
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 February 2025
Sec. Cytokines and Soluble Mediators in Immunity
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1544137
This article is part of the Research TopicCytokines, and biomarkers involved in the immunomodulation of pediatric cancersView all 3 articles
 Enrico Maggi1
Enrico Maggi1 Nadine Landolina1
Nadine Landolina1 Enrico Munari2
Enrico Munari2 Francesca Romana Mariotti1
Francesca Romana Mariotti1 Nicola Tumino3
Nicola Tumino3 Paola Vacca3
Paola Vacca3 Bruno Azzarone1
Bruno Azzarone1 Lorenzo Moretta1*
Lorenzo Moretta1*Neuroblastoma (NB) is an immunologically “cold” tumor with poor or no inflamed substrates as most of solid pediatric tumors (SPT). Consistent data indicate that NB tumor microenvironment (TME) is dominated by myeloid cells, with little (but variable) T cell infiltration. The obstacles to lymphocyte infiltration and to their anti-tumor activity are due to different tumor immune evasion strategies, including loss of HLA Class I molecules, high expression of immune checkpoint molecular ligands leading to exhaustion of T effector (and NK) cells, induction of T regulatory, myeloid and stromal cells and secretion of immunosuppressive mediators. In odds with adult solid tumors, NB displays weak immunogenicity caused by intrinsic low mutational burden and scant expression of neoepitopes in the context of MHC-class I antigens which, in turn, are particularly poorly expressed on NB cells, thus inducing low anti-tumor T cell responses. In addition, NB is generated from embryonal cells and is the result of transcriptional abnormalities and not of the accumulation of genetic mutations over time, thus further explaining the low immunogenicity. The poor expression of immunogenic molecules on tumor cells is associated with the high production of immunosuppressive factors which further downregulate lymphocyte infiltration and activity, thus explaining the limited efficacy of new drugs in NB, as immune checkpoint inhibitors. This review is focused on examining the role of T effector and regulatory cells infiltrating TME of NB, taking into account their repertoire, phenotype, function, plasticity and, importantly, predictive value for defining novel targets for therapy.
Tumor-infiltrating lymphocytes (TILs) of solid tumors, and in particular T cells, influence tumorigenesis, cancer progression, metastasis, response to immune checkpoint inhibitors (ICIs) and may be predictive of clinical outcomes (1–6).
While the actual scenario of adult solid tumors highlights the coordination of circulating (cytolytic T lymphocytes -CTL, T helper -Th- and T regulatory - Treg-cells) and tissue-resident (T resident memory -Trm-, Mucosal associated invariant T – MAIT- γδ T and natural killer T-NKT-) cells (7–11), the T cell landscape of solid pediatric tumors (SPT) and in particular of Neuroblastoma (NB) is currently poorly explored and the results are often not definitive or even conflicting.
NB is the most common SPT, accounting for about 6% of all tumors in childhood, with a prevalence of 1/70,000 in children under 15yrs (12–14). Patients are usually categorized into low-, intermediate- and high-risk (LR, IR, HR) groups based on clinical stage, age at diagnosis, tumor histology, MYCN oncogene amplification and chromosomal ploidy. The great majority of LR patients with less one year displays spontaneous regression. More than 90% of LR tumors survives 5yrs or more, while only 40–50% of patients with HR NB achieves 5yrs survival: the survival outcomes drop to less than 10% for patients with relapsed metastatic disease (15–18).
Due to the poor availability of fresh tumor samples and the practical challenges of in-depth immune analyses, most studies evaluate a limited number of patients. Immunotherapy (IT) which has demonstrated successful in adult cancers, however, has low efficacy in SPT due to unsatisfactory prognostic value obtained even with new drugs (ie. anti-di-sialo-ganglioside (GD)-2 monoclonal antibody – mAb -, Dinutuximab) (12, 13). This is because NB displays low immunogenicity, due to low mutational burden and, in turn, scarce expression of neoepitopes in the context of low density (if any) of MHC-class I molecules (14, 19). Furthermore, tumor immune evasion strategies, including expression of immune checkpoint molecules, induction of immunosuppressive Treg, myeloid- and stromal cells as well as secretion of immunoregulatory factors effective, both in TME and systemically, can further impair lymphocyte infiltration and activity (20). In this review, we will analyze the features of T cells infiltrating the TME of NB, their function, plasticity, and novel targets for therapeutical interventions.
Although NB has been considered an immunological “cold” tumor, the role of T cell infiltrating the TME has been recently reevaluated in several reports (21–23).
