 Zhi Li1†
Zhi Li1† Xiaoxia Zhang
Xiaoxia Zhang Zejiao Da
Zejiao Da
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 March 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1555910
This article is part of the Research Topic Neuroinflammation: Mechanisms and Therapeutic Interventions View all 7 articles
Hypobaric hypoxia is widely recognized as a prominent risk factor for high-altitude cerebral edema (HACE), which contributes to the exacerbation of multiple pathological mechanisms, including oxidative stress, mitochondrial dysfunction, disruption of blood−;brain barrier integrity, neuroinflammation, and neuronal apoptosis. Among these mechanisms, abnormalities in oxygen metabolism, including hypoxia, oxidative stress, and mitochondrial dysfunction, play pivotal roles in the pathophysiology of HACE. In this review, our objective is to enhance our comprehension of the underlying molecular mechanisms implicated in HACE by investigating the potential involvement of oxygen metabolism. Addressing aberrations in oxygen metabolism holds promise for providing innovative therapeutic strategies for managing HACE.
High altitude, defined as an elevation of 2500 m above sea level, encompasses approximately one-fifth of the Earth’s surface. According to a report in The Lancet, more than 140 million individuals resided at high altitudes above 2500 m as early as 2001 (1). Inadequately acclimatized individuals exposed to high altitudes may experience various high-altitude illnesses, including acute mountain sickness, high-altitude cerebral edema (HACE), and high-altitude pulmonary edema (2, 3). Despite its low incidence rate, HACE is a rapidly progressive and potentially fatal condition. Without prompt and effective treatment, patients can deteriorate rapidly within hours, leading to coma or even death (4). The pathophysiology of HACE remains incompletely understood; however, accumulating evidence suggests that it involves multiple factors. These include oxidative stress (5), cerebral blood flow disorders, disruption of blood−;brain barrier (BBB) integrity (6), inflammation (7, 8), REDOX homeostasis imbalance (9), mitochondrial dysfunction and neuronal apoptosis (10).
Hypobaric hypoxia (HH), the primary characteristic of a high-altitude environment, leads to reduced oxygen delivery to tissues (11). Owing to the heavy reliance of the brain on a consistent oxygen supply, it becomes vulnerable to fluctuations in oxygen levels. Consequently, abnormalities in oxygen metabolism play a pivotal role in the pathophysiology of HACE. The intricate interplay of molecular events governing oxygen metabolism may significantly contribute to the pathogenesis and progression of HACE. Therefore, this review places particular emphasis on elucidating the molecular mechanisms underlying recent discoveries related to abnormalities in oxygen metabolism associated with HACE, encompassing hypoxia, oxidative stress, and mitochondria. This comprehensive analysis has the potential to enhance our understanding of the correlation between HACE and oxygen metabolism while facilitating therapeutic approaches targeting these metabolic abnormalities for effective management.
The process of oxygen metabolism, which is vital for maintaining normal physiological functions in cells and tissues, involves a series of intricate mechanisms involved in the transfer, transportation, and utilization of oxygen. This metabolic pathway converts oxygen into adenosine triphosphate (ATP) to provide energy, while a small fraction is transformed into reactive oxygen species (ROS) (11). However, the aberrant accumulation of ROS can disrupt organismal functionality. The mitochondrion plays a crucial role in cellular oxygen consumption, exerting an influence on redox potential and governing the levels of ROS.
Aberrations in oxygen metabolism predominantly include hypoxia, excessive ROS accumulation, and mitochondrial dysfunction. These factors can disturb physiological processes or initiate disease development (11). Upon exposure to a high-altitude hypoxic environment, diminished ambient oxygen availability induces an increase in ROS production within brain tissue. This phenomenon is positively associated with altitude. ROS exhibit strong reactivity and have the ability to oxidize lipids, proteins, and DNA, leading to alterations in cellular structure and function (12). The ROS target polyunsaturated fatty acids within biofilms, initiating a cascade of lipid peroxidation reactions. This leads to biofilm dysfunction and damage to membrane-bound enzymes (13). The excessive generation of ROS further exacerbates the oxidative stress response and disrupts the redox balance, ultimately resulting in cerebral injury (14). The presence of ROS in the brain can contribute to neuronal loss and cerebral edema (15). Elevated levels of ROS can also result in mitochondrial impairment, DNA alterations, augmented cytokine production, and even cellular apoptosis. (16, 17). Conversely, hypoxia-induced mitochondrial autophagy, which is regulated by the expression of BCL2-interacting protein 3-like (BNIP3L) via hypoxia-inducible factor (HIF), functions as an adaptive metabolic response to prevent excessive levels of ROS and maintain cellular survival (18). Therefore, abnormal oxygen metabolism is a significant factor in the development of HACE. Figure 1 illustrates the contribution of oxygen metabolism abnormalities to HACE pathology.
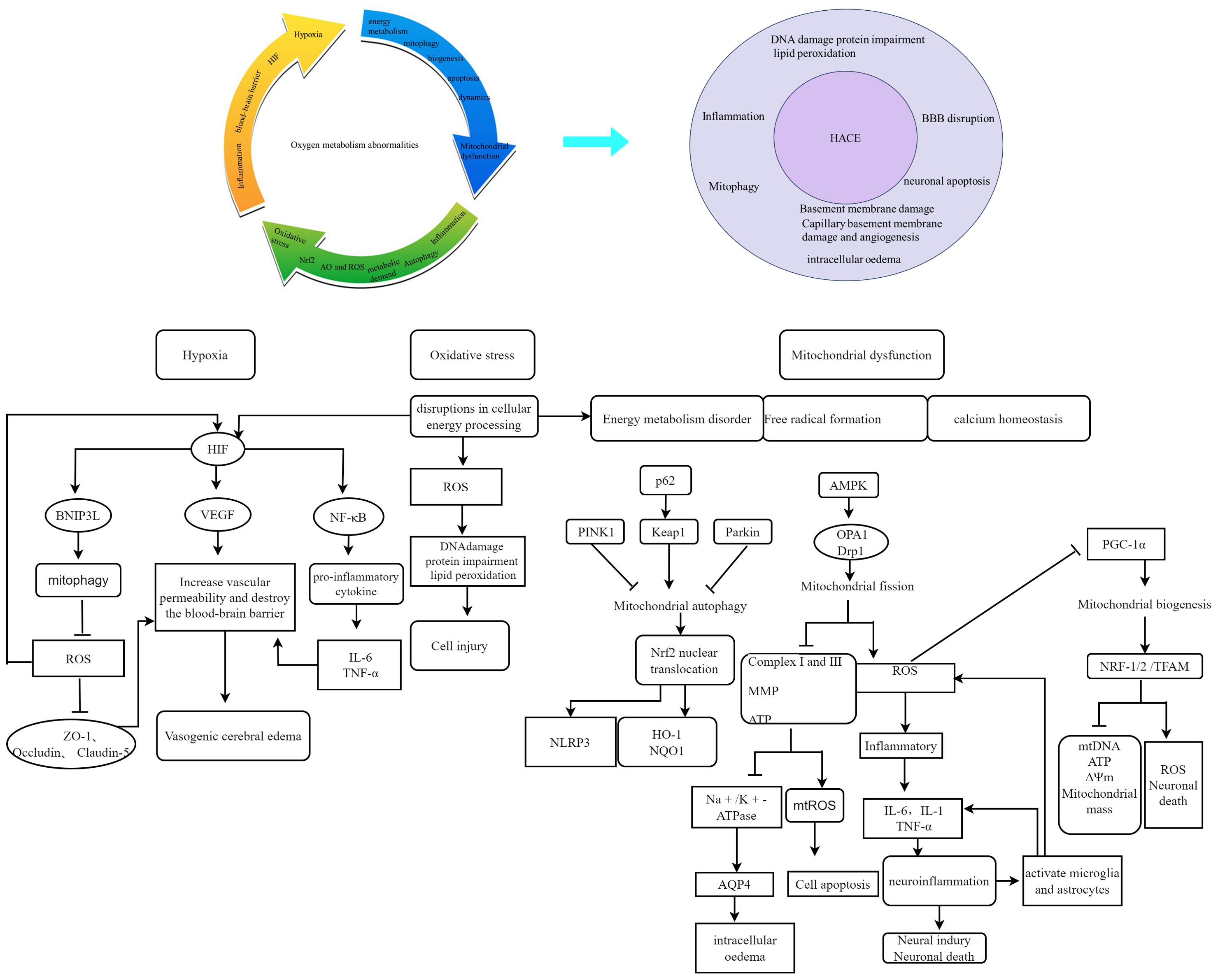
Figure 1. Schematic representation of the contribution of oxygen metabolism abnormalities to HACE pathology. Oxygen metabolism abnormalities constitute a significant risk factor in the pathogenesis and progression of HACE. Recent studies have highlighted hypoxia, oxidative stress, and mitochondrial dysfunction as pivotal components in both HACE and oxygen metabolism research. The interplay between oxygen metabolism disturbances and HACE pathology significantly contributes to disease deterioration.
The limited availability of oxygen at high altitudes triggers modifications in gene expression through the HIF pathway, which plays a crucial role in the cellular response to hypoxia. HIFs are heterodimers composed of one of three hypoxia-inducible α subunits (HIF-1α, HIF-2α, or HIF-3α) and a constitutively expressed β subunit (HIF-β). Under normal oxygen conditions, HIF-α undergoes hydroxylation facilitated by interactions with prolyl hydroxylase domain (PHD) proteins, which provide a binding site for the von Hippel−;Lindau (VHL) protein and lead to degradation of HIF-α. However, under low-oxygen conditions, hydroxylation is inhibited, resulting in stabilized dimerization of HIF-α with HIF-β and subsequent binding to numerous target genes to initiate the transcriptional activation of key factors, such as erythropoietin (EPO), hemoglobin (HB), and vascular endothelial growth factor (VEGF) (19). Figure 2 illustrates the regulation of HIF in response to hypoxic conditions.
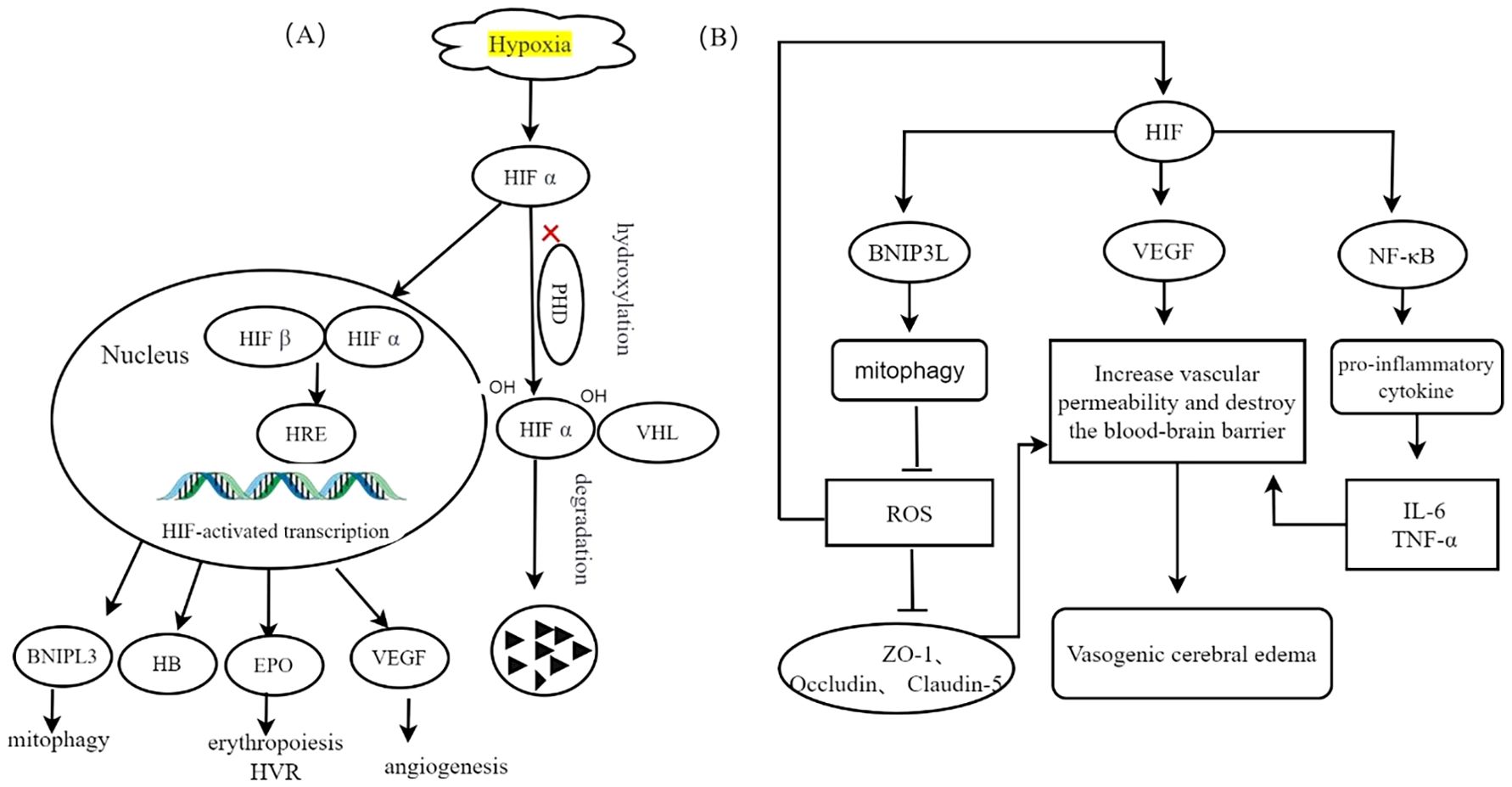
Figure 2. Transcriptional regulation of HIF induced by hypoxia, adapted from (3). (A) Hypoxia inhibits the proteasomal degradation of HIF-α and promotes its translocation into the nucleus. Following interaction with HIF-β, this complex synergistically activates the transcription of target genes, thereby enhancing cellular adaptation to hypoxic conditions. (B) The specific role of HIF-1α in different cell types is discussed. In mitochondria, elevated levels of HIF enhance the upregulation of BNIP3L, resulting in increased mitophagy and decreased ROS production. Elevated ROS levels compromise the integrity of cellular tight junction proteins, including ZO-1, Occludin, and Claudin-5, which in turn increases vascular permeability and contributes to vasogenic cerebral edema. In endothelial cells, the accumulation of HIF-1 results in the upregulation of VEGF, consequently increasing vascular permeability, compromising the integrity of the blood-brain barrier, and inducing vasogenic cerebral edemaIn microglia, HIF activation of the NF-κB signaling pathway facilitates the release of proinflammatory cytokines, such as IL-6, IL-1β, and TNF-α, increases vascular permeability, and contributes to cerebral edema.
