 Janki Bhatt
Janki Bhatt Alessandra Ghigo1,2
Alessandra Ghigo1,2 Emilio Hirsch
Emilio Hirsch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 31 March 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1549277
This article is part of the Research TopicUnraveling the Molecular Web of Inflammation and Fibrosis: Pathways, Immune Interactions, Epigenetics, and Therapeutic FrontiersView all 3 articles
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive lung disease characterized by abnormal epithelial repair, persistent inflammation, and excessive extracellular matrix deposition, leading to irreversible scarring and respiratory failure. Central to its pathogenesis is the dysregulation of the PI3K/Akt signaling pathway, which drives fibroblast activation, epithelial-mesenchymal transition, apoptosis resistance, and cellular senescence. Senescent cells contribute to fibrosis through the secretion of pro-inflammatory and profibrotic factors in the senescence-associated secretory phenotype (SASP). Current antifibrotic therapies, Nintedanib and Pirfenidone, only slow disease progression and are limited by side effects, highlighting the need for novel treatments. This review focuses on the role of PI3K/Akt signaling in IPF pathogenesis, its intersection with inflammation and fibrosis, and emerging therapeutic approaches targeting molecules along this pathway.
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive fibrosing interstitial pneumonia of unknown origin, characterized by a progressively worsening dyspnea and declining lung function (1, 2). It is a fatal age-related disorder, predominantly occurring in males, with a median survival age of 2-5 years after diagnosis (3, 4). Although considered rare, existing data indicates that the global occurrence of IPF rivals several cancer types, including stomach, liver, testicular, and cervical cancers (5), with approximately 40,000 new cases diagnosed annually in Europe alone (6), and its incidence doubling with each successive decade after the age of 50. The fatality of the disease, combined with its increasing prevalence and the subsequent strain on healthcare resources, highlights the urgency of addressing IPF treatment.
Historically, IPF was understood to be an inflammation driven disease that eventually progressed to fibrosis. However, this concept was challenged by two pivotal clinical trials. The first, INSPIRE, found that patients treated with Interferon-γ did not experience significant clinical benefits. The second, PANTHER, evaluated a combination of three immunosuppressive agents (prednisone, azathioprine, and N-acetylcysteine) and revealed increased risks of death and hospitalization compared to placebo. (1, 7). These trials marked a paradigm shift in our understanding of IPF, suggesting that fibrosis, rather than inflammation, is central to IPF pathophysiology. Fibrosis in IPF is characterized by fibroblast activation, excessive collagen deposition, leading to the distortion of lung architecture and impaired gas exchange (Figure 1). This fibrotic process is driven by the aberrant activation of fibroblasts and myofibroblasts, which secrete collagen and other extracellular matrix proteins, resulting in the thickening and stiffening of the lung tissue. In the context of these findings, two antifibrotic therapies, Nintedanib and Pirfenidone, have been developed and approved for the treatment of IPF (8, 9). However, these drugs only slow down disease progression (10) and up to 40% patients discontinue treatment citing gastrointestinal, dermatological or liver-associated adverse drug reactions (11). Hence, lung transplantation remains the last resort for IPF patients, which is impractical for a majority of cases, considering factors such as age and comorbidities (4).
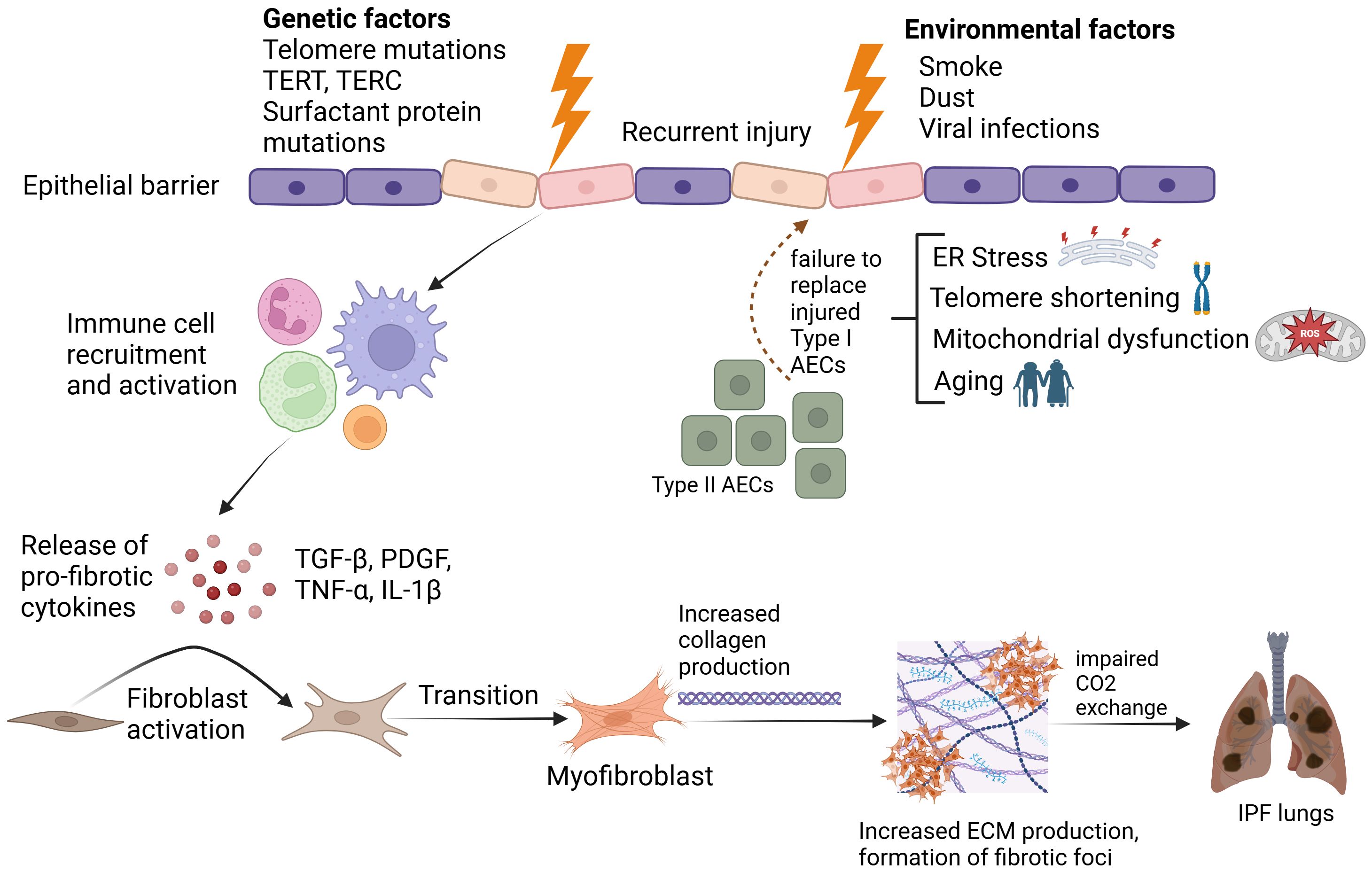
Figure 1. Pathophysiological mechanisms underlying Idiopathic Pulmonary Fibrosis. Created in BioRender. Ghigo, A. (2025) https://BioRender.com/2jn4ve5.
