
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 April 2025
Sec. Microbial Immunology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1549293
This article is part of the Research TopicModulation of Pulmonary Immunity and Function by Bacterial and Host MetabolitesView all 4 articles
 Gaurav Kumar Lohia
Gaurav Kumar Lohia Sebastián A. Riquelme*
Sebastián A. Riquelme*Pulmonary diseases, arising from infections caused by bacteria, fungi, and viruses, or stemming from underlying genetic factors are one of the leading causes of mortality in humans, accounting for millions of deaths every year. At the onset of pulmonary diseases, crucial roles are played by phagocytic immune cells, particularly tissue-resident macrophages, in regulating the immune response at the mucosal barrier. Recent strides have illuminated the pivotal role of host bioenergetics modulated by metabolites derived from both pathogens and hosts in influencing the pathophysiology of major organs. Their influence extends to processes such as the infiltration of immune cells, activation of macrophages, and the polarization phenomenon. Furthermore, host-derived metabolites, such as itaconate, contribute to the promotion of anti-inflammatory responses, thereby preventing immunopathology and facilitating the preservation of mucosal niches to thrive for the long-term. This review explores recent advancements in the field of immunometabolism, with a particular emphasis on the intricacies of disease progression in pulmonary infections caused by bacteria such as P. aeruginosa, M. tuberculosis and S. aureus and fungi like C. albicans.
According to recent epidemiology studies, pulmonary infections caused by pathogens such as Pseudomonas aeruginosa, Mycobacterium tuberculosis, Staphylococcus aureus and Candida albicans represent significant contributors to global mortality (1, 2). These infections are frequently observed in individuals with co-morbidities, such as cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), and primary ciliary dyskinesia (PCD), as well as during ventilator-associated pneumonia (VAP), which complicate prognosis and disease management (3–9). In the lung, these pathogens trigger a brisk inflammatory response that often aids in infection clearance. However, in specific contexts, this immune response is either insufficient or aberrant, leading to tissue damage, pathogen persistence, and chronic colonization. Multiple immunomodulatory platforms, including metabolites, cytokines, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) influence the outcome of the immune response (10). Despite advances in understanding these mediators, their role in modulating mucosal integrity through host bioenergetics remains an emerging area of study. This review examines recent progress in immunometabolism research, with particular emphasis on host bioenergetics during pulmonary infection by these pathogens.
Host cell primarily rely on two pathways for producing energy; namely, glycolysis and oxidative phosphorylation (OXPHOS). These routes provide cells with ATP and substrates for various cellular processes. However, different cell types specialize in specific pathways to maintain their bioenergetic integrity. For example, neurons rely heavily on OXPHOS as they lack key components of the glycolytic pathway (11). Similarly, heart generates ~90% of its energy from this same mitochondrial platform (12). Red blood cells, on other hand, use glycolysis to synthesize ATP, as they lack mitochondria (13). Brown adipose tissue (BAT), which is involved in thermogenesis, uses the energy generated from fatty acid oxidation (FAO) in mitochondria to produce heat instead of ATP (14, 15). In contrast, immune cells, such as macrophages exhibit remarkable plasticity in their bioenergetic pathways. This bioenergetic dynamics plays a crucial role in regulating their immunological properties. Pro-inflammatory responses in macrophages are predominantly associated with enhanced glycolysis and impaired OXPHOS (16). Consistently, anti-inflammatory responses are marked by functional OXPHOS and many networks that fuel this platform, such as FAO and the tricarboxylic acid (TCA) cycle (17). During infection, balanced pro- and anti-inflammatory responses are critical in macrophage effector activity, as it helps in appropriate disease resolution, thereby maintaining tissue integrity (18, 19). Thus, glycolysis and OXPHOS are major coordinators of host cell bioenergetics, and their modulation during infection might define the outcome of diseases.
The lung serves as the primary site of infections caused by P. aeruginosa, M. tuberculosis, and S. aureus. This mucosal environment is guarded by immune cells, including resident subsets like alveolar macrophages and other infiltrating phagocytes, such as neutrophils and monocytes. During infection, these myeloid cells undergo metabolic reprogramming to bolster their inflammatory responses. This process is coordinated by many PAMPs, such as lipopolysaccharide (LPS), flagella, and peptidoglycan, which, respectively, interact with Pattern Recognition Receptors (PRRs), like Toll-like receptor 4 (TLR4), TLR5, and hexokinase (HK) (10, 20). These PAMP-PRR interactions alter host bioenergetics, particularly by disrupting OXPHOS. OXPHOS dysfunction is further exacerbated by the downregulation of key TCA cycle enzymes, like isocitrate dehydrogenase (IDH), which is essential for the generation of metabolites that sustain OXPHOS activity, such as αketoglutarate (21). To overcome this bioenergetic deficit, infected cells switch to glycolysis (22, 23). This shift not only provides ATP to maintain cell viability but also stabilizes hypoxia-inducible factor 1-alpha (HIF1α). Once stabilized, HIF1α translocates to the nucleus and orchestrate the expression of a range of pro-inflammatory cytokines, which are critical for confronting the infection (24). However, if not adequately controlled, excessive inflammation can result in tissue damage, a condition often observed in inflammatory pathologies like sepsis, COVID-19, influenza, lupus, and arthritis (3, 25, 26). In such situations, tight regulation of bioenergetic pathways becomes crucial to balance inflammation and prevent organ oxidation. Thus, during infection, appropriate bioenergetic reprogramming of myeloid cells ensures effective immunity compatible with host health.
In this review, we will examine evidence on the impact of bioenergetics on immune regulation and bacterial persistence during infection caused by (A) intracellular pathogen such as M. tuberculosis and (B) extracellular pathogen such as P. aeruginosa and S. aureus. Furthermore, (C) we will cover emerging information pertinent to fungal pathologies, and how specific the survival of these organisms is modulated by metabolic cues.
M. tuberculosis (herein Mtb), the causative agent of tuberculosis (TB), remains a major global health challenge, with nearly a quarter of the world’s population harboring latent infections (Global tuberculosis report 2024) (27). Mtb transmission primarily occurs through the inhalation of aerosolized droplets containing the pathogen, which subsequently deposit in the pulmonary alveoli (28). Mtb primarily infects alveolar macrophages, which are expected to be the primary cellular barrier against the infection (28, 29). Mtb can also subsist within a range of other different phagocytes, including neutrophils and dendritic cells, as well as non-immune cells such as fibroblasts, endothelial cells, and hematopoietic stem cells (30, 31). The infection triggers immune cell recruitment, leading to the formation of granulomas (32), where Mtb evades clearance by suppressing antigen presentation and autophagy (33, 34). The granuloma environment determines whether the infection remains contained or progresses to systemic disease.
Mtb survival relies on host macrophage glycolysis. Emerging evidence indicates that different Mtb strains influence macrophage glycolysis to persist. For example, many strains like H37Ra (avirulent strain) and H37Rv (virulent strain) trigger glycolysis, which seems to be linked to TLR2 stimulation (35). This TLR2 activation drives the expression of the glucose transporters GLUT6 and GLUT1/3 (36) (Figure 1). ESAT-6, a 6kDa early secretory antigenic target protein, enhances macrophage glucose uptake by facilitating GLUT3-mediated transport (36, 37). Multidrug-resistant (MDR) strains exhibit elevated ESAT-6 expression, further augmenting host cell glucose uptake (38). Mtb uses the glycolytic byproduct lactate as a major carbon source to fuels its TCA and gluconeogenesis. This is essential for the bacterium to proliferate, as ΔlldD2 strains, which cannot oxidize lactate, fail to grow intracellularly in human macrophages (39). However, other groups have reported opposite results, where lactate enhances Mtb clearance (40). To avoid their eradication, these Mtb strains inhibit host glycolysis through microRNA-21 (miR-21), which targets phosphofructokinase-M (PFK-M) (41) (Figure 1). Furthermore, other Mtb strains differentially regulate downstream glycolytic genes, such as 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (Pfkfb3) (42). Differential regulation of glycolysis by Mtb is mediated by strain-specific virulence factors. For instance, less virulent strains, such as Mtb CDC1551, induce significantly higher levels of Pfkfb3 compared to the hypervirulent Mtb HN878 (42, 43). Consistently, drug-resistant strains like W_7642 exhibit even lower Pfkfb3 expression than HN878. This regulation is linked to variations in cell wall lipid composition, particularly PDIMs (phthiocerol dimycocerosates) and other lipids that are highly expressed in MDR strains (42, 44). HIF-1α, which regulates the transcription of glycolytic genes, requires nitric oxide (NO) for its activity (45). However, MDR and extensively drug-resistant strains of Mtb induce an augmented IL-10 response, which inhibits NO production. This disruption impairs glycolysis induction (46, 47). These findings highlight the adaptability of Mtb strains in calibrating host glycolysis to sustain survival.
