 Minghui Lin
Minghui Lin Cunxin Zhang
Cunxin Zhang Haiming Li
Haiming Li Kang Li
Kang Li Shuao Gou
Shuao Gou Xiao He
Xiao He Chaoliang Lv
Chaoliang Lv Kai Gao
Kai Gao- 1Second College of Clinical Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Department of Orthopedics, Jining No.1 People’s Hospital, Jining, China
- 3Jining No.1 People's Hospital, affiliated with Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
- 4Medical Integration and Practice Center, Shandong University, Jinan, China
Osteoarthritis (OA) is a widely prevalent chronic degenerative disease often associated with significant pain and disability. It is characterized by the deterioration of cartilage and the extracellular matrix (ECM), synovial inflammation, and subchondral bone remodeling. Recent studies have highlighted pyroptosis—a form of programmed cell death triggered by the inflammasome—as a key factor in sustaining chronic inflammation. Central to this process are the inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18), which play crucial roles mediating intra-articular pyroptosis through the NOD-like receptor protein 3 (NLRP3) inflammasome. This paper investigates the role of the pyroptosis pathway in perpetuating chronic inflammatory diseases and its linkage with OA. Furthermore, it explores the mechanisms of pyroptosis, mediated by nuclear factor κB (NF-κB), the purinergic receptor P2X ligand-gated ion channel 7 (P2X7R), adenosine monophosphate (AMP)-activated protein kinase (AMPK), and hypoxia-inducible factor-1α (HIF-1α). Additionally, it examines the interactions among various cellular components in the context of OA. These insights indicate that targeting the regulation of pyroptosis presents a promising therapeutic approach for the prevention and treatment of OA, offering valuable theoretical perspectives for its effective management.
1 Introduction
Osteoarthritis (OA) is a widely prevalent degenerative disease characterized by the deterioration of intra-articular tissues and cells, frequently accompanied by inflammation. Epidemiological data indicate that approximately 240 million individuals globally suffer from OA (1). The incidence of OA escalates with age, and women are particularly more susceptible, experiencing higher rates and earlier onset compared to men, which imposes a significant economic burden on individuals and society (2, 3). OA manifests symptoms such as joint pain, swelling, stiffness, and restricted movement (4). Over time, the expansion of local inflammatory processes within the joint leads to structural changes in the articular cartilage, synovial tissues, and subchondral bone (5). Significantly affecting patients’ lives of through pain, OA is a primary cause of reduced mobility and diminished quality of life among the elderly, highlighting the need for more effective prevention and treatment strategies (6). Current treatments primarily focus on symptom relief rather than disease prevention. Conservative treatment options include pharmacotherapy and non-drug interventions such as education, self-management, exercise, and weight management (7). However, while pharmacotherapy may temporarily alleviate pain, it can also lead to adverse reactions such as gastrointestinal bleeding, edema, hypertension, and heart failure, potentially hastening the need for joint replacement surgery (8, 9). Moreover, there is a lack of medications that effectively prevent joint synovial inflammation, which can promote cell death in joint tissues and contribute to the progression of OA. Therefore, understanding the underlying mechanisms of OA and developing effective treatment strategies are urgently needed.
The pathogenesis of OA is multifaceted, characterized by irreversible cartilage destruction and uncontrolled chronic inflammation. Current research focuses intensely on deciphering the complex mechanisms behind inflammation-mediated cell death in OA joint tissue, aiming to develop targeted and effective therapeutic interventions that curb this pathogenic process (10). However, the specific mechanisms and therapeutic targets related to inflammation and histocyte dysfunction remain elusive, with a notable deficiency in targeted therapies to halt OA progression. Recent studies have highlighted that the overactivation of pyroptosis, a type of programmed cell death mediated by inflammasomes, plays a significant role in chronic aseptic inflammation associated with OA (11–13). Additionally, mounting evidence suggests that pyroptosis frequently triggers excessive inflammation and secondary damage to essential tissues, leading severe infections and exacerbating the progression of various diseases, including neuroinflammatory, cardiovascular, and inflammatory diseases affecting vital organs (14–16). Consequently, anti-inflammation interventions that effectively modulate the pyroptosis pathway have shown considerable promise as a strategic approach to treating treatment of inflammatory diseases, notably in the case of OA.
This review synthesizes the fundamental molecular mechanisms controlling pyroptosis, examining the intricate signaling cascades that initiate this process. Special attention is given to pyroptosis in the context of OA, detailing current research advancements related to the primary intra-articular tissues such as the synovium, cartilage, and subchondral bone. We also investigate the impact of these signaling pathways on OA progression, with the ultimate goal of improving therapeutic and management strategies for the disease.
2 Pyroptosis
2.1 Research process of pyroptosis
Pyroptosis was initially identified in macrophages of mice infected with Salmonella typhi, which was initially mistaken for apoptosis (17). Subsequent research by Hilbi et al. (18) demonstrated that mice lacking cysteine-aspartic acid protease-1 (Caspase-1) and infected with Shigella flexneri did not exhibit apoptosis in their macrophages. This discovery prompted a reevaluation of the distinctions between apoptosis and pyroptosis. Further investigations by Bergsbaken et al. (19) revealed that Salmonella could induce rapid macrophage death, attributed to Caspase-1-mediated cytotoxicity, thus defining this process as pyroptosis. Additional studies have established a significant link between the Caspase family and pyroptosis, with findings by Matikainen et al. (20) indicating that Caspase-4/5/11 are also involved in this cell death process. Notably, Caspase-11 indirectly influences interleukin-1β (IL-1β) through the Caspase-1 pathway (21), suggesting a regulatory interplay among Caspases in pyroptosis.
Pyroptosis is a distinct form of programmed cell death, differentiated from apoptosis and necrosis. It is characterized by inflammasome activation and the release of pro-inflammatory cytokines such as IL-1β and interleukin-18 (IL-18). In contrast, apoptosis is driven by apoptotic proteins such as Caspase-3, and necrosis typically results from external insults such as ischemia or toxins (22–24). Pyroptosis leads to cell swelling, lysis, and membrane rupture, which release cellular contents that provoke inflammation. Although apoptosis and pyroptosis share certain features, pyroptosis is uniquely inflammatory and regulated. Apoptosis, on the other hand, involves cell shrinkage, nuclear condensation, DNA fragmentation, and apoptotic body formation, all while maintaining membrane integrity (25–27). Pyroptosis plays a crucial role in combating infections and managing immune responses; apoptosis helps maintain tissue homeostasis by removing damaged or unnecessary cells; and necrosis is an uncontrolled response to cell injury (28). Since its discovery, this uniquely programmed Caspase activity-dependent pro-inflammatory process has attracted significant attention, particularly following the identification of pyroptosis in innate immune cells, highlighting its critical biological significance (29). Moreover, recent evidence suggests that pyroptosis also occurs in non-immune cells. As a key component of innate immunity, pyroptosis is essential in defending against infections and alarmins. It is widely implicated in the development and progression of tumors (30), infectious diseases (31), and degenerative inflammatory disorders (32).
Notably, pyroptosis appears to function as a double-edged sword within histocytes. During the inflammatory phase, moderate pyroptosis inhibits intracellular pathogen replication, removes damaged cells, and induces a controlled inflammatory response to counteract danger signals (33). However, excessive pyroptosis can lead to widespread cellular destruction, ultimately exacerbating the progression of multiple diseases (34). Consequently, increasing attention has been directed toward the role of pyroptosis, with growing expectations that its regulation may serve as a pivotal therapeutic strategy in managing inflammatory diseases.
2.2 Pathways involved in pyroptosis
Pyroptosis is a programmed inflammatory form of cell death initiated by the inflammasome and executed by the gasdermin family (22). The inflammasome is a cytoplasmic multiprotein complex composed of pattern recognition receptors (PRRs), apoptosis-associated speck-like protein containing a Caspase recruitment domain (CARD) (ASC), and Caspase-1. It plays a crucial role in detecting and responding to both exogenous pathogens and endogenous danger signals (35). ASCs serves as an adaptor protein, bridging sensor proteins with effector components (36). Gasdermins (GSDMs), including GSDM (A–E) and Pejvakin, constitute a family of membrane pore-forming proteins (37). Among these, gasdermin D (GSDMD) is the primary executor, possessing the ability to initiate pore formation, disrupt cell membranes, and release inflammatory cytokines, while Caspases are crucial components (38, 39). Based on the type of Caspase involved, pyroptosis is classified into classical, non-classical, and other alternative pathways (40) (Figure 1).
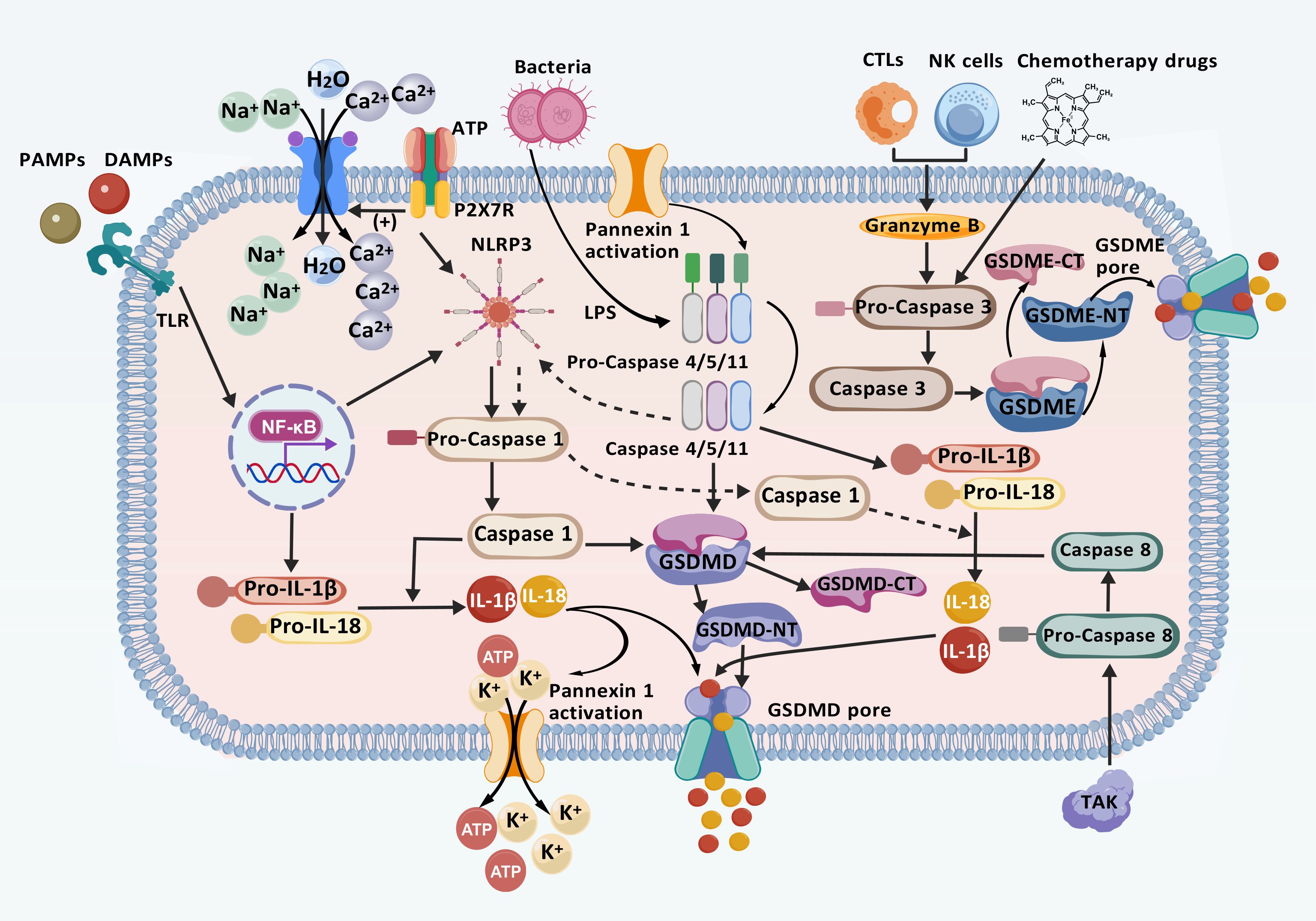
Figure 1. Mechanisms of classical, non-classical, and other pathways involved in pyroptosis. In the classical pathway, PRRs detect intracellular and extracellular signals, initiating the NF-κB/NLRP3 signaling cascade and activating the inflammasome. This activation leads to the cleavage of Caspase-1, which cleaved GSDMD. GSDMD then aggregates and inserts into the plasma membrane to form a membrane pore. Subsequently, Caspase-1 facilitates the release of inflammatory factors such as IL-1β and IL-18, promoting ion flow, water influx, cell swelling, and eventual cell rupture through the GSDMD pore, leading to pyroptosis. In the atypical pathway, LPS activates Pro Caspase-4/5/11, promoting the production of IL-1β and IL-18, production and inducing pyroptosis. Furthermore, Caspase-11 activation leads to intracellular K+ efflux and ATP release through the cleavage and modification of Pannexin-1. When ATP binds to P2X7R, it opens Ca2+ and Na+ influx channels, leading to rapid depolarization and pyroptosis. Additionally, in other pathways, TKA1 enhances Caspase-8 activation, initiating the GSDMD pore formation. Similarly, CTLs, NK cells, and the chemotherapeutic agent Caspase-3 activate the GSDME pore. These processes promote the production of inflammatory factors that ultimately contribute to pyroptosis. (Created with BioGDP.com).