At a molecular level, the transition from inflammation to fibrosis as the focus of IPF research was further substantiated by studies that identified key signaling pathways involved in fibrogenesis (12). Among these, the PI3K/Akt pathway is involved in vital cellular processes such as survival, growth, proliferation, metabolism, apoptosis and angiogenesis. PI3Ks are lipid kinases divided into three classes – Class I, Class II and Class III. In mammalian cells, Class I PI3K catalytic subunits catalyze the phosphorylation of PtdIns-4,5-P2 (PIP2) to generate PtdIns-3,4,5-P3 (PIP3). Upon phosphorylation, PIP3 recruits two pleckstrin homology domain-containing kinases: the serine threonine kinase, Akt, and phosphoinositide-dependent kinase 1 (PDK-1). Akt undergoes conformational changes on direct binding to PIP3, exposing two of its amino acid sites, serine 473 and threonine 308, for phosphorylation by mammalian target of rapamycin complex 2 (mTORC2) and PDK1, respectively (13, 14). Once fully activated, Akt readily phosphorylates its downstream effectors such as mammalian target of rapamycin (mTOR), nuclear factor-κB (NF-κB), and p70 ribosomal protein S6 kinase (p70S6K), contributing to various cellular processes. PI3K/Akt signaling mediates key cellular functions such as proliferation and survival, bridging the mechanistic overlap between tumorigenesis and fibrotic progression of IPF. Understanding its regulation and potential for therapeutic targeting is critical to advancing IPF research and treatment strategies. Therefore, this review focuses on the role of the PI3K/Akt pathway in IPF pathogenesis, examining its multifaceted interactions and elucidating how dysregulation within this signaling axis contributes to the progression of fibrotic lung disease. Additionally, the review explores existing and emerging therapeutic avenues targeting key molecules along the PI3K pathway, discussing novel exploratory strategies for IPF treatment.
The PI3K/Akt signaling axis integrates key processes of cellular senescence, linking hallmark features of aging—such as telomere attrition, mitochondrial dysfunction, and impaired autophagy—to the molecular pathways underlying IPF pathogenesis. The association between IPF and aging is evidenced by the characteristic features of aging that are also observed in IPF, including impaired autophagy, DNA damage, mitochondrial dysfunction and telomere attrition. Telomere shortening triggers senescence, and is also an important risk factor for IPF, particularly familial IPF (15). Mutations in hTERT or hTR result in shorter telomeres and this telomere attrition in murine alveolar epithelial type 2 cells (AEC), but not mesenchymal cells, has been shown to promote fibrosis (16, 17). Lung fibroblasts from IPF patients also exhibit a senescent phenotype characterized by shorter telomeres and mitochondrial dysfunction (18). However, cellular senescence typically serves as a protective mechanism by limiting excessive cell proliferation, hence its role in promoting fibrosis in IPF appears paradoxical. Studies have shed light on the mechanisms through which senescence contributes to fibrosis. (19, 20). Telomere shortening exacerbates AEC dysfunction, while mitochondrial impairment and increased reactive oxygen species (ROS) drive the activation of pathways such as PI3K/mTOR. In the context of aging and IPF, reduced or lost PTEN expression induces Akt-dependent alveolar epithelial senescence (21). Akt hyperactivation drives apoptosis resistance and mTOR activity leads to inhibition of autophagy, factors which are necessary for fibrogenesis (22). This process leads to the upregulation of senescence markers including senescence-associated β-galactosidase, p21, p16, and p53, along with the emergence of a senescence-associated secretory phenotype (SASP). SASP is characterized by the secretion of pro-inflammatory and pro-fibrotic cytokines, including interleukin-6 (IL-6), interleukin-8 (IL-8), and TGF-β.
The emergence of SASP, driven by upregulated senescence markers, has been closely linked to PTEN loss and Akt hyperactivation to senescence, as demonstrated in bleomycin-induced models where Akt2 knockdown mitigated the senescence phenotype (21). In IPF, AEC are particularly prone to this senescent phenotype due to persistent PI3K/Akt activation in the absence of adequate PTEN function (21, 23, 24). Although SASP production occurs in both fibroblasts and AECs, it is the SASP from AECs that appears to be critical for the progression of fibrosis. This dysfunctional feedback loop of senescence, SASP production, PI3K signaling mediated fibroproliferation and apoptosis resistance, ultimately accelerates fibrosis, making the PI3K/Akt axis a critical target for therapeutic intervention in IPF by addressing the underlying mechanisms of senescence and fibrosis (15, 24, 25).
Upstream and downstream effectors of the PI3K/Akt signaling pathway drive IPF progression, mediating the interplay between pro-fibrotic stimuli and cellular responses. Upstream, growth factors and cytokines such as TGF-β, PDGF, and VEGF aberrantly activate the pathway, driving processes like fibroblast proliferation and extracellular matrix production. Downstream, PI3K/Akt signaling regulates key effector mechanisms, including myofibroblast differentiation, epithelial-mesenchymal transition (EMT), and metabolic reprogramming, which together perpetuate the fibrotic environment. Understanding these interactions reveals how dysregulated PI3K signaling orchestrates IPF pathogenesis.
In IPF, damaged alveolar epithelial cells, alveolar macrophages and fibroblasts release profibrotic cytokines such as TGF-β, Platelet-Derived growth factor (PDGF), Connective Tissue Growth Factor (CTGF) Vascular Endothelial Growth Factor (VEGF), and fibroblast growth factor (FGF) which aberrantly stimulate the PI3K/Akt pathway (26–28), hence perpetuating the cycle of injury and dysregulated repair. PDGF, a key target of Nintedanib, plays a critical role in stimulating fibroblast proliferation and collagen deposition via the PI3K/Akt pathway (29). TGF-β is the primary orchestrator of lung fibrosis, driving fibroblast activation, differentiation, and Extra Cellular Matrix (ECM) deposition. TGF-β also activates CTGF expression by primary AECs (30), which further stimulates EMT and upregulates ECM deposition. CTGF in turn induces activation of TGF-β as well as VEGF in a positive feedback loop (30, 31). This loop can be broken by CTGF inhibition, but CTGF expression by type 2 AECs and active fibroblasts is more prominent in the early stages of fibrosis and reduced in the later stages (32). This could explain why a phase 3 study of a CTGF inhibitor, Pamrevlumab, showed no clinical benefits compared to placebo (33). Apart from its direct effect on CTGF, TGF-β, increases hypoxia-inducible factor-1α (HIF-1α) expression via Smad signaling, thereby upregulating VEGF gene expression (34, 35). Akt activation also induces the upregulation of HIF-1α expression, which subsequently increases VEGF levels, promoting angiogenesis (36). Growth factors play a complex and sometimes isoform-specific role in fibrosis, with certain variants promoting fibrotic responses while others exhibit antifibrotic properties. For instance, VEGF165b has been shown to inhibit fibrosis, in contrast to VEGF165a, which promotes fibrotic processes (37, 38), implying that a more isoform-specific approach is required to yield substantial clinical benefits. Similarly, FGF was traditionally considered profibrotic, but newer findings suggest a preventative and therapeutic role. FGF1 attenuates TGF-β1 induced fibrosis by decreasing e-cadherin expression and inhibiting myofibroblast differentiation (39), FGF2 suppresses collagen production and fibroblast differentiation (40) while FGF7 and 10 improve lung repair and prolong survival in mouse models (41).
Growth factor activation of PI3K promotes cell survival, but PTEN counterbalances it by dephosphorylating PIP3 to inhibit Akt (42). Myofibroblasts extracted from IPF biopsies show reduced PTEN expression alongside Akt upregulation, and in bleomycin-induced mouse models, PTEN deficiency has been shown to exacerbate fibrosis, resulting in a 55% increase in collagen content compared to wild-type mice (43, 44). PTEN suppression and aberrant activation of Akt can result in decreased autophagic activity, creating an environment that promotes fibroblast proliferation and apoptosis resistance. When IPF fibroblasts interact with type I collagen—a major component of the ECM in fibrotic tissues— Akt activity remains high while autophagy markers, such as LC3-II, are suppressed (45–47).
