral infections and the immune system. Influenza infection can result in a spectrum of clinical outcomes, ranging from mild illness to severe disease, including mortality. Annual influenza vaccination remains the most effective strategy for preventing infection and its associated complications. The complications arising from acute influenza infection are attributable not only to the direct effects of the viral infection but also to the dysregulated immune response it elicits. Notably, associations between influenza and various autoimmune diseases, such as Guillain-Barré Syndrome (GBS), Type 1 Diabetes (T1D), and antiphospholipid syndrome, have been reported. While viral infections have long been recognized as potential triggers of autoimmunity, the underlying mechanisms remain to be elucidated. Here, we described the pathophysiology caused by influenza infection and the influenza-associated autoimmune manifestations. Current advances on the understanding of the underlying immune mechanisms that lead to the potential strategies were also summarized.
Influenza viruses (IV), classified under the Orthomyxoviridae family, encompass three types—A, B, and C—that are pathogenic to humans (1). In the outbreak seasons subsequent to the 2009 H1N1 pandemic, the incidence of influenza-related hospitalizations has varied between 10 and 375 per 100,000 individuals per season, with the highest hospitalization rates observed in infants under 6 months of age (2). Influenza exerts a substantial disease burden on the pediatric population globally, characterized by elevated rates of hospitalization, morbidity, and mortality (3). Influenza infection typically manifests with upper respiratory tract symptoms, including fever, headache, cough, pharyngitis, and nasal congestion, as well as systemic symptoms such as malaise, myalgia, and muscle fatigue. Severe cases of influenza can lead to pneumonia and extrapulmonary complications, affecting the cardiovascular, nervous, musculoskeletal, and renal systems, and may result in multiple organ damage, shock, and sepsis (4). An increasing body of research has recently identified correlations between influenza infection and autoimmune diseases, such as Guillain–Barré syndrome (GBS) (5) and Type 1 diabetes (T1D) mellitus (6). Given the high incidence of influenza which can act as a trigger for autoreactivity and is implicated in the initiation of autoimmune manifestations, investigating the relationship between autoimmunity and influenza infection is of significant interest. The immune response plays a crucial role in both infectious and autoimmune diseases. Understanding the molecular mechanisms associated with influenza infection and autoimmunity could, if translated into clinical applications, expedite the development of diagnostic and therapeutic strategies. In this review, we will discuss the impact of the immune response on tissue damage induced by influenza infection, as well as the influenza-associated autoimmune diseases and various underlying mechanisms driving these autoimmune responses.
Influenza is an acute respiratory viral disease caused by viruses of the Orthomyxoviridae family, with three primary types affecting humans: influenza A, B, and C. These viruses are the predominant pathogens responsible for respiratory infections, leading to epidemics and pandemics that impose significant financial burdens, morbidity, and mortality globally (1). The upper respiratory tract symptoms, fever, headache and cough, are common manifestations of influenza infection in the majority of the population, with most cases resolving completely within 7-10 days. The spectrum of disorders resulting from influenza infection varies across different age groups, from infants to the elderly. Individuals aged 65 years or older, children under 5 years, pregnant women, those who are immunocompromised (7–12) (e.g., individuals with HIV, leukemia, or those taking immunosuppressants), and persons with chronic comorbidities (e.g., asthma, heart disease, liver disease, kidney disease, obesity) are at an elevated risk for severe illnesses such as tracheobronchitis and pharyngitis (13). Additionally, a small subset of these high-risk individuals may rapidly develop serious complications, including pneumonia and acute respiratory distress syndrome (ARDS), and even death with undefined reasons (14). In addition to respiratory injuries, extrapulmonary injuries were observed, including encephalitis (15), hepatic spotty necrosis accompanied by fatty degeneration in some hepatocytes (16), and focal infiltration of lymphocytes and phagocytes in the ileum or rectum (17). A portion of hospitalized influenza patients had developed acute kidney injury (18–20). Additionally, focal myocyte injury was found in the heart (21) (Figure 1). Previous studies have reported that influenza infection can result in autoimmune manifestations such as elevated autoantibodies against lung surfactant proteins and brain proteins, suggesting that the implications of influenza exposure may extend beyond the immediate effects of the virus (22). Thus, understanding these links is critical for optimizing patient outcomes and managing complications arising from influenza infections, particularly among vulnerable populations such as the children and the elderly.
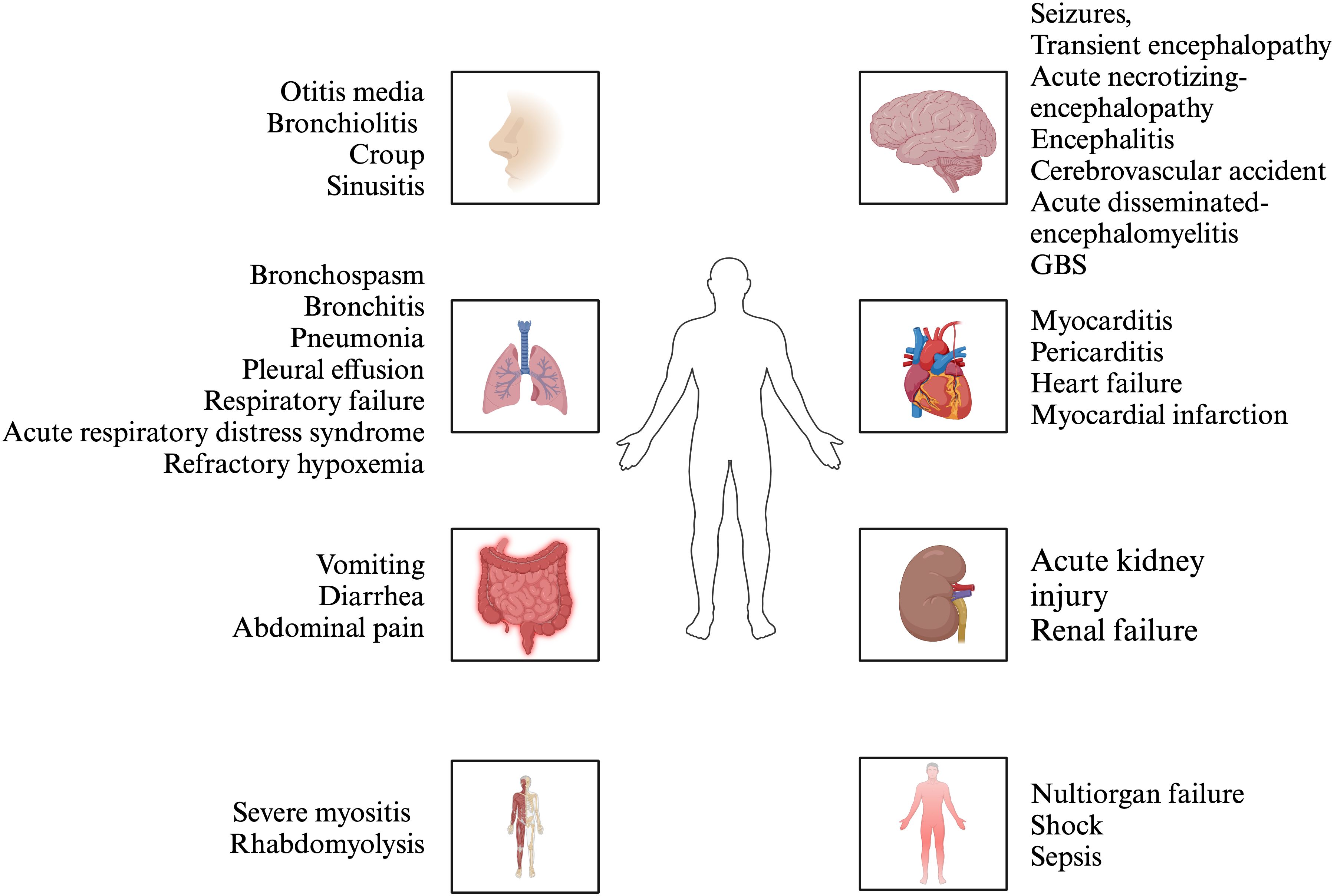
Figure 1. The manifestations of influenza. Influenza infection can affect multiple organs, including upper respiratory tract, lung, nervous system, cardiovascular system, digestive system, kidney, musculoskeletal system, etc. The manifestation of upper respiratory tract and lung is the predominant symptom.
In addition to the factors mentioned above that cause severe influenza-associated diseases, immune hyperactivation is considered as a driving factor of severe influenza and influenza-associated diseases. A prominent histopathological feature of influenza infection show congestion, inflammation, and epithelia necrosis of larger airways with lesser extension of the inflammatory process to alveoli (23). Severe influenza infection manifests atypical pneumonia with diffuse alveolar damage accompanied by intrapulmonary hemorrhage and edema, varying degrees of inflammatory infiltration including lymphocytes, phagocytes and mononuclear inflammatory cells (24–26). Monique et al. compared lung tissues from patients of diffuse alveolar damage due to influenza and non-pulmonary causes and found more significant infiltration of natural killer cells, granzyme A+ CD4+ and CD8+ T lymphocytes, and CD83+ dendritic cells, in influenza patients (27). Patients infected with influenza virus progressing to severe disease had higher concentration of cytokines and chemokines comparing to mild patients and healthy controls, such as interleukin (IL-6), IL-15, IL-8, monocyte chemoattractant protein (MCP)-1 and tumor necrosis factor (TNF)-α (28–31). In mouse model of influenza virus infection, combination of antivirals and etanercept which impairs TNF signaling evidently alleviate clinical symptoms and improve lung pathology (32). Profiles of host chemokine and cytokine responses to infections with different strains of influenza viruses were described. Highly pathogenic H5N1 infection was prone to induce CXCL-10/IP-10,TNF-α and CCL-5/RANTES compared with avian influenza H9N2 and seasonal human influenza H1N1 (33). The production profiles of the inflammatory cytokines TNF and IL-6 are comparable between influenza A and B viruses, while IFN-γ and IL-4 levels in influenza A patients were significantly higher than those in influenza B patients (34). Chemokines and cytokines exhibit prognostic potential as predictive indicators of disease progression and clinical outcomes. In addition, complement was activated manifested by the terminal C5b-9 complement complex (TCC) formation (35) in severe H1N1 pandemic influenza infections. C4d deposition, a marker of immune complex-mediated complement activation, was found in lung sections of deceased cases (36). It is suggested that hyperactive immune response may play a fundamental role in the pathogenesis of severe influenza and could be a predisposing factor in autoimmune diseases.
