 Chenxiao Liu1†
Chenxiao Liu1† Nan Liu
Nan Liu Yanyang Tu
Yanyang Tu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 April 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1557906
Colorectal cancer (CRC) is a major cause of cancer-related morbidity and mortality worldwide, with limited options for patients at advanced stages. Immunotherapy, particularly immune cell-based therapies, has gained significant attention as an innovative approach for targeting CRC. This review summarizes the progress in various immune cell therapies, including DC vaccine, CAR/TCR-T cells, CAR-NK cells et al, each engineered to recognize and attack cancer cells expressing specific antigens. CAR-T cell therapy, which has been successful in hematologic cancers, faces challenges in CRC due to the solid tumor microenvironment, which limits cell infiltration and persistence. CAR-NK cells, CAR-M and CAR-γδ T cells, however, offer alternative strategies due to their unique properties, such as the ability to target tumor cells without prior sensitization and a lower risk of inducing severe cytokine release syndrome. Recent advances in lentiviral transduction have enabled effective expression of CARs on NK and γδ T cells, providing promising preclinical results in CRC models. This review explores the mechanisms, tumor targets, preclinical studies, and early-phase clinical trials of these therapies, addressing key challenges such as enhancing specificity to tumor antigens and overcoming the immunosuppressive tumor microenvironment. The potential of combination therapies, including immune checkpoint inhibitors and cytokine therapy, is also discussed some as a means to improve the effectiveness of immune cell-based treatments for CRC. Continued research is essential to translate these promising approaches into clinical settings, offering new hope for CRC patients.
According to the latest global cancer burden data for 2020 released by the World Health Organization, there were 19.29 million new cancer cases worldwide in 2020, including 1.93 million cases of colorectal cancer, ranking third. There were 9.96 million cancer deaths worldwide, and colorectal cancer deaths were 940,000, ranking second (1). Colorectal cancer (CRC) is difficult to detect in its early stages, and current treatment options for advanced colorectal cancer include surgery, radiotherapy, chemotherapy (such as fluoropyrimidines, irinotecan and oxiplatin) and targeted therapy (e.g., bevacizumab, EGFR inhibitors and multikinase inhibitors). Even with multiple treatment options, the 5-year survival rate for patients with advanced colorectal cancer is only 20%-30% (2).
Immunotherapy represents a promising class of novel therapeutic strategies in oncology. These therapies include the administration of cytokines to stimulate immune responses, monoclonal antibodies targeting immune checkpoint inhibitors, dendritic cell (DC)-based vaccines, and other immunomodulatory approaches. Among these, adoptive cell therapy (ACT) has emerged as one of the most promising interventions. ACT involves the infusion of ex vivo-expanded and/or genetically modified immune cells, such as T cells or natural killer (NK) cells, which are specifically targeted to tumor sites to enhance anti-tumor immunity. This approach has demonstrated significant potential in the treatment of various malignancies (Figure 1).
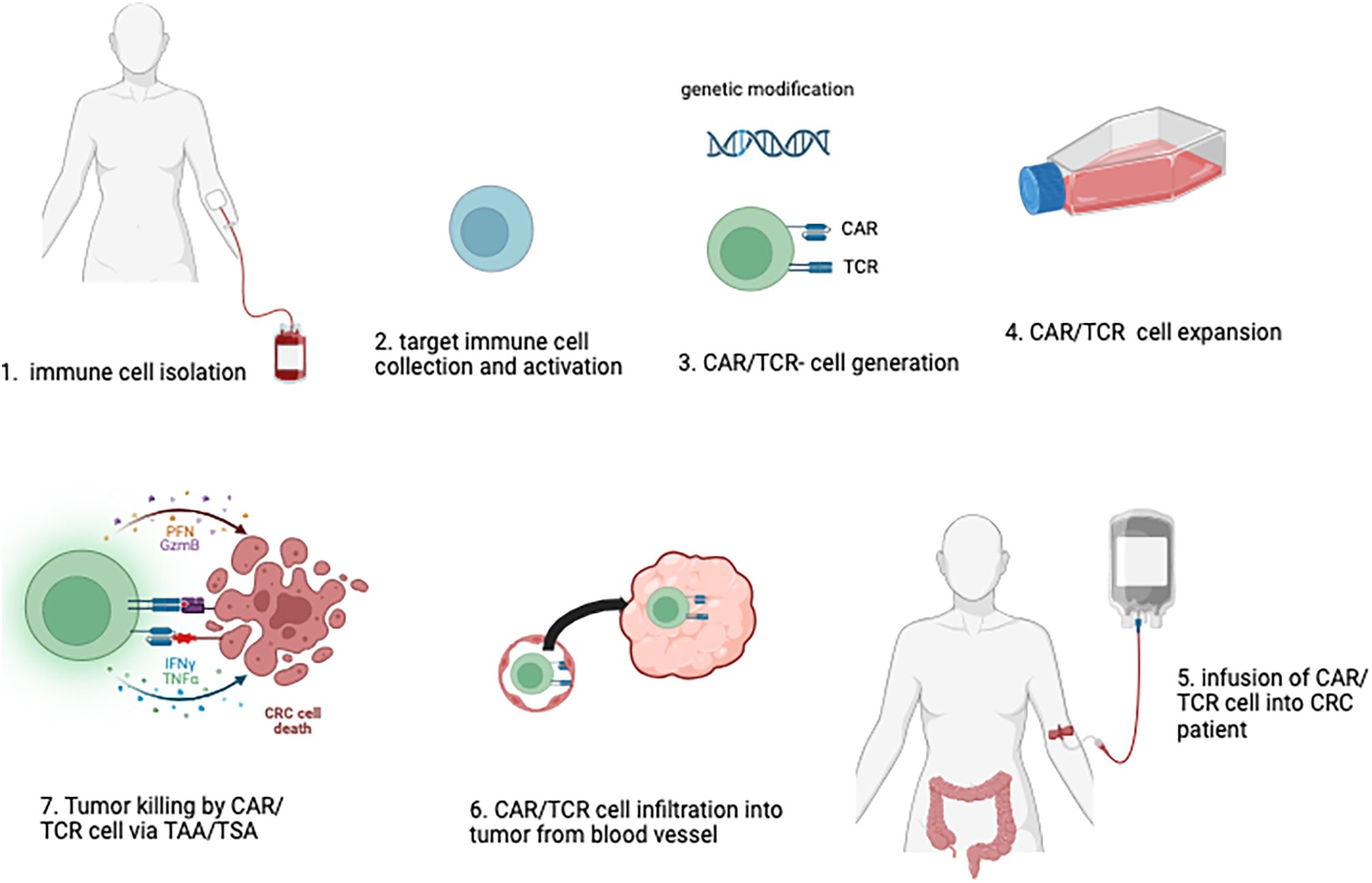
Figure 1. Adoptive Immune Cell Therapy Approach Against Colorectal Cancer. In the adoptive immune cell therapy process, human immune cells are initially harvested (1), selected and activated (2), in vitro. If necessary, genetic modifications are introduced to enable the expression of CAR (Chimeric Antigen Receptor) or TCR (T-cell Receptor) on the cells (3). These modified immune cells are then expanded and formulated into a therapeutic product, with rigorous quality control measures in place (4). The final cell product is subsequently administered to the patient (5), where it is designed to target and eliminate tumor cells by tumor homing, and recognizing specific antigens associated with colorectal cancer (6, 7).
ACT has evolved significantly since the 1960s. Early research demonstrated that T-cells from cancer patients, especially melanoma, could recognize and kill tumor cells. Steven Rosenberg et al. ‘s pioneering work in the 1980s led to the discovery that lymphokine-activated killer cell (LAK) and tumor-infiltrating lymphocytes (TILs) could be expanded ex vivo and used effectively in therapy (3), and in 1990s, his team significantly advanced the molecular understanding of T-cell responses to cancer and laid the foundation for antigen-targeted immunotherapies (4). These contributions also spanned the development of gene-engineered T-cells, which led to the emergence of chimeric antigen receptors (CARs)-T therapy in 2000s. Later, immune checkpoint inhibitors, such as PD-1/PD-L1 and CTLA-4 inhibitors, enhanced T-cell responses by relieving functional suppression, improving the efficacy of immune cell therapies. By the 2020s, CAR-T cell therapy demonstrated notable success in treating hematologic cancers and began to be extended to solid tumors, with ongoing efforts to overcome immunosuppressive tumor microenvironments and improve treatment strategies (5).
ACT involves the infusion of various immune cell types into patients, including tumor-infiltrating lymphocytes (TILs), peripheral blood-derived immune cells, induced pluripotent stem cell (iPSC)-derived immune cells, and others. The early milestones in the development of ACT were marked by the adoptive transfer of TILs and ex vivo-expanded lymphokine-activated killer (LAK/CIK) cells. Since then, the use of peripheral blood T cells genetically engineered to express transgenic antigen receptors specific to tumor-associated antigens has become a common practice in ACT. Typically, autologous T cells derived from peripheral blood are activated ex vivo and subsequently modified to express transgenic receptors, such as chimeric antigen receptors (CARs) or T cell receptors (TCRs). In recent advancements, gene-modified immune cells undergo a brief in vitro expansion phase before being infused into patients, further enhancing their therapeutic potential (6–8). ACT for colorectal cancer has been extensively explored in both basic and clinical research, with several studies demonstrating promising outcomes (9). However, the therapeutic application of ACT remains limited due to numerous challenges encountered in the treatment of colorectal cancer.
In addition to addressing the limitations and the need for further research to make this novel therapeutic approach more widely accessible in clinical settings, our review aims to provide a comprehensive overview of the current state of knowledge regarding adoptive immune cell therapies for CRC, including insights from both preclinical studies and clinical trials.
Before any therapeutic intervention is considered, cancer staging serves as a foundational tool for assessing the anatomical extent of the disease. The clinical stage, determined based on the patient’s medical history, physical examination, diagnostic biopsies, and imaging conducted prior to the initiation of treatment, reflects the severity of the disease at the time of diagnosis (10). In addition to the clinical stage, a distinct pathological stage—based on post-surgical histological analysis—is assigned to patients undergoing surgery as a treatment option. At the various time points outlined above, both the clinical and pathological stages involve characterizing the extent of the primary tumor (T category), the involvement of regional lymph nodes (N category), and the presence of metastasis to distant sites (M category). Stage groups I, II, III, and IV are determined by combining the T, N, and M categories once they have been assessed. Colon cancer typically grows intraepithelial, initially invading the mucosal lamina propria and submucosa before spreading throughout the entire colon and potentially metastasizing to other organs. The TNM classification system also incorporates tumor deposits in its assessment. Currently, colon cancer is staged into four phases based on the extent of primary tumor invasion (T), the number of regional lymph nodes (N) involved with tumor spread, and the presence of distant metastases (M) (11, 12).
Chromosomal instability, CpG (cytosine nucleotide phosphate guanine nucleotide) island methylator phenotype, and microsatellite instability are major causes of CRC at the genomic level in general. RAS and BRAF mutations in colorectal cancer are often driven by genetic and environmental factors. KRAS mutations typically arise due to chromosomal instability, whereas BRAF mutations, particularly V600E, are commonly associated with MSI and defective DNA repair mechanisms. Both mutations result from accumulated genetic alterations over time, influenced by carcinogens, inflammation, and hereditary predispositions (13). These mutations analyses are usually important, with RAS being a predictive marker for anti-EGFR therapy and BRAF p.V600E mutation serving as a prognostic marker. High-throughput sequencing technologies have introduced novel biomarkers and testing strategies, such as tumor mutation burden (TMB) and liquid biopsy, which can predict immunotherapy response and monitor therapy response when tissue samples are unavailable. However, standardization of these assays and guidelines for their use are needed (14). Commonly, genomic changes related to colorectal tumorigenesis also include a loss of at least one wild-type copy of a tumor-suppressor gene such as APC, P53 or SMAD4 related to chromosomal instability. These genetic losses contribute to the malignant transformation of colorectal cells by disrupting key regulatory pathways involved in cell growth and apoptosis. In fact, the APC gene is often mutated early in CRC progression, leading to the initiation of adenomatous polyps. Similarly, mutations in TP53 and SMAD4 play critical roles in later stages, promoting tumor progression and metastasis sequencing technologies continue to evolve, they are poised to transform clinical practice by offering more precise and personalized treatment options for CRC patients (15, 16).
