 Sergio A. Jimenez1*
Sergio A. Jimenez1* Fabian A. Mendoza
Fabian A. Mendoza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 April 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1551911
Systemic Sclerosis (SSc) is a systemic autoimmune disease of unknown etiology characterized by the development of frequently progressive cutaneous and internal organ fibrosis accompanied by severe vascular alterations. The pathogenesis of SSc is highly complex and, despite extensive investigation, has not been fully elucidated. Numerous studies have suggested that unknown etiologic factors cause multiple alterations in genetically receptive hosts, leading to SSc development and progression. These events may be functionally and pathologically interconnected and include: 1) Structural and functional microvascular and endothelial cell abnormalities; 2) Severe oxidative stress and high reactive oxygen species (3); Frequently progressive cutaneous and visceral fibrosis; 4) Transdifferentiation of various cell types into activated myofibroblasts, the cells ultimately responsible for the fibrotic process; 5) Establishment of a chronic inflammatory process in various affected tissues; 6) Release of cytokines, chemokines, and growth factors from the inflammatory cells; 7) Abnormalities in humoral and cellular immunity with the production of specific autoantibodies; and 8) Epigenetic alterations including changes in multiple non-coding RNAs. These events manifest with different levels of intensity in the affected organs and display remarkable individual variability, resulting in a wide heterogeneity in the extent and severity of clinical manifestations. Here, we will review some of the recent studies related to SSc pathogenesis.
● Systemic Sclerosis (SSc) is a systemic autoimmune disease characterized by a severe fibrotic process affecting the skin and multiple internal organs associated with a generalized obstructive vasculopathy of small arteries and arterioles.
● Despite extensive genetic, biochemical, and molecular studies, the exact pathogenetic mechanisms of SSc have not been fully elucidated, although several affected molecular pathways have been identified.
● The discovery of these molecular pathway alterations has led to a marked improvement in the understanding of SSc pathogenesis.
● Although there is no curative therapy for the disease, and it may progress, causing severe disabilities and high mortality, the recent identification of novel molecular therapeutic targets should be of value to optimize the treatment and reduce the mortality caused by the disease.
Systemic Sclerosis (SSc) is a clinically heterogeneous systemic autoimmune disease of unknown etiology characterized by a frequently progressive fibrotic process affecting the skin and various internal organs. The fibrotic process in SSc is usually accompanied by vasculopathy of small arteries and arterioles, the presence of a chronic inflammatory process in the affected tissues, and the occurrence of humoral and cellular immune abnormalities resulting in the production of multiple autoantibodies, some with high specificity for the disease and the SSc clinical phenotype (1–4).
The molecular mechanisms involved in the clinical and pathologic manifestations of the disease are highly complex, and although numerous studies have provided substantial information about its intricate picture and clarified some of its early events, the precise altered regulatory pathways involved have not been completely elucidated. However, it has been well recognized that SSc involves multiple alterations in various molecular pathways (5–9) that may occur simultaneously or may develop sequentially. These events include: 1) Fibroproliferative lesions of small arteries and arterioles accompanied by severe structural and functional endothelial cell alterations; 2) Severe oxidative and high reactive oxygen species; 3) Excessive and often progressive deposition of collagen and other extracellular matrix (ECM) macromolecules in skin and various internal organs; 4) Alterations of cellular and humoral immunity with the production of numerous autoantibodies, some with high disease and clinical phenotype specificity; 5) Establishment of a chronic inflammatory process in affected tissues; 6) Cellular transdifferentation resulting in the phenotypic conversion of various cell types including resting fibroblasts, endothelial cells, epithelial cells, adipocytes, and other cells into activated myofibroblasts, the cellular elements ultimately involved in the exaggerated and excessive production and accumulation of fibrotic tissue; 7) Production and release of increased levels of various cytokines and growth factors causing profibrotic and inflammatory effects; and 8) Epigenetic alterations including numerous changes mediated by non-coding RNAs. However, despite extensive investigation of the numerous pathogenetic events in SSc, it has not been established which of these processes is of primary importance or how they are temporally related during the development and progression of the disease.
The current hypothesis of SSc pathogenesis proposes that the disease is initiated by unknown etiologic factors that may include toxic or chemical exposures, viral infections, microbial pathogens, or other still not identified mechanisms (10, 11). More specifically, the resemblance of SSc to fibrotic syndromes associated to toxic exposure such as polyvinyl chloride (12), contaminated rapeseed oil (Spanish toxic oil syndrome) (13), L-tryptophane contained products (eosinophilia myalgia syndrome) (14), and gadolinium (nephrogenic systemic fibrosis) (15), favors the implication of an environmental trigger, whereas the presence of an early type I interferon signature and activation of TLR8 by EBV genes found in monocytes from patients with SSc, suggest a possible viral etiology (16). In both scenarios, the activation of macrophages, monocytes and T-Cells are proposed to be the main mediators for the abnormal activation of the immune system (17). Another theory, states that under proper conditions and acting on a genetically receptive host, the causative initial mechanism(s) induces severe microvascular injury with profound structural and functional endothelial cell abnormalities and cause the development of frequently progressive cutaneous and internal organ tissue fibrosis. Although it is well recognized that SSc is not a genetic or genetically transmitted disease, the role of genetic factors is of crucial importance, as discussed extensively in numerous recent publications (18–21).
The prominent vascular involvement in SSc was initially described in early reports of SSc pathologic alterations that demonstrated the presence of intimal sclerosis and vessel wall hyperplasia in the arterioles of the kidneys and other affected visceral organs (22–24). Numerous subsequent studies confirmed the occurrence of vascular alterations as the earliest clinical manifestations of the disease, leading to the hypothesis that SSc was a vascular disease (25–27). The vascular alterations are clinically manifested as Raynaud’s Phenomenon, small artery vasculopathy, digital ulcers, multiple cutaneous and mucosal telangiectasias, and the development of severe fibroproliferative vasculopathy in multiple internal organs causing alterations in their function and their eventual failure (28–31). A second component of SSc pathogenesis is the recruitment of specific cellular elements resulting in the establishment of a chronic inflammatory process in the affected tissues. The participating inflammatory cells include dendritic cells, macrophages, T- and B- lymphocytes, and mast cells (32–36). The inflammatory cells initiate the production and release of numerous cytokines, chemokines, growth factors, and other molecular mediators that allow the establishment of the fibrotic process through multiple cellular interactions and cell-to-cell communications (37–41). The molecular effects induced by the cytokines and growth factors also cause remarkable cellular transdifferentiation events, including the phenotypic conversion of quiescent fibroblasts, epithelial cells, endothelial cells, and other cells such as pericytes and adipocytes into activated myofibroblasts, the cells ultimately responsible for the establishment and progression of the fibrotic process (42–48). This sequence of events (illustrated in Figure 1) results in the development of a severe and often progressive fibroproliferative vasculopathy, exaggerated and widespread accumulation of fibrotic tissue in the skin and numerous internal organs, and the establishment of a chronic inflammatory process in affected organs.
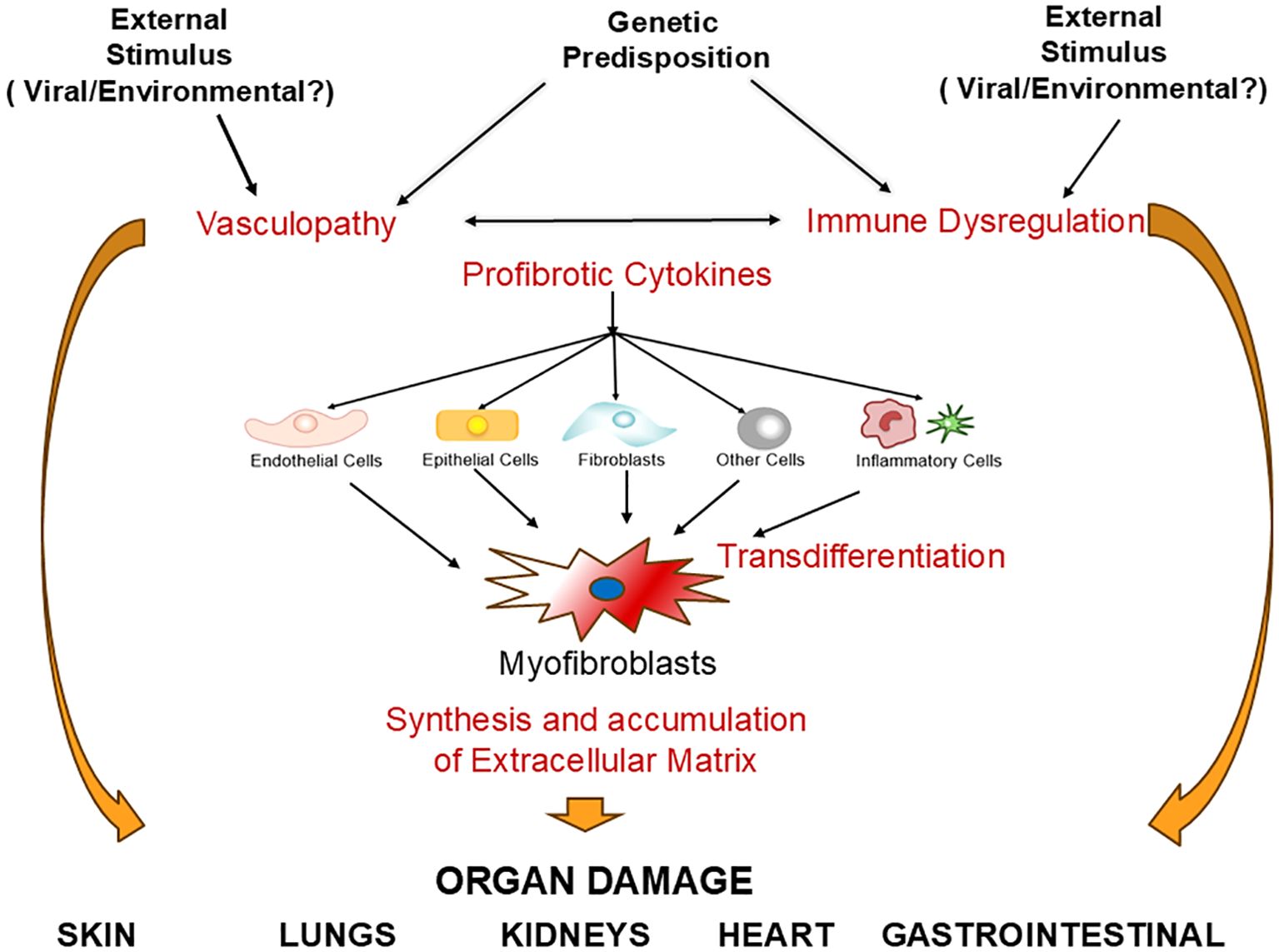
Figure 1. Systemic sclerosis pathogenesis. An unknown etiologic event (such as a virus or chemical substance) in a genetically predisposed host causes activation of multiple cell types including endothelial cells, inflammatory cells and fibroblasts. This activation process triggers abnormalities of the vascular tone causing vasospastic alterations, increases chemoattraction and adherence of monocytes/macrophages, promoting tissue inflammation and abnormal oxidative stress. An additional effect is the conversion of endothelial cells into myofibroblasts through Endothelial Mesenchymal Transdifferentiation (EndoMT) and the activation of other cells, including resident fibroblasts, circulating fibrocytes, epithelial cells (EMT), and adipocytes (AMT) into myofibroblasts. Increased myofibroblast numbers and metabolic activation cause increased production and accumulation of extracellular matrix molecules (ECM), which, coupled with microvasculopathy in multiple organs, and chronic inflammation are responsible for the most prominent SSc clinical manifestations in multiple target organs.
