 Xiaoqi Wu
Xiaoqi Wu Jingyuan Zhang
Jingyuan Zhang Min Shen
Min Shen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 18 March 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1556372
Phospholipase C gamma 2 (PLCG2) gene mutations might cause PLCG2-associated antibody deficiency and immune dysregulation (PLAID)/autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) syndrome. They are two forms of autosomal-dominant immune dysregulation (ID). APLAID patients are usually characterized by skin lesions, pulmonary involvement, and musculoskeletal, ophthalmic, and gastrointestinal tract symptoms, but unlike PLAID patients, these patients do not present with cold urticaria or autoimmunity. Here, we report a 25-year-old man with B-cell lymphopenia, pulmonary bullae, recurrent sinopulmonary infections, and cutis laxa but without cold-induced urticaria. Anti-nuclear antibodies were negative. Trio whole-genome sequencing revealed a de novo heterozygous PLCG2 gene (NM_002661.5) variant c.3417_3419del, p.E1139del, located on chromosome chr16-81973600-81973602. Our findings expand the variety of clinical and genetic phenotypes for APLAID and suggest that this variant would be meaningful.
Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) is a rare autoinflammatory disorder caused by PLCG2 gene variants. PLCγ2 is a pleiotropic signaling molecule that controls cellular responses in many hematopoietic cells, including B lymphocytes and natural killer (NK) cells (1). PLCγ2 can hydrolyze the substrate phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 functions as a second messenger to increase the intracellular calcium concentration, inducing downstream cell activities (2–4). Pathogenic variants of the PLCG2 gene can cause PLCG2-associated antibody deficiency and immune dysregulation (PLAID) or APLAID (5–7). Both are rare disorders inherited in an autosomal dominant pattern. Notably, their pathogenesis, along with clinical phenotypes and genotypes, are still being investigated.
Here, we report a rare case of APLAID that manifested primarily as pulmonary bullae and cutis laxa, with a de novo and novel in-frame deletion in the PLCG2 gene.
A 25-year-old Chinese Han man occasionally experienced chest distress and shortness of breath since the age of 3 years old. Pulmonary bullae were found through X-ray when he was 17 years old, and then, he experienced severe dyspnea exacerbations requiring hospitalization several times annually. He developed pulmonary Aspergillus infection when he was 20 years old and COVID-19 pneumonitis when he was 23 years old, leading to reduced exercise tolerance and frequent sinopulmonary infections requiring hospitalization and ICU (Intensive Care Unit) admission more than five times every year. At age 15, he underwent surgery for cryptorchidism and experienced fat liquefaction at the wound site, which persisted for more than a month. He also had cutis laxa ever since the age of 18, initially on the face and then subsequently spread to the trunk and extremities over 4–5 years. No cold-induced urticaria, abdominal pain, conjunctivitis, hearing loss, headache, prolonged fever unrelated to infection, or joint involvement was observed. The patient’s history of food and drug allergies was negative. He had no history of smoking or drinking. Family history was unremarkable. Physical examination revealed premature aging and obvious cutis laxa on the face, neck, and abdomen. Red papules can be seen on the back, with pus-filled pustules in the center (Figure 1). Lung examination revealed clear breath sounds bilaterally, with no dry or wet rales.