As mentioned before, below, we describe common cell therapeutic approaches directed concretely against CRC (Table 1).
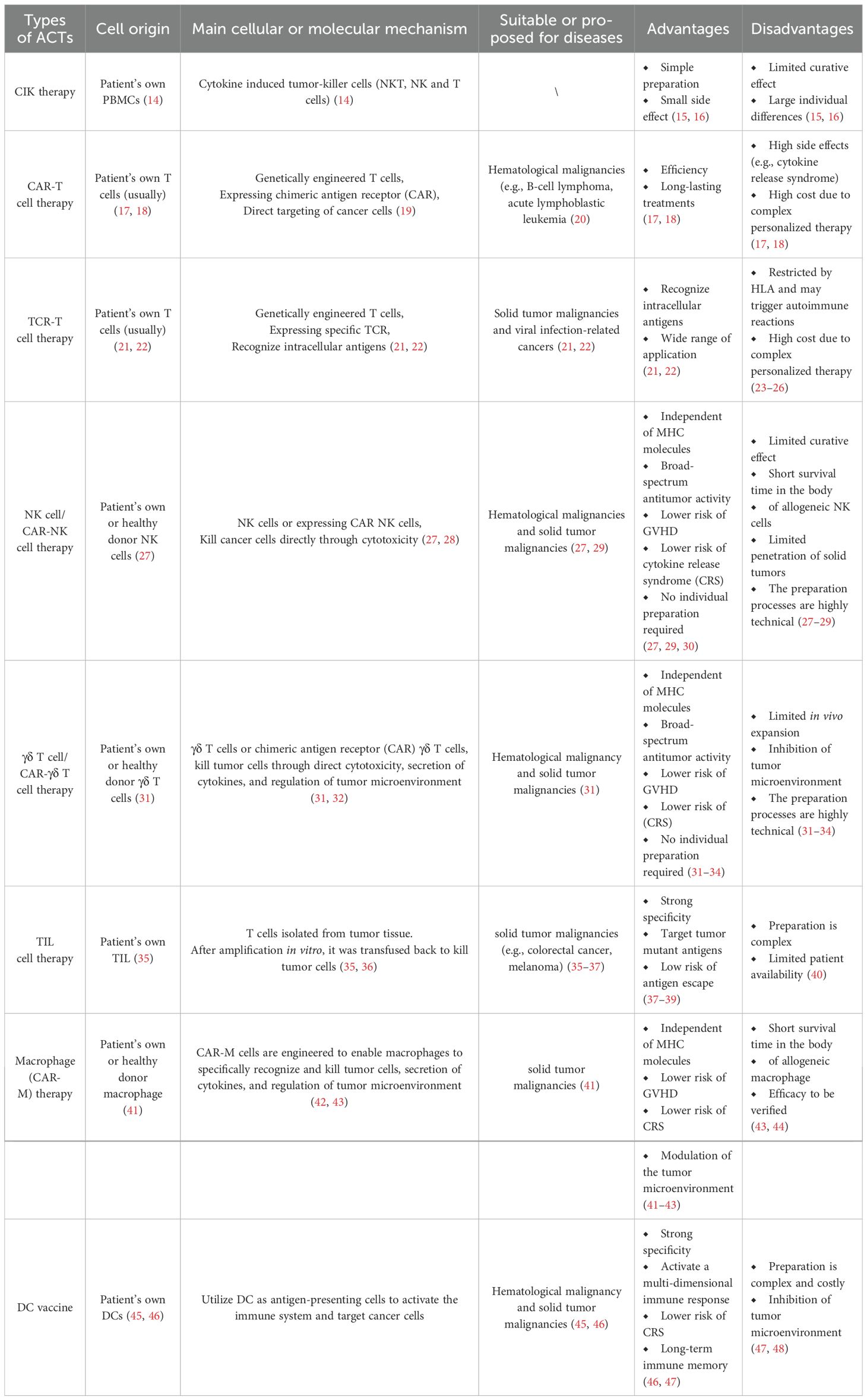
Table 1. Summary of ACTs.
The cytokine-induced killer (CIK) cells, a frequently studied cell immunotherapy in CRC, are a heterogeneous group of cells obtained from peripheral blood mononuclear cells (PBMCs) stimulated ex vivo with an anti-CD3 antibody and a cocktail of cytokines (17). Briefly, these cells share functional and phenotypic properties with NKT, NK and T cells and are characterized by rapid expansion ex vivo, non-major histocompatibility complex (MHC)-restricted tumor-killing activity, strong antitumour activity and minimal toxicity. It has been observed that the combination of adjuvant chemotherapy with sequential infusions of CIK cells significantly improved the progression-free survival (PFS), disease-free survival (DFS) and OS rates of CRC patients, especially in those with high-risk T4 stage and insufficient duration of chemotherapy (DFS and OS). Moreover, this combination therapy has also been used on mCRC patients, showing good tolerability and a significant increase in OS (18, 19).
Chimeric Antigen Receptor T-cell (CAR-T) therapy is an advanced immunotherapy that involves genetically engineering a patient’s own T cells to target and kill cancer cells. The process starts with collecting T cells from the patient, which are then modified in the laboratory to express a synthetic receptor, known as the CAR (20, 49). This receptor is designed to specifically recognize antigens present on the surface of cancer cells. Chimeric Antigen Receptors (CARs) consist of an extracellular single-chain variable fragment (scFv) that serves as an antigen-binding domain, an intracellular signaling domain (typically CD3ζ chain) for T cell activation, and co-stimulatory molecules that enhance T cell responses. T cells genetically engineered to express CARs, which facilitate tumor-associated antigen (TAA) recognition via the scFv and subsequent T cell activation through the intracellular signaling domain, are referred to as CAR-T cells. Once modified, the CAR-T cells are expanded and infused back into the patient’s body. Upon encountering cancer cells, the CAR on the T cells binds to the tumor antigen, activating the T cells to attack and destroy the cancer cells. CAR-T therapy has shown remarkable success in treating certain types of blood cancers, including leukemia and lymphoma, by leveraging the body’s immune system to achieve targeted tumor destruction (21).
CAR-T cell therapies have demonstrated significant promise in the treatment of hematological malignancies over the past decade, with ongoing efforts to expand these therapies to solid tumors (22). However, further research is needed to validate the efficacy of adoptive cell therapy (ACT) in colorectal cancer (CRC) and to optimize treatment strategies, particularly in areas such as tumor-associated antigen (TAA) selection and CAR-T cell dosing (23). Several CAR-T cell therapies targeting antigens such as EGFR (NCT03152435, NCT03542799), NKG2D and NKG2D ligands (NCT03370198, NCT03310008, NCT03692429), CEA (NCT02959151, NCT03682744, NCT02850836), c-Met (NCT03638206), and EpCAM (NCT03013712) have been investigated in patients with metastatic colorectal cancer. These trials currently enroll CRC patients irrespective of their microsatellite instability (MSI) status. Further research is required to assess whether CAR-T cell therapies could benefit patients with either MSI or microsatellite stable (MSS) tumors. These trials aim to provide new therapeutic options for patients with colorectal cancer by leveraging the specificity and potency of -T cell therapies.
In combination therapy, MSS colorectal cancer (CRC) responds poorly to immunotherapy due to an immunosuppressive tumor microenvironment (TME). Combining CAR-T cells with immune checkpoint inhibitors (ICIs) like anti-PD-1 enhances CAR-T persistence and anti-tumor activity (24). Pre-clinical CRC models show improved T-cell infiltration with PD-1 blockade (25). Ongoing trials (e.g., NCT05089266) are evaluating this strategy, though challenges like toxicity remain. Chemotherapy enhances CAR-T efficacy by reducing tumor burden, promoting immune infiltration, and modulating the TME. Agents like oxaliplatin and 5-FU induce immunogenic cell death (ICD), boosting CAR-T recognition. Low-dose chemotherapy also reduces immunosuppressive cells, further improving CAR-T function (26, 27). Radiotherapy enhances CAR-T efficacy by inducing tumor apoptosis, increasing antigen release, and improving TME accessibility. It promotes CAR-T recruitment, upregulates MHC-I and tumor antigens, and enhances targeting (28, 29). CAR-T combined with ICIs, chemotherapy, and radiotherapy offers new strategies for colorectal cancer treatment. However, more preclinical and clinical studies are needed to explore various aspects of mechanism and efficacy.
More details on CAR-T therapy are stated in next chapter (in antigens/targets introduction part).
TCR-T cell therapy is an emerging cancer immunotherapy that involves genetically engineering T cells with tumor-specific T cell receptors (TCRs) to recognize naturally occurring antigens and attack malignant cells. Unlike CAR-T therapy, TCR-T targets peptide-MHC complexes, enabling it to address intracellular antigens (tumor cells produce mutant or overexpressed proteins, which are degraded into peptide fragments. These peptides are presented by antigen-presenting cells (APCs) or tumor cells themselves via MHC molecules). And expanding its potential applications, particularly for solid tumors. While promising results have been observed in melanoma and certain other solid cancers, TCR-T faces challenges such as immunogenicity, MHC restriction, and off-target effects (30, 31). Current research focuses on optimizing TCR affinity, developing non-viral transduction methods, and exploring combination therapies with immune checkpoint inhibitors (31). By enhancing specificity and safety, TCR-T holds significant promise for advancing precision medicine and establishing itself as a critical tool in solid tumor treatment. However, the MHC restriction of TCR-T therapy arises from its reliance on T cell receptors (TCRs) recognizing tumor-associated peptides presented by specific MHC molecules. Due to the high polymorphism of HLA genes, a single TCR-T construct is typically limited to patients expressing a compatible MHC allele, such as HLA-A*02:01. This restriction significantly narrows the eligible patient population (32–35). More details on TCR-T therapy are stated in next chapter (in antigens/targets introduction part).
NK cell therapy leverages the natural cytotoxicity of NK cells to identify and kill cancer cells based on stress signals or the absence of “self” markers without prior antigen exposure. In detail, NK cells recognize tumor antigens through a diverse family of receptors, enabling immune surveillance and tumor clearance. NK cell receptors are categorized into activating and inhibitory receptors, with their balance determining NK cell activation. Inhibitory receptors, such as KIRs and NKG2A, recognize MHC-I molecules on normal cells, preventing unintended attacks. However, tumor cells often evade immune detection by downregulating the MHC-Is expression. Activating receptors, including NKG2D, NKp30, and NKp46, recognize stress-induced ligands (e.g., MICA, MICB) or tumor-specific proteins upregulated on the tumor cell surface. When activating signals outweigh inhibitory ones, NK cells become activated, releasing perforin and granzymes to kill tumor cells directly. They also secrete cytokines like IFN-γ to amplify the anti-tumor immune response (36–38). CAR-NK cell therapy enhances this process by engineering NK cells with Chimeric Antigen Receptors (CARs) that precisely target specific cancer antigens, improving their ability to locate and destroy tumor cells (39). This combined approach not only boosts the cancer-killing efficiency of NK cells but also lowers the risk of severe side effects, making it a promising option for cancer immunotherapy.