The EPAS1 gene, which encodes HIF-2α, has been identified as the predominant isoform involved in high-altitude adaptation on the basis of recent genome-wide studies (20). EPAS1 has a tissue- and cell-specific distribution and is expressed primarily in organs such as the brain (21). Hypoxic stabilization of HIF-2α at high altitudes leads to the formation of the HIF-2 complex, which activates EPO expression. EPO is a glycoprotein hormone primarily produced by the kidney that stimulates erythropoiesis in the bone marrow, thereby increasing the oxygen-carrying capacity of the blood. In the brain, EPO modulates both central and peripheral neural respiratory centers, leading to an augmented hypoxic ventilatory response (HVR) and improved tissue oxygenation.
VEGF is an endothelial-specific mitogen that plays a crucial role in promoting angiogenesis through various mechanisms, including migration, proliferation, differentiation, and extracellular matrix proteolysis. It acts as a downstream effector of HIF-1. After 24 hours of acute exposure to HH, the expression of VEGF in the brain increased. Additionally, prolonged exposure to elevated levels of VEGF can lead to disruption of endothelial barrier integrity and increased vascular permeability, resulting in cerebral damage and the development of high-altitude cerebral edema (22). Furthermore, the activity of VEGF closely correlates with that of HIF-1α under hypoxic conditions, which further exacerbates inflammatory responses (23). Hypoxia-induced upregulation of HIF-1α serves as a pivotal regulatory mechanism that enhances subsequent VEGF expression, thereby promoting vasogenic cerebral edema.
The BNIP3L protein, known as BCL2-interacting protein 3-like, functions as a specific receptor responsible for recognizing mitophagy while being localized on the outer membrane of mitochondria. Under hypoxic conditions, BNIP3L plays an essential role in promoting autophagy induced by low oxygen levels and ensuring cell survival (24). Hypoxia significantly increases BNIP3 expression through direct transcriptional targeting by HIF-1α. Elevated levels of HIF intensify the upregulation of BNIP3L, consequently leading to enhanced mitophagy induction and reduced production of ROS (25).
HIF-a, hypoxia inducible factor α-subunit; HIF-β, hypoxia inducible factor β-subunit; PHD, prolyl hydroxylase; VHL, Van Hippel–Lindau proteins; HRE, hypoxia response element; VEGF, vascular endothelial growth factor; EPO, erythropoietin; HVR, hypoxic ventilatory response; HB, hemoglobin;BNIF3, BCL2 interacting protein
The BBB functions as a crucial protective shield for the central nervous system, ensuring the stability of the brain microenvironment (26). The disruption of the BBB induced by hypoxia is considered a primary contributing factor to the development of HACE (27, 28). This disruption results in elevated BBB permeability, and its severity directly correlates with the degree of cerebral edema observed on T2-weighted MR images in both HACE patients and animal models (29, 30). The occurrence of white matter edema in HACE signifies impaired function or disruption of the blood−;brain barrier, potentially resulting from membrane destabilization and inflammation induced by ROS or localized activation of HIF and VEGF (6).
HH stimulation disrupts blood−;brain barrier integrity by compromising the levels of the cellular tight junction proteins ZO-1, Occludin and Claudin-5 and increasing vascular permeability, resulting in vasogenic cerebral edema (6). Additionally, this stimulation induces the production of VEGF via HIF-1α, resulting in vascular injury (6, 7). Consequently, an inflammatory response is triggered, ultimately culminating in cytotoxic brain edema. Inflammation plays a pivotal role in the pathogenesis of cerebral edema, as inflammatory mediators (ET-1, TNF-α, and IL-1β) act on angiotropic endothelial cells within brain tissue to increase BBB permeability (31). The exposure of animals to HH combined with lipopolysaccharide (LPS) treatment likely synergistically affects BBB integrity and function, which is characterized by a compromised oxygen supply and exacerbated inflammatory response (32). Reinforcing the integrity of the BBB has thus emerged as a potent strategy for pharmacological intervention in cerebral edema.
Microglia, which are a type of tissue-resident macrophage in the brain, serve dual functions in preserving the integrity of the BBB. Resting microglia support BBB maintenance, whereas activated microglia compromise BBB integrity by migrating toward blood vessels and phagocytosing the tight junctions of vascular endothelial cells, thereby increasing vascular permeability and inducing vasogenic cerebral edema (33, 34). Moreover, the release of proinflammatory factors from activated microglia and subsequent astrocyte swelling contribute to cytotoxic cerebral edema in HACE (33, 35). Consequently, inhibiting microglial activation may represent a promising therapeutic strategy for HACE (33). Figure 3 elucidates the proposed mechanisms that are hypothesized to underlie HACE.
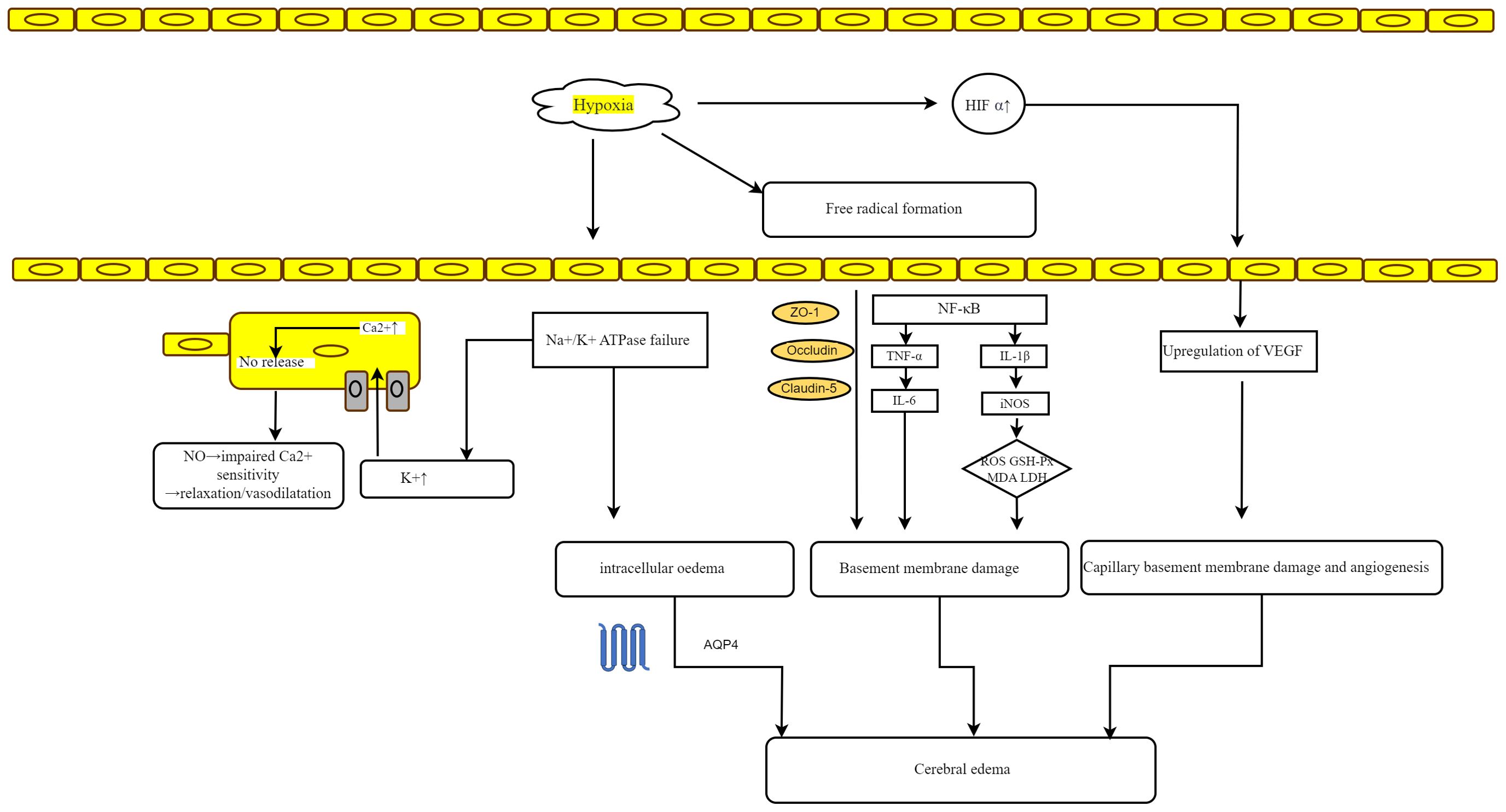
Figure 3. Mechanisms hypothesized to underlie HACE. Hypoxia-induced failure of Na+/K+ ATPase results in local hyperkalemia, which may trigger calcium-mediated nitric oxide release, leading to vasodilation in vascular smooth muscle. Additionally, ATPase failure under hypoxic conditions can cause intracellular edema. The formation of free radicals disrupts cellular tight junction proteins (ZO-1, Occludin, and Claudin-5) and triggers the release of inflammatory mediators, thereby increasing vascular permeability and damaging vessel basement membranes, ultimately resulting in vasogenic edema. The accumulation of HIF-1α and subsequent upregulation of VEGF further contribute to basement membrane damage and edema. Moreover, alterations in endothelial cells, including increased pinocytotic vesicles and disassembly of interendothelial tight junction proteins, as well as swelling of astrocyte endfeet, may also increase capillary permeability.
Hypoxia can induce a cerebral inflammatory response and facilitate the initiation and progression of cerebral edema in mice exposed to high-altitude conditions. A growing body of evidence emphasizes the pivotal role of inflammation in the progression of HACE (7, 28). A study by Song et al. demonstrated that exposure of volunteers to an altitude of 3860 m resulted in a significant increase in the plasma levels of TNF-α, IL-1β, and IL-6 (7). Proinflammatory cytokines, namely, IL-6 and IL-1β, were induced from the first day of high-altitude exposure onward according to Chauhan et al. (36).