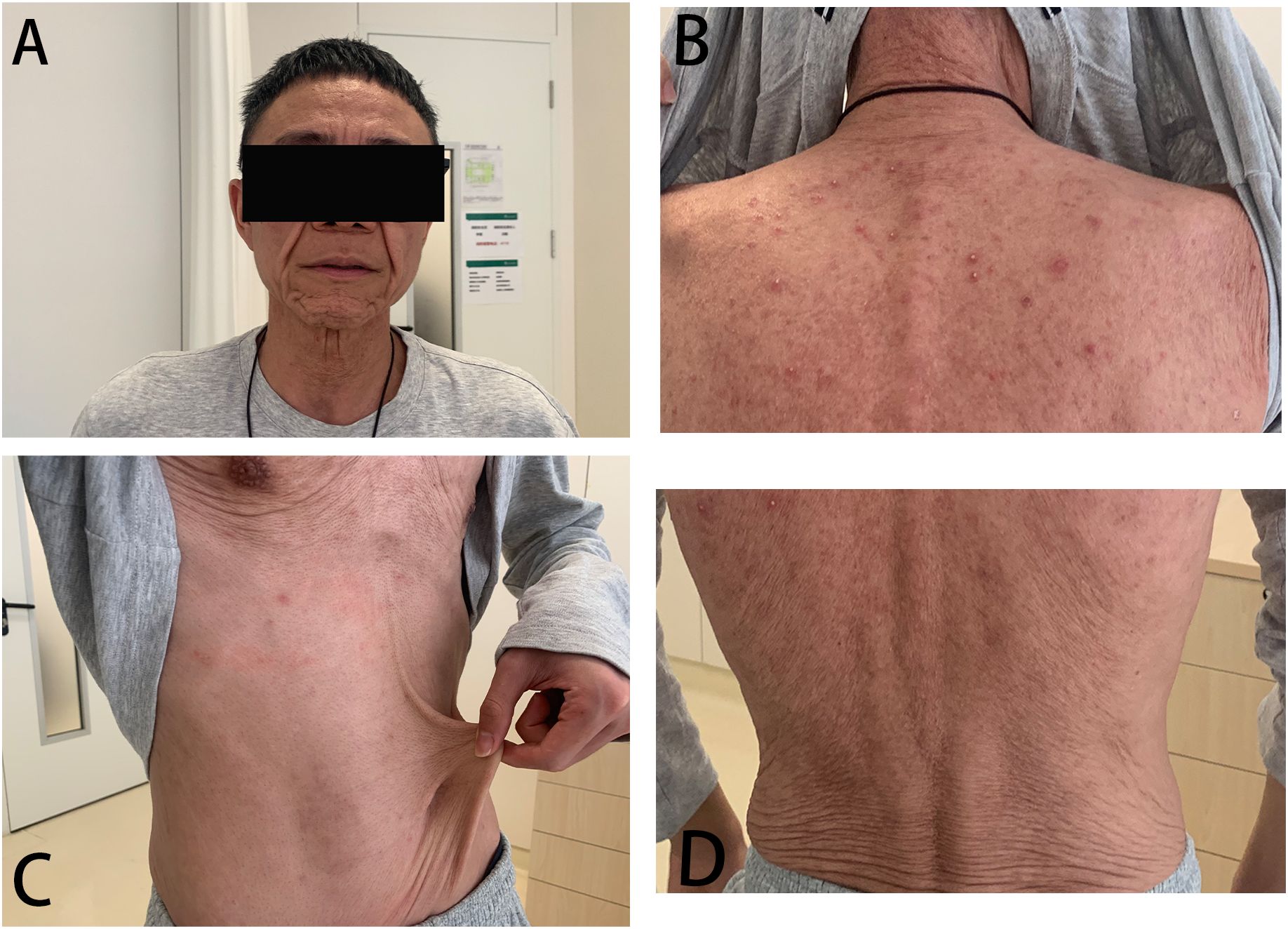
Figure 1. (A-D) Description of cutaneous manifestations in the patient. Cutis laxa on his face, neck, and abdomen. Red papules can be seen on his back, with pus-filled pustules in the center.
The laboratory results revealed normal T-cell counts, reduced circulating B cells 80/μL (normal range: 180–324/μL) and NK cells 160/μL (175–567/μL). The serum immunoglobulin (Ig) levels, including those of the IgG, IgA, IgM, IgE, and IgG subclasses, were within the normal limits. The neutrophil, eosinophil, and platelet counts and the serum levels of C-reactive protein (CRP) and complement were within the reference ranges. Serum protein electrophoresis and liver function tests were within normal parameters. Antinuclear antibodies (ANAs), lung cancer-related tumor markers and EBV-DNA were negative. Arterial blood gas analysis was normal. Skin biopsy revealed extensive infiltration of lymphocytes and neutrophils within the dermis, accompanied by loose dermal tissue and collagen fibers exhibiting curling and rupture upon elastic fiber staining, which was consistent with folliculitis and cutis laxa. A chest CT revealed bilateral emphysema and pulmonary bullae (Figure 2). The lung function measurements revealed extremely severe obstructive ventilation disorder, decreased diffusion capacity, and a negative bronchial dilation test. Trio whole-genome sequencing revealed a de novo heterozygous PLCG2 gene variant (NM_002661.5): c.3417_3419del, p.E1139del, located at chr: 81973600 -81973602.