Lingyu Li’s clinical study demonstrates that combining natural killer (NK) cell therapy with chemotherapy significantly improves 5-year progression-free and overall survival rates compared to chemotherapy alone. Several studies (ect.al. NCT03319459, NCT04616196) are ongoing to utilize the combination of natural killer (NK) therapy with cetuximab, a first-line treatment for EGFR-positive mCRC that also interacts with NK cells, triggering antibody-dependent cell-mediated cytotoxicity (ADCC) at clinical level. Another two NKG2D CAR-NK Cell Therapy studies (NCT05213195 and NCT05248048) is recruiting patients with refractory metastatic colorectal cancer. These therapies involve using NKG2D CAR-modified NK cells to target cancer cells and aim to evaluate the safety and efficacy of this treatment.
Adoptive transfer of γδ T cells has emerged as a promising immunotherapy for CRC. Unlike conventional αβ T cells, γδ T cells possess innate-like properties, enabling them to recognize and target a broad spectrum of tumor-associated antigens without prior sensitization. γδ T cells expressed multiple anti-tumor receptors NKG2D, NKp30 and NKp46 et al. like CD8+ T cells and NK cells. Interesting, the γ9δ2 TCR recognizes over-synthesized phosphoantigens presented by BTN3A1/BTN2A1, while the γδ1 TCR targets lipid antigens presented by CD1c/d in certain cancer cells. These over-synthesized phosphoantigens and lipid antigens result from lipid metabolism disorders, which commonly occur in cancer cells. This recognition might allow γδ T cells to identify and potentially eliminate tumor cells exhibiting such metabolic aberrations. Recent studies have shown that γδ T cells can effectively induce tumor cell cytotoxicity and modulate the tumor microenvironment, enhancing anti-tumor immunity in CRC. Clinical trials (ACT therapies and non-ACT therapies) have explored the safety and efficacy of γδ T cell-based treatments in CRC, demonstrating encouraging results (40–43). By engineering γδ T cells to express CARs targeting specific tumor antigens, this therapy enhances their tumor-homing ability and cytotoxicity, providing a dual mechanism to recognize and destroy cancer cells. One notable study is a phase I/II trial evaluating the safety and efficacy of allogeneic CAR γδ T cells (CAR001) in subjects with relapsed/refractory solid tumors, including CRC. This study involves dose-escalation to determine the maximum tolerated dose and a dose-expansion phase to assess efficacy, including objective response rate, progression-free survival, and overall survival.
Recently, TIL therapy has shown remarkable success in treating melanoma, with response rates as high as 50-70% (44). Tumor-Infiltrating Lymphocyte (TIL) therapy is a very personalized cancer treatment that uses a patient’s own immune cells to target cancer. TILs are T cells naturally found within a tumor, indicating they can recognize cancer cells. In this therapy, TILs are extracted from the patient’s tumor, expanded in large numbers in the laboratory, and then re-infused into the patient. Once back in the body, these TILs target and attack cancer cells more effectively (45). In a pilot study, Sentinel-node-based adoptive immunotherapy using expanded CD4(+) Th1 lymphocytes showed feasibility, no side effects, and improved survival and tumor regression in advanced CRC patients (46). While some applications in CRC is still under investigation (47, 48) (NCT05902520, NCT06530303, NCT05576077).
microsatellite instability-high (MSI-H) tumors are characterized by a high mutation burden, which increases the likelihood of generating neoantigens that TILs can recognize. As a result, TIL therapy TIL therapy may be suitable for treating colorectal cancer (CRC) patients with MSI-H (50). Despite its potential, TIL therapy for CRC faces several special challenges, optimizing culture conditions and selecting the most reactive TIL subsets are critical areas of research. Identifying patients who are most likely to benefit from TIL therapy is crucial. Biomarkers such as MSI status and PD-L1 expression are being studied to refine patient selection criteria. While there are significant challenges to overcome, ongoing research and clinical trials are continually improving the efficacy and feasibility of TIL therapy.
In recent years, CAR-M (CAR-macrophages) therapy has emerged as a promising immunotherapy for solid tumors (51). CAR-M cells are engineered to enable macrophages to specifically recognize and kill tumor cells. Studies have shown that CAR-M cells can enhance anti-tumor immune responses by targeting immunosuppressive factors within the tumor microenvironment. Compared to traditional CAR-T therapies, CAR-M cells can provide broader anti-tumor effects by eliminating tumor-associated macrophages and overcoming immune tolerance mechanisms. Current research focuses on improving the persistence of CAR-M cells, enhancing their cytotoxicity, and overcoming the immunosuppressive environment of solid tumors (51–53).
The self-developed humanized anti-HER2 chimeric antigen receptor macrophage (human anti-HER2 CAR-M) has been developed to use an adenoviral vector system to express CAR molecules, which specifically bind to human HER2 antigens to recognize and kill tumor cells (53, 54). Cellular and animal experiments showed that anti-human HER2 CAR M cells have significant anti-tumor efficacy and good safety (53, 54). Anti-HER2 CAR-M cell therapy is currently undergoing phase I clinical trial targeting colorectal cancer and other solid tumors (NCT04660929).
Dendritic cell (DC) vaccines utilize dendritic cells as antigen-presenting cells to activate the immune system and target cancer cells. This immunotherapy involves isolating the patient’s own DCs, culturing, expanding, and inducing their differentiation, followed by loading them with tumor-specific antigens derived from the patient’s tumor tissue lysate. The activated DCs are then re-infused into the patient to increase the number of functional DCs in the body and stimulate a T-cell-mediated immune response. This enables precise recognition and elimination of cancer cells carrying specific antigens (55, 56). Even in patients with stage III/IV colorectal cancer, this approach has been used in combination with CIK therapy to enhance quality of life (QOL), minimize toxic side effects, and achieve significant improvements in overall survival (OS), disease-free survival (DFS), and progression-free survival (PFS) (57, 58). Additionally, Ishii et al. demonstrated tumor growth suppression in mouse models by combining this immunotherapy with interferon (IFN)-α therapy, using IFN-α gene-transduced tumor cells (59). One Phase II study (NCT00103142) assesses the survival benefits of two cancer DC vaccines in patients with resected colorectal cancer (CRC) metastases. Vaccinated patients demonstrated improved survival compared to an unvaccinated contemporary group (60). Another Phase I trial (NCT02692976) focuses specifically on the feasibility and safety of this approach (60, 61). A third Phase II study (NCT00558051) explores the feasibility and safety of long-term intralymphatic (IL) infusion of autologous dendritic cells (DCs) using implantable delivery ports in nine stage IV colorectal cancer patient’s post-resection. Eight of nine port implantations were successful (62). Together, these trials aim to determine the potential of DC-based immunotherapy to improve treatment outcomes for patients with advanced colorectal cancer (63). This set of clinical trials investigates the safety, feasibility, and efficacy of dendritic cell (DC) vaccination combined with chemotherapy in patients with metastatic CRC (63, 64).
A critical challenge in developing Immunity cell therapies for cancer treatment is identifying target antigens that are specific to each cancer type. The choice of targets influences the specificity, efficacy, and toxicity of the therapy, ultimately determining its success. Cancer-derived antigens are classified into tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs), also known as neoantigens, and tumor non-peptide antigens (TNAs) (Figure 2).
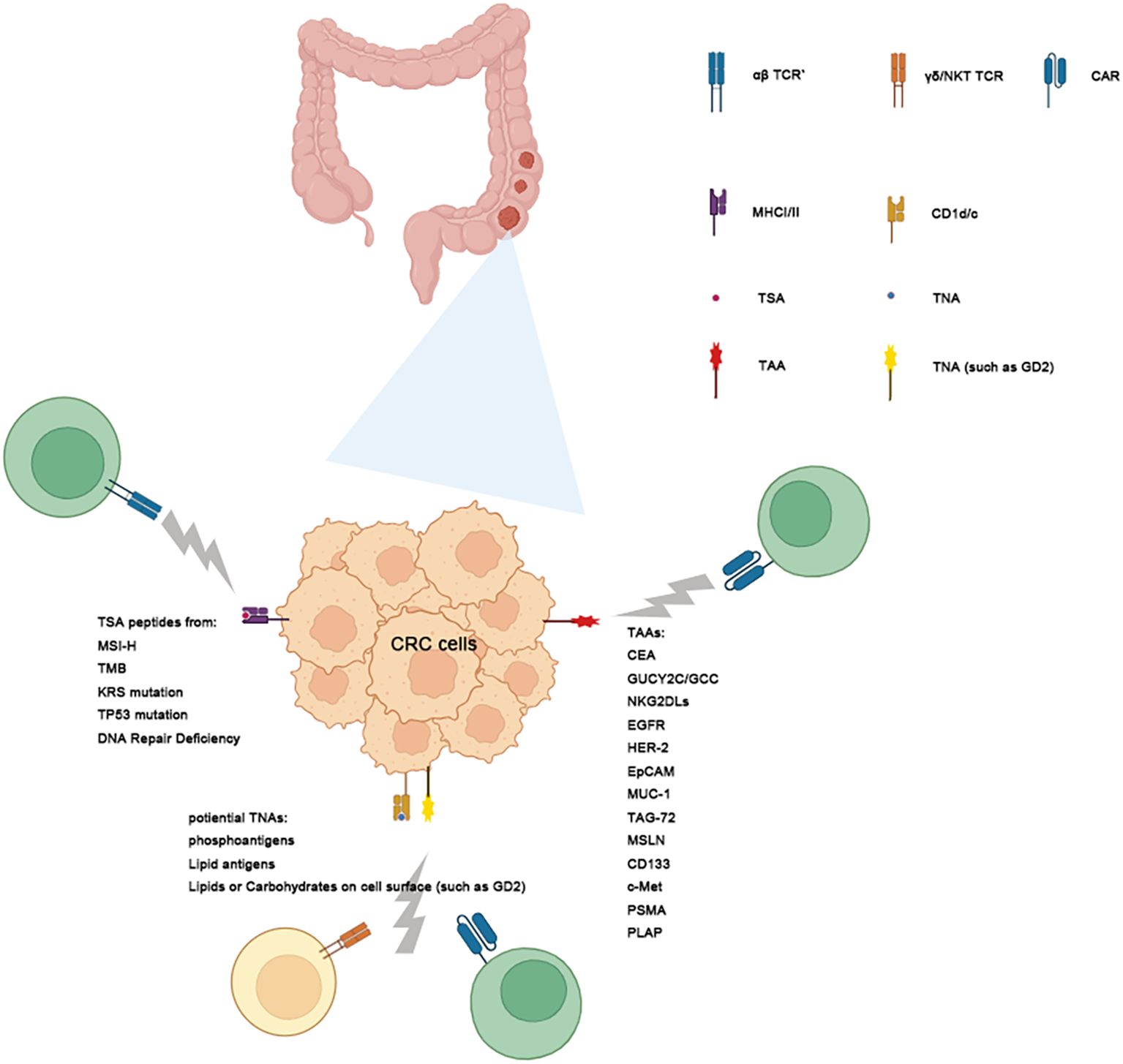
Figure 2. Targeting Tumor Antigens with immunity Cell Therapy. This schematic illustrates the classification of tumor antigens and their role. Tumor antigens are categorized into 3 groups: Tumor-Associated Antigens (TAAs), Tumor-Specific Antigens (TSAs) and Tumor-Non-peptide Antigens (TNAs) in CRC. TAAs: These antigens are expressed in both normal and tumor cells but are overexpressed in tumors. Examples include MUA-1, KKAG-1, and KRAS. While TAAs are common targets for CAR-T therapy, their presence on normal tissues may result in off-target toxicities. Tumor-Specific Antigens (TSAs): TSAs are exclusively expressed in tumor cells and antigen-presented by MCH system, often arising from somatic mutations such as those in KRAS, TP53, and TPAS. These antigens exhibit high specificity, making them ideal targets for TCR-T therapy or DC vaccine with reduced risk of harming healthy tissues. TNAs, including lipids and carbohydrates, are presented by CD1 molecules to activate immune cells like NKT and γδ1 T cells. Gray lightens emanating from the CAR/αβ TCR/γδ TCR/NKTCR indicate its ability to target these antigens, with distinct pathways leading to TAAs, TSAs and TNAs.