HH triggers the activation of the NF-κB signaling pathway and facilitates the release of proinflammatory cytokines, such as IL-6, IL-1β, and TNF-α, leading to detrimental inflammatory responses within the central nervous system (35, 37, 38). The NF-κB signaling pathway plays a crucial regulatory role in the development of cerebral edema induced by high-altitude exposure (39)]. First, the expression of aquaporin 4 (AQP4) is directly controlled by the NF-κB signaling pathway. Wang suggested that puerarin exerts an anti-inflammatory effect by inhibiting the activation of the NF-κB pathway, thereby reducing AQP levels in the cerebrum (40). Second, the NF-κB signaling pathway significantly regulates the inflammatory response mediated through IL-1β, IL-6, and TNF-α generation during high-altitude exposure (41). Activation of the NF-κB signaling pathway was observed in the cerebral cortex of mice exposed to simulated high-altitude conditions as well as in astrocytes or microglia exposed to hypoxia. This activation was characterized by increased P65 phosphorylation and subsequent nuclear translocation. Pretreatment with mdivi-1 and quercetin effectively inhibited hypoxia-induced activation of the NF-κB signaling pathway (7, 42). Hyperoside inhibits the p38 and NF-κB pathways in microglia (43). Finally, the relationship between NF-κB and HIF-1α is strongly interdependent (44). Numerous studies have demonstrated that the inhibition of inflammatory cytokines mediated by NF-κB and HIF-1α, such as AP and curcumin, significantly alleviates cerebral edema (45, 46). Conversely, NF-κB stimulates the upregulation of p62, which negatively regulates caspase-1 activation through mitophagic clearance of mitochondria that release activating signals for the NLRP3 inflammasome (47). NLRP3 inflammatory corpuscles, which contain the pyrin domain of NLR family 3 (NLRP3), play a pivotal and extensive role in orchestrating the inflammatory response (48). Neuroinflammation induced by hypoxia triggers the activation of the NLRP3 inflammasome (8). The activation of the NLRP3 inflammasome has been demonstrated to serve as a major catalyst for neuroinflammation, stimulating the secretion of proinflammatory cytokines and subsequent inflammatory responses. Research indicates that the suppression of the NLRP3 inflammasome can alleviate hypoxic–ischemic brain damage (49).
Hypoxia also promotes an inflammatory environment and triggers an inflammatory cascade in CNS tissue, which is further exacerbated by glial cells releasing proinflammatory cytokines (50). Exposure to HH leads to the activation of microglia via the p38 MAPK pathway, subsequently triggering the secretion of proinflammatory cytokines within 24 hours. This cascade ultimately results in astrocyte activation at three and seven days postexposure (36, 51). Concurrently, preexisting inflammation exacerbates hypoxia-induced injury by increasing astrocyte permeability and augmenting AQP4 activity through the Toll-like receptor 4 (TLR4), mitogen-activated protein kinase (MAPK), and NF-κB signaling pathways (52). Additionally, Song et al. demonstrated that preexisting systemic inflammation worsens hypoxic cerebral edema in a rat model via the activation of inflammatory signaling pathways through astrocyte−;microglia interactions (7).
The activation of glial cells and the release of cytokines have been demonstrated to initiate and contribute to cognitive impairments in individuals with HH (36, 53). Hypoxia-induced neuroinflammation represents a potential therapeutic target for the treatment of HACE (8). Microglia, characterized by their extensively branched morphology, represent the predominant resident immune cells in the brain and serve as the primary site for NLRP3 inflammatory body expression. Upon activation, microglia undergo functional and morphological alterations (54). Exposure to HH results in a decrease in the branching complexity of microglia in the hippocampus (36). During the early stages, hypoxia induces microglial activation through the upregulation of nuclear respiratory factor 1 (NRF1), which in turn activates NF-κB p65 and TFAM, leading to an inflammatory response. Moreover, hypoxia upregulates AP2B1 and CAV-1, enhancing microglial phagocytosis of cellular debris and attenuating inflammatory bursts. However, excessive microglial activation subsequently triggers microglial migration toward blood vessels, resulting in the release of proinflammatory factors and the phagocytosis of endothelial cells, which induce endothelial damage and disrupt the integrity of the BBB, ultimately leading to astrocyte swelling. The presence of hypoxia triggers the activation of microglia, which can manifest distinct yet overlapping phenotypes characterized by proinflammatory (M1) and anti-inflammatory (M2) properties (55). M1-type microglia lead to oligodendrocyte and neuronal death by promoting the production and release of inflammatory mediators such as cytokines, chemokines, ROS, NO, and matrix metalloproteinases (56). In contrast, M2 microglia exhibit both immunosuppressive and neuroprotective properties. On the one hand, they release anti-inflammatory factors as well as neurotrophic factors (such as IGF, TGF-β, GDNF, and IL-10) to promote angiogenesis, tissue remodeling, and neurorepair (57, 58). Therefore, the regulation of microglia during brain injury is a complex process in which the beneficial or detrimental effects of microglia depend on their activation status and environmental cues (33).The polarization of microglia is a reversible process that plays a critical role in the progression, reversal, and treatment of diseases. Consequently, modulating microglial polarization to inhibit inflammation and promote tissue repair has emerged as a promising therapeutic strategy for the treatment of HACE. Wang et al. reported that stem cells derived from human exfoliated deciduous teeth (SHEDs) exhibit significant preventive and therapeutic effects on HACE by inhibiting M1-type polarization and promoting M2-type polarization of microglia via the HIF/ERK pathway (59). Studies have shown that inhibiting the NF-kB signaling pathway can effectively inhibit M1 polarization of microglia (42, 60). Figure 4 illustrates the proposed inflammation mechanisms hypothesized to underlie HACE. Research progress on neuroinflammation in HACE patients and HACE animal models is summarized in Table 1.
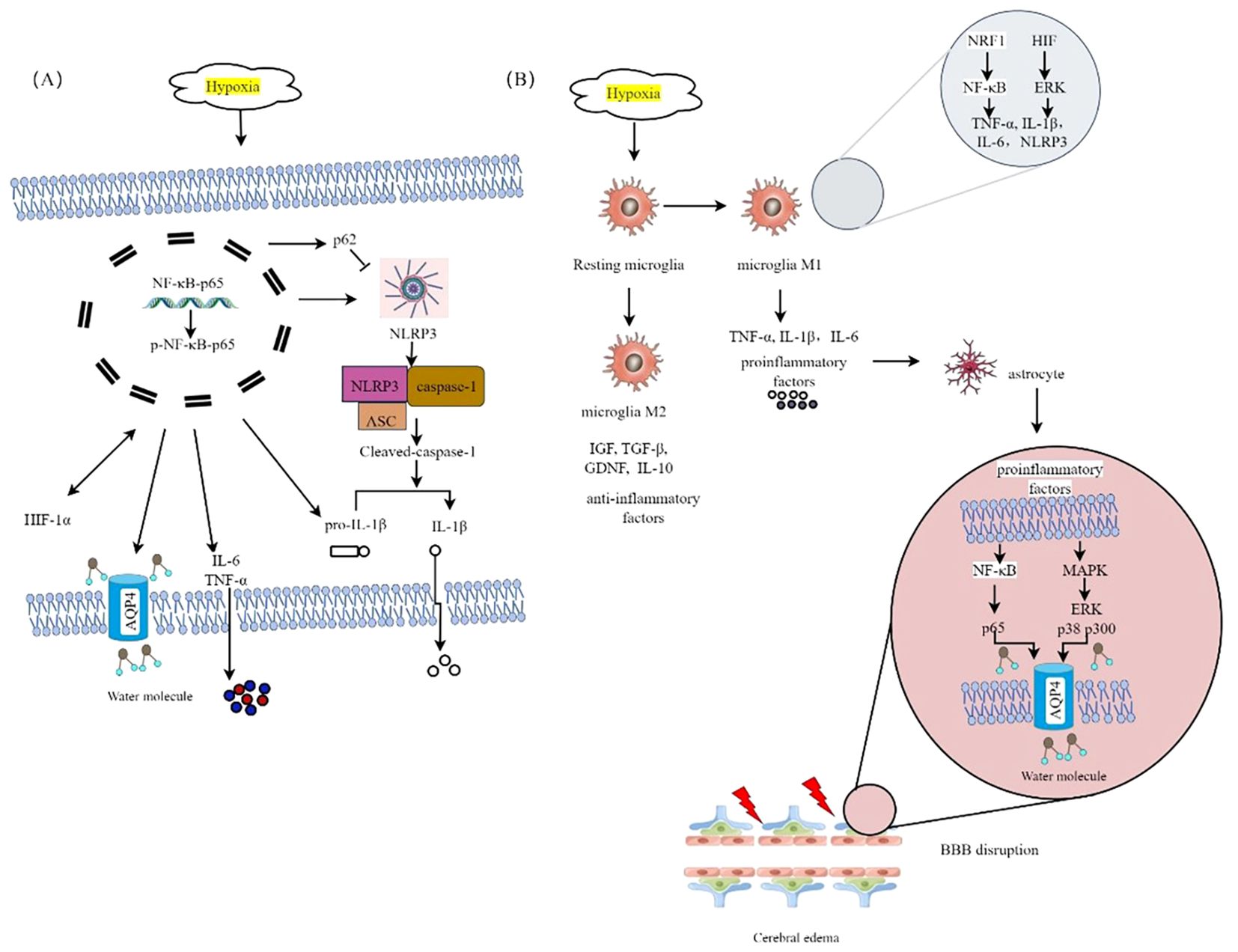
Figure 4. The proposed inflammatory mechanisms hypothesized to underlie HACE. (A) HH triggers the activation of the NF-κB signaling pathway. Hypoxia-induced NF-κB activation. Hypoxia triggers the activation of the NF-κB signaling pathway. Hypoxia-induced NF-κB activation can upregulate AQP4 expression, trigger the release of inflammatory cytokines such as IL-6 and TNF-α, activate the NLRP3 inflammasome, increase p62 expression to inhibit caspase-1 activation, and increase HIF-1α levels, which subsequently further activate NF-κB. (B) HACE is mediated by microglia in a hypoxic environment at high altitude. Hypoxia triggers the activation of microglia, which can exhibit distinct yet overlapping phenotypes. Specifically, the M1 phenotype is characterized by the release of proinflammatory mediators, whereas the M2 phenotype is associated with the secretion of anti-inflammatory factors. Hypoxia can promote M1 polarization of microglia by upregulating the expression of Nrf1, releasing many proinflammatory factors to activate the MAPK and NF-κB signaling pathways in astrocytes, upregulating the expression of AQP4, destroying the integrity of the BBB, and inducing HACE.
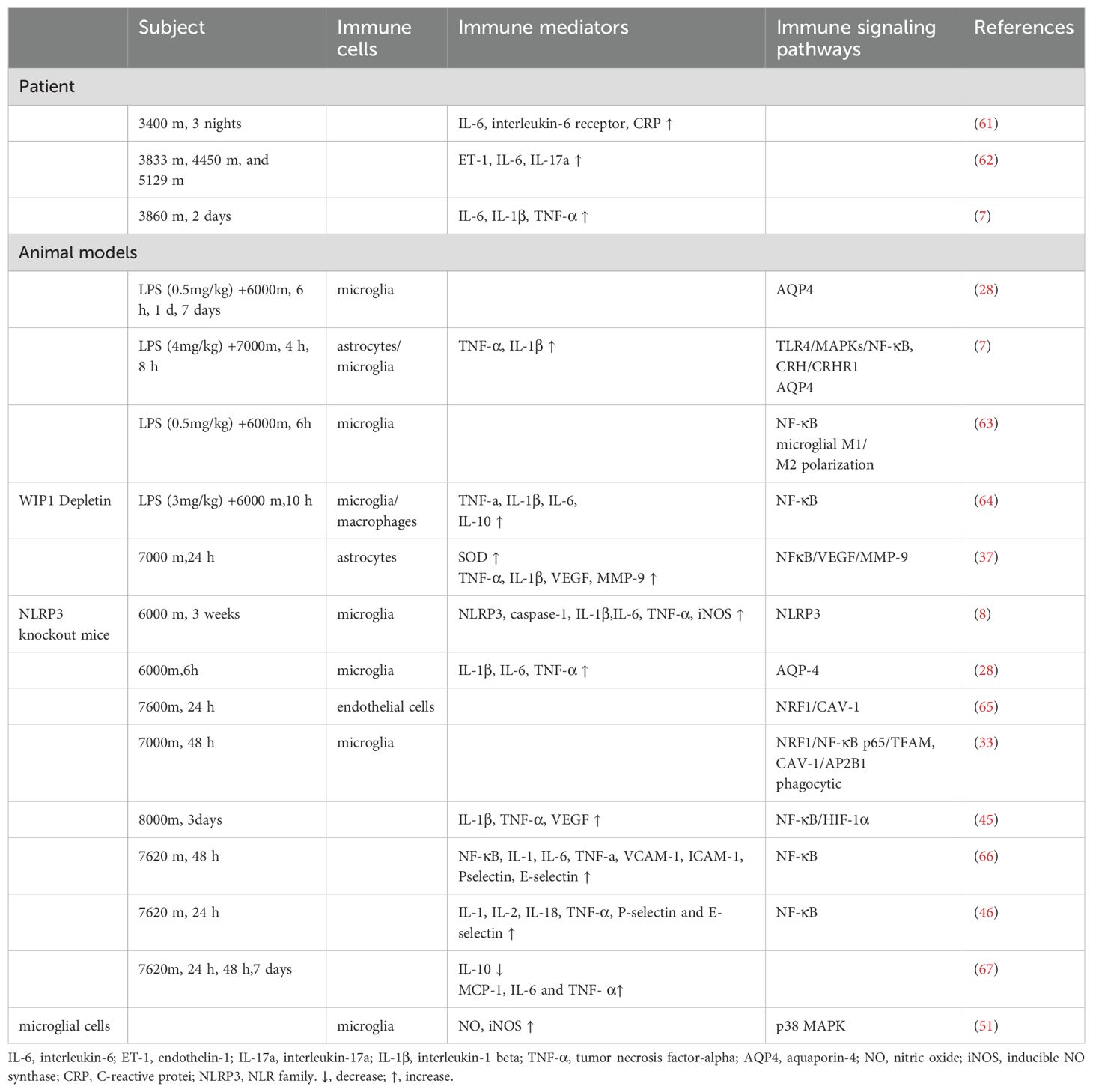
Table 1. Research progress on neuroinflammation in HACE patients and HACE animal models.