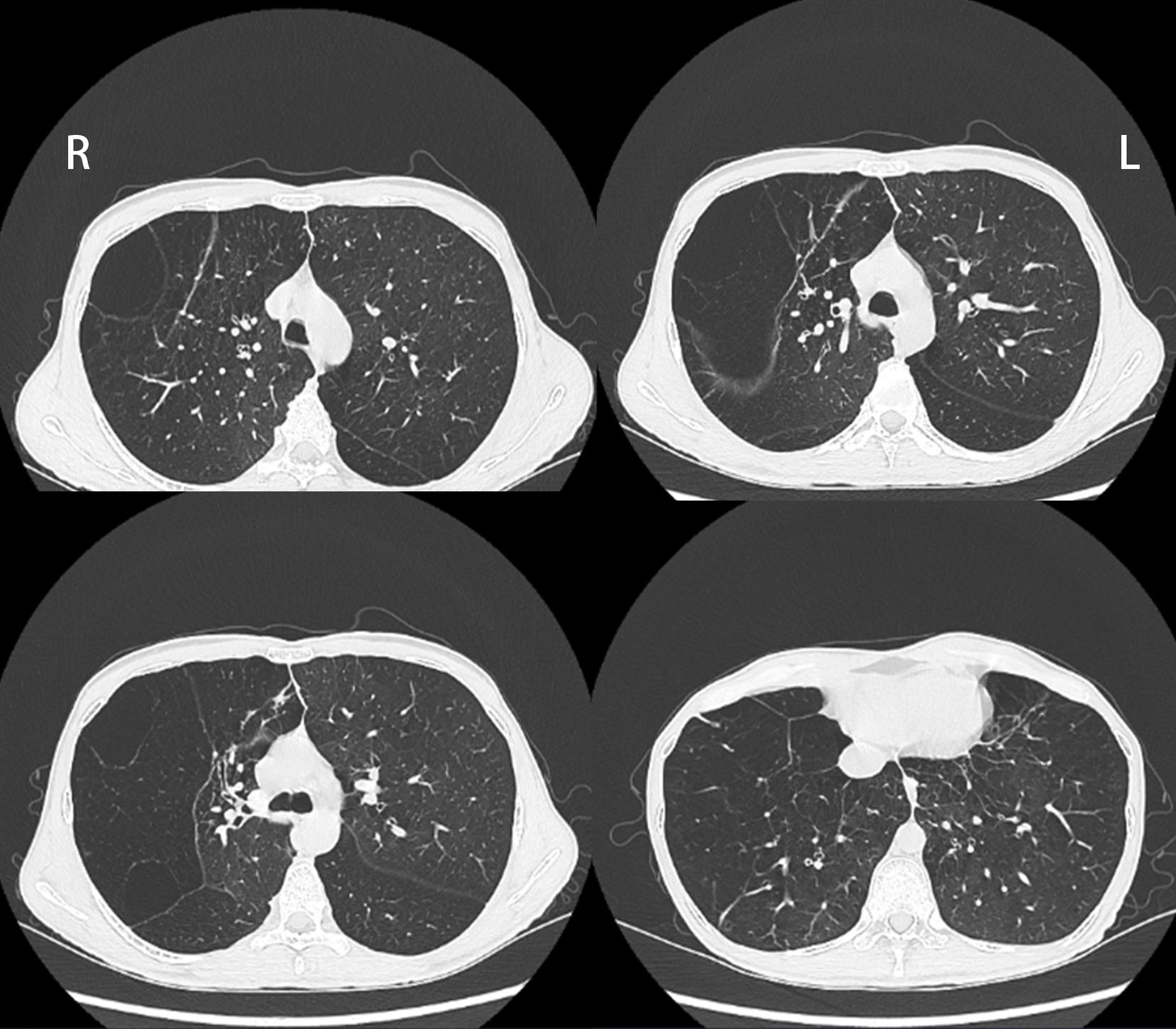
Figure 2. Chest CT showed bilateral emphysema and pulmonary bullae.