The profile of immune cells infiltrating NB has been reported in few studies usually performed on a small number of specimens (24, 25). In these studies, the presence of TILs varies significantly in individual patients and tumor samples (depending on the cutoffs used): from 28% to virtually all tumors were considered positive as assessed by immunohistochemistry (IHC), even though often few lymphoid cells are detectable (26, 27). Both αβ- and γδ T cells as well as type1/2 NKT and NK cells have been identified in NB TILs, where B cells are rare or undetectable (28–33). In this regard, T cells were consistently less than 5% of total cells in the NB TME (24, 32), consisting of both CD4+ and CD8+ T cells, more frequently detectable in septa rather than in tumor cell nests (25, 34), CD8+ being predominant over CD4+T cells. Notably, recent reports utilizing computational methods identified different signatures of immune infiltration by deconvoluting bulk RNA sequencing data of tumor samples (22, 35–38). According to these studies, surprisingly, at least 50% of TME cell components were predicted to be immune cells, half of which were myeloid (DC and a small fraction of tumor- associated macrophages -TAMs-) and half lymphoid cells (37). The lymphoid compartment was mainly represented by CD4+T helper (Th) cells, while B cells and cytotoxic CD8+T cells (CTLs) were present in a small fraction, thus greatly differing from IHC-based studies (21). NKT, γδ T and NK cells were predicted to be present as well (29, 30, 36, 37, 39), while no data are available on Trm and MAIT cells. T cell infiltration has been evidenced not only in primary but also in relapsed tumors (34): a single-cell RNA sequencing (scRNA-seq) study evidenced that T cell infiltration increased in paired patient samples (diagnostic biopsy and surgical material) after neo-adjuvant chemotherapy, suggesting lymphocytes recruitment into tumor (33). Both CD4+ and CD8+ T cells showed an activated (CD25+ and HLA-DR+) profile, often with effector memory (CCR7-CD45RA-) phenotype (25). Results on freshly- or IL-2-cultured TILs from NB indicate that: i. CD4/CD8 T cell ratio ranged from 0.5 to 5; ii. rIL-2-cultured TILs show increased CD56+CD8+ T cells with cytolytic activity; iii. cultured TILs express IFN-γ, IL-4, IL-5, TNFα, IL-8, IL-10- and, to a lesser extent, IL-2 mRNAs while, in the corresponding tumor samples only TNF-α, IL-8 and IL-10- transcripts were expressed (40). Such cytokine pattern suggests a mixed CD4+ population of both Th1 and Th2 cells (31). The presence of Th17 cells in NB TILs has not been studied: only γδ T cells producing IL-17 have been described. The main characteristics of T effector/regulatory cell subsets, their master regulators, associated pathological conditions and anti-tumor activity are summarized in Figure 1.
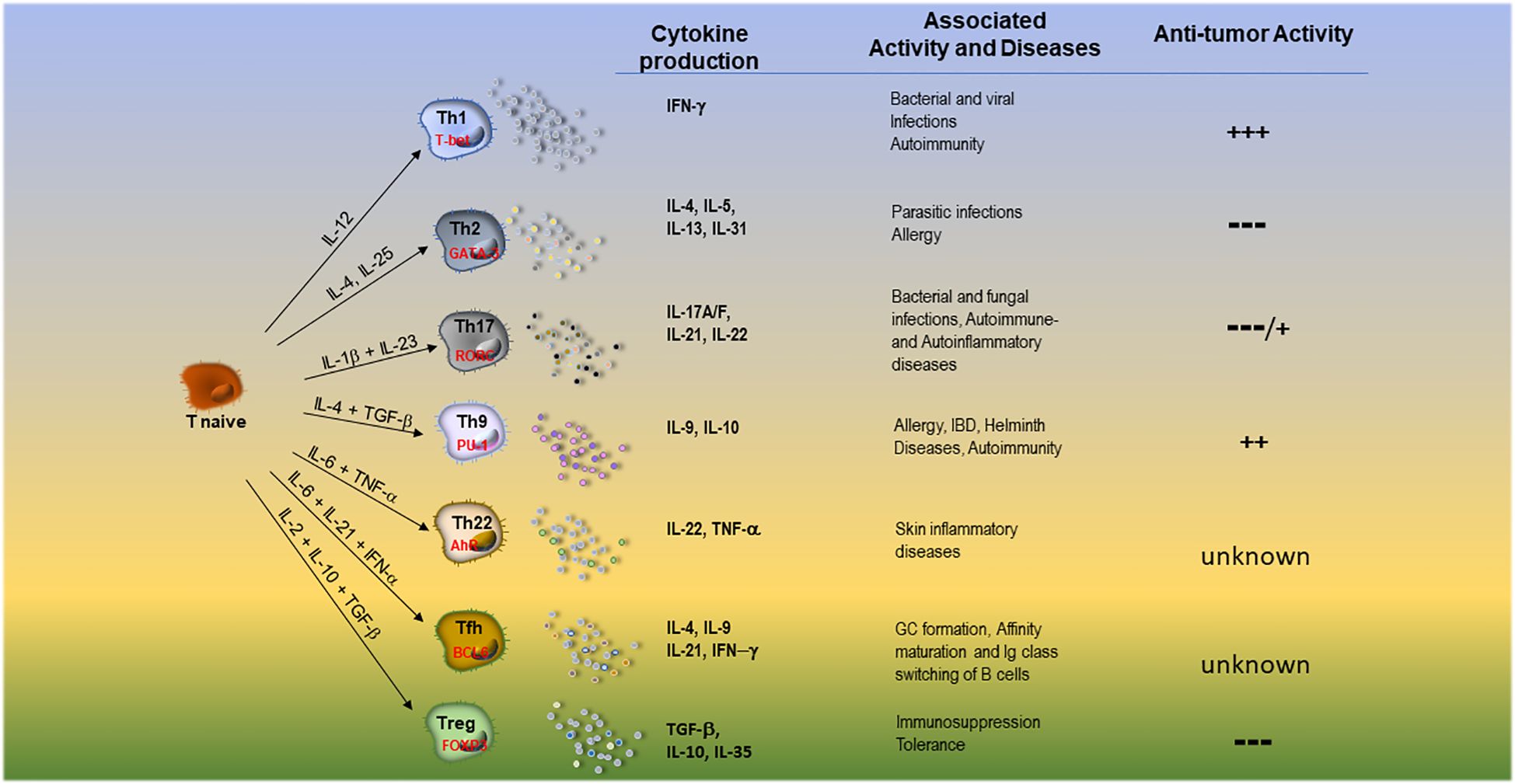
Figure 1. Main features of T effector cell subsets and T regulatory cells. DC-derived cytokines able to orient the development, the main transcription factors, the released groups of cytokines, the associated pathological conditions and anti-tumor activity of different T cell subsets are summarized.