Influenza viruses infect respiratory epithelial cells, triggering innate immune response via pathogen-associated molecular pattern recognition receptor (PAMP) recognized by host pattern recognition receptor (PRRs) (37). Viral RNA replication activates pathways such as NF-κB and IRF3, leading to the release of early cytokines like IFN-α/β and TNF-α (38). These cytokines recruit immune cells, including macrophages (39), neutrophils (40) and T cells (41), which further amplify the inflammatory cascade. However, in some individuals, these responses become uncontrolled due to genetic predispositions, comorbidities, or viral virulence factors, resulting in a cytokine storm (14). The cytokine storm is marked by elevated levels of IL-6, IFN-γ, IL-1β, and TNF-α, which promote endothelial damages and vascular leakage (42, 43). IL-6, in particular, activates the JAK-STAT3 pathway, driving further cytokine production and immune cell infiltration. Paradoxically, studies have shown that early upregulation of SOCS3 (a negative regulator of IL-6/STAT3 signaling) occurs independently of IL-6 during influenza infection. SOCS3 deficiency in murine models exacerbates cytokine storm, suggesting its role in modulating excessive inflammation (44). Additionally, IFN-γ enhance recruitment of monocytes, contributing to alveolar epithelial injury and pulmonary edema (45). Elevated circulating cytokines such as IL-6, TNF-α, and IL-1β disrupt vascular endothelial integrity across multiple organs (46–48). IL-6 activates the JAK-STAT3 pathway in endothelial cells, upregulating adhesion molecules (e.g., ICAM-1, VCAM-1) and promoting leukocyte adhesion (49). TNF-α induces apoptosis via caspase-8 activation and increases vascular permeability by degrading tight junction proteins (e.g., claudins, occludins) (50). This allows inflammatory cells and cytotoxic mediators to infiltrate tissues, causing microvascular leakage and ischemia. In the heart, this process contributes to myocarditis and arrhythmias (51), while in the kidneys, it exacerbates acute kidney injury through tubular epithelial cell death (52). Influenza virus infection also results in the formation of immune complexes (ICs) through the binding of viral antigens to host antibodies (53, 54). Although ICs aid in viral neutralization, their excessive deposition in extrapulmonary tissues may contribute to systemic inflammation and multi-organ damage.
Interferons (IFNs) contribute to cell-intrinsic antiviral immunity through inducing hundreds of interferon-stimulated genes (ISGs) and enhance antiviral immune responses to facilitate viral clearance. In some patients, influenza virus could not induced sufficient type I and type III IFN responses, leading to uncontrolled influenza replication (55, 56). However, other studies demonstrated that type I interferons (Type I IFNs) also contribute to influenza virus-induced alveolar epithelial damages and lung injury via inducing expression of the pro-apoptotic factor tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (57). During Influenza A virus (IAV) infection, the IFN-γ is mainly derived from CD8+ T cells and regulates the recruitment of CCR2+ monocytes which mediate the lung tissue damage (58). Excessive IFN signaling induced the pathogenesis of lung during influenza virus infection (59). Some studies also suggested that IFN signaling disturbs with lung repairment after influenza infection in mice (45, 60). While type I IFNs perform a protective role in early stages of infection, further work is required to determine the roles of type I IFNs in different stages of influenza virus-mediated pathogenesis.
A series of cases about impaired type I IFNs immunity resulted from inborn error have been reported. It has been reported that patients with autosomal recessive (AR) interferon regulatory factor 7 (IRF7) deficiency suffer from life-threatening influenza pneumonia (55, 56). IRF7 is a transcription factor that is required for the production of IFNs in response to viruses (61). Plasmacytoid dendritic cells (pDCs) are the major cells of type I and III IFNs production with high levels of constitutive IRF7 expression (62). pDCs from these patients barely produced type I and III IFNs in response to IAV 24 hours post infection except IFN-β. Furthermore, dermal fibroblasts and induced pluripotent stem cell (iPSC)-derived pulmonary epithelial cells displayed increased influenza virus replication because of reduced production of type I IFNs. Inherited IRF9 deficiency manifests as severe pulmonary influenza, which depends on impaired IRF9 and ISGF3-dependent type I and type III IFN induction. The dermal fibroblasts from those patients can not sufficiently restrict influenza A virus replication (63). Giorgia Bucciol and colleagues reported that two children with AR signal transducer and activator of transcription (STAT) 2 deficiency suffered from influenza A infection developing to ARDS at 9 months of age, one of two patients died of overwhelming infection at 5 years old (64). The patients displayed hyperinflammation attributing to uncontrolled viral infection in the absence of STAT2-dependent type I and III IFN immunity. The cells from those patients and transfected with mutant STAT2 alleles conformed the phenomenon of impaired virus control (64). Three children with AR STAT1 deficiency suffered from severe influenza at 1 month and 6 months after birth was reported by Tom Le Voyer and colleagues. STAT1 is a transcription factor critical for mediation of types I, II, and III IFN responses in cells, the deficiency of the gene impairs these responses (65). Hye Kyung Lim and colleagues reported three children with inherited TLR3 deficiency developed ARDS. The pulmonary epithelial cells (PECs) and fibroblasts from these patients are susceptible to IAV attributing to lower levels of IFN-β and -λ, whereas TLR3-mutated leukocytes can produce normal level of IFNs (66). In summary, these cases demonstrate a pivotal role of intrinsic and innate type I and III IFN immunity in host defense against influenza and the associated genetic deficiency in impaired IFNs responses leading to severe influenza.
Interferon-neutralizing antibodies were discovered in a patient treated with human leukocyte interferon since 1981 (67). In viral diseases, neutralizing autoantibodies against type I IFN were firstly identified in a woman with disseminated shingles (68). It has long been thought that these autoantibodies have no pathological consequences until the COVID-19 pandemic. Patients with autoimmune polyendocrine syndrome type 1 (APS-1), which arises due to mutations in the autoimmune regulator gene (AIRE), exhibit antibodies against type I IFNs (IFN-α and IFN-ω) (69). The majority of patients with APS-1 infected with SARS-CoV-2 developed severe COVID-19 pneumonia, further confirmed that preexisting autoantibodies against type I IFN are related with life-threatening COVID-19 pneumonia (70). Moreover, autoantibodies against type I IFNs also contributed to live attenuated yellow fever virus vaccine associated life-threatening disease (71). In a cohort of 279 patients aged 6 to 73 years diagnosed with critical influenza pneumonia, neutralizing autoantibodies against type I IFNs were identified in 4.7% of the patients. This prevalence indicates a notable enrichment in severe influenza pneumonia compared to the general population. Individuals under the age of 70 who possess autoantibodies exhibit a heightened risk of developing severe influenza pneumonia compared to those who are negative for autoantibodies (72). The autoantibodies in patients with critical influenza pneumonia primarily targeted IFN-α2 and IFN-ω, but not IFN-β (72). Altogether, current data indicate a role of type I IFNs immune disruption in viral infection-induced tissue pathogenesis (Figure 2).
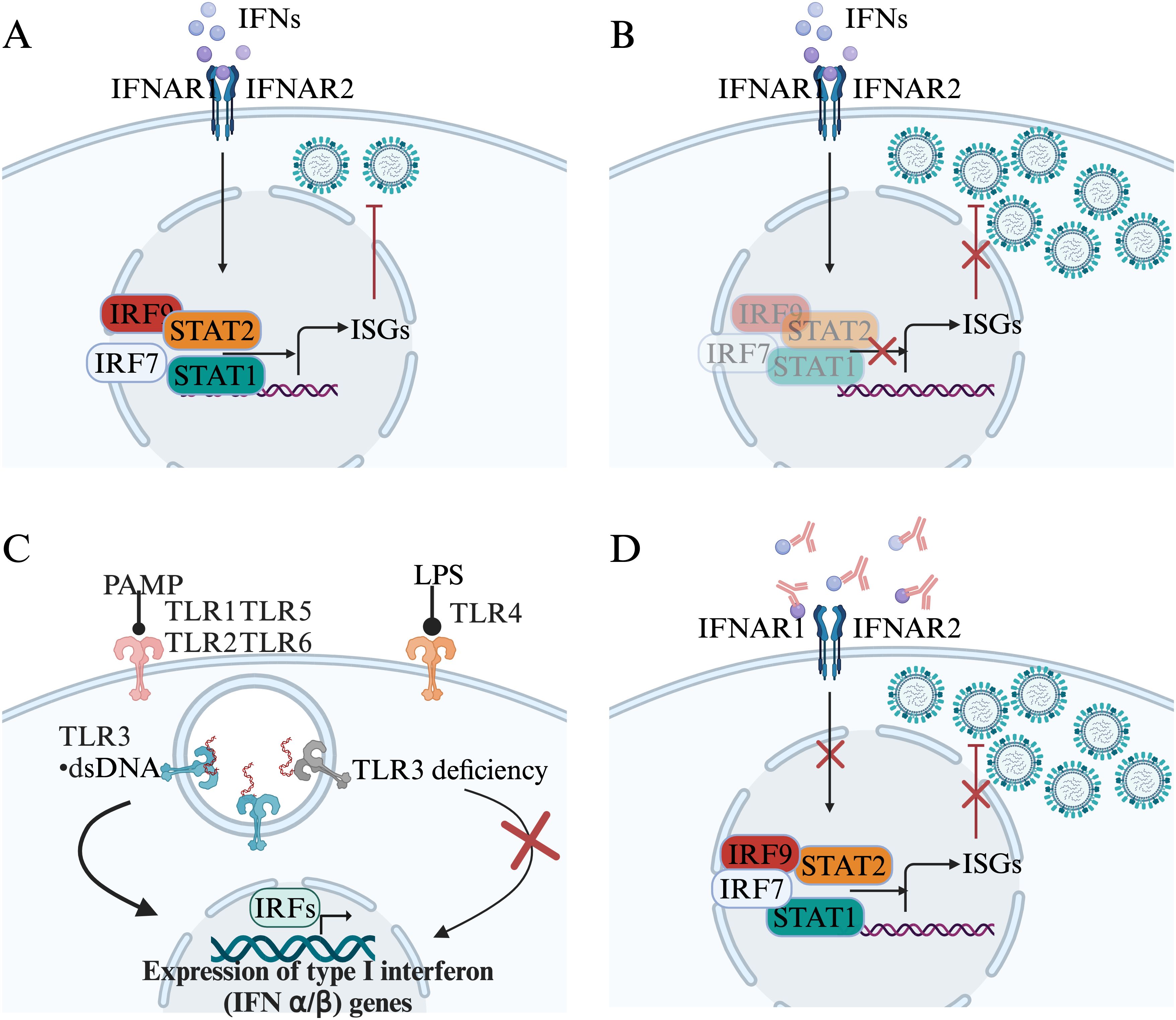
Figure 2. Genetics and autoantibodies associates with type I IFNs immunity disruption. (A). Interferons (IFNs) contribute to cell-intrinsic antiviral immunity. (B). The deficiency of IRF9, IRF7, STAT1, or STAT2 disturbs IFNs responses. (C). The deficiency of TLR3 impairs the expression of IFNs. (D). Autoantibodies target IFNs disturb the type I IFN response and induce uncontrol virus replication.