The presence of B-cell lymphopenia and recurrent infections, signaled underlying immune dysregulation, alongside skin lesions and a PLCG2 gene mutation, leading to a diagnosis of APLAID in the patient. Budegolide aerosol or indacaterol combined with glycopyrronium had been previously utilized but was ineffective. He was given fluticasone inhalation powder once daily, 0.25 g azithromycin once daily for 3 days per week, regular bacterial lysates and vaccination (influenza/pneumonia vaccine). After the 6-month follow-up, the patient still had recurrent upper respiratory infections, but his overall condition was stable. The 6-minute walk test distance was approximately 400 m. The results of the reexamination of lung function were similar to those of the previous study. No obvious abnormalities were observed on the echocardiogram, and the systolic pulmonary artery pressure was 24 mmHg. The patient was suggested to continue with current treatment and to undergo lung transplantation if necessary.
We identified a novel in-frame deletion in PLCG2 (c.3417_3419del, p.E1139del) within the C2 domain of an APLAID patient. While this specific variant (g.81973600_81973602delAGA) remains unreported, a proximal genomic deletion (g.81973597_81973599delAGA) resulting in the identical glutamic acid residue loss at position 1139 has been described (8). The reference sequence at chr16:81973594-81973606 is TGAAGAAGATATG. In vitro experiments have revealed that the E1139del mutation in PLCγ2 induces constitutive activation of the PLCγ2 signaling pathway in B cells, characterized by enhanced intracellular calcium flux and sustained ERK phosphorylation upon B-cell receptor (BCR) stimulation (8). It is a gain-of-function (GOF) mutation. The phred-scaled combined annotation-dependent depletion (CADD) of this variant is 22.8, and a score > 20 is in the top 1% of the predicted deleteriousness. Therefore, this variant has been predicted to be highly damaging. The patient with the PLCG2 E1139 mutation, included in the previous study, presented with granulomas, recurrent sinopulmonary infections, hypogammaglobulinemia (low IgG levels), B-cell lymphopenia, and mildly reduced NK cell counts, but did not exhibit cold-induced urticaria. These findings showed similarities with our patient, suggesting the novel PLCG2 c.3417_3419del mutation may contribute to this clinical phenotype.
Through a literature review, we found that most APLAID patients carried PLCG2 missense variants, whereas in-frame deletions are always observed in PLAID (5, 6, 9). However, with continuous improvements in understanding the disease, some studies have described the p.Leu845-Leu848del variant in APLAID, whereas missense variants such as c.3249C>G (p.P1083=), c.3244T>C (p.C1082R), c.3524T>A (p.I1175K), and c.415C>T (p.P139S) are related to PLAID. In addition, the PLCG2 variant c.77C>T, p.Thr26Met might cause phenotypical overlap of the PLAID and APLAID patterns (10–12). For the mutation region, the autoinhibitory C-terminal Src homology 2 (cSH2) domain is a critical site for the restriction of intrinsic enzyme activity, conferring GOF activity. Additionally, a key region linked to Ibrutinib resistance mutations in the PLCγ2 protein is located between amino acids 1139 and 1141 within the C2 domain, which is crucial for calcium binding and membrane attachment of PLCγ2. The PLCG2 c.3417_3419del identified in our patient is also located in the C2 domain. Novice et al. (13) reported three patients with a novel heterozygous missense mutation in the C2 domain of PLCG2 (c. 3422 T>A, p.Met1141Lys), which have been shown to cause GOF effects, including hyperactive B-cell receptor (BCR) signaling and elevated calcium influx and two of the patients shared clinical features with APLAID. Thus the mutations in the C2 domain of PLCG2 region may disrupt autoinhibitory mechanisms or calcium binding, leading to altered signaling. These data support an accurate diagnosis of APLAID in our patient, especially at the genetic level. As the E1139del mutation had been shown to cause GOF effects, so we did not conduct functional studies experiments in our patient.