The term “oxidative stress” refers to a temporary or prolonged increase in the stable concentration of ROS, which leads to the oxidative alteration of biomolecules, necrosis or apoptosis, and ultimately, cell death (68). Under normal oxygen conditions, the generation and elimination of ROS are typically maintained in a state of dynamic equilibrium. However, under HH conditions, this delicate balance is disrupted, leading to an increase in ROS levels (69). The elevation of ROS levels can lead to the accumulation of reducing equivalents in the mitochondrial electron transport chain, thereby augmenting the generation of superoxide anion radicals within the respiratory chain (70). The brain, which is highly susceptible to oxidative stress, possesses a limited capacity to endure such stress owing to its high rate of oxygen utilization, abundant iron content, substantial levels of unsaturated fatty acids, and deficiency in antioxidant defense mechanisms. Under hypoxic conditions, cerebral transvascular leakage is mediated by free radicals (5), disruption of the structural integrity of the blood−;brain barrier, cellular edema, and the initiation of an inflammatory response within the brain (6, 66, 71, 72).
The redox-sensitive transcriptional regulator nuclear factor E2-related factor-2 (Nrf2) plays a crucial role in conferring resistance against intracellular ROS by activating the transcription of antioxidant genes to modulate cellular redox homeostasis (73). When there is an adequate supply of oxygen, Nrf2 forms a complex with Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm to facilitate its degradation. However, under conditions of hypoxia-induced ROS, Nrf2 dissociates from Keap1, translocates into the nucleus, and binds to musculoaponeurotic fibrosarcoma protein (Maf). This protein subsequently interacts with antioxidant response elements and initiates the transcriptional activation of antioxidation genes, including crucial enzymes such as glutathione reductase (GR), glutathione peroxidase (GPx), superoxide dismutase (SOD) and heme oxygenase-1 (HO-1) (74). The regulatory mechanism of Nrf2 expression in response to hypoxia-induced ROS has been demonstrated (Figure 5).
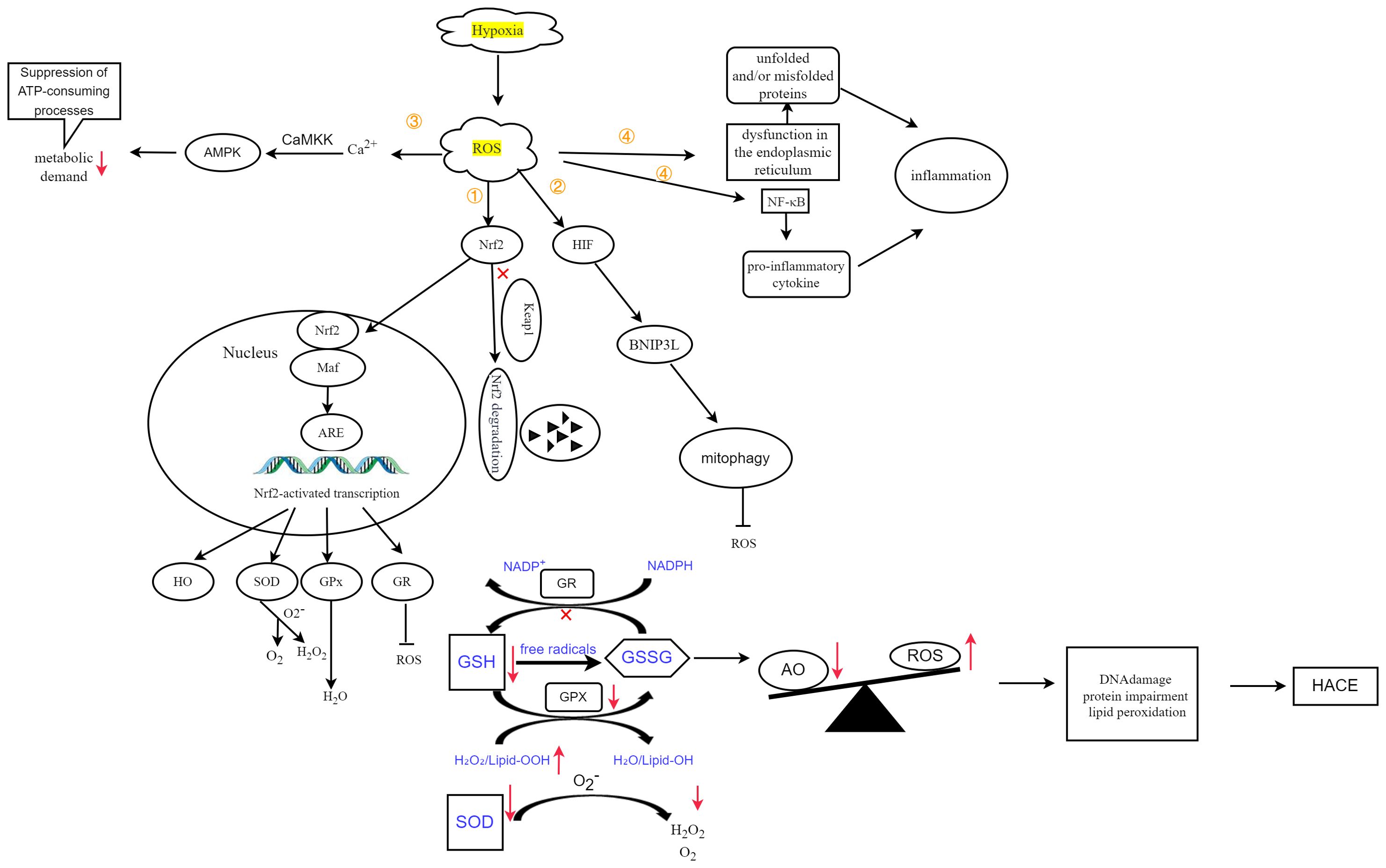
Figure 5. Oxidative stress, defined by an imbalance between antioxidants (AOs) and ROS, ultimately leads to DNA damage, protein dysfunction, and lipid peroxidation, thereby contributing to the development of HACE. Figure adapted from Lee et al. (25) and Burtscher et al. (3). ① Hypoxia-induced ROS inhibit the degradation of Nrf2, thereby promoting its translocation into the nucleus. Following interaction with Maf proteins, this complex subsequently activates the expression of target genes. ② Hypoxia-induced ROS activate HIF, leading to increased expression of BNIP3L and promoting mitophagy. ③ ROS can also induce Ca2+ accumulation, which in turn activates the CaMKK-AMPK pathway, thereby reducing metabolic demand. ④ ROS induce endoplasmic reticulum (ER) stress, resulting in ER dysfunction and the accumulation of unfolded or misfolded proteins, initiating inflammatory responses. Additionally, it increases NF-κB levels and stimulates the expression of proinflammatory cytokines, further triggering inflammatory responses.
In the context of hypoxia associated with high altitude, the dysregulation of antioxidants (AOs) and ROS, as evidenced by numerous studies in both human and animal models, either due to augmented ROS production or diminished AO capacity, collectively culminates in DNA damage, protein impairment, and lipid peroxidation. To counteract the detrimental impact of ROS, superoxide dismutase (SOD) facilitates the dismutation process, transforming superoxide radicals (O2-) into oxygen molecules (O2) and hydrogen peroxide (H2O2), whereas glutathione peroxidase (GPx) converts H2O2 to H2O. Glutathione peroxidase 4 (GPX4) facilitates the reduction and detoxification of phospholipid hydroperoxides (PLOOHs) to protect against oxidative damage (75). Glutathione (GSH), an essential constituent of the cellular antioxidant system, plays a crucial role in effectively regulating the cellular levels of ROS and maintaining redox homeostasis. GSH can directly quench free radicals and ROS while also serving as a cosubstrate for GPX in the conversion of hydrogen peroxide (H2O2) and lipid peroxide (Lipid-OOH) into water and lipid alcohol (Lipid-OH), respectively. Upon reacting with oxidizing substances through either direct or enzymatic pathways, GSH undergoes oxidation to form glutathione disulfide (GSSG), which can potentially be toxic to cells. Excess GSSG can be efficiently reduced back to GSH under the catalytic action of glutathione reductase (GR) (76). The measurement of MDA, a byproduct resulting from the process of lipid peroxidation, can serve as an indicative marker for assessing lipid peroxidation levels within brain tissue. When exposed to HH, brain tissue presented increased levels of ROS and MDA, accompanied by decreased levels of the antioxidants SOD, GSH and GPx (66, 77). Antioxidant pretreatment has the potential to mitigate or even reverse oxidative stress-induced damage (77). Oxidative stress and antioxidants in HACE are summarized in Table 2.
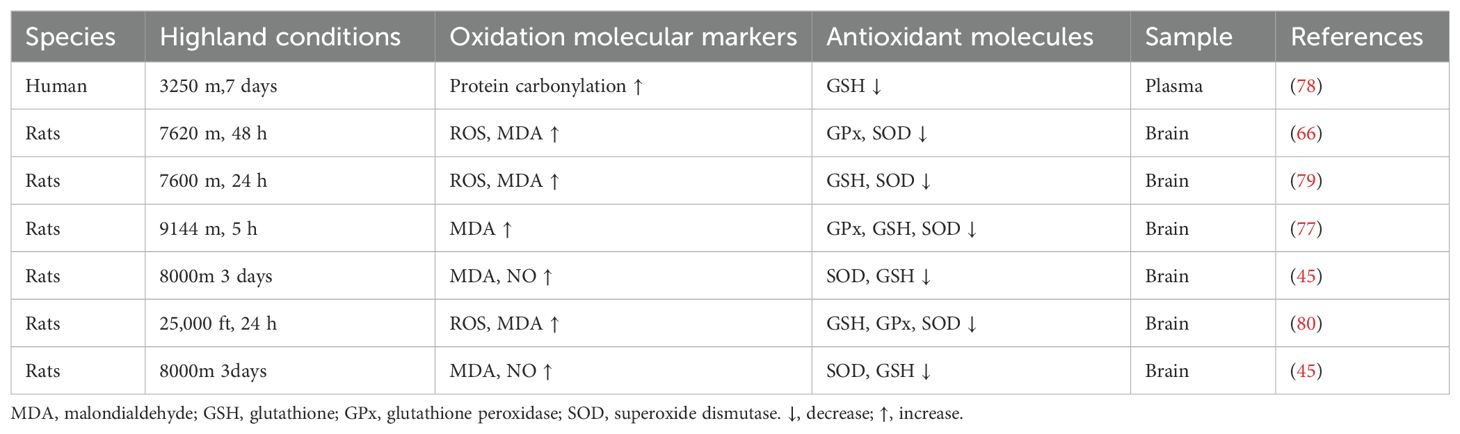
Table 2. Oxidative stress and antioxidants in HACE.
Additionally, the development of HACE pathologies is facilitated by oxidative stress-mediated activation of inflammatory pathways (5). Hypoxia induces oxidative stress, thereby causing dysfunction in the endoplasmic reticulum (81, 82), resulting in the accumulation of unfolded and/or misfolded proteins. This accumulation further disrupts inflammatory pathway regulation and accelerates various associated processes (83). Moreover, acute HH exposure in rat brains may increase oxidative stress and subsequently upregulate NF-κB levels (46, 66), inducing the expression of proinflammatory cytokines (IL-6, IL-1β, and TNF-α), as well as cell adhesion molecules (ICAM-1, VCAM-1, E-selectin, and P-selectin), thus contributing to transvascular leakage (66). The compound Mdivi-1 may mitigate the inflammatory response by modulating the ROS/NF-κB signaling pathway and suppressing AQP4 expression to alleviate cerebral edema (42).