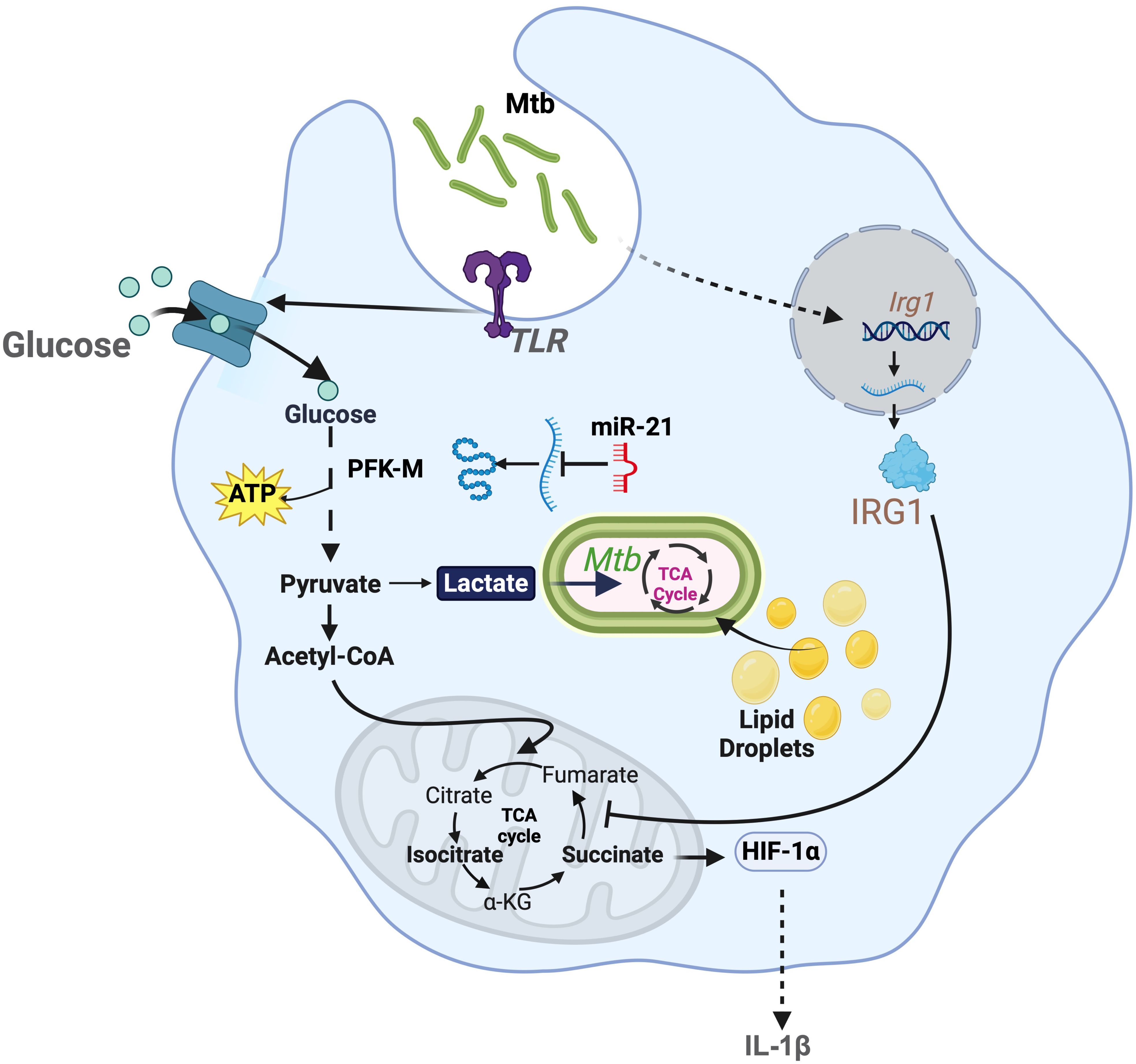
Figure 1. Modulation of bioenergetic pathways by M. tuberculosis: M. tuberculosis induces robust glycolysis in myeloid cells by increasing the uptake of glucose via TLR signaling. This also promotes accumulation of lactate, which is used by the pathogen to generate energy. Lactate also hinders M. tuberculosis growth. In response to this, M. tuberculosis induces expression of miR-21, that regulates the expression of phosphofructokinase-M (PFK-M). M. tuberculosis regulates host TCA cycle by promoting the expression of Irg1, which inhibits SDH. This metabolic control attenuates the expression of pro-inflammatory cytokine, such as IL-1β.
Mtb also disrupts the macrophage TCA cycle to persist (43). Mtb infection downregulates the enzymes IDH2 and succinate dehydrogenase (SDH). While it still remains unclear how Mtb suppresses SDH function, this pathogen limits IDH2 activity by stimulating TLR2 signaling and downregulating the deacetylase SIRT3 (48). Collectively, these interferences break the TCA cycle, resulting in the accumulation of isocitrate and succinate, respectively (21, 49) (Figure 1). This process repurposes mitochondria from producing energy to manufacture ROS (3). During early phase of infection, Mtb exploits ROS to enhance its replication and induce macrophage necrosis (50). This is facilitated by the generation of reactive nitrogen species (RNS), which act as potent inhibitors of the electron transport chain (ETC), further aggravating ROS signaling (51–53).
Mtb infection drives itaconate synthesis, which also contributes to pathogenesis (Figure 1). By stimulating the TLR2-STING axis, Mtb facilitates host upregulation of the enzyme Irg1 (Immunoresponsive Gene 1, also known as Aconitate decarboxylase 1; ACOD1) (54). This enzyme diverts TCA cycle aconitate to generate itaconate (17). Itaconate drives immunosuppression, maintaining host survival and thus providing Mtb with a functional niche to thrive for the long-term. Indeed, mice lacking Irg1 rapidly succumb to the infection, process mediated by neutrophil-driven inflammation (55). Furthermore, Mtb prospers by exploiting lipids and fatty acids as key nutritional sources. Mtb induces abnormal lipid accumulation in macrophages, leading to the formation of foamy cells (56). These metabolically altered cells exhibit elevated expression of genes involved in lipid synthesis and uptake, while showing reduced expression of genes responsible for lipid efflux (56). Mtb worsens this setting by inhibiting lipid droplet degradation, particularly by suppressing lipophagy, thereby creating a favorable environment for bacterial growth (56–58).
Collectively, these findings suggests that Mtb survival hinges on its ability to manipulate host macrophage metabolism. By reprogramming glycolysis, disrupting the TCA cycle, and exploiting lipid metabolism, Mtb outcompetes immune responses, sustains its growth, and establishes a persistent niche within the host.
