
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 24 March 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1555415
 Paola Faggioli1
Paola Faggioli1 Marianna Galeazzi1*Carlotta Ferrari1Francesca Capelli1Chiara Marchesi1Lucia Marchionni1
Marianna Galeazzi1*Carlotta Ferrari1Francesca Capelli1Chiara Marchesi1Lucia Marchionni1 Laura Castelnovo1Antonio Tamburello1Eugenio Capparelli1Cristina Campidelli2Antonino Mazzone1*
Laura Castelnovo1Antonio Tamburello1Eugenio Capparelli1Cristina Campidelli2Antonino Mazzone1*Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome, often referred to as macrophage activation syndrome (MAS) in the context of autoimmune disease-induced forms. We report the case of a 41-year-old woman with a previous diagnosis of Crohn’s disease complicated by dermatomyositis, who was admitted in our hospital for the acute onset of fever, pancytopenia, and disseminated intravascular coagulation (DIC). The laboratory findings documented hyperferritinemia, hypertransaminasemia, increased lactate-dehydrogenase (LDH), hypertriglyceridemia, and elevation of inflammatory indices, along with complement consumption. MAS was confirmed by examination of the bone marrow. Consequently, the patient was treated with high doses of glucocorticoids, subcutaneous anakinra, and intravenous immunoglobulin (IVIg). Due to the persistence of signs of thrombotic microangiopathy, we started therapy with eculizumab which stabilized the patient without improvement, so we added emapalumab, resulting in clinical improvement and normalization of blood tests.
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome characterized by dysregulated immune activation. When HLH arises in the context of a well-defined autoimmune condition, it is more specifically referred to as macrophage activation syndrome (MAS). Both HLH and MAS are life-threatening systemic hyperinflammatory syndromes characterized by fever, elevated markers of systemic inflammation, pancytopenia, hyperferritinemia, disseminated intravascular coagulopathy, liver dysfunction, splenomegaly, and central nervous system (CNS) dysfunction (1).
Certain estimates of the prevalence and distribution of HLH within the population are difficult to establish. So far, several studies have been published on HLH incidence. Meeths M et al. published in 2015 the results from a Swedish national registry that collected data with regard to primary HLH from 1987 to 2006. They demonstrated a yearly incidence of roughly 1.5 per million (2). It is the most comprehensive data with regard to primary HLH. Interestingly, the incidence rate seems to have increased in the last years, and a recent study published by West J. et al. shows an incidence of 14.6 per million in children below 1 year, 2.2 per million in older patients (≥75 years), and a lowest incidence of 0.8 per million in those aged 15–44 years (3, 4).
There are several criteria to identify patients with HLH or MAS (5, 6). When HLH and MAS are suspected, it is essential to identify the most likely contributors, such as genetic causes, predisposing conditions, and acute triggers. HLH and MAS therapy is based on immunomodulatory drugs and on the treatment of contributing factors.
Here we describe the case of a 41-year-old woman suffering from Crohn’s disease complicated by dermatomyositis. The patient was diagnosed with Crohn’s disease, predominantly involving the ileum at the age of 16, and she was initially treated with mesalazine and prednison. At the age of 20, mesalazine was discontinued following a concurrent diagnosis of dermatomyositis, which was subsequently managed with azathioprine, cyclosporine, and methotrexate. However, the new treatments proved ineffective, and prednisone was reintroduced. At the age of 41, in September and October 2023, the patient was prescribed her first biological disease-modifying antirheumatic drug (DMARD), infliximab, at a dose of 5 mg/kg. The 300-mg drug was administered twice, resulting in the successful management of Crohn’s disease.
In December 2023, she was hospitalized for 10 days long due to a persistent fever despite the use of azithromycin and cefixime. No other symptoms were present.