Tumor-associated antigens (TAAs) are often overexpressed proteins found on tumor cells. While these antigens are relatively easy to identify and are commonly shared among patients through pathological examination, targeting them with CAR-T cells can lead to severe adverse effects due to their limited specificity. In CRC, TAAs include carcinoembryonic antigen (CEA), mucin-1 (MUC-1) et al. Additionally, cancer/testis antigens (CTAs) are highly immunogenic, as they are typically expressed only in testicular cells. Melanoma-associated antigen (MAGE)-A, NY-ESO-1, PASD1, LAGE-1, OIP5, TTK, PLU1, DKKL1, and FBXO39 are among CTAs that are overexpressed in CRC (Figure 2, Table 2). These antigens are potential targets for immunotherapy due to their selective expression in tumor cells (9).
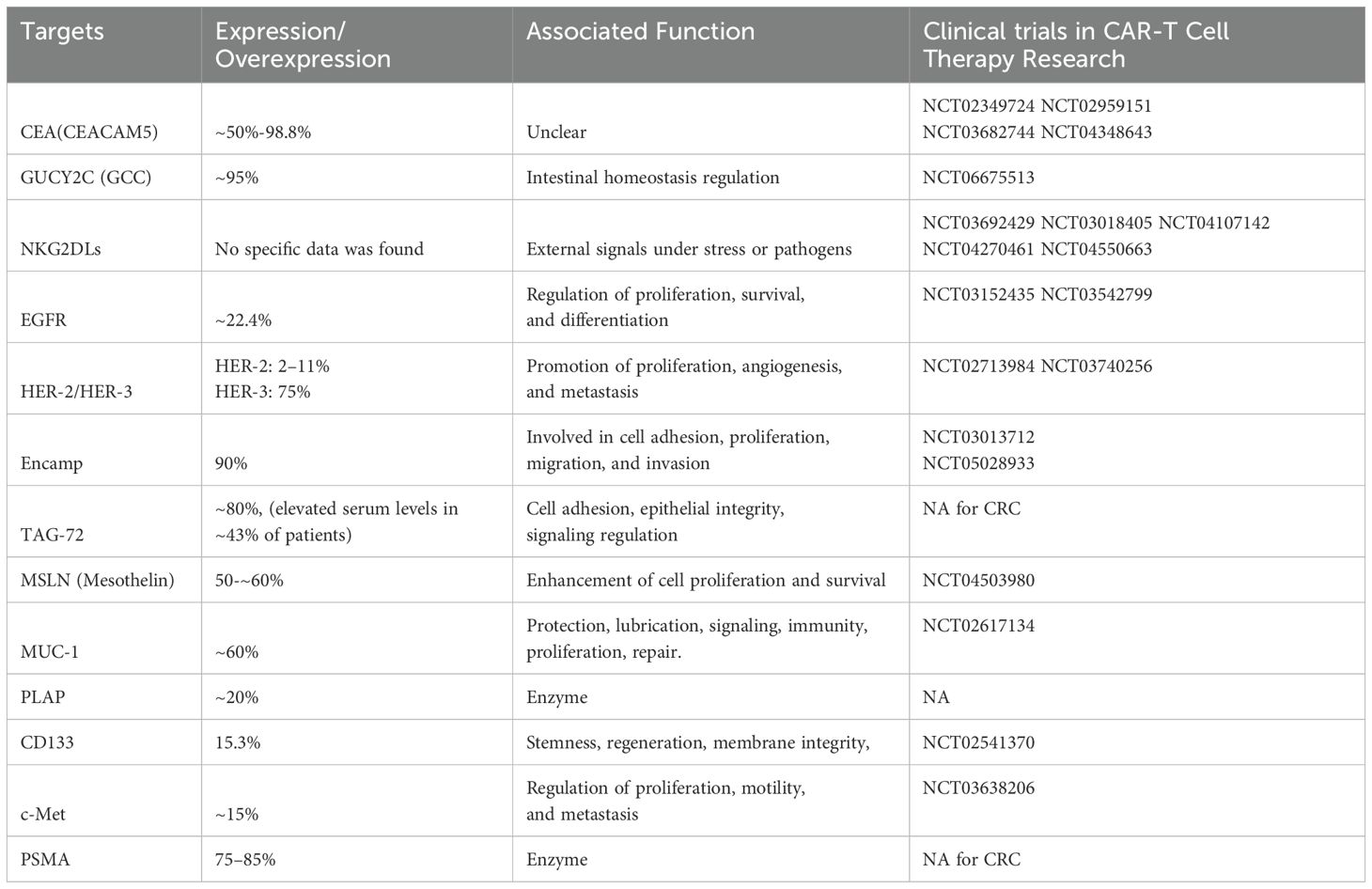
Table 2. TAA summary in CRC.
Conversely, non-synonymous somatic mutations, including single-nucleotide variations, insertions/deletions, structural alterations, frameshift mutations, and fusion genes, lead to the production of defective peptides (9), referred to as tumor-specific antigens (TSAs) or neoantigens. These neoantigens are potential targets for immune recognition (65). These antigens are often presented by MHC system on surface of tumor cells and recognized by TCR of αβ T cells, but they are difficult to be identified with routine pathological examination. Since TSAs are exclusively expressed in tumor cells, they have high therapeutic value due to the low risk of on-target off-tumor effects (65) (Figure 2).
TNA are non-protein molecules synthesized by tumor cells that play a critical role in immune recognition and cancer immunotherapy. Unlike peptide antigens, which are derived from protein fragments, non-peptide antigens primarily encompass lipids, glycolipids, carbohydrates, and certain small molecules unique to or over synthesized in cancer cells. These antigens often arise due to alterations in cellular metabolism and biosynthetic pathways that are characteristic of malignant transformation. The presentation of tumor non-peptide antigens to the immune system typically occurs through specialized antigen-presenting molecules such as the CD1 family, which can present lipid-based antigens to natural killer T (NKT) cells, γδ1 T cells and other immune effectors (66–68). Recent research on tumor non-peptide antigens in colorectal cancer (CRC) highlights promising advancements in immunotherapy approaches (69–73). But most current gene modified T cells therapies focus on CAR-T therapy targeting TAAs due to the low prevalence of these specific mutations and among CRC patients. And some gene modified immune cells therapies focus on TCR-T therapy or DC vaccines therapy targeting TSAs (Figure 2).
Tumor-associated antigen (TAA) is usually highly expressed on tumor cells, which is an important target for adoptive immune cell therapy of colorectal cancer (Table 2). The fetal glycosylphosphatidylinositol-anchored glycoprotein CEA, also known as CEACAM5, is a member of the immunoglobulin superfamily. Although the exact function and signaling mechanisms of CEA remain unclear, it is typically undetectable in normal adult tissues, except in the gastrointestinal system, where it is expressed at low levels during the early stages of human embryonic and fetal development (74). It is overexpressed in ~50%- 98.8% of colorectal cancer (CRC) tissues, making it a valuable diagnostic and prognostic tumor marker as well as a promising target for novel CRC treatments (75, 76).
Guanylyl cyclase 2C (GUCY2C or GCC), a member of the mucosal cyclase receptor family, is predominantly expressed in intestinal epithelial cells ranging from the duodenum to the rectum, with the exception of certain hypothalamic neurons (77). It converts GTP to cGMP upon activation by guanylin and uroguanylin. This cGMP signaling regulates fluid secretion, intestinal barrier integrity, and epithelial cell proliferation, differentiation, and apoptosis. GUCY2C also influences systemic energy balance and appetite control. Dysregulation of this signaling pathway can contribute to conditions such as cancer, bowel transit disorders, and inflammatory bowel disease. During the early stages of tumorigenesis, the loss of GUCY2C-binding ligands (guanylin or uroguanylin) leads to the silencing of this pathway, thereby promoting intestinal remodeling (78, 79). GUCY2C is overexpressed in nearly 95% of CRC cases, including metastatic CRC (mCRC), as well as in pancreatic and some gastroesophageal tumors (80).
NKG2D (Natural killer group 2 member D) ligands (NKG2DL) include the cytomegalovirus UL16-binding proteins (ULBP1-6) and MHC-I Chain-related molecules (MIC-A and MIC-B) in humans. These ligands are expressed response to external signals such as stress or pathogens and during neoplastic cell transformation, but are rarely present in healthy tissues (80). NKG2D is a C-type lectin-like transmembrane receptor primarily expressed on NK cells, as well as on CD8+ T cells, γδ T cells, and certain autoreactive or immunosuppressive CD4+ T cells (81, 82). This receptor activates immune cells through the adaptor molecule DAP10/12 and its binding to NKG2DLs, stimulating proliferation, production of pro-inflammatory cytokines, and cytotoxic function. NKG2DLs are expressed in various tumors, including carcinomas (CRC et al), leukemia, lymphoma, multiple myeloma, melanoma, glioma, osteosarcoma, and neuroblastoma (83). Immune cells are capable of recognizing and eliminating CRC cells through the interaction between NKG2D and NKG2D ligands (NKG2DLs) (83). However, the data regarding the overexpression of NKG2DLs on cancer cells have not been found by the authors.
Epidermal Growth Factor (EGFR) is a transmembrane glycoprotein and a member of the protein kinase superfamily. Ligand binding induces receptor dimerization, which subsequently activates signaling pathways that promote cell division, survival, and proliferation (84, 85). Recent studies have identified novel roles for EGFR in regulating autophagy and cellular metabolism, which are activated in response to environmental and cellular stresses in cancer cells (86–88). EGFR is often overexpressed and/or mutated in most solid tumors, including colorectal, brain, renal, ovarian, breast, head-and-neck, and non-small-cell lung cancers (89, 90). Oncogenic mutations, including EGFR variant III (EGFRvIII), which seems to be expressed only in tumor tissue, are the specific target of some immunotherapies. This mutation allows for ligand-independent activation and is found in the extracellular domain of EGFR (91, 92). It has been reported that EGFR expression was detected in 44.7% of cancer tissues compared to 21.4% in normal tissues. Notably, high expression of EGFR was observed in 22.4% of cancer tissues, while no high expression (0%) was found in normal tissues (93).
Human epidermal growth factor receptor 2 (HER-2), also known as ERBB2, a member of the EGFR family, is a transmembrane receptor tyrosine kinase involved in cell growth and differentiation. It is overexpressed in various cancers (including CRC), particularly breast cancer. HER-2 activation through dimerization with other EGFR family members (HER-3) promotes signaling pathways that drive tumor progression, including proliferation, survival, and metastasis (94, 95). HER-2 is overexpressed in CRC with expression rates ranging from 2% to 11%, and both HER-2 and HER-3 are overexpressed in liver metastases of CRC patients (8% and 75%, respectively), making them promising targets for CAR-T therapies (96, 97).
The epithelial cell adhesion molecule (EpCAM) is a type I transmembrane glycoprotein primarily found on the basolateral membrane of normal epithelial cells, where it mediates cell-cell adhesion and regulates epithelial integrity (98, 99). Under normal conditions, EpCAM not only participates in cell-cell adhesion but also regulates differentiation in progenitor and embryonic stem cells (100). However, in cancer, EpCAM overexpression is associated with increased proliferation, migration, invasion, and tumor metastasis (101). In CRC, EpCAM is reported that overexpressed in more than 90% of tumor cells (102).