Here, we will review recent developments in the understanding of SSc pathogenesis and the involvement of microRNA (miRNA) alterations in SSc pathogenesis. Studies describing the role of inflammatory, humoral and cellular immunological abnormalities in SSc pathogenesis will not be discussed in detail here owing to the large number of publications and extensive reviews about these topics (18–21, 37, 44–50). We wish to emphasize, however, that the numerous and extensive publications related to SSc pathogenesis, preclude us from including in this review all the relevant studies on this important subject and we truly wish to express our deepest apologies to the investigators whose work was not specifically cited or discussed.
Vascular dysfunction is one of the earliest and most prominent clinical manifestations of SSc, as indicated by the occurrence of Raynaud’s Phenomenon and nailfold capillary microvascular alterations often preceding the appearance of other signs or symptoms of the disease. These early alterations are followed by the development of a systemic vasculopathy that results in abnormally dilated capillaries, microhemorrhages, vascular rarefaction and capillary loss, tissue injury caused by ischemia and hypoxia, cutaneous and mucosal telangiectasias, and fibroproliferative/occlusive vascular lesions in multiple organs (26–31). The visceral vasculopathy causes a spectrum of severe and even fatal clinical manifestations including scleroderma renal crisis (SRC), pulmonary arterial hypertension (PAH), interstitial lung disease, gastric antral vascular ectasia (GAVE), gastrointestinal dysmotility, myocardial dysfunction, erectile failure, and central retinal artery occlusion (51–59).
Following the vascular hypothesis to explain SSc pathogenesis proposed by Norton and Nardo (24) and by Campbell and LeRoy (25), this concept has been subsequently endorsed by numerous investigators (26–29), and it has become generally accepted that regardless of the putative SSc etiologic agent or event, microvascular alterations and endothelial cell injury and activation are central to the pathogenesis and the development of most of SSc clinical manifestations (60–62).
The initial events responsible for the vascular and endothelial cell injury in SSc are not fully known, although numerous putative factors have been suggested (Reviewed in Refs 62,63). Among these, the following have been most commonly considered: chemical and toxic agents, vasculotropic viral pathogens (10, 11), endothelial cell dysfunction (62), anti-endothelial cell antibodies (63, 64), and oxidative stress caused by reactive oxygen species (ROS) generated during episodes of ischemia/reperfusion (65). These factors either alone or in combination result in the development of crucial vascular and endothelial cell abnormalities that include: 1) Activation of endothelial cells; 2) Endothelial cell transdifferentiation into activated myofibroblasts, the cells responsible for the production of elevated amounts of collagens and other fibrotic macromolecules (48); 3) Production of increased levels of the potent profibrotic and vasoconstrictor polypeptide, endothelin-1, that besides its vascular effects, is a potent inducer of proliferation and ECM production by fibroblastic cells that also causes an exaggerated vasoconstrictor response resulting in further vascular hypoxia and endothelial injury (66–68); and 4) A complex imbalance of pro- and anti-angiogenic factors resulting in marked impairment of angiogenesis (69–71). The activated endothelial cells may also directly stimulate the profibrotic activities of various target cells such as vascular smooth muscle cells and fibroblasts and participate in the recruitment of other cells that may induce fibrotic alterations in the affected tissues. Collectively, the extensive endothelial cell and vascular alterations induce subsequent pathological events that maintain the vicious cycle of vascular injury, inflammatory response and tissue fibrosis that are the crucial components of SSc pathogenesis (5–9).
Thirty years ago, Murrell proposed the hypothesis that elevated reactive oxygen species (ROS) caused abnormally increased systemic oxidative stress, which subsequently triggered cellular and molecular alterations that were responsible for the development of tissue fibrosis in SSc (65). Numerous subsequent studies have pursued this hypothesis and have provided strong support to the crucial role of oxidative stress in SSc pathogenesis. Indeed, it has been shown that elevated oxidative stress caused by ROS and reactive nitrogen species can stimulate the production of pro-fibrotic cytokines and growth factors (such as PDGF and TGF-β), induce proliferation and activation of fibroblasts, increase the expression and synthesis of type I collagen, and promote inflammatory changes and vascular dysfunction (72–77). Furthermore, it has been demonstrated that fibroblasts from affected SSc skin contain higher ROS levels and display markedly elevated activity of NADPH oxidases (NOX), particularly of NOX2 and NOX4 compared to fibroblasts cultured from skin from normal controls (78–81). It has also been shown that hypoxia induces alterations in the expression of several genes that participate in the phenotypic conversion of endothelial cells into activated myofibroblasts through a process of endothelial to mesenchymal transition (82).
Several in vitro studies have shown that reversal or reduction of the increased oxidative stress employing specific antioxidants or a NOX4 small molecule inhibitor abrogated these effects (78, 83). Furthermore, in vitro studies found a marked reduction in collagen expression levels in SSc dermal fibroblasts and in lung tissues from patients with SSc-associated ILD following treatment with antioxidant compounds (84, 85). These observations have been supported by several in vivo studies demonstrating that a decrease of ROS-generation caused suppression of fibroblast activation and abrogation of experimentally induced skin fibrosis (85). Several studies examined the effects of the anti-oxidant generator N-acetyl cysteine in SSc patients. One of these studies described a retrospective analysis of pulmonary function tests in SSc patients with pulmonary fibrosis following 24 months of intravenous N-acetyl cysteine. The treatment resulted in significant improvement in lung function, and the beneficial effects were greater in patients with early SSc-lung involvement (86). Another study was a randomized placebo-controlled trial on 25 patients with diffuse SSc without lung involvement (87). In contrast to the previous study (86), the results of this trial did not show any beneficial effects after 24 months of N-acetyl cysteine therapy (87).
Extensive experimental evidence has confirmed the occurrence of increased oxidative stress in SSc including the demonstration of elevated serum and plasma levels of various oxidative stress metabolic products such as 8-isoprostane, F2-iosprostane, malondialdehyde (MDA), and asymmetric dimethylarginine (ADMA); as well as elevated concentrations of DNA oxidation markers in the urine of SSc patients (Reviewed in Ref. 83). Furthermore, elevated levels of F2-isoprostane have been shown to correlate with the extent and severity of the SSc fibrotic process, particularly with lung fibrosis (88). A detailed mechanistic study assessed the molecular markers of oxidative stress in the serum of 36 diffuse SSc patients in comparison with 26 healthy controls performing quantitative measurement of reactive oxidative metabolites, including MDA and ADMA, total antioxidant capacity, lipid peroxidation, and evaluation of DNA oxidative damage (89). The results confirmed that total oxidative capacity and oxidative stress index were significantly increased in SSc patients as compared to healthy control subjects and demonstrated a correlation between these oxidative stress abnormalities and the presence of SSc pulmonary and gastrointestinal involvement. A remarkable observation in this study was that the oxidative stress abnormalities were also associated with the presence of anti-topoisomerase antibodies. Thus, this study confirmed the presence of molecular alterations indicative of excessive oxidative stress in the serum of SSc patients. However, it should be emphasized that the results did not show evidence of extensive lipid or DNA molecular oxidative damage and, therefore, it was suggested that SSc patients may have increased antioxidant capacity, although this possibility should require confirmation and validation in further studies (89).
Despite the published evidence of a strong association between fibrosis and oxidative stress/ROS, the pathways responsible are highly complex and have not been clearly delineated although there has been extensive investigation to elucidate the mechanisms involved. Among these studies, it was recently demonstrated that a novel oxidative stress pathway results in the induction of premature fibroblast senescence causing a marked increase in the production of inflammatory cascade mediators (90). Another recently identified mechanism was the demonstration that oxidative stress caused stabilization of specific kinase-phosphatase complexes that mediate activation of the SSc fibrotic process (91).
The progressive fibrotic process affecting the skin and numerous internal organs is one of the most distinctive clinical and pathological features of SSc. It has been generally accepted that this crucial pathologic alteration results from the accumulation in skin and other affected tissues of activated myofibroblasts. Myofibroblasts are mesenchymal cells first described in granulation tissue and were considered to be modified fibroblasts (92, 93). These cells display a markedly fibrogenic phenotype that is characterized by a persistent and exaggerated increase in the expression of genes encoding various interstitial collagens and other ECM proteins, the downregulation of genes for matrix-degrading enzymes, and the initiation of expression of contractile proteins such as α-SMA (43, 94). Myofibroblasts are considered the crucial cellular elements responsible for developing tissue fibrosis in SSc (95). The accumulation of activated myofibroblasts in affected tissues and the uncontrolled persistence of their elevated biosynthetic functions are considered to be crucial determinants of the extent, severity, and rate of progression of the fibrotic process in SSc (96–100).
There are multiple cellular sources responsible for originating the activated myofibroblasts present in affected SSc tissues. These include: 1) Proliferation and activation of tissue resident fibroblasts or perivascular and vascular adventitial fibroblasts and the selection of ECM over-producer and apoptosis resistant cells in response to specific signals from infiltrating inflammatory cells (101–103); 2) Recruitment of bone marrow fibrocytes, a unique class of fibroblast precursor cells expressing the CD34 hematopoietic/stem cell surface marker and able to produce type I procollagen and other ECM proteins typically expressed by fibroblastic cells (104, 105); 3) Transdifferentiation of endothelial cells to myofibroblasts, a process known as endothelial to mesenchymal transition (EndoMT) in which endothelial cells lose their phenotypic characteristics and acquire a myofibroblast phenotype and play a crucial role in the vasculopathy and fibrotic process in SSc (106–111); 4) Epithelial to mesenchymal transition (EMT), is another cellular phenotypic transition process (112–114) that has been postulated to contribute to the accumulation of myofibroblasts in SSc affected tissues (115–117); and 5) An additional source of myofibroblasts in affected SSc tissues are adipocytes and adipocyte precursor cells that may undergo a reverse differentiation process and acquire fibroblastic or myofibroblastic phenotype (118, 119). This process has been named “adipocyte myofibroblast transition” (AMT). The most important sources of activated myofibroblasts in SSc will be briefly described in the following sections.
An increased number of cells capable of expression and production of ECM and other profibrotic macromolecules is a very important mechanism involved in the development and extension of the SSc fibrotic process. The crucial molecular pathways responsible cause an increase in the rate of proliferation of these cells and an increase in their viability and survival (95–97). Soluble mediators, either transported through the circulation or released by activated inflammatory cells present within the affected tissue infiltrates, induce a marked increase in the number of cells capable of producing excessive amounts of the molecular components of the fibrotic tissue. These effects may result from a direct action of the soluble mediators on the target cells or may be mediated by an autocrine stimulation of the production of profibrotic mediators from other cells. However, there is also an important, although less extensively studied, contribution of a reduction of cellular apoptosis and other cell death pathways, resulting in the prolongation of the cellular lifespan and in increased cellular survival (120–123).
Fibrocytes are another important source of myofibroblasts. Fibrocytes are fibroblast precursor cells that play an important role in the development of tissue fibrosis (104, 105). These cells migrate from the bone marrow in response to specific chemokines released from the infiltrating inflammatory cells present in the affected tissues. These circulating bone marrow fibroblast precursor cells represent a unique cell population that is characterized by the expression of hematopoietic/stem cell surface markers, such as CD34 protein, coupled with the ability to synthesize type I procollagen and other ECM molecules typically produced and secreted by fibroblastic cells (103, 105).
Cellular transdifferentiation is a highly complex biological process that results in the phenotypic conversion of fully differentiated somatic cell types into cells of another lineage. The molecular alterations involved in this highly complex transdifferentiation process have not been fully elucidated, although extensive investigation has shown that it is mediated by distinct mechanisms. These include changes in the expression and activity of specific protein transcription factors, involvement of complex protein interaction cascades, and the regulatory effects of various species of RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). The main cellular transdifferentiation processes that may play a role in SSc-associated tissue fibrosis have been recently reviewed (48) and include: 1) Epithelial-Mesenchymal Transition, 2) Endothelial-Mesenchymal Transition, and 3) Adipocyte-Myofibroblast Transition. These processes will be briefly reviewed in the following sections.