A recent groundbreaking study in fibroblast lineage tracing identified a fibrotic fibroblast subset and two distinct inflammatory fibroblast subsets that drive both fibrosis and chronic inflammation. The fibrotic fibroblasts, marked by high Collagen Triple Helix Repeat Containing 1 (CTHRC1) expression, are central to active fibrotic foci and serve as the predominant producers of collagen and extracellular matrix (ECM) components, thereby driving fibrosis. The inflammatory fibrotic subset 1 localizes near the fibrotic foci, while inflammatory fibrotic subset 2 resides within the fibrotic foci, toward the interstitial side (48, 49). The PI3K/Akt signaling pathway has been shown to promote persistent fibrosis by driving apoptosis resistance and fibroblast proliferation, suggesting that this pathway may contribute to the maintenance and expansion of the fibrotic fibroblast population. Downstream of PI3K/Akt, Akt phosphorylates and inactivates FOXO3a, impairing its ability to regulate apoptosis, thereby allowing fibroblast accumulation and transition into myofibroblasts, further exacerbating fibrosis (50–52). While CTHRC1-expressing fibrotic fibroblasts are central to fibrosis progression, Tsukui et al. demonstrated that eliminating this population alone was insufficient to attenuate fibrosis, highlighting the involvement of additional fibroblast subsets. Beyond fibroblast proliferation, fibrosis progression also involves epithelial remodeling through epithelial-mesenchymal transition (EMT), a key process in which epithelial cells lose their identity and acquire mesenchymal traits, further contributing to the expansion of pro-fibrotic fibroblast populations. EMT is characterized by the loss of epithelial markers like E-cadherin and upregulation of mesenchymal markers such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin (53). Akt promotes EMT by activating transcription factors like Snail, ZEB1/2, Twist, and LEF-1, which repress E-cadherin and induce mesenchymal marker expression (54). Akt also phosphorylates GSK3-β, preventing β-catenin degradation. This stabilizes and translocates β-catenin to the nucleus, driving EMT-associated gene transcription (55, 56). In vivo, Akt1−/− mice show resistance to hypoxia-induced pulmonary fibrosis, directly linking Akt inhibition to reduced fibroblast activity and apoptosis resistance (57). Considering that EMT-derived fibroblasts may also contribute to the fibrotic fibroblast pool, the PI3K/Akt axis appears to play a central role in sustaining fibrotic fibroblast populations through both direct fibroblast proliferation and EMT-driven mesenchymal transition.
Over the past two decades, extensive clinical trials have aimed to identify effective treatments for IPF. However, most investigational therapies have failed to demonstrate significant efficacy, highlighting the complex and challenging nature of this disease (58, 59). This challenge arises in part from the poorly understood mechanisms driving IPF progression and the difficulty of reversing established fibrosis. Current approved therapies, Pirfenidone and Nintedanib, offer some benefit in slowing disease progression but are far from optimal in halting or reversing the fibrotic processes.
IPF and non-small cell lung cancer (NSCLC) share molecular, cellular, and genetic similarities, including mutations in genes like p53, KRAS, and p21. Shared genetic predispositions, such as mutations in SFTPA1 and SFTPA2, lead to endoplasmic reticulum stress, impaired protein secretion, and apoptosis, driving fibrosis in IPF and tumorigenesis in lung cancer. Both diseases exhibit epigenetic changes, such as deregulated noncoding RNAs like miR-21, and involve common processes like EMT and apoptosis resistance. Abnormal activation of pathways like Wnt/β-catenin and PI3K/Akt further contributes to metaplasia, hyperproliferation, fibrosis, and tumor growth (60–62).The PI3K/Akt pathway is central to both diseases, promoting cell survival, proliferation, and resistance to apoptosis in NSCLC, and driving fibroblast proliferation, ECM deposition, and apoptosis resistance in IPF. Its influence on EMT, a shared process in fibrosis and cancer, facilitates epithelial marker loss and mesenchymal trait acquisition, fueling fibrosis and tumor invasion.
Despite these overlaps, IPF differs from cancer as it lacks monoclonality and metastatic potential. Cancer often begins in one organ and spreads, while IPF is a bilateral disease from early stages (63, 64). These differences necessitate tailored treatment strategies for IPF. Drugs like Nintedanib, originally developed for cancer, target pathways common to fibrosis and tumorigenesis, slowing disease progression in both NSCLC and IPF. Similarly, Pirfenidone inhibits EMT and tumor stroma, addressing fibrosis and cancer metastasis. However, both treatments have dose-limiting toxicities, highlighting the urgent need for new therapies in IPF.
Based on the overlapping metabolic profiles of IPF and lung cancer, Omipalisib was the first PI3K/mTOR inhibitor originally developed for solid tumors, and later repurposed for IPF. Pharmacokinetic studies with Omipalisib showed a decrease in glucose uptake and an acceptable tolerability profile; however, patients also experienced adverse events like gastrointestinal disturbances and hyperglycemia, which are consistent with the expected toxicities of PI3K/mTOR inhibition (65, 66). These findings highlight the therapeutic promise of PI3K/mTOR inhibitors for IPF while emphasizing the need for safer alternatives.
Prior to Omipalisib, Everolimus, a selective mTORc1 inhibitor, was tested in 89 IPF patients but worsened disease progression, possibly due to its partial inhibition of 4E-BP1, a key driver of TGFβ1-induced collagen production (67, 68). Newer dual mTORc1/mTORc2 inhibitors, such as Sapanisertib and Vistusertib, have shown promise in reducing TGF-β1-induced EMT, fibrosis markers, and proinflammatory cytokines like tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β) and interleukin-6 (IL-6)in preclinical models (69–71). Their clinical success, however, depends on balancing efficacy and safety, a challenge for PI3K/mTOR inhibitors. Current limitations of mTOR inhibitors suggest a need for better selectivity and targeted delivery. A key question that remains unaddressed in IPF is whether isoform selective PI3K inhibition, such as with drugs like Alpelisib that selectively target PI3Kα, could provide improved clinical benefits. If isoform-specific inhibition is sufficient to halt fibrosis progression, a PI3Kα degrader like WJ112-14, which selectively tags the alpha isoform for degradation, could offer greater efficacy over reversible kinase inhibition by ensuring sustained depletion of PI3Kα levels, although this remains to be clinically validated (72). Notably, a dual PI3K-δ/γ inhibitor, Duvelisib, indicated for Chronic Lymphocytic Leukemia, was recently shown to attenuate fibrosis in vivo and in vitro (73). Although these findings are clinically relevant, delivery, dosage and toxicity remain key considerations given the heterogenous nature of this disease. In contrast to isoform-specific inhibition, a first-in-class pan-PI3K and dual mTORc1/mTORc2 inhibitor, Gedatolisib, was shown to have superior potency and efficacy over alpelisib, capivasertib (AKT inhibitor), and everolimus in breast cancer cell lines. Currently, a global combinatorial phase 3 study with Gedatolisib is underway for patients with HR+/HER2− ABC mutations (74). The results of this trial may inform future studies exploring the potential of pan-PI3K/dual mTOR inhibitors in the treatment of IPF.
While drug repurposing remains a promising strategy, significant advancements have also been made in developing novel PI3K/mTOR inhibitors designed specifically for IPF pathophysiology. These drugs provide unique mechanisms of action and modes of administration, which could potentially leading to greater clinical benefits over existing drugs. Campa et al. developed KITCL27, as an inhalation-based prodrug pan-PI3K inhibitor designed to be activated only after hydrolysis inside target cells. Preclinical studies of KITCL27 in bleomycin-induced mouse models showed that inhaled CL27c effectively reduced lung fibrosis by decreasing collagen deposition and downregulated key pro-fibrotic markers, including TGF-β1, CTGF, Collagen type I (Col1a1), and matrix metalloproteinase-2 (MMP2) (75). Another innovative approach to PI3K inhibition was the development of FAPL-PI3Ki1 by Hettiarachchi et. al., which is a PI3K inhibitor conjugated to a ligand that targets fibroblast activation protein (FAP), a protein exclusively expressed by collagen-producing myofibroblasts. This FAPL-PI3Ki1 demonstrated reduced collagen deposition, decreased the expression of fibrotic markers such as α-SMA and Col1A1, and improved survival in bleomycin-induced mice (76).