Additionally, under prolonged hypoxic conditions, the increase in ROS leads to an increase in calcium (Ca2+) levels. This subsequently triggers the activation of the CaMKK-AMPK pathway, resulting in a subsequent reduction in metabolic demand (25).
The increase in ROS results in the upregulation of HIF, which intensifies the induction of BNIP3L, leading to increased mitophagy and reduced ROS production (25).
ROS, reactive oxygen species; Nrf2, nuclear factor E2-related factor-2; Keap1, Kelch-like ECH-associated protein 1; Maf, musculoaponeurotic fibrosarcoma protein; ARE, antioxidant response elements; HO, heme oxygenase; SOD, superoxide dismutase; O2, oxygen; H2O2, hydrogen peroxide; GPx, glutathione peroxidase; GR, glutathione reductase; HIF, hypoxia inducible factor; BNIP3L, BCL2-interacting protein 3-like; CaMKK, Ca2+/calmodulin-dependent protein kinase; AMPK, AMP-activated protein kinase. AO, antioxidants; HACE, high-altitude cerebral edema; GSH, glutathione; GSSG, glutathione disulfide; NADPH, nicotinamide adenine dinucleotide phosphate; GR, glutathione reductase; Lipid-OOH, lipid peroxide; Lipid-OH, lipid alcohol; H2O2, hydrogen peroxide; SOD, superoxide dismutase; O2, oxygen.
Mitochondria, the powerhouses of cells, are highly susceptible to oxygen deficiency. Hypoxia impairs mitochondrial components, mass, and dynamics, compromising cellular energy availability, altering mitochondrial function, and even leading to mitochondrial cell death (72). Consequently, mitochondria have emerged as crucial focal points in high-altitude hypoxia research (10).
Mitochondrial dysfunction is a severe adverse consequence of abnormal oxygen metabolism. During hypoxia associated with high altitudes, the mitochondrial electron transport chain (ECT) generates an excessive amount of ROS, leading to detrimental effects on cellular function and integrity. The overproduction of ROS can induce mutations in mitochondrial DNA, initiate lipid peroxidation, and trigger the opening of mitochondrial membrane channels. Consequently, these events result in the collapse of the mitochondrial membrane potential, further exacerbating ROS accumulation and disrupting mitochondrial permeability. As a consequence, impaired mitochondrial function ensues. Figure 6 illustrates the molecular mechanisms linking mitochondrial dysfunction and HACE.
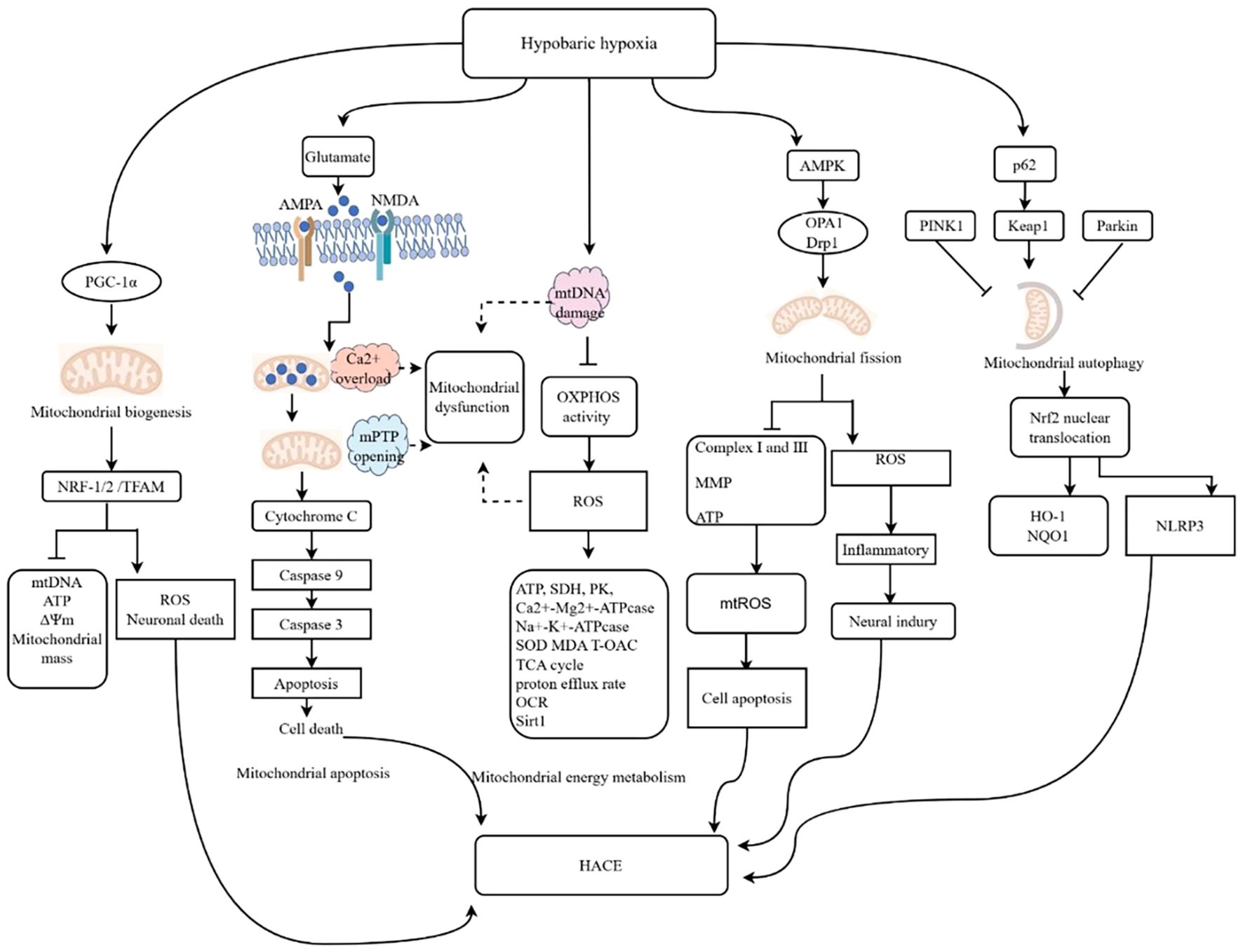
Figure 6. Molecular mechanisms linking mitochondrial dysfunction and HACE. The illustrated diagram delineates the five primary pathways through which mitochondrial dysfunction influences the cellular environment in the context of HH. These pathways include energy metabolism, mitochondrial biogenesis, mitochondrial apoptosis, mitochondrial dynamics, and mitophagy. Additionally, the diagram elucidates the specific molecular mechanisms underlying each pathway. The activation of AMPA and NMDA receptors leads to mitochondrial calcium (Ca2+) overload, resulting in the opening of the mitochondrial permeability transition pore (mPTP), which subsequently activates the caspase cascade and induces cell death. Damage to mitochondrial DNA (mtDNA) results in decreased oxidative phosphorylation (OXPHOS) activity and increased ROS levels. Calcium overload, mPTP opening, mtDNA damage, and oxidative stress are potential contributors to mitochondrial dysfunction following HACE. ↑, promotional effect; ⊥, inhibitive effect. OCR, oxygen consumption rate MCU, mitochondrial calcium uniporter; MPTP, mitochondrial permeability transition pore.
Increasing evidence has provided substantial support for the critical involvement of mitochondrial energy metabolism in HH-induced brain injury. HH leads to the downregulation of specific mitochondrial proteins that play a regulatory role in brain energy production, such as the F1-ATPase beta-subunit and electron transfer flavoprotein alpha-subunit (84). Concurrently, disruption of mitochondrial energy metabolism, including failure of membrane ion pumps and efflux of neuronal K+ (38, 85), occurs.
Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), a pivotal regulator that governs mitochondrial biogenesis, initiates the transcription and replication of mtDNA by activating nuclear respiratory factor (NRF-1/2) and mitochondrial transcription factor A (TFAM). Consequently, this induces the interaction between TFAM and crucial mitochondrial enzymes to fulfill energy requirements in response to environmental changes (86, 87).
Under physiological conditions, the mitochondrial permeability transition pore (mPTP) plays a crucial role in facilitating ATP synthesis through oxidative phosphorylation, thereby maintaining the mitochondrial membrane potential and ensuring both the intracellular and the extracellular ion balance. In the context of brain injury, excessive release of glutamate activates NMDA and AMPA receptors, which leads to a significant increase in the cytoplasmic Ca2+ concentration. To counteract this, mitochondria absorb excess Ca2+, aiming to preserve cytoplasmic Ca2+ homeostasis. However, this absorption results in mitochondrial Ca2+ overload, which subsequently triggers opening of the mPTP. The prolonged opening of the mPTP impairs the mitochondrial respiratory chain and increases the production of ROS. Over time, this condition causes morphological changes in mitochondria, such as swelling, and compromises their functionality. Ultimately, it initiates the endogenous apoptotic pathway by promoting the release of cytochrome C, thereby activating the caspase cascade (87). Sal treatment can mitigate HACE by reducing Ca2+ levels, preventing opening of the mPTP, and inhibiting cellular apoptosis (88). Bax, a proapoptotic protein, increases mitochondrial membrane permeability and facilitates the release of caspase-9 and caspase-3. Conversely, Bcl-2, an antiapoptotic protein, reduces mitochondrial membrane permeability and inhibits mitochondrial apoptosis. The ratio of Bax/Bcl-2 is considered a crucial marker for pro- or antiapoptotic activity, and it increases following exposure to HH (10, 85). Therefore, targeting mitochondrial apoptosis could be a potential therapeutic strategy for high-altitude cerebral hypoxia (10).
The process of mitochondrial dynamics, which is characterized by coordinated cycles of fission and fusion, significantly impacts mitochondrial morphology, biogenesis, and subcellular localization and distribution, as well as cellular bioenergetics, injury, and apoptosis. In high-altitude cerebral hypoxia, electron microscopy revealed the presence of abnormal mitochondria with distinct swelling, unclear or disrupted membranes, and reduced cristae (89–91). Furthermore, when exposed to high-altitude hypoxia, mouse hippocampal neurons exhibit decreased mitochondrial length, increased fragmentation of mitochondria, and decreased membrane potential and oxidative phosphorylation, resulting in increased mitochondrial fission and dysfunction (10). High-altitude exposure induces increased phosphorylation of Dynamin-related protein-1 (Drp1), a key regulator of mitochondrial division, resulting in excessive mitochondrial fission and subsequent fragmentation. Treatment with mdivi-1 effectively inhibits Drp1 phosphorylation, thereby preventing mitochondrial fragmentation and glial cell activation and ultimately alleviating cerebral edema in mice (42). The inhibition of the mitochondrial fusion/fission-related proteins Drp1 and Fis1 plays a crucial role in significantly mitigating cerebral edema after HACE (86). The proteins Drp1 and OPA1 play crucial roles in maintaining mitochondrial dynamics homeostasis and preventing mitochondrial damage in HACE (88).
Mitochondrial autophagy, or mitophagy, is a specific mechanism that selectively eliminates damaged mitochondria. This process is crucial for maintaining mitochondrial homeostasis and cellular survival, particularly in postmitotic neurons. PINK1 and Parkin are the two most representative proteins involved in mitophagy (25).
The prevention of HACE is of paramount importance for military operations and economic progress. A recommended approach to prevent HACE involves a gradual ascent with sufficient time allocated for acclimatization. However, prophylactic medication remains the predominant method. Research has revealed that acute HH-induced oxidative stress can contribute to the development of HACE. The primary therapeutic modalities against oxidative stress include preventing oxidant production, inhibiting redox signaling that leads to inflammation or cell death, and increasing the levels of antioxidant enzymes and their substrates. Antioxidants play crucial roles in combating oxidative stress and are being explored as promising therapeutic strategies to alleviate hypoxia-induced HACE (5). It has been documented that various antioxidants such as flavones naringenin and quercetin (80, 86, 92, 93) 5,6,7,8-trtrahydroxyflavon (5,6,7,8-THF), synthetic derivatives of flavones (94), withania somnifera root extract (95), sea buckthorn seed oil (77), ginkgolide B (96), acetyl-L-carnitine (97, 98), polysaccharide derived from Potentilla anserina L (PAP) (45), alga Spirogyra porticalis (99), and dihydromyricetin (100) have the potential to alleviate cerebral dysfunction induced by HH.