Epithelial-mesenchymal transition (EMT) is a complex cellular trans-differentiation process in which stationary and fully differentiated epithelial cells undergo profound changes in their cellular phenotype. These changes are characterized by the acquisition of mesenchymal cell features including the loss of cell-cell adhesion and apical-basal polarity, profound modifications in gene expression levels, marked morphological and cell shape alterations, and development of migratory capacity (112–114). Although EMT was extensively studied as an important mechanistic pathway involved in wound healing and cancer development and progression, extensive experimental evidence has demonstrated that this phenotypic cellular conversion participates in a large number of pathological processes, including a prominent role in the pathogenesis of SSc (115, 116), and of other autoimmune diseases (123).
The relevance of EMT to the pathogenesis of the SSc fibrotic process has been extensively demonstrated (115–117). Additional evidence includes the presence of increased nuclear translocation of myocardin-related transcription factor-A (MRTF-A), a factor responsive to mechanical stimuli that are key for EMT transformation in SSc patients (124). Furthermore, the secreted frizzled receptor protein 4 (SFRP4), a protein recently associated with EMT development, has been shown to be increased in the epidermis of SSc patients, and its serum concentration appeared to correlate with the degree of skin and lung fibrosis. These observations have suggested that SFRP4 should be considered as a biomarker of skin and lung fibrosis in SSc (123).
EndoMT is a complex biological process in which endothelial cells lose their specific phenotype and progressively evolve into cells expressing mesenchymal characteristics acquiring a fusiform and elongated cell shape, cellular motility capabilities, and contractile properties (106, 107). At the molecular level, EndoMT results in the loss of endothelial cell-specific proteins including von Willebrand factor (vWF), CD31/platelet-endothelial cell adhesion molecule-1 (CD31/PECAM-1), and vascular-endothelial cadherin (VE-cadherin). There is a concomitant initiation of expression and production of mesenchymal cell-specific proteins including α-smooth muscle actin (α-SMA), extra domain A (EDA) fibronectin, N-cadherin, vimentin, fibroblast-specific protein-1 (FSP-1; also known as S100A4 protein), fibroblast activating protein (FAP), and fibrillar collagens type I and type III (106, 107). There has been extensive recent interest in the role of EndoMT in the pathogenesis of various human malignant, vascular, inflammatory and fibrotic disorders, and it has been demonstrated that it may play a very important role in the pathogenesis and in the development of the SSc-associated fibrotic process (108, 109) as well as in the vascular manifestations of numerous diseases (Reviewed in Ref. 110). Although most studies on EndoMT are related to the phenotypic conversion of arterial, venous, or capillary endothelial cells, recent studies have shown that lymphatic endothelial cells may also be able to trans-differentiate into myofibroblasts (125).
Extensive studies have shown that TGF-β1 and other TGF-β related growth factors, are the main inducers of EndoMT (126–129) and that these effects may be mediated by the nuclear translocation of the transcription factor Snail1 (127). Other studies have identified additional molecules that participate in the initiation, development and progression of fibrotic processes. One of these molecules is SOX9, a multifunctional transcription factor that plays a crucial role in chondrogenesis and skeletal tissue development and differentiation (130). SOX9 has also been shown to participate in numerous other regulatory functions in various tissues and organs and to play a role in various fibrotic diseases including idiopathic and SSc-associated pulmonary fibrosis (131). The mechanisms mediating the pro-fibrotic SOX9 effects have been recently examined. One extensive analysis provided evidence that SOX9 expression in human endothelial cells induced these cells to undergo EndoMT (132). It was further shown that these effects were mediated by alterations in the chromatin structure of the cells allowing histone modifications at previously silent binding sites causing a phenotypic change of the endothelial cells resulting in the initiation of expression of mesenchymal genes (132). These results allow to suggest the hypothesis that the initiation of EndoMT mediated by SOX9 expression in endothelial cells may play a crucial role in the development of various SOX9-mediated pathological fibrotic processes including pulmonary fibrosis, and SSc.
It should be emphasized, however, that given the great complexity of EndoMT and of the multiple and often redundant molecular mechanisms involved in its regulation, the overall effects of SOX9 may be quite variable, and highly dependant on the tissues affected as well as on the various pathophysiological conditions responsible for initiation and development of this process.
A highly relevant mechanism for the induction of EndoMT is tissue hypoxia (82, 133, 134). Although the molecular alterations involved have not been studied in detail, it has been shown that endothelial cells cultured under hypoxic conditions loose the expression of molecules that regulate the endothelial cell phenotype and acquire typical mesenchymal cell morphology and initiate the production of interstitial fibroblast macromolecules.
Adipocyte-mesenchymal transition (AMT) is another cellular phenotype change that causes the conversion of mature adipocytes into pro-fibrotic myofibroblasts (135, 136). This process can be induced by exposure of adipocytes to TGF-β in vitro, resulting in the loss of the adipose specific markers (including PPAR-γ, perilipin, and FABP4), and promoting expression of fibrotic and mesenchymal proteins characteristic of the myofibroblast phenotype. Of remarkable importance to SSc pathogenesis were the observations that serum from SSc patients was able to induce the conversion of adipose tissue-derived stem cells into profibrotic myofibroblasts (118).
An additional cellular component related to ATM involves lipofibroblasts. Lipofibroblasts are fibroblast-like cells characterized by the content of abundant lipid storage deposits and the expression of adipose differentiation-related protein markers. These cells are abundant in the lungs and they also may undergo transdifferentiation into myofibroblasts, contributing to the activated profibrotic myofibroblast population during the initiation and development of SSc-associated lung fibrosis (136).
Pericytes have an important role in vessel stabilization during angiogenesis (137), and have shown an increased expression of RGS5, an inhibitor of vessel maturation (138), and PDGFRβ along with decrease expression of αSMA. These activated immature pericytes are more abundant in the dermis of SSc patients (139) and can explain the defective angiogenesis seen in SSc despite of the presence of pro-angiogenic factors such as VEGF. In addition, it has been postulated that pericytes expressing PDGFβ+, NG2+, ADAM12+ can be precursors of myofibroblasts in skin from SSc patients (140).
Besides tissue fibrosis, the presence of chronic inflammatory cells in affected SSc skin is one of the earliest and most consistent pathological alteration of the disease (32–34, 47). Numerous studies have shown that these cells, especially monocytes and macrophages, are capable of production and release of numerous profibrotic macromolecules including various cytokines and growth factors. These profibrotic cellular products induce quiescent fibroblasts in affected tissues to become active myofibroblasts and initiate their production of exaggerated amounts of interstitial collagens and other ECM macromolecules that result in pathologic and excessive fibrotic tissue accumulation (49, 50, 141, 142). Indeed, a detailed study employing coculture experiments of B cells and fibroblasts demonstrated that B cells were capable of direct stimulation of collagen and ECM protein synthesis by fibroblasts in vitro (143). This study also showed that B cell stimulation was further enhanced by BAFF (B cell activating factor), and it was suggested that these effects may be mediated by two distinct mechanisms, one resulting from cell-cell contact and the other induced by the B cell production of profibrotic molecules including IL-6 and TGF-β (143). Of substantial relevance to SSc pathogenesis are recent studies demonstrating that the products released by inflammatory cells also participate in the fibroproliferative vasculopathy characteristic of the disease (144).
Other studies have demonstrated that T cells secrete numerous cytokines, including IL-4, IL-6, IL-13, IL-17, and IL-31, that play an important role in SSc pathogenesis, including the stimulation of exaggerated synthesis and deposition of collagens and other fibrotic proteins by fibroblasts (46–50). IL-13 is an important product of CD8+ T cells, and its serum levels in SSc-affected patients were found to correlate with the presence and extent of skin fibrosis. Furthermore, it has been shown that IL-13 was able to stimulate collagen production in cultured SSc fibroblasts in vitro, and, therefore, it has been suggested to play a pathogenetic role in SSc-associated tissue fibrosis (145, 146). IL-31, is another cytokine that is also elevated in the serum of SSc patients. Recent studies have shown that IL-31 also stimulates collagen synthesis in dermal fibroblasts isolated from patients with SSc, and it has been strongly implicated in the development of SSc-associated Pulmonary Fibrosis (147–149). Indeed, it has been described that there is a highly significant negative correlation between serum IL-31 levels and the reduction of DLCO values in SSc patients (149).
Numerous polypeptide growth factors play a crucial role in developing and extending the fibrotic process in SSc. The experimental evidence supporting the role of these growth factors in the pathogenesis of tissue fibrotic responses and the molecular mechanisms involved is very large and has been reviewed extensively in numerous publications (150–153). Therefore, we will only briefly discuss a few relevant recent studies related to the pathogenetic role of growth factors in the fibrotic process of SSc.
It has become well recognized that TGF-β plays a crucial role in the initiation and progression of a large number of fibrotic processes (153, 154), and extensive studies have shown that it is of great relevance to the fibrotic process associated with SSc (151–153). These effects are mediated by multiple mechanisms including a potent stimulation of expression of genes encoding various interstitial collagens and other ECM macromolecules by fibroblasts and other mesenchymal cells (155–157), and the induction of cellular transdifferentiation of various types of cells into cells displaying very high expression of various collagens (Reviewed in Ref. 48).
The production and activation of TGF-β are highly regulated and involve numerous complex pathways. TGF-β is initially produced in a latent form in which the polypeptide is bound to a latency-associated peptide (LAP) and is subsequently secreted to the extracellular compartment as a large complex with a carrier protein called latent TGF-β binding protein (LTBP). Once released from the LTBP, the TGF-β molecules become a bioactive dimeric complex that is able to bind to different isoforms of TGF-β receptors (TβR) located in the cell membrane of TGF-β-responsive cells and initiate intracellular signaling pathways that are context dependent and transduce the signal from the cell surface to the nucleus resulting in a potent modulation of expression of multiple genes (158–164). The intracellular molecular cascades initiated following TGF-β binding and receptor activation have been extensively described in multiple publications (155–161) and will not be discussed here in further detail.
Extensive research studies have shown that the main signal transducer of TGF-β fibrotic signaling is a family of proteins known as Smad proteins that play an essential role in the regulation of the TGF-β-induced fibrotic response (164–166). Other regulatory pathways of TGF-β effects involve various non-receptor tyrosine kinases (167, 168), including the cytoplasmic Abelson kinase (c-Abl) and protein kinase C-δ (PKC-δ), to contribute to the fibrosis and vasculopathy of the skin and internal organs in SSc. These effects are most likely mediated by the crucial participation of c-Abl in TGF-β-induced EndoMT, an effect mediated by the cooperative interaction with PKC-δ (169, 170).
CTGF, also known as CCN2, is a cysteine-rich protein with pleotropic effects that has recently been recognized as an important mediator of normal and pathological tissue fibrotic responses (171–173). Numerous studies have demonstrated the potent profibrotic effects of CTGF (172), and it has been considered to play a crucial role in the SSc fibrotic process and to correlate with the extent and severity of tissue fibrosis (174–177). Some of these effects are of substantial relevance to SSc pathogenesis owing to the fact that vascular wall smooth muscle cells are the main targets of these effects (178), and therefore, it is possible that CTGF may contribute to the development of Raynaud’s Phenomenon and other vascular alterations characteristic of SSc.
The PDGF family of growth factors plays an important role in the development and maintenance of normal connective tissue, and abnormalities of their signaling pathways may be involved in the pathogenesis of multiple diseases (Reviewed in Ref. 183). Numerous studies have described the involvement of PDGF and related molecules in the pathogenesis of fibrotic diseases, including SSc (179–182). Elevated expression of PDGF and its receptors has been found in SSc skin and lung tissues and there is evidence that TGF-β stimulates the expression of the PDGF receptor, PDGFRα, in SSc cells suggesting that cross-talk between TGF-β and PDGF pathways may regulate tissue fibrosis in SSc (180) and it has been suggested that PDGF/PDGF receptor may represent a novel molecular target for SSc (182). Indeed, pharmacologic inhibition of these pathways has been shown to halt the SSc associated fibrotic process (182).