Mucin-1 (MUC-1) is a heavily glycosylated transmembrane glycoprotein primarily expressed on the surface of epithelial cells. It plays a critical role in protecting mucosal surfaces, mediating cell signaling, and facilitating cell adhesion (103). In cancer, MUC-1 is often overexpressed and aberrantly glycosylated, contributing to tumor progression, metastasis, and immune evasion. Its altered expression in various cancers, including breast, ovarian, and lung cancers, makes MUC-1 a significant biomarker for cancer diagnosis and prognosis (104, 105). MUC-1 expression was observed in 61% of colorectal cancer (CRC) tissues; however, no significant overexpression was detected (106).
The membrane-bound glycoprotein known as tumor-associated glycoprotein (TAG)-72 is normally absent from most normal tissues, with the exception of fetal tissue and the endometrium during the secretory phase (107, 108). It is a mucin-like protein, which contributes to its role in tumor progression, metastasis, and immune evasion and expressed in various epithelial tumors, including colorectal, ovarian, and breast cancers. Compared to normal mucosa, TAG-72 is overexpressed in 80% of colorectal cancers (CRCs), and serum levels are 43% higher in CRC patients (109, 110).
Mesothelin (MSLN) is a glycosylphosphatidylinositol-anchored cell surface protein involved in various aspects of cancer biology, including promoting cell adhesion, proliferation, and survival. However, its precise biological functions are not yet fully understood (111). Normally, MSLN is normally expressed in limited tissues, primarily in the mesothelial cells lining the pleura, peritoneum, and pericardium. It is also expressed in various normal epithelial tissues, including the pancreas, ovaries, and some cells of the lung (112). In cancer, MSLN is overexpressed not only CRC but also in mesothelioma, ovarian cancer, pancreatic cancer, as well as in cervical, endometrial, biliary cancers, uterine serous carcinoma, cholangiocarcinoma, and pediatric acute myeloid leukemia (112, 113). In CRC, up to 50%-60% of malignancies over express MSLN (106, 114).
Cluster of differentiation 133 (CD133), or prominin-1, is a pentaspan transmembrane glycoprotein expressed on membrane protrusions. It serves as a stem cell marker, playing crucial roles in cancer progression, tumorigenesis, and cell differentiation, particularly in hematopoietic and neural stem cells (115). CD133 is expressed by several progenitor and stem cells and plays a role in plasma membrane architecture. In cancers of the kidney, brain, colorectal, lung, pancreatic, and ovarian tissues, it has been proposed as a surface marker for cancer stem cells (116). Higher rates of distant metastasis, metastatic recurrence, and chemoresistance are all linked to CD133 expression in colorectal cancer (117). A study showed 15.3% positive rate in CRC tissues (118).
A receptor tyrosine kinase called mesenchymal–epithelial transition factor (c-Met) is essential for cellular functions like growth, differentiation, and survival. It is expressed in a variety of cell types, including fibroblasts, hematological cells, keratinocytes, endothelium, liver cells, and epithelial cells. It is activated by its ligand, hepatocyte growth factor (HGF) (119) (120). Colorectal, gastric, renal, lung, pancreatic, ovarian, prostate, and breast malignancies all overexpress c-Met. About 15% of individuals with colorectal cancer have overexpressed c-Met, which is linked to cell proliferation, differentiation, cancer progression, and metastasis (119).
Usually found on prostate epithelial cells, prostate-specific membrane antigen (PSMA), also referred to as folate hydrolase I or glutamate carboxypeptidase II, is a transmembrane protein that is also expressed in the duodenal mucosa, salivary glands, some renal tubular cells, and a subset of neuroendocrine cells in colonic crypts (121). In CRC, PSMA is expressed in approximately 75–85% of primary tumors and metastases (122, 123).
Phosphoric acid monoesters are hydrolyzed by the metalloenzyme placental alkaline phosphatase (PLAP). While predominantly expressed in the placenta, PLAP is also detected in trace amounts in other tissues, including the lung, testis, fallopian tubes, and uterine cervix (124, 125). PLAP is overexpressed in CRC and is detected in over 20% of colorectal adenocarcinomas (126).
Advances in next-generation sequencing (NGS) have enabled the identification of CRC neoantigens, which are predominantly generated by DNA mismatch repair (MMR) deficiencies and microsatellite instability (MSI)tumors exhibit a high mutation burden, increasing the likelihood of neoantigen formation (127, 128). Neoantigen-based therapies, such as personalized cancer vaccines and adoptive T-cell therapies, have demonstrated potential in preclinical studies and early-phase clinical trials. Moreover, immunity inhibitors (e.g., anti-PD-1/PD-L1 therapies) show enhanced efficacy in MSI-high CRC due to the immunogenic tumor microenvironment fostered by neoantigens (129, 130). However, challenges neoantigen heterogeneity and immune escape mechanisms. Efforts to improve computational algorithms for neoantigen prediction and the integration of combinatory therapies are underway to enhance clinical outcomes.
Linnebacher M et al. have successfully developed cytotoxic T lymphocytes (CTLs) capable of recognizing HLA-A2.1-restricted frameshift peptides through the study of specific frameshift-derived antigens. Among the 16 predicted frameshift peptides, 3 demonstrated the ability to specifically lyse target cells loaded with their corresponding peptides (131). Yu et al. identified various neoantigen-containing peptides, such as SEC11A-R11L and ULK1-S248L, through cytotoxicity assays using neoantigen-reactive T cells (NRTs) in HLA-A0201+PW11 cell models. These peptides demonstrated superior efficacy in eliciting antigen-specific cytotoxic T lymphocyte (CTL) responses compared to their natural peptide counterparts (132). Moreover, Mutations at codon 12 of the Ki-ras gene are frequently observed in pancreatic and colorectal cancers. Analysis of T-cell immune responses revealed that 75% of pancreatic cancer patients and 35% of colorectal cancer patients exhibited specific responses to mutated Ki-ras-derived peptides. Although T cells in individual patients may not recognize the mutated Ki-ras peptides expressed in their own tumors, these peptides show potential as targets for cancer immunotherapy (133).
The tumor microenvironment (TME) in CRC presents various challenges that can reduce the effectiveness of immune cell therapies, including CAR-T and other APC immune cell treatments. The main immunosuppressive mechanisms, including hypoxia, tumor immune microenvironment (TIME), immunosuppressive cytokines, and the tumor stromal barrier, and we discuss potential strategies to counteract these challenges and improve the therapeutic outcomes for CRC.
Hypoxia is a common feature in solid tumors due to rapid cell proliferation and inadequate blood supply. In CRC, hypoxia is associated with an increase in hypoxia-inducible factors (HIFs), which drive metabolic reprogramming and support an immunosuppressive environment (134). Impact on Immune Cells, Hypoxia reduces the function and persistence of cytotoxic immune cell. For instance, low oxygen levels reduce the effectiveness of glycolysis-dependent T cells, weakening their cytotoxic response (135). Approaches to mitigate hypoxia include using HIF inhibitors, such as acriflavine (136), to block HIF activity, as well as engineering CAR/TCR immune cells to resist hypoxic stress. Genetic modifications enabling CAR/TCR cells to express enzymes that mitigate hypoxia-induced acidity, such as carbonic anhydrase IX, have shown promise (137, 138).
The tumor immune microenvironment (TIME) in CRC can be classified into three distinct subtypes based on cytotoxic T cell landscape: inflamed (“hot”), immune-altered (excluded and immunosuppressed), and immune-desert (“cold”) tumors (139). Inflamed CRC, commonly associated with MSI-H) tumors, is characterized by a robust infiltration of CD8+ T cells, Th1 cells, and dendritic cells (DCs), along with high levels of pro-inflammatory cytokines such as IFN-γ and TNF-α. These tumors typically exhibit strong responses to immune checkpoint inhibitors (ICIs) which widely are expressed on tumor cells as well as tumor immune cells (140–142). Immune- altered CRC displays a substantial presence of T cells; however, these cells are largely confined to the tumor periphery and fail to infiltrate the tumor core. A significant proportion of microsatellite stable (MSS) CRCs fall into this category. Immune-desert CRC, which comprises the majority of MSS CRC cases, is characterized by a paucity of T cells within the tumor due to low tumor mutational burden (TMB), impaired antigen presentation, and an immunosuppressive tumor microenvironment (139). In both immune-excluded and immune-desert tumors, M2 macrophages, recruiting regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) play key roles in immune suppression. They play a significant role in creating an immunosuppressive microenvironment by secreting cytokines such as TGF-β, IL-10, and VEGF (143). Moreover, M2 macrophages contribute to immune suppression by enhancing tumor angiogenesis and promoting fibrosis, further inhibiting T cell infiltration (142). Tregs and MDSCs inhibit the activation of cytotoxic T cells and reduce the overall anti-tumor immune response.
CRC tumors (tumor cells, Cancer-Associated Fibroblasts (CAFs) and the immune cells) release a range of immunosuppressive cytokines, including TGF-β, IL-10, and VEGF. These cytokines further contribute to an immunosuppressive environment by recruiting regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which inhibit the cytotoxic function of effector immune cells (130–132). Immunosuppressive cytokines reduce engineering CAR/TCR immune cells efficacy by decreasing their proliferation, persistence, and cytokine release (133). To improve the effectiveness of CAR-T therapy/other ACT in the tumor immune microenvironment (TIME) of CRC, key strategies can be employed. These include modifying CAR-T cells to resist the suppressive effects of immunosuppressive cells and cytokines. One effective approach is to make CAR-T cells resistant to immunosuppressive cytokines such as TGF-β and IL-10. Alternatively, combining CAR-T therapy with TGF-β inhibitors or other drugs may help enhance the infiltration and activity of CAR-T cells within the tumor, thereby improving treatment efficacy (137, 138). Moreover, combining engineering CAR immune cells therapies with checkpoint inhibitors (e.g., anti-PD-1/PD-L1 antibodies) can help counteract the suppressive cytokine environment (134–136).
The tumor stroma in CRC is a dense structure composed of extracellular matrix (ECM), fibroblasts, and various stromal cells. This dense ECM physically restricts immune cell infiltration and limits the access of CAR-T cells to tumor cells (144, 145). The stromal barrier prevents effective immune cell infiltration and reduces the efficacy of immune cell therapies. Cancer-associated fibroblasts (CAFs) contribute to this barrier by producing ECM components like collagen and fibronectin (146). Stromal remodeling strategies, such as targeting CAFs with fibroblast activation protein (FAP) inhibitors or using hyaluronidase to degrade hyaluronic acid, can enhance immune cell penetration (147–149). Another approach involves modifying CAR cells or other APC immune cells to express enzymes that degrade ECM proteins, which has shown promise in increasing APC cell infiltration in solid tumors (149, 150).
In summary, the immunosuppressive tumor microenvironment in CRC is a major obstacle to effective immune cell therapies. Strategies such as genetic modification of immune cells (151), combination therapies with checkpoint inhibitors (134–136), and tumor stromal remodeling hold promise for overcoming these barriers (142–144). Advances in these approaches could significantly improve the therapeutic outcomes for CRC patients undergoing immune cell therapies.
In parallel, rapid advancements in biomedical engineering, synthetic biology, and artificial intelligence (AI), ACT is entering a new era of enhanced precision, efficacy, and safety. Cutting-edge biomaterials and tissue engineering technologies are optimizing immune cells delivery and persistence in vivo, improving their functionality within the immunosuppressive tumor microenvironment. Synthetic biology enables fine-tuned regulation of gene-modified immune cells, such as through logic-gated CARs (152) or gene circuit-based controls (153), allowing activation only under specific conditions while mitigating off-target toxicity and immune-related side effects (154, 155). Moreover, artificial intelligence technology (AI) is revolutionizing TCR/CAR therapy by facilitating target selection and leveraging data-driven approaches to refine patient stratification and treatment response prediction (156). Additionally, the combination of CRISPR/Cas9 and AI, particularly in data analysis, optimizing gene editing, studying tumor heterogeneity, CAR-T cell therapy, and clinical trials, can greatly enhance the efficiency of precision medicine and cancer treatment (157).