P. aeruginosa, a Gram-negative ESKAPE pathogen, induces a brisk inflammatory response in the respiratory tract. Instead of clearing the infection, this inflamed milieu aggravates tissue destruction, providing P. aeruginosa with cell debris and metabolites to flourish. This environment supports bacterial growth, with the pathogen existing as planktonic cells or biofilms (59). Macrophage metabolic reprogramming plays a central role in this process, as the succinate released from this cell, a pro-OXPHOS nutrient, fuels P. aeruginosa’s TCA cycle and bioenergetics (59). As directed by the catabolite repressor locus (Crc), succinate is the preferred carbon source for P. aeruginosa, which is consumed before any other nutrient available (59–61). Thus, the survival of P. aeruginosa in the alveolar space is tightly linked to host OXPHOS, primarily to succinate metabolite activities.
P. aeruginosa is a prominent pathogen in people with CF (pwCF) (19). CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) (62). In pwCF, lack of CFTR compromises the function of PTEN, which is an essential metabolic checkpoint that directs mitochondrial bioenergetics. At baseline, CFTR forms a complex with PTEN at the cell membrane, which directs mitochondrial OXPHOS (62). In pwCF, lack of the CFTR-PTEN complex drives mitochondrial OXPHOS disruption, leading to succinate accumulation and release (62). During infection, this succinate supports P. aeruginosa bioenergetics, promoting bacterial proliferation, stimulation of inflammation, and massive infiltration of myeloid cells (59, 62). This succinate-rich milieu worsens IL-1β-driven inflammation, facilitating alveolar oxidation and progressive pulmonary decline (62).
P. aeruginosa exacerbates host bioenergetic reprogramming and succinate release by exposing LPS on its surface through the transporter lptD (63). By stimulating TLR4, membrane-attached LPS disrupts lung mitochondrial OXPHOS, impairing host bioenergetics (10, 64) (Figure 2). Mechanistically, LPS depletes pulmonary ATP synthase, compromising OXPHOS integrity. In response, respiratory cells upregulate glycolysis, fueling the release of pro-inflammatory cytokines that accelerate tissue damage, such as IL-1β and elevated bacterial burden (3, 19). Furthermore, this inflamed environment permeabilizes the alveolar space, enabling the infiltration of different nutrients from circulation that the pathogen exploits to reduce LPS exposure and thus adapt to the host to be better tolerated, like ketone bodies (19) (Figure 2).
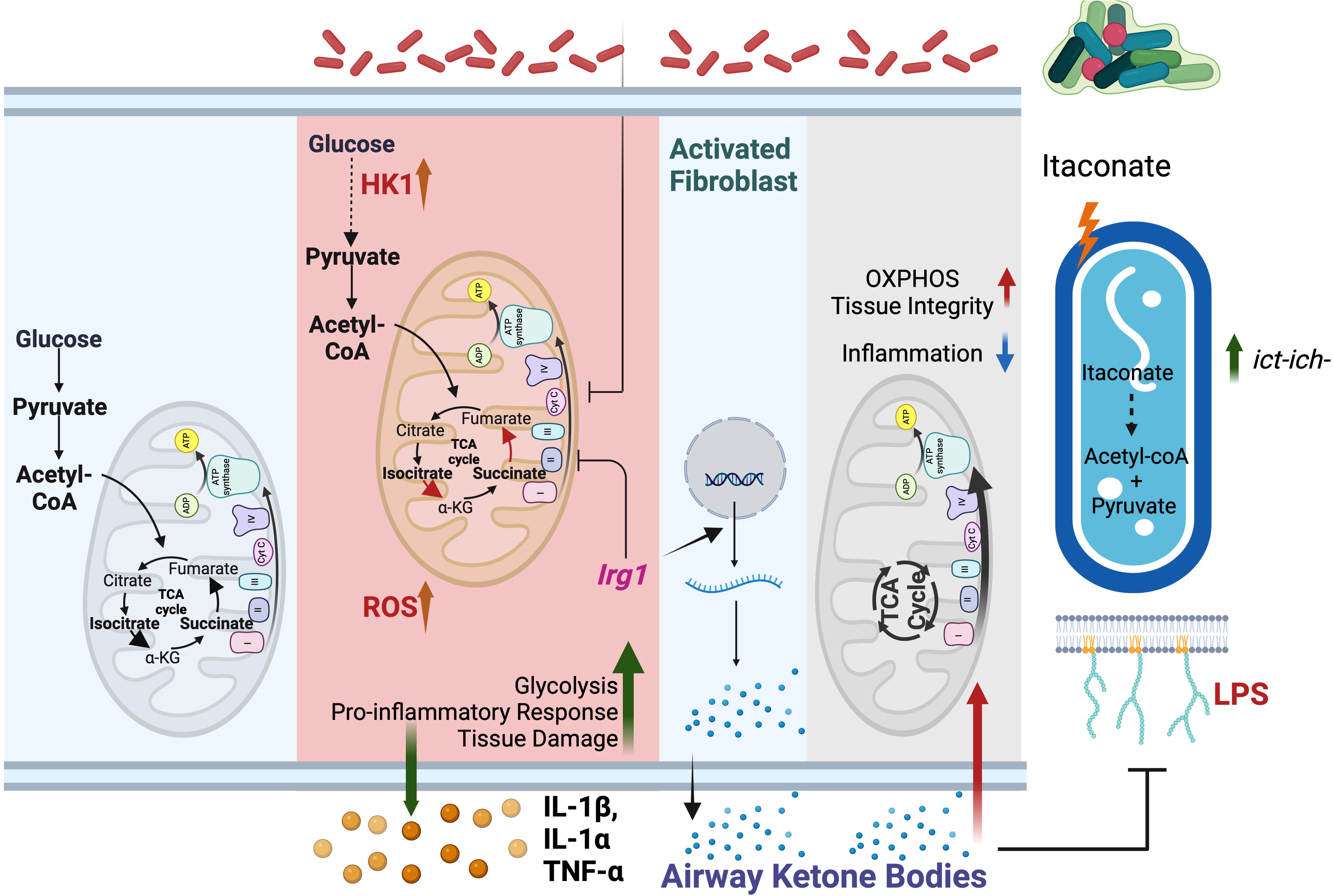
Figure 2. Mitochondrial metabolites drive P. aeruginosa infection. Left panel: At baseline, macrophage bioenergetics is primarily fueled by OXPHOS. Glycolysis contribution to ATP synthesis is limited. Middle panel: Upon P. aeruginosa infection, OXPHOS is impaired, and glycolysis is increased. Glycolysis drives release of tissue-damaging cytokines. To mitigate immunopathology, mitochondria express Irg1 and produces itaconate. Right panel: P. aeruginosa adapts to itaconate by increasing the ict-ich-ccl locus, selecting for strains that exploit the immunometabolite to generate energy. Furthermore, itaconate-adapted P. aeruginosa also favor local enrichment of ketone bodies, particularly from fibroblasts. These ketone bodies maintain tissue integrity by suppressing P. aeruginosa expression of surface-exposed LPS and by fueling host OXPHOS. This milieu favors the progressive establishment of infection tolerance.
Macrophage succinate metabolism also drives ROS production through SDH and IDH (65). However, by sensing this ROS, P. aeruginosa activates the bacterial antioxidant response, limiting planktonic growth and fostering the formation of biofilms (62, 66). This lifestyle shift permits the bacterial community to persist in harsh environments, such as the imparted by antibiotics (67–69). This setting has been observed in CF, COPD, PCD and VAP, validating the clinical significance of succinate and ROS metabolism during P. aeruginosa pulmonary pathology.