The FGFs comprise a family of twenty two polypeptide growth factors grouped in seven subfamilies collectively characterized by their ability to induce potent mitogenic effects that play important roles in development, angiogenesis and wound healing (183, 184). Numerous studies have demonstrated the mitogenic effects of FGF during inflammatory and fibrotic responses often potentiating the pro-fibrotic effects of TGF-β, although some recent studies have described that some of the members of the FGF family may cause antifibrotic effects that may be mediated by inhibition of TGF-β pathways (185, 186). Regarding the role of FGF in SSc pathogenesis, it has been shown that basic FGF is increased in the skin of SSc patients (187). However, the precise role of FGFs in the initiation of progression of the fibrotic process in SSc has not been completely elucidated and further studies will be required to conclusively determine the contribution of these potent growth factors to the pathogenesis of fibrosis in SSc.
VEGF is an endothelial cell specific growth factor mediating key signals for angiogenesis including stimulation of endothelial cell proliferation and differentiation, and modulation of endothelial permeability (188–190). Quantitative analysis of serum levels of VEGF in patients with SSc and healthy controls showed that serum VEGF levels were significantly higher in SSc patients and correlated with the extent and severity of skin fibrosis and nailfold capillary loss, suggesting that high VEGF levels may promote in the capillary damage in SSc and may correlate with the extent and severity of the fibrotic process and with disturbed angiogenesis (191–193).
Although early studies identified IGFs as the main stimulators of sulfate incorporation into cartilage (194, 195), several recent studies have shown that IGFs display strong profibrotic effects. Studies related to their role in the SSc fibrotic process have shown elevated levels of serum IGF-I and IGFBP-3 in SSc patients that correlated with the extent of skin involvement and the presence of pulmonary fibrosis (196). Furthermore, IGF-I mRNA was found to be upregulated in the affected skin tissues of patients with SSc. Subsequent extensive studies demonstrated that IGF-II is involved in SS-associated pulmonary fibrosis and is a potent inducer of collagen production and other fibrotic pathways and that these effects are mediated by multiple mechanisms, including an increase in the expression of pro-fibrotic signaling molecules, a decrease in the expression of several collagen degradation enzymes, and an increase in the differentiation of fibroblasts into myofibroblasts (197, 198).
MicroRNAs (miRNAs) are small (~22 nucleotides), evolutionarily conserved non-coding RNA, which play important roles in the regulation of the expression of a large number of protein-coding genes at the post-transcriptional level (199). The mechanisms involved in post-transcriptional miRNA regulation of gene expression are complex and require the sequence-specific complementary binding to the 3’ untranslated region (UTR) of target mRNAs, suppressing their expression by either inhibiting mRNA translation or facilitating mRNA degradation of their corresponding mRNA (199, 200). Non-coding RNAs can also induce potent cellular transdifferentiation effects (201).
Several non-coding RNAs have been shown to be involved in SSc tissue fibrosis, displaying either profibrotic or antifibrotic effects (202–204). Among the most extensively studied miRNAs in the pathogenesis of SSc are miR-21 and miR-29 (201–203) and it has been shown that elevated expression of miR-21 is associated with stimulation of fibroblast proliferation and increased production and accumulation of various ECM proteins. Furthermore, it has been demonstrated that miR- 21 expression is upregulated by TGF-β1 and may represent one of the profibrotic molecular effects of the growth factor on dermal fibroblasts. The miR-29 family comprises several distinct miRNAs that play a crucial role in the regulation of the fibrotic process and are capable of potent anti-fibrotic effects including regulatory modulation of several fibrosis-related genes such as the genes encoding collagens type I, II and IV, fibronectin, and laminin, as well as various enzymes involved in tissue remodeling, including TIMP and other matrix metalloproteinases. Indeed, it has been shown that miR-29 inhibits the TGF-β1/Smad signaling pathway and suppresses the TGF- β1-induced pro-fibrotic process. The expression of several members of the miR-29 family is reduced in SSc and in other tissue fibrosis diseases and the extent of their reduction has been shown to inversely correlate with the extent and severity of the fibrotic process (205, 206).
Recent studies have found the scaffold long non-coding RNA (lncRNA), HOTAIR, was found to be overexpressed in SSc in dermal fibroblasts, inducing a histone EZH2-dependent increase in collagen production and the expression of the myofibroblast marker α-SMA in vitro. In addition, histone-mediated repression of miRNA-34A expression was also observed, with the subsequent activation of the NOTCH pathway (207).
Another lncRNA that has been associated with the strong IFN-I signature in SSc is the X-inactive specific transcript (XIST), which is involved in X chromosome inactivation (XCI) in female mammals. Plasmocytic dendritic cells (pDC) in SSc overexpress TLR8, contributing to keeping a strong IFN-I signature in skin and lung tissues. Escaping from silencing by incomplete XCI in inflammatory cells from women may affect TLR7/8 signaling. The decreased expression of XIST and of the transcriptional repressor SPEN in SSc pDCs, suggests that an altered XCI at the TLR7/8 locus may, indeed, contribute to IFN-I chronic activation mediated by pDCs (208). In addition to that effect, TSIX, the XIST antisense, can also play a more direct role in SSc fibroblasts, activating endogenous TGF-β signaling and may play a role in stabilizing collagen RNA and upregulating collagen in these cells (209).
Upregulation and downregulation of multiple non-coding RNAs have been described in SSc (205–234), and it is very likely that although the evidence is not conclusive, these RNAs may be shown to be highly relevant to the SSc fibrotic process. A list of the non-coding RNA associated with SSc fibrosis is shown in Table 1. Multiple other miRNAs have been implicated in SSc vasculopathy and immune dysregulation. Although the scope of this review is focused on fibrosis, this subject has been recently reviewed (202, 235).

Table 1. MicroRNA implicated in SSc fibrosis.
SSc is a highly complex autoimmune disease of unknown etiology, causing severe clinical and pathological alterations in affected individuals that often manifest as progressive cutaneous and multiple organ fibrosis and associated generalized obstructive vasculopathy. Despite the severity of the disease, there is no curative therapy at the present time, however, it is expected that further knowledge about its pathogenesis may provide effective therapeutic approaches. This review outlines the current understanding of the disease pathogenesis and may be of value to the development of effective treatment for this serious autoimmune pathologic condition.
SJ: Writing – original draft, Writing – review & editing. FM: Writing – original draft, Writing – review & editing. SP-V: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. Supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number AM19606.
The authors wish to thank and acknowledge the expert assistance of Carol Kelly in the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
1. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. (2015) 1:15002. doi: 10.1038/nrdp.2015.2
2. Denton CP, Khanna D. Systemic sclerosis. Lancet. (2017) 390:1685–99. doi: 10.1016/S0140-6736(17)30933-9
3. Lazzaroni MG, Piantoni S, Angeli F, Bertocchi S, Franceschini F, Airò P. A narrative review of pathogenetic and histopathologic aspects, epidemiology, classification systems, and disease outcome measures in systemic sclerosis. Clin Rev Allergy Immunol. (2023) 64:358–77. doi: 10.1007/s12016-022-08929-x
4. Romanowska-Próchnicka K, Dziewit M, Lesiak A, Reich A, Olesińska M. Scleroderma and scleroderma-like syndromes. Front Immunol. (2024) 15:1351675. doi: 10.3389/fimmu.2024.1351675
5. Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of Systemic Sclerosis. Ann Int Med. (2004) 140:37–50. doi: 10.7326/0003-4819-140-1-200401060-00010
6. Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: Recent insights of molecular and cellular mechanisms and therapeutic opportunities. J Scleroderma Relat Disord. (2017) 2:137–52. doi: 10.5301/jsrd.5000249
7. Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol. (2019) 15:753–64. doi: 10.1080/1744666X.2019.1614915
8. Thoreau B, Chaigne B, Renaud A, Mouthon L. Pathophysiology of systemic sclerosis. Presse Med. (2021) 50:104087. doi: 10.1016/j.lpm.2021.104087
9. Ko J, Noviani M, Chellamuthu VR, Albani S, Low AHL. The pathogenesis of systemic sclerosis: the origin of fibrosis and interlink with vasculopathy and autoimmunity. Int J Mol Sci. (2023) 24:14287. doi: 10.3390/ijms241814287
10. Ferri C, Arcangeletti MC, Caselli E, Zakrzewska K, Maccari C, Calderaro A, et al. Insights into the knowledge of complex diseases: Environmental infectious/toxic agents as potential etiopathogenetic factors of systemic sclerosis. J Autoimmun. (2021) 124:102727. doi: 10.1016/j.jaut.2021.102727
11. Alahmari H, Ahmad Z, Johnson SR. Environmental risks for systemic sclerosis. Rheum Dis Clin North Am. (2022) 48:845–60. doi: 10.1016/j.rdc.2022.06.006
12. Black CM, Welsh KI, Walker AE, Bernstein RM, Catoggio LJ, McGregor AR, et al. Genetic susceptibility to scleroderma-like syndrome induced by vinyl chloride. Lancet. (1983) 1:53–5. doi: 10.1016/S0140-6736(83)91578-7
13. Tabuenca JM. Toxic-allergic syndrome caused by ingestion of rapeseed oil denatured with aniline. Lancet. (1981) 2:567–8. doi: 10.1016/S0140-6736(81)90949-1
14. Centers for Disease Control (CDC). Eosinophilia-myalgia syndrome and L-tryptophan-containing products–New Mexico, Minnesota, Oregon, and New York, 1989. MMWR Morb Mortal Wkly Rep. (1989) 38:785–8.
15. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheumatol. (2006) 35:238–49. doi: 10.1016/j.semarthrit.2005.08.002
16. Farina A, Peruzzi G, Lacconi V, Lenna S, Quarta S, Rosato E, et al. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res Ther. (2017) 19:39. doi: 10.1186/s13075-017-1237-9
17. Sakkas LI, Platsoucas CD. Is systemic sclerosis an antigen-driven T cell disease? Arthritis Rheum. (2004) 50:1721–33. doi: 10.1002/art.20315
18. Agarwal SK, Tan FK, Arnett FC. Genetics and genomic studies in scleroderma (systemic sclerosis). Rheum Dis Clin North Am. (2008) 34:17–40. doi: 10.1016/j.rdc.2007.10.001
19. Bossini-Castillo L, López-Isac E, Mayes MD, Martín J. Genetics of systemic sclerosis. Semin Immunopathol. (2015) 37:443–51. doi: 10.1007/s00281-015-0499-z
20. Ramos PS, Silver RM, Feghali-Bostwick CA. Genetics of systemic sclerosis: Recent advances. Curr Opin Rheumatol. (2015) 27:521–9. doi: 10.1097/BOR.0000000000000214
21. Angiolilli C, Marut W, van der Kroef M, Chouri E, Reedquist KA, Radstake TR. New insights into the genetics and epigenetics of systemic sclerosis. Nat Rev Rheumatol. (2018) 14:657–73. doi: 10.1038/s41584-018-0099-0
22. Rodnan GP, Schreiner GE, Black RL. Renal involvement in progressive systemic sclerosis (generalized scleroderma). Am J Med. (1957) 23:445–62. doi: 10.1016/0002-9343(57)90324-8
23. D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study fifty-eight autopsy cases fifty-eight matched controls. Am J Med. (1969) 46:428–40. doi: 10.1016/0002-9343(69)90044-8
24. Norton WL, Nardo JM. Vascular disease in progressive systemic sclerosis (scleroderma). Ann Intern Med. (1970) 73:317–24. doi: 10.7326/0003-4819-73-2-317
25. Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: a vascular hypothesis. Semin Arthritis Rheumatol. (1975) 4:351–68. doi: 10.1016/0049-0172(75)90017-7
26. Kahaleh B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am. (2008) 34:57–71. doi: 10.1016/j.rdc.2007.12.004
27. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheumatol. (2013) 65:1953–62. doi: 10.1002/art.37988
28. Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev. (2007) 6:520–3. doi: 10.1016/j.autrev.2006.12.006
29. Pattanaik D, Brown M, Postlethwaite AE. Vascular involvement in systemic sclerosis (scleroderma). J Inflammation Res. (2011) 4:105–25. doi: 10.2147/JIR.S18145
30. Hughes M, Allanore Y, Chung L, Pauling JD, Denton CP, Matucci-Cerinic M. Raynaud phenomenon and digital ulcers in systemic sclerosis. Nat Rev Rheumatol. (2020) 16:208–21. doi: 10.1038/s41584-020-0386-4
31. Ren H, Liu L, Xiao Y, Shi Y, Zeng Z, Ding Y, et al. Further insight into systemic sclerosis from the vasculopathy perspective. BioMed Pharmacother. (2023) 166:115282. doi: 10.1016/j.biopha.2023.115282
32. Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheumatol. (1977) 20:975–84. doi: 10.1002/art.1780200410
33. Roumm AD, Whiteside TL, Medsger TA Jr, Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification subtyping Clin correlations. Arthritis Rheumatol. (1984) 6):645–53. doi: 10.1002/art.1780270607
34. Kräling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology. (1995) 63:48–56. doi: 10.1159/000163933
35. Kalogerou A, Gelou E, Mountantonakis S, Settas L, Zafiriou E, Sakkas L. Early T cell activation in the skin from patients with systemic sclerosis. Ann Rheum Dis. (2005) 64:1233–5. doi: 10.1136/ard.2004.027094
36. Bosello S, Angelucci C, Lama G, Alivernini S, Proietti G, Tolusso B. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res Ther. (2018) 20:75. doi: 10.1186/s13075-018-1569-0
37. Gu YS, Kong J, Cheema GS, Keen CL, Wick G, Gershwin ME. The immunobiology of systemic sclerosis. Semin Arthritis Rheumatol. (2008) 38:132–60. doi: 10.1016/j.semarthrit.2007.10.010
38. Raja J, Denton CP. Cytokines in the immunopathology of systemic sclerosis. Semin Immunopathol. (2015) 37:543–57. doi: 10.1007/s00281-015-0511-7
39. King J, Abraham D, Stratton R. Chemokines in systemic sclerosis. Immunol Lett. (2018) 195:68–75. doi: 10.1016/j.imlet.2017.12.001
40. Brown M, O’Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin Exp Immunol. (2019) 195:310–21. doi: 10.1111/cei.13238
41. Worrell JC, O’Reilly S. Bi-directional communication: conversations between fibroblasts and immune cells in systemic sclerosis. J Autoimmun. (2020) 113:102526. doi: 10.1016/j.jaut.2020.102526
42. Karasek MA. Does transformation of microvascular endothelial cells into myofibroblasts play a key role in the etiology and pathology of fibrotic disease? Med Hypotheses. (2007) 68:650–5. doi: 10.1016/j.mehy.2006.07.053
43. Krieg T, Abraham D, Lafyatis R. Fibrosis in connective tissue disease: the role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res Ther. (2007) 9 Suppl 2:S4. doi: 10.1186/ar2188
44. Varga JA, Trojanowska M. Fibrosis in systemic sclerosis. Rheum Dis Clin North Am. (2008) 34:115–43. doi: 10.1016/j.rdc.2007.11.002
45. Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol. (2004) 16:733–8. doi: 10.1097/01.bor.0000139310.77347.9c
46. Wei J, Bhattacharyya S, Tourtellotte WG, Varga J. Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmun Rev. (2011) 10:267–75. doi: 10.1016/j.autrev.2010.09.015
47. Ebmeier S, Horsley V. Origin of fibrosing cells in systemic sclerosis. Curr Opin Rheumatol. (2015) 27:555–62. doi: 10.1097/BOR.0000000000000217
48. Jimenez SA, Piera-Velazquez S. Cellular transdifferentiation: A crucial mechanism of fibrosis in systemic sclerosis. Curr Rheumatol Rev. (2024) 20:388–404. doi: 10.2174/0115733971261932231025045400
49. Haynes DC, Gershwin ME. The immunopathology of progressive systemic sclerosis (PSS). Semin Arthritis Rheumatol. (1982) 11:331–51. doi: 10.1016/0049-0172(82)90055-5
50. Jiménez SA. Cellular immune dysfunction and the pathogenesis of scleroderma. Semin Arthritis Rheumatol. (1983) 13:104–13. doi: 10.1016/0049-0172(83)90029-X
51. Scheen M, Dominati A, Olivier V, Nasr S, De Seigneux S, Mekinian A, et al. Renal involvement in systemic sclerosis. Autoimmun Rev. (2023) 22:103330. doi: 10.1016/j.autrev.2023.103330
52. Cole A, Ong VH, Denton CP. Renal disease and systemic sclerosis: an update on scleroderma renal crisis. Clin Rev Allergy Immunol. (2023) 64:378–91. doi: 10.1007/s12016-022-08945-x
53. Denton CP, Black CM. Pulmonary hypertension in systemic sclerosis. Rheum Dis Clin North Am. (2003) 29:335–49. doi: 10.1016/S0889-857X(03)00024-3
54. Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. (2020) 8:304–20. doi: 10.1016/S2213-2600(19)30480-1
55. Liakouli V, Ciancio A, Del Galdo F, Giacomelli R, Ciccia F. Systemic sclerosis interstitial lung disease: unmet needs and potential solutions. Nat Rev Rheumatol. (2024) 20:21–32. doi: 10.1038/s41584-023-01044-x
56. McMahan ZH, Kulkarni S, Chen J, Chen JZ, Xavier RJ, Pasricha PJ, et al. Systemic sclerosis gastrointestinal dysmotility: risk factors, pathophysiology, diagnosis and management. Nat Rev Rheumatol. (2023) 19:166–81. doi: 10.1038/s41584-023-00929-1
57. Moysidou GS, Dara A, Arvanitaki A, Skalkou A, Pagkopoulou E, Daoussis D, et al. Understanding and managing cardiac involvement in systemic sclerosis. Expert Rev Clin Immunol. (2023) 19:293–304. doi: 10.1080/1744666X.2023.2171988
58. Lally EV, Jimenez SA. Erectile failure in systemic sclerosis. N Engl J Med. (1990) 322:1398–9. doi: 10.1056/NEJM199005103221918
59. Busquets J, Lee Y, Santamarina L, Federman JL, Abel A, Del Galdo F, et al. Acute retinal artery occlusion in systemic sclerosis: a rare manifestation of systemic sclerosis fibroproliferative vasculopathy. Semin Arthritis Rheumatol. (2013) 43:204–8. doi: 10.1016/j.semarthrit.2012.12.025
60. Müller-Ladner U, Distler O, Ibba-Manneschi L, Neumann E, Gay S. Mechanisms of vascular damage in systemic sclerosis. Autoimmunity. (2009) 42:587–95. doi: 10.1080/08916930903002487
61. Mostmans Y, Cutolo M, Giddelo C, Decuman S, Melsens K, Declercq H, et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun Rev. (2017) 16:774–86. doi: 10.1016/j.autrev.2017.05.024
62. Patnaik E, Lyons M, Tran K, Pattanaik D. Endothelial dysfunction in systemic sclerosis. Int J Mol Sci. (2023) 24:14385. doi: 10.3390/ijms241814385
63. Corallo C, Franci B, Lucani B, Montella A, Chirico C, Gonnelli S, et al. From microvasculature to fibroblasts: Contribution of anti-endothelial cell antibodies in systemic sclerosis. Int J Immunopathol Pharmacol. (2015) 28:93–103. doi: 10.1177/0394632015572750
64. Akbarzadeh R, Müller A, Humrich JY, Riemekasten G. When natural antibodies become pathogenic: autoantibodies targeted against G protein-coupled receptors in the pathogenesis of systemic sclerosis. Front Immunol. (2023) 14:1213804. doi: 10.3389/fimmu.2023.1213804
65. Murrell DF. A radical proposal for the pathogenesis of scleroderma. J Am Acad Dermatol. (1993) 28:78–85. doi: 10.1016/0190-9622(93)70014-K
66. Kahaleh MB. Endothelin, an endothelial-dependent vasoconstrictor in scleroderma. Enchanced production and profibrotic action. Arthritis Rheumatol. (1991) 34:978–83. doi: 10.1002/art.1780340807
67. Shi-Wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD, et al. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. (2007) 26:625–32. doi: 10.1016/j.matbio.2007.06.003
68. Argentino G, Barbieri A, Beri R, Bason C, Ruzzenente A, Olivieri O, et al. Profibrotic effects of endothelin-1 on fibroblasts are mediated by aldosterone in vitro: relevance to the pathogenesis and therapy of systemic sclerosis and pulmonary arterial hypertension. Biomedicines. (2022) 10:2765. doi: 10.3390/biomedicines10112765
69. Manetti M, Guiducci S, Ibba-Manneschi L, Matucci-Cerinic M. Mechanisms in the loss of capillaries in systemic sclerosis: angiogenesis versus vasculogenesis. J Cell Mol Med. (2010) 14:1241–54. doi: 10.1111/j.1582-4934.2010.01027.x
70. Sun X, Nkennor B, Mastikhina O, Soon K, Nunes SS. Endothelium-mediated contributions to fibrosis. Semin Cell Dev Biol. (2020) 101:78–86. doi: 10.1016/j.semcdb.2019.10.015
71. Rokni M, Sadeghi Shaker M, Kavosi H, Shokoofi S, Mahmoudi M, Farhadi E. The role of endothelin and RAS/ERK signaling in immunopathogenesis-related fibrosis in patients with systemic sclerosis: an updated review with therapeutic implications. Arthritis Res Ther. (2022) 24:108. doi: 10.1186/s13075-022-02787-w
72. Sambo P, Baroni SS, Luchetti M, Paroncini PM, Dusi S, Orlandini G, et al. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheumatol. (2001) 44:2653–64. doi: 10.1002/1529-0131(200111)44:11<2653::AID-ART445>3.0.CO;2-1
73. Gabrielli A, Svegliati S, Moroncini G, Luchetti M, Tonninic C, Avvedimento EV. Oxidative stress and the pathogenesis of scleroderma; The Murrell’s hypothesis revisited. Semin Immunopathol. (2008) 30:329–37. doi: 10.1007/s00281-008-0125-4
74. Beyer C, Schett G, Gay S, Distler O, Distler JH. Hypoxia. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Res Ther. (2009) 11:220. doi: 10.1186/ar2598