2.2.1 The classical pathway of pyroptosis
In the classical activation pathway, the activation of NOD-like receptor thermal protein domain- associated protein-3 (NLRP3) is regulated by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) (41). The activation process of NLRP3 occurs in two distinct phases: “activation” and “assembly”. Although the precise mechanisms underlying NLRP3-induced pyroptosis remains unclear, it is widely hypothesized that the formation of inflammasome vesicles plays a central role in the activation process, ultimately leading to Caspase-1 (42). NLRP3 activation is triggered by the exposure to various activators, including microparticles, toxins, adenosine triphosphate (ATP), pathogens, crystals, and protein aggregates, which induce the accumulation of mitochondrial reactive oxygen species (ROS) (43). The activation process of NLRP3 involves two steps: “activation” and “assembly”. The activation phase begins when PAMPs bind to Toll-like receptors (TLRs), activating the nuclear factor-κB (NF-κB) pathway (44). This process is further modulated by regulatory non-coding RNAs, including circular RNAs (circRNAs) and microRNAs (miRNAs). Specifically, miR-665 has been identified as a key regulator of circRNF121 and myeloid differentiation primary response protein 88 (MyD88) (44, 45). Through the circRNF121/MyD88/NF-κB signaling pathway, miR-665 promotes the transcription of downstream genes, including NLRP3, Caspase family proteins, interleukin-1β precursor (pro-IL-1β), and interleukin-18 precursor (pro-IL-18), leading to the production of these inflammatory mediators (45, 46). During the execution phase, the assembly of NLRP3 depends on mitochondrial DNA (mtDNA), as an increase in advanced oxidation protein products (AOPP) leads to a decline in mitochondrial membrane potential, thereby elevating mitochondrial reactive oxygen species (mtROS). This increase in mtROS oxidizes, releasing oxidized mitochondrial DNA (Ox-mtDNA), which binds to cytosolic NLRP3, ultimately triggering inflammasome assembly (47–49). Cytidine/uridine monophosphate kinase-2 (CMPK2) is an enzyme that promotes mtDNA synthesis, resulting in the formation of Ox-mtDNA fragments, and its activity is regulated in an interferon regulatory factor-1 (IRF1)-dependent manner (50, 51). Once the inflammasome is assembled, NLRP3 oligomerization occurs through interactions among its homologous Nacht domains, forming a high-molecular-weight complex that induces Caspase-1 autoactivation (35). Caspase-1 activation subsequently cleaves its precursor, leading to the release and activation of Caspase-1. This enzyme then converts the pro-IL-1β and pro-IL-18 precursors into their mature cytokine forms, IL-1β and IL-18, through proteolysis (52). Concurrently, GSDMD remains inhibited, with its N-terminal and C-terminal domains directly linked (53). During the process of pyroptosis, the Caspase-1-mediated cleavage at ASP275 initiate the proteolysis of GSDMD, producing a 31 kDa GSDMD-NT fragment and a 22 kDa GSDMD-CT fragment (54). The GSDMD-NT protein binds to acidic phospholipids on the cell membrane, forming a pore composed of 16 identical proteins. This pore formation disrupts the ion concentration balance across the cell membrane, causing water influx, cell swelling, and the leakage of cellular contents (39). Ultimately, cell lysis occurs, releasing inflammatory mediators such as IL-1β, IL-18, and high mobility group box 1 (HMGB1), which stimulate the inflammatory response and contribute to pyroptosis (55).
2.2.2 Non-classical pyroptosis pathway
The non-canonical pathway pyroptosis primarily involves human Caspase-4/5 and their murine counterparts, Caspase-11 (39). Lipopolysaccharides (LPS), either intracellular or from Gram-negative bacteria, binds to the CARD domain of Caspase-4/5/11, thereby activating these Caspases (56). This activation leads to the cleavage of GSDMD at Asp276, generating GSDMD-NT, which oligomerizes and moves to the plasma membrane to form pores, ultimately inducing pyroptosis. This process can be inhibited by the chemotherapeutic drug oxidized 1-Palmitoyl-2-Arachidonoyl-sn-Glycero-3-Phosphocholine (oxPAPC) (57, 58). This distinctive mechanism may promote the selective recognition and interaction of Caspase with GSDMD, subsequently triggering its activation process (59). It is noteworthy that, unlike the direct effect of Caspase-1, the activation of Caspase-4/5/11 does not directly lead to the maturation of IL-1β and IL-18. However, the NLRP3 inflammasome can be activated by Caspase-11, promoting the maturation and release of IL-1β and IL-18 (60). This non-canonical pathway, by bypassing the inflammasome and not relying on its activation stimuli, provides an alternative regulatory mechanism for the inflammatory response. Additionally, Caspase-11 activation can trigger the canonical pyroptosis pathway by cleaving and modifying Pannexin-1, which crucially affects intracellular K+ efflux and ATP release. When ATP binds to the purinergic receptor P2X ligand-gated ion channel 7 (P2X7R), it opens the channel for Ca2+ and Na+ influx, causing rapid depolarization and subsequently triggering cellular pyroptosis (61, 62).
2.2.3 Other pathways involved in pyroptosis
In their in-depth study of the mechanisms underlying pyroptosis, researchers identified granzymes as a class of serine proteases located in the cytosolic granules of cytotoxic T lymphocytes (CTLs) and natural killer cells (NK) cclls (63). Unlike the classical and non-classical pathways, Caspase-3, activated by granzyme B secreted by chimeric antigen receptor T cells and chemotherapy drugs, cleaves the GSDME protein, causing N-GSDME to migrate to the cell membrane and form pores, thereby initiating pyroptosis (64–66). Additionally, TAK1-activated Caspase-8 cleaves GSDMD, and granzyme A, released by lymphocytes, cleaves GSDMB within cancer cells; both processes culminate in pyroptosis (67, 68).
3 Pyroptosis and OA
OA is a chronic, aseptic inflammatory condition marked by degenerative alterations in cartilage and subsequent osteophyte formation. This disease clinically presents as joint pain, stiffness, swelling, and restricted mobility (69). Emerging research links the etiology and progression of OA to various components of the pyroptosis pathway, including inflammasomes, cytokines, and the Caspase family. This linkage suggests that inflammation and fibrosis, induced by pyroptosis in chondrocytes (70), are significant contributors to the disease mechanism. Further, current studies provide evidence of pyroptosis in both chronic aseptic inflammation and tissue fibrosis, highlighted by instances such as microglial pyroptosis inducing neuroinflammation and fibrosis (71) and renal macrophage pyroptosis leading to renal inflammation and fibrosis (72). The cytokines IL-1β and IL-18, produced during synoviocyte pyroptosis, exacerbate synovial inflammation and extracellular matrix (ECM) degradation. Conversely, inhibiting pyroptosis can mitigate synovitis and fibrosis in OA (73), revealing the pivotal role of chondrocyte and synoviocyte pyroptosis in the onse and progression of the disease.
3.1 Synovium and pyroptosis in OA
The synovium of OA exhibits a complex environment rich in various metabolites and soluble factors, which significantly contribute to the inflammatory responses and cartilage damage characteristic of OA (74). The intricate interaction between synovial tissue inflammation and pyroptosis mechanisms reveals that synovial tissue comprises fibroblast-like synoviocytes (FLS) and macrophages, exhibiting which substantial biological activity (75). These cells not only regulate the trafficking of small molecules but also play a pivotal role in mediating pyroptosis and inflammatory responses.
3.1.1 FLS in the OA synovium
Within the joint space, synovial fluid is primarily produced by synovial tissue and secreted by FLS and macrophage. This fluid contains inflammatory mediators and cytokines, serving as a medium for intercellular material exchange (76). The synovial fluid facilitates interactions among released pro-inflammatory factors and proteases with various cellular components, triggering pyroptosis and contributing to progression changes in joint inflammation (73, 77). Notably, FLS are identified as the primary effector cells in synovial fibrosis. Further research indicates that multiple calcium salt crystals in the synovial fluid of OA patients may alter synovial permeability and decrease hyaluronic acid concentration. This alteration prompts FLS to secrete pro-inflammatory cytokines, thereby intensifying joint inflammation and damage (76, 78, 79).
During the development of synovitis, an extensive array of pro-inflammatory cytokines is released and transported into the synovial cavity. These cytokines serve not only as signaling molecules affecting the activities of various cells within the joint but also induce excessive and sustained inflammatory responses, which are primary contributors to OA and cartilage degeneration (70). HMGB1 is highly expressed in various inflammatory and autoimmune diseases, such as including sepsis (80), rheumatoid arthritis (RA) (81), and systemic lupus erythematosus (SLE) (82), acting as a DAMP to trigger inflammatory responses and exacerbatee disease progression. Research by Xiao et al. (83) demonstrates that in a rat model of knee osteoarthritis (KOA), significant increases in HMGB1 and fibrosis markers within the synovial tissue are due to pyroptosis in FLS, which in turn stimulates further production of these markers. Intriguingly, HMGB1 not only arises from pyroptosis but also feedbacks on FLS, enhancing the aggregation of pro-inflammatory factors and thereby exacerbating synovitis symptoms (84). The NLRP3 inflammasome plays a critical role in the synovial fluid of OA patients by facilitating the secretion of inflammatory cytokines that induce FLS pyroptosis (85). Under the combined influence of LPS and ATP, the expression of NLRP3-related proteins in FLS is significantly increased. In the KOA model, effectively inhibiting the NLRP3 signaling pathway reduces the expression of IL-1β, IL-18, and Caspase-1 (86). Activated IL-1β initiates pyroptosis in FLS and chondrocytes (CC), and stimulates the secretion of matrix-degrading enzymes crucial for the degradation of articular cartilage and cartilage damage (87). Zhang et al. (73) found that the number of NLRP3 inflammasomes increased in the synovial cells of OA rats. Knockout of Caspase-1 or the use of the Caspase-1 inhibitor Ac-YVAD-cmk could inhibit LPS-induced pyroptosis, as well as the expression of inflammatory markers and fibrosis. The NLRP3 inhibitor also reduces the expression of IL-1β, GSDMD, and Caspase-1 in synovial cells, indicating that both inhibitors significantly affect pyroptosis-related factors (88). Additionally, extracellular vesicles similar to exosomes play a crucial role in intercellular communication within joint tissues, impacting both the metabolism of the ECM and inflammatory processes (89). Wang et al. (90) discovered that miR-25-3p within fibroblast-like synoviocytes-extracellular vesicles (FLS-EVs), by binding to the cytoplasmic polyadenylation element-binding protein 1 (CPEB1), inhibits CPEB1 transcription. This inhibition leads to decreased expression of NLRP3, Caspase-1, GSDMD-NT, IL-1β, IL-18, and matrix metalloproteinases (MMPs), reducing CC pyroptosis and inflammation in mouse knee joints.
In summary, genetic engineering targeting or knocking out specific miRNAs in FLS-EVs, combined with the use of NLRP3 and Caspase-1 inhibitors to block interactions between inflammatory mediators such as HMGB1, IL-1β, IL-18, and matrix-degrading enzymes in synovial fluid, holds promise as a therapeutic approach for OA. This strategy is likely to become a focal point in future clinical research.
3.1.2 Macrophage in the OA synovium
The macrophages possess phagocytic and immunomodulatory capabilities, which are responsible for producing chemotactic factors and cytokines that regulate inflammatory responses and promote tissue repair (91, 92). Synovial inflammation mediated by activated macrophages plays a significant role in the progression of OA. Upon the onset of OA, macrophages are exposed to stimulatory factors and exhibit distinct inflammatory properties. Pro-inflammatory macrophages exacerbate OA progression by infiltrating synovium and releasing excessive pro-inflammatory cytokines, which activate synovial fibrosis and further promote cartilage degradation (75). Thus, macrophages are pivotal in mediating intra-articular inflammation and maintaining tissue homeostasis, significantly influencing the pathogenesis of OA.
The functional plasticity of macrophages spans a polarization spectrum from pro-inflammatory M1 to anti-inflammatory M2 phenotypes, crucial for shaping the synovial inflammatory environment in OA (93–95). M1 macrophages promote inflammatory tissue damage by secretion of pro-inflammatory mediators such as IL-1β and TNF-α. Conversely, M2 macrophages mitigate immune hyperactivation and facilitate the resolution of inflammation through anti-inflammatory cytokines such as IL-10 and TGF-β, thus managing the balance between destructive and reparative phases in inflammatory diseases (96, 97). Research by Wang et al. (98) suggests that the elevated miR-146a levels in synovial exosomes of OA rates, compared to healthy counterparts, likely modulate the tumor necrosis factor receptor associated factor 6 (TRAF6)/NF-κB signaling pathway, thereby reducing CC degradation and affecting phenotypic transformation in synovial macrophage polarization. Additionally, macrophage supernatants stimulated by pentraxin 3 (PTX3) induces an OA phenotype in cartilage, demonstrating PTX3’s anti-chondrogenic effects mediated by macrophages. In the absence of miR-224-5p, increased PTX3 levels activate the p65/NF-κB pathway and promote M1 macrophage polarization. PTX3 also activates CD32 on macrophages, releasing pro-inflammatory mediators and further joint cartilage damage (99). The paracrine interactions between macrophages and chondrocytes create a feedback loop that exacerbates synovitis and cartilage damage. Therefore, modulating the polarization from M1 to M2 may represent a viable therapeutic strategy.
Emerging evidence emphasizes pyroptosis as critical mechanism in the functional transformation of macrophages, closely linked to intra-articular inflammation and the progression of OA. A study confirms that Caspase-1 siRNA transfected macrophages show decreased cell death in an LPS+ATP-induced pyroptosis model and co-culture with GSDMD siRNA transfected macrophages results in a significant reduction in FLS fibrosis marker levels (73). Additionally, research has pointed out that elevated chondrocyte inflammation in OA leads to cartilage degradation, with free cartilage fragments and released inflammatory mediators acting as DAMPs to trigger synovial macrophage pyroptosis, further exacerbating the inflammatory microenvironment and creating a vicious cycle (100). Moreover, a study by Xie et al. (101) demonstrated that Degrasyn, used in treating synovitis, reduces OA by inhibiting macrophage pyroptosis by suppressing the NLRP3/GSDMD signaling pathway.
In conclusion, M2 macrophages support chondrocytes in OA, revealing their therapeutic value (102). Future treatments may leverage exosomes or miRNA regulation to mitigate OA-related pyroptosis. These interventions aim to suppress the NLRP3 inflammasome and Caspase-1 activation in macrophages, inhibiting pyroptosis and M1 polarization while promoting M2 polarization. This strategy is anticipated to reduce IL-1β/IL-18 production, ultimately alleviating the progression OA.
3.2 Cartilage and pyroptosis in OA
Cartilage is a vital connective tissue in the human body, composed primarily of chondrocytes and the ECM. Encased within the ECM they themselves produce, CC are the sole cells capable of synthesizing and secreting both matrix and fibers, making them indispensable for sustaining a steady cycle of collagen metabolism (103). The ECM’s solid components predominantly include type II collagen (COL-II), proteoglycans, and minor amounts of other collagens and non-collagenous proteins (104). These elements are intricately woven to form a robust network, providing resistance to external mechanical pressures.
OA is primarily linked to chondrocyte necrosis, driven by various factors that disrupt the metabolic imbalance within chondrocytes and hinder the damaged cartilage’s self-repair capabilities. Several risk factors for OA, such as obesity (105), unhealthy lifestyles (106), lipopolysaccharides (107), hydroxyapatite crystals (108), and oxidized low-density lipoprotein (109), can initiate PAMPs or DAMPs. These, in turn, activate the NLRP3 inflammasome, leading to the release of proteolytic enzymes and inflammatory cytokines, including MMPs, a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), IL-1β, IL-18, and tumor necrosis factor-α (TNF-α) (110). MMP13 and ADAMTS4/5 are recognized as key factors in ECM degradation, partially due to their upregulation by IL-1β (111).