On examination, her temperature was 38.6°C, heart rate was 110 beats/minute with regular rhythm, blood pressure was 110/70 mmHg, and oxygen saturation was 96%. The Glasgow Coma Scale score was 15. The cardiovascular and pulmonary examinations were normal. We found hepatomegaly at liver palpation and ecchymosis on the limbs. The blood tests documented the following results: white blood cell count of 0.9 × 103/μL (normal range, 4–10 × 103/μL), hemoglobin level of 79 g/L (normal range, 120–160 g/L), and platelet count of 39 × 103/μL (normal range, 150–440 × 103/μL). The sodium level was 129 mmol/l (normal range, 135–145 mmol/L). The other serum electrolyte and renal function tests had results in the normal range. Proteinuria was absent. The liver function tests showed an aspartate aminotransferase level of 476 U/L (normal range, ≤40 U/L), an alanine aminotransferase level of 175 U/L (normal range, ≤45 U/L), a lactate dehydrogenase level of 1,800 U/L (normal range, 135–214 U/L), a ferritin level of 9,308 ng/mL (normal range, 10–150 ng/mL), a D-dimer level of 27,662 ng/mL (normal range, ≤270 ng/mL), a fibrinogen level of 39 mg/dL (normal range, 180–400 mg/dL), a serum level of triglycerides of 419 mg/dL (normal range, ≤200 mg/dL), a C-reactive protein level of 6.30 mg/dL (normal range, ≤0.5 mg/dL), a complement component C3 level of 53 mg/dL (normal range, 90–180 mg/dL), and a complement component C4 level of 8 mg/dL (normal range, 10–40 mg/dL).
The blood and urine cultures were negative. The chest X-ray showed normal findings. The test for detection of SARS-CoV2 was negative.
To investigate possible septic foci, the patient underwent a computer tomography (CT) of the chest and abdomen. The CT showed ground glass opacity in the lungs (in particular, bilaterally in the lung bases) and inhomogeneity of the rectus abdominal muscles.