75. Vona R, Giovannetti A, Gambardella L, Malorni W, Pietraforte D, Straface E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J Cell Mol Med. (2018) 22:3308–14. doi: 10.1111/jcmm.2018.22.issue-7
76. Abdulle AE, Diercks GFH, Feelisch M, Mulder DJ, van Goor H. The role of oxidative stress in the development of systemic sclerosis related vasculopathy. Front Physiol. (2018) 9:1177. doi: 10.3389/fphys.2018.01177
77. Doridot L, Jeljeli M, Chêne C, Batteux F. Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol. (2019) 25:101122. doi: 10.1016/j.redox.2019.101122
78. Piera-Velazquez S, Makul A, Jiménez SA. Increased expression of NAPDH oxidase 4 in systemic sclerosis dermal fibroblasts: regulation by transforming growth factor β. Arthritis Rheumatol. (2015) 67:2749–58. doi: 10.1002/art.v67.10
79. Spadoni T, Svegliati Baroni S, Amico D, Albani L, Moroncini G, Avvedimento EV, et al. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. (2015) 67:1611–22. doi: 10.1002/art.39084
80. Svegliati S, Spadoni T, Moroncini G, Gabrielli A. NADPH oxidase, oxidative stress and fibrosis in systemic sclerosis. Free Radic Biol Med. (2018) 125:90–7. doi: 10.1016/j.freeradbiomed.2018.04.554
81. Piera-Velazquez S, Jimenez SA. Oxidative stress induced by reactive oxygen species (ROS) and NADPH oxidase 4 (NOX4) in the pathogenesis of the fibrotic process in systemic sclerosis: A promising therapeutic target. J Clin Med. (2021) 10:4791. doi: 10.3390/jcm10204791
82. Li K, Wang Q, Bian B, Xu J, Bian H. Exploration and validation of the hub genes involved in hypoxia-induced endothelial-mesenchymal transition of systemic sclerosis. Clin Exp Rheumatol. (2023) 41:1618–31. doi: 10.55563/clinexprheumatol/j7ema8
83. Dooley A, Shi-Wen X, Aden N, Tranah T, Desai N, Denton CP, et al. Modulation of collagen type I, fibronectin and dermal fibroblast function and activity, in systemic sclerosis by the antioxidant epigallocatechin-3-gallate. Rheumatol (Oxford). (2010) 49:2024–36. doi: 10.1093/rheumatology/keq208
84. Dosoki H, Stegemann A, Taha M, Schnittler H, Luger TA, Schröder K, et al. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp Dermatol. (2017) 26:73–81. doi: 10.1111/exd.2017.26.issue-1
85. Furst DE, Clements PJ, Harris R, Ross M, Levy J, Paulus HE. Measurement of clinical change in progressive systemic sclerosis: A 1 year double-blind placebo-controlled trial of N-acetylcysteine. Ann Rheum Dis. (1979) 38:356–61. doi: 10.1136/ard.38.4.356
86. Rosato E, Rossi C, Molinaro I, Giovannetti A, Pisarri S, Salsano F. Long-term N-acetylcysteine therapy in systemic sclerosis interstitial lung disease: A retrospective study. Int J Immunopathol Pharmacol. (2011) 24:727–33. doi: 10.1177/039463201102400319
87. Mehrabi S, Moradi MM, Khodamoradi Z, Nazarinia MA. Effects of N-acetylcysteine on pulmonary functions in patients with systemic sclerosis: A randomized double blind, placebo controlled study. Curr Rheumatol Rev. (2020) 16:149–57. doi: 10.2174/1573397115666191212092608
88. Volpe A, Biasi D, Caramaschi P, Mantovani W, Bambara LM, Canestrini S, et al. Levels of F2-isoprostanes in systemic sclerosis: correlation with clinical features. Rheumatol (Oxford). (2006) 45:314–20. doi: 10.1093/rheumatology/kei151
89. Brezovec N, Perdan-Pirkmajer K, Burja B, Rotar Ž, Osredkar J, Sodin-Šemrl S, et al. Disturbed antioxidant capacity in patients with systemic sclerosis associates with lung and gastrointestinal symptoms. Biomedicines. (2023) 11:2110. doi: 10.3390/biomedicines11082110
90. Kizilay Mancini O, Acevedo M, Fazez N, Cuillerier A, Fernandez Ruiz A, Huynh DN, et al. Oxidative stress-induced senescence mediates inflammatory and fibrotic phenotypes in fibroblasts from systemic sclerosis patients. Rheumatol (Oxford). (2022) 61:1265–75. doi: 10.1093/rheumatology/keab477
91. Zhang R, Kumar GS, Hansen U, Zoccheddu M, Sacchetti C, Holmes ZJ, et al. Oxidative stress promotes fibrosis in systemic sclerosis through stabilization of a kinase-phosphatase complex. JCI Insight. (2022) 7:e155761. doi: 10.1172/jci.insight.155761
92. Gabbiani G, Ryan GB, Majno G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. (1971) 127:549–50. doi: 10.1007/BF02147594
93. Ryan GB, Cliff WJ, Gabbiani G, Irlé C, Montandon D, Statkov PR, et al. Myofibroblasts in human granulation tissue. Hum Pathol. (1974) 5:55–67. doi: 10.1016/S0046-8177(74)80100-0
94. McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. (2007) 39:666–71. doi: 10.1016/j.biocel.2006.11.005
95. Kirk TZ, Mark ME, Chua CC, Chua BH, Mayes MD. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J Biol Chem. (1995) 270:3423–8. doi: 10.1074/jbc.270.7.3423
96. Abraham DJ, Eckes B, Rajkumar V, Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr Rheumatol Rep. (2007) 9:136–43. doi: 10.1007/s11926-007-0008-z
97. Gyftaki-Venieri DA, Abraham DJ, Ponticos M. Insights into myofibroblasts and their activation in scleroderma: opportunities for therapy? Curr Opin Rheumatol. (2018) 30:581–87. doi: 10.1097/BOR.0000000000000543
98. van Caam A, Vonk M, van den Hoogen F, van Lent P, van der Kraan P. Unraveling SSc pathophysiology; the myofibroblast. Front Immunol. (2018) 9:2452. doi: 10.3389/fimmu.2018.02452
99. Chadli L, Sotthewes B, Li K, Andersen SN, Cahir-McFarland E, Cheung M, et al. Identification of regulators of the myofibroblast phenotype of primary dermal fibroblasts from early diffuse systemic sclerosis patients. Sci Rep. (2019) 9:4521. doi: 10.1038/s41598-019-41153-w
100. Bellando-Randone S, Del Galdo F, Matucci-Cerinic M. Insights into molecular and clinical characteristics of very early systemic sclerosis. Curr Opin Rheumatol. (2022) 34:351–6. doi: 10.1097/BOR.0000000000000903
101. Kahaleh MB, LeRoy EC. Effect of scleroderma serum on human fibroblast collagen production: possible selection through proliferation. J Rheumatol. (1986) 13:99–102.
102. Perlish JS, Fleischmajer R. Immunological modulation of dermal fibroblasts in scleroderma. Immunol Ser. (1989) 46:605–24.
103. Kähäri VM. Activation of dermal connective tissue in scleroderma. Ann Med. (1993) 25:511–8. doi: 10.1080/07853890.1993.12088576
104. Chesney J, Bucala R. Peripheral blood fibrocytes: mesenchymal precursor cells and the pathogenesis of fibrosis. Curr Rheumatol Rep. (2000) 2:501–5. doi: 10.1007/s11926-000-0027-5
105. Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. (2006) 8:145–50. doi: 10.1007/s11926-006-0055-x
106. Bischoff J. Endothelial-to-mesenchymal transition. Circ Res. (2019) 124:1163–65. doi: 10.1161/CIRCRESAHA.119.314813
107. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. (2019) 99:1281–324. doi: 10.1152/physrev.00021.2018
108. Di Benedetto P, Ruscitti P, Berardicurti O, Vomero M, Navarini L, Dolo V, et al. Endothelial-to-mesenchymal transition in systemic sclerosis. Clin Exp Immunol. (2021) 205:12–27. doi: 10.1111/cei.13599
109. Cipriani P, Di Benedetto P, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol. (2015) 42:1808–16. doi: 10.3899/jrheum.150088
110. Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. (2017) 76:924–34. doi: 10.1136/annrheumdis-2016-210229
111. Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA, et al. Potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol. (2018) 9:1985. doi: 10.3389/fimmu.2018.01985
112. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. (2003) 112:1776–84. doi: 10.1172/JCI20530
113. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
114. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
115. Nikitorowicz-Buniak J, Denton CP, Abraham D, Stratton R. Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased TGFβ Signaling within lesional scleroderma skin. PloS One. (2015) 10:e0134092. doi: 10.1371/journal.pone.0134092
116. Asano Y, Takahashi T, Saigusa R. Systemic sclerosis: Is the epithelium a missing piece of the pathogenic puzzle? J Dermatol Sci. (2019) 94:259–65. doi: 10.1016/j.jdermsci.2019.04.007
117. Sarrand J, Soyfoo MS. Involvement of epithelial-mesenchymal transition (EMT) in autoimmune diseases. Int J Mol Sci. (2023) 24:14481. doi: 10.3390/ijms241914481
118. Manetti M, Romano E, Rosa I, Fioretto BS, Praino E, Guiducci S, et al. Systemic sclerosis serum steers the differentiation of adipose-derived stem cells toward profibrotic myofibroblasts: pathophysiologic implications. J Clin Med. (2019) 8:1256. doi: 10.3390/jcm8081256
119. Marangoni RG, Korman B, Varga J. Adipocytic progenitor cells give rise to pathogenic myofibroblasts: adipocyte-to-mesenchymal transition and its emerging role in fibrosis in multiple organs. Curr Rheumatol Rep. (2020) 22:79. doi: 10.1007/s11926-020-00957-w
120. Jelaska A, Korn JH. Role of apoptosis and transforming growth factor β1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheumatol. (2000) 43:2230–9. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8
121. Santiago B, Galindo M, Rivero M, Pablos JL. Decreased susceptibility to Fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis Rheumatol. (2001) 44:1667–76. doi: 10.1002/1529-0131(200107)44:7<1667::AID-ART291>3.0.CO;2-Y
122. Kozlova A, Pachera E, Maurer B, Jüngel A, Distler JHW, Kania G, et al. Regulation of fibroblast apoptosis and proliferation by microRNA-125b in systemic sclerosis. Arthritis Rheumatol. (2019) 71:2068–80. doi: 10.1002/art.v71.12
123. Chiu YH, Spierings J, van Laar JM, de-Vries-Bouwstra JK, van Dijk M, Goldschmeding R. Association of endothelial to mesenchymal transition and cellular senescence with fibrosis in skin biopsies of systemic sclerosis patients: a cross-sectional study. Clin Exp Rheumatol. (2023) 41:1612–17. doi: 10.55563/clinexprheumatol/i49d3o
124. Shiwen X, Stratton R, Nikitorowicz-Buniak J, Ahmed-Abdi B, Ponticos M, Denton C, et al. A role of myocardin related transcription factor-A (MRTF-A) in scleroderma related fibrosis. PloS One. (2015) 10:e0126015. doi: 10.1371/journal.pone.0126015
125. Rosa I, Romano E, Fioretto BS, El Aoufy K, Bellando-Randone S, Matucci-Cerinic M, et al. Lymphatic endothelial-to-myofibroblast transition: A potential new mechanism underlying skin fibrosis in systemic sclerosis. Cells. (2023) 12:2195. doi: 10.3390/cells12172195
126. Jimenez SA, Castro SV, Piera-Velazquez M. Role of Growth factors in the pathogenesis of tissue fibrosis in Systemic Sclerosis. Curr Rheumatol Rev. (2010) 6:283–94. doi: 10.2174/157339710793205611
127. Medici D, Potenta S, Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. (2011) 437:515–20. doi: 10.1042/BJ20101500
128. Wermuth PJ, Li Z, Mendoza FA, Jimenez SA. Stimulation of transforming growth factor-β1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): A novel profibrotic effect of ET-1. PloS One. (2016) 11:e0161988. doi: 10.1371/journal.pone.0161988
129. Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P. TGF-beta-induced endothelial-mesenchymal transition in fibrotic diseases. Int J Mol Sci. (2017) 18:2157. doi: 10.3390/ijms18102157
130. Ming Z, Vining B, Bagheri-Fam S, Harley V. SOX9 in organogenesis: shared and unique transcriptional functions. Cell Mol Life Sci. (2022) 79:522. doi: 10.1007/s00018-022-04543-4
131. Waldrep KM, Rodgers JI, Garrett SM, Wolf BJ, Feghali-Bostwick CA. The role of SOX9 in IGF-II-mediated pulmonary fibrosis. Int J Mol Sci. (2023) 24:11234. doi: 10.3390/ijms241411234
132. Fuglerud BM, Drissler S, Lotto J, Stephan TL, Thakur A, Cullum R, et al. SOX9 reprograms endothelial cells by altering the chromatin landscape. Nucleic Acids Res. (2022) 50:8547– 65. doi: 10.1093/nar/gkac652
133. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. (2006) 99:675–91. doi: 10.1161/01.RES.0000243584.45145.3f
134. Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, et al. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol. (2006) 148:714–23. doi: 10.1038/sj.bjp.0706779
135. Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. (2015) 67:1062–73. doi: 10.1002/art.38990
136. Brezovec N, Burja B, Lakota K. Adipose tissue and adipose secretome in systemic sclerosis. Curr Opin Rheumatol. (2021) 33:505–13. doi: 10.1097/BOR.0000000000000838
137. Cho H, Kozasa T, Bondjers C, Betsholtz C, Kehrl JH. Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation. FASEB J. (2003) 17:440–2. doi: 10.1096/fj.02-0340fje
138. Manzur M, Ganss R. Regulator of G protein signaling 5: a new player in vascular remodeling. Trends Cardiovasc Med. (2009) 1):26–30. doi: 10.1016/j.tcm.2009.04.002
139. Helmbold P, Fiedler E, Fischer M, WCh M. Hyperplasia of dermal microvascular pericytes in scleroderma. J Cutan Pathol. (2004) 31:431–40. doi: 10.1111/j.0303-6987.2004.00203.x
140. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. (2012) 18:1262–70. doi: 10.1038/nm.2848
141. Lescoat A, Lecureur V, Varga J. Contribution of monocytes and macrophages to the pathogenesis of systemic sclerosis: recent insights and therapeutic implications. Curr Opin Rheumatol. (2021) 33:463–70. doi: 10.1097/BOR.0000000000000835
142. Fang D, Chen B, Lescoat A, Khanna D, Mu R. Immune cell dysregulation as a mediator of fibrosis in systemic sclerosis. Nat Rev Rheumatol. (2022) 18:683–93. doi: 10.1038/s41584-022-00864-7
143. François A, Chatelus E, Wachsmann D, Sibilia J, Bahram S, Alsaleh G, et al. B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthritis Res Ther. (2013) 15:R168. doi: 10.1186/ar4352
144. Kawaguchi Y, Kuwana M. Pathogenesis of vasculopathy in systemic sclerosis and its contribution to fibrosis. Curr Opin Rheumatol. (2023) 35:309–16. doi: 10.1097/BOR.0000000000000959
145. Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA Jr. Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheumatol. (2013) 65:236–46. doi: 10.1002/art.37706
146. Roeb E. Interleukin-13 (IL-13)-A pleiotropic cytokine involved in wound healing and fibrosis. Int J Mol Sci. (2023) 24:12884. doi: 10.3390/ijms241612884
147. Yaseen B, Lopez H, Taki Z, Zafar S, Rosario H, Abdi BA, et al. Interleukin-31 promotes pathogenic mechanisms underlying skin and lung fibrosis in scleroderma. Rheumatol (Oxford). (2020) 59:2625–36. doi: 10.1093/rheumatology/keaa195
148. Kuzumi A, Yoshizaki A, Matsuda KM, Kotani H, Norimatsu Y, Fukayama M, et al. Interleukin-31 promotes fibrosis and T helper 2 polarization in systemic sclerosis. Nat Commun. (2021) 12:5947. doi: 10.1038/s41467-021-26099-w
149. Pellicano C, Vantaggio L, Colalillo A, Pocino K, Basile V, Marino M, et al. Type 2 cytokines and scleroderma interstitial lung disease. Clin Exp Med. (2023) 23:3517–25. doi: 10.1007/s10238-023-01125-x
150. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
151. Dees C, Chakraborty D, Distler JHW. Cellular and molecular mechanisms in fibrosis. Exp Dermatol. (2021) 30:121–31. doi: 10.1111/exd.14193
152. Lurje I, Gaisa NT, Weiskirchen R, Tacke F. Mechanisms of organ fibrosis: Emerging concepts and implications for novel treatment strategies. Mol Aspects Med. (2023) 92:101191. doi: 10.1016/j.mam.2023.101191
153. Frangogiannis NG. Transforming growth factor–β in tissue fibrosis. J Exp Med. (2020) 217:e20190103. doi: 10.1084/jem.20190103
154. Lodyga M, Hinz B. TGF-β1-a truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol. (2020) 101:123–39. doi: 10.1016/j.semcdb.2019.12.010
155. Varga J, Jimenez SA. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem Biophys Res Commun. (1986) 138:974–80. doi: 10.1016/S0006-291X(86)80591-5
156. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U.S.A. (1986) 83:4167–71. doi: 10.1073/pnas.83.12.4167
157. Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. (1987) 247:597–604. doi: 10.1042/bj2470597
158. Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. (2005) 280:7409–12. doi: 10.1074/jbc.R400029200
159. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature. (2011) 474:343–9. doi: 10.1038/nature10152
160. Rifkin D, Sachan N, Singh K, Sauber E, Tellides G, Ramirez F. The role of LTBPs in TGF beta signaling. Dev Dyn. (2022) 251:95–104. doi: 10.1002/dvdy.331
161. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. (2003) 113:685–700. doi: 10.1016/s0092-8674(03)00432-x
162. Budi EH, Duan D, Derynck R. Transforming growth factor-β Receptors and smads: regulatory complexity and functional versatility. Trends Cell Biol. (2017) 27:658–72. doi: 10.1016/j.tcb.2017.04.005
163. Vander Ark A, Cao J, Li X. TGF-β receptors: In and beyond TGF-β signaling. Cell Signal. (2018) 52:112–20. doi: 10.1016/j.cellsig.2018.09.002
164. Aashaq S, Batool A, Mir SA, Beigh MA, Andrabi KI, Shah ZA. TGF-β signaling: A recap of SMAD-independent and SMAD-dependent pathways. J Cell Physiol. (2022) 237:59–85. doi: 10.1002/jcp.v237.1
165. Varga J. Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheumatol. (2002) 46:1703–13. doi: 10.1002/art.10413
166. Yan X, Liao H, Cheng M, Shi X, Lin X, Feng XH, et al. Smad7 protein interacts with receptor-regulated smads (R-smads) to inhibit transforming growth factor-β (TGF-β)/smad signaling. J Biol Chem. (2016) 291:382–92. doi: 10.1074/jbc.M115.694281
167. Finnson KW, Almadani Y, Philip A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Semin Cell Dev Biol. (2020) 101:115–22. doi: 10.1016/j.semcdb.2019.11.013
168. Bale S, Verma P, Yalavarthi B, Scarneo SA, Hughes P, Amin MA, et al. Pharmacological inhibition of TAK1 prevents and induces regression of experimental organ fibrosis. JCI Insight. (2023) 8:e165358. doi: 10.1172/jci.insight.165358
169. Karimizadeh E, Motamed N, Mahmoudi M, Jafarinejad-Farsangi S, Jamshidi A, Faridani H, et al. Attenuation of fibrosis with selective inhibition of c-Abl by siRNA in systemic sclerosis dermal fibroblasts. Arch Dermatol Res. (2015) 307:135–42. doi: 10.1007/s00403-014-1532-0
170. Piera-Velazquez S, Jimenez SA. Simultaneous inhibition of c-Abl and Src kinases abrogates the exaggerated expression of profibrotic genes in cultured systemic sclerosis dermal fibroblasts. Clin Exp Rheumatol. (2018) 36 Suppl 113:36–44.
171. Igarashi A, Bradham DM, Okochi H, Grotendorst GR. Connective tissue growth factor. J Dermatol. (1992) 19:642–43. doi: 10.1111/j.1346-8138.1992.tb03748.x
172. Chen Z, Zhang N, Chu HY, Yu Y, Zhang ZK, Zhang G, et al. Connective tissue growth factor: from molecular understandings to drug discovery. Front Cell Dev Biol. (2020) 8:593269. doi: 10.3389/fcell.2020.593269
173. Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. (1997) 8:171–9. doi: 10.1016/S1359-6101(97)00010-5
174. Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Grotendorst GR, et al. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J Invest Dermatol. (1995) 105:280–4. doi: 10.1111/1523-1747.ep12318465
175. Sato S, Nagaoka T, Hasegawa M, Tamatani T, Nakanishi T, Takigawa M, et al. Serum levels of connective tissue growth factor are elevated in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. J Rheumatol. (2000) 27:149–54.
176. Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol. (2004) 122:1–6. doi: 10.1046/j.0022-202X.2003.22133.x
177. Henrot P, Truchetet ME, Fisher G, Taïeb A, Cario M. CCN proteins as potential actionable targets in scleroderma. Exp Dermatol. (2019) 28:11–8. doi: 10.1111/exd.2019.28.issue-1
178. Tejera-Muñoz A, Marquez-Exposito L, Tejedor-Santamaría L, Rayego-Mateos S, Orejudo M, Suarez-Álvarez B, et al. CCN2 increases TGF-β Receptor type II expression in vascular smooth muscle cells: essential role of CCN2 in the TGF-β Pathway regulation. Int J Mol Sci. (2021) 23:375. doi: 10.3390/ijms23010375
179. Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatol (Oxford). (2008) 47 Suppl 5:v2–4. doi: 10.1093/rheumatology/ken265
180. Ludwicka A, Ohba T, Trojanowska M, Yamakage A, Strange C, Smith EA, et al. Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol. (1995) 22:1876–83.
181. Iwayama T, Olson LE. Involvement of PDGF in fibrosis and scleroderma: recent insights from animal models and potential therapeutic opportunities. Curr Rheumatol Rep. (2013) 15:304. doi: 10.1007/s11926-012-0304-0
182. Paolini C, Agarbati S, Benfaremo D, Mozzicafreddo M, Svegliati S, Moroncini G. PDGF/PDGFR: A possible molecular target in scleroderma fibrosis. Int J Mol Sci. (2022) 23:3904. doi: 10.3390/ijms23073904
183. Thomas KA. Fibroblast growth factors. FASEB J. (1987) 1:434–40. doi: 10.1096/fasebj.1.6.3315806
184. Gospodarowicz D, Neufeld G, Schweigerer L. Fibroblast growth factor: structural and biological properties. J Cell Physiol Suppl. (1987) S5:15–26. doi: 10.1002/(ISSN)1097-4652
185. Ichiki Y, Smith EA, LeRoy EC, Trojanowska M. Basic fibroblast growth factor inhibits basal and transforming growth factor-beta induced collagen α 2(I) gene expression in scleroderma and normal fibroblasts. J Rheumatol. (1997) 24:90–5.
186. Shimbori C, Bellaye PS, Xia J, Gauldie J, Ask K, Ramos C, et al. Fibroblast growth factor-1 attenuates TGF-β1-induced lung fibrosis. J Pathol. (2016) 240:197–210. doi: 10.1002/path.4768
187. Lawrence A, Khanna D, Misra R, Aggarwal A. Increased expression of basic fibroblast growth factor in skin of patients with systemic sclerosis. Dermatol Online J. (2006) 12:2. doi: 10.5070/D36S6582VR
188. Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. (1989) 161:851–8. doi: 10.1016/0006-291X(89)92678-8
189. Gospodarowicz D, Abraham JA, Schilling J. Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary-derived folliculo stellate cells. Proc Natl Acad Sci U S A. (1989) 86:7311–5. doi: 10.1073/pnas.86.19.7311
190. Pérez-Gutiérrez L, Ferrara N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat Rev Mol Cell Biol. (2023) 24:816–34. doi: 10.1038/s41580-023-00631-w
191. Choi JJ, Min DJ, Cho ML, Min SY, Kim SJ, Lee SS, et al. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. (2003) 30:1529–33.
192. Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. (2004) 95:109–16. doi: 10.1161/01.RES.0000134644.89917.96
193. Flower VA, Barratt SL, Ward S, Pauling JD. The role of vascular endothelial growth factor in systemic sclerosis. Curr Rheumatol Rev. (2019) 15:99–109. doi: 10.2174/1573397114666180809121005
194. Salmon WD Jr., Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. (1957) 49:825–36.
195. LeRoith D, Holly JMP, Forbes BE. Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol Metab. (2021) 52:101245. doi: 10.1016/j.molmet.2021.101245
196. Hsu E, Feghali-Bostwick CA. Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via jun N-terminal kinase- and phosphatidylinositol-3 kinase-dependent pathways. Am J Pathol. (2008) 172:1580–90. doi: 10.2353/ajpath.2008.071021
197. Garrett SM, Hsu E, Thomas JM, Pilewski JM, Feghali-Bostwick C. Insulin-like growth factor (IGF)-II- mediated fibrosis in pathogenic lung conditions. PloS One. (2019) 14:e0225422. doi: 10.1371/journal.pone.0225422
198. Hamaguchi Y, Fujimoto M, Matsushita T, Hasegawa M, Takehara K, Sato S. Elevated serum insulin-like growth factor (IGF-1) and IGF binding protein-3 levels in patients with systemic sclerosis: possible role in development of fibrosis. J Rheumatol. (2008) 35:2363–71. doi: 10.3899/jrheum.080340
199. Luo Y, Wang Y, Shu Y, Lu Q, Xiao R. Epigenetic mechanisms: An emerging role in pathogenesis and its therapeutic potential in systemic sclerosis. Int J Biochem Cell Biol. (2015) 67:92–100. doi: 10.1016/j.biocel.2015.05.023
200. Liu Y, Cheng L, Zhan H, Li H, Li X, Huang Y, et al. The roles of noncoding RNAs in systemic sclerosis. Front Immunol. (2022) 13:856036. doi: 10.3389/fimmu.2022.856036
201. Zhang H, Zhou Y, Wen D, Wang J. Noncoding RNAs: master regulator of fibroblast to myofibroblast transition in fibrosis. Int J Mol Sci. (2023) 24:1801. doi: 10.3390/ijms24021801
202. Henry TW, Mendoza FA, Jimenez SA. Role of microRNA in the pathogenesis of systemic sclerosis tissue fibrosis and vasculopathy. Autoimmun Rev. (2019) 18:102396. doi: 10.1016/j.autrev.2019.102396
203. Truchetet ME, Brembilla NC, Chizzolini C. Current concepts on the pathogenesis of systemic sclerosis. Clin Rev Allergy Immunol. (2023) 64:262–83. doi: 10.1007/s12016-021-08889-8
204. Wajda A, Walczyk M, Dudek E, Stypińska B, Lewandowska A, Romanowska-Próchnicka K, et al. Serum microRNAs in systemic sclerosis, associations with digital vasculopathy and lung involvement. Int J Mol Sci. (2022) 23:10731. doi: 10.3390/ijms231810731
205. Peng WJ, Tao JH, Mei B, Chen B, Li BZ, Yang GJ, et al. MicroRNA-29: a potential therapeutic target for systemic sclerosis. Expert Opin Ther Targets. (2012) 16:875–9. doi: 10.1517/14728222.2012.708339
206. Gallant-Behm CL, Piper J, Lynch JM, Seto AG, Hong SJ, Mustoe TA, et al. A microRNA-29 mimic (Remlarsen) represses extracellular matrix expression and fibroplasia in the skin. J Invest Dermatol. (2019) 139:1073–81. doi: 10.1016/j.jid.2018.11.007
207. Wasson CW, Abignano G, Hermes H, Malaab M, Ross RL, Jimenez SA, et al. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of NOTCH. Ann Rheum Dis. (2020) 79:507–17. doi: 10.1136/annrheumdis-2019-216542
208. Du Y, Faz-Lopez B, Ah Kioon MD, Cenac C, Pierides M, Lakin KS, et al. Altered X-chromosome inactivation of the TLR7/8 locus and heterogeneity of pDCs in systemic sclerosis. J Exp Med. (2025) 222:e20231809. doi: 10.1084/jem.20231809
209. Wang Z, Jinnin M, Nakamura K, Harada M, Kudo H, Nakayama W, et al. Long non-coding RNA TSIX is upregulated in scleroderma dermal fibroblasts and controls collagen mRNA stabilization. Exp Dermatol. (2016) 25:131–6. doi: 10.1111/exd.2016.25.issue-2
210. Shumnalieva R, Kachakova D, Kaneva R, Kolarov Z, Monov S. Serum miR-21 and miR-29a expression in systemic sclerosis patients. Clin Exp Rheumatol. (2023) 41:1688–94. doi: 10.55563/clinexprheumatol/165gj5
211. Ciechomska M, Zarecki P, Merdas M, Swierkot J, Morgiel E, Wiland P, et al. The role of microRNA-5196 in the pathogenesis of systemic sclerosis. Eur J Clin Invest. (2017) 47:555–64. doi: 10.1111/eci.12776
212. Rossato M, Affandi AJ, Thordardottir S, Wichers C, Cossu M, Broen J, et al. Association of microRNA-618 expression with altered frequency and activation of plasmacytoid dendritic cells in patients with systemic sclerosis. Arthritis Rheumatol. (2017) 69:1891–902. doi: 10.1002/art.40163
213. Chouri E, Wang M, Hillen MR, Angiolilli C, Silva-Cardoso SC, Wichers C, et al. Implication of miR-126 and miR-139-5p in Plasmacytoid Dendritic Cell Dysregulation in Systemic Sclerosis. J Clin Med. (2021) 10:491. doi: 10.3390/jcm10030491
214. Chouri E, Servaas NH, Bekker C, Affandi AJ, Cossu M, Hillen MR, et al. Serum microRNA Screening and Functional Studies Reveal miR-483-5p as a Potential Driver of Fibrosis in Systemic Sclerosis. J Autoimmun. (2018) 89:162–70. doi: 10.1016/j.jaut.2017.12.015
215. Shi X, Liu Q, Li N, Tu W, Luo R, Mei X, et al. MiR-3606-3p inhibits systemic sclerosis through targeting TGF-beta type II receptor. Cell Cycle. (2018) 17:1967–78. doi: 10.1080/15384101.2018.1509621
216. Yao Q, Xing Y, Wang Z, Liang J, Lin Q, Huang M, et al. MiR-16-5p suppresses myofibroblast activation in systemic sclerosis by inhibiting NOTCH signaling. Aging (Albany NY). (2020) 13:2640–54. doi: 10.18632/aging.202308
217. Luo H, Zhu H, Zhou B, Xiao X, Zuo X. MicroRNA-130b regulates scleroderma fibrosis by targeting peroxisome proliferator-activated receptor gamma. Mod Rheumatol. (2015) 25:595–602. doi: 10.3109/14397595.2014.1001311
218. Li Y, Huang J, Hu C, Zhou J, Xu D, Hou Y, et al. MicroRNA-320a: an important regulator in the fibrotic process in interstitial lung disease of systemic sclerosis. Arthritis Res Ther. (2021) 23:21. doi: 10.1186/s13075-020-02411-9
219. Zhu H, Luo H, Li Y, Zhou Y, Jiang Y, Chai J, et al. MicroRNA-21 in scleroderma fibrosis and its function in TGF-β-regulated fibrosis-related genes expression. J Clin Immunol. (2013) 33:1100–9. doi: 10.1007/s10875-013-9896-z
220. Li H, Yang R, Fan X, Gu T, Zhao Z, Chang D, et al. MicroRNA array analysis of microRNAs related to systemic scleroderma. Rheumatol Int. (2012) 32:307–13. doi: 10.1007/s00296-010-1615-y
221. Cheng Q, Chen M, Wang H, Chen X, Wu H, Du Y, et al. MicroRNA-27a-3p inhibits lung and skin fibrosis of systemic sclerosis by negatively regulating SPP1. Genomics. (2022) 114:110391. doi: 10.1016/j.ygeno.2022.110391
222. Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheumatol. (2010) 62:1733–43. doi: 10.1002/art.27443
223. Tanaka S, Suto A, Ikeda K, Sanayama Y, Nakagomi D, Iwamoto T, et al. Alteration of circulating miRNAs in SSc: miR-30b regulates the expression of PDGF receptor β. Rheumatol Oxf Engl. (2013) 52:1963–72. doi: 10.1093/rheumatology/ket254
224. Zhou B, Zuo XX, Li YS, Gao SM, Dai XD, Zhu HL, et al. Integration of microRNA and mRNA expression profiles in the skin of systemic sclerosis patients. Sci Rep. (2017) 7:42899. doi: 10.1038/srep42899
225. Henderson J, Pryzborski S, Stratton R, O’Reilly S. Wnt antagonist DKK-1 levels in systemic sclerosis are lower in skin but not in blood and are regulated by microRNA33a-3p. Exp Dermatol. (2021) 30:162–8. doi: 10.1111/exd.14136
226. Sing T, Jinnin M, Yamane K, Honda N, Makino K, Kajihara I, et al. microRNA-92a expression in the sera and dermal fibroblasts increases in patients with scleroderma. Rheumatol Oxf. Engl. (2012) 51:1550–6. doi: 10.1093/rheumatology/kes120
227. Nakashima T, Jinnin M, Yamane K, Honda N, Kajihara I, Makino T, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. (2012) 188:3573–83. doi: 10.4049/jimmunol.1100591
228. Makino K, Jinnin M, Kajihara I, Honda N, Sakai K, Masuguchi S, et al. Circulating miR- 142-3p levels in patients with systemic sclerosis. Clin Exp Dermatol. (2012) 37:34–9. doi: 10.1111/j.1365-2230.2011.04158.x
229. Honda N, Jinnin M, Kira-Etoh T, Makino K, Kajihara I, Makino T, et al. miR-150 down- regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin β3. Am J Pathol. (2013) 182:206–16. doi: 10.1016/j.ajpath.2012.09.023
230. Alivernini S, Bosello SL, Luca GD, Canestri S, Mario CD, Gigante MR, et al. A3.21 MicroRNA-34a and microRNA-155 in systemic sclerosis: possible epigenetic bio-markers of endothelial dysfunction in VEDOSS and long-standing disease. . Ann Rheumatol Dis. (2014) 73:A50.
231. Honda N, Jinnin M, Kajihara I, Makino T, Makino K, Masuguchi S, et al. TGF-β-mediated downregulation of microRNA-196a contributes to the constitutive up regulated type I collagen expression in scleroderma dermal fibroblasts. J Immunol. (2012) 188:3323–31. doi: 10.4049/jimmunol.1100876
232. Zhou B, Zhu H, Luo H, Gao S, Dai X, Li Y, et al. MicroRNA-202-3p regulates scleroderma fibrosis by targeting matrix metalloproteinase 1. BioMed Pharmacother. (2017) 87:412–418.241. doi: 10.1016/j.biopha.2016.12.080
233. Nakayama W, Jinnin M, Tomizawa Y, Nakamura K, Kudo H, Inoue K, et al. Dysregulated interleukin-23 signalling contributes to the increased collagen production in scleroderma fibroblasts via balancing microRNA expression. Rheumatol (Oxford). (2017) 56:145–155.243. doi: 10.1093/rheumatology/kew336
234. Makino K, Jinnin M, Hirano A, Yamane K, Eto M, Kusano T, et al. The down-regulation of microRNA let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J Immunol. (2013) 190:3905–15. doi: 10.4049/jimmunol.1200822
Keywords: Systemic Sclerosis, pathogenesis, myofibroblast, endothelial cell, miRNA - microRNA, lncRNA - long non-coding RNA, fibrosis, TGF
Citation: Jimenez SA, Mendoza FA and Piera-Velazquez S (2025) A review of recent studies on the pathogenesis of Systemic Sclerosis: focus on fibrosis pathways. Front. Immunol. 16:1551911. doi: 10.3389/fimmu.2025.1551911
Received: 26 December 2024; Accepted: 07 March 2025;
Published: 16 April 2025.
Edited by:
Lazaros Ignatios Sakkas, University of Thessaly, GreeceReviewed by:
Raffaele De Palma, University of Genoa, ItalyCopyright © 2025 Jimenez, Mendoza and Piera-Velazquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio A. Jimenez, c2VyZ2lvLmppbWVuZXpAamVmZmVyc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.