To balance the harmful effects of succinate, LPS, and ROS, macrophages produce itaconate as a protective immunometabolite (70). However, P. aeruginosa turns this host defense into an advantage. In contrast with most opportunist, P. aeruginosa’s genome harbors the ict-ich-ccl locus (71). This platform helps the pathogen to breakdown itaconate into less toxic metabolites, like acetyl-CoA and pyruvate, which enter the bacterial central metabolism to drive bioenergetics (59, 71). Furthermore, P. aeruginosa adapts to the outer membrane stress caused by itaconate by forming biofilms, gaining protection against diverse threats, such as antibiotics, ROS, antibodies, and phagocytosis (59) (Figure 2). As reported in pwCF, throughout the course of the infection, P. aeruginosa adapts to this itaconate-rich environment, selecting for communities that co-evolve with the immunometabolite (59). These strains further exploit itaconate to enrich pulmonary ketone bodies, which promote bacterial populations that are better tolerated by the host immune system (19) (Figure 2). By inducing infection tolerance, these ketone-adapted P. aeruginosa communities attenuate immunopathology, preserving both host survival and the lung as a functional niche to thrive for the long-term (19).
The extraordinary ability of P. aeruginosa to survive in mucosal tissues synchronized with the bioenergetic changes experience by infected macrophages is strong evidence of co-evolution with host defenses. This principle is supported by the development of infection tolerance, where P. aeruginosa readily adapts to the airway environment integrating into the local microbiome as discrete communities to thrive. These findings highlight the clinical significance of host bioenergetics directing chronic pathogen activities in mucosal tissues.
S. aureus is a Gram-positive ESKAPE pathogen that normally resides in the human microbiome (72). Under still unclear conditions, it can invade mucosal tissues, causing severe and life-threatening infections such as endocarditis, bacteremia, and pneumonia (72). The risk escalates when medical devices become contaminated, providing a gateway for infection (72, 73). In infected subjects, S. aureus drives both acute and chronic infections, characterized by intense neutrophil-mediated inflammation (74, 75). To enhance tissue damage and neutrophil infiltration, S. aureus produces toxins like leukocidins and hemolysins, which induce host cell death and create an inflammatory, oxidative environment (76, 77) (Figure 3). Instead of being cleared, S. aureus thrives in this detrimental milieu, feeding off nutrients derived from damaged tissue.
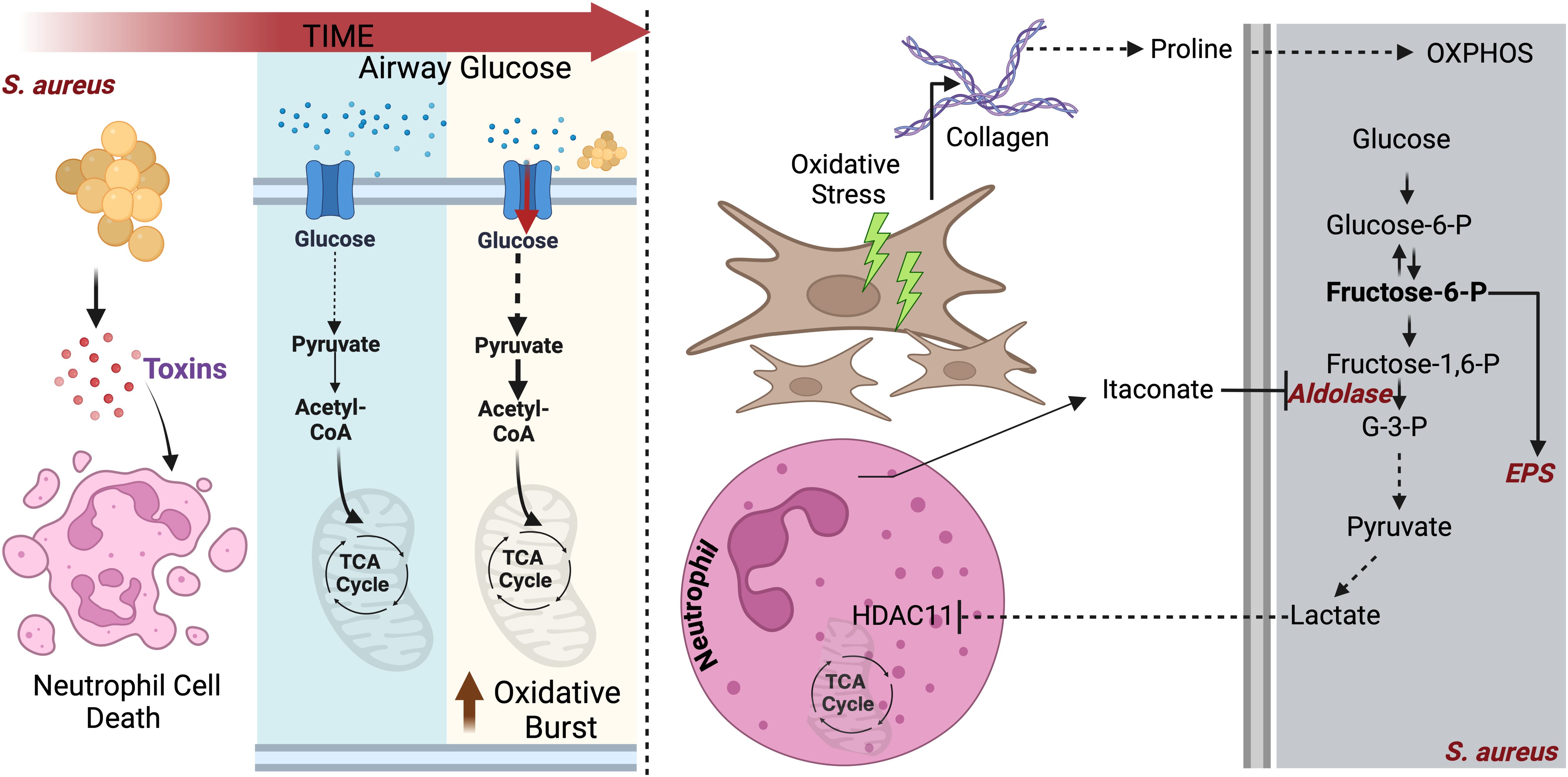
Figure 3. S. aureus adapts to lung bioenergetic changes: Left panel: During initial infection, S. aureus causes pulmonary pathology by secreting various toxins that kills neutrophils. To maintain survival, neutrophils deplete glucose to fuel their glycolysis, which is required to eliminate the pathogen via oxidative burst. Right panel: In addition to consume glucose, infected neutrophils block S. aureus glycolysis by inhibiting aldolase. Glycolysis interference by neutrophils promotes S. aureus adaptation to proline, released from activated fibroblast. Proline fuels S. aureus OXPHOS.
The preferred S. aureus carbon source is glucose, which is used to drive glycolysis (78, 79). However, in the host lung, glucose is limited, as myeloid cells consume it through their own glycolysis in response to S. aureus sensing (78–80). This glucose limitation forces S. aureus to adapt to alternative carbon sources to persist. During pneumonia, tissue repair mechanisms activate fibroblasts to release proline, a key component of collagen (81, 82). S. aureus adapts to utilize proline, particularly in diseases like CF, where chronic infection and progressive tissue scarring result in extensive collagen deposition (82). By capitalizing on proline availability during tissue remodeling, S. aureus generates energy and biofilms to sustain infection, promoting inflammation and persistent infection (82) (Figure 3). The high metabolic plasticity harbored by S. aureus provides the pathogen with essentials advantages to coexist with metabolically reprogrammed phagocytes.
Host cells respond to S. aureus infection by releasing itaconate (83). However, unlike P. aeruginosa, S. aureus lacks the ict-ich-ccl locus and hence cannot degrade itaconate (83). To persist in the itaconate-rich airway, S. aureus forms biofilms. To generate these communities, S. aureus specializes in the manufacture of extracellular polysaccharides (84, 85) (Figure 3). Furthermore, this pathogen exploits this same itaconate response to abrogate neutrophil function, particularly by blocking the oxidative burst required to kill the bacteria. In tissues different than the lung, S. aureus releases lactate, which contributes to immune evasion by altering the activity of Histone deacetylase 11 (HDAC11), redirecting host cells to produce IL-10 and thus skew immune responses towards an immunosuppressed phenotype (86) (Figure 3).