Looking ahead, the integration of these cutting-edge technologies will drive the development of next-generation ACT with superior therapeutic efficacy and minimized adverse effects. By harnessing precise targeting, programmable regulation, and optimized manufacturing, ACTs hold the potential to overcome existing challenges, achieve durable anti-tumor responses, and provide safer, more effective treatment options for CRC patients.
CL: Writing – original draft, Writing – review & editing. NL: Writing – original draft, Writing – review & editing. TZ: Writing – review & editing. YT: Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. Provincial Science and Technology Expert Workstation Grant for Tongcun Zhang from Huizhou Central People’s Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J For Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Chen CH, Hsieh MC, Lao WT, Lin EK, Lu YJ, Wu SY. Multidisciplinary team intervention associated with improved survival for patients with colorectal adenocarcinoma with liver or lung metastasis. Am J Cancer Res. (2018) 8:1887–98.
3. Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. (1989) 142:3714–25. doi: 10.4049/jimmunol.142.10.3714
4. Rosenberg SA, Kawakami Y, Robbins PF, Wang R. Identification of the genes encoding cancer antigens: implications for cancer immunotherapy. Adv Cancer Res. (1996) 70:145–77. doi: 10.1016/S0065-230X(08)60874-X
5. Yang JC, Rosenberg SA. Adoptive T-cell therapy for cancer. Adv Immunol. (2016) 130:279–94. doi: 10.1016/bs.ai.2015.12.006
6. Girard-Gagnepain A, Amirache F, Costa C, Lévy C, Frecha C, Fusil F, et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood. (2014) 124:1221–31. doi: 10.1182/blood-2014-02-558163
7. Radek C, Bernadin O, Drechsel K, Cordes N, Pfeifer R, Sträßer P, et al. Vectofusin-1 improves transduction of primary human cells with diverse retroviral and Lentiviral pseudotypes, enabling robust, automated closed-system manufacturing. Hum Gene Ther. (2019) 30:1477–93. doi: 10.1089/hum.2019.157
8. Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and robust NK-cell transduction with baboon envelope Pseudotyped lentivector. Front Immunol. (2019) 10:2873. doi: 10.3389/fimmu.2019.02873
9. Aparicio C, Belver M, Enríquez L, Espeso F, Núñez L, Sánchez A, et al. Cell therapy for colorectal cancer: the promise of chimeric antigen receptor (CAR)-T cells. Int J Mol Sci. (2021) 22:11781. doi: 10.3390/ijms222111781
10. Burke HB. Outcome prediction and the future of the TNM staging system. JNCI: J Natl Cancer Institute. (2004) 96:1408–9. doi: 10.1093/jnci/djh293
11. Gunderson LL, Jessup JM, Sargent DJ, Greene FL, Stewart AK. Revised TN categorization for colon cancer based on national survival outcomes data. J Clin Oncol. (2010) 28:264–71. doi: 10.1200/JCO.2009.24.0952
12. Weiser MR. AJCC 8th edition: colorectal cancer. Ann Surg Oncol. (2018) 25:1454–5. doi: 10.1245/s10434-018-6462-1
13. Lee DW, Han SW, Cha Y, Bae JM, Kim HP, Lyu J, et al. Association between mutations of critical pathway genes and survival outcomes according to the tumor location in colorectal cancer. Cancer. (2017) 123:3513–23. doi: 10.1002/cncr.v123.18
14. Harada S, Morlote D. Molecular pathology of colorectal cancer. Adv Anat Pathol. (2020) 27:20–6. doi: 10.1097/PAP.0000000000000247
15. Michel M, Kaps L, Maderer A, Galle PR, Moehler M. The role of p53 dysfunction in colorectal cancer and its implication for therapy. Cancers (Basel). (2021) 13:2296. doi: 10.3390/cancers13102296
16. Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. (2015) 149:1204–1225.e12. doi: 10.1053/j.gastro.2015.07.011
17. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front Immunol. (2017) 8:774. doi: 10.3389/fimmu.2017.00774
18. Pan QZ, Zhao JJ, Yang CP, Zhou YQ, Lin JZ, Tang Y, et al. Efficacy of adjuvant cytokine-induced killer cell immunotherapy in patients with colorectal cancer after radical resection. Oncoimmunology. (2020) 9:1752563. doi: 10.1080/2162402X.2020.1752563
19. Zhang J, Zhu L, Zhang Q, He X, Yin Y, Gu Y, et al. Effects of cytokine-induced killer cell treatment in colorectal cancer patients: a retrospective study. BioMed Pharmacother. (2014) 68:715–20. doi: 10.1016/j.biopha.2014.07.010
20. Fischer JW, Bhattarai N. CAR-T cell therapy: mechanism, management, and mitigation of inflammatory toxicities. Front Immunol. (2021) 12:693016. doi: 10.3389/fimmu.2021.693016
21. Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. (2023) 12:14. doi: 10.1186/s40164-023-00373-7
22. Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-cell therapy in hematological Malignancies. Int J Mol Sci. (2021) 22:8996. doi: 10.3390/ijms22168996
23. Uslu U, June CH. Beyond the blood: expanding CAR T cell therapy to solid tumors. Nat Biotechnol. (2024) 12. doi: 10.1038/s41587-024-02446-2
24. Kaviyarasan V, Das A, Deka D, Saha B, Banerjee A, Sharma NR, et al. Advancements in immunotherapy for colorectal cancer treatment: a comprehensive review of strategies, challenges, and future prospective. Int J Colorectal Dis. (2024) 40:1. doi: 10.1007/s00384-024-04790-w
25. Chen J, Zhu T, Jiang G, Zeng Q, Li Z, Huang X. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol Cancer. (2023) 22:131. doi: 10.1186/s12943-023-01830-x
26. Zheng Z, Wieder T, Mauerer B, Schäfer L, Kesselring R, Braumüller H. T cells in colorectal cancer: unravelling the function of different T cell subsets in the tumor microenvironment. Int J Mol Sci. (2023) 24:11673. doi: 10.3390/ijms241411673
27. Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. (2019) 10:1486. doi: 10.1038/s41467-019-09415-3
28. Szlasa W, Sztuder A, Kaczmar-Dybko A, Maciejczyk A, Dybko J. Efficient combination of radiotherapy and CAR-T - A systematic review. BioMed Pharmacother. (2024) 174:116532. doi: 10.1016/j.biopha.2024.116532
29. Qin VM, Haynes NM, D’Souza C, Neeson PJ, Zhu JJ. CAR-T plus radiotherapy: A promising combination for immunosuppressive tumors. Front Immunol. (2021) 12:813832. doi: 10.3389/fimmu.2021.813832
30. Klebanoff CA, Chandran SS, Baker BM, Quezada SA, Ribas A. T cell receptor therapeutics: immunological targeting of the intracellular cancer proteome. Nat Rev Drug Discov. (2023) 22:996–1017. doi: 10.1038/s41573-023-00809-z
31. Mullard A. TCR-boosted T cell therapy yields proof-of-principle clinical data in solid cancer. Nat Rev Drug Discov. (2024) 23:651. doi: 10.1038/d41573-024-00121-4
32. Gaissmaier L, Elshiaty M, Christopoulos P. Breaking bottlenecks for the TCR therapy of cancer. Cells. (2020) 9:2095. doi: 10.3390/cells9092095
33. Crowther MD, Svane IM, Met Ö. T-cell gene therapy in cancer immunotherapy: why it is no longer just CARs on the road. Cells. (2020) 9:1588. doi: 10.3390/cells9071588
34. Sun Z, Chen F, Meng F, Wei J, Liu B. MHC class II restricted neoantigen: A promising target in tumor immunotherapy. Cancer Lett. (2017) 392:17–25. doi: 10.1016/j.canlet.2016.12.039
35. Shafer P, Kelly LM, Hoyos V. Cancer therapy with TCR-engineered T cells: current strategies, challenges, and prospects. Front Immunol. (2022) 13:835762. doi: 10.3389/fimmu.2022.835762
36. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
37. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. (2021) 18:85–100. doi: 10.1038/s41571-020-0426-7
38. Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. (2022) 22:557–75. doi: 10.1038/s41568-022-00491-0
39. Du N, Guo F, Wang Y, Cui J. NK cell therapy: A rising star in cancer treatment. Cancers (Basel). (2021) 13:4129. doi: 10.3390/cancers13164129
40. Izumi T, Kondo M, Takahashi T, Fujieda N, Kondo A, Tamura N, et al. Ex vivo characterization of γδ T-cell repertoire in patients after adoptive transfer of Vγ9Vδ2 T cells expressing the interleukin-2 receptor β-chain and the common γ-chain. Cytotherapy. (2013) 15:481–91. doi: 10.1016/j.jcyt.2012.12.004
41. Bennouna J, Levy V, Sicard H, Senellart H, Audrain M, Hiret S, et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vgamma9Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol Immunother. (2010) 59:1521–30. doi: 10.1007/s00262-010-0879-0
42. Noguchi A, Kaneko T, Kamigaki T, Fujimoto K, Ozawa M, Saito M, et al. Zoledronate-activated Vγ9γδ T cell-based immunotherapy is feasible and restores the impairment of γδ T cells in patients with solid tumors. Cytotherapy. (2011) 13:92–7. doi: 10.3109/14653249.2010.515581
43. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer. (2011) 105:778–86. doi: 10.1038/bjc.2011.293
44. Friese C, Heeke C, Kirketerp-Møller N, Færch S, Nielsen MJ, Hey A, et al. 175 High-dose TIL product with improved phenotype and functionality. J ImmunoTherapy Cancer. (2021) 9:A187–7. doi: 10.1136/jitc-2021-SITC2021.175
45. Wang Z, Ahmed S, Labib M, Wang H, Hu X, Wei J, et al. Efficient recovery of potent tumour-infiltrating lymphocytes through quantitative immunomagnetic cell sorting. Nat BioMed Eng. (2022) 6:108–17. doi: 10.1038/s41551-021-00820-y
46. Karlsson M, Marits P, Dahl K, Dagöö T, Enerbäck S, Thörn M, et al. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann Surg Oncol. (2010) 17:1747–57. doi: 10.1245/s10434-010-0920-8
47. Amaria R, Knisely A, Vining D, Kopetz S, Overman MJ, Javle M, et al. Efficacy and safety of autologous tumor-infiltrating lymphocytes in recurrent or refractory ovarian cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. J Immunother Cancer. (2024) 12:e006822. doi: 10.1136/jitc-2023-006822
48. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
49. Yu WL, Hua ZC. Chimeric antigen receptor T-cell (CAR T) therapy for hematologic and solid Malignancies: efficacy and safety-A systematic review with meta-analysis. Cancers (Basel). (2019) 11:47. doi: 10.3390/cancers11010047
50. Wankhede D, Yuan T, Kloor M, Halama N, Brenner H, Hoffmeister M. Clinical significance of combined tumour-infiltrating lymphocytes and microsatellite instability status in colorectal cancer: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. (2024) 11:609–19. doi: 10.1016/S2468-1253(24)00091-8
51. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. (2021) 81:1201–8. doi: 10.1158/0008-5472.CAN-20-2990
52. Yang S, Wang Y, Jia J, Fang Y, Yang Y, Yuan W, et al. Advances in engineered macrophages: A new frontier in cancer immunotherapy. Cell Death Dis. (2024) 15:238. doi: 10.1038/s41419-024-06616-7
53. Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol. (2021) 12:783305. doi: 10.3389/fimmu.2021.783305
54. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
55. Filin IY, Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV. Recent advances in experimental dendritic cell vaccines for cancer. Front Oncol. (2021) 11:730824. doi: 10.3389/fonc.2021.730824
56. Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized dendritic cell vaccines-recent breakthroughs and encouraging clinical results. Front Immunol. (2019) 10:766. doi: 10.3389/fimmu.2019.00766
57. Xie Y, Huang L, Chen L, Lin X, Chen L, Zheng Q. Effect of dendritic cell-cytokine-induced killer cells in patients with advanced colorectal cancer combined with first-line treatment. World J Surg Oncol. (2017) 15:209. doi: 10.1186/s12957-017-1278-1
58. Zhu H, Yang X, Li J, Ren Y, Zhang T, Zhang C, et al. Immune response, safety, and survival and quality of life outcomes for advanced colorectal cancer patients treated with dendritic cell vaccine and cytokine-induced killer cell therapy. BioMed Res Int. (2014) 2014:603871. doi: 10.1155/2014/603871
59. Ishii S, Hiroishi K, Eguchi J, Hiraide A, Imawari M. Dendritic cell therapy with interferon-alpha synergistically suppresses outgrowth of established tumors in a murine colorectal cancer model. Gene Ther. (2006) 13:78–87. doi: 10.1038/sj.gt.3302608
60. Westdorp H, Creemers JHA, van Oort IM, Schreibelt G, Gorris MAJ, Mehra N, et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J Immunother Cancer. (2019) 7:302. doi: 10.1186/s40425-019-0787-6
61. Westdorp H, Creemers JHA, van Oort IM, Mehra N, Hins-de-Bree SM, Figdor CG, et al. High health-related quality of life during dendritic cell vaccination therapy in patients with castration-resistant prostate cancer. Front Oncol. (2020) 10:536700. doi: 10.3389/fonc.2020.536700
62. Radomski M, Zeh HJ, Edington HD, Pingpank JF, Butterfield LH, Whiteside TL, et al. Prolonged intralymphatic delivery of dendritic cells through implantable lymphatic ports in patients with advanced cancer. J Immunother Cancer. (2016) 4:24. doi: 10.1186/s40425-016-0128-y
63. Wooster AL, Girgis LH, Brazeale H, Anderson TS, Wood LM, Lowe DB. Dendritic cell vaccine therapy for colorectal cancer. Pharmacol Res. (2021) 164:105374. doi: 10.1016/j.phrs.2020.105374
64. Barth RJ Jr., Fisher DA, Wallace PK, Channon JY, Noelle RJ, Gui J, et al. A randomized trial of ex vivo CD40L activation of a dendritic cell vaccine in colorectal cancer patients: tumor-specific immune responses are associated with improved survival. Clin Cancer Res. (2010) 16:5548–56. doi: 10.1158/1078-0432.CCR-10-2138
65. Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discov. (2022) 21:261–82. doi: 10.1038/s41573-021-00387-y
66. Hakomori S. Tumor-associated carbohydrate antigens defining tumor Malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. (2001) 491:369–402. doi: 10.1007/978-1-4615-1267-7_24
67. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. (2007) 448:44–9. doi: 10.1038/nature05907
68. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature. (1995) 375:155–8. doi: 10.1038/375155a0
69. Wang D, Madunić K, Mayboroda OA, Lageveen-Kammeijer GSM, Wuhrer M. (Sialyl)Lewis antigen expression on glycosphingolipids, N-, and O-glycans in colorectal cancer cell lines is linked to a colon-like differentiation program. Mol Cell Proteomics. (2024) 23:100776. doi: 10.1016/j.mcpro.2024.100776
70. Shi M, Kleski KA, Trabbic KR, Bourgault JP, Andreana PR. Sialyl-tn polysaccharide A1 as an entirely carbohydrate immunogen: synthesis and immunological evaluation. J Am Chem Soc. (2016) 138:14264–72. doi: 10.1021/jacs.6b05675
71. Kakiuchi Y, Tsuji S, Tsujii M, Murata H, Kawai N, Yasumaru M, et al. Cyclooxygenase-2 activity altered the cell-surface carbohydrate antigens on colon cancer cells and enhanced liver metastasis. Cancer Res. (2002) 62:1567–72.
72. Minami S, Furui J, Kanematsu T. Role of carcinoembryonic antigen in the progression of colon cancer cells that express carbohydrate antigen. Cancer Res. (2001) 61:2732–5.
73. von Gerichten J, Lamprecht D, Opálka L, Soulard D, Marsching C, Pilz R, et al. Bacterial immunogenic α-galactosylceramide identified in the murine large intestine: dependency on diet and inflammation. J Lipid Res. (2019) 60:1892–904. doi: 10.1194/jlr.RA119000236
74. Xiang W, Lv Q, Shi H, Xie B, Gao L. Aptamer-based biosensor for detecting carcinoembryonic antigen. Talanta. (2020) 214:120716. doi: 10.1016/j.talanta.2020.120716
75. Khaled YS, Khot MI, Aiyappa-Maudsley R. Photoactive imaging and therapy for colorectal cancer using a CEA-Affimer conjugated Foslip nanoparticle. Nanoscale. (2024) 16:7185–99. doi: 10.1039/D3NR04118B
76. Wang W, Li Y, Zhang X, Jing J, Zhao X, Wang Y, et al. Evaluating the significance of expression of CEA mRNA and levels of CEA and its related proteins in colorectal cancer patients. J Surg Oncol. (2014) 109:440–4. doi: 10.1002/jso.v109.5
77. Prasad H, Visweswariah SS. Impaired intestinal sodium transport in inflammatory bowel disease: from the passenger to the driver’s seat. Cell Mol Gastroenterol Hepatol. (2021) 12:277–92. doi: 10.1016/j.jcmgh.2021.03.005
78. Arora K, Huang Y, Mun K, Yarlagadda S, Naren AP. Guanylate cyclase 2C agonism corrects CFTR mutants. JCI Insight. (2017) 2:e93686. doi: 10.1172/jci.insight.93686
79. Romi H, Cohen I, Landau D, Alkrinawi S, Yerushalmi B, Hershkovitz R, et al. Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am J Hum Genet. (2012) 90:893–9. doi: 10.1016/j.ajhg.2012.03.022
80. Lisby AN, Flickinger JC, Bashir B, Weindorfer M, Waldman SA. GUCY2C as a biomarker to target precision therapies for patients with colorectal cancer. Expert Rev Precis Med Drug Dev. (2021) 6:1–13. doi: 10.1080/23808993.2021.1876518
81. Alessandra Z, Rosa M, Cinzia F, Alessandra S, Rossella P, Marco C, et al. NKG2D and its ligands: “One for all, all for one. Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
82. Pereira BI, Maeyer RPHD, Covre LP, Nehar-Belaid D, Akbar AN. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat Immunol. (2020) 22:1–11. doi: 10.1038/s41590-020-0643-3
83. Liu H, Wang S, Xin J, Wang J, Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am J Cancer Res. (2019) 9:2064–78.
84. Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspect Biol. (2014) 6:a020768–a020768. doi: 10.1101/cshperspect.a020768
85. Jin N, An Y, Tian Y, Zhang Z, He K, Chi C, et al. Multispectral fluorescence imaging of EGFR and PD-L1 for precision detection of oral squamous cell carcinoma: a preclinical and clinical study. BMC Med. (2024) 22:1–16. doi: 10.1186/s12916-024-03559-w
86. Jutten B, Rouschop KM. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. (2014) 13:42–51. doi: 10.4161/cc.27518
87. Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. (2015) 57:207–18. doi: 10.1016/j.molcel.2014.11.013
88. Oh SJ, Lim J, Son MK, Ahn JH, Song KH, Lee HJ, et al. TRPV1 inhibition overcomes cisplatin resistance by blocking autophagy-mediated hyperactivation of EGFR signaling pathway. Nat Commun. (2023) 14:2691. doi: 10.1038/s41467-023-38318-7
89. Zubair T, Bandyopadhyay D. Small molecule EGFR inhibitors as anti-cancer agents: discovery, mechanisms of action, and opportunities. Int J Mol Sci. (2023) 24:2651. doi: 10.3390/ijms24032651
90. Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. (2013) 19:1389–400. doi: 10.1038/nm.3388
91. Youri H, Ghisai SA, Maurice DW, Iris DH, Kaspar D, Job VR, et al. The EGFRvIII transcriptome in glioblastoma: A meta-omics analysis. Neuro-Oncology. (2022) 24:429–41. doi: 10.1093/neuonc/noab231
92. Tong F, Zhao J-x, Fang Z-y, Cui X-t, Su D-y, Liu X, et al. MUC1 promotes glioblastoma progression and TMZ resistance by stabilizing EGFRvIII. Pharmacol Res. (2023) 187:106606. doi: 10.1016/j.phrs.2022.106606
93. Guo GF, Cai YC, Zhang B, Xu RH, Qiu HJ, Xia LP, et al. Overexpression of SGLT1 and EGFR in colorectal cancer showing a correlation with the prognosis. Med Oncol. (2011) 28:197–203. doi: 10.1007/s12032-010-9696-8
94. Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. (2016) 11:414–9. doi: 10.1016/j.jtho.2015.10.025
95. Pal B, Chen Y, Vaillant F, Capaldo BD, Joyce R, Song X, et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. (2021) 40:e107333. doi: 10.15252/embj.2020107333
96. Siena S, Sartore-Bianchi A, Marsoni S, Hurwitz HI, Mccall SJ, Penault-Llorca F, et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann Oncol. (2018) 29:1108–19. doi: 10.1093/annonc/mdy100
97. Styczen H, Nagelmeier I, Beissbarth T, Nietert M, Homayounfar K, Sprenger T, et al. HER-2 and HER-3 expression in liver metastases of patients with colorectal cancer. Oncotarget. (2015) 6:15065. doi: 10.18632/oncotarget.v6i17
98. Park DJ, Sung PS, Kim JH, Lee GW, Jang JW, Jung ES, et al. EpCAM-high liver cancer stem cells resist natural killer cell–mediated cytotoxicity by upregulating CEACAM1. J Immunotherapy Cancer. (2020) 8:e000301. doi: 10.1136/jitc-2019-000301
99. Muñoz-Hernández R, Gato S, Gil-Gómez A, Aller R, Rojas A, Morán L, et al. Role of EpCAM+ CD133+ extracellular vesicles in steatosis to steatohepatitis transition in NAFLD. Liver Int. (2023) 43:1909–19. doi: 10.1111/liv.15604
100. Sarrach S, Huang Y, Niedermeyer S, Hachmeister M, Fischer L, Gille S, et al. Spatiotemporal patterning of EpCAM is important for murine embryonic endo- and mesodermal differentiation. Sci Rep. (2018) 8:1801. doi: 10.1038/s41598-018-20131-8
101. Liang K-H, Tso H-C, Hung S-H, Kuan II, Lai J-K, Ke F-Y, et al. Extracellular domain of EpCAM enhances tumor progression through EGFR signaling in colon cancer cells. Cancer Lett. (2018) 433:165–75. doi: 10.1016/j.canlet.2018.06.040
102. Shirin E, Safar F, Siavoush D, Farzad K, Habib Z, Mojghan B. Antibody based epCAM targeted therapy of cancer, review and update. Curr Cancer Drug Targets. (2018) 18:857–68. doi: 10.2174/1568009618666180102102311
103. Nath S, Mukherjee P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. (2014) 20:332–42. doi: 10.1016/j.molmed.2014.02.007
104. Nabavinia MS, Gholoobi A, Charbgoo F, Nabavinia M, Ramezani M, Abnous K. Anti-MUC1 aptamer: A potential opportunity for cancer treatment. Medicinal Res Rev. (2017) 37:1518–39. doi: 10.1002/med.2017.37.issue-6