Clinical and laboratory evidence indicate critical participation of immune reaction in severe cases of influenza. A study compared the concentration of inflammatory cytokines in severe and mild patients with IAV infection. IL-6, IL-10, IL-15, IP-10, IL-2R, HGF, ST2 and MIG were detected at higher levels in the plasma of severe patients (29). Daniel et al. had examined cases including severe, moderate influenza and healthy controls with bronchoalveolar lavage (BAL) samples. They reported a significantly elevated level of IL-6, IL-8, MCP-1, MIG, IP-10, IL-12, MIP-1ß and IL-1, which enhance pro-inflammatory T help 1 (Th1) immune responses (73). Moreover, total immune cells were increased in severe cases compared to moderate influenza and controls (73). They also found the elevation of CD14+ monocytes and plasmablasts, which indicated that autoantibodies might contribute to the lung injury. Although the number of total T cells and CD4/CD8+ subpopulations showed no differences from controls, the activated CD8+ and CD4+ T cells were elevated markedly in severe cases (73). Early secretion of Th17 (IL-8, IL-9, IL-17, IL-6) and Th1 cytokines (TNF-α, IL-15, IL-12p70) were detected in severe influenza patients, which were involved in cell-mediated immunity, may be associated with pathogenesis and autoimmune diseases induced by influenza (74). Hemophagocytic lymphohistiocytosis (HLH) is a syndrome characterized by severe systemic hyperinflammation presented by fevers, pancytopenia and hepatosplenomegaly (75). Hemophagocytic lymphohistiocytosis (HLH) is reported in critically ill patients with influenza virus infection (76–78). This further demonstrated that activation and proliferation of lymphocytes accompanied by excessive production of cytokines induced by influenza may lead to self-tissue damages. Taking into account the above findings regarding immune dysregulation in patients with severe influenza, the autoimmune process in the course of influenza viruses infection deserves an increasing attention.
Viral infections have been proposed as potential triggers of autoimmunity, and an increasing body of evidence indicates a significant association between specific viral infections and the development of various autoimmune diseases, such as type 1 diabetes mellitus with coxsackievirus (79), cytomegalovirus (CMV) (80), enteroviruses (81), as well as systemic lupus erythematosus (SLE) with hepatitis C virus (HCV) (82), CMV (83), dengue virus (84), and parvovirus B19 (85). Although with the popularity of influenza vaccination, while nearly 10% of the world’s population with all ages is still affected by influenza annually (86). Influenza-associated immune diseases may affect a huge crowd worldwide and cause a tremendous socio-economic burden. Here we review some autoimmune diseases that reported are related to influenza infection (Figure 3).
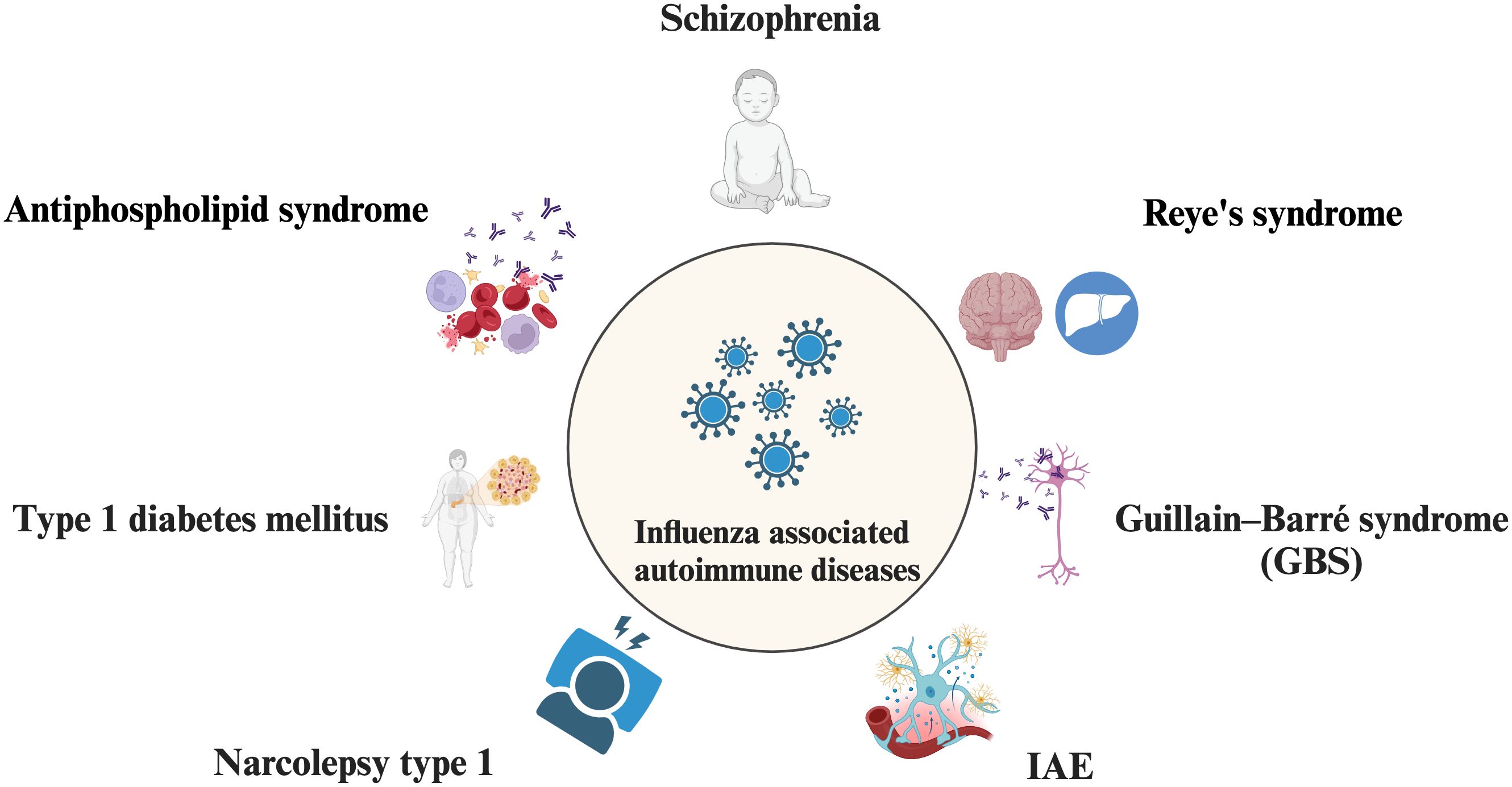
Figure 3. Influenza-associated autoimmune diseases. Influenza infection is involved in autoimmune disease such as Schizophrenia, Reye’s syndrome, Guillain–Barré syndrome (GBS), Influenza‐associated encephalopathy or encephalitis (IAE), Narcolepsy type 1 (NT1), Type 1 diabetes mellitus (T1D), Antiphospholipid syndrome.
Schizophrenia is a functional psychotic disorder characterized by a spectrum of symptoms, including hallucinations, delusions, disorganized thought processes and behavior, as well as diminished cognitive and emotional capacities (87). Many studies reported the link between influenza and psychosis since 1900s. Many reports suggested that the increased risk of psychosis in offspring was associated with maternal influenza infection (88). Comparing schizophrenic patients with healthy controls, 15% of the patients had influenza antibodies in the central nervous system (89). Guglielmo et al. reported axon guidance molecules have a vast pentapeptide overlap with Influenza hemagglutinin (HA), immune cross-reactivity of axon guidance molecules and virus proteins is one potential mechanism by which influenza could contribute to autoimmunity (90).
Reye’s syndrome is distinguished by hepatic pathology and non-inflammatory encephalopathy (91). The incidence of Reye’s syndrome following influenza B was between 0.03-0.06% in population of influenza B infection. 86% cases were reported associated with the outbreak of influenza B in America (92). The recent study reported a cohort including 29,676 influenza-associated hospitalizations, which assessed the incidence of Reye’s syndrome about 0.01% in US (93). The decreased incidence might attribute to reduced use of aspirin (94). Experiments on mouse model have shown that the excessive proliferation of lymphocytes is dose-dependent on aspirin, and it is believed that Reye’s syndrome may be caused by aspirin inducing aberrant immune responses to viral infection (95). In the meanwhile, case of Reye’s syndrome caused by influenza A without the administration of aspirin has also been reported, suggesting other mechanisms leading to this disease and immune responses may contribute to the development of the disease (96).
Guillain–Barré syndrome (GBS), an immune-mediated disorder impacting the peripheral nervous system and affecting approximately 100,000 individuals annually worldwide, represents the most prevalent and severe form of acute paralytic neuropathy (97). The clinical manifestations of GBS are heterogeneous, and several different clinical variants exist, but patients with GBS typically present with weakness and sensory symptoms in the legs, progressing to the arm and head muscles (98).Valérie et al. reported a cohort of 405 patients, 234 cases of which caused by an unidentified agent had a positive association with influenza-like illnesses (99). Some reports yielded a similar conclusion that a positive correlation of hospitalization rate was found between influenza and GBS (100, 101). The recent study manifested that 53% patients of GBS were confirmed recent infection by serology, of which influenza viruses accounting for 33% (102). Masaki et al. investigated 63 patients of Guillain-Barré syndrome (GBS)-related diseases (GBSRDs) after influenza virus infection, detecting anti-GQ1b and anti-GT1a which are grouped to anti-glycolipid antibodies accounting for 24% of cases (103). Debprasad et al. reported that the levels of antibodies targeting GM1, GM2, GD1a, GD1b, GT1b, and GQ1b were significantly elevated in patients with GBS compared to healthy controls, and these elevated antibody levels were correlated with immunoreactivities against influenza viruses (104). Influenza infection may be a trigger for GBS, mediated by immune responses. The underlying mechanisms between GBS and influenza will require further studies.
Influenza‐associated encephalopathy or encephalitis (IAE) is a disorder characterized by consciousness disturbance with a few days after influenza infection (105). Its clinical characteristics are rapid progressive brain damage after viral infection, and pathologically by cerebral edema without direct invasion of viruses and inflammatory cells (106). The majority of IAE cases are reported among children worldwide. Hideo et al. reported that during 2010-2015 in Japan, the incidence of IAE among children and adults was 2.83 and 0.19 cases per 100,0000 population, respectively, whereas the morality was higher in adults (107). Pierre et al. reported a cohort of 41 children admitted for influenza-associated encephalopathy between 2010 and 2019 in France, of which 17% patients died in hospital, 49% had neurologic sequelae and 27% had severe disabilities according to modified Rankin Score (108). Ayukawa et al. reported that in patients with IAE, there was a significant increase in CTLA-4+ CD4+ T cells compared to influenza patients without encephalopathy. This increase was correlated with the down-regulation of antigen-activated immune responses, suggesting that CTLA-4+ CD4+ T cells may play a role in the pathogenesis of IAE (109). Shunji et al. reported that the cytokines of IL-6, TNF-α, and IL-10 were higher in patients with 2009 pandemic H1N1 influenza-associated encephalopathy than those without neurological sequelae (110). All of the above indicated that immune responses might participate in the progression of IAE.