The activation and assembly of the NLRP3 inflammasome also depend on a second signal, where ROS play a crucial role (112, 113). Studies have demonstrated that the activation of peroxisome proliferator-activated receptor gamma (PPAR-γ), primarily via the use of pioglitazone as an activator, markedly alleviates cellular oxidative damage induced by ROS, thus providing significant protection against chondrocyte damage and pyroptosis in OA (114, 115). Bai et al. have shown that activation of the adenosine A3 receptor (A3AR) with its agonist (CF101) effectively inhibits Caspase-1, offering protection to CC stimulated by hydrogen peroxide (H2O2). This protective effect is primarily achieved by blocking the ROS-induced activation of the NLRP3 inflammasome, highlighting the involvement of the NLRP3/Caspase-1 axis OA pathogenesis (116). Additionally, Zhang et al. (117) noted that the downregulation of miR-17 in OA chondrocytes of OA exacerbates pyroptosis progression. Conversely, supplementing exogenous miR-17 or inducing its endogenous expression through growth differentiation factor 5 can protect against OA by effectively inhibiting the activity of catabolic factors, including MMP3/13, ADAMTS5, and nitric oxide synthase 2 (NOS2). Similarly, the downregulation of miR-155 inhibits KOA in mice by targeting Smad2 and suppressing the NLRP3/Caspase-1 pathway, thus reducing pyroptosis in mouse KOA chongrocytes (118). Another study showed that exosomes from bone marrow mesenchymal stem cells (BMSC-Exos) specifically target histone deacetylase 3 (HDAC3) and the signal transducer and activator of transcription 1 (STAT1)/NF-κB pathway to inhibit pyroptosis in chondrocytes and cartilage tissue, while they also delivering miR-326 to chondrocytes, thus promoting the amelioration of OA (119).
Therefore, exploring the potential to inhibit chondrocyte pyroptosis presents a promising therapeutic strategy for halting ECM degradation and mitigating inflammatory responses in OA. This section summarizes the various inflammatory mediators released within the joint cavity during OA progression.
3.3 Subchondral bone and pyroptosis in OA
Subchondral bone, consisting of both bone matrix and bone cells, incorporates organic components such as collagen fibers and an amorphous matrix synthesized by osteoblasts. It also includes inorganic components, predominantly hydroxyapatite crystals, which provid hardness and toughness to the bone structure (120).
Studies on OA models have demonstrated that changes in subchondral bone are dynamic and often precede cartilage degeneration. These changes involve thickening the subchondral bone plate and trabecular bone remodeling, likely as an adaptation to altered joint loading (121). In mouse and rat OA models, early bone alterations such as trabecular reduction and osteosclerosis have been linked to the pyroptosis of articular chondrocytes (121, 122). Microstructural modifications in the subchondral bone influences its structure and integrity chondrocyte behavior, impacting biomechanical properties at various stages of OA. Microcomputed tomography (μCT) has revealed significant differences in the mechanical properties of subchondral bone between OA patients and healthy individuals, potentially exacerbating joint dysfunction (123). Initially, Chen et al. (124) observed reduced subchondral bone mass, increased trabecular separation, more osteoclasts, and decreased osteoprotegerin (OPG), indicating bone structure deterioration. Sun et al. (125) discovered that in a unilateral anterior crossbite mouse model, overexpression of miR-29b specifically in BMSCs, significantly alleviated cartilage degradation cartilage subchondral bone loss while reducing osteoclast overactivity. OPG plays a vital role in bone metabolism by inhibiting osteoclasts through binding to the NF-κB activating factor ligand (RANKL), and low levels OPG can disrupt bone balance, leading to increased resorption (126). Dihydroartemisinin (DHA), a derivative of artemisinin, effectively reduces osteoclast activity by targeting the NF-κB/MAPK/RANKL signaling pathway, thereby alleviating subchondral bone remodeling and associated cartilage degeneration in OA (127). Zheng et al. (128) conducted an experimental study on mice with destabilized medial meniscus (DMM) to demonstrate that paroxetine can inhibit pyroptosis and reduce osteoclast formation by suppressing the NF-κB/RANKL signaling pathway, suggesting its potential as a key therapeutic pathway in treating OA. The interplay between pyroptosis and osteoclast activity reveals the intricate mechanisms underlying OA pathology, where inflammatory cytokines released during pyroptosis may intensify bone degradation and contribute to the disease’s overall progression (100).
Indeed, a rigorous exploration of the interactions between macrophages, FLS, CC, and pyroptosis is vital for advancing our comprehension of the pathophysiological mechanisms underlying OA. Such studies are poised to inform the creation of innovative therapeutic strategies and methodologies. The interactions among macrophages, FLS, and CC, closely tied to pyroptosis, are likely crucial drivers of OA progression at various disease stages—whether early, intermediate, or late—and may impact the disease trajectory collectively or independently. The role of the NLRP3 inflammasome and its downstream factors in disrupting the microenvironments of different tissue cells during the OA progression has garnered increasing attention (Figure 2). Unfortunately, comprehensive studies investigating the specific roles of pyroptosis at different stages of OA are lacking. Current research primarily utilizes imaging technologies and histological techniques to detail histopathological changes and inflammasome-related molecular features in end-stage OA. These studies do not capture the dynamic changes throughout the disease course and fail to offer definitive conclusions regarding the controll of cell pyroptosis at various stages. Consequently, it is critically important to thoroughly understand the specific roles and intricate mechanisms of pyroptosis in the interactions among different cell types at various OA stages. Such knowledge could accelerate the development of more targeted interventions, aiming to halt or even reverse the pathological processes of OA from onset.
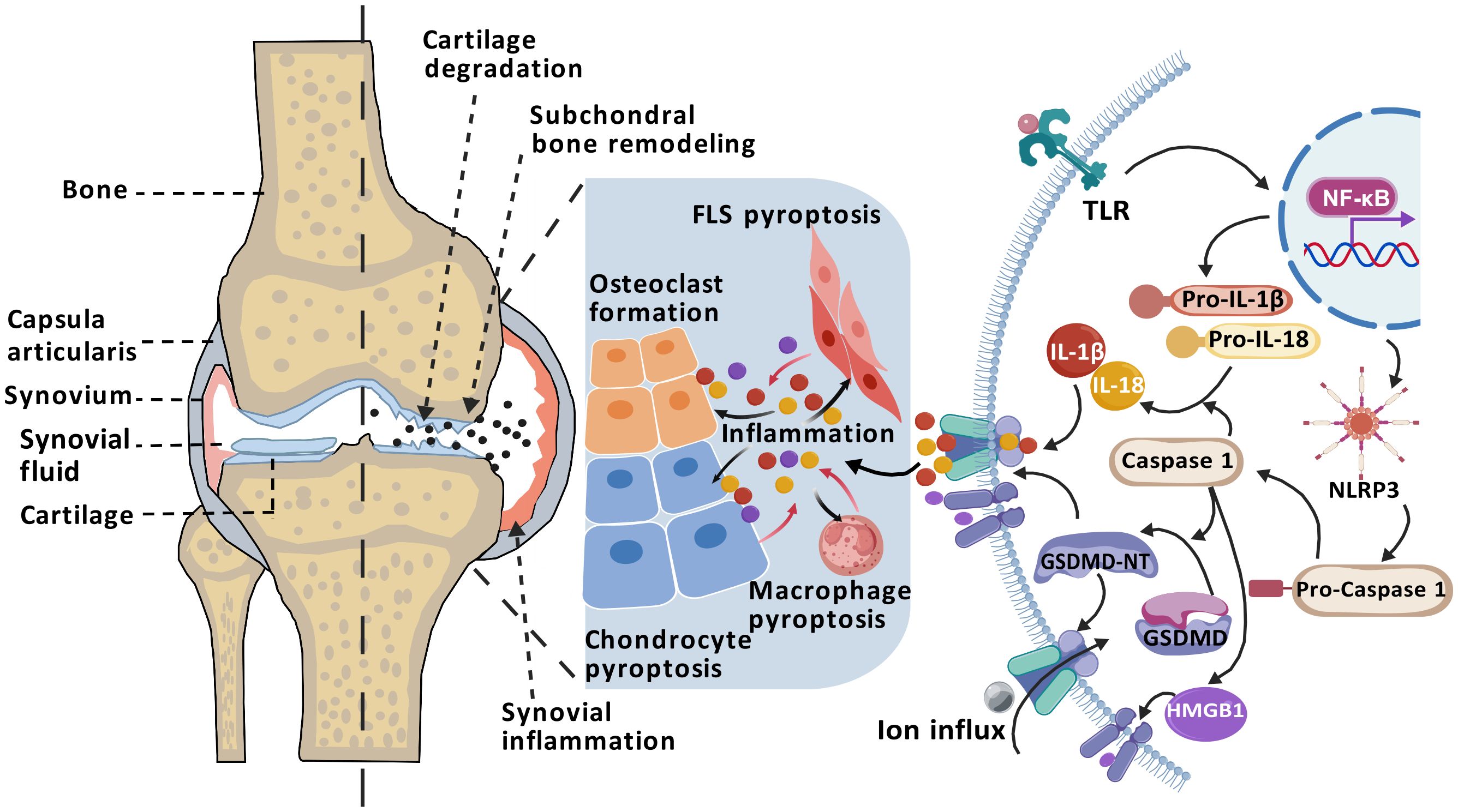
Figure 2. Mechanism of pyroptosis in OA pathogenesis. The progression of OA is closely associated with NLRP3 inflammasome-mediated pyroptosis. Activation of the TLR/NF-κB signaling pathway promotes the assembly of the NLRP3 inflammasome and activates Caspase-1, which cleaves GSDMD to generate the pore-forming GSDMD-NT, releasing inflammatory cytokines such as IL-1β, IL-18, and HMGB1. The accumulation of inflammatory mediators in the joint cavity—including IL-1β, IL-18, and HMGB1—promote FLS pyroptosis, macrophage pyroptosis, chondrocyte pyroptosis and osteoclast formation. Consequently, in a persistent chronic inflammatory environment, it can cause synovial inflammation, osteophyte formation, cartilage degradation, leading to OA. (Created with BioGDP.com).
Furthermore, miRNAs play a crucial role not only in regulating the pyroptosis of macrophages, FLS, and CC, but also in influencing the pyroptosis of subchondral bone cells during the progression of OA (129, 130). Recent evidence suggests that inappropriate macrophage polarization responses are pivotal in the damage to bone and joint tissues. Given this, miRNAs are increasingly recognized as important upstream regulators of pyroptosis, and numerous new studies have confirmed that miRNAs can modulate the polarization between macrophages phenotypes in such contexts. Additionally, the balance between pyroptosis of non-immune cells and the inflammatory microenvironment in joints results from regulation by miRNAs. This understanding has led to the strategy of using miRNAs within vesicles secreted by intra-articular tissue cells to precisely regulate the activation pathways of NLRP3 inflammasomes, regarded as a promising therapeutic approach for treating osteoarthritic diseases. These miRNA regulatory pathways in the pyroptosis mechanism are summarized in Table 1.
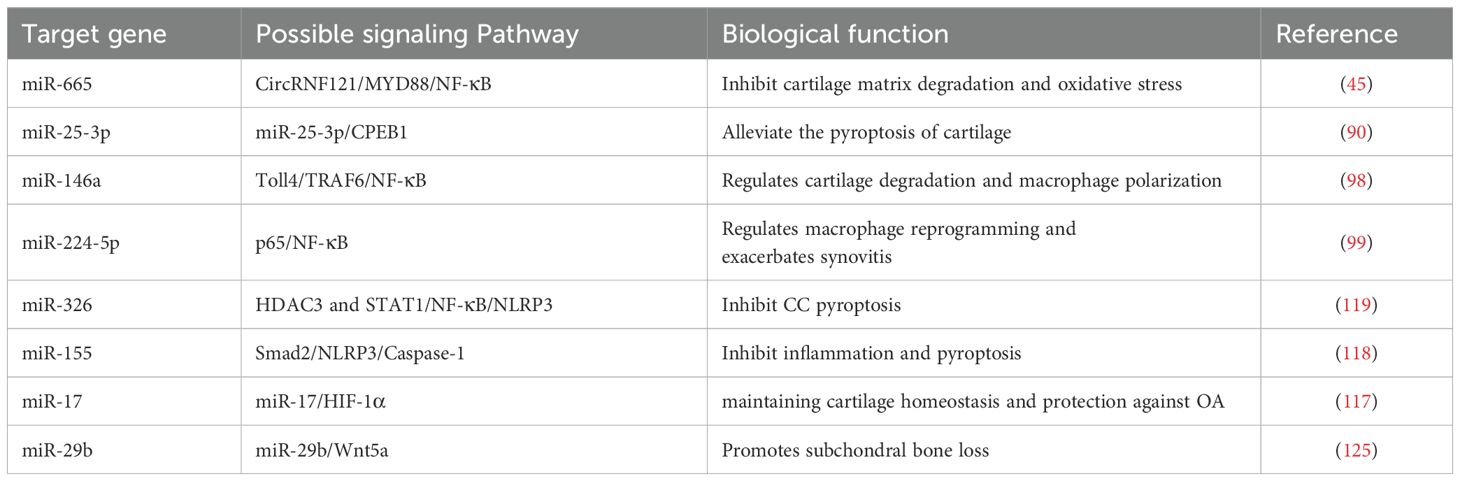
Table 1. Summary the targeting miRNAs to regulate mediated pyroptosis in OA.
4 The treatment of OA pyroptosis
4.1 Targeting NLRP3 for the treatment of pyroptosis
The inflammasome, a complex structure integral to the immune response, consists of core proteins such as NLRP1, NLRP3, NLRC4, AIM2, pyrin, and IFI16 (131), with NLRP3 being particularly significant due to its involvement in numerous pathophysiological responses. The NLRP3 inflammasome can be activated through various stimuli, including LPS, ROS, ATP, Ca2+ inflow, K+ outflow, inflammatory factors, lysosomal instability, mitochondrial dysfunction, and diverse pathogens (46, 132, 133). Research has highlighted an increased activation of NLRP3-related proteins in OA patients, and LPS-induced pyroptosis is alleviated by NLRP3 siRNA (134).
In cases of temporomandibular joint osteoarthritis (TMJOA), the use of Caspase-1 inhibitor Ac-YVAD-cmk and the NLRP3 inhibitor MCC950 has shown effectiveness in inhibiting LPS-induced pyroptosis and reducing inflammation and fibrosis (88). Moreover, the ubiquitination of thioredoxin-interacting protein (TXNIP) by the USP family leads to ROS accumulation, activating the NLRP3 inflammasome, increasing IL-1β and IL-18 production, and promoting GSDMD-N-dependent pyroptosis and ECM remodeling (135). The role of guanylate binding protein 5 (GBP5), a member of the Interferon-gamma-induced guanosine triphosphatase family, is also noteworthy. In TNF-α-induced models, GBP5 expression is upregulated, inhibiting the synthesis of ECM and COL-II and promoting the expression of NLRP3, Caspase-1, GSDMD, and MMP13. Additionally, IRF1 is found to bind to the promoter region of GBP5, thereby enhancing its expression (136). The involvement of O-GlcNAc transferase (OGT)-induced O-linked N-acetylglucosamine (O-GlcNAcylation) in many human diseases has been documented, though its specific role in OA remains unclear. Research by He et al. (137) indicates that the silencing or knockout of OGT increases the phosphorylation of (Never in Mitosis Gene A)-related kinase 7 (NEK7), particularly at the S260 residue. This phosphorylation impedes the interaction between NEK7 and NLRP3, effectively suppressing LPS-induced chondrocyte pyroptosis. Furthermore, pharmacological agents such as the NLRP3 inhibitor CY-09 (138), icariin (139) and Sipeimine (Sip) (140) have demonstrated efficacy in decreasing NLRP3 inflammasome expression, inhibiting chondrocyte pyroptosis, reducing catabolic and inflammatory responses, and ameliorating cartilage damage.