Considering the ineffectiveness of existing drugs, the toxicity of PI3K inhibitors and the role of Akt hyperactivation in fibrosis progression, targeting Akt through pharmacological inhibition offers a new therapeutic approach for IPF. Given the extensive research and development that has already been conducted on Akt inhibitors for cancers, their pharmacokinetics, safety profiles, and molecular interactions are well understood, making them strong candidates for repurposing in IPF (Table 1) (57, 77). Notably, Akt inhibitors like Triciribine and MK-2206 have already been tested in bleomycin-induced mouse models of IPF (78, 79) supporting their use and development for IPF.
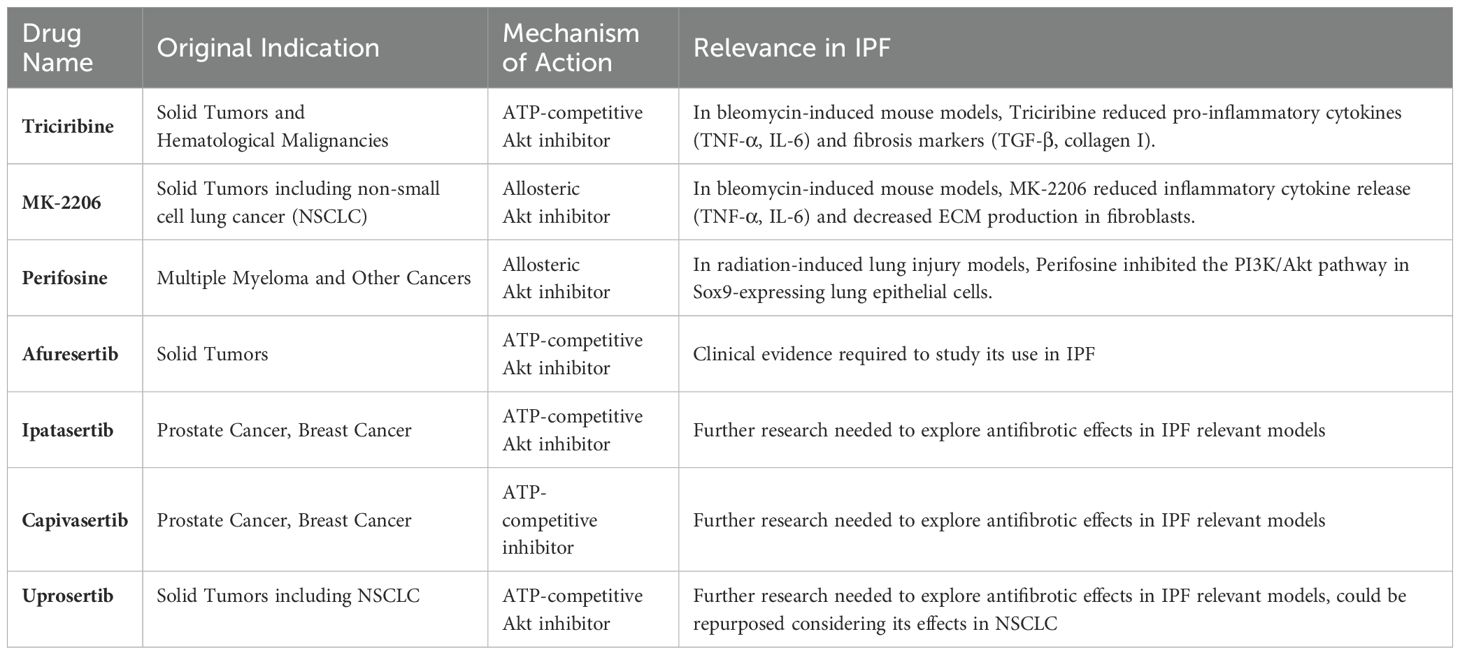
Table 1. Summary of Akt inhibitors used as monotherapies for cancers, with the potential for repurposing in IPF.
Similar to oral PI3K inhibitors, the development of Akt inhibitors faces challenges due to adverse effects such as diarrhea, nausea, and skin rashes, as observed in clinical trials for solid tumors. ATP-competitive inhibitors like ipatasertib and capivasertib struggle with selectivity issues within the AGC kinase family, leading to off-target effects. In contrast, allosteric inhibitors that bind to the Pleckstrin Homology domain of Akt offer greater specificity for Akt. However, these allosteric inhibitors have shown limited clinical efficacy and require optimization before they can be considered viable options for IPF treatment.
A novel alternative to pharmacological inhibition of Akt is to reduce intracellular Akt levels via protein degradation. This strategy has led to the development of Akt degraders, which are proteolysis targeting chimeras linked to Akt inhibitors. INY-03-041 is a pan-Akt degrader that combines GDC-0068, a pan-Akt inhibitor, with Lenalidomide, an immunomodulatory drug that recruits Cereblon (CRBN) and brings it into close proximity to Akt. CRBN acts as a substrate adaptor, guiding Akt to the E3 ubiquitin ligase complex, where Akt is tagged with ubiquitin and subsequently degraded by the proteasome (80, 81). To date, there are only a handful of Akt degraders currently under development for solid tumors, including INY-03-041, MS21, MS143 and MS5033. Although they are yet to be repurposed for clinical studies in IPF, Akt degraders exhibit a longer half-life and a more prolonged pharmacological effect compared to their constituent Akt inhibitors alone (81), which could provide added benefits in terms of dose reduction. However, the on-target toxicities already observed with pharmacological AKT inhibitors, combined with the increased potency of AKT degraders suggest that AKT degraders may have a smaller therapeutic window. Therefore, it will be critical to explore dosages to achieve a balance between safety and efficacy for both, cancers and IPF.
Since the approval of Nintedanib, several tyrosine kinase inhibitors have been repurposed for IPF with mixed outcomes. Imatinib, a PDGFR and TGF-β inhibitor, was discontinued due to lack of clinical benefits (82). Pamufetinib, a VEGFR, PDGFR, and HGFR inhibitor, showed acceptable safety in a phase 2 trial (83). Similar to Nintedanib, Pamufetinib also inhibits both PDGFR-α and PDGFR-β (29), however, the targeted inhibition of PDGFR-β but not PDGFR-α attenuates fibrosis (84) in bleomycin-treated mice. Additionally, some tyrosine kinases like VEGFR and FGFR exhibit isoform-specific, context-dependent profibrotic or antifibrotic effects which make growth factor inhibition increasingly complex.
TGF-β remains a key therapeutic target upstream of PI3K/Akt, though its inhibition poses challenges due to adverse effects, including heart valve lesions and skin cancers (85–87). Indirect approaches like integrin inhibition with Bexotegrast, a dual αvβ6/αvβ1 inhibitor, have shown promise, reducing lung function decline in a phase 2 study with no severe adverse events (88). Longer and larger clinical trials are awaited to conclude positive clinical outcomes. Another novel strategy to indirectly inhibit TGF-β signaling involves targeting lysyl oxidase–like 2 (LOXL2), an enzyme restricted to fibroblasts. Inhibition of LOXL2 enzymatic activity induces its auto-oxidation, leading to the creation of a metabolite that directly inhibits TGF-β receptor I (TβRI) kinase (89). Although TGF-β is the main driver of fibrosis, in vivo deletion of TGF-β is shown to exacerbate inflammation, hence it is crucial to consider drug design, dosage and strategies for targeting TGF-β. Downstream of PI3K/Akt/mTOR, drugs targeting NFκB, GSK-3β (90) and p706Sk have been developed, though not all have been tested in IPF models (91). Specifically, ACT001, an NFκB inhibitor, was the latest of NFκB inhibitors that demonstrated anti-fibrotic activity in fibroblasts from IPF patients (92).