Narcolepsy type 1 (NT1) is a chronic sleep disorder characterized by the degeneration of a specific subset of hypothalamic neurons responsible for the production of hypocretin (HCRT; also referred to as orexin, peptides) that promote wakefulness (111). Narcolepsy is highly correlated with H1N1 influenza independent of H1N1 vaccination (112). The estimated incidence was at least 10 per 100,000 individuals per year (113). NT1 is considered as autoimmunity associated with human leukocyte antigen (HLA) DQB1*06:02/DQA1*01:02 heterodimer (DQ0602). Guo et al. proposed a mechanism of molecular mimicry with influenza antigens modulated by genetic components in the pathogenesis of NT1 (114). T cells specific to tribbles homologue 2, an additional self-antigen of hypocretin neurons, were identified in patients with narcolepsy, thereby reinforcing the autoimmune etiology of the disorder (115).
Type 1 diabetes mellitus (T1D) is a chronic autoimmune disorder marked by insulin deficiency and hyperglycemia, resulting from the destruction of pancreatic β cells (116). Paz et al. reported that a twofold higher risk of subsequent T1D was detected in the group of pandemic influenza A infection (117). Yuichi et al. also reported the similar conception that the risk of T1D increased after influenza in the Japanese population-based cohort (118). Lijun et al. found a cross-reactive antibody between H1N1 influenza virus HA and human pancreatic tissue, suggesting the immune-mediated tissue damage caused by influenza (119).
Antiphospholipid syndrome is an autoimmune disorder characterized by the presence of pathogenic autoantibodies that target cell surface phospholipids and phospholipid-binding proteins (120). Melonie et al. reported a case of catastrophic antiphospholipid syndrome (CAPS), suggesting a link between CAPS and influenza (121). Further studies are required to elucidate the underlying mechanisms of autoimmunity induced by influenza.
Autoimmune responses arise when the immune system erroneously targets and damages healthy cells (122). These conditions are believed to stem from a multifaceted interaction of genetic susceptibilities, environmental influences, and dysregulated immune responses (123). Current research suggests that autoimmune disorders impact approximately 10% of the global population, encompassing over 80 identified types, such as rheumatoid arthritis, type 1 diabetes, and multiple sclerosis (124). Notably, autoimmune diseases tend to co-occur more frequently than would be expected by chance, indicating that some autoimmune diseases may share common risk factors. This co-occurrence was particularly evident among rheumatic and endocrine diseases (123). As mentioned above, viral infections are related to the onset or development of some autoimmune diseases. Here, we explore a few possible mechanisms of infection associated autoimmune diseases (Figure 4).
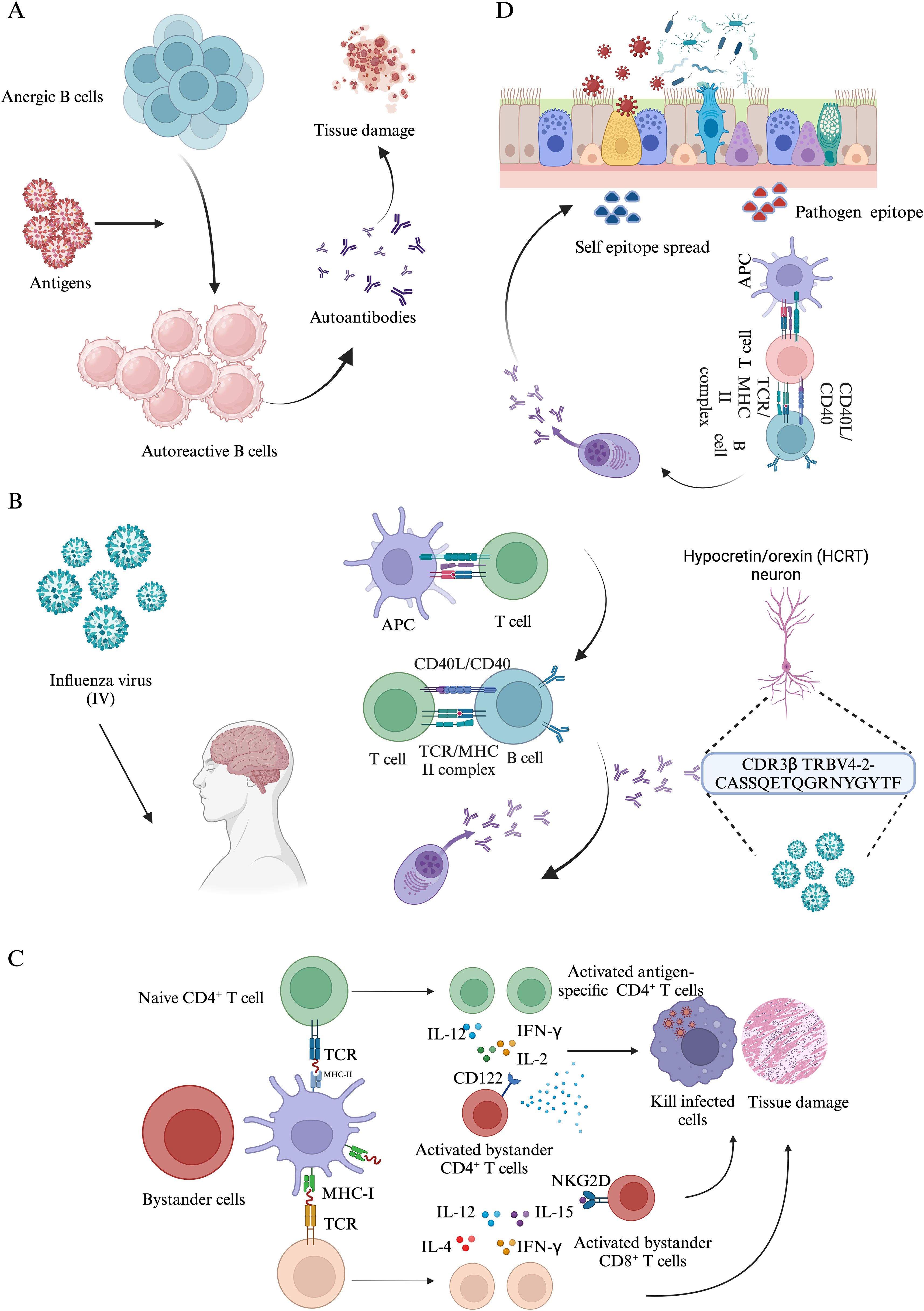
Figure 4. The mechanisms of autoimmune response induced by pathogen infections. (A). Anergic B cells reversion. (B). The molecular mimicry between Influenza virus (IV) and Narcolepsy type 1 (NT1). (C). Bystander activation mechanisms induced by pathogen infections. (D). During infections, self-epitope spreading distinct from the epitopes of initial pathogen contributes to tissue damages.
Autoantibodies are considered as the most prominent immunological manifestation of autoimmune diseases, which serve as biomarker for diagnosis, classification and the progression of disease (125). Allan et al. detected autoantibodies against antigens such as IL-6, IL-7, IL-12p70, and IL-22 in 48% of hospitalized patients with influenza, whereas none were found in the healthy control group. Furthermore, antibodies linked to rare connective tissue diseases (CTDs) were prevalent in 25% of the influenza patients in America (126). Autoantibodies were prevalent in SARS-CoV-2 patients with acute respiratory distress syndrome as well as non-SARS-CoV-2 infections of patients with severe pneumonia (127, 128). Moreover, the presence of autoantibodies in patients with influenza and COVID-19 has been correlated with the severity of these diseases. These autoantibodies specifically target Type I IFN, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-6 (127, 129). B cells play an essential role in host defense by secreting antibodies and presenting antigens from foreign microorganisms to T cells (130). Depletion of B cells tends to be favorable in autoimmune diseases such as systemic lupus erythematosus (SLE) (131) and rheumatoid arthritis (132), probably through reduced production of autoantibodies as well as antigen presentation to autoreactive T cells. After central tolerance mediated clonal deletion, 15%-20% of mature B cells have a capacity reactive with self-antigens, especially in newly formed B cells, of which the proportion up to 50% (133). In healthy individuals, the underlying autoreactive B cells are functionally silenced by the mechanism of anergy. However, the anergic state could be reversible. In pathological settings, the anergic B cells reactivate and produce autoantibodies against host tissue such as dsDNA and ssDNA. Noorchashm et al. reported that the anergic B cells are partially reversible upon stimulation of IL-4 and CD40 ligands derived from T cells associated factors (134). Peter et al. reached a similar conclusion that the anergic state of B cells have the potential to transfer to autoantibodies-secreting plasma cells and contribute to autoimmunity (135). Altogether, anergy reversion contributes to the transfer of anergy B cells to auto-reactive B cells, which secrete autoantibodies to damage tissues. Anergy reversion of B cells contributes but not sufficient to explain viral infections implicated in the development or exacerbation of autoimmune disease (136). Further studies may provide new insights in understanding the role of B cells during the development of infection-induced autoimmunity.
The term “molecular mimicry” was formally introduced by Damian in 1964 to describe the phenomenon wherein infectious organism express antigens that are structurally similar to those of their host. This similarity may confer an advantage to the microbes by enabling them to evade the host’s immune response (137). Molecular mimicry occurs when viral antigens share structural similarities with host-antigens, leading to the cross-reactive immune response. This phenomenon has been noted in cases involving varicella zoster virus (VZV) (138) and Epstein-Barr virus (EBV) (139), which were thought to play roles in the pathogenesis of diseases like multiple sclerosis. There are four types of molecular mimicry that have been reported (140–143). Type 1 molecular mimicry: “microorganisms and their hosts have complete protein identity” (e.g., in chronic graft-versus-host disease, CMV hijacks CD13 and presents it to antigen-presenting cells to trigger autoimmune responses against the autoantigen); Type 2 molecular mimicry: “molecular homology between microorganisms and their hosts, based on a protein encoded by the bacteria” (e.g., human carbonic anhydrase II and alpha-carbonic anhydrase of Helicobacter pylori share a homologous binding motif with the HLA molecule DRB1*0405, which is linked to autoimmune pancreatitis.); Type3 molecular mimicry: microorganisms or environmental agents that share similar amino acid sequences or epitopes with its host; Type 4 molecular mimicry: “microbes or environmental agents have structural similarities to their hosts”. Among them, type 3 is the most prevalent type in eliciting autoimmune responses. However, molecular mimicry mediated autoimmunity is rare and complex, which is not sufficient to breach host immune tolerance mechanism (144), suggesting other factors are involved in the autoimmunity.