By adapting to glucose scarcity and other pulmonary amino acids involved in repair, like proline, S. aureus generates energy to survive. Furthermore, by exploiting the own host metabolic response to preserve tissue integrity, itaconate, S. aureus disrupt neutrophil function, skewing immune responses towards a state of infection tolerance. These findings, phenocopying the behavior of other pathogens like Mtb and P. aeruginosa, showcase the ability of S. aureus to thrive in bioenergetically altered environments, like the inflamed lung.
Fungal infections, primarily caused by Candida albicans, Cryptococcus neoformans, and Aspergillus sp. are responsible for high mortality especially in immunocompromised individuals and pwCF (87, 88). However, the impact of bioenergetic pathways during fungal infection is still an emerging field, and how it associates with clinical outcomes remains unclear.
Recent findings indicate that glycolysis is central to the immune response against fungal infection (89, 90). Anti-fungal neutrophils rely heavily on glycolysis, particularly on expression of key glucose transporters, like GLUT1 (90). GLUT1 perturbation in these cells blunt phagocytosis, ROS, and formation of extracellular traps, facilitating fungi survival (89, 90). During C. albicans infection, in the yeast phase, host cells respond by promoting glycolysis, OXPHOS, and glutamine metabolism. This supports higher energy demands required for a robust oxidative burst and cytokine response. However, at later stages, as C. albicans transitions from yeast to hyphal forms, the induction of glycolysis and OXPHOS is reduced leading to an attenuated oxidative burst and a diminished cytokine response (91). These distinctive programs are attributed to the difference in the cell wall composition, such as the presence of β-glucans, that acts as a major PAMP for the PRR Dectin-1 (92). To curb host immunity, C. albicans reduces the surface expression of β-glucans, particularly when exposed to the glycolysis byproduct lactate (93) (Figure 4). This adjustment also promotes active glycolysis in C. albicans, depleting glucose form the milieu and starving immune cells to death (94). Inhibition of host glycolysis by 2-DG or by dichloroacetate, which is an enhancer of pyruvate dehydrogenase, perturbs the host response against C. albicans. This phenotype is associated with reduced expression of IL-1β, TNFα, and IL-6 in human monocytes (91, 95). Additionally, these treatments impair the production of IL-17, IL-22, IFN-γ and IL-10, cytokines regulated by Th1/Th17 T cells and crucial for host protection during C. albicans infection (91). In line with this, activation of host glycolysis by nutritional supplementation of glucose facilitates health and C. albicans eradication (94). Thus, during C. albicans infection, there is a dynamic host-pathogen glycolytic competition that determines the outcome of the disease.
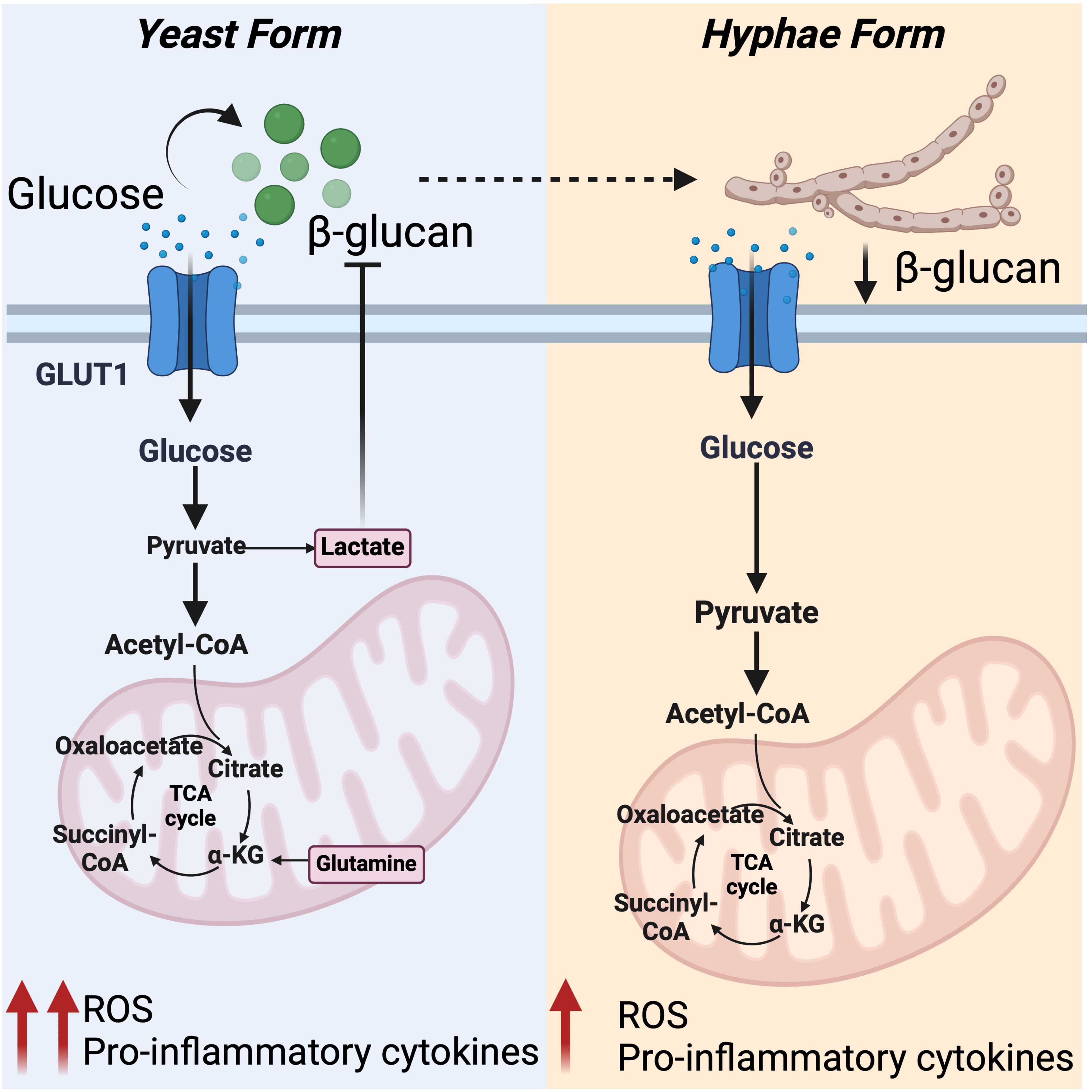
Figure 4. Differential regulation of host metabolic pathway by C. albicans: Left panel: In response to C. albicans β-glucans, primarily exposed on yeast form, host cell responds by enhancing glycolysis, OXPHOS, and glutamine metabolism. This metabolic shift triggers anti-fungal responses by supporting ROS production and cytokine response. Enhanced glycolysis results in elevated lactate levels, which limits β -glucans expression and drives glycolysis in C. albicans, depleting glucose from extracellular milieu. Right panel: Reduced exposure of β -glucans on hyphae form of C. albicans fails to activate glycolysis and the pentose phosphate pathway, leading to reduced mitochondrial ROS production and cytokine response. This setting favors C. albicans survival.
In contrast to C. albicans, melanin is a major component of the cell wall in A. fumigatus (96). This melanin serves as a key driver of metabolic reprogramming in immune cells (97). Mechanistically, within the phagosome, melanin released from the spores promotes the sequestering of calcium, which triggers the recruitment of mTOR on the phagosome (97). mTOR is a major regulator of glycolysis. It regulates the expression level of HIF1α and other genes involved in glycolysis (98). By promoting mTOR and HIF1α activity, A. fumigatus induces glycolysis in the infected macrophage, leading to the release of cytokines involved in tissue oxidation (97). Additionally, melanin also aids in the consolidation of the pathology by modulating calcium signaling. Melanin effectively blocks secretion of chemokines such as CXCL1 and CXCL8, reducing the infiltration of anti-fungal immune cells (99). These studies show how fungal pathogens use specific PAMPS to modulate glycolysis, cytokine release, and myeloid cell recruitment. By stimulating glycolysis, fungal organisms trigger inadequate immune responses, leading to tissue disruption and organ disease.