Despite the pervasive role of senescence in IPF, current standard-of-care drugs have no effect on senescence markers in IPF tissues (93), highlighting the possibility of a more direct approach. Senolytics, a class of drugs targeting senescent cells, showed promising preclinical and early clinical trial results in IPF patients (20). Quercetin attenuated bleomycin-induced pulmonary fibrosis, by reducing senescence markers like p21 and SASP, and inhibiting apoptosis resistance via modulating Akt activity (94). A first-in-human study with Quercetin and Dasatinib demonstrated selective senescent cell elimination, reduced markers, and modest functional improvement, supporting their potential as IPF therapies (95). Although, larger, randomized clinical trials are necessary to further validate this data, given that mTOR inhibitors have also directly demonstrated reduction in senescence markers, it would be interesting to evaluate the effects of more diverse “senolytic cocktails” targeting both senescence and fibrosis. Senolytics may also complement PI3K inhibitors, offering a synergistic approach to disrupt the profibrotic cycle. Furthermore, other classes of anti-senescence drugs, including SASP inhibitors and drugs targeting senescent pathways such as Navitoclax could be included in these drug cocktails to develop newer therapies. Considering that shortened telomeres are linked to higher risks of IPF, telomerase activators such as TA-65 (96), which counteract telomere shortening linked to IPF progression could be studied in IPF, alone and in combination with inhibitors of PI3K/Akt.
The systemic toxicities of oral PI3K/Akt inhibitors, including gastrointestinal effects, hyperglycemia, and skin rashes, have driven next-generation approaches against the PI3K/Akt pathway such as Akt degraders, inhalation-based drug delivery, prodrugs, and fibroblast-specific conjugates to enhance target selectivity and reduce adverse effects. However, whether such next-generation drugs will not face the same challenges as older ones still needs further clinical experimentation. Despite the promising improvements in safety and efficacy offered by the new class of mTOR inhibitors like Gedatolisib over existing drugs in breast cancer (74), if repurposed to IPF, chronic PI3K inhibition may still lead to unexpected compensatory activation of alternative pro-fibrotic pathways such as Mitogen-Activated Protein Kinase (MAPK) signaling (97). These pathway redundancies could be addressed by Akt degraders like INY-03-041, which was initially developed for oncology, and leverages short-term, high-intensity therapy to achieve rapid systemic Akt depletion, suppressing tumor growth. However, while cancer patients may tolerate these effects during short-term therapy, IPF patients require long-term treatment. These obstacles could be overcome by considering newer routes of administration, intermittent dosing strategies and biomarker detection to monitor early signs of toxicity and resistance to treatment.
In addition to pre-existing issues, these therapeutics will also face new challenges arising from their novelty, such as the inhaled route of administration, prodrug formulations, or unique structures like fibroblast-specific conjugates. Inhaled formulations could enhance local drug bioavailability while minimizing systemic toxicity, as shown by a clinical trial with inhaled pirfenidone (98) but this would bring about inhalation-based challenges such as limited biodistribution due to reduced airflow in areas of severe fibrosis and reduced efficacy due to poor lung penetration. Next, prodrug formulations are a promising approach for minimizing toxicities, but they could generate highly reactive intermediates, increasing the risk of adverse effects, such as DNA damage or mitochondrial dysfunction. Drugs with unique conjugates in their structures like FAPL-PI3Ki1, aim to restrict drug action to fibrotic lung tissue by binding to FAP, whose expression is limited to lungs, thus improving precision and reducing systemic toxicity. However, FAP expression varies across different fibrotic foci and is not uniformly upregulated in all myofibroblasts (99), potentially leading to incomplete pathway inhibition, and subsequently, in residual fibrosis progression despite treatment.
These challenges pose significant hurdles that could delay drug development, but rather than obstacles, they should be seen as critical checkpoints that refine the drug development strategy. Major concerns such as target engagement, response to treatment, resistance mechanisms, and toxicities related to PI3K inhibitors could be better addressed with biomarker-driven patient monitoring. In this regard, response and resistance biomarkers are integral to future clinical trial design. IPF is a highly heterogeneous disease with multiple pathogenic drivers, and the absence of PI3K mutations suggests that its activation in IPF is more likely due to signaling dysregulation rather than genetic alterations. Moreover, disease progression is highly variable and some patients experience acute lung function decline with rapid fibrosis, while others may remain stable for years without significant deterioration. Thus IPF-specific molecular biomarkers would facilitate classification of patient subgroups most likely to benefit from therapies like PI3K inhibitors, Akt degraders or senolytic cocktails. Considering trials with current standard of care, for monitoring Pirfenidone efficacy, serum surfactant protein (SP)-D, a protein released by damaged AECs is a validated pharmacodynamic biomarker with limited prognostic value, while SP-A and KL-6 have been tested as efficacy biomarkers for both Nintedanib and Pirfenidone in a single-center retrospective study (100, 101). However, these biomarkers reflect alveolar epithelial injury but do not capture PI3K/Akt-specific pathway activity or therapeutic response. This makes it even more crucial to establish biomarkers that specifically reflect PI3K/Akt pathway activation and response to inhibition.
Some biomarkers used to assess PI3K inhibitors in cancer could potentially be translated to IPF, given their shared mechanisms. The Omipalisib trial has already demonstrated the feasibility of using p-Akt levels in bronchoalveolar lavage (BAL) fluid macrophages to track target engagement in IPF patients, indicating its relevance as a transferable biomarker (65). Pharmacodynamic biomarkers of PI3K inhibition relevant in tumors—such as phosphorylated proteins like p-PRAS40, p-S6 kinase, and p-4EB, metabolic markers like glucose metabolism, GLUT1, and GLUT4, or imaging biomarkers like 18F-FDG-PET (102, 103) —could also have relevance in IPF. Additionally, computational analyses of large IPF datasets could identify new biomarkers, although thus far they have primarily been used to identify diagnostic or predictive markers, rather than prognostic biomarkers tailored for pathway-specific therapies. To date, no single biomarker readout has clear prognostic value over the treatment course. Since a standard-of-care has been established for IPF, new clinical trials cannot ethically incorporate a true placebo. Thus, a composite biomarker framework for monitoring PI3K inhibitor efficacy in IPF, combining endpoint markers like Forced Vital Capacity with transcriptional biomarkers (like FOXO3A), senescence-associated markers (p21, SA-β-gal), and phosphorylation-based readouts would enable dynamic monitoring of patient response, drug efficacy and disease progression. Despite the added complexity of standard-of-care, as more PI3K inhibitors are tested in clinical settings, integrating computational analyses of large-scale patient data in the study design will progressively address the gap in efficacy biomarkers specific to IPF. This strategy will enhance biomarker discovery and trial design efficacy, accelerating the translation of PI3K inhibitors from bench to bedside.
IPF is a chronic, interstitial lung disease marked by significant patient heterogeneity and a lack of relevant biomarkers to guide drug development. Over the years, considerable progress has been made in understanding the mechanisms driving IPF, particularly the dysregulation of the PI3K/Akt pathway. While recent advances in selective PI3K inhibitors, Akt degraders, and dual PI3K/mTOR inhibitors offer promising therapeutic potential, challenges such as selectivity and systemic toxicities remain significant barriers to their clinical application. Innovative strategies, such as inhalation-based prodrugs [e.g., KITCL27 (75)] and fibroblast-specific conjugates [e.g., FAPL-PI3Ki1 (76)], represent important steps toward reducing systemic toxicity while enhancing therapeutic efficacy.