Bystander activation was first described by Tough et al. in 1996. They found that a massive expansion of T cells after viral infection was mediated by cytokines other than T cell receptor (TCR) and type I IFN was a robust inducer (145). Zarozinski et al. and Murali-Krishna et al. also found that the majority of clonally expanded T cells were virus-specific but of which include a small proportion of non-virus-specific clones (146–148). Memory CD4+ and CD8+ T could be bystander activated and proliferate in IFN-γ or IL-12-dependent but TCR-independent manner (149). The bystander activation of naïve CD8+ T also occurs during the early phase of infection and displays an innate anti-viral feature (150). The bystander activation of naïve CD4+ T also could be induced by high-dose IL-2 but independent on TCR activation (151). However, bystander activation is a double-edged sword. Martin et al. reported that bystander CD8+ T cells were activated by inflammatory cytokines following infection and provided protective roles in host defenses (152). Preexisting non-specific memory CD8+ T cells are activated rapidly and display cytotoxic features following infections, which target infectious cells and contribute to pathogen clearance in an NKG2D-dependent manner (153). Rolot et al. made a similar finding that IL-4 rapidly expands non-specific CD8+ T cells, which are essential to the activation of antigen-specific CD8+ T cells for controlling virus load (154). On another hand, Bergamaschi et al. reported that hospitalized individuals of COVID-19 manifested delayed bystander CD8+ T cells activation which might drive lung pathology, suggesting a role of immunopathogenesis in severe pneumonia (155). Zhang et al. found that high level of IL-15 drove bystander activation of CD8+ T cells through NKG2D, which mediated endothelium injury in the Hantana virus (HTNV) infection model (156). Ohya et al. analyzed 23 autopsy specimens come from patients of diffuse alveolar damage, in which large amount of non-antigen-activated bystander CD8+ T cells were observed. These CD8+ T cells expressed unique granzyme B marker and might involve in the tissue injury in the progression of diffuse alveolar damage (157). Lee et al. reported bystander-activated CD4+ T cells also contributed to the progression of autoimmune diseases (158). Bystander activation of T cells plays an essential role in antigens clearance and also causes tissues injuries. Further studies are required to elucidate molecular mechanisms involved in different immune responses to onset and/or exacerbations of infectious diseases.
Epitope spreading comprises intramolecular and intermolecular spreading. Intramolecular spreading refers to the phenomenon where an immune response extends from the initially targeted epitope to additional epitopes within the same molecule. In contrast, intermolecular spreading describes the process by which an immune response broadens to include epitopes on a different antigenic molecule (159). The etiology of autoimmune diseases remains incompletely elucidated, with numerous factors influencing the development of autoimmunity. The involvement of the epitope spreading mechanism is implicated in human diseases such as autoimmune hepatitis and primary biliary cholangitis (160, 161). This study provides evidence that viral infections can induce autoimmune diseases through epitope spreading. Specifically, chronic infection with Theiler’s murine encephalomyelitis virus (TMEV) in mice resulted in demyelination, which was initiated by a TMEV-specific CD4+ T cell response. The subsequent T-cell response to multiple myelin autoepitopes was secondary to the virus-specific T cell response and arose from epitope spreading rather than molecular mimicry (162). Epitope spreading following viral infection may play an important role in the development of autoimmune pathogenesis.
Influenza exerts a significant impact on global health through its annual epidemics and occasional pandemics. Autoimmune pathogenesis, characterized as chronic and disabling conditions, impose a considerable burden on individuals, families, and society. While infections have frequently been implicated in the development of autoimmune diseases, they are not sufficient on their own to trigger these conditions. Factors such as genetic predispositions, defects in the innate and adaptive immune systems, and gender are known to increase the risk of autoimmune diseases. Both influenza and other viral infections such as COVID-19 have been reported to be associated with autoimmune responses and the onset of autoimmune diseases. The underlying mechanisms remain to be elucidated. Further clinical and basic investigations are necessary to uncover the mechanisms underlying autoimmune diseases following viral infections. Such research will be crucial for the prevention of post-infection autoimmunity and may offer insights for the development of immune cell-based therapeutic strategies for the treatment of autoimmune diseases.
SX: Data curation, Formal Analysis, Writing – original draft. JW: Writing – review & editing. XW: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82201946) to XW, Natural Science Foundation of Guangdong Province (2314050000724) to XW, Guangzhou Municipal Science and Technology Bureau (SL2022A03J01156) to XW, Plan on Enhancing Scientific Research in Guangzhou Medical University (02-410-2302152XM) to XW. Jointly funded project by Guangzhou National Laboratory - State Key Laboratory of Respiratory Disease (GZNL2024B01006) to XW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Uyeki TM, Hui DS, Zambon M, Wentworth DE, Monto AS. Influenza. Lancet. (2022) 400:693–706. doi: 10.1016/S0140-6736(22)00982-5
2. Kamidani S, Garg S, Rolfes MA, Campbell AP, Cummings CN, Haston JC, et al. Epidemiology, clinical characteristics, and outcomes of influenza-associated hospitalizations in US children over 9 seasons following the 2009 H1N1 pandemic. Clin Infect Dis. (2022) 75:1930–9. doi: 10.1093/cid/ciac296
3. Nayak J, Hoy G, Gordon A. Influenza in children. Csh Perspect Med. (2021) 11:a038430. doi: 10.1101/cshperspect.a038430
5. Grimaldi-Bensouda L, Alpérovitch A, Besson G, Vial C, Cuisset J-M, Papeix C, et al. Lucien Abenhaim for the GBS-PGRx Study Group. Guillain-Barre syndrome, influenzalike illnesses, and influenza vaccination during seasons with and without circulating A/H1N1 viruses. Am J Epidemiol. (2011) 174:326–35. doi: 10.1093/aje/kwr072
6. Karaoglan M, Eksi F. The coincidence of newly diagnosed type 1 diabetes mellitus with igM antibody positivity to enteroviruses and respiratory tract viruses. J Diabetes Res. (2018) 2018:8475341. doi: 10.1155/2018/8475341
7. Ramos-Rincón J-M, Pinargote-Celorio H, González-de-la-Aleja P, Sánchez-Payá J, Reus S, Rodríguez-Díaz J-C, et al. Impact of influenza related hospitalization in Spain: characteristics and risk factor of mortality during five influenza seasons (2016 to 2021). Front Public Health. (2024) 12:1360372. doi: 10.3389/fpubh.2024.1360372
8. Louie JK, Acosta M, Samuel MC, Schechter R, Vugia DJ, Harriman K, et al. California Pandemic (H1N1) Working Group. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1). Clin Infect Dis. (2011) 52:301–12. doi: 10.1093/cid/ciq152
9. Vitoratou D-I, Milas G-P, Korovessi P, Kostaridou S, Koletsi P. Obesity as a risk factor for severe influenza infection in children and adolescents: a systematic review and meta-analysis. Eur J Pediatr. (2023) 182:363–74. doi: 10.1007/s00431-022-04689-0
10. Mertz D, Geraci J, Winkup J, Gessner BD, Ortiz JR, Loeb M. Pregnancy as a risk factor for severe outcomes from influenza virus infection: A systematic review and meta-analysis of observational studies. Vaccine. (2017) 35:521–8. doi: 10.1016/j.vaccine.2016.12.012
11. Dupont A, Couffignal C, Arias C, Salah K, Phillips-Houlbraq M, Le Brun M, et al. Outcomes and risk factors with COVID-19 or influenza in hospitalized asthma patients. Resp Res. (2022) 23:342. doi: 10.1186/s12931-022-02265-6
12. Mertz D, Lo CK-F, Lytvyn L, Ortiz JR, Loeb M, FLURISK-INVESTIGATORS. Pregnancy as a risk factor for severe influenza infection: an individual participant data meta-analysis. BMC Infect Dis. (2019) 19:683. doi: 10.1186/s12879-019-4318-3
14. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol. (2021) 19:425–41. doi: 10.1038/s41579-021-00542-7
15. Ghaderi S, Størdal K, Gunnes N, Bakken IJ, Magnus P, Håberg SE. Encephalitis after influenza and vaccination: a nationwide population-based registry study from Norway. Int J Epidemiol. (2017) 46:1618–26. doi: 10.1093/ije/dyx149
16. Nonaka K, Matsuda Y, Kakizaki M, Takakuma S, Hamamatsu A, Sakashita Y, et al. Acute liver failure associated with influenza A virus infection: an autopsy case report. Jpn J Infect Dis. (2019) 72:347–9. doi: 10.7883/yoken.JJID.2018.494
17. Li H, Liu X, Chen F, Zuo K, Wu C, Yan Y, et al. Avian influenza virus subtype H9N2 affects intestinal microbiota, barrier structure injury, and inflammatory intestinal disease in the chicken ileum. Viruses. (2018) 10:270. doi: 10.3390/v10050270
18. Bhasin B, Veitla V, Dawson AZ, Garacci Z, Sturgill D, Ozieh MN, et al. AKI in hospitalized patients with COVID-19 and seasonal influenza: A comparative analysis. Kidney360. (2021) 2:619–28. doi: 10.34067/KID.0007322020
19. Sood MM, Rigatto C, Zarychanski R, Komenda P, Sood AR, Bueti J, et al. Acute kidney injury in critically ill patients infected with 2009 pandemic influenza A(H1N1): report from a Canadian Province. Am J Kidney Dis. (2010) 55:848–55. doi: 10.1053/j.ajkd.2010.01.011
20. Pettilä V, Webb SAR, Bailey M, Howe B, Seppelt IM, Bellomo R. Acute kidney injury in patients with influenza A (H1N1) 2009. Intensive Care Med. (2011) 37:763–7. doi: 10.1007/s00134-011-2166-8
21. Kenney AD, Aron SL, Gilbert C, Kumar N, Chen P, Eddy A, et al. Influenza virus replication in cardiomyocytes drives heart dysfunction and fibrosis. Sci Adv. (2022) 8:eabm5371. doi: 10.1126/sciadv.abm5371
22. Needham EJ, Ren AL, Digby RJ, Norton EJ, Ebrahimi S, Outtrim JG, et al. Brain injury in COVID-19 is associated with dysregulated innate and adaptive immune responses. Brain. (2022) 145:4097–107. doi: 10.1093/brain/awac321
23. Guarner J, Falcón-Escobedo R. Comparison of the pathology caused by H1N1, H5N1, and H3N2 influenza viruses. Arch Med Res. (2009) 40:655–61. doi: 10.1016/j.arcmed.2009.10.001
24. Shieh W-J, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol. (2010) 177:166–75. doi: 10.2353/ajpath.2010.100115
25. Skálová H, Povýšil C, Hofmanová J, Goldová B, Jakša R, Jandová K, et al. Histopathological autoptic findings in 8 patients with pandemic influenza A (H1N1) pneumonia. Cesk Patol. (2012) 48:161–4.