In this review, we highlight the impact of metabolism on host-pathogen interactions in the lung, particularly through modulation of cellular bioenergetics. We examine recent evidence suggesting how pulmonary pathogens exploit host bioenergetic platforms to persist, and how this environment influences inflammatory responses and disease progression. Bacterial pathogens like P. aeruginosa, Mtb, and S. aureus alter lung cell bioenergetics - inducing shifts like increased glycolysis, impaired OXPHOS, and the release of metabolites like succinate, proline, and itaconate - to evade immune responses and promote survival. This metabolic disruption weakens antimicrobial defenses, exacerbates tissue damage, and drives chronic inflammation. Although similar findings have been reported during fungal diseases, the influence of bioenergetics and metabolism on mycoses remains underexplored. Metabolism plays a critical role in many physiological processes, which can be co-opted by opportunists to drive tissue disruption via a range of mechanisms, including autophagy, cell death pathways – i.e., apoptosis and ferroptosis - and epigenetic modifications, shaping immune responses to fuel pathology evolution instead of organ repair. Given the impact of bioenergetic platforms on infection outcomes, these metabolic pathways emerge as potential therapeutic targets for drug design. Key players such as GLUT1/3, HK, and SDH have been considered to prevent exacerbated inflammatory response and promote anti-inflammatory routes, especially during sepsis and other immunopathologies (17). Similarly, inhibition of PKM2 (Pyruvate Kinase M2) during sepsis reduces inflammation via NLRP3 (100). This also provides implication of nutrition in bioenergetics dynamics. However, further investigation is needed to uncover how dietary interventions, or metabolic modulators could influence infectious disease progression. Interfering pathogens from utilizing immunometabolites such as succinate, itaconate, and lactate could also aid in clearing the infection and resolution of inflammation. On the host side, major challenges associated with immunometabolism-based therapy are the so-called off-target effects, impacting homeostasis of “healthy” cells. In such scenarios, delivering the drugs to the specific cells by utilizing nanoparticle formulation would improve treatment outcome (17, 101). These processes may open new avenues for metabolo-therapies aimed at restoring immune balance, mitigating mucosal damage, and limiting pathogen persistence in diseases such as pneumonia.
SR: Writing – original draft, Writing – review & editing. GL: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by grants R35GM146776 (NIGMS, NIH) and R21AI181780 (NIAID, NIH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. CDC. Data and Statistics on Fungal Diseases | Fungal Diseases. Available online at: https://www.cdc.gov/fungal/data-research/facts-stats/index.html (Accessed December 13, 2024).
2. Ikuta KS, Swetschinski LR, Aguilar GR, Sharara F, Mestrovic T, Gray AP, et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. (2022) 400:2221–48. doi: 10.1016/S0140-6736(22)02185-7
3. Chen YT, Lohia GK, Chen S, Riquelme SA. Immunometabolic regulation of bacterial infection, biofilms, and antibiotic susceptibility. J Innate Immun. (2024) 16:143. doi: 10.1159/000536649
4. Rodrigo-Troyano A, Suarez-Cuartin G, Peiró M, Barril S, Castillo D, Sanchez-Reus F, et al. Pseudomonas aeruginosa resistance patterns and clinical outcomes in hospitalized exacerbations of COPD. Respirology. (2016) 21:1235–42. doi: 10.1111/resp.12825
5. Patil N, Marco A, Montales MT, Bhaskas N, Mittadodla P, Mukasa LN, et al. Pulmonary tuberculosis in a patient with cystic fibrosis. N Am J Med Sci. (2015) 7:233. doi: 10.4103/1947-2714.157494
6. Sershen CL, Salim T, May EE. Investigating the comorbidity of COPD and tuberculosis, a computational study. Front Syst Biol. (2023) 3:940097. doi: 10.3389/fsysb.2023.940097
7. Kanemitsu Y, Fukumitsu K, Kurokawa R, Akamatsu T, Fukuda S, Ito Y, et al. Staphylococcus aureus enterotoxin sensitization is associated with declined capsaicin cough sensitivity in chronic obstructive pulmonary disease. Sci Rep. (2024) 14:1–10. doi: 10.1038/s41598-024-81297-y
8. Rumpf C, Lange J, Schwartbeck B, Kahl BC. Staphylococcus aureus and cystic fibrosis—A close relationship. What can we learn from sequencing studies? Pathogens. (2021) 10:1177. doi: 10.3390/pathogens10091177
9. Haiko J, Saeedi B, Bagger G, Karpati F, Özenci V. Coexistence of Candida species and bacteria in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis. (2019) 38:1071. doi: 10.1007/s10096-019-03493-3
10. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduction Targeted Ther. (2021) 6:1–24. doi: 10.1038/s41392-021-00687-0
11. Strope TA, Birky CJ, Wilkins HM. The role of bioenergetics in neurodegeneration. Int J Mol Sci. (2022) 23:9212. doi: 10.3390/ijms23169212
12. Porter GA, Hom JR, Hoffman DL, Quintanilla RA, Bentley KL de M, Sheu SS, et al. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog Pediatr Cardiol. (2011) 31:75. doi: 10.1016/j.ppedcard.2011.02.002
13. Van Wijk R, Van Solinge WW. The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. (2005) 106:4034–42. doi: 10.1182/blood-2005-04-1622
14. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
15. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winthers S, Vidoni S, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature. (2018) 560:102–6:7716. doi: 10.1038/s41586-018-0353-2
16. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633. doi: 10.1016/j.immuni.2013.04.005
17. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
18. Rodríguez-Morales P, Franklin RA. Macrophage phenotypes and functions: resolving inflammation and restoring homeostasis. Trends Immunol. (2023) 44:986–98. doi: 10.1016/j.it.2023.10.004
19. Tomlinson KL, Chen YT, Junker A, Urso AC, Wong Fok Lung T, Ahn D, et al. Ketogenesis promotes tolerance to Pseudomonas aeruginosa pulmonary infection. Cell Metab. (2023) 35:1767–1781.e6. doi: 10.1016/j.cmet.2023.09.001
20. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell. (2016) 166:624–36. doi: 10.1016/j.cell.2016.05.076
21. Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Longinicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
22. Blagih J, Jones RG. Polarizing macrophages through reprogramming of glucose metabolism. Cell Metab. (2012) 15:793–5. doi: 10.1016/j.cmet.2012.05.008
23. Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
24. Tannahill GM, Curtis GM, Adamik GM, Palsson-Mcdermott GM, McGettrick GM, Goel GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
25. Jeljeli MM, Adamopoulos IE. Innate immune memory in inflammatory arthritis. Nat Rev Rheumatol. (2023) 19:627–39. doi: 10.1038/s41584-023-01009-0
26. Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun. (2020) 110:102374. doi: 10.1016/j.jaut.2019.102374
27. Global Tuberculosis Report. (2024). Available online at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024 (Accessed December 13, 2024).
28. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
29. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci Immunol. (2019) 4. doi: 10.1126/sciimmunol.aaw6693
30. Mayito J, Andia I, Belay M, Jolliffe DA, Kateete DP, Reece ST, et al. Anatomic and cellular niches for mycobacterium tuberculosis in latent tuberculosis infection. J Infect Dis. (2019) 219:685–94. doi: 10.1093/infdis/jiy579
31. Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. (2019) 43:341–61. doi: 10.1093/femsre/fuz006
32. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. (2009) 136:37–49. doi: 10.1016/j.cell.2008.11.014
33. Holla S, Kurowska-Stolarska M, Bayry J, Balaji KN. Selective inhibition of IFNG-induced autophagy by Mir155- and Mir31-responsive WNT5A and SHH signaling. Autophagy. (2014) 10:311–30. doi: 10.4161/auto.27225
34. Noss EH, Harding CV, Boom WH. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell Immunol. (2000) 201:63–74. doi: 10.1006/cimm.2000.1633
35. Cumming BM, Addicott KW, Adamson JH, Steyn AJC. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife. (2018) 7:e39169. doi: 10.7554/eLife.39169
36. Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, et al. Pathogenicity of mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PloS Pathog. (2014) 10:e1004265. doi: 10.1371/journal.ppat.1004265
37. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KVX. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. (2012) 12:669–81. doi: 10.1016/j.chom.2012.09.012
38. Jhingan GD, Kumari S, Jamwal SV, Kalam H, Arora D, Jain N, et al. Comparative proteomic analyses of avirulent, virulent, and clinical strains of mycobacterium tuberculosis identify strain-specific patterns. J Biol Chem. (2016) 291:14257–73. doi: 10.1074/jbc.M115.666123
39. Billig S, Schneefeld M, Huber C, Grassl GA, Eisenreich W, Bange FC, et al. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci Rep. (2017) 7. doi: 10.1038/s41598-017-05916-7
40. Maoldomhnaigh CÓ, Cox DJ, Phelan JJ, Mitermite M, Murphy DM, Leisching G, et al. Lactate alters metabolism in human macrophages and improves their ability to kill mycobacterium tuberculosis. Front Immunol. (2021) 12:663695. doi: 10.3389/fimmu.2021.663695
41. Hackett EE, Charles-Messance H, O’Leary SM, Gleeson LE, Muñoz-Wolf N, Case S, et al. Mycobacterium tuberculosis Limits Host Glycolysis and IL-1β by Restriction of PFK-M via MicroRNA-21. Cell Rep. (2020) 30:124–136.e4. doi: 10.1016/j.celrep.2019.12.015
42. Howard NC, Khader SA. Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. (2020) 28:832–50. doi: 10.1016/j.tim.2020.04.010
43. Shi L, Jiang Q, Bushkin Y, Subbian S, Tyagi S. Biphasic Dynamics of Macrophage Immunometabolism during Mycobacterium tuberculosis Infection. mBio. (2019) 10. doi: 10.1128/mBio.02550-18
44. Howard NC, Marin ND, Ahmed M, Rosa BA, Martin J, Bambouskova M, et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat Microbiol. (2018) 3:1099–108. doi: 10.1038/s41564-018-0245-0
45. Braverman J, Stanley SA. Nitric Oxide Modulates Macrophage Responses to Mycobacterium tuberculosis Infection through Activation of HIF-1α and Repression of NF-κB. J Immunol. (2017) 199:1805–16. doi: 10.4049/jimmunol.1700515
46. Basingnaa A, Antwi-Baffour S, Nkansah DO, Afutu E, Owusu E. Plasma levels of cytokines (IL-10, IFN-γ and TNF-α) in multidrug resistant tuberculosis and drug responsive tuberculosis patients in Ghana. Diseases. (2018) 7:2. doi: 10.3390/diseases7010002
47. Baseler WA, Davies LC, Quigley L, Ridnour LA, Weiss JM, Hussian SP, et al. Autocrine IL-10 functions as a rheostat for M1 macrophage glycolytic commitment by tuning nitric oxide production. Redox Biol. (2016) 10:12. doi: 10.1016/j.redox.2016.09.005
48. Smulan LJ, Martinez N, Kiritsy MC, Kativhu C, Cavallo K, Sassetti CM, et al. Sirtuin 3 downregulation in mycobacterium tuberculosis-infected macrophages reprograms mitochondrial metabolism and promotes cell death. mBio. (2021) 12:e03140–20. doi: 10.1128/mBio.03140-20
49. Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. (2007) 282:4524–32. doi: 10.1074/jbc.M610415200
50. Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MRG, Herbst S, et al. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol. (2017) 216:583–94. doi: 10.1083/jcb.201603040
51. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. (2015) 5. doi: 10.1038/srep18176
52. Everts B, Amiel E, Van Der Windt GJW, Freitas TC, Chott R, Yarashesk KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. (2012) 120:1422–31. doi: 10.1182/blood-2012-03-419747
53. Beltrán B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc Natl Acad Sci U.S.A. (2000) 97:14602–7. doi: 10.1073/pnas.97.26.14602
54. Bomfim CCB, Fisher L, Amaral EP, Mittereder L, McCann K, Correa AAS, et al. Mycobacterium tuberculosis induces irg1 in murine macrophages by a pathway involving both TLR-2 and STING/IFNAR signaling and requiring bacterial phagocytosis. Front Cell Infect Microbiol. (2022) 12. doi: 10.3389/fcimb.2022.862582
55. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med. (2018) 215:1035–45. doi: 10.1084/jem.20180118
56. Holla S, Prakhar P, Singh V, Karnam A, Mukherjee T, Mahadik K, et al. MUSASHI-mediated expression of JMJD3, a H3K27me3 demethylase, is involved in foamy macrophage generation during mycobacterial infection. PloS Pathog. (2016) 12. doi: 10.1371/journal.ppat.1005814
57. Prakhar P, Bhatt B, Lohia GK, Shah A, Mukherjee T, Kolthur-Seetharam U, et al. G9a and Sirtuin6 epigenetically modulate host cholesterol accumulation to facilitate mycobacterial survival. PloS Pathog. (2023) 19. doi: 10.1371/journal.ppat.1011731
58. Mukherjee T, Bhatt B, Prakhar P, Lohia GK, Rajmani RS, Balaji KN, et al. Epigenetic reader BRD4 supports mycobacterial pathogenesis by co-modulating host lipophagy and angiogenesis. Autophagy. (2022) 18:391–408. doi: 10.1080/15548627.2021.1936355
59. Riquelme SA, Liimatta K, Wong Fok Lung T, Fields B, Ahn D, Chen D, et al. Pseudomonas aeruginosa utilizes host-derived itaconate to redirect its metabolism to promote biofilm formation. Cell Metab. (2020) 31:1091–1106.e6. doi: 10.1016/j.cmet.2020.04.017
60. Rojo F. Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol Rev. (2010) 34:658–84. doi: 10.1111/j.1574-6976.2010.00218.x
61. Görke B, Stülke J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol. (2008) 6:613–24. doi: 10.1038/nrmicro1932
62. Riquelme SA, Lozano C, Moustafa AM, Liimatta K, Tomlinson KL, Britto C, et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aav4634
63. Balibar CJ, Grabowicz M. Mutant Alleles of lptD Increase the Permeability of Pseudomonas aeruginosa and Define Determinants of Intrinsic Resistance to Antibiotics. Antimicrob Agents Chemother. (2015) 60:845–54. doi: 10.1128/AAC.01747-15
64. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LAB, et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. (2016) 2:1–10. doi: 10.1038/nmicrobiol.2016.246
65. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064
66. Groitl B, Dahl JU, Schroeder JW, Jakob U. Pseudomonas aeruginosa defense systems against microbicidal oxidants. Mol Microbiol. (2017) 106:335–50. doi: 10.1111/mmi.2017.106.issue-3
67. Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. (1999) 284:1318–22. doi: 10.1126/science.284.5418.1318