Making a diagnosis of APLAID or PLAID is challenging because only 11 APLAID individuals with 7 variants of PLCG2 have been reported thus far (5, 7, 9, 10, 13–15). It is not clear to what extent their clinical manifestations could differ from or overlap with those of other immune disorders. Similarly, the range of genetic changes has not been defined. APLAID patients always present with skin lesions, including cutaneous granulomas, vesiculopustular rashes and cutis laxa, posterior uveitis, inflammatory bowel disease (IBD) and recurrent sinopulmonary infections caused by a humoral defect, but with no circulating autoantibodies and have no cold-induced urticaria, in contrast to PLAID patients. On the basis of the above description combined with the patient’s clinical manifestations and genetic variation information, we diagnosed this patient with APLAID. The most common skin feature in APLAID is being vesiculopustular. However, our patient had rare cutis laxa, which has been previously reported in only three patients with APLAID (9, 10). Pulmonary bullae was the other major manifestation in our patient and have not yet been reported in APLAID. We ruled out infectious factors (such as tuberculosis) and lung cancer, as the patient’s serum protein electrophoresis was normal, the liver was not involved, and the genetic results did not indicate any mutations related to alpha-1 antitrypsin. Therefore, alpha-1 antitrypsin deficiency was also excluded from consideration. The patient experienced shortness of breath from childhood and pulmonary bullae were found before recurrent pulmonary infection, we think that this was probably related to autoinflammation in the lungs and might be one of the phenotypes of APLAID. But more case validation and further mechanistic research are needed to enhance our understanding of APLAID.
In APLAID, a disorder linked to pathogenic variants in the PLCG2 gene, the phenomenon of B - cell lymphopenia is a key aspect of its immunological profile (5, 9).
PLC-γ2 is needed for efficient generation of memory B cells especially switched memory B- cell and that has no direct significant role in differentiation of germinal center B cells to plasma cells (16, 17).The relatively normal immunoglobulin (Ig) levels in APLAID can be attributed to the fact that the unaffected memory B cells and plasma cells can still contribute to immunoglobulin production. We regret that we did not perform a more detailed analysis of B lymphocyte subsets. Although the patient’s IgG levels were not low, he got recurrent sinopulmonary infections, so we recommended intravenous immunoglobulin (IVIG) therapy. However, he declined our recommendation due to personal reasons. Respiratory tract management, vaccination, and routine anti-infection treatment were provided to reduce infections while awaiting lung transplantation.
Overall, we suggest that the in-frame deletion mutation of the PLCG2 variant c.3417_3419del p.E1139del might be causative for the APLAID phenotype in this patient. We hope that the present report adds more clinical and genetic evidence to the profile of APLAID.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Institutional Review Board of Peking Union Medical College Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XW: Writing – original draft, Data curation, Investigation, Writing – review & editing. JZ: Writing – review & editing. MS: Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by CAMS Innovation Fund for Medical Sciences(CIFMS) (2024-I2M-C&T-B-014) and National High Level Hospital Clinical Research Funding (2022-PUMCH-D-002).