26. Gao R, Pan M, Li X, Zou X, Zhao X, Li T, et al. Post-mortem findings in a patient with avian influenza A (H5N6) virus infection. Clin Microbiol Infect. (2016) 22:574.e1–5. doi: 10.1016/j.cmi.2016.03.017
27. Buttignol M, Pires-Neto RC, Rossi E Silva RC, Albino MB, Dolhnikoff M, Mauad T. Airway and parenchyma immune cells in influenza A(H1N1)pdm09 viral and non-viral diffuse alveolar damage. Respir Res. (2017) 18:147. doi: 10.1186/s12931-017-0630-x
28. Hagau N, Slavcovici A, Gonganau DN, Oltean S, Dirzu DS, Brezoszki ES, et al. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit Care. (2010) 14:R203. doi: 10.1186/cc9324
29. Bradley-Stewart A, Jolly L, Adamson W, Gunson R, Frew-Gillespie C, Templeton K, et al. Cytokine responses in patients with mild or severe influenza A(H1N1)pdm09. J Clin Virol. (2013) 58:100–7. doi: 10.1016/j.jcv.2013.05.011
30. Dou M, Song W, Lin Y, Chen Q, Lu C, Liu Z. Clinical characteristics and viral analysis of severe influenza A [H1N1]pdm09 in Guangzhou, 2019. J Med Virol. (2022) 94:2568–77. doi: 10.1002/jmv.27642
31. Karaba AH, Zhou W, Hsieh LL, Figueroa A, Massaccesi G, Rothman RE, et al. Differential cytokine signatures of severe acute respiratory syndrome coronavirus 2 (SARS-coV-2) and influenza infection highlight key differences in pathobiology. Clin Infect Dis. (2022) 74:254–62. doi: 10.1093/cid/ciab376
32. Pandey P, Al Rumaih Z, Kels MJT, Ng E, Kc R, Malley R, et al. Therapeutic targeting of inflammation and virus simultaneously ameliorates influenza pneumonia and protects from morbidity and mortality. Viruses. (2023) 15:318. doi: 10.3390/v15020318
33. Lam WY, Yeung ACM, Chu IMT, Chan PKS. Profiles of cytokine and chemokine gene expression in human pulmonary epithelial cells induced by human and avian influenza viruses. Virol J. (2010) 7:344. doi: 10.1186/1743-422X-7-344
34. Sato M, Hosoya M, Wright PF. Differences in serum cytokine levels between influenza virus A and B infections in children. Cytokine. (2009) 47:65–8. doi: 10.1016/j.cyto.2009.05.003
35. Berdal J-E, Mollnes TE, Wæhre T, Olstad OK, Halvorsen B, Ueland T, et al. Excessive innate immune response and mutant D222G/N in severe A (H1N1) pandemic influenza. J Infect. (2011) 63:308–16. doi: 10.1016/j.jinf.2011.07.004
36. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. (2011) 17:195–9. doi: 10.1038/nm.2262
37. Herold S, Becker C, Ridge KM, Budinger GRS. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J. (2015) 45:1463–78. doi: 10.1183/09031936.00186214
38. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
39. Ruscitti C, Radermecker C, Marichal T. Journey of monocytes and macrophages upon influenza A virus infection. Curr Opin Virol. (2024) 66:101409. doi: 10.1016/j.coviro.2024.101409
40. Tang BM, Shojaei M, Teoh S, Meyers A, Ho J, Ball TB, et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat Commun. (2019) 10:3422. doi: 10.1038/s41467-019-11249-y
41. Kim TS, Sun J, Braciale TJ. T cell responses during influenza infection: getting and keeping control. Trends Immunol. (2011) 32:225–31. doi: 10.1016/j.it.2011.02.006
42. Liu Q, Zhou Y, Yang Z. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. (2016) 13:3–10. doi: 10.1038/cmi.2015.74
43. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. (2011) 146:980–91. doi: 10.1016/j.cell.2011.08.015
44. Liu S, Yan R, Chen B, Pan Q, Chen Y, Hong J, et al. Influenza virus-induced robust expression of SOCS3 contributes to excessive production of IL-6. Front Immunol. (2019) 10:1843. doi: 10.3389/fimmu.2019.01843
45. Schmit T, Guo K, Tripathi JK, Wang Z, McGregor B, Klomp M, et al. Interferon-γ promotes monocyte-mediated lung injury during influenza infection. Cell Rep. (2022) 38:110456. doi: 10.1016/j.celrep.2022.110456
46. Zhang P, Fleming P, Andoniou CE, Waltner OG, Bhise SS, Martins JP, et al. IL-6-mediated endothelial injury impairs antiviral humoral immunity after bone marrow transplantation. J Clin Invest. (2024) 134:e174184. doi: 10.1172/JCI174184
47. Zuniga M, Gomes C, Chen Z, Martinez C, Devlin JC, Loke P, et al. Plasmodium falciparum and TNF-α Differentially regulate inflammatory and barrier integrity pathways in human brain endothelial cells. mBio. (2022) 13:e0174622. doi: 10.1128/mbio.01746-22
48. Ferreira AM, McNeil CJ, Stallaert KM, Rogers KA, Sandig M. Interleukin-1beta reduces transcellular monocyte diapedesis and compromises endothelial adherens junction integrity. Microcirculation. (2005) 12:563–79. doi: 10.1080/10739680500253493
49. Martino N, Ramos RB, Lu S, Leyden K, Tomaszek L, Sadhu S, et al. Endothelial SOCS3 maintains homeostasis and promotes survival in endotoxemic mice. JCI Insight. (2021) 6:e147280. doi: 10.1172/jci.insight.147280
50. Koizumi K, Poulaki V, Doehmen S, Welsandt G, Radetzky S, Lappas A, et al. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis. vivo Invest Ophthalmol Vis Sci. (2003) 44:2184–91. doi: 10.1167/iovs.02-0589
51. To KKW, Hung IFN, Li IWS, Lee K-L, Koo C-K, Yan W-W, et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. (2010) 50:850–9. doi: 10.1086/650581
52. Huangfu Y, Wang J, Feng J, Zhang Z-L. Distal renal tubular system-on-a-chip for studying the pathogenesis of influenza A virus-induced kidney injury. Lab Chip. (2023) 23:4255–64. doi: 10.1039/d3lc00616f
53. Krammer F. The human antibody response to influenza A virus infection and vaccination. Nat Rev Immunol. (2019) 19:383–97. doi: 10.1038/s41577-019-0143-6
54. Jegaskanda S, Vandenberg K, Laurie KL, Loh L, Kramski M, Winnall WR, et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity in intravenous immunoglobulin as a potential therapeutic against emerging influenza viruses. J Infect Dis. (2014) 210:1811–22. doi: 10.1093/infdis/jiu334
55. Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. (2015) 348:448–53. doi: 10.1126/science.aaa1578
56. Campbell TM, Liu Z, Zhang Q, Moncada-Velez M, Covill LE, Zhang P, et al. Respiratory viral infections in otherwise healthy humans with inherited IRF7 deficiency. J Exp Med. (2022) 219:e20220202. doi: 10.1084/jem.20220202
57. Högner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PloS Pathog. (2013) 9:e1003188. doi: 10.1371/journal.ppat.1003188
58. Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. (2008) 205:3065–77. doi: 10.1084/jem.20080201
59. Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. (2020) 369:712–7. doi: 10.1126/science.abc2061
60. Liu S, Liu S, Yu Z, Zhou W, Zheng M, Gu R, et al. STAT3 regulates antiviral immunity by suppressing excessive interferon signaling. Cell Rep. (2023) 42:112806. doi: 10.1016/j.celrep.2023.112806
61. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. (2005) 434:772–7. doi: 10.1038/nature03464
62. Webster B, Werneke SW, Zafirova B, This S, Coléon S, Décembre E, et al. Plasmacytoid dendritic cells control dengue and Chikungunya virus infections via IRF7-regulated interferon responses. Elife. (2018) 7:e34273. doi: 10.7554/eLife.34273
63. Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med. (2018) 215:2567–85. doi: 10.1084/jem.20180628
64. Bucciol G, Moens L, Ogishi M, Rinchai D, Matuozzo D, Momenilandi M, et al. Human inherited complete STAT2 deficiency underlies inflammatory viral diseases. J Clin Invest. (2023) 133:e168321. doi: 10.1172/JCI168321
65. Le Voyer T, Sakata S, Tsumura M, Khan T, Esteve-Sole A, Al-Saud BK, et al. Genetic, immunological, and clinical features of 32 patients with autosomal recessive STAT1 deficiency. J Immunol. (2021) 207:133–52. doi: 10.4049/jimmunol.2001451
66. Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. (2019) 216:2038–56. doi: 10.1084/jem.20181621
67. Vallbracht A, Treuner J, Flehmig B, Joester KE, Niethammer D. Interferon-neutralizing antibodies in a patient treated with human fibroblast interferon. Nature. (1981) 289:496–7. doi: 10.1038/289496a0
68. Pozzetto B, Mogensen KE, Tovey MG, Gresser I. Characteristics of autoantibodies to human interferon in a patient with varicella-zoster disease. J Infect Dis. (1984) 150:707–13. doi: 10.1093/infdis/150.5.707
69. Bjørklund G, Pivin M, Hangan T, Yurkovskaya O, Pivina L. Autoimmune polyendocrine syndrome type 1: Clinical manifestations, pathogenetic features, and management approach. Autoimmun Rev. (2022) 21:103135. doi: 10.1016/j.autrev.2022.103135
70. Bastard P, Orlova E, Sozaeva L, Lévy R, James A, Schmitt MM, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med. (2021) 218:e20210554. doi: 10.1084/jem.20210554
71. Bastard P, Michailidis E, Hoffmann H-H, Chbihi M, Le Voyer T, Rosain J, et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J Exp Med. (2021) 218:e20202486. doi: 10.1084/jem.20202486
72. Zhang Q, Pizzorno A., Miorin L, Bastard P, Gervais A, Voyer TL, et al. Autoantibodies against type I IFNs in patients with critical influenza pneumonia. J Exp Med. (2022) 219:e20220514. doi: 10.1084/jem.20220514
73. Reynolds D, Vazquez Guillamet C, Day A, Borcherding N, Vazquez Guillamet R, Choreño-Parra JA, et al. Comprehensive immunologic evaluation of bronchoalveolar lavage samples from human patients with moderate and severe seasonal influenza and severe COVID-19. J Immunol. (2021) 207:1229–38. doi: 10.4049/jimmunol.2100294
74. Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramírez P, et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care. (2009) 13:R201. doi: 10.1186/cc8208
75. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 135:1332–43. doi: 10.1182/blood.2019000936
76. Beutel G, Wiesner O, Eder M, Hafer C, Schneider AS, Kielstein JT, et al. Virus-associated hemophagocytic syndrome as a major contributor to death in patients with 2009 influenza A (H1N1) infection. Crit Care. (2011) 15:R80. doi: 10.1186/cc10073
77. von Bahr Greenwood T, Holzgraefe B, Chiang SCC, Wang Y, Tesi B, Bryceson YT, et al. Clinical and laboratory signs of haemophagocytic lymphohistiocytosis associated with pandemic influenza A (H1N1) infection in patients needing extracorporeal membrane oxygenation: A retrospective observational study. Eur J Anaesth. (2021) 38:692–701. doi: 10.1097/EJA.0000000000001386
78. Asai N, Ohkuni Y, Matsunuma R, Iwama K, Otsuka Y, Kawamura Y, et al. A case of novel swine influenza A (H1N1) pneumonia complicated with virus-associated hemophagocytic syndrome. J Infect Chemother. (2012) 18:771–4. doi: 10.1007/s10156-011-0366-3
79. Carré A, Vecchio F, Flodström-Tullberg M, You S, Mallone R. Coxsackievirus and type 1 diabetes: diabetogenic mechanisms and implications for prevention. Endocr Rev. (2023) 44:737–51. doi: 10.1210/endrev/bnad007
80. Yoneda S, Imagawa A, Fukui K, Uno S, Kozawa J, Sakai M, et al. A histological study of fulminant type 1 diabetes mellitus related to human cytomegalovirus reactivation. J Clin Endocrinol Metab. (2017) 102:2394–400. doi: 10.1210/jc.2016-4029
81. Lloyd RE, Tamhankar M, Lernmark Å. Enteroviruses and type 1 diabetes: multiple mechanisms and factors? Annu Rev Med. (2022) 73:483–99. doi: 10.1146/annurev-med-042320-015952
82. Stölzel U, Schuppan D, Tillmann HL, Manns MP, Tannapfel A, Doss MO, et al. Autoimmunity and HCV infection in porphyria cutanea tarda: a controlled study. Cell Mol Biol (Noisy-le-grand). (2002) 48:43–7.