68. Lebeaux D, Ghigo J-M, Beloin C. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol Mol Biol Rev. (2014) 78:510. doi: 10.1128/MMBR.00013-14
69. Moser C, Jensen PØ, Thomsen K, Kolpen M, Rybtke M, Lauland AS, et al. Immune responses to pseudomonas aeruginosa biofilm infections. Front Immunol. (2021) 12:625597. doi: 10.3389/fimmu.2021.625597
70. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
71. Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA. Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol. (2014) 10:371–7. doi: 10.1038/nchembio.1482
72. Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. (2015) 28:603–61. doi: 10.1128/CMR.00134-14
73. Brown AF, Leech JM, Rogers TR, McLoughlin RM. Staphylococcus aureus colonization: modulation of host immune response and impact on human vaccine design. Front Immunol. (2014) 4. doi: 10.3389/fimmu.2013.00507
74. Marciano BE, Leech JM, Rogers TR, McLoughlin RM. Common severe infections in chronic granulomatous disease. Clin Infect Dis. (2015) 60:1176–83. doi: 10.1093/cid/ciu1154
75. Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of Staphylococcus aureus. Virulence. (2021) 12:547. doi: 10.1080/21505594.2021.1878688
76. Spaan AN, Vrieling M, Wallet P, Badiou C, Reyes-Robles T, Ohneck EA, et al. The staphylococcal toxins γ-hemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat Commun. (2014) 5:5438. doi: 10.1038/ncomms6438
77. Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U.S.A. (2018) 115:7416–21. doi: 10.1073/pnas.1721949115
78. Vitko NP, Grosser MR, Khatri D, Lance TR, Richardson AR. Expanded Glucose Import Capability Affords Staphylococcus aureus Optimized Glycolytic Flux during Infection. mBio. (2016) 7. doi: 10.1128/mBio.00296-16
79. Vitko NP, Spahich NA, Richardson AR. Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. mBio. (2015) 6. doi: 10.1128/mBio.00045-15
80. Gabryszewski SJ, Lung TWF, Annavajhala MK, Tomlinson KL, Riquelme SA, Khan IN, et al. Metabolic adaptation in methicillin-resistant staphylococcus aureus pneumonia. Am J Respir Cell Mol Biol. (2019) 61:185–97. doi: 10.1165/rcmb.2018-0389OC
81. Ghonim MA, Boyd DF, Flerlage T, Thomas PG. Pulmonary inflammation and fibroblast immunoregulation: from bench to bedside. J Clin Invest. (2023) 133:e170499. doi: 10.1172/JCI170499
82. Urso A, Monk IR, Cheng YT, Predella C, Wong Fok Lung T, Theiller EM, et al. Staphylococcus aureus adapts to exploit collagen-derived proline during chronic infection. Nat Microbiol. (2024) 9:2506–21. doi: 10.1038/s41564-024-01769-9
83. Tomlinson KL, Riquelme SA, Baskota SU, Drikic M, Monk IR, Stinear TP, et al. Staphylococcus aureus stimulates neutrophil itaconate production that suppresses the oxidative burst. Cell Rep. (2023) 42:112064. doi: 10.1016/j.celrep.2023.112064
84. Peng Q, Tang X, Dong W, Sun N. amp]]amp; Yuan, W. A review of biofilm formation of staphylococcus aureus and its regulation mechanism. Antibiotics. (2022) 12:12. doi: 10.3390/antibiotics12010012
85. Tomlinson KL, Lung TWF, Dach F, Annavajhala MK, Gabryszewski SJ, Groves RA, et al. Staphylococcus aureus induces an itaconate-dominated immunometabolic response that drives biofilm formation. Nat Commun. (2021) 12:1–13. doi: 10.1038/s41467-021-21718-y
86. Heim CE, Bosch ME, Yamada KJ, Aldrich AL, Chaudhari SS, Klinkebiel D, et al. Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat Microbiol. (2020) 5:1271–84. doi: 10.1038/s41564-020-0756-3
87. Mukherjee B, Paul P, Dutta L, Chakraborty S, Dhara M, Mondal L, et al. Pulmonary administration of biodegradable drug nanocarriers for more efficacious treatment of fungal infections in lungs: insights based on recent findings. Multifunctional Syst Combined Delivery Biosensing Diagnostics. (2017), 261–80. doi: 10.1016/B978-0-323-52725-5.00014-9
88. Li Z, Lu G, Meng G. Pathogenic fungal infection in the lung. Front Immunol. (2019) 10:1524. doi: 10.3389/fimmu.2019.01524
89. Gonçalves SM, Ferreira AV, Cunha C, Carvalho A. Targeting immunometabolism in host-directed therapies to fungal disease. Clin Exp Immunol. (2022) 208:158–66. doi: 10.1093/cei/uxab014
90. Li DD, Jawale C V, Zhou C, Lin L, Trevejo-Nunez GJ, Rahman SA, et al. Fungal sensing enhances neutrophil metabolic fitness by regulating antifungal Glut1 activity. Cell Host Microbe. (2022) 30:530–544.e6. doi: 10.1016/j.chom.2022.02.017
91. Domínguez-Andrés J, Arts RJW, ter Horst R, Gresnigt MS, Smeekens SP, Ratter JM, et al. Rewiring monocyte glucose metabolism via C-type lectin signaling protects against disseminated candidiasis. PloS Pathog. (2017) 13. doi: 10.1371/journal.ppat.1006632
92. Silva-Gomes R, Caldeira I, Fernandes R, Cunha C, Carvalho A. Metabolic regulation of the host–fungus interaction: from biological principles to therapeutic opportunities. J Leukoc Biol. (2024) 116:469–86. doi: 10.1093/jleuko/qiae045
93. Ballou ER, Verma J, Harrison PF, Snelgrove SL, Lo TL, Scherer AK, et al. Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat Microbiol. (2016) 2. doi: 10.1038/nmicrobiol.2016.238
94. Tucey TM, et al. Glucose homeostasis is important for immune cell viability during candida challenge and host survival of systemic fungal infection. Cell Metab. (2018) 27:988–1006.e7. doi: 10.1016/j.cmet.2018.03.019
95. Weerasinghe H, Traven A. Immunometabolism in fungal infections: the need to eat to compete. Curr Opin Microbiol. (2020) 58:32–40. doi: 10.1016/j.mib.2020.07.001
96. Heinekamp T, Thywißen A, Macheleidt J, Keller S, Valiante V, Brakhage AA, et al. Aspergillus fumigatus melanins: Interference with the host endocytosis pathway and impact on virulence. Front Microbiol. (2012) 3:36675. doi: 10.3389/fmicb.2012.00440
97. Gonçalves SM, Duarte-Oliveira C, Campos CF, Aimanianda V, ter Horst R, Leite L, et al. Phagosomal removal of fungal melanin reprograms macrophage metabolism to promote antifungal immunity. Nat Commun. (2020) 11:1–15. doi: 10.1038/s41467-020-16120-z
98. Fan H, Wu Y, Yu S, Li X, Wang A, Wang S, et al. Critical role of mTOR in regulating aerobic glycolysis in carcinogenesis (Review). Int J Oncol. (2021) 58:9–19. doi: 10.3892/ijo.2020.5152
99. Reedy JL, Jensen KN, Crossen AJ, Basham KJ, Ward RA, Reardon CM, et al. Fungal melanin suppresses airway epithelial chemokine secretion through blockade of calcium fluxing. Nat Commun. (2024) 15:1–16. doi: 10.1038/s41467-024-50100-x
100. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. (2016) 7:1–13. doi: 10.1038/ncomms13280
Keywords: immunometabolism, MACROPHAGE METABOLISM, itaconate, ESKAPE bacteria, fungal infection, host-pathogen interaction, bioenergetics, pneumonia (infectious disease)
Citation: Lohia GK and Riquelme SA (2025) Influence of cell bioenergetics on host-pathogen interaction in the lung. Front. Immunol. 16:1549293. doi: 10.3389/fimmu.2025.1549293
Received: 20 December 2024; Accepted: 19 March 2025;
Published: 03 April 2025.
Edited by:
Laurence Rahme, Harvard Medical School, United StatesReviewed by:
Carlos Villarreal, National Autonomous University of Mexico, MexicoCopyright © 2025 Lohia and Riquelme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastián A. Riquelme, c3IzMzAyQGN1bWMuY29sdW1iaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.