Therefore, inhibition of NLRP3 activity or blockade of its upstream signaling pathway may reduce the joint inflammatory response, protect chondrocytes, and delay the progression of OA. Further investigation into the mechanism of NLRP3 in OA is expected to provide novel targets and approaches for OA treatment (Table 2).
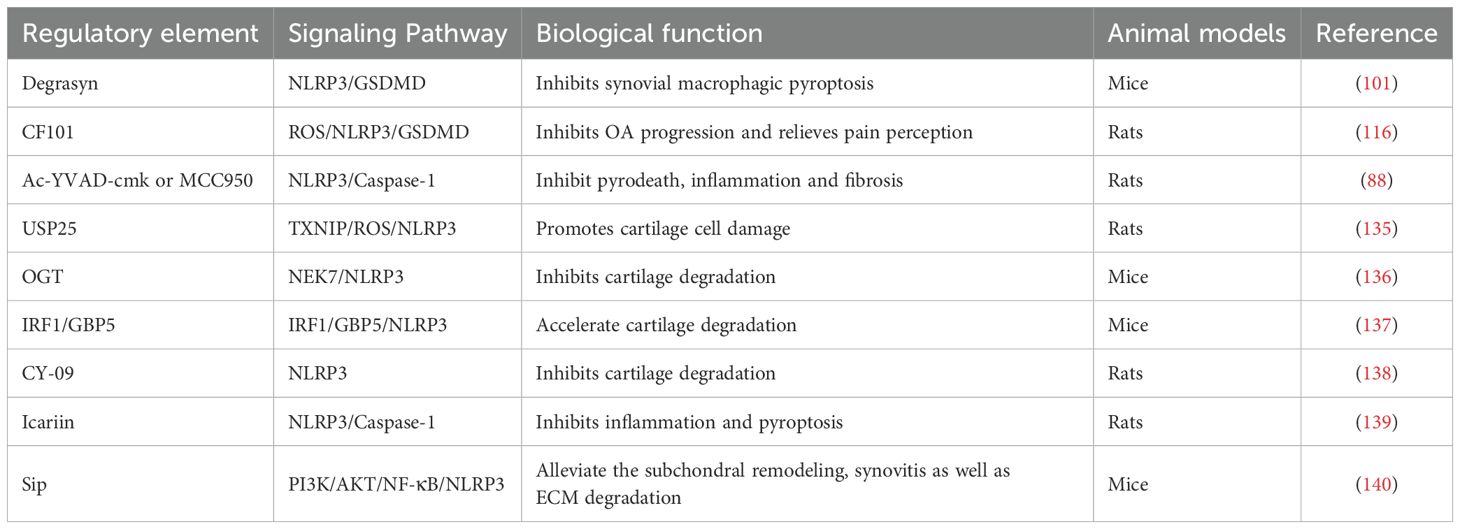
Table 2. Summarize the regulatory factors involved in NLPR3-mediated pyroptosis in OA.
4.2 Targeting NF-κB for the treatment of pyroptosis
Nuclear factor kappa-B (NF-κB) is an essential family of nuclear transcription factors that includes RelA, RelB, c-Rel, p52/p100, and p50/p105. These factors are vital for processes such as inflammation, immune response, and cell proliferation (141). They regulate gene expression by forming dimers with dynamic compositions. Notably, the RelA-p52 heterodimer plays a crucial role in late NF-κB activation, bridging classical and non-classical pathways (142).
The activation of the NF-κB signaling pathway generally depends on a diverse array of intracellular stimuli in articular tissues, such as cytokines, oxidative stress, I-κB phosphorylation, and motor stimulation, which typically leads to pyroptosis (143). These stimuli activate the inhibitor of the kappa B kinase (IKK) complex, resulting in IκB degradation and enabling NF-κB to enter the nucleus, bind DNA, and regulate gene transcription. NF-κB also collaborates with other transcription factors to refine its regulatory role, and it activates NLRP3, which promotes inflammasome assembly, inflammation, and pyroptosis (144). Numerous studies (145–148) have demonstrated that specific small molecule drugs, including Ursolic acid, GYY4137, Loganin, and α-Solanine, can reduce Caspase-1 and MMP protein levels and enhance COL-II expression by decreasing IκBα phosphorylation and blocking p65 nuclear translocation. Furthermore, inhibiting Caspase-1 and GSDMD activity, along with preventing ECM degradation and pyroptosis, effectively alleviates LPS-induced OA symptoms. Additionally, compounds such as Resolvin D1, derived from omega-3 fatty acids, and spermidine (SPD), exhibit anti-inflammatory effects in chondrocytes by inhibiting the NLRP3/Caspase-1/GSDMD pathway (149, 150). This restoration of COL-II expression, reduction in MMP13 and ADAMTS5 levels, improvement in chondrocyte viability, and decrease in pyroptosis contribute significantly to therapeutic outcomes.
In bone metabolism research, NF-κB plays a dual role by influencing both osteoclast formation and osteoblast function. The MEK inhibitor PD0325901 has demonstrated significant effectiveness in inhibiting the increase in NF-κB and NLRP3 levels that result from subchondral destruction (151). Moreover, small mechanical stress can inhibit pyroptosis by activating transforming growth factor-β (TGF-β) and Smad2/3 while concurrently suppressing the NF-κB/NLRP3 pathway. This dual action reduce inflammation and osteoclas activity, thereby aiding in the reconstruction of subchondral bone. Similarly, the knockdown of miR-155 achieves comparable effects through the Smad2/NF-κB/NLRP3 pathway (118, 143, 152).
In summary, the NF-κB/NLRP3 signaling pathway is central to numerous biological processes and pathologies. An enhanced understanding of its mechanisms could provide novel insights and strategies for treating OA, as outlined in Table 3.

Table 3. Summarize the regulatory factors involved in NF-κB-mediated pyroptosis in OA.
4.3 Targeting P2X7R for the treatment of pyroptosis
P2X7R is an ATP-sensitive ion channel characterized by an intracellular amino terminus, a carboxyl terminus, and two hydrophobic transmembrane segments separated by glycosylated extracellular ATP-binding domains (153). In the pathological context of OA, P2X7R also mediates the influx of Na+ and Ca2+ and the efflux of K+, contributing to various inflammatory responses and playing a significant role in different mechanisms of cell death (61, 154).
Due to cell damage and inflammatory responses in OA, the release of ATP is crucial in activating P2X7R. This activation enhances NLRP3 inflammasome activity, fostering inflammatory responses and potentially inducing pyroptosis (155, 156). P2X7R acts as a key regulator of inflammation by modulating the ionic environment within and outside the cell, promoting the assembly and activation of the NLRP3 inflammasome. This activation results in the release of pro-inflammatory cytokines such as IL-1β and IL-18, thereby intensifying joint inflammation and damage (156–158). Research indicates that the activation of the NLRP3 inflammasome is closely linked to the modulation of the adenylate-activated protein kinase (AMPK)/mechanistic target of the rapamycin (mTOR) signaling pathway. Decreased AMPK activity as a cellular energy sensor may lead to NLRP3 overactivation. Conversely, linear ubiquitinated LKB1 can activate the AMPK pathway, reversing the NLRP3 inflammasome response and potentially halting the progression of pyroptosis (159, 160). Additionally, Liang et al. found that piperine, a pharmacologically active phytochemical found in black pepper with anti-inflammatory properties, can significantly inhibit ATP-induced AMPK activation, thereby markedly suppressing pyroptosis and reducing IL-1β levels (161). Activation of AMPK inhibits the mTOR signaling pathway, which plays a critical role in regulating cell growth, promoting autophagy, eliminating excess or damaged cellular components, and maintaining cellular health (162). Experimental evidence indicates that high expression of mTOR suppresses autophagy in articular CC, accelerating cartilage degeneration. Conversely, inhibition of mTOR activity can trigger autophagy, reducing pyroptosis and cartilage loss (163). Research has demonstrated that mTOR inhibitors, such as rapamycin and the traditional Chinese medicine Gu yan xiao tincture, as well as the P2X7R inhibitor A740003, effectively inhibit mTOR, induce autophagy, and reduce pyroptosis and cartilage loss. On the other hand, injection of mTOR agonists, such as MHY1485, or P2X7R agonists, such as Bz-ATP, promotes pyroptosis and ECM degradation (163–166). Additionally, during moderate-intensity exercise, P2X7R activation facilitates ion flux, which activates the AMPK/mTOR signaling pathway. This promotes autolysosomal targeting and degradation of the inflammasome component NLRP3, thereby inhibiting autophagy and alleviating pyroptosis (167).
Consequently, the P2X7R functions as a pivotal ion channel, modifying the ionic milieu upon ATP stimulation and facilitating the production of NLRP3. The P2X7R can initiate both autophagy and pyroptosis. Engaging in early and appropriate physical activity can stimulate autophagy through the P2X7R, potentially slowing the progression of OA, though as the condition evolve, it may towards pyroptosis (167). The extent of P2X7R activation closely correlated with cellular destiny, and suggesting that judicious activation may hold the key to effective OA treatment, as outlined in Table 4.
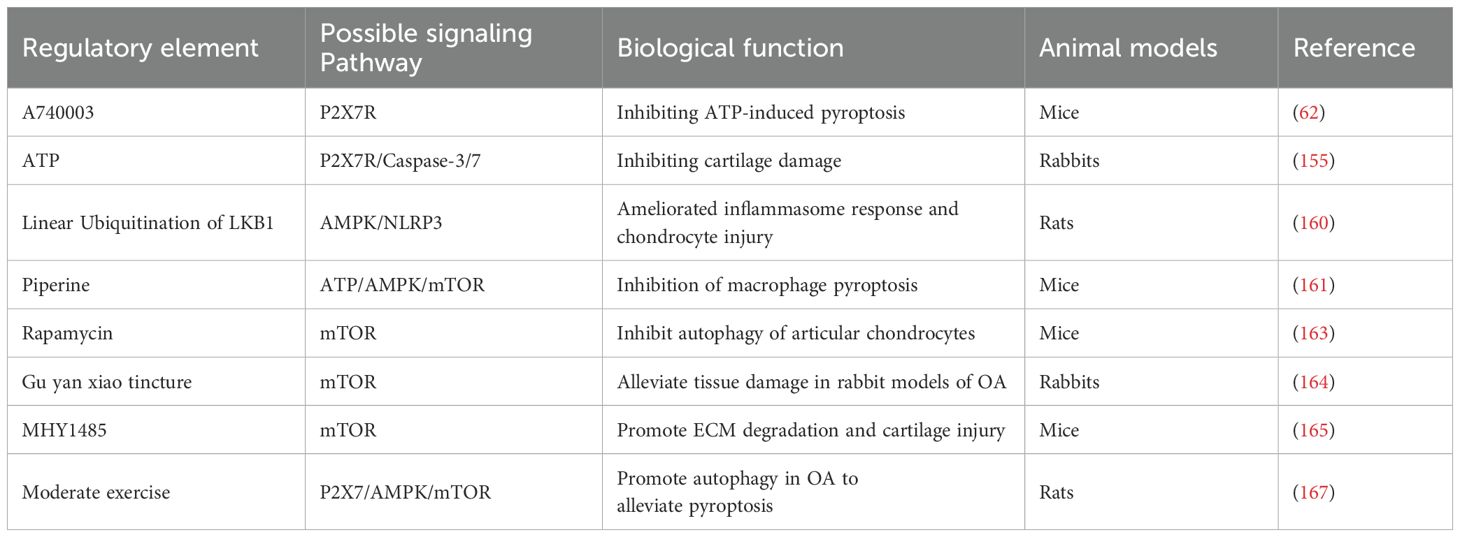
Table 4. Summarize the regulatory factors involved in P2X7R-mediated pyroptosis in OA.
4.4 Targeting HIF-1α and Nrf2/HO-1 for the treatment of pyroptosis
HO-1 is critical for heme catabolism, providing both antioxidant and anti-inflammatory protection. Nrf2 is a key transcription factor that upregulates the expression of HO-1, enhancing Nrf2 activity and creating a positive feedback loop. During hypoxia, the activation of HIF-1α promotes the expression of HO-1, thereby strengthening the cellular antioxidant capacity and forming a robust feedback mechanism under hypoxia and oxidative stress (168, 169). The role of HIF-1α in the tumor and inflammatory microenvironments is intricately linked to the Nrf2/HO-1 pathway, exerting a complex influence on cell proliferation and survival (170). Research has shown that compounds such as PD184352 and Myrislignan activate the Nrf2/HO-1 signaling pathway, promoting the expression of antioxidant proteins, reducing the accumulation of ROS, and ultimately inhibiting the production of IL-1β-induced nitric oxide (NO), inducible nitric oxide synthase (iNOS), and Prostaglandin E2 (PGE2), while also attenuating pyroptosis (171, 172).
The bone-cartilage interface, particularly the calcified cartilage, connects firmly to the subchondral bone, preventing blood vessels from invading the hyaline cartilage (173, 174). Activation of HIF-1α upregulates the expression of VEGF, promoting angiogenesis and infiltration into calcified cartilage areas, which alters the microenvironment of articular cartilage and leads to tissue damage (175). Feng et al. (176) conducted a study in which human synovial fibroblasts were stimulated with HMGB1, resulting in elevated expression levels of HIF-1α and VEGF. This finding further substantiates the pivotal role of HIF-1α in the pathogenesis of OA. Similarly, Zhang et al. (177) observed a significant increase in HIF-1α activity within the synovial tissue of OA rat models. Furthermore, silencing HIF-1α reduced the expression of fibrosis-related markers, including TGF-β, type 1A1 collagen (COL-1A1), and tissue inhibitor of metalloproteinases-1 (TIMP1). Further studies have demonstrated that the natural small molecule AGN, derived from verbena family extracts, inhibits the HIF-1α/NLRP3 signaling pathway. This inhibition reduces LPS-induced levels of NLRP3, IL-1β, and IL-18, contributing to reduced hypoxia, inflammation, and fibrosis in synovial tissue (178). Zhang et al. (130) discovered that lymphocyte cytosolic protein 1 (Lcp1) knockout in ACLT mice inhibited osteoclast activation in subchondral bone. This inhibition subsequently decreased HIF-1α levels, reduced subchondral bone remodeling, and slowed cartilage degeneration, alongside an increase in H-type vessels and oxygen concentration. Additionally, they also found that Oroxylin A, an inhibitor of the Lcp1-encoded protein l-plastin (LPL), could alleviate the progression of OA.