83. Zhang T, Yu Z, Gao S, Zhang Y, Wang C, Jian S, et al. Clinical phenotypes and prognosis of cytomegalovirus infection in the pediatric systemic lupus erythematosus: a longitudinal analysis. Pediatr Rheumatol Online J. (2023) 21:25. doi: 10.1186/s12969-023-00807-w
84. Harris VK, Danda D, Murali NS, Das PK, Abraham M, Cherian AM, et al. Unusual association of Kikuchi’s disease and dengue virus infection evolving into systemic lupus erythematosus. J Indian Med Assoc. (2000) 98:391–3.
85. Chabert P, Kallel H. Simultaneous presentation of parvovirus B19 infection and systemic lupus erythematosus in a patient: description and review of the literature. Eur J Case Rep Intern Med. (2020) 7:1729. doi: 10.12890/2020_001729
86. Javanian M, Barary M, Ghebrehewet S, Koppolu V, Vasigala V, Ebrahimpour S. A brief review of influenza virus infection. J Med Virol. (2021) 93:4638–46. doi: 10.1002/jmv.26990
87. Hany M, Rehman B, Rizvi A, Chapman J. Schizophrenia, in: StatPearls (2024). Treasure Island (FL: StatPearls Publishing. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK539864/ (Accessed June 16, 2024).
88. Kępińska AP, Iyegbe CO, Vernon AC, Yolken R, Murray RM, Pollak TA. Schizophrenia and influenza at the centenary of the 1918-1919 spanish influenza pandemic: mechanisms of psychosis risk. Front Psychiatry. (2020) 11:72. doi: 10.3389/fpsyt.2020.00072
89. Albrecht P, Torrey EF, Boone E, Hicks JT, Daniel N. Raised cytomegalovirus-antibody level in cerebrospinal fluid of schizophrenic patients. Lancet. (1980) 2:769–72. doi: 10.1016/s0140-6736(80)90386-4
90. Lucchese G, Capone G, Kanduc D. Peptide sharing between influenza A H1N1 hemagglutinin and human axon guidance proteins. Schizophr Bull. (2014) 40:362–75. doi: 10.1093/schbul/sbs197
91. Casteels-Van-Daele M, Van Geet C, Wouters C, Eggermont E. Reye’s syndrome. Lancet. (2001) 358:334. doi: 10.1016/S0140-6736(01)05512-X
92. Corey L, Rubin RJ, Thompson TR, Noble GR, Cassidy E, Hattwick MA, et al. Influenza B-associated Reye’s syndrome: incidence in Michigan and potential for prevention. J Infect Dis. (1977) 135:398–407. doi: 10.1093/infdis/135.3.398
93. Antoon JW, Hall M, Herndon A, Johnson DP, Brown CM, Browning WL, et al. Prevalence, risk factors, and outcomes of influenza-associated neurologic complications in children. J Pediatr. (2021) 239:32–38.e5. doi: 10.1016/j.jpeds.2021.06.075
94. Pugliese A, Beltramo T, Torre D. Reye’s and reye’s-like syndromes. Cell Biochem Funct. (2008) 26:741–6. doi: 10.1002/cbf.1465
95. Bailey JM, Low CE, Pupillo MB. Reye’s syndrome and aspirin use: a possible immunological relationship. Prostaglandins Leukot Med. (1982) 8:211–8. doi: 10.1016/0262-1746(82)90044-0
96. Noor A, Gradidge E. A case of reye syndrome caused by influenza A virus. Ochsner J. (2018) 18:425–7. doi: 10.31486/toj.18.0098
97. Willison HJ, Jacobs BC, van Doorn PA. Guillain-barré syndrome. Lancet. (2016) 388:717–27. doi: 10.1016/S0140-6736(16)00339-1
98. Leonhard SE, Mandarakas MR, Gondim FAA, Bateman K, Ferreira MLB, Cornblath DR, et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. (2019) 15:671–83. doi: 10.1038/s41582-019-0250-9
99. Sivadon-Tardy V, Orlikowski D, Porcher R, Sharshar T, Durand M-C, Enouf V, et al. Guillain-Barré syndrome and influenza virus infection. Clin Infect Dis. (2009) 48:48–56. doi: 10.1086/594124
100. Iqbal S, Li R, Gargiullo P, Vellozzi C. Relationship between Guillain-Barré syndrome, influenza-related hospitalizations, and influenza vaccine coverage. Vaccine. (2015) 33:2045–9. doi: 10.1016/j.vaccine.2015.02.064
101. Ghaderi S, Gunnes N, Bakken IJ, Magnus P, Trogstad L, Håberg SE. Risk of Guillain-Barré syndrome after exposure to pandemic influenza A(H1N1)pdm09 vaccination or infection: a Norwegian population-based cohort study. Eur J Epidemiol. (2016) 31:67–72. doi: 10.1007/s10654-015-0047-0
102. Hao Y, Wang W, Jacobs BC, Qiao B, Chen M, Liu D, et al. Antecedent infections in Guillain-Barré syndrome: a single-center, prospective study. Ann Clin Transl Neur. (2019) 6:2510–7. doi: 10.1002/acn3.50946
103. Yamana M, Kuwahara M, Fukumoto Y, Yoshikawa K, Takada K, Kusunoki S. Guillain-Barré syndrome and related diseases after influenza virus infection. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e575. doi: 10.1212/NXI.0000000000000575
104. Dutta D, Debnath M, Seshagiri DV, Nair BVS, Das SK, Wahatule R, et al. Impact of antecedent infections on the antibodies against gangliosides and ganglioside complexes in guillain-barré Syndrome: A correlative study. Ann Indian Acad Neurol. (2022) 25:401–6. doi: 10.4103/aian.aian_121_22
105. Torisu H, Hara T. Influenza-associated encephalopathy. Brain Nerve. (2015) 67:859–69. doi: 10.11477/mf.1416200224
107. Okuno H, Yahata Y, Tanaka-Taya K, Arai S, Satoh H, Morino S, et al. Characteristics and outcomes of influenza-associated encephalopathy cases among children and adults in Japan, 2010-2015. Clin Infect Dis. (2018) 66:1831–7. doi: 10.1093/cid/cix1126
108. Cleuziou P, Renaldo F, Renolleau S, Javouhey E, Tissieres P, Léger P-L, et al. Mortality and neurologic sequelae in influenza-associated encephalopathy: retrospective multicenter PICU cohort in France. Pediatr Crit Care Med. (2021) 22:e582–7. doi: 10.1097/PCC.0000000000002750
109. Ayukawa H, Matsubara T, Kaneko M, Hasegawa M, Ichiyama T, Furukawa S. Expression of CTLA-4 (CD152) in peripheral blood T cells of children with influenza virus infection including encephalopathy in comparison with respiratory syncytial virus infection. Clin Exp Immunol. (2004) 137:151–5. doi: 10.1111/j.1365-2249.2004.02502.x
110. Hasegawa S, Matsushige T, Inoue H, Shirabe K, Fukano R, Ichiyama T. Serum and cerebrospinal fluid cytokine profile of patients with 2009 pandemic H1N1 influenza virus-associated encephalopathy. Cytokine. (2011) 54:167–72. doi: 10.1016/j.cyto.2011.01.006
111. Liblau RS, Latorre D, Kornum BR, Dauvilliers Y, Mignot EJ. The immunopathogenesis of narcolepsy type 1. Nat Rev Immunol. (2024) 24:33–48. doi: 10.1038/s41577-023-00902-9
112. Han F, Lin L, Warby SC, Faraco J, Li J, Dong SX, et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann Neurol. (2011) 70:410–7. doi: 10.1002/ana.22587
113. Heier MS, Gautvik KM, Wannag E, Bronder KH, Midtlyng E, Kamaleri Y, et al. Incidence of narcolepsy in Norwegian children and adolescents after vaccination against H1N1 influenza A. Sleep Med. (2013) 14:867–71. doi: 10.1016/j.sleep.2013.03.020
114. Luo G, Ambati A, Lin L, Bonvalet M, Partinen M, Ji X, et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. P Natl Acad Sci USA. (2018) 115:E12323–32. doi: 10.1073/pnas.1818150116
115. Latorre D, Kallweit U, Armentani E, Foglierini M, Mele F, Cassotta A, et al. T cells in patients with narcolepsy target self-antigens of hypocretin neurons. Nature. (2018) 562:63–8. doi: 10.1038/s41586-018-0540-1
116. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
117. Ruiz PLD, Tapia G, Bakken IJ, Håberg SE, Hungnes O, Gulseth HL, et al. Pandemic influenza and subsequent risk of type 1 diabetes: a nationwide cohort study. Diabetologia. (2018) 61:1996–2004. doi: 10.1007/s00125-018-4662-7
118. Nishioka Y, Noda T, Okada S, Myojin T, Kubo S, Higashino T, et al. Association between influenza and the incidence rate of new-onset type 1 diabetes in Japan. J Diabetes Investig. (2021) 12:1797–804. doi: 10.1111/jdi.13540
119. Sun L, Li H, Sun J, Guo C, Feng Y, Li Y, et al. Antibodies against H1N1 influenza virus hemagglutinin cross-react with prohibitin. Biochem Biophys Res Commun. (2019) 513:446–51. doi: 10.1016/j.bbrc.2019.03.188
120. Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ. (2023) 380:e069717. doi: 10.1136/bmj-2021-069717
121. Durkin ML, Marchese D, Robinson MD, Ramgopal M. Catastrophic antiphospholipid syndrome (CAPS) induced by influenza A virus subtype H1N1. BMJ Case Rep. (2013) 2013:bcr2013200474. doi: 10.1136/bcr-2013-200474
122. Li S-J, Wu Y-L, Chen J-H, Shen S-Y, Duan J, Xu HE. Autoimmune diseases: targets, biology, and drug discovery. Acta Pharmacol Sin. (2024) 45:674–85. doi: 10.1038/s41401-023-01207-2