Furthermore, we documented possibly recent Epstein–Barr virus (EBV) and Mycoplasma pneumoniae infections due to the presence of anti-VCA IgG, EBNA antibodies IgG, and anti-Mycoplasma pneumoniae IgM. EBV DNA was undetectable. The patient received antibiotic therapy with cephalosporin (piperacillin/tazobactam) and teicoplanin as prescibed by infectivologists. Initially, the clinical presentation could subtend a sepsis. A rheumatology evaluation on the admission day concluded for MAS (laboratory tests met the criteria: fever, elevated level of C-reactive protein, elevated level of lactate dehydrogenase, pancytopenia, hyperferritinemia, elevated level of aspartate aminotransferase and alanine aminotransferase, and elevated serum level of triglycerides), and disseminated intravascular coagulation in patients with suspected infections that could contribute to the MAS relapse (5, 6). MAS was confirmed by examination of the bone marrow (Figure 1).
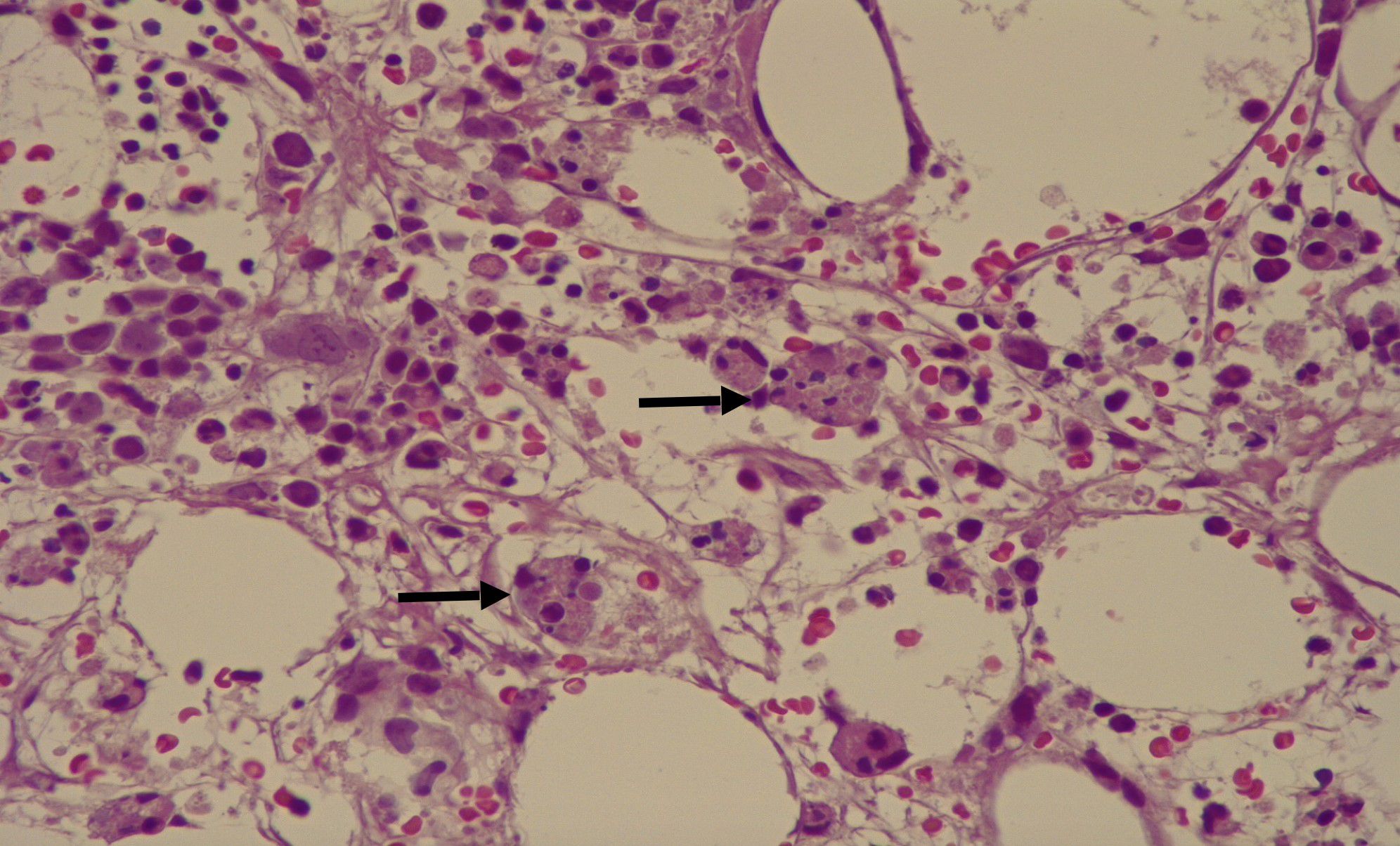
Figure 1. Bone marrow biopsy with macrophages (CD68+) containing numerous red blood cells in their cytoplasm (arrow).
An initial treatment with a high dose of glucocorticoids (methylprednisolone, 2 mg/kg daily), subcutaneous anti-IL1R antagonist anakinra (1,200 mg/day), and intravenous immunoglobulins (IVIg) at a dosage of 1 g/kg/day for two consecutive days was established.
For fever relapse, the antibiotic therapy was changed: piperacillin/tazobactam was discontinued, and meropenem, levofloxacin, and caspofungin were added as prescribed by infectivologists.
Due to the persistence of fever and signs of thrombotic microangiopathy (TMA), in particular, hypocomplementemia, thrombocytopenia, anemia, red blood cell fragmentation (schistocytes) on peripheral blood smear, and fever, an add-on therapy with eculizumab was prescribed. We reached stabilization but no improvement so that the patient was consequently switched to emapalumab. Emapalumab, an anti-IFNγ monoclonal antibody, required the following dosages: 6 mg/kg on the first day, 3 mg/kg every 3 days until the 15th day, and 3 mg/kg twice a week until the 28th day. The therapy should be stopped at disease remission but not before three administrations have taken place. Emapalumab was subsequently discontinued due to clinical improvement and normalization of LDH, complement levels, and fibrinogen.