We appreciate all supports from the patient and his families.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jackson JT, Mulazzani E, Nutt SL, Masters SL. The role of PLCgamma2 in immunological disorders, cancer, and neurodegeneration. J Biol Chem. (2021) 297:100905. doi: 10.1016/j.jbc.2021.100905
2. Hurley JH, Grobler JA. Protein kinase C and phospholipase C: bilayer interactions and regulation. Curr Opin Struct Biol. (1997) 7:557–65. doi: 10.1016/S0959-440X(97)80122-4
3. Hiller G, Sundler R. Regulation of phospholipase C-gamma 2 via phosphatidylinositol 3-kinase in macrophages. Cell Signal. (2002) 14:169–73. doi: 10.1016/S0898-6568(01)00252-2
4. Everett KL, Bunney TD, Yoon Y, Rodrigues-Lima F, Harris R, Driscoll PC, et al. Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J Biol Chem. (2009) 284:23083–93. doi: 10.1074/jbc.M109.019265
5. Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. (2012) 91:713–20. doi: 10.1016/j.ajhg.2012.08.006
6. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366:330–8. doi: 10.1056/NEJMoa1102140
7. Wu N, Zhang B, Wang T, Shen M, Zeng X. Case report: A rare case of autoinflammatory phospholipase Cγ2 (PLCγ2)-associated antibody deficiency and immune dysregulation complicated with gangrenous pyoderma and literature review. Front Immunol. (2021) 12:667430. doi: 10.3389/fimmu.2021.667430
8. Baysac K, Sun G, Nakano H, Schmitz EG, Cruz AC, Fisher C, et al. PLCG2-associated immune dysregulation (PLAID) comprises broad and distinct clinical presentations related to functional classes of genetic variants. J Allergy Clin Immunol. (2024) 153:230–42. doi: 10.1016/j.jaci.2023.08.036
9. Neves JF, Doffinger R, Barcena-Morales G, Martins C, Papapietro O, Plagnol V, et al. Novel PLCG2 mutation in a patient with APLAID and cutis laxa. Front Immunol. (2018) 9:2863. doi: 10.3389/fimmu.2018.02863
10. Martín-Nalda A, Fortuny C, Rey L, Bunney TD, Alsina L, Esteve-Sole A, et al. Severe autoinflammatory manifestations and antibody deficiency due to novel hypermorphic PLCG2 mutations. J Clin Immunol. (2020) 40:987–1000. doi: 10.1007/s10875-020-00794-7
11. Welzel T, Oefelein L, Holzer U, Müller A, Menden B, Haack TB, et al. Variant in the PLCG2 gene may cause a phenotypic overlap of APLAID/PLAID: case series and literature review. J Clin Med. (2022) 11:4369. doi: 10.3390/jcm11154369
12. Wang W, Yu Z, Gou L, Zhong L, Li J, Ma M, et al. Single-center overview of pediatric monogenic autoinflammatory diseases in the past decade: a summary and beyond. Front Immunol. (2020) 11:565099. doi: 10.3389/fimmu.2020.565099
13. Novice T, Kariminia A, Del Bel KL, Lu H, Sharma M, Lim CJ, et al. A germline mutation in the C2 domain of PLCγ2 associated with gain-of-function expands the phenotype for PLCG2-related diseases. J Clin Immunol. (2020) 40:267–76. doi: 10.1007/s10875-019-00731-3
14. Moran-Villasenor E, Saez-de-Ocariz M, Torrelo A, Arostegui JI, Yamazaki-Nakashimada MA, Alcantara-Ortigoza MA, et al. Expanding the clinical features of autoinflammation and phospholipase Cgamma2-associated antibody deficiency and immune dysregulation by description of a novel patient. J Eur Acad Dermatol Venereol. (2019) 33:2334–9. doi: 10.1111/jdv.15918
15. Khabbazi A, Rahbar Kafshboran H, Nasiri Aghdam M, Nouri Nojadeh J, Dsghagh H, Daneshmandpour Y, et al. A new report of autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) with a homozygous pattern from Iran. Immunol Lett. (2020) 221:27–32. doi: 10.1016/j.imlet.2020.01.008
16. Hikida M, Casola S, Takahashi N, Kaji T, Takemori T, Rajewsky K, et al. PLC-gamma2 is essential for formation and maintenance of memory B cells. J Exp Med. (2009) 206:681–9. doi: 10.1084/jem.20082100
Keywords: PLCG2, APLAID, B-cell lymphopenia, pulmonary bullae, cutis laxa
Citation: Wu X, Zhang J and Shen M (2025) Case Report: de novo in-frame deletion in PLCG2 gene: a case report of B-cell lymphopenia, pulmonary bullae, and cutis laxa. Front. Immunol. 16:1556372. doi: 10.3389/fimmu.2025.1556372
Received: 06 January 2025; Accepted: 26 February 2025;
Published: 18 March 2025.
Edited by:
Chris Wincup, King’s College Hospital NHS Foundation Trust, United KingdomReviewed by:
Panfeng Tao, Zhejiang University, ChinaCopyright © 2025 Wu, Zhang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Shen, c2hlbm1wdW1jaEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.