Senescence is increasingly recognized as a key driver of IPF, with PTEN loss and PI3K/Akt hyperactivation exacerbating senescence in alveolar epithelial cells and fibroblasts, forming a feedback loop that accelerates disease progression. Senolytics, such as Dasatinib and Quercetin, offer a transformative approach by selectively eliminating senescent cells, reducing fibrosis and inflammation in preclinical studies. Addressing IPF requires a multifaceted strategy combining targeted therapies like PI3K inhibitors with interventions tackling aging-related mechanisms, including mitochondrial dysfunction and telomere attrition. Exploring such combination therapies may provide greater benefits by addressing multiple aspects of IPF pathogenesis. While current treatments remain limited, optimizing drug formulations, developing better biomarker-driven patient selection strategies, and combining these therapies with other antifibrotic agents may help overcome these hurdles and improve treatment outcomes in IPF.
JB: Writing – original draft, Writing – review & editing. AG: Writing – review & editing. EH: Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 955534.
EH is the co-founder and scientific advisor of Kither Biotech, a pharmaceutical company developing PI3K inhibitors for the treatment of respiratory diseases. JB works as a research associate at Kither Biotech. Author AG was employed by the company Kither Biotech.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that Generative AI was used in the creation of this manuscript.
Generative AI was used to tweak or enhance sentences and summarize reference citations.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 366:1968–77. doi: 10.1056/NEJMoa1113354
2. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. (2011) 183:788–824. doi: 10.1164/rccm.2009-040GL
3. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. (2022) 17:515–46. doi: 10.1146/annurev-pathol-042320-030240
4. Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T, et al. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol Ther. (2021) 222:107798. doi: 10.1016/j.pharmthera.2020.107798
5. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. (2015) 46:795–806. doi: 10.1183/09031936.00185114
6. Leslie KO. Idiopathic pulmonary fibrosis may be a disease of recurrent, tractional injury to the periphery of the aging lung: a unifying hypothesis regarding etiology and pathogenesis. Arch Pathol Lab Med. (2012) 136:591–600. doi: 10.5858/arpa.2011-0511-OA
7. Behr J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 367:869; author reply 870–871. doi: 10.1056/NEJMc1207471
8. Galli JA, Pandya A, Vega-Olivo M, Dass C, Zhao H, Criner GJ. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: Tolerability and adverse drug reactions. Respirol Carlton Vic. (2017) 22:1171–8. doi: 10.1111/resp.13024
9. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
10. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2083–92. doi: 10.1056/NEJMoa1402582
11. Ogura T, Inoue Y, Azuma A, Homma S, Kondoh Y, Tanaka K, et al. Real-world safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: interim report of a post-marketing surveillance in Japan. Adv Ther. (2023) 40:1474–93. doi: 10.1007/s12325-022-02411-y
12. Kulkarni YM, Dutta S, Iyer AKV, Venkatadri R, Kaushik V, Ramesh V, et al. A proteomics approach to identifying key protein targets involved in VEGF inhibitor mediated attenuation of bleomycin-induced pulmonary fibrosis. Proteomics. (2016) 16:33–46. doi: 10.1002/pmic.201500171
13. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. (2014) 46:372–83. doi: 10.3109/07853890.2014.912836
14. Sarris EG, Saif MW, Syrigos KN. The biological role of PI3K pathway in lung cancer. Pharm Basel Switz. (2012) 5:1236–64. doi: 10.3390/ph5111236
15. Alder JK, Chen JJ-L, Lancaster L, Danoff S, Su S, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. (2008) 105:13051–6. doi: 10.1073/pnas.0804280105
16. Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. (2012) 730:52–8. doi: 10.1016/j.mrfmmm.2011.10.013
17. Armanios MY, Chen JJ-L, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. (2007) 356:1317–26. doi: 10.1056/NEJMoa066157
18. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1164–73. doi: 10.1152/ajplung.00220.2017
19. Hashimoto M, Asai A, Kawagishi H, Mikawa R, Iwashita Y, Kanayama K, et al. Elimination of p19ARF-expressing cells enhances pulmonary function in mice. JCI Insight. (2016) 1:e87732. doi: 10.1172/jci.insight.87732
20. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
21. Qiu T, Tian Y, Gao Y, Ma M, Li H, Liu X, et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging. (2019) 11:7492–509. doi: 10.18632/aging.102262
22. Romero Y, Bueno M, Ramirez R, Álvarez D, Sembrat JC, Goncharova EA, et al. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell. (2016) 15:1103–12. doi: 10.1111/acel.12514
23. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L391–401. doi: 10.1152/ajplung.00097.2010
24. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. (2021) 203:707–17. doi: 10.1164/rccm.202004-1274OC
25. Tu M, Wei T, Jia Y, Wang Y, Wu J. Molecular mechanisms of alveolar epithelial cell senescence and idiopathic pulmonary fibrosis: a narrative review. J Thorac Dis. (2023) 15:186–203. doi: 10.21037/jtd-22-886
26. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. (2000) 275:36803–10. doi: 10.1074/jbc.M005912200
27. Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev Off J Eur Respir Soc. (2011) 20:85–97. doi: 10.1183/09059180.00001111
28. Wilkes MC, Mitchell H, Penheiter SG, Doré JJ, Suzuki K, Edens M, et al. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. (2005) 65:10431–40. doi: 10.1158/0008-5472.CAN-05-1522
29. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. (2014) 349:209–20. doi: 10.1124/jpet.113.208223
30. Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L786–796. doi: 10.1152/ajplung.00243.2013
31. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. (2012) 5:S24. doi: 10.1186/1755-1536-5-S1-S24
32. Pan LH, Yamauchi K, Uzuki M, Nakanishi T, Takigawa M, Inoue H, et al. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur Respir J. (2001) 17:1220–7. doi: 10.1183/09031936.01.00074101
33. Raghu G, Richeldi L, Fernández Pérez ER, De Salvo MC, Silva RS, Song JW, et al. Pamrevlumab for idiopathic pulmonary fibrosis: the ZEPHYRUS-1 randomized clinical trial. JAMA. (2024) 332:380–9. doi: 10.1001/jama.2024.8693
34. Epstein Shochet G, Bardenstein-Wald B, McElroy M, Kukuy A, Surber M, Edelstein E, et al. Hypoxia inducible factor 1A supports a pro-fibrotic phenotype loop in idiopathic pulmonary fibrosis. Int J Mol Sci. (2021) 22:3331. doi: 10.3390/ijms22073331
35. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. (2006) 281:24171–81. doi: 10.1074/jbc.M604507200
36. Lu Y, Azad N, Wang L, Iyer AKV, Castranova V, Jiang B-H, et al. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. (2010) 42:432–41. doi: 10.1165/rcmb.2009-0002OC