In addition, the clinical course was complicated by Saprochaete capitata sepsis that required treatment with amphotericin b and voriconazole. Moreover, the patient also experienced an episode of severe rectal bleeding that needed surgery intervention with hemicolectomy and subsequent ileostomy. In Figures 2–4, a flow chart is reported in order to summarize the clinical course.
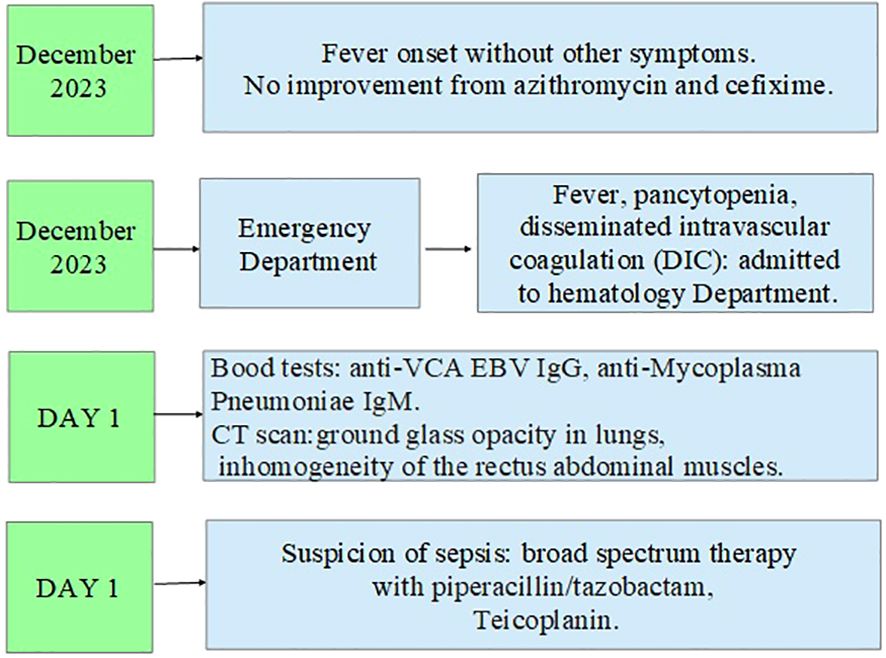
Figure 2. First part of the clinical course.
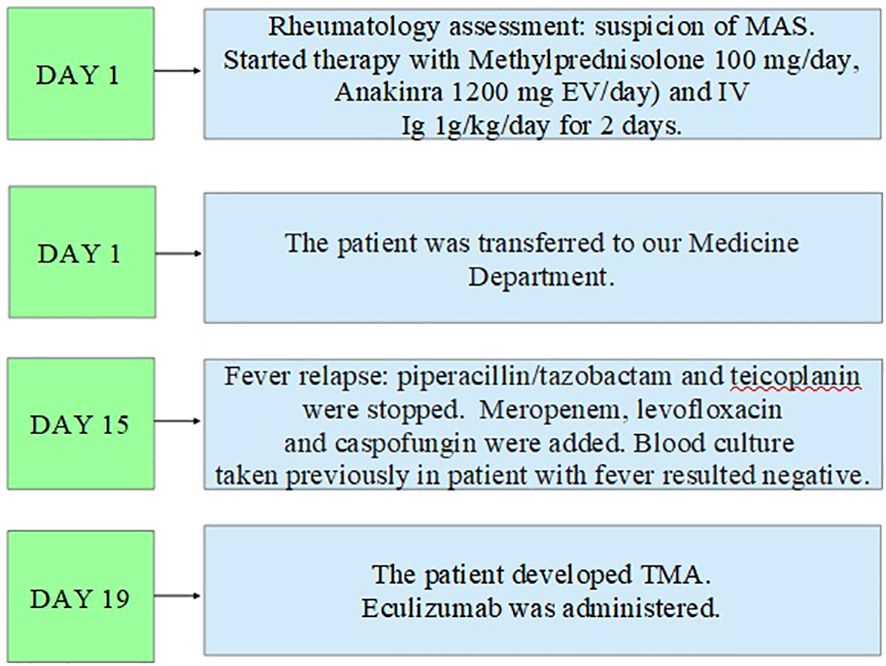
Figure 3. Second part of the clinical course.