Nrf2 antioxidants can activate the Nrf2 pathway, which plays a crucial role in regulating oxidative stress and neuroinflammation. Substantial advancements have been made in the field of research on Nrf2 activators and drugs associated with antioxidant mechanisms, as they are considered promising pharmaceutical agents for enhancing antioxidant capacity and mitigating the progression of HACE. Gong et al. demonstrated that ganglioside GM1, a neuroprotective sphingolipid, alleviates the severity of HACE by attenuating oxidative stress and the inflammatory response through activation of the PI3K/AKT-Nrf2 pathway in HACE patients (79). Additionally, acetyl-L-carnitine (ALCAR) mitigates Nrf2 degradation and enhances Nrf2 nuclear translocation, thereby facilitating the transcription of Nrf2-regulated antioxidant genes through the TrkA/ERK1/2/Nrf2 pathway, which confers neuroprotective effects against oxidative stress on hippocampal neurons (98). A preliminary, underpowered study suggested potential benefits of antioxidants in preventing acute mountain sickness (101); however, these findings were not supported by a subsequent larger randomized controlled trial (102). The limited body of research currently available restricts the ability to draw definitive conclusions (5, 103).
The involvement of inflammation in the pathogenesis of HACE suggests that targeting inflammatory processes could be a viable strategy for mitigating its development. Animals preconditioned with sphingosine-1phosphate (S1P), a bioactive lipid, demonstrated protection against inflammation, vascular permeability, oxidative damage, and brain tissue injury during acute and subchronic HH (67). Furthermore, the NF-κB signaling pathway plays a crucial regulatory role in the pathogenesis of HACE, making it a promising therapeutic target. Pretreatment with curcumin significantly attenuated hypoxia-induced cerebral transvascular leakage, accompanied by a concurrent decrease in brain NF-κB levels (66). Subsequently, Sarada et al. proposed that curcumin prophylaxis preserves the integrity of tight junction proteins and enhances ion channel expression in the brains of rats exposed to hypoxia through the modulation of oxygen-dependent NF-κB and HIF-1α transcripts, which regulate adaptive responses such as Na+/K+-ATPase and ENaC (46). Furthermore, THC, a major bioactive metabolite of curcumin, may exert a prophylactic effect through inhibition of the NF-κB/VEGF/MMP-9 pathway (37). Studies have documented the potential of Puerarin (40), PhGCs (104), polysaccharides derived from Potentilla anserina L. (PAP) (45), GP-14, a newly identified dammarane-type saponin (63), salidroside (105), Rhodiola crenulate extract (38), epicatechin gallat (60) and exendin-4 (106) for preventing and/or treating HACE by inhibiting the NF-κB signaling pathway. The inhibition of microglial NF-κB signaling effectively suppresses M1-type polarization, offering novel insights into the prevention of HACE through the modulation of microglial polarization (60, 63).
The maintenance of mitochondrial functional stability and activation of the HIF-1 signaling pathway are attractive and feasible targets for the prevention or alleviation of HACE pathology (9, 107). Rhodiola crenulata can regulate HIF-1α-mediated processes related to mitochondrial energy to attenuate neuronal apoptosis, thereby protecting rats from brain damage at high altitudes (85). The dynamin-related protein-1 (Drp1) inhibitor Mdivi-1 directly reduces mitochondrial fission, decreases AQP4 expression, decreases IL-6 and TNF-α secretion, and alleviates cerebral edema in mice. It shows promise as a potential molecule for treating HACE (42). Quercetin inhibits microglial activation to alleviate neurotoxicity through the interplay between the NLRP3 inflammasome and mitophagy (108). Dihydromyricetin is a flavonoid derived from natural sources that attenuates oxidative stress while promoting mitochondrial biogenesis and enhancing synaptic function through activation of the SIRT3/FOXO3 signaling pathway (100). The administration of Rhodiola crenulata and its active ingredient Sal preserves blood−;brain barrier integrity and enhances energy metabolism by activating the AMPK/Sirt1 pathway, inhibiting Drp1 activation, and promoting OPA1 expression (88). The HIF-1α/microRNA 210/ISCU1/2 (COX10) signaling pathway is involved in the regulation of apoptosis and mitochondrial energy metabolism by Rhodiola crenulata (85). Treatment strategies for HACE patients and HACE animal models are summarized in Table 3.
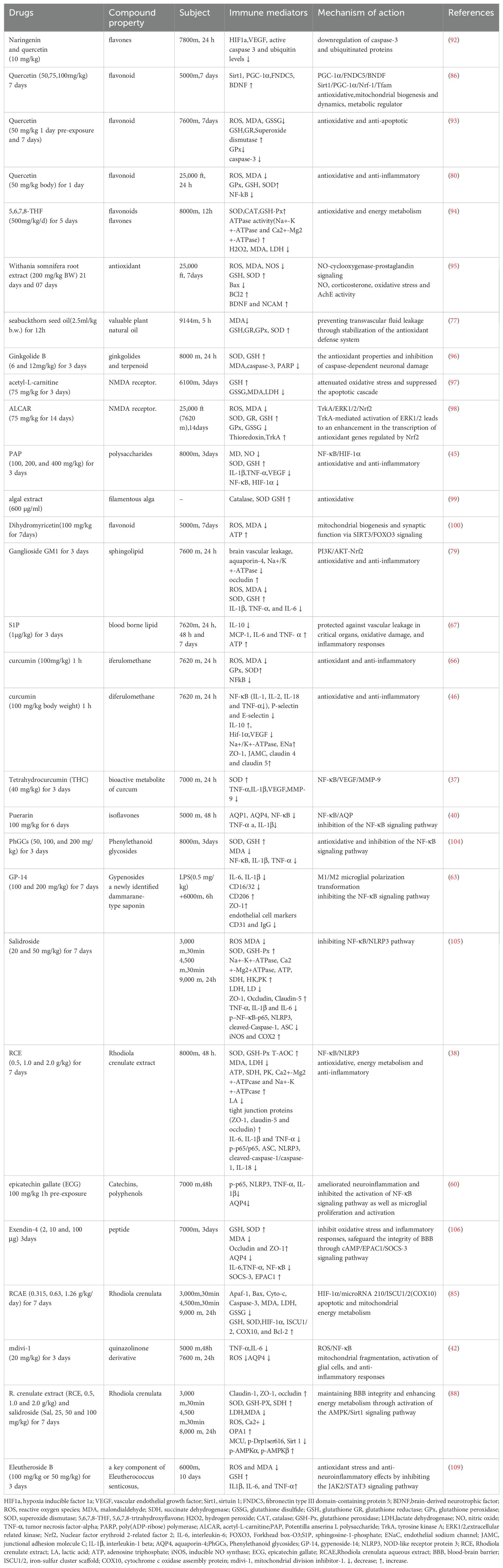
Table 3. Treatment HACE patients and HACE animal models.
In this study, we systematically elucidated the mechanisms of oxygen metabolism in HACE, including hypoxia, oxidative stress, and mitochondrial dysfunction. This comprehensive analysis may enhance our understanding of the disease. Abnormal oxygen metabolism plays a pivotal role in influencing the occurrence and progression of HACE. Therefore, in-depth research into key molecular mechanisms associated with oxygen metabolism could uncover new diagnostic and therapeutic targets for HACE. An integrated approach may be necessary to address the multiple pathogenic mechanisms of HACE. Disordered oxygen metabolism serves as a bridge connecting various pathogenic factors of HACE and is an integral part of these mechanisms. We examined alterations in oxygen metabolism during the pathogenesis of HACE, in which the activation and expression levels of ROS, HIF-1α, Nrf2, NF-κB, NLRP3 and AQP4 are critical factors. Several potential therapeutic agents targeting these pathways have been investigated. These treatment strategies demonstrate feasibility and provide novel insights into the prevention of HACE, showcasing promising clinical application prospects. However, further validation through larger randomized controlled trials and a more extensive body of research is necessary. Numerous factors affect oxygen metabolism, including environmental and individual factors. Further exploration of oxygen metabolism-related treatments is essential to correct metabolic abnormalities in HACE. The central role of oxygen metabolism in HACE provides a novel avenue for discovering new anti-HACE treatments.
ZL: Writing – original draft, Writing – review & editing. JZ: Writing – original draft. XZha: Data curation, Software, Writing – review & editing. QJ: Methodology, Writing – original draft. XZhe: Software, Writing – original draft. LM: Formal analysis, Writing – review & editing. ZD: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This review was supported by funding from the Key Research and Development Program of Gansu (Grant No. 23YFFA0037), the Natural Science Foundation of Gansu Province (Grant No. 24JRRA1092 and 23JRRA0966), the Cuiying Scientific and Technological Innovation Program of The Second Hospital & Clinical Medical School, Lanzhou University (Grant No. CY2023-QN-B15), and the Dingxi City Science and Technology Plan (Grant No. DX2024BY047).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
HACE, high-altitude cerebral edema; HH, hypobaric hypoxia; ATP, adenosine triphosphate; ROS, reactive oxygen species; BNIP3L,BCL2-interacting protein 3-like;
HIF, hypoxia-inducible factor; EPO, erythropoietin; VEGF, vascular endothelial growth factor; BBB, blood−;brain barrier; NLRP3, pyrin domain of NLR family 3; AQP4, aquaporin 4; GR, glutathione reductase; GPx, glutathione peroxidase; SOD, superoxide dismutase; GSH, glutathione.