In summary, using HIF-1α-specific inhibitors or gene knockout can alleviate OA induced by various fibrotic markers, whereas the Nrf2/HO-1 pathway can also inhibit the inflammatory progression of OA and reduce fibrotic indicators. However, HIF-1α can also exacerbate OA by promoting the release of fibrotic indicators such as TGF-β, COL-1A1, and TIMP1, which are activated by VEGF. Therefore, the dual roles of HIF-1α need to be utilized judiciously, and more extensive and precise research is required to elucidate the relationship between these pathways, as outlined in Table 5.
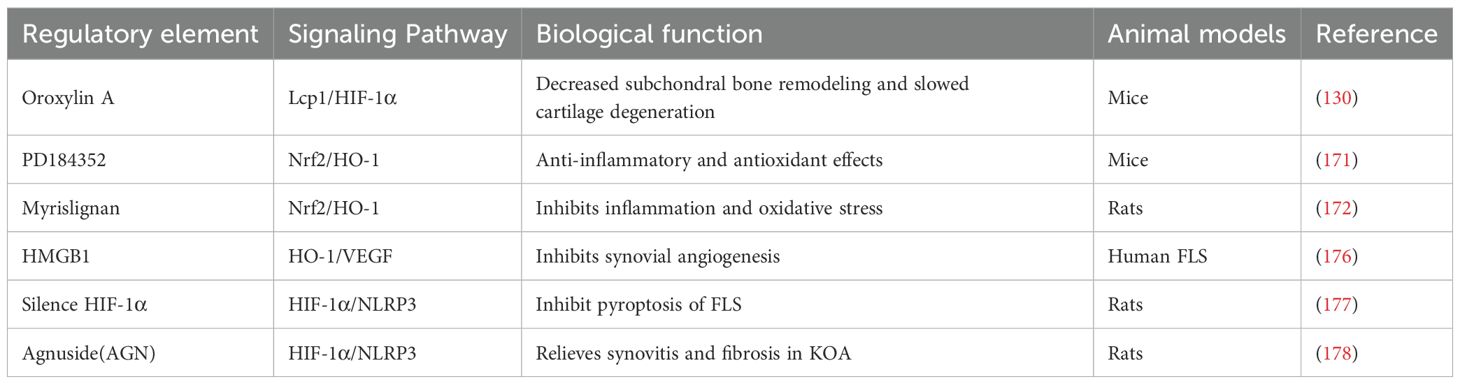
Table 5. Summarize the regulatory factors involved in Nrf2/HO-1 and HIF-1α-mediated pyroptosis in OA.
4.5 Cellular therapy
Cellular therapy, a therapeutic strategy utilizing specific cells and their functions for tissue repair, has shown promising prospects in treating OA recently. The core mechanisms of this approach include the inhibition of inflammation, regulation of cell death, and promotion of tissue regeneration. Recent research has demonstrated that human adipose-derived mesenchymal stem cells (hAD-MSCs) can significantly delay in rats and improve joint pathology by suppressing the expression of NLRP3, Caspase-1, GSDMD, and TNF-α receptor 1 (TNFR1). Further in vitro experiments have validated that the abundant soluble TNF-α receptor 1 (sTNFR1) secreted by hAD-MSCs competes with TNFR1 on the surface of chondrocytes for TNF-α binding. This competition inhibits chondrocyte pyroptosis (179), highlighting the therapeutic potential of hAD-MSCs, particularly in targeting chondrocyte pyroptosis. By modulating the NLRP3 pyroptosis pathway, cellular therapy effectively inhibits the release of inflammatory cytokines and cartilage destruction, offering a novel perspective for OA treatment and infusing new vitality into its clinical application.
Future research may explore pretreatment strategies to enhance the therapeutic potential of seed cells, aiming to achieve efficient intervention in pyroptosis and rapidly restore the balance of pyroptosis in OA.
4.6 Exercise and diet therapy
OA is a degenerative joint disorder characterized by the progressive loss of cartilage, resulting in pain and functional impairment. Recent research has highlighted the potential of dietary therapies, such as the ketogenic diet (180), and physical therapies, including exercise therapy (181) and reducing mechanical load on the knee joint (152), in influencing the pathogenic mechanisms of CC pyroptosis in OA. This connection raises important questions about how interventions such as exercise and dietary adjustments can affect cellular processes involved in the management of OA.
Exercise has been shown to play a dual role in OA management. Moderate physical activity generally benefits joint health by activating various cell signaling pathways, promoting cartilage health and reducing inflammation. Conversely, excessive or inappropriate exercise may increase cell pyroptosis, exacerbating cartilage degeneration (143, 181, 182). A systematic review and meta-analysis have revealed the benefits of therapeutic exercise, demonstrating significant improvements in pain and physical function compared to non-exercise controls. The findings from these studies suggest that the effects of exercise therapy are particularly beneficial for individuals with shorter symptom durations, indicating a critical window for intervention (183). Controlled exercise interventions that normalize joint movement have been shown to mitigate cartilage degeneration, suggesting that tailored exercise regimens could enhance the effectiveness of treatment for OA (184). Similarly, dietary interventions play a crucial role in managing OA symptoms. Research indicates that the intake of specific polyunsaturated fatty acids (PUFAs) can modulate inflammation and improve cartilage health. For instance, a study found that a diet low in n-6/n-3 PUFA significantly improved cartilage structure and inhibited articular cartilage polysaccharide loss in osteoporotic mice, highlighting the potential of dietary strategies in OA management (185). Furthermore, dietary therapy affects CC pyroptosis within the joint, which is a key mediator of pain and functional limitation in OA. Jin et al. (186) discovered that dietary fatty acids (FAs) are intimately linked to CC pyroptosis in obese mouse models of post-traumatic OA and LPS-stimulated CC, potentially by modulating the TLR4 signaling pathway to affect the NLRP3/Caspase-1/GSDMD pathway. Notably, a diet abundant in PUFAs can alleviate OA and reduce pyroptosis. Therefore, the cell pyroptosis pathway may be a crucial mechanism underlying the therapeutic effects of exercise and dietary interventions for OA. This provides a theoretical foundation for such treatment interventions.
In conclusion, integrating of therapeutic medications targeting the pyroptosis signaling pathway in OA with exercise and dietary interventions constitutes a robust foundation for holistic OA management. It is anticipated that these strategies will undergo continuous refinement and expansion, ultimately benefiting a broader spectrum of OA patients (Figure 3).
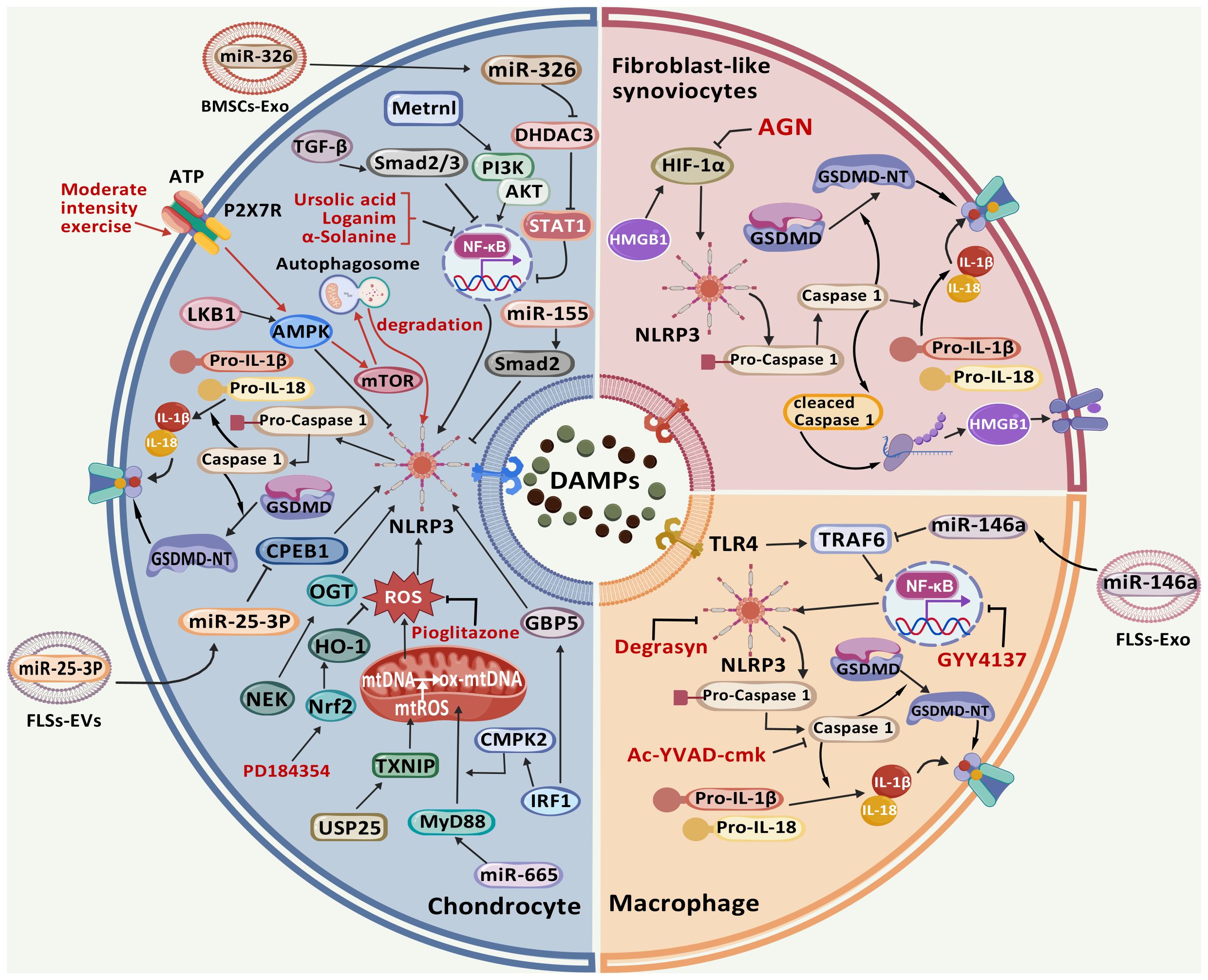
Figure 3. The signaling pathways involved in the pathophysiological development of OA, along with their corresponding targeted therapeutic drugs. During OA progression, pyroptosis in articular tissue cells is regulated through multiple molecular targets. Exposure to PAMPs or DAMPs initiates TLR-mediated activation of the NLRP3 inflammasome and Caspase-1. Subsequently, Caspase-1 and Caspase-4/5/11 cleave GSDMD to generate GSDMD-NT that oligomerize on plasma membranes, forming transmembrane pores. This process facilitates the proteolytic maturation and release of pro-inflammatory cytokines IL-1β and IL-18, ultimately triggering pyroptosis. The NLRP3 inflammasome activation is potentiated through ROS accumulation mediated by USP25/TXNIP and miR-665/MyD88 axis, which can be pharmacologically suppressed by pioglitazone and PD184352 via Nrf2/HO-1 signaling. Multiple regulatory pathways including IRF1/GBP5, NEK/OGT, miR-25-3P/CPEB1, miR-155/Smad2, and LKB1/AMPK converge on NLRP3 to modulate CC pyroptosis. Additional modulators such as miR-326/HDAC3/STAT1, Metrnl/PI3K/AKT, TGF-β/Smad2/3 signaling, along with pharmacological agents ursolic acid, loganin, and α-solanine, exert regulatory effects through NF-κB/NLRP3 pathway manipulation. Mechanical and biochemical stimuli demonstrate protective potential: ATP and moderate intensity exercise activate P2X7R ion channels, witch subsequently activate AMPK/mTOR-mediated autophagy for NLRP3 inflammasome component degradation. HMGB1 and AGN regulate FLS pyroptosis through HIF-1α/NLRP3 signaling. Macrophage pyroptosis is modulated by GYY4137 via NF-κB/NLRP3 axis, while pharmacological inhibitors Degrasyn and Ac-YVAD-cmk specifically target NLRP3 and Caspase-1 respectively to suppress this process. (Created with BioGDP.com).
5 Summary and outlook
In recent years, an increasing body of research has established a significant link between pyroptosis, a pro-inflammatory programmed cell death pathway, and the pathogenesis of OA, particularly concerning articular cartilage and synovial tissue lesions. Consequently, this has led to a detailed exploration of various signaling pathways and targeted therapeutic drugs that may influence the development of OA. Extensive documentation has revealed changes in inflammatory mediators during the pathological progression of OA, including members of the Caspase family, IL-1β, IL-18, TNF-α, HMGB1, and proteins such as MMP and ADAMTS. A notable finding is a positive correlation between the levels of NLRP3-mediated pyroptosis in OA patients’ cartilage and the severity of the disease (187). In vitro and animal model studies have shown a significant correlation between the suppression of molecules associated with pyroptosis, and the subsequent alleviation of OA symptoms. However, despite the mounting evidence of pyroptosis’s critic role in OA, there is still a shortage of OA-specific regulatory mechanisms identified, and more specific drugs targeting pyroptosis are needed. Moreover, evaluating the mechanisms of the potential adverse effects of these treatments is essential to avoid osteolytic inflammatory responses and enhance the therapeutic effectiveness. Specifically, drug application strategies must balance defensive pyroptosis to prevent antagonistic interference between different factors.
Research has highlighted the central role of the NLRP3 inflammasome in the pathogenesis of osteoarthritic diseases. Inhibiting its activation can significantly reduce the inflammatory response in OA and improve the local pathological condition of the joint. Given the complex interactions among multiple cell types within the joint, treating of OA should not be limited to targeting a single area or cell type. Indeed, adopting a comprehensive treatment approach and emphasizing the synergistic effects among cells is crucial. Investigating the interactions between cells and pyroptosis within joint tissues will enhance our understanding of the pathophysiological processes of OA. Whether through regulating CC, macrophages, FLS, cellular therapy, or adopting exercise and dietary therapies, most current methods likely alleviate or inhibit pyroptosis in OA by activating or inhibiting various signaling pathways such as NF-κB, MAPK, P2X7R, Nrf2/HO-1, and HIF-1α, which are activated by the NLRP3 inflammasome. Notably, while numerous studies indicate that the occurrence of pyroptosis accelerates the progression of OA, a few studies have expressed skepticism about the positive effects of inhibiting pyroptosis in the synovium, highlighting the need for further research to clarify these potential discrepancies and develop more targeted and effective treatments (188). More extensive research is needed to elucidate the upstream or downstream pathways and key targets of the NLRP3 inflammasome, further enhancing our understanding of its precise regulation and deeper pathological significance, which will contribute to providing more options for the prevention and treatment of human OA. On the other hand, as elaborated in previous sections, the spatiotemporal regulation of pyroptosis across various OA stages may present a promising therapeutic potential. By integrating single-cell spatial transcriptomics to map pyroptosis-associated gene networks within joint microenvironments, this approach could identify stage-specific biomarkers, enabling precision therapies that are dynamically aligned with OA progression. Such strategies may address current limitations in temporally adaptive interventions, shifting the paradigm from broad inflammasome inhibition to context-dependent regulation of pyroptosis.