Figure 4. Third part of the clinical course.
At discharge, the patient was asymptomatic, the laboratory tests were normal as reported in Table 1, and the therapy included prednisone at a dosage of 50 mg per day, with plans for gradual tapering.

Table 1. Laboratory tests at admission, after 14 days of treatment with anakinra and steroids, at development of TMA (day 19), at the end of eculizumab treatment, at initiation of emapalumab (day 47), and at discharge.
The patient felt a sense of relief at discharge. During hospitalization, she experienced moments of deep concern for her situation.
At the last follow-up in September 2024, the patient was maintained on prednisone at a reduced dose of 10 mg per day, which corresponds to the typical maintenance dose for CD. The patient is currently in clinical remission and is being evaluated for potential stoma reversal surgery. The laboratory tests indicated significant improvements, with CRP and erythrocyte sedimentation rate (ESR) levels within normal limits and complement components and hematological parameters restituted to normal values.
At first, we started to treat MAS using glucocorticoids (methylprednisolone at 100 mg per day), anakinra (1,200 mg per day), and IVIg (1 g/kg for 2 days) as defined by the guidelines (1). We also treated the underlying disease with cephalosporin (piperacillin/tazobactam), teicoplanin, and antifungals as recommended (7).
Unfortunately, the patient developed worsening TMA. The laboratory tests showed lower levels of hemoglobin, platelet count, and fibrinogen and a higher level of lactate dehydrogenase and D-dimer. Hypocomplementemia persisted as concurrent clinical and serological findings in the context of MAS.
As regard to the latter, in 2011, Gorelik M et al. described hypocomplementemia associated with MAS in two patients with systemic juvenile idiopathic arthritis (sJIA) and in one patient with adult-onset Still’s disease (AOSD) (8). More recently, some authors have suggested that the control of the complement pathway may lead to the successful control of complement-mediated TMA in the context of MAS (9, 10).
The patient did not present kidney and central nervous system involvement as TMA complication, but she developed rectal bleeding that needed surgery. The surgeons performed hemicolectomy and ileostomy.
To investigate other potential causes of TMA, we measured the activity of ADAMTS13, a von Willebrand factor-cleaving protease that cuts von Willebrand factor multimers secreted from vascular endothelial cells. ADAMTS13 activity was 93% (normal range 61–131). The result could rule out a diagnosis of thrombotic thrombocytopenic purpura.
The association between TMA and MAS has rarely been assessed, but it is reported with increased frequency (9–11). The coexistence of TMA and MAS has been outlined as a complication of hematopoietic stem cell transplantation (12, 13), renal transplantation (14), autoimmune disease in children (9), and complement-mediated TMA (15) and, recently, associated to systemic lupus erythematosus (SLE) (11).
In the study “Thrombotic Microangiopathy Associated with Macrophage Activation Syndrome: A Multinational Study of 23 Patients”, the authors reported the therapies used for MAS (cyclosporine, anakinra, etoposide, and intravenous immunoglobulins) and for TMA (plasma exchange, eculizumab, rituximab, cyclophosphamide, and mycophenolate) (9).
In 2023, Yamaguchi M et al. published a case report of TMA concomitant with MAS in systemic lupus erythematosus, refractory to conventional treatment, and successfully treated with eculizumab. In the two abovementioned studies, emapalumab was not used (11).
In 2020, Gloude NJ et al. published data regarding 16 patients with HLH and TMA treated with emapalumab and eculizumab. They hypothesized that high levels of interferon gamma in HLH might contribute directly to endothelial damage or injure the endothelium through complement system activation (10). Considering that also in systemic juvenile idiopathic arthritis-associated MAS a prominent pathogenetic role of interferon gamma has been demonstrated, the same authors suggested that the pathophysiology of TMA in MAS is similar to the one hypothesized for the other forms of HLH (10). Our case report could confirm their hypothesis.