37. Atamas SP. Vascular endothelial growth factor in idiopathic pulmonary fibrosis. Imbalancing Act Am J Respir Crit Care Med. (2017) 196:409–11. doi: 10.1164/rccm.201703-0605ED
38. Barratt SL, Blythe T, Jarrett C, Ourradi K, Shelley-Fraser G, Day MJ, et al. Differential expression of VEGF-axxx isoforms is critical for development of pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 196:479–93. doi: 10.1164/rccm.201603-0568OC
39. Shimbori C, Bellaye P-S, Xia J, Gauldie J, Ask K, Ramos C, et al. Fibroblast growth factor-1 attenuates TGF-β1-induced lung fibrosis. J Pathol. (2016) 240:197–210. doi: 10.1002/path.4768
40. Koo HY, El-Baz LM, House S, Cilvik SN, Dorry SJ, Shoukry NM, et al. Fibroblast growth factor 2 decreases bleomycin-induced pulmonary fibrosis and inhibits fibroblast collagen production and myofibroblast differentiation. J Pathol. (2018) 246:54–66. doi: 10.1002/path.5106
41. MacKenzie B, Korfei M, Henneke I, Sibinska Z, Tian X, Hezel S, et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir Res. (2015) 16:83. doi: 10.1186/s12931-015-0242-2
42. Cantley LC. The phosphoinositide 3-kinase pathway. Science. (2002) 296:1655–7. doi: 10.1126/science.296.5573.1655
43. White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, et al. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med. (2006) 173:112–21. doi: 10.1164/rccm.200507-1058OC
44. Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. (2008) 205:1659–72. doi: 10.1084/jem.20080001
45. Nho RS, Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PloS One. (2014) 9:e94616. doi: 10.1371/journal.pone.0094616
46. Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PloS One. (2012) 7:e41394. doi: 10.1371/journal.pone.0041394
47. Yue Y-L, Zhang M-Y, Liu J-Y, Fang L-J, Qu Y-Q. The role of autophagy in idiopathic pulmonary fibrosis: from mechanisms to therapies. Ther Adv Respir Dis. (2022) 16:17534666221140972. doi: 10.1177/17534666221140972
48. Cabrera-Fuentes HA, Barreto G, Al-Suhaimi EA, Liehn EA. Fibroblast plasticity in pulmonary and cardiovascular fibrosis: Mechanistic insights and emerging therapeutic targets. Arch Med Res. (2025) 56:103173. doi: 10.1016/j.arcmed.2024.103173
49. Tsukui T, Wolters PJ, Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature. (2024) 631:627–34. doi: 10.1038/s41586-024-07660-1
50. Nho RS, Peterson M, Hergert P, Henke CA. FoxO3a (Forkhead Box O3a) deficiency protects Idiopathic Pulmonary Fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas. PloS One. (2013) 8:e61017. doi: 10.1371/journal.pone.0061017
51. Wan H, Xie T, Xu Q, Hu X, Xing S, Yang H, et al. Thy-1 depletion and integrin β3 upregulation-mediated PI3K-Akt-mTOR pathway activation inhibits lung fibroblast autophagy in lipopolysaccharide-induced pulmonary fibrosis. Lab Investig J Tech Methods Pathol. (2019) 99:1636–49. doi: 10.1038/s41374-019-0281-2
52. Xie T, Xu Q, Wan H, Xing S, Shang C, Gao Y, et al. Lipopolysaccharide promotes lung fibroblast proliferation through autophagy inhibition via activation of the PI3K-Akt-mTOR pathway. Lab Investig J Tech Methods Pathol. (2019) 99:625–33. doi: 10.1038/s41374-018-0160-2
53. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. (2014) 7:re8. doi: 10.1126/scisignal.2005189
54. Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. (2007) 26:7445–56. doi: 10.1038/sj.onc.1210546
55. Zheng H, Yang Z, Xin Z, Yang Y, Yu Y, Cui J, et al. Glycogen synthase kinase-3β: a promising candidate in the fight against fibrosis. Theranostics. (2020) 10:11737–53. doi: 10.7150/thno.47717
56. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. (2004) 6:931–40. doi: 10.1038/ncb1173
57. Abdalla M, Sabbineni H, Prakash R, Ergul A, Fagan SC, Somanath PR. The Akt inhibitor, triciribine, ameliorates chronic hypoxia-induced vascular pruning and TGFβ-induced pulmonary fibrosis. Br J Pharmacol. (2015) 172:4173–88. doi: 10.1111/bph.13203
58. Raghu G. Idiopathic pulmonary fibrosis: lessons from clinical trials over the past 25 years. Eur Respir J. (2017) 50:1701209. doi: 10.1183/13993003.01209-2017
59. Zamora AC, Ortega VE, Carmona EM. When the third time is not the charm-trial outcomes in idiopathic pulmonary fibrosis. JAMA. (2024) 332:374–6. doi: 10.1001/jama.2024.8776
60. Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PloS One. (2012) 7:e33770. doi: 10.1371/journal.pone.0033770
61. Tzouvelekis A, Gomatou G, Bouros E, Trigidou R, Tzilas V, Bouros D. Common pathogenic mechanisms between idiopathic pulmonary fibrosis and lung cancer. Chest. (2019) 156:383–91. doi: 10.1016/j.chest.2019.04.114
62. Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Respir Rev Off J Eur Respir Soc. (2013) 22:265–72. doi: 10.1183/09059180.00003613
63. Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med. (2006) 174:654–8. doi: 10.1164/rccm.200602-205OC
64. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. (2010) 35:496–504. doi: 10.1183/09031936.00077309
65. Lukey PT, Harrison SA, Yang S, Man Y, Holman BF, Rashidnasab A, et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 53:1801992. doi: 10.1183/13993003.01992-2018
66. Mercer PF, Woodcock HV, Eley JD, Platé M, Sulikowski MG, Durrenberger PF, et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax. (2016) 71:701–11. doi: 10.1136/thoraxjnl-2015-207429
67. Malouf MA, Hopkins P, Snell G, Glanville AR, Everolimus in IPF Study Investigators. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirol Carlton Vic. (2011) 16:776–83. doi: 10.1111/j.1440-1843.2011.01955.x
68. Platé M, Guillotin D, Chambers RC. The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur Respir Rev Off J Eur Respir Soc. (2020) 29:200269. doi: 10.1183/16000617.0269-2020
69. Basu B, Krebs MG, Sundar R, Wilson RH, Spicer J, Jones R, et al. Vistusertib (dual m-TORC1/2 inhibitor) in combination with paclitaxel in patients with high-grade serous ovarian and squamous non-small-cell lung cancer. Ann Oncol Off J Eur Soc Med Oncol. (2018) 29:1918–25. doi: 10.1093/annonc/mdy245
70. Shaikh TB, Chandra Y, Andugulapati SB, Sistla R. Vistusertib improves pulmonary inflammation and fibrosis by modulating inflammatory/oxidative stress mediators via suppressing the mTOR signalling. Inflamm Res Off J Eur Histamine Res Soc Al. (2024) 73:1223–37. doi: 10.1007/s00011-024-01894-5
71. Xu Z, Lv Y, Kong D, Jiang W. Sapanisertib attenuates pulmonary fibrosis by modulating Wnt5a/mTOR signalling. Basic Clin Pharmacol Toxicol. (2023) 133:226–36. doi: 10.1111/bcpt.13924
72. Jauslin WT, Schild M, Schaefer T, Borsari C, Orbegozo C, Bissegger L, et al. A high affinity pan-PI3K binding module supports selective targeted protein degradation of PI3Kα. Chem Sci. (2024) 15:683–91. doi: 10.1039/D3SC04629J
73. Li X, Ma X, Miao Y, Zhang J, Xi B, Li W, et al. Duvelisib attenuates bleomycin-induced pulmonary fibrosis via inhibiting the PI3K/Akt/mTOR signalling pathway. J Cell Mol Med. (2023) 27:422–34. doi: 10.1111/jcmm.17665