In summary, current research on OA primarily focuses on cellular and animal models, with relatively few studies involving clinical patients. The inherent limitations of these models in replicating the complex human microenvironment reduce the persuasiveness of these studies. Before progressing to the clinical trial phase, a thorough assessment of the treatment’s safety and potential risks must be conducted to ensure that patient health and safety are not compromised. Alternatively, there is growing interest in the emergence of targeted molecular biomaterials in the therapeutic landscape of OA, which have shown exceptionally promising prospects for clinical translation and application. These materials, including extracellular vesicles, hydrogels, and scaffolds, highlight the significant potential in advancing OA treatment strategies (189). Looking forward, it is crucial to further explore the molecular mechanisms of pyroptosis and its role in OA, seeking effective therapeutic strategies and potential targets. The combination of pyroptosis-targeting drugs with targeted biomaterials aims to achieve precision treatment and ultimately cure patients, marking an exciting direction in the field of OA treatment.
Author contributions
ML: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review & editing. CZ: Investigation, Validation, Writing – review & editing. HL: Validation, Writing – original draft, Investigation. KL: Funding acquisition, Investigation, Supervision, Validation, Writing – original draft. SG: Investigation, Validation, Writing – original draft. XH: Formal Analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. CL: Conceptualization, Formal Analysis, Methodology, Resources, Supervision, Writing – review & editing. KG: Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from Natural Science Foundation of Shan dong Province, No. ZR2021LZY008; Shandong Provincial Medical and Health Science and Technology Projects, No, 202404070779; Shandong Province Natural Science Foundation Joint Fund for Innovative Development, No. ZR2023LZY025; Jining city focus on research and development projects, No. 2021YXNS137, No. 2024YXNS143 and 2022YXNS114; Doctoral Fund of Jining No.1 People’s Hospital, No. 2023-BS-001.
Acknowledgments
Thanks to the editors and reviewers for their hard work and important comments. Thanks to BioGDP.com for the drawing material.
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Weng Q, Chen Q, Jiang T, Zhang Y, Zhang W, Doherty M, et al. Global burden of early-onset osteoarthritis, 1990-2019: results from the Global Burden of Disease Study 2019. Ann Rheum Dis. (2024) 83:915–25. doi: 10.1136/ard-2023-225324
3. Cao F, Xu Z, Li XX, Fu ZY, Han RY, Zhang JL, et al. Trends and cross-country inequalities in the global burden of osteoarthritis, 1990-2019: A population-based study. Ageing Res Rev. (2024) 99:102382. doi: 10.1016/j.arr.2024.102382
4. Gelber AC. Knee osteoarthritis. Ann Intern Med. (2024) 177:Itc129–itc44. doi: 10.7326/annals-24-01249
6. Moradi-Lakeh M, Forouzanfar MH, Vollset SE, El Bcheraoui C, Daoud F, Afshin A, et al. Burden of musculoskeletal disorders in the Eastern Mediterranean Region, 1990-2013: findings from the Global Burden of Disease Study 2013. Ann Rheum Dis. (2017) 76:1365–73. doi: 10.1136/annrheumdis-2016-210146
7. Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: A review. Jama. (2021) 325:568–78. doi: 10.1001/jama.2020.22171
8. Palma C, Piazza S, Visone R, Ringom R, Björklund U, Bermejo Gómez A, et al. An advanced mechanically active osteoarthritis-on-chip model to test injectable therapeutic formulations: the SYN321 case study. Adv Healthc Mater. (2024) 13(32):e2401187. doi: 10.1002/adhm.202401187
9. Salis Z, Sainsbury A. Association of long-term use of non-steroidal anti-inflammatory drugs with knee osteoarthritis: a prospective multi-cohort study over 4-to-5 years. Sci Rep. (2024) 14:6593. doi: 10.1038/s41598-024-56665-3
10. Meng J, Lu J, Jiang C, Deng L, Xiao M, Feng J, et al. Collagen hydrogel-driven pyroptosis suppression and combined microfracture technique delay osteoarthritis progression. Biomaterials. (2025) 314:22817. doi: 10.1016/j.biomaterials.2024.122817
11. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:128. doi: 10.1038/s41392-021-00507-5
12. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
13. Li Z, Cheng W, Gao K, Liang S, Ke L, Wang M, et al. Pyroptosis: A spoiler of peaceful coexistence between cells in degenerative bone and joint diseases. J Adv Res. (2024) 13:S2090-1232(24)00247-9. doi: 10.1016/j.jare.2024.06.010
14. Ding R, Li H, Liu Y, Ou W, Zhang X, Chai H, et al. Activating cGAS-STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J Neuroinflammation. (2022) 19:137. doi: 10.1186/s12974-022-02511-0
15. Toldo S, Abbate A. The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat Rev Cardiol. (2024) 21:219–37. doi: 10.1038/s41569-023-00946-3
16. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. (2018) 15:721–37. doi: 10.1038/s41575-018-0054-1
17. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A. (1996) 93:9833–8. doi: 10.1073/pnas.93.18.9833
18. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. (1998) 273:32895–900. doi: 10.1074/jbc.273.49.32895
19. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
20. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol. (2020) 204:3063–9. doi: 10.4049/jimmunol.2000373
21. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:1117–21. doi: 10.1038/nature10558
22. Du G, Healy LB, David L, Walker C, El-Baba TJ, Lutomski CA, et al. ROS-dependent S-palmitoylation activates cleaved and intact gasdermin D. Nature. (2024) 630:437–46. doi: 10.1038/s41586-024-07373-5
23. Hentzen NB, Mogaki R, Otake S, Okuro K, Aida T. Intracellular photoactivation of caspase-3 by molecular glues for spatiotemporal apoptosis induction. J Am Chem Soc. (2020) 142:8080–4. doi: 10.1021/jacs.0c01823
24. Tkachenko A, Havranek O. Erythronecroptosis: an overview of necroptosis or programmed necrosis in red blood cells. Mol Cell Biochem. (2024) 479:3273–91. doi: 10.1007/s11010-024-04948-8
25. Zhang S, Wang D, Ding Y, Li Y, Wang Y, Zeng J. Inhibition of calpain reduces oxidative stress and attenuates pyroptosis and ferroptosis in Clostridium perfringens Beta-1 toxin-induced macrophages. Microbiol Res. (2024) 289:127916. doi: 10.1016/j.micres.2024.127916
26. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. (2007) 14:1590–604. doi: 10.1038/sj.cdd.4402194
27. Hadian K, Stockwell BR. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discovery. (2023) 22:723–42. doi: 10.1038/s41573-023-00749-8
28. Liu K, Wang M, Li D, Duc Duong NT, Liu Y, Ma J, et al. PANoptosis in autoimmune diseases interplay between apoptosis, necrosis, and pyroptosis. Front Immunol. (2024) 15:1502855. doi: 10.3389/fimmu.2024.1502855
29. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. (2024) 25:5379–95. doi: 10.1038/s41580-023-00689-6
30. Liu Y, Pan R, Ouyang Y, Gu W, Xiao T, Yang H, et al. Pyroptosis in health and disease: mechanisms, regulation and clinical perspective. Signal Transduct Target Ther. (2024) 9:1245. doi: 10.1038/s41392-024-01958-2
31. Jiang Y, Gao S, Chen Z, Zhao X, Gu J, Wu H, et al. Pyroptosis in septic lung injury: Interactions with other types of cell death. BioMed Pharmacother. (2023) 169:115914. doi: 10.1016/j.biopha.2023.115914
32. Chen D, Jiang X, Zou H. hASCs-derived exosomal miR-155-5p targeting TGFβR2 promotes autophagy and reduces pyroptosis to alleviate intervertebral disc degeneration. J Orthop Translat. (2023) 39:163–76. doi: 10.1016/j.jot.2023.02.004
33. Zheng X, Chen W, Gong F, Chen Y, Chen E. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: A review. Front Immunol. (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
34. Li J, Wang M, Zhou H, Jin Z, Yin H, Yang S. The role of pyroptosis in the occurrence and development of pregnancy-related diseases. Front Immunol. (2024) 15:1400977. doi: 10.3389/fimmu.2024.1400977
35. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. (2021) 18:92114–27. doi: 10.1038/s41423-021-00740-6
36. Zhan X, Li Q, Xu G, Xiao X, Bai Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front Immunol. (2022) 1109938:1109938. doi: 10.3389/fimmu.2022.1109938
37. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol. (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
38. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
39. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
40. Zhang WJ, Li KY, Lan Y, Zeng HY, Chen SQ, Wang H. NLRP3 Inflammasome: A key contributor to the inflammation formation. Food Chem Toxicol. (2023) 174:113683. doi: 10.1016/j.fct.2023.113683
41. Zhao C, Zhao W. NLRP3 inflammasome-A key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
42. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. (2023) 69:101781. doi: 10.1016/j.smim.2023.101781
43. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
44. Velasco WV, Khosravi N, Castro-Pando S, Torres-Garza N, Grimaldo MT, Krishna A, et al. Toll-like receptors 2, 4, and 9 modulate promoting effect of COPD-like airway inflammation on K-ras-driven lung cancer through activation of the MyD88/NF-ĸB pathway in the airway epithelium. Front Immunol. (2023) 141:1118721. doi: 10.3389/fimmu.2023.1118721
45. Wang T, Hao Z, Liu C, Yuan L, Li L, Yin M, et al. LEF1 mediates osteoarthritis progression through circRNF121/miR-665/MYD88 axis via NF-кB signaling pathway. Cell Death Dis. (2020) 11:598. doi: 10.1038/s41419-020-02769-3
46. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
47. Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C, et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation. (2020) 17:90. doi: 10.1186/s12974-020-01751-2
48. Li W, Jiang WS, Su YR, Tu KW, Zou L, Liao CR, et al. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis. (2023) 14:88. doi: 10.1038/s41419-023-05595-5
49. Huang B, Zhang N, Qiu X, Zeng R, Wang S, Hua M, et al. Mitochondria-targeted SkQ1 nanoparticles for dry eye disease: Inhibiting NLRP3 inflammasome activation by preventing mitochondrial DNA oxidation. J Control Release. (2024) 365:1–15. doi: 10.1016/j.jconrel.2023.11.021
50. Guan X, Zhu S, Song J, Liu K, Liu M, Xie L, et al. Microglial CMPK2 promotes neuroinflammation and brain injury after ischemic stroke. Cell Rep Med. (2024) 5:101522. doi: 10.1016/j.xcrm.2024.101522
51. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
52. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
53. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Erratum: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. (2016) 540:150. doi: 10.1038/nature20106
54. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
55. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun. (2020) 11:14561. doi: 10.1038/s41467-020-18443-3
56. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
57. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
58. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun. (2018) 9:996. doi: 10.1038/s41467-018-03409-3
59. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. (2020) 180:941–55.e20. doi: 10.1016/j.cell.2020.02.002
60. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med. (2018) 215:2279–88. doi: 10.1084/jem.20180589
61. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
62. Sawai Y, Suzuki Y, Asagiri M, Hida S, Kondo R, Zamponi GW, et al. Caveolin-1 forms a complex with P2X7 receptor and tunes P2X7-mediated ATP signaling in mouse bone marrow-derived macrophages. Am J Physiol Cell Physiol. (2024) 326(1):C125–42. doi: 10.1152/ajpcell.00303.2023
63. Ritter AT, Shtengel G, Xu CS, Weigel A, Hoffman DP, Freeman M, et al. ESCRT-mediated membrane repair protects tumor-derived cells against T cell attack. Science. (2022) 376:377–82. doi: 10.1126/science.abl3855
64. Pan J, Li Y, Gao W, Jiang Q, Geng L, Ding J, et al. Transcription factor Sp1 transcriptionally enhances GSDME expression for pyroptosis. Cell Death Dis. (2024) 15:66. doi: 10.1038/s41419-024-06455-6
65. Jiao C, Zhang H, Li H, Fu X, Lin Y, Cao C, et al. Caspase-3/GSDME mediated pyroptosis: A potential pathway for sepsis. Int Immunopharmacol. (2023) 124:111022. doi: 10.1016/j.intimp.2023.111022
66. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
67. Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int J Cancer. (2021) 149:408–20. doi: 10.1002/ijc.33698
68. Oltra SS, Colomo S, Sin L, Pérez-López M, Lázaro S, Molina-Crespo A, et al. Distinct GSDMB protein isoforms and protease cleavage processes differentially control pyroptotic cell death and mitochondrial damage in cancer cells. Cell Death Differ. (2023) 30:366–81. doi: 10.1038/s41418-023-01143-y
69. Mei X, Villamagna IJ, Nguyen T, Beier F, Appleton CT, Gillies ER. Polymer particles for the intra-articular delivery of drugs to treat osteoarthritis. BioMed Mater. (2021) 16:4. doi: 10.1088/1748-605X/abee62
70. An S, Hu H, Li Y, Hu Y. Pyroptosis plays a role in osteoarthritis. Aging Dis. (2020) 11:1146–57. doi: 10.14336/ad.2019.1127
71. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. (2018) 115:E6065–e74. doi: 10.1073/pnas.1722041115
72. Islamuddin M, Qin X. Renal macrophages and NLRP3 inflammasomes in kidney diseases and therapeutics. Cell Death Discovery. (2024) 10:229. doi: 10.1038/s41420-024-01996-3
73. Zhang L, Xing R, Huang Z, Zhang N, Zhang L, Li X, et al. Inhibition of synovial macrophage pyroptosis alleviates synovitis and fibrosis in knee osteoarthritis. Mediators Inflamm. (2019) 2019:2165918. doi: 10.1155/2019/2165918
74. Anderson JR, Chokesuwattanaskul S, Phelan MM, Welting TJM, Lian LY, Peffers MJ, et al. (1)H NMR metabolomics identifies underlying inflammatory pathology in osteoarthritis and rheumatoid arthritis synovial joints. J Proteome Res. (2018) 17:3780–90. doi: 10.1021/acs.jproteome.8b00455
75. Chen Y, Zeng D, Wei G, Liao Z, Liang R, Huang X, et al. Pyroptosis in osteoarthritis: molecular mechanisms and therapeutic implications. J Inflammation Res. (2024) 17:791–803. doi: 10.2147/jir.S445573
76. Sanchez-Lopez E, Coras R, Torres A, Lane NE, Guma M. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. (2022) 18:258–75. doi: 10.1038/s41584-022-00749-9
77. Zhang X, Wang Q, Cao G, Luo M, Hou H, Yue C. Pyroptosis by NLRP3/caspase-1/gasdermin-D pathway in synovial tissues of rheumatoid arthritis patients. J Cell Mol Med. (2023) 27:2448–56. doi: 10.1111/jcmm.17834
78. Niessink T, Stassen R, Kischkel B, Vuscan P, Emans PJ, van den Akker GGH, et al. Discovery of calcite as a new pro-inflammatory calcium-containing crystal in human osteoarthritic synovial fluid. Osteoarthritis Cartilage. (2024) 32:1261–72. doi: 10.1016/j.joca.2024.05.004
79. Han M, Russo MJ, Desroches PE, Silva SM, Quigley AF, Kapsa RMI, et al. Calcium ions have a detrimental impact on the boundary lubrication property of hyaluronic acid and lubricin (PRG-4) both alone and in combination. Colloids Surf B Biointerfaces. (2024) 234:113741. doi: 10.1016/j.colsurfb.2023.113741
80. Li L, Lu YQ. The regulatory role of high-mobility group protein 1 in sepsis-related immunity. Front Immunol. (2020) 11:601815. doi: 10.3389/fimmu.2020.601815
81. Taniguchi N, Kawakami Y, Maruyama I, Lotz M. HMGB proteins and arthritis. Hum Cell. (2018) 31:11–9. doi: 10.1007/s13577-017-0182-x
82. Liu T, Son M, Diamond B. HMGB1 in systemic lupus erythematosus. Front Immunol. (2020) 11:1057. doi: 10.3389/fimmu.2020.01057
83. Xiao Y, Ding L, Yin S, Huang Z, Zhang L, Mei W, et al. Relationship between the pyroptosis of fibroblast−like synoviocytes and HMGB1 secretion in knee osteoarthritis. Mol Med Rep. (2021) 23:2. doi: 10.3892/mmr.2020.11736
84. García-Arnandis I, Guillén MI, Gomar F, Pelletier JP, Martel-Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1β in osteoarthritic synoviocytes. Arthritis Res Ther. (2010) 12:R165. doi: 10.1186/ar3124
85. Zheng SC, Zhu XX, Xue Y, Zhang LH, Zou HJ, Qiu JH, et al. Role of the NLRP3 inflammasome in the transient release of IL-1β induced by monosodium urate crystals in human fibroblast-like synoviocytes. J Inflammation (Lond). (2015) 12:30. doi: 10.1186/s12950-015-0070-7
86. Wang Q, Huang PY, Wu JG, Zhang TQ, Li LF, Huang LD, et al. miR-219a-5p inhibits the pyroptosis in knee osteoarthritis by inactivating the NLRP3 signaling via targeting FBXO3. Environ Toxicol. (2022) 37:2673–82. doi: 10.1002/tox.23627
87. Serrano RL, Chen LY, Lotz MK, Liu-Bryan R, Terkeltaub R. Impaired proteasomal function in human osteoarthritic chondrocytes can contribute to decreased levels of SOX9 and aggrecan. Arthritis Rheumatol. (2018) 70:1030–41. doi: 10.1002/art.40456
88. Xin Y, Wang W, Mao E, Yang H, Li S. Targeting NLRP3 inflammasome alleviates synovitis by reducing pyroptosis in rats with experimental temporomandibular joint osteoarthritis. Mediators Inflamm. (2022) 2022:2581151. doi: 10.1155/2022/2581151
89. Liu B, Xian Y, Chen X, Shi Y, Dong J, Yang L, et al. Inflammatory fibroblast-like synoviocyte-derived exosomes aggravate osteoarthritis via enhancing macrophage glycolysis. Adv Sci (Weinh). (2024) 11:e2307338. doi: 10.1002/advs.202307338
90. Wang J, Sun T. Mir-25-3p in extracellular vesicles from fibroblast-like synoviocytes alleviates pyroptosis of chondrocytes in knee osteoarthritis. J Bioenerg Biomembr. (2023) 55:365–80. doi: 10.1007/s10863-023-09964-9
91. Chou CH, Jain V, Gibson J, Attarian DE, Haraden CA, Yohn CB, et al. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep. (2020) 10:10868. doi: 10.1038/s41598-020-67730-y
92. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/jci124615
93. Bogie JF, Stinissen P, Hellings N, Hendriks JJ. Myelin-phagocytosing macrophages modulate autoreactive T cell proliferation. J Neuroinflammation. (2011) 8:85. doi: 10.1186/1742-2094-8-85
94. Kloosterman DJ, Erbani J, Boon M, Farber M, Handgraaf SM, Ando-Kuri M, et al. Macrophage-mediated myelin recycling fuels brain cancer Malignancy. Cell. (2024) 187:5336–56.e30. doi: 10.1016/j.cell.2024.07.030
95. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
96. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/circresaha.115.306256
97. Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev. (2017) 280:102–11. doi: 10.1111/imr.12603
98. Wang H, Zhang Y, Zhang C, Zhao Y, Shu J, Tang X. Exosomes derived from miR-146a-overexpressing fibroblast-like synoviocytes in cartilage degradation and macrophage M1 polarization: a novel protective agent for osteoarthritis? Front Immunol. (2024) 15:1361606. doi: 10.3389/fimmu.2024.1361606
99. Yin J, Zeng H, Fan K, Xie H, Shao Y, Lu Y, et al. Pentraxin 3 regulated by miR-224-5p modulates macrophage reprogramming and exacerbates osteoarthritis associated synovitis by targeting CD32. Cell Death Dis. (2022) 13:567. doi: 10.1038/s41419-022-04962-y
100. Kuang S, Sheng W, Meng J, Liu W, Xiao Y, Tang H, et al. Pyroptosis-related crosstalk in osteoarthritis: Macrophages, fibroblast-like synoviocytes and chondrocytes. J Orthop Translat. (2024) 47:223–34. doi: 10.1016/j.jot.2024.06.014
101. Xie S, Wang L, Lu C, Chen H, Ding Y, Jian X, et al. Degrasyn alleviates osteoarthritis by blocking macrophagic pyroptosis via suppressing NLRP3/GSDMD signaling pathway and protecting chondrocytes. Cell Signal. (2024) 120:111220. doi: 10.1016/j.cellsig.2024.111220
102. Li K, Wu L, Jiang J. Apigenin accelerates wound healing in diabetic mice by promoting macrophage M2-type polarization via increasing miR-21 expression. Mol Cell Biochem. (2024) 479:3119–27. doi: 10.1007/s11010-023-04885-y
103. Bhosale AM, Richardson JB. Articular cartilage: structure, injuries and review of management. Br Med Bull. (2008) 87:77–95. doi: 10.1093/bmb/ldn025
104. Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS, et al. A guide to the composition and functions of the extracellular matrix. FEBS J. (2021) 288:6850–912. doi: 10.1111/febs.15776
105. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
106. Ho J, Mak CCH, Sharma V, To K, Khan W. Mendelian randomization studies of lifestyle-related risk factors for osteoarthritis: A PRISMA review and meta-analysis. Int J Mol Sci. (2022) 23(19):11906. doi: 10.3390/ijms231911906
107. Huang Z, Kraus VB. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat Rev Rheumatol. (2016) 12:123–9. doi: 10.1038/nrrheum.2015.158
108. Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci U S A. (2011) 108:14867–72. doi: 10.1073/pnas.1111101108
109. Ma W, Chen H, Zhang Z, Xiong Y. Association of lipid-lowering drugs with osteoarthritis outcomes from a drug-target Mendelian randomization study. PloS One. (2024) 19:e0293960. doi: 10.1371/journal.pone.0293960
110. McAllister MJ, Chemaly M, Eakin AJ, Gibson DS, McGilligan VE. NLRP3 as a potentially novel biomarker for the management of osteoarthritis. Osteoarthritis Cartilage. (2018) 26:5612–9. doi: 10.1016/j.joca.2018.02.901
111. Zhang L, Ma S, Su H, Cheng J. Isoliquiritigenin inhibits IL-1β-induced production of matrix metalloproteinase in articular chondrocytes. Mol Ther Methods Clin Dev. (2018) 9:153–9. doi: 10.1016/j.omtm.2018.02.006
112. Mourmoura E, Papathanasiou I, Trachana V, Konteles V, Tsoumpou A, Goutas A, et al. Leptin-depended NLRP3 inflammasome activation in osteoarthritic chondrocytes is mediated by ROS. Mech Ageing Dev. (2022) 208:111730. doi: 10.1016/j.mad.2022.111730
113. Li Q, Zhao Y, Guo H, Li Q, Yan C, Li Y, et al. Impaired lipophagy induced-microglial lipid droplets accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy. (2023) 19:102639–56. doi: 10.1080/15548627.2023.2213984
114. Chen J, Xu W, Dai T, Jiao S, Xue X, Jiang J, et al. Pioglitazone-loaded cartilage-targeted nanomicelles (Pio@C-HA-DOs) for osteoarthritis treatment. Int J Nanomedicine. (2023) 18:5871–90. doi: 10.2147/ijn.S428938
115. Ni S, Yi N, Yuan H, Li D, Chen X, Zhuang C. Angelica sinensis polysaccharide improves mitochondrial metabolism of osteoarthritis chondrocytes through PPARγ/SOD2/ROS pathways. Phytother Res. (2023) 37:5394–406. doi: 10.1002/ptr.7979
116. Bai H, Zhang Z, Liu L, Wang X, Song X, Gao L. Activation of adenosine A3 receptor attenuates progression of osteoarthritis through inhibiting the NLRP3/caspase-1/GSDMD induced signalling. J Cell Mol Med. (2022) 26:4230–43. doi: 10.1111/jcmm.17438
117. Zhang Y, Li S, Jin P, Shang T, Sun R, Lu L, et al. Dual functions of microRNA-17 in maintaining cartilage homeostasis and protection against osteoarthritis. Nat Commun. (2022) 13:2447. doi: 10.1038/s41467-022-30119-8
118. Li G, Xiu L, Li X, Ma L, Zhou J. miR-155 inhibits chondrocyte pyroptosis in knee osteoarthritis by targeting SMAD2 and inhibiting the NLRP3/Caspase-1 pathway. J Orthop Surg Res. (2022) 17:48. doi: 10.1186/s13018-021-02886-5
119. Xu H, Xu B. BMSC-Derived Exosomes Ameliorate Osteoarthritis by Inhibiting Pyroptosis of Cartilage via Delivering miR-326 Targeting HDAC3 and STAT1//NF-κB p65 to Chondrocytes. Mediators Inflamm. (2021) 2021:9972805. doi: 10.1155/2021/9972805
120. Deng C, Chang J, Wu C. Bioactive scaffolds for osteochondral regeneration. J Orthop Translat. (2019) 17:15–25. doi: 10.1016/j.jot.2018.11.006
121. Colyn W, Azari F, Bellemans J, van Lenthe GH, Scheys L. Microstructural adaptations of the subchondral bone are related to the mechanical axis deviation in end stage varus oa knees. Eur Cell Mater. (2023) 45:60–71. doi: 10.22203/eCM.v045a05
122. Oláh T, Cucchiarini M, Madry H. Temporal progression of subchondral bone alterations in OA models involving induction of compromised meniscus integrity in mice and rats: A scoping review. Osteoarthritis Cartilage. (2024) 32:1220–34. doi: 10.1016/j.joca.2024.06.002
123. He Z, Chu L, Liu X, Han X, Zhang K, Yan M, et al. Differences in subchondral trabecular bone microstructure and finite element analysis-based biomechanical properties between osteoporosis and osteoarthritis. J Orthop Translat. (2020) 24:39–45. doi: 10.1016/j.jot.2020.05.006
124. Chen W, Wang Q, Tao H, Lu L, Zhou J, Wang Q, et al. Subchondral osteoclasts and osteoarthritis: new insights and potential therapeutic avenues. Acta Biochim Biophys Sin (Shanghai). (2024) 56:499–512. doi: 10.3724/abbs.2024017
125. Sun JL, Yan JF, Yu SB, Zhao J, Lin QQ, Jiao K. MicroRNA-29b promotes subchondral bone loss in TMJ osteoarthritis. J Dent Res. (2020) 99:1469–77. doi: 10.1177/0022034520937617
126. Robling AG, Bonewald LF. The osteocyte: new insights. Annu Rev Physiol. (2020) 82:485–506. doi: 10.1146/annurev-physiol-021119-034332
127. Ding D, Yan J, Feng G, Zhou Y, Ma L, Jin Q. Dihydroartemisinin attenuates osteoclast formation and bone resorption via inhibiting the NF−κB, MAPK and NFATc1 signaling pathways and alleviates osteoarthritis. Int J Mol Med. (2022) 49:1. doi: 10.3892/ijmm.2021.5059
128. Zheng X, Qiu J, Gao N, Jiang T, Li Z, Zhang W, et al. Paroxetine attenuates chondrocyte pyroptosis and inhibits osteoclast formation by inhibiting NF-κB pathway activation to delay osteoarthritis progression. Drug Des Devel Ther. (2023) 17:2383–99. doi: 10.2147/dddt.S417598
129. Hu Y, Chen X, Wang S, Jing Y, Su J. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res. (2021) 9:20. doi: 10.1038/s41413-021-00147-z
130. Zhang H, Wang L, Cui J, Wang S, Han Y, Shao H, et al. Maintaining hypoxia environment of subchondral bone alleviates osteoarthritis progression. Sci Adv. (2023) 9:eabo7868. doi: 10.1126/sciadv.abo7868
131. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
132. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. (2010) 10:210–5. doi: 10.1038/nri2725
133. Yan Z, He Z, Jiang H, Zhang Y, Xu Y, Zhang Y. TRPV4-mediated mitochondrial dysfunction induces pyroptosis and cartilage degradation in osteoarthritis via the Drp1-HK2 axis. Int Immunopharmacol. (2023) 123:110651. doi: 10.1016/j.intimp.2023.110651
134. Zhao LR, Xing RL, Wang PM, Zhang NS, Yin SJ, Li XC, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP−induced pyroptosis in knee osteoarthritis. Mol Med Rep. (2018) 17:5463–9. doi: 10.3892/mmr.2018.8520
135. Sui J, Dai F, Shi J, Zhou C. Ubiquitin-specific peptidase 25 exacerbated osteoarthritis progression through facilitating TXNIP ubiquitination and NLRP3 inflammasome activation. J Orthop Surg Res. (2023) 18:762. doi: 10.1186/s13018-023-04083-y