The patient underwent eculizumab treatment at 900 mg/week for 4 weeks as suggested by the guidelines (16). The patient also received blood product or component transfusion. After 4 weeks, we obtained stabilization without improvement. In particular, the blood tests showed a low level of fibrinogen and a higher level of D-dimer, while the white blood cell count and hemoglobin levels rose without reaching normal values.
In the light of MAS relapse, the patient was treated with emapalumab, with clinical improvement and normalization of LDH, complement, and fibrinogen. Emapalumab is the first targeted therapy approved by the US FDA for primary pediatric HLH (17). Emapalumab is a monoclonal antibody that binds and neutralizes interferon gamma. It binds free and receptor-bound interferon gamma and neutralizes its biologic activity, reducing hyperinflammation.
The choice of emapalumab rather than etoposide or JAK2 inhibitors stems from several reasons. First of all, as mentioned earlier, emapalumab is the first targeted therapy approved by the US FDA for primary pediatric HLH. Secondly, the absence of EBV DNA and underlying hematological malignancies allowed us to rule out etoposide (1). Thirdly, with regard to the use of JAK2 inhibitors, there are a few reports in literature of patients receiving JAKi treatment (18).
Furthermore, it has been demonstrated that patients affected by TMA and HLH who received the complement blocker eculizumab in addition to the interferon gamma inhibitor emapalumab had complete resolution of TMA and HLH (10).
Ours is one of the first reports of the use of emapalumab and eculizumab in adult patients with MAS and TMA: the drug emapalumab has proven to be effective in adults for a pathology burdened with a very high mortality rate.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
PF: Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. MG: Writing – original draft. CF: Investigation, Methodology, Supervision, Writing – original draft. FC: Methodology, Supervision, Writing – original draft. CM: Investigation, Methodology, Supervision, Writing – original draft. LM: Investigation, Supervision, Writing – original draft. LC: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft. AT: Investigation, Software, Writing – original draft. EC: Writing – review & editing. CC: Investigation, Writing – original draft. AM: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. The funding for the publication came from the hospital (ASST Ovest Milanese, Legnano, MI), Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Shakoory B, Geerlinks A, Wilejto M, Kernan K, Hines M, Romano M, et al. The 2022 EULAR/ACR points to consider at the early stages of diagnosis and management of suspected haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS). Ann Rheum Dis. (2023) 82:1271–85. doi: 10.1136/ard-2023-224123
2. Konkol S, Rai M. Lymphohistiocytosis. StatPearls. Treasure Island (FL: StatPearls Publishing (2025).
3. West J, Stilwell P, Liu H, Ban L, Bythell M, Card TR, et al. Temporal trends in the incidence of hemophagocytic lymphohistiocytosis: A nationwide cohort study from England 2003-2018. Hemasphere. (2022) 6:e797. doi: 10.1097/HS9.0000000000000797
4. West J, Card TR, Bishton MJ, et al. Incidence and survival of haemophagocytic lymphohistiocytosis: A population-based cohort study from England. J Intern Med. (2022) 291:493–504. doi: 10.1111/joim.13432
5. Schulert GS, Grom AA. Macrophage activation syndrome and cytokine-directed therapies. Best Pract Res Clin Rheumatol. (2014) 28:277–92. doi: 10.1016/j.berh.2014.03.002