1. West JB. High-altitude medicine. Lancet Respir Med. (2015) 3:12–3. doi: 10.1016/S2213-2600(14)70238-3
2. Hackett PH, Roach RC. High-altitude illness. N Engl J Med. (2001) 345:107–14. doi: 10.1056/NEJM200107123450206
3. Burtscher M, Hefti U, Hefti JP. High-altitude illnesses: Old stories and new insights into the pathophysiology, treatment and prevention. Sports Med Health Sci. (2021) 3:59–69. doi: 10.1016/j.smhs.2021.04.001
4. Sawicka M, Szymczak RK. A fatal case of high-altitude cerebral oedema on a climbing expedition to Karakoram. Travel Med Infect Dis. (2023) 51:102493. doi: 10.1016/j.tmaid.2022.102493
5. Pena E, El Alam S, Siques P, Brito J. Oxidative stress and diseases associated with high-altitude exposure. Antioxidants (Basel). (2022) 11:267. doi: 10.3390/antiox11020267
6. Lafuente JV, Bermudez G, Camargo-Arce L, Bulnes S. Blood-brain barrier changes in high altitude. CNS Neurol Disord Drug Targets. (2016) 15:1188–97. doi: 10.2174/1871527315666160920123911
7. Song TT, Bi YH, Gao YQ, Huang R, Hao K, Xu G, et al. Systemic pro-inflammatory response facilitates the development of cerebral edema during short hypoxia. J Neuroinflamm. (2016) 13:63. doi: 10.1186/s12974-016-0528-4
8. Zhu X, Liu H, Wang D, Guan R, Zou Y, Li M, et al. NLRP3 deficiency protects against hypobaric hypoxia induced neuroinflammation and cognitive dysfunction. Ecotoxicology Environ Saf. (2023) 255:114828. doi: 10.1016/j.ecoenv.2023.114828
9. Li Y, Li C, Luo T, Yue T, Xiao W, Yang L, et al. Progress in the treatment of high altitude cerebral edema: targeting REDOX homeostasis. J Inflammation Res. (2023) 16:2645–60. doi: 10.2147/JIR.S415695
10. Huan Y, Quan H, Jia B, Hao G, Shi Z, Zhao T, et al. High-altitude cerebral hypoxia promotes mitochondrial dysfunction and apoptosis of mouse neurons. Front Mol Neurosci. (2023) 16:1216947. doi: 10.3389/fnmol.2023.1216947
11. Liu G, Yang C, Wang X, Chen X, Wang Y, Le W. Oxygen metabolism abnormality and Alzheimer’s disease: An update. Redox Biol. (2023) 68:102955. doi: 10.1016/j.redox.2023.102955
12. Zhang J, Zhao T, Zhang P, Shi Z, Da Q, Ma H, et al. Moslosooflavone protects against brain injury induced by hypobaric hypoxic via suppressing oxidative stress, neuroinflammation, energy metabolism disorder, and apoptosis. J Pharm Pharmacol. (2024) 76:44–56. doi: 10.1093/jpp/rgad109
13. Palabıyık E, Sulumer AN, Uguz H, Avcı B, Askın S, Askın H, et al. Assessment of hypolipidemic and anti-inflammatory properties of walnut (Juglans regia) seed coat extract and modulates some metabolic enzymes activity in triton WR-1339-induced hyperlipidemia in rat kidney, liver, and heart. J Mol Recognit. (2023) 36:e3004. doi: 10.1002/jmr.3004
14. Gaur P, Prasad S, Kumar B, Sharma SK, Vats P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int J Biometeorol. (2021) 65:601–15. doi: 10.1007/s00484-020-02037-1
15. Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. (2018) 592:692–702. doi: 10.1002/feb2.2018.592.issue-5
16. Juan CA, Pérez de la Lastra JM, Plou FJ, Pérez-Lebeña E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int J Mol Sci. (2021) 22:4642. doi: 10.3390/ijms22094642
17. Mrakic-Sposta S, Gussoni M, Dellanoce C, Marzorati M, Montorsi M, Rasica L, et al. Effects of acute and sub-acute hypobaric hypoxia on oxidative stress: a field study in the Alps. Eur J Appl Physiol. (2021) 121:297–306. doi: 10.1007/s00421-020-04527-x
18. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. (2009) 29:2570–81. doi: 10.1128/MCB.00166-09
19. Zhao P, He Z, Xi Q, Sun H, Luo Y, Wang J, et al. Variations in HIF-1α Contributed to high altitude hypoxia adaptation via affected oxygen metabolism in tibetan sheep. Anim (Basel). (2021) 12:58. doi: 10.3390/ani12010058
20. Li C, Li X, Xiao J, Liu J, Fan X, Fan F, et al. Genetic changes in the EPAS1 gene between Tibetan and Han ethnic groups and adaptation to the plateau hypoxic environment. PeerJ. (2019) 7:e7943. doi: 10.7717/peerj.7943
21. Kleszka K, Leu T, Quinting T, Jastrow H, Pechlivanis S, Fandrey J. Hypoxia-inducible factor-2α is crucial for proper brain development. Sci Rep. (2020) 10:19146. doi: 10.1038/s41598-020-75838-4
22. Zhou Y, Yan F, Han X, Huang X, Cheng X, Geng Y, et al. NB-3 expression in endothelial cells contributes to the maintenance of blood brain barrier integrity in a mouse high-altitude cerebral edema model. Exp Neurol. (2022) 354:114116. doi: 10.1016/j.expneurol.2022.114116
23. Yan HT, Su GF. Expression and significance of HIF-1 α and VEGF in rats with diabetic retinopathy. Asian Pac J Trop Med. (2014) 7:237–40. doi: 10.1016/S1995-7645(14)60028-6
24. Brennan LA, McGreal-Estrada R, Logan CM, Cvekl A, Menko AS, Kantorow M. BNIP3L/NIX is required for elimination of mitochondria, endoplasmic reticulum and Golgi apparatus during eye lens organelle-free zone formation. Exp Eye Res. (2018) 174:173–84. doi: 10.1016/j.exer.2018.06.003
25. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
26. Al Rihani SB, Batarseh YS, Kaddoumi A. The Blood-Brain Barrier in Health and Disease. Int J Mol Sci. (2023) 24(11). doi: 10.3390/ijms24119261
27. Baneke A. What role does the blood brain barrier play in acute mountain sickness? Travel Med Infect Dis. (2010) 8:257–62. doi: 10.1016/j.tmaid.2010.04.006
28. Zhou Y, Huang X, Zhao T, Qiao M, Zhao X, Zhao M, et al. Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav Immun. (2017) 64:266–75. doi: 10.1016/j.bbi.2017.04.013
29. Huang X, Zhou Y, Zhao T, Han X, Qiao M, Ding X, et al. A method for establishing the high-altitude cerebral edema (HACE) model by acute hypobaric hypoxia in adult mice. J Neurosci Methods. (2015) 245:178–81. doi: 10.1016/j.jneumeth.2015.02.004
30. Sagoo RS, Hutchinson CE, Wright A, Handford C, Parsons H, Sherwood V, et al. Magnetic Resonance investigation into the mechanisms involved in the development of high-altitude cerebral edema. J Cereb Blood Flow Metab. (2017) 37:319–31. doi: 10.1177/0271678X15625350
31. Didier N, Romero IA, Créminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1beta by astrocytes mediates endothelin-1 and tumour necrosis factor-alpha effects on human brain microvascular endothelial cell permeability. J Neurochem. (2003) 86:246–54. doi: 10.1046/j.1471-4159.2003.01829.x
32. Shi Z, Jiang X, Geng Y, Yue X, Gao J, Cheng X, et al. Expression profile of cytokines and chemokines in a mouse high-altitude cerebral edema model. Int J Immunopathology Pharmacol. (2023) 37:3946320231177189. doi: 10.1177/03946320231177189
33. Wang X, Chen G, Wan B, Dong Z, Xue Y, Luo Q, et al. NRF1-mediated microglial activation triggers high-altitude cerebral edema. J Mol Cell Biol. (2022) 14:mjac036. doi: 10.1093/jmcb/mjac036
34. Liu FY, Cai J, Wang C, Ruan W, Guan GP, Pan HZ, et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: a possible role for the regulation of TLR4/MyD88/NF-κB signaling pathway. J Neuroinflamm. (2018) 15:347. doi: 10.1186/s12974-018-1388-x
35. Wang C, Yan M, Jiang H, Wang Q, He S, Chen J, et al. Mechanism of aquaporin 4 (AQP 4) up-regulation in rat cerebral edema under hypobaric hypoxia and the preventative effect of puerarin. Life Sci. (2018) 193:270–81. doi: 10.1016/j.lfs.2017.10.021
36. Chauhan G, Roy K, Kumar G, Kumari P, Alam S, Kishore K, et al. Distinct influence of COX-1 and COX-2 on neuroinflammatory response and associated cognitive deficits during high altitude hypoxia. Neuropharmacology. (2019) 146:138–48. doi: 10.1016/j.neuropharm.2018.11.026
37. Pan Y, Zhang Y, Yuan J, Ma X, Zhao Y, Li Y, et al. Tetrahydrocurcumin mitigates acute hypobaric hypoxia-induced cerebral oedema and inflammation through the NF-κB/VEGF/MMP-9 pathway. Phytother Res. (2020) 34:2963–77. doi: 10.1002/ptr.v34.11
38. Xie N, Fan F, Jiang S, Hou Y, Zhang Y, Cairang N, et al. Rhodiola crenulate alleviates hypobaric hypoxia-induced brain injury via adjusting NF-κB/NLRP3-mediated inflammation. Phytomedicine. (2022) 103:154240. doi: 10.1016/j.phymed.2022.154240
39. Ito H, Yamamoto N, Arima H, Hirate H, Morishima T, Umenishi F, et al. Interleukin-1beta induces the expression of aquaporin-4 through a nuclear factor-kappaB pathway in rat astrocytes. J Neurochem. (2006) 99:107–18. doi: 10.1111/j.1471-4159.2006.04036.x
40. Wang C, Yan M, Jiang H, Wang Q, Guan X, Chen J, et al. Protective effects of puerarin on acute lung and cerebrum injury induced by hypobaric hypoxia via the regulation of aquaporin (AQP) via NF-κB signaling pathway. Int Immunopharmacol. (2016) 40:300–9. doi: 10.1016/j.intimp.2016.09.010
41. Zheng J, Lu J, Mei S, Wu H, Sun Z, Fang Y, et al. Ceria nanoparticles ameliorate white matter injury after intracerebral hemorrhage: microglia-astrocyte involvement in remyelination. J Neuroinflamm. (2021) 18:43. doi: 10.1186/s12974-021-02101-6
42. Lu Y, Chang P, Ding W, Bian J, Wang D, Wang X, et al. Pharmacological inhibition of mitochondrial division attenuates simulated high-altitude exposure-induced cerebral edema in mice: Involvement of inhibition of the NF-κB signaling pathway in glial cells. Eur J Pharmacol. (2022) 929:175137. doi: 10.1016/j.ejphar.2022.175137
43. Fan HH, Zhu LB, Li T, Zhu H, Wang YN, Ren XL, et al. Hyperoside inhibits lipopolysaccharide-induced inflammatory responses in microglial cells via p38 and NFκB pathways. Int Immunopharmacol. (2017) 50:14–21. doi: 10.1016/j.intimp.2017.06.004
44. Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. (2008) 586:4055–9. doi: 10.1113/jphysiol.2008.157669
45. Shi J, Wang J, Zhang J, Li X, Tian X, Wang W, et al. Polysaccharide extracted from Potentilla anserina L ameliorate acute hypobaric hypoxia-induced brain impairment in rats. Phytother Res. (2020) 34:2397–407. doi: 10.1002/ptr.v34.9
46. Sarada SK, Titto M, Himadri P, Saumya S, Vijayalakshmi V. Curcumin prophylaxis mitigates the incidence of hypobaric hypoxia-induced altered ion channels expression and impaired tight junction proteins integrity in rat brain. J Neuroinflamm. (2015) 12:113. doi: 10.1186/s12974-015-0326-4
47. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. (2016) 164:896–910. doi: 10.1016/j.cell.2015.12.057
48. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. (2017) 214:1351–70. doi: 10.1084/jem.20150237
49. Yang L, Zhang Y, Yu X, Li D, Liu N, Xue X, et al. Periventricular microglia polarization and morphological changes accompany NLRP3 inflammasome-mediated neuroinflammation after hypoxic-ischemic white matter damage in premature rats. J Immunol Res. (2023) 2023:5149306. doi: 10.1155/2023/5149306
50. Arias C, Sepulveda P, Castillo RL, Salazar LA. Relationship between hypoxic and immune pathways activation in the progression of neuroinflammation: role of HIF-1alpha and th17 cells. Int J Mol Sci. (2023) 24:3073. doi: 10.3390/ijms24043073
51. Park SY, Lee H, Hur J, Kim SY, Kim H, Park JH, et al. Hypoxia induces nitric oxide production in mouse microglia via p38 mitogen-activated protein kinase pathway. Brain Res Mol Brain Res. (2002) 107:9–16. doi: 10.1016/S0169-328X(02)00421-7
52. Pham K, Parikh K, Heinrich EC. Hypoxia and inflammation: insights from high-altitude physiology. Front Physiol. (2021) 12:676782. doi: 10.3389/fphys.2021.676782
53. Chen PZ, He WJ, Zhu ZR, Xu G, Chen DW, Gao YQ. Adenosine A(2A) receptor involves in neuroinflammation-mediated cognitive decline through activating microglia under acute hypobaric hypoxia. Behav Brain Res. (2018) 347:99–107. doi: 10.1016/j.bbr.2018.02.038
54. Garaschuk O, Verkhratsky A. Physiology of microglia. Methods Mol Biol. (2019) 2034:27–40. doi: 10.1007/978-1-4939-9658-2
55. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. (2009) 4:399–418. doi: 10.1007/s11481-009-9164-4
56. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
57. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. (2007) 8:57–69. doi: 10.1038/nrn2038
58. Ziemka-Nalecz M, Jaworska J, Zalewska T. Insights into the neuroinflammatory responses after neonatal hypoxia-ischemia. J Neuropathology Exp Neurol. (2017) 76:644–54. doi: 10.1093/jnen/nlx046
59. Wang YM, Zhou YK, Han CS, Chen LJ, Zhuang ZM, Yang RL, et al. Stem cells from human exfoliated deciduous teeth alleviate high-altitude cerebral oedema by shifting microglial M1/M2 polarisation. Chin J Dent Res. (2023) 26:153–62. doi: 10.3290/j.cjdr.b4330807