136. Tang H, Gong X, Dai J, Gu J, Dong Z, Xu Y, et al. The IRF1/GBP5 axis promotes osteoarthritis progression by activating chondrocyte pyroptosis. J Orthop Translat. (2024) 44:47–59. doi: 10.1016/j.jot.2023.11.005
137. He C, Wu Q, Zeng Z, Yang Y, He H, Hu M, et al. OGT-induced O-GlcNAcylation of NEK7 protein aggravates osteoarthritis progression by enhancing NEK7/NLRP3 axis. Autoimmunity. (2024) 57:319202. doi: 10.1080/08916934.2024.2319202
138. Zhang Y, Lin Z, Chen D, He Y. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. (2021) 55:119–25. doi: 10.1016/j.bbrc.2021.03.055
139. Zu Y, Mu Y, Li Q, Zhang ST, Yan HJ. Icariin alleviates osteoarthritis by inhibiting NLRP3-mediated pyroptosis. J Orthop Surg Res. (2019) 14:307. doi: 10.1186/s13018-019-1307-6
140. Fang Y, Lou C, Lv J, Zhang C, Zhu Z, Hu W, et al. Sipeimine ameliorates osteoarthritis progression by suppression of NLRP3 inflammasome-mediated pyroptosis through inhibition of PI3K/AKT/NF-κB pathway: An in vitro and in vivo study. J Orthop Translat. (2024) 46:1–17. doi: 10.1016/j.jot.2024.04.004
141. Msweli S, Pakala SB, Syed K. NF-κB transcription factors: their distribution, family expansion, structural conservation, and evolution in animals. Int J Mol Sci. (2024) 25(18):9793. doi: 10.3390/ijms25189793
142. Dhaka N, Misra S, Sahoo S, Mukherjee SP. Biophysical characterization of RelA-p52 NF-κB dimer-A link between the canonical and the non-canonical NF-κB pathway. Protein Sci. (2024) 33:e5184. doi: 10.1002/pro.5184
143. Wang Y, Jin Z, Jia S, Shen P, Yang Y, Huang Y. Mechanical stress protects against chondrocyte pyroptosis through TGF-β1-mediated activation of Smad2/3 and inhibition of the NF-κB signaling pathway in an osteoarthritis model. BioMed Pharmacother. (2023) 159:114216. doi: 10.1016/j.biopha.2023.114216
144. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
145. Wang C, Gao Y, Zhang Z, Chen C, Chi Q, Xu K, et al. Ursolic acid protects chondrocytes, exhibits anti-inflammatory properties via regulation of the NF-κB/NLRP3 inflammasome pathway and ameliorates osteoarthritis. BioMed Pharmacother. (2020) 130:110568. doi: 10.1016/j.biopha.2020.110568
146. Ma J, Yang P, Zhou Z, Song T, Jia L, Ye X, et al. GYY4137-induced p65 sulfhydration protects synovial macrophages against pyroptosis by improving mitochondrial function in osteoarthritis development. J Adv Res. (2024) 4:S2090-1232(24)00223-6. doi: 10.1016/j.jare.2024.05.033
147. Hu J, Zhou J, Wu J, Chen Q, Du W, Fu F, et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-κB activity and pyroptosis in chondrocytes. J Ethnopharmacol. (2020) 247:112261. doi: 10.1016/j.jep.2019.112261
148. Zhou J, Wu J, Fu F, Yao S, Zheng W, Du W, et al. [amp]]alpha;-Solanine attenuates chondrocyte pyroptosis to improve osteoarthritis via suppressing NF-κB pathway. J Cell Mol Med. (2024) 28:e18132. doi: 10.1111/jcmm.18132
149. Wang X, Yang M, Yu G, Qi J, Jia Q, Liu S, et al. Promoting the proliferation of osteoarthritis chondrocytes by resolvin D1 regulating the NLRP3/caspase-1 signaling pathway. Cell Signal. (2024) 113:110960. doi: 10.1016/j.cellsig.2023.110960
150. Guo X, Feng X, Yang Y, Zhang H, Bai L. Spermidine attenuates chondrocyte inflammation and cellular pyroptosis through the AhR/NF-κB axis and the NLRP3/caspase-1/GSDMD pathway. Front Immunol. (2024) 15:1462777. doi: 10.3389/fimmu.2024.1462777
151. Jiang T, Gong Y, Zhang W, Qiu J, Zheng X, Li Z, et al. PD0325901, an ERK inhibitor, attenuates RANKL-induced osteoclast formation and mitigates cartilage inflammation by inhibiting the NF-κB and MAPK pathways. Bioorg Chem. (2023) 132:106321. doi: 10.1016/j.bioorg.2022.106321
152. He Z, Nie P, Lu J, Ling Y, Guo J, Zhang B, et al. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Joint Res. (2020) 9:10731–41. doi: 10.1302/2046-3758.910.Bjr-2019-0368.R2
153. Karasawa A, Kawate T. Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife. (2016) 5:e22153. doi: 10.7554/eLife.22153
154. Li Z, Huang Z, Bai L. The P2X7 receptor in osteoarthritis. Front Cell Dev Biol. (2021) 9:628330. doi: 10.3389/fcell.2021.628330
155. Tanigawa H, Toyoda F, Kumagai K, Okumura N, Maeda T, Matsuura H, et al. P2X7 ionotropic receptor is functionally expressed in rabbit articular chondrocytes and mediates extracellular ATP cytotoxicity. Purinergic Signal. (2018) 14:3245–58. doi: 10.1007/s11302-018-9611-x
156. Xiao J, Li Y, Zhang J, Xu G, Zhang J. Pannexin 3 activates P2X7 receptor to mediate inflammation and cartilage matrix degradation in temporomandibular joint osteoarthritis. Cell Biol Int. (2023) 47:71183–97. doi: 10.1002/cbin.12010
157. Franceschini A, Capece M, Chiozzi P, Falzoni S, Sanz JM, Sarti AC, et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. (2015) 29:62450–61. doi: 10.1096/fj.14-268714
158. Novak I, Solini A. P2X receptor-ion channels in the inflammatory response in adipose tissue and pancreas-potential triggers in onset of type 2 diabetes? Curr Opin Immunol. (2018) 52:1–7. doi: 10.1016/j.coi.2018.02.002
159. Chen Y, Wu YY, Si HB, Lu YR, Shen B. Mechanistic insights into AMPK-SIRT3 positive feedback loop-mediated chondrocyte mitochondrial quality control in osteoarthritis pathogenesis. Pharmacol Res. (2021) 166:105497. doi: 10.1016/j.phrs.2021.105497
160. Chen Y, Liu Y, Jiang K, Wen Z, Cao X, Wu S. Linear ubiquitination of LKB1 activates AMPK pathway to inhibit NLRP3 inflammasome response and reduce chondrocyte pyroptosis in osteoarthritis. J Orthop Translat. (2023) 39:1–11. doi: 10.1016/j.jot.2022.11.002
161. Liang YD, Bai WJ, Li CG, Xu LH, Wei HX, Pan H, et al. Piperine suppresses pyroptosis and interleukin-1β Release upon ATP triggering and bacterial infection. Front Pharmacol. (2016) 7:390. doi: 10.3389/fphar.2016.00390
162. Liu W, Zhao Y, Wang G, Feng S, Ge X, Ye W, et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. (2022) 53:102344. doi: 10.1016/j.redox.2022.102344
163. Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. (2015) 74:71432–40. doi: 10.1136/annrheumdis-2013-204599
164. Cai H, Zheng Y, Chen Y, Lu Q, Hong W, Guo Q, et al. Miao medicine Gu Yan Xiao tincture inhibits mTOR to stimulate chondrocyte autophagy in a rabbit model of osteoarthritis. J Ethnopharmacol. (2024) 328:118095. doi: 10.1016/j.jep.2024.118095
165. Kitami M, Kaku M, Thant L, Maeda T. A loss of primary cilia by a reduction in mTOR signaling correlates with age-related deteriorations in condylar cartilage. Geroscience. (2024) 46:65995–6007. doi: 10.1007/s11357-024-01143-x
166. Li Z, Huang Z, Zhang H, Lu J, Wei Y, Yang Y, et al. IRE1-mTOR-PERK axis coordinates autophagy and ER stress-apoptosis induced by P2X7-mediated ca(2+) influx in osteoarthritis. Front Cell Dev Biol. (2021) 9:695041. doi: 10.3389/fcell.2021.695041
167. Li Z, Huang Z, Zhang H, Lu J, Tian Y, Piao S, et al. Moderate-intensity exercise alleviates pyroptosis by promoting autophagy in osteoarthritis via the P2X7/AMPK/mTOR axis. Cell Death Discovery. (2021) 7:1346. doi: 10.1038/s41420-021-00746-z
168. Luo W, Li J, Li Z, Lin T, Zhang L, Yang W, et al. HO-1 nuclear accumulation and interaction with NPM1 protect against stress-induced endothelial senescence independent of its enzymatic activity. Cell Death Dis. (2021) 12:8738. doi: 10.1038/s41419-021-04035-6
169. Burczyk G, Cichon I, Kolaczkowska E. Itaconate suppresses formation of neutrophil extracellular traps (NETs): involvement of hypoxia-inducible factor 1α (Hif-1α) and heme oxygenase (HO-1). Front Immunol. (2022) 13:864638. doi: 10.3389/fimmu.2022.864638
170. Toth RK, Warfel NA. Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants (Basel). (2017) 6:2. doi: 10.3390/antiox6020027
171. Zheng X, Qiu J, Zhang H, Gao N, Jiang T, Gong Y, et al. PD184352 exerts anti-inflammatory and antioxidant effects by promoting activation of the Nrf2/HO-1 axis. Biochem Pharmacol. (2023) 211:115542. doi: 10.1016/j.bcp.2023.115542
172. Wang L, Fu X, Xia T, Yang Z, Zhao R. Myrislignan ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Int Immunopharmacol. (2024) 140:112887. doi: 10.1016/j.intimp.2024.112887
173. Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Fransès RE, Mapp PI, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatol (Oxford). (2010) 49:101852–61. doi: 10.1093/rheumatology/keq188
174. Pan J, Wang B, Li W, Zhou X, Scherr T, Yang Y, et al. Elevated cross-talk between subchondral bone and cartilage in osteoarthritic joints. Bone. (2012) 51:2212–7. doi: 10.1016/j.bone.2011.11.030
175. Short B. VEGF tips its hand in angiogenesis. J Cell Biol. (2015) 209:4474. doi: 10.1083/jcb.2094fta
176. Feng Y, Hu S, Liu L, Ke J, Long X. HMGB1 contributes to osteoarthritis of temporomandibular joint by inducing synovial angiogenesis. J Oral Rehabil. (2021) 48:5551–9. doi: 10.1111/joor.13129
177. Zhang L, Zhang L, Huang Z, Xing R, Li X, Yin S, et al. Increased HIF-1α in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev. (2019) 2019:6326517. doi: 10.1155/2019/6326517
178. Zhang L, Li X, Zhang H, Huang Z, Zhang N, Zhang L, et al. Agnuside alleviates synovitis and fibrosis in knee osteoarthritis through the inhibition of HIF-1α and NLRP3 inflammasome. Mediators Inflamm. (2021) 2021:5534614. doi: 10.1155/2021/5534614
179. Xu L, Zhang F, Cheng G, Yuan X, Wu Y, Wu H, et al. Attenuation of experimental osteoarthritis with human adipose-derived mesenchymal stem cell therapy: inhibition of the pyroptosis in chondrocytes. Inflammation Res. (2023) 72:189–05. doi: 10.1007/s00011-022-01655-2
180. Kong G, Wang J, Li R, Huang Z, Wang L. Ketogenic diet ameliorates inflammation by inhibiting the NLRP3 inflammasome in osteoarthritis. Arthritis Res Ther. (2022) 24:1113. doi: 10.1186/s13075-022-02802-0
181. Marriott KA, Birmingham TB. Fundamentals of osteoarthritis. Rehabilitation: Exercise, diet, biomechanics, and physical therapist-delivered interventions. Osteoarthritis Cartilage. (2023) 31:101312–26. doi: 10.1016/j.joca.2023.06.011
182. Jia S, Yang Y, Bai Y, Wei Y, Zhang H, Tian Y, et al. Mechanical stimulation protects against chondrocyte pyroptosis through irisin-induced suppression of PI3K/akt/NF-κB signal pathway in osteoarthritis. Front Cell Dev Biol. (2022) 10:797855. doi: 10.3389/fcell.2022.797855
183. van Middelkoop M, Schiphof D, Hattle M, Simkins J, Bennell KL, Hinman RS, et al. People with short symptom duration of knee osteoarthritis benefit more from exercise therapy than people with longer symptom duration: An individual participant data meta-analysis from the OA trial bank. Osteoarthritis Cartilage. (2024) 32:121620–7. doi: 10.1016/j.joca.2024.07.007
184. Oka Y, Murata K, Kano T, Ozone K, Arakawa K, Kokubun T, et al. Impact of controlling abnormal joint movement on the effectiveness of subsequent exercise intervention in mouse models of early knee osteoarthritis. Cartilage. (2021) 13:1334s–44s. doi: 10.1177/1947603519885007
185. Dai Y, Zhang L, Yan Z, Li Z, Fu M, Xue C, et al. A low proportion n-6/n-3 PUFA diet supplemented with Antarctic krill (Euphausia superba) oil protects against osteoarthritis by attenuating inflammation in ovariectomized mice. Food Funct. (2021) 12:156766–79. doi: 10.1039/d1fo00056j
186. Jin X, Dong X, Sun Y, Liu Z, Liu L, Gu H. Dietary fatty acid regulation of the NLRP3 inflammasome via the TLR4/NF-κB signaling pathway affects chondrocyte pyroptosis. Oxid Med Cell Longev. (2022) 2022:3711371. doi: 10.1155/2022/3711371
187. Feng Z, Huang Q, Zhang X, Xu P, Li S, Ma D, et al. PPAR-γ Activation alleviates osteoarthritis through both the nrf2/NLRP3 and PGC-1α/Δψ (m) pathways by inhibiting pyroptosis. PPAR Res. (2023) 2023:2523536. doi: 10.1155/2023/2523536
188. Bougault C, Gosset M, Houard X, Salvat C, Godmann L, Pap T, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheumatol. (2012) 64:123972–81. doi: 10.1002/art.34678
189. Liu W, Liu A, Li X, Sun Z, Sun Z, Liu Y, et al. Dual-engineered cartilage-targeting extracellular vesicles derived from mesenchymal stem cells enhance osteoarthritis treatment via miR-223/NLRP3/pyroptosis axis: Toward a precision therapy. Bioact Mater. (2023) 30:169–83. doi: 10.1016/j.bioactmat.2023.06.012
Keywords: pyroptosis, osteoarthritis, chondrocytes, synoviocytes, NLRP3, inflammatory cytokines
Citation: Lin M, Zhang C, Li H, Li K, Gou S, He X, Lv C and Gao K (2025) Pyroptosis for osteoarthritis treatment: insights into cellular and molecular interactions inflammatory. Front. Immunol. 16:1556990. doi: 10.3389/fimmu.2025.1556990
Received: 07 January 2025; Accepted: 13 March 2025;
Published: 01 April 2025.
Edited by:
Daniela Novick, Weizmann Institute of Science, IsraelReviewed by:
Shangxue Yan, Anhui Medical University, ChinaXuchang Zhou, Beijing Sport University, China
Copyright © 2025 Lin, Zhang, Li, Li, Gou, He, Lv and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao He, eGlhb2hlaHhAMTYzLmNvbQ==; Chaoliang Lv, bHZjaGFvbGlhbmdna0AxNjMuY29t; Kai Gao, Z2FvaGFpa2FpODhAMTI2LmNvbQ==
†These authors share first authorship