123. Conrad N, Misra S, Verbakel JY, Verbeke G, Molenberghs G, Taylor PN, et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: a population-based cohort study of 22 million individuals in the UK. Lancet. (2023) 401:1878–90. doi: 10.1016/S0140-6736(23)00457-9
124. Habibi MA, Nezhad Shamohammadi F, Rajaei T, Namdari H, Pashaei MR, Farajifard H, et al. Immunopathogenesis of viral infections in neurological autoimmune disease. BMC Neurol. (2023) 23:201. doi: 10.1186/s12883-023-03239-x
125. Pisetsky DS. Pathogenesis of autoimmune disease. Nat Rev Nephrol. (2023) 19:509–24. doi: 10.1038/s41581-023-00720-1
126. Feng A, Yang EY, Moore AR, Dhingra S, Chang SE, Yin X, et al. Autoantibodies are highly prevalent in non-SARS-CoV-2 respiratory infections and critical illness. JCI Insight. (2023) 8:e163150. doi: 10.1172/jci.insight.163150
127. Feng A, Yang E, Moore A, Dhingra S, Chang S, Yin X, et al. Autoantibodies targeting cytokines and connective tissue disease autoantigens are common in acute non-SARS-CoV-2 infections. Res Sq. (2022), rs.3.rs–1233038. doi: 10.21203/rs.3.rs-1233038/v1
128. Cabral-Marques O, Halpert G, Schimke LF, Ostrinski Y, Vojdani A, Baiocchi GC, et al. Autoantibodies targeting GPCRs and RAS-related molecules associate with COVID-19 severity. Nat Commun. (2022) 13:1220. doi: 10.1038/s41467-022-28905-5
129. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse functional autoantibodies in patients with COVID-19. Nature. (2021) 595:283–8. doi: 10.1038/s41586-021-03631-y
130. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
131. Jin X, Xu Q, Pu C, Zhu K, Lu C, Jiang Y, et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. (2021) 18:1896–903. doi: 10.1038/s41423-020-0472-1
132. Hsue PY, Scherzer R, Grunfeld C, Imboden J, Wu Y, Del Puerto G, et al. Depletion of B-cells with rituximab improves endothelial function and reduces inflammation among individuals with rheumatoid arthritis. J Am Heart Assoc. (2014) 3:e001267. doi: 10.1161/JAHA.114.001267
133. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. (2003) 301:1374–7. doi: 10.1126/science.1086907
134. Noorchashm H, Bui A, Li HL, Eaton A, Mandik-Nayak L, Sokol C, et al. Characterization of anergic anti-DNA B cells: B cell anergy is a T cell-independent and potentially reversible process. Int Immunol. (1999) 11:765–76. doi: 10.1093/intimm/11.5.765
135. Szodoray P, Stanford SM, Molberg Ø, Munthe LA, Bottini N, Nakken B. T-helper signals restore B-cell receptor signaling in autoreactive anergic B cells by upregulating CD45 phosphatase activity. J Allergy Clin Immunol. (2016) 138:839–851.e8. doi: 10.1016/j.jaci.2016.01.035
136. Laron Z, Shulman L, Hampe C, Blumenfeld O. Hypothesis: Viral infections of pregnant women may be early triggers of childhood type 1 diabetes and other autoimmune disease. J Autoimmun. (2023) 135:102977. doi: 10.1016/j.jaut.2022.102977
137. Damian RT. Molecular mimicry: antigen sharing by parasite and host and its consequences. Am Nat. (1964) 98:129–49. doi: 10.1086/282313
138. Neidhart S, Vlad B, Hilty M, Högelin KA, Ziegler M, Berenjeno-Correa E, et al. HLA associations of intrathecal igG production against specific viruses in multiple sclerosis. Ann Neurol. (2024) 95:1112–26. doi: 10.1002/ana.26921
139. Lanz TV, Brewer RC, Ho PP, Moon J-S, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. (2022) 603:321–7. doi: 10.1038/s41586-022-04432-7
140. Rojas M, Herrán M, Ramírez-Santana C, Leung PSC, Anaya J-M, Ridgway WM, et al. Molecular mimicry and autoimmunity in the time of COVID-19. J Autoimmun. (2023) 139:103070. doi: 10.1016/j.jaut.2023.103070
141. Soderberg C, Larsson S, Rozell BL, Sumitran-Karuppan S, Ljungman P, Moller E. Cytomegalovirus-induced CD13-specific autoimmunity–a possible cause of chronic graft-vs-host disease. Transplantation. (1996) 61:600–9. doi: 10.1097/00007890-199602270-00015
142. Guarneri F, Guarneri C, Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J Cell Mol Med. (2005) 9:741–4. doi: 10.1111/j.1582-4934.2005.tb00506.x
143. Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. (2002) 3:940–3. doi: 10.1038/ni835
144. Martins YC, Jurberg AD, Daniel-Ribeiro CT. Visiting molecular mimicry once more: pathogenicity, virulence, and autoimmunity. Microorganisms. (2023) 11:1472. doi: 10.3390/microorganisms11061472
145. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon. vivo Sci. (1996) 272:1947–50. doi: 10.1126/science.272.5270.1947
146. Zarozinski CC, Welsh RM. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J Exp Med. (1997) 185:1629–39. doi: 10.1084/jem.185.9.1629
147. Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. (1998) 8:177–87. doi: 10.1016/s1074-7613(00)80470-7
148. Ehl S, Hombach J, Aichele P, Hengartner H, Zinkernagel RM. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J Exp Med. (1997) 185:1241–51. doi: 10.1084/jem.185.7.1241
149. Eberl G, Brawand P, MacDonald HR. Selective bystander proliferation of memory CD4+ and CD8+ T cells upon NK T or T cell activation. J Immunol. (2000) 165:4305–11. doi: 10.4049/jimmunol.165.8.4305
150. Holay N, Kennedy BE, Murphy JP, Konda P, Giacomantonio M, Brauer-Chapin T, et al. After virus exposure, early bystander naïve CD8 T cell activation relies on NAD+ salvage metabolism. Front Immunol. (2022) 13:1047661. doi: 10.3389/fimmu.2022.1047661
151. Chakir H, Lam DKY, Lemay A-M, Webb JR. Bystander polarization” of CD4+ T cells: activation with high-dose IL-2 renders naive T cells responsive to IL-12 and/or IL-18 in the absence of TCR ligation. Eur J Immunol. (2003) 33:1788–98. doi: 10.1002/eji.200323398
152. Martin MD, Badovinac VP. Antigen-dependent and -independent contributions to primary memory CD8 T cell activation and protection following infection. Sci Rep-uk. (2015) 5:18022. doi: 10.1038/srep18022
153. Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. (2013) 3:701–8. doi: 10.1016/j.celrep.2013.02.020
154. Rolot M, Dougall AM, Chetty A, Javaux J, Chen T, Xiao X, et al. Helminth-induced IL-4 expands bystander memory CD8+ T cells for early control of viral infection. Nat Commun. (2018) 9:4516. doi: 10.1038/s41467-018-06978-5
155. Bergamaschi L, Mescia F, Turner L, Hanson AL, Kotagiri P, Dunmore BJ, et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity. (2021) 54:1257–1275.e8. doi: 10.1016/j.immuni.2021.05.010
156. Zhang X, Zhang Y, Liu H, Tang K, Zhang C, Wang M, et al. IL-15 induced bystander activation of CD8+ T cells may mediate endothelium injury through NKG2D in Hantaan virus infection. Front Cell Infect Mi. (2022) 12:1084841. doi: 10.3389/fcimb.2022.1084841
157. Ohya M, Tateishi A, Matsumoto Y, Satomi H, Kobayashi M. Bystander CD8 + T cells may be involved in the acute phase of diffuse alveolar damage. Virchows Arch. (2023) 482:605–13. doi: 10.1007/s00428-023-03521-w
158. Lee H-G, Lee J-U, Kim D-H, Lim S, Kang I, Choi J-M. Pathogenic function of bystander-activated memory-like CD4+ T cells in autoimmune encephalomyelitis. Nat Commun. (2019) 10:709. doi: 10.1038/s41467-019-08482-w
159. Johnson D, Jiang W. Infectious diseases, autoantibodies, and autoimmunity. J Autoimmun. (2023) 137:102962. doi: 10.1016/j.jaut.2022.102962
160. Hintermann E, Holdener M, Bayer M, Loges S, Pfeilschifter JM, Granier C, et al. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J Autoimmun. (2011) 37:242–53. doi: 10.1016/j.jaut.2011.06.005
161. Yang Y, Choi J, Chen Y, Invernizzi P, Yang G, Zhang W, et al. E. coli and the etiology of human PBC: Antimitochondrial antibodies and spreading determinants. Hepatology. (2022) 75:266–79. doi: 10.1002/hep.32172
Keywords: influenza virus, infection, cytokine, autoimmunity, autoantibody
Citation: Xie S, Wei J and Wang X (2025) The intersection of influenza infection and autoimmunity. Front. Immunol. 16:1558386. doi: 10.3389/fimmu.2025.1558386
Received: 10 January 2025; Accepted: 17 March 2025;
Published: 03 April 2025.
Edited by:
Rajeev K. Tyagi, Institute of Microbial Technology (CSIR), IndiaReviewed by:
A. Raj Kumar Patro, Kalinga Institute of Medical Sciences (KIMS), IndiaCopyright © 2025 Xie, Wei and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohui Wang, eGlhb2h1aXdhbmcyMDIxNjIxODE4QGd6aG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.