105. Gao T, Cen Q, Lei H. A review on development of MUC1-based cancer vaccine. Biomedicine Pharmacotherapy. (2020) 132:110888. doi: 10.1016/j.biopha.2020.110888
106. Malla M, Deshmukh SK, Wu S, Samec T, Olevian DC, El Naili R, et al. Mesothelin expression correlates with elevated inhibitory immune activity in patients with colorectal cancer. Cancer Gene Ther. (2024) 31:1547–58. doi: 10.1038/s41417-024-00816-1
107. Cho J, Kim K-M, Kim HC, Lee WY, Kang WK, Park YS, et al. The prognostic role of tumor associated glycoprotein 72 (TAG-72) in stage II and III colorectal adenocarcinoma. Pathol - Res Pract. (2019) 215:171–6. doi: 10.1016/j.prp.2018.10.024
108. Xu Y, Chen Y, Wei L, Lai S, Zheng W, Wu F. Serum tumor-associated glycoprotein 72, a helpful predictor of lymph nodes invasion in esophagogastric junction adenocarcinoma. Biochem Biophys Res Commun. (2019) 509:133–7. doi: 10.1016/j.bbrc.2018.12.083
109. Jelski W, Mroczko B. Biochemical markers of colorectal cancer - present and future. Cancer Manage Res. (2020) 12:4789–97. doi: 10.2147/CMAR.S253369
110. Cho J, Kim K-M, Lee HC, Kang WY, Park WKi, Ha YS, et al. The prognostic role of tumor associated glycoprotein 72 (TAG-72) in stage II and III colorectal adenocarcinoma. Pathol Res Pract. (2019) 215:171–6. doi: 10.1016/j.prp.2018.10.024
111. Haas AR, Golden RJ, Litzky LA, Engels B, Zhao L, Xu F, et al. Two cases of severe pulmonary toxicity from highly active mesothelin-directed CAR T&xa0;cells. Mol Ther. (2023) 31:2309–25. doi: 10.1016/j.ymthe.2023.06.006
112. Chu Q. Targeting mesothelin in solid tumours: anti-mesothelin antibody and drug conjugates. Curr Oncol Rep. (2023) 25:309–23. doi: 10.1007/s11912-023-01367-8
113. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. (2016) 6:133–46. doi: 10.1158/2159-8290.CD-15-0583
114. Inoue S, Tsunoda T, Riku M, Ito H, Inoko A, Murakami H, et al. Diffuse mesothelin expression leads to worse prognosis through enhanced cellular proliferation in colorectal cancer. Oncol Lett. (2020) 19:1741–50. doi: 10.3892/ol.2020.11290
115. Park J, Kim SK, Hallis SP, Choi BH, Kwak M. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front Oncol. (2022) 11:808300. doi: 10.3389/fonc.2021.808300
116. Kazama S, Kishikawa J, Tanaka T, Hata K, Ishihara S. Immunohistochemical expression of CD133 and LGR5 in ulcerative colitis-associated colorectal cancer and dysplasia. In Vivo (Athens Greece). (2019) 33:1279–84. doi: 10.21873/invivo.11600
117. Kim H, Ju JH, Son S, Shin I. Silencing of CD133 inhibits GLUT1-mediated glucose transport through downregulation of the HER3/Akt/mTOR pathway in colon cancer. FEBS Lett. (2020) 594:1021–35. doi: 10.1002/1873-3468.13686
118. Kojima M, Ishii G, Atsumi N, Fujii S, Saito N, Ochiai A. Immunohistochemical detection of CD133 expression in colorectal cancer: a clinicopathological study. Cancer Sci. (2008) 99:1578–83. doi: 10.1111/j.1349-7006.2008.00849.x
119. Lee SJ, Lee J, Park SH, Park JO, Lim HY, Kang WK, et al. c-MET overexpression in colorectal cancer: A poor prognostic factor for survival. Clin Colorectal Cancer. (2018) 17:165–9. doi: 10.1016/j.clcc.2018.02.013
120. Raj S, Kesari KK, Kumar A, Rathi B, Sharma A, Gupta PK, et al. Molecular mechanism(s) of regulation(s) of c-MET/HGF signaling in head and neck cancer. Mol Cancer. (2022) 21:31. doi: 10.1186/s12943-022-01503-1
121. Werner RA, Hartrampf PE, Fendler WP, Serfling SE, Derlin T, Higuchi T, et al. Prostate-specific membrane antigen reporting and data system version 2.0. Eur Urol. (2023) 84:491–502. doi: 10.1016/j.eururo.2023.06.008
122. Cuda TJ, Riddell AD, Liu C, Whitehall VL, Borowsky J, Wyld DK, et al. PET imaging quantifying ga-68-PSMA-11 uptake in metastatic colorectal cancer. J Nucl Med. (2020) 61:1576–9. doi: 10.2967/jnumed.119.233312
123. Haffner MC, Kronberger IE, Ross JS, Sheehan CE, Zitt M, Mühlmann G, et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum Pathol. (2009) 40:1754–61. doi: 10.1016/j.humpath.2009.06.003
124. Reiswich V, Gorbokon N, Luebke AM, Burandt E, Krech T. Pattern of placental alkaline phosphatase (PLAP) expression in human tumors: a tissue microarray study on 12,381 tumors. J Pathol Clin Res. (2021) 7:577–89. doi: 10.1002/cjp2.v7.6
125. Borosky GL. Alkaline phosphatases: in silico study on the catalytic effect of conserved active site residues using human placental alkaline phosphatase (PLAP) as a model protein. J Chem Inf Modeling. (2020) 60:6228–41. doi: 10.1021/acs.jcim.0c00860
126. Li X, Berahovich R, Zhou H, Liu X, Li F, Xu S, et al. PLAP -CAR Tcells mediate high specific cytotoxicity against colon cancer cells. FBL. (2020) 25:1765–86. doi: 10.2741/4877
127. Westcott PMK, Sacks NJ, Schenkel JM, Ely ZA, Smith O, Hauck H, et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat Cancer. (2021) 2:1071–85. doi: 10.1038/s43018-021-00247-z
128. Wagner S, Mullins CS, Linnebacher M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J Gastroenterol. (2018) 24:5418–32. doi: 10.3748/wjg.v24.i48.5418
129. Zhu YJ, Li X, Chen TT, Wang JX, Zhou YX, Mu XL, et al. Personalised neoantigen-based therapy in colorectal cancer. Clin Transl Med. (2023) 13:e1461. doi: 10.1002/ctm2.v13.11
130. Zheng Y, Fu Y, Wang PP, Ding ZY. Neoantigen: A promising target for the immunotherapy of colorectal cancer. Dis Markers. (2022) 2022:8270305. doi: 10.1155/2022/8270305
131. Linnebacher M, Gebert J, Rudy W, Woerner S, Yuan YP, Bork P, et al. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. (2001) 93:6–11. doi: 10.1002/(ISSN)1097-0215
132. Yu Y, Zhang J, Ni L, Zhu Y, Yu H, Teng Y, et al. Neoantigen-reactive T cells exhibit effective anti-tumor activity against colorectal cancer. Hum Vaccin Immunother. (2022) 18:1–11. doi: 10.1080/21645515.2021.1891814
133. Shono Y, Tanimura H, Iwahashi M, Tsunoda T, Tani M, Tanaka H, et al. Specific T-cell immunity against Ki-ras peptides in patients with pancreatic and colorectal cancers. Br J Cancer. (2003) 88:530–6. doi: 10.1038/sj.bjc.6600697
134. Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, et al. The HIF-1α hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov. (2012) 2:608–23. doi: 10.1158/2159-8290.CD-11-0314
135. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
136. Zhang X, He C, He X, Fan S, Ding B, Lu Y, et al. HIF-1 inhibitor-based one-stone-two-birds strategy for enhanced cancer chemodynamic-immunotherapy. J Control Release. (2023) 356:649–62. doi: 10.1016/j.jconrel.2023.03.026
137. Pastorek J, Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol. (2015) 31:52–64. doi: 10.1016/j.semcancer.2014.08.002
138. Fragai M, Comito G, Di Cesare Mannelli L, Gualdani R, Calderone V, Louka A, et al. Lipoyl-homotaurine derivative (ADM_12) reverts oxaliplatin-induced neuropathy and reduces cancer cells Malignancy by inhibiting carbonic anhydrase IX (CAIX). J Medicinal Chem. (2017) 60:9003–11. doi: 10.1021/acs.jmedchem.7b01237
139. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
140. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. (2005) 353:2654–66. doi: 10.1056/NEJMoa051424
141. Koi M, Carethers JM. The colorectal cancer immune microenvironment and approach to immunotherapies. Future Oncol. (2017) 13:1633–47. doi: 10.2217/fon-2017-0145
142. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
143. Bhat AA, Nisar S, Singh M, Ashraf B, Masoodi T, Prasad CP, et al. Cytokine-and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. (2022) 42:689–715. doi: 10.1002/cac2.12295
144. Zhang X, Zhao Y, Chen X. Collagen extracellular matrix promotes gastric cancer immune evasion by activating IL4I1-AHR signaling. Trans Oncol. (2024) 49:102113. doi: 10.1016/j.tranon.2024.102113
145. Zaninelli S. Development of innovative CAR molecules to be transduced in Cytokine Induced Killer cells for the treatment of different neoplasia. (2023).
146. Belhabib I, Zaghdoudi S, Lac C, Bousquet C, Jean C. Extracellular matrices and cancer-associated fibroblasts: targets for cancer diagnosis and therapy? Cancers. (2021) 13:3466. doi: 10.3390/cancers13143466
147. Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. (2006) 5:1026–33. doi: 10.1038/nrd2086
148. Qin X, Wu F, Chen C, Li Q. Recent advances in CAR-T cells therapy for colorectal cancer. Front Immunol. (2022) 13:904137. doi: 10.3389/fimmu.2022.904137
149. Shakhpazyan N, Mikhaleva L, Bedzhanyan A, Gioeva Z, Sadykhov N, Mikhalev A, et al. Cellular and molecular mechanisms of the tumor stroma in colorectal cancer: insights into disease progression and therapeutic targets. Biomedicines. (2023) 11:2361. doi: 10.3390/biomedicines11092361
150. Mai Z, Lin Y, Lin P, Zhao X, Cui L. Modulating extracellular matrix stiffness: a strategic approach to boost cancer immunotherapy. Cell Death Dis. (2024) 15:307. doi: 10.1038/s41419-024-06697-4
151. Albarrán-Fernández V, Angelats L, Delgado J, Gros A, Urbano-Ispizua Á, Guedan S, et al. Unlocking the potential of engineered immune cell therapy for solid tumors. Nat Commun. (2025) 16:1144. doi: 10.1038/s41467-025-56527-0
152. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. (2016) 167:419–432.e16. doi: 10.1016/j.cell.2016.09.011
153. Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science. (2022) 378:eaba1624. doi: 10.1126/science.aba1624
154. Irvine DJ, Maus MV, Mooney DJ, Wong WW. The future of engineered immune cell therapies. Science. (2022) 378:853–8. doi: 10.1126/science.abq6990
155. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
156. Boretti A. Improving chimeric antigen receptor T-cell therapies by using artificial intelligence and internet of things technologies: A narrative review. Eur J Pharmacol. (2024) 974:176618. doi: 10.1016/j.ejphar.2024.176618
Keywords: adoptive immune cell therapy (ACT), immune cells, colorectal cancer (CRC), tumor antigens, clinical trials
Citation: Liu C, Liu N, Zhang T and Tu Y (2025) Adoptive immune cell therapy for colorectal cancer. Front. Immunol. 16:1557906. doi: 10.3389/fimmu.2025.1557906
Received: 09 January 2025; Accepted: 28 February 2025;
Published: 01 April 2025.
Edited by:
Jun Wang, Dalhousie University, CanadaReviewed by:
Anna Maria Corsale, University of Palermo, ItalyCopyright © 2025 Liu, Liu, Zhang and Tu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongcun Zhang, emhhbmd0b25nY3VuQHd1c3QuZWR1LmNu; Yanyang Tu, dHVmbW11QDE4OC5jb20=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.