74. Rossetti S, Broege A, Sen A, Khan S, MacNeil I, Molden J, et al. Gedatolisib shows superior potency and efficacy versus single-node PI3K/AKT/mTOR inhibitors in breast cancer models. NPJ Breast Cancer. (2024) 10:40. doi: 10.1038/s41523-024-00648-0
75. Campa CC, Silva RL, Margaria JP, Pirali T, Mattos MS, Kraemer LR, et al. Inhalation of the prodrug PI3K inhibitor CL27c improves lung function in asthma and fibrosis. Nat Commun. (2018) 9:5232. doi: 10.1038/s41467-018-07698-6
76. Hettiarachchi SU, Li Y-H, Roy J, Zhang F, Puchulu-Campanella E, Lindeman SD, et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci Transl Med. (2020) 12:eaay3724. doi: 10.1126/scitranslmed.aay3724
77. Chen M, Yu Y, Mi T, Guo Q, Xiang B, Tian X, et al. MK-2206 alleviates renal fibrosis by suppressing the akt/mTOR signaling pathway in vivo and in vitro. Cells. (2022) 11:3505. doi: 10.3390/cells11213505
78. Cui H, Cheng Y, He Y, Cheng W, Zhao W, Zhao H, et al. The AKT inhibitor MK2206 suppresses airway inflammation and the pro−remodeling pathway in a TDI−induced asthma mouse model. Mol Med Rep. (2020) 22:3723–34. doi: 10.3892/mmr.2020.11450
79. Gou M, Wang M. Effects of akt inhibition on the regulatory and proteomic profiles of alveolar macrophages enriched from lung lavages of pulmonary fibrosis patients. Front Med Case Rep. (2022) 03. doi: 10.47746/FMCR.2022.3101
80. You I, Erickson EC, Donovan KA, Eleuteri NA, Fischer ES, Gray NS, et al. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem Biol. (2020) 27:66–73.e7. doi: 10.1016/j.chembiol.2019.11.014
81. Yu X, Xu J, Shen Y, Cahuzac KM, Park K-S, Dale B, et al. Discovery of potent, selective, and in vivo efficacious AKT kinase protein degraders via structure-activity relationship studies. J Med Chem. (2022) 65:3644–66. doi: 10.1021/acs.jmedchem.1c02165
82. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. (2004) 114:1308–16. doi: 10.1172/JCI19603
83. Nishioka Y, Homma S, Ogura T, Sato S, Arai N, Tomii K, et al. Exploratory phase 2 study of the novel oral multi-kinase inhibitor TAS-115 in patients with idiopathic pulmonary fibrosis. Respir Investig. (2023) 61:498–507. doi: 10.1016/j.resinv.2023.04.008
84. Kishi M, Aono Y, Sato S, Koyama K, Azuma M, Abe S, et al. Blockade of platelet-derived growth factor receptor-β, not receptor-α ameliorates bleomycin-induced pulmonary fibrosis in mice. PloS One. (2018) 13:e0209786. doi: 10.1371/journal.pone.0209786
85. Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol Off J Eur Soc Med Oncol. (2020) 31:1336–49. doi: 10.1016/j.annonc.2020.07.009
86. Massagué J, Sheppard D. TGF-β signaling in health and disease. Cell. (2023) 186:4007–37. doi: 10.1016/j.cell.2023.07.036
87. Mitra MS, Lancaster K, Adedeji AO, Palanisamy GS, Dave RA, Zhong F, et al. A potent pan-TGFβ Neutralizing monoclonal antibody elicits cardiovascular toxicity in mice and cynomolgus monkeys. Toxicol Sci Off J Soc Toxicol. (2020) 175:24–34. doi: 10.1093/toxsci/kfaa024
88. Lancaster L, Cottin V, Ramaswamy M, Wuyts WA, Jenkins RG, Scholand MB, et al. Bexotegrast in patients with idiopathic pulmonary fibrosis: the INTEGRIS-IPF clinical trial. Am J Respir Crit Care Med. (2024) 210:424–34. doi: 10.1164/rccm.202403-0636OC
89. Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest. (2017) 127:3675–88. doi: 10.1172/JCI94624
90. Rizzieri DA, Cooley S, Odenike O, Moonan L, Chow KH, Jackson K, et al. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk Lymphoma. (2016) 57:1800–6. doi: 10.3109/10428194.2015.1122781
91. Cheng Y, Liu B, Qian H, Yang H, Wang Y, Wu Y, et al. BAY11-7082 inhibits the expression of tissue factor and plasminogen activator inhibitor-1 in type-II alveolar epithelial cells following TNF-α stimulation via the NF-κB pathway. Exp Ther Med. (2021) 21:177. doi: 10.3892/etm.2020.9608
92. Jaffar J, Glaspole I, Symons K, Westall G. Inhibition of NF-κB by ACT001 reduces fibroblast activity in idiopathic pulmonary fibrosis. Biomed Pharmacother Biomed Pharmacother. (2021) 138:111471. doi: 10.1016/j.biopha.2021.111471
93. Zhang Y, Jones KD, Achtar-Zadeh N, Green G, Kukreja J, Xu B, et al. Histopathological and molecular analysis of idiopathic pulmonary fibrosis lungs from patients treated with pirfenidone or nintedanib. Histopathology. (2019) 74:341–9. doi: 10.1111/his.13745
94. Hohmann MS, Habiel DM, Coelho AL, Verri WA, Hogaboam CM. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am J Respir Cell Mol Biol. (2019) 60:28–40. doi: 10.1165/rcmb.2017-0289OC
95. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
96. Tiendrébéogo AJF, Soumagne T, Pellegrin F, Dagouassat M, Tran Van Nhieu J, Caramelle P, et al. The telomerase activator TA-65 protects from cigarette smoke-induced small airway remodeling in mice through extra-telomeric effects. Sci Rep. (2023) 13:25. doi: 10.1038/s41598-022-25993-7
97. Butler DE, Marlein C, Walker HF, Frame FM, Mann VM, Simms MS, et al. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget. (2017) 8:56698–713. doi: 10.18632/oncotarget.18082
98. West A, Chaudhuri N, Barczyk A, Wilsher ML, Hopkins P, Glaspole I, et al. Inhaled pirfenidone solution (AP01) for IPF: a randomised, open-label, dose–response trial. Thorax. (2023) 78:882–9. doi: 10.1136/thorax-2022-219391
99. Prior TS, Hoyer N, Davidsen JR, Shaker SB, Hundahl MP, Lomholt S, et al. Fibroblast activation protein and disease severity, progression, and survival in idiopathic pulmonary fibrosis. Scand J Immunol. (2024) 100:e13392. doi: 10.1111/sji.13392
100. Ikeda K, Hirofumi C, Nishikiori H, Azuma A, Kondoh Y, Ogura T, et al. Serum surfactant protein D as a predictive biomarker for the efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis: a post-hoc analysis of the phase 3 trial in Japan. Respir Res. (2020) 21:316. doi: 10.1186/s12931-020-01582-y
101. Yoshikawa T, Otsuka M, Chiba H, Ikeda K, Mori Y, Umeda Y, et al. Surfactant protein A as a biomarker of outcomes of anti-fibrotic drug therapy in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. (2020) 20:27. doi: 10.1186/s12890-020-1060-y
102. Josephs DH, Sarker D. Pharmacodynamic biomarker development for PI3K pathway therapeutics. Transl Oncogenomics. (2016) 7 (Suppl. 1):33–49. doi: 10.4137/TOG.S30529
Keywords: idiopathic pulmonary fibrosis, PI3K/AKT, inflammation, fibrosis, senescence, PI3K inhibitor, PI3K inhibition
Citation: Bhatt J, Ghigo A and Hirsch E (2025) PI3K/Akt in IPF: untangling fibrosis and charting therapies. Front. Immunol. 16:1549277. doi: 10.3389/fimmu.2025.1549277
Received: 20 December 2024; Accepted: 13 March 2025;
Published: 31 March 2025.
Edited by:
Guillermo Barreto, UMR 7365 CNRS-Université de Lorraine, FranceReviewed by:
Hector A. Cabrera-Fuentes, Imam Abdulrahman bin Faisal University, Saudi ArabiaCopyright © 2025 Bhatt, Ghigo and Hirsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janki Bhatt, amFua2kuYmhhdHRAdW5pdG8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.