60. Chen G, Cheng K, Niu Y, Zhu L, Wang X. (-)-Epicatechin gallate prevents inflammatory response in hypoxia-activated microglia and cerebral edema by inhibiting NF-κB signaling. Arch Biochem Biophys. (2022) 729:109393. doi: 10.1016/j.abb.2022.109393
61. Hartmann G, Tschöp M, Fischer R, Bidlingmaier C, Riepl R, Tschöp K, et al. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine (2000) 12(3):246–252. doi: 10.1006/cyto.1999.0533
62. Boos CJ, Woods DR, Varias A, Biscocho S, Heseltine P, Mellor AJ. High altitude and acute mountain sickness and changes in circulating endothelin-1, interleukin-6, and interleukin-17a. High Alt Med Biol. (2016) 17:25–31. doi: 10.1089/ham.2015.0098
63. Geng Y, Yang J, Cheng X, Han Y, Yan F, Wang C, et al. A bioactive gypenoside (GP-14) alleviates neuroinflammation and blood brain barrier (BBB) disruption by inhibiting the NF-κB signaling pathway in a mouse high-altitude cerebral edema (HACE) model. Int Immunopharmacol. (2022) 107:108675. doi: 10.1016/j.intimp.2022.108675
64. Li D, Zhang L, Huang X, Liu L, He Y, Xu L, et al. WIP1 phosphatase plays a critical neuroprotective role in brain injury induced by high-altitude hypoxic inflammation. Neurosci Bull. (2017) 33:292–8. doi: 10.1007/s12264-016-0095-9
65. Xue Y, Wang X, Wan B, Wang D, Li M, Cheng K, et al. Caveolin-1 accelerates hypoxia-induced endothelial dysfunction in high-altitude cerebral edema. Cell Commun Signal. (2022) 20:160. doi: 10.1186/s12964-022-00976-3
66. Himadri P, Kumari SS, Chitharanjan M, Dhananjay S. Role of oxidative stress and inflammation in hypoxia-induced cerebral edema: a molecular approach. High Alt Med Biol. (2010) 11:231–44. doi: 10.1089/ham.2009.1057
67. Chawla S, Rahar B, Tulswani R, Saxena S. Preventive preclinical efficacy of intravenously administered sphingosine-1-phosphate (S1P) in strengthening hypoxia adaptive responses to acute and sub-chronic hypobaric hypoxia. Eur J Pharmacol. (2020) 870:172877. doi: 10.1016/j.ejphar.2019.172877
68. Lushchak VI, Storey KB. Oxidative stress concept updated: Definitions, classifications, and regulatory pathways implicated. Excli J. (2021) 20:956–67. doi: 10.17179/excli2021-3596
69. Bakonyi T, Radak Z. High altitude and free radicals. J Sports Sci Med. (2004) 3:64–9. doi: 10.1016/j.comgeo.2010.12.004
70. Schild L, Reinheckel T, Reiser M, Horn TF, Wolf G, Augustin W. Nitric oxide produced in rat liver mitochondria causes oxidative stress and impairment of respiration after transient hypoxia. Faseb j (2003) 17(15):2194–201. doi: 10.1096/fj.02-1170com
71. Li X, Zhang J, Liu G, Wu G, Wang R, Zhang J. High altitude hypoxia and oxidative stress: The new hope brought by free radical scavengers. Life Sci. (2024) 336:122319. doi: 10.1016/j.lfs.2023.122319
72. Mallet RT, Burtscher J, Pialoux V, Pasha Q, Ahmad Y, Millet GP, et al. Molecular mechanisms of high-altitude acclimatization. Int J Mol Sci. (2023) 24:1698. doi: 10.3390/ijms24021698
73. Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K. Regulation of nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules. (2020) 10:320. doi: 10.3390/biom10020320
74. Xin X, Li Y, Liu H. Hesperidin ameliorates hypobaric hypoxia-induced retinal impairment through activation of Nrf2/HO-1 pathway and inhibition of apoptosis. Sci Rep. (2020) 10:19426. doi: 10.1038/s41598-020-76156-5
75. Dang R, Wang M, Li X, Wang H, Liu L, Wu Q, et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J Neuroinflamm. (2022) 19:41. doi: 10.1186/s12974-022-02400-6
76. Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan X, et al. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials. (2021) 277:121110. doi: 10.1016/j.biomaterials.2021.121110
77. Purushothaman J, Suryakumar G, Shukla D, Malhotra AS, Kasiganesan H, Kumar R, et al. Modulatory effects of seabuckthorn (Hippophae rhamnoides L.) in hypobaric hypoxia induced cerebral vascular injury. Brain Res Bull. (2008) 77:246–52. doi: 10.1016/j.brainresbull.2008.08.026
78. Sharma S, Singh Y, Sandhir R, Singh S, Ganju L, Kumar B, et al. Mitochondrial DNA mutations contribute to high altitude pulmonary edema via increased oxidative stress and metabolic reprogramming during hypobaric hypoxia. Biochim Biophys Acta Bioenerg. (2021) 1862:148431. doi: 10.1016/j.bbabio.2021.148431
79. Gong G, Yin L, Yuan L, Sui D, Sun Y, Fu H, et al. Ganglioside GM1 protects against high altitude cerebral edema in rats by suppressing the oxidative stress and inflammatory response via the PI3K/AKT-Nrf2 pathway. Mol Immunol. (2018) 95:91–8. doi: 10.1016/j.molimm.2018.02.001
80. Patir H, Sarada SK, Singh S, Mathew T, Singh B, Bansal A. Quercetin as a prophylactic measure against high altitude cerebral edema. Free Radic Biol Med. (2012) 53:659–68. doi: 10.1016/j.freeradbiomed.2012.06.010
81. Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol. (2015) 308:C415–25. doi: 10.1152/ajpcell.00334.2014
82. Amodio G, Moltedo O, Faraonio R, Remondelli P. Targeting the endoplasmic reticulum unfolded protein response to counteract the oxidative stress-induced endothelial dysfunction. Oxid Med Cell Longev. (2018) 2018:4946289. doi: 10.1155/2018/4946289
83. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. (2006) 86:1133–49. doi: 10.1152/physrev.00015.2006
84. Li J, Qi Y, Liu H, Cui Y, Zhang L, Gong H, et al. Acute high-altitude hypoxic brain injury: Identification of ten differential proteins. Neural Regener Res. (2013) 8:2932–41. doi: 10.3969/j.issn.1673-5374.2013.31.006
85. Wang X, Hou Y, Li Q, Li X, Wang W, Ai X, et al. Rhodiola crenulata attenuates apoptosis and mitochondrial energy metabolism disorder in rats with hypobaric hypoxia-induced brain injury by regulating the HIF-1α/microRNA 210/ISCU1/2(COX10) signaling pathway. J Ethnopharmacol. (2019) 241:111801. doi: 10.1016/j.jep.2019.03.028
86. Liu P, Zou D, Yi L, Chen M, Gao Y, Zhou R, et al. Quercetin ameliorates hypobaric hypoxia-induced memory impairment through mitochondrial and neuron function adaptation via the PGC-1α pathway. Restor Neurol Neurosci. (2015) 33:143–57. doi: 10.3233/RNN-140446
87. Li Y, Liu H, Tian C, An N, Song K, Wei Y, et al. Targeting the multifaceted roles of mitochondria in intracerebral hemorrhage and therapeutic prospects. BioMed Pharmacother. (2022) 148:112749. doi: 10.1016/j.biopha.2022.112749
88. Hou Y, Fan F, Xie N, Zhang Y, Wang X, Meng X. Rhodiola crenulata alleviates hypobaric hypoxia-induced brain injury by maintaining BBB integrity and balancing energy metabolism dysfunction. Phytomedicine. (2024) 128:155529. doi: 10.1016/j.phymed.2024.155529
89. Zhang XY, Zhang XJ, Xv J, Jia W, Pu XY, Wang HY, et al. Crocin attenuates acute hypobaric hypoxia-induced cognitive deficits of rats. Eur J Pharmacol. (2018) 818:300–5. doi: 10.1016/j.ejphar.2017.10.042
90. Chen C, Li B, Chen H, Qin Y, Cheng J, He B, et al. Epigallocatechin-3-gallate ameliorated iron accumulation and apoptosis and promoted neuronal regeneration and memory/cognitive functions in the hippocampus induced by exposure to a chronic high-altitude hypoxia environment. Neurochem Res. (2022) 47:2254–62. doi: 10.1007/s11064-022-03611-2
91. Choudhary R, Kumar M, Katyal A. 12/15-Lipoxygenase debilitates mitochondrial health in intermittent hypobaric hypoxia induced neuronal damage: An in vivo study. Redox Biol. (2022) 49:102228. doi: 10.1016/j.redox.2021.102228
92. Sarkar A, Angeline MS, Anand K, Ambasta RK, Kumar P. Naringenin and quercetin reverse the effect of hypobaric hypoxia and elicit neuroprotective response in the murine model. Brain Res. (2012) 1481:59–70. doi: 10.1016/j.brainres.2012.08.036
93. Prasad J, Baitharu I, Sharma AK, Dutta R, Prasad D, Singh SB. Quercetin reverses hypobaric hypoxia-induced hippocampal neurodegeneration and improves memory function in the rat. High Alt Med Biol. (2013) 14:383–94. doi: 10.1089/ham.2013.1014
94. Jing L, Shao J, Zhao T, He L, Ma H. Protective effect of 5,6,7,8-trtrahydroxyflavone against acute hypobaric hypoxia induced-oxidative stress in mice. Pak J Pharm Sci. (2021) 34:513–9. doi: 10.36721/PJPS.2021.34.2.REG.513-519.1
95. Baitharu I, Jain V, Deep SN, Hota KB, Hota SK, Prasad D, et al. Withania somnifera root extract ameliorates hypobaric hypoxia induced memory impairment in rats. J Ethnopharmacol. (2013) 145:431–41. doi: 10.1016/j.jep.2012.10.063
96. Botao Y, Ma J, Xiao W, Xiang Q, Fan K, Hou J, et al. Protective effect of ginkgolide B on high altitude cerebral edema of rats. High Alt Med Biol. (2013) 14:61–4. doi: 10.1089/ham.2012.1080
97. Barhwal K, Singh SB, Hota SK, Jayalakshmi K, Ilavazhagan G. Acetyl-L-carnitine ameliorates hypobaric hypoxic impairment and spatial memory deficits in rats. Eur J Pharmacol. (2007) 570:97–107. doi: 10.1016/j.ejphar.2007.05.063
98. Barhwal K, Hota SK, Jain V, Prasad D, Singh SB, Ilavazhagan G. Acetyl-l-carnitine (ALCAR) prevents hypobaric hypoxia-induced spatial memory impairment through extracellular related kinase-mediated nuclear factor erythroid 2-related factor 2 phosphorylation. Neuroscience. (2009) 161:501–14. doi: 10.1016/j.neuroscience.2009.02.086
99. Kumar J, Khan S, Mandotra SK, Dhar P, Tayade AB, Verma S, et al. Nutraceutical profile and evidence of alleviation of oxidative stress by Spirogyra porticalis (Muell.) Cleve inhabiting the high altitude Trans-Himalayan Region. Sci Rep. (2019) 9:4091. doi: 10.1038/s41598-018-35595-x
100. Liu P, Zou D, Chen K, Zhou Q, Gao Y, Huang Y, et al. Dihydromyricetin improves hypobaric hypoxia-induced memory impairment via modulation of SIRT3 signaling. Mol Neurobiol. (2016) 53(10):7200–12. doi: 10.1007/s12035-015-9627-y
101. Bailey DM, Davies B. Acute mountain sickness; prophylactic benefits of antioxidant vitamin supplementation at high altitude. High Alt Med Biol. (2001) 2:21–9. doi: 10.1089/152702901750067882
102. Baillie JK, Thompson AA, Irving JB, Bates MG, Sutherland AI, Macnee W, et al. Oral antioxidant supplementation does not prevent acute mountain sickness: double blind, randomized placebo-controlled trial. Qjm. (2009) 102:341–8. doi: 10.1093/qjmed/hcp026
103. Gatterer H, Villafuerte FC, Ulrich S, Bhandari SS, Keyes LE, Burtscher M. Altitude illnesses. Nat Rev Dis Primers. (2024) 10(1):43. doi: 10.1038/s41572-024-00526-w
104. Luan F, Li M, Han K, Ma Q, Wang J, Qiu Y, et al. Phenylethanoid glycosides of Phlomis younghusbandii Mukerjee ameliorate acute hypobaric hypoxia-induced brain impairment in rats. Mol Immunol. (2019) 108:81–8. doi: 10.1016/j.molimm.2019.02.002
105. Jiang S, Fan F, Yang L, Chen K, Sun Z, Zhang Y, et al. Salidroside attenuates high altitude hypobaric hypoxia-induced brain injury in mice via inhibiting NF-κB/NLRP3 pathway. Eur J Pharmacol. (2022) 925:175015. doi: 10.1016/j.ejphar.2022.175015
106. Sun ZL, Jiang XF, Cheng YC, Liu YF, Yang K, Zhu SL, et al. Exendin-4 inhibits high-altitude cerebral edema by protecting against neurobiological dysfunction. Neural Regener Res. (2018) 13:653–63. doi: 10.4103/1673-5374.230291
107. Aboouf MA, Thiersch M, Soliz J, Gassmann M, Schneider Gasser EM. The brain at high altitude: from molecular signaling to cognitive performance. Int J Mol Sci. (2023) 24:10179. doi: 10.3390/ijms241210179
108. Han X, Xu T, Fang Q, Zhang H, Yue L, Hu G, et al. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. (2021) 44:102010. doi: 10.1016/j.redox.2021.102010
Keywords: oxygen metabolism, hypobaric hypoxia, oxidative stress, mitochondrial dysfunction, high-altitude cerebral edema
Citation: Li Z, Zhang J, Zhang X, Jin Q, Zheng X, Mo L and Da Z (2025) Oxygen metabolism abnormalities and high-altitude cerebral edema. Front. Immunol. 16:1555910. doi: 10.3389/fimmu.2025.1555910
Received: 05 January 2025; Accepted: 26 February 2025;
Published: 19 March 2025.
Edited by:
Ma. Cecilia Opazo, Universidad de Las Américas, ChileReviewed by:
Xiaokang Wang, Guangdong Medical University, ChinaCopyright © 2025 Li, Zhang, Zhang, Jin, Zheng, Mo and Da. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zejiao Da, ZGF6ZWppYW9AMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.