6. Tada Y, Inokuchi S, Maruyama A, Suematsu R, Sakai M, Sadanaga Y, et al. Are the 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome applicable to patients with adult-onset Still’s disease? Rheumatol Int. (2019) 39:97–104. doi: 10.1007/s00296-018-4114-1
7. Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. (2020) 2:e358–67. doi: 10.1016/S2665-9913(20)30096-5
8. Gorelik M, Torok KS, Kietz DA, Hirsch R. Hypocomplementemia associated with macrophage activation syndrome in systemic juvenile idiopathic arthritis and adult onset still’s disease: 3 cases. J Rheumatol. (2011) 38:396–7. doi: 10.3899/jrheum.100833
9. Minoia F, Tibaldi J, Muratore V, Gallizzi R, Bracaglia C, Arduini A, et al. Thrombotic microangiopathy associated with macrophage activation syndrome: A multinational study of 23 patients. J Pediatr. (2021) 235:196–202. doi: 10.1016/j.jpeds.2021.04.004
10. Gloude NJ, Dandoy CE, Davies SM, Myers KC, Jordan MB, Marsh RA, et al. Thinking beyond HLH: clinical features of patients with concurrent presentation of hemophagocytic lymphohistiocytosis and thrombotic microangiopathy. J Clin Immunol. (2020) 40:699–707. doi: 10.1007/s10875-020-00789-4
11. Yamaguchi M, Mizuno M, Kitamura F, Iwagaitsu S, Nobata H, Kinashi H, et al. Case report: Thrombotic microangiopathy concomitant with macrophage activation syndrome in systemic lupus erythematosus refractory to conventional treatment successfully treated with eculizumab. Front Med (Lausanne). (2023) 9:1097528. doi: 10.3389/fmed.2022.1097528
12. Lerkvaleekul B, Vilaiyuk S. Macrophage activation syndrome: early diagnosis is key. Open Access Rheumatol. (2018) 10:117–28. doi: 10.2147/OARRR.S151013
13. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. (2019) 10:119. doi: 10.3389/fimmu.2019.00119
14. Jodele S, Dandoy CE, Myers KC, El-Bietar J, Nelson A, Wallace G, et al. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transfus Apher Sci. (2016) 54:181–90. doi: 10.1016/j.transci.2016.04.007
15. Chiurchiu C, Ruggenenti P, Remuzzi G. Thrombotic microangiopathy in renal transplantation. Ann Transplant. (2002) 7:28–33.
16. Leisring J, Brodsky SV, Parikh SV. Clinical evaluation and management of thrombotic microangiopathy. Arthritis Rheumatol. (2024) 76:153–65. doi: 10.1002/art.42681
17. De Benedetti F, Grom AA, Brogan PA, Bracaglia C, Pardeo M, Marucci G, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis. (2023) 82:857–65. doi: 10.1136/ard-2022-223739
18. Bindoli S, De Matteis A, Mitrovic S, Fautrel B, Carmona L, De Benedetti F. Efficacy and safety of therapies for Still’s disease and macrophage activation syndrome (MAS): a systematic review informing the EULAR/PReS guidelines for the management of Still’s disease. Ann Rheum Dis. (2024) 83:1731–47. doi: 10.1136/ard-2024-225854
Keywords: hemophagocytic lymphohistiocytosis (HLH), macrophage activation syndrome (MAS), eculizumab, emapalumab, thrombotic microangiopathy (TMA)
Citation: Faggioli P, Galeazzi M, Ferrari C, Capelli F, Marchesi C, Marchionni L, Castelnovo L, Tamburello A, Capparelli E, Campidelli C and Mazzone A (2025) Macrophage activation syndrome successfully treated with eculizumab and emapalumab: a case report. Front. Immunol. 16:1555415. doi: 10.3389/fimmu.2025.1555415
Received: 04 January 2025; Accepted: 20 February 2025;
Published: 24 March 2025.
Edited by:
Chris Wincup, King’s College Hospital NHS Foundation Trust, United KingdomReviewed by:
Tomas Escobar Gil, University of New Mexico, United StatesCopyright © 2025 Faggioli, Galeazzi, Ferrari, Capelli, Marchesi, Marchionni, Castelnovo, Tamburello, Capparelli, Campidelli and Mazzone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianna Galeazzi, bWFyaWFubmEuZ2FsZWF6emlAYXNzdC1vdmVzdG1pLml0; Antonino Mazzone, YW50b25pbm8ubWF6em9uZUBhc3N0LW92ZXN0bWkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.