As previously mentioned, NB is a non-immunogenic tumor due to low/absent MHC Class I and II antigen expression and reduced adhesion molecules (41, 42). MHC-class I expression on tumor cells correlated directly with the density of tumor-infiltrating T cells (25) and with the overall survival (OS): stages 3 and 4 display lower MHC-class I expression than stages 1, 2 and 4S (25, 43). The growth of NB cells in the presence of T cells, however, suggests that these cells do not recognize and/or kill tumor cells, suggesting that T cells are not tumor antigen -TA- specific or exhausted lymphocytes. Indeed, T cells infiltrating NB are polyclonal, characterized by a diverse Vα and Vβ usage (44) and poor recognition of autologous tumor cells (ATC). However, in some patients a higher degree of limited clonality was observed in TILs (vs peripheral blood lymphocytes -PBL-), suggesting that, even though rarely, TA-specific T cell expansion may occur in vivo. Ex vivo expanded blood-derived CD8+ CTL from patients stimulated with IFN-γ-treated ATC are cytolytic for these cells, suggesting some tumor specificity (45). Anyway, the great majority of ex vivo expanded TILs were non-reactive to ATC, even though they maintained the capacity to home towards NB cells (46).
Importantly, the MHC-Class I low expression can be reverted by culturing NB cells with IFN-γ and TNF-α produced by activated NK cells, allowing recognition by CTLs. For instance, oncoprotein PRAME serves as immunodominant TA since NK-modulated NB cells are recognized by PRAME-specific CTL clones (47). Notably, some activated T cells can kill NB cells in an MHC-unrestricted fashion (48, 49) in a caspase-dependent- or independent manner. Soluble factors released by activated CTLs skew the phenotype of NB cells, activate caspase-8, and increase their susceptibility to lysis by TRAIL and FAS agonist antibodies (49). The recruitment of activated CTLs into the tumor may constitute a strategy for NB treatment, even in the absence of a specific TA recognition.
T cell exhaustion occurs when cells become hyporesponsive as the consequence of persistent TCR stimulation with TA (50). The exhaustion genes resulted upregulated in NB, namely CD244, HAVCR2, CTLA4, TIGIT and PDCD1 were found to be expressed in tumors with high cytotoxic signatures, indicating that chronic stimulation of T cells with their cognate antigens may lead to an exhausted state (44). Preclinical and clinical data revealed that anti-GD2 CAR-T cells may progressively lose their function in vivo (50). Such exhausted T cells currently express immune checkpoints (IC) as PD-1, CTLA-4, and LAG-3, thus contributing to impaired anti-tumor immune response (51–53). In this context, ICI in combination with CAR-T cells, which are beneficial in adult tumors (40, 54), display low efficacy in SPT mainly in HR NB essentially for: i. the poor or no expression of immune checkpoints on NB TME cells, ii. the effective and persistent immunosuppressive signals from the NB microenvironment on immune cells, iii.T (also CAR-T) cell exhaustion not allowing any modulation or activation with biologicals.
Other IC molecules expressed in NB are B7-H3 and CD200/CD200R. High B7-H3 expression in NB correlated with poor event-free survival (EFS), while its blockade resulted in enhanced NK cell-mediated killing of NB cells in vitro (55, 56). CD200 is overexpressed on NB cells and CD200bright tumors have few numbers of CD4+ and CD8+ T cells producing low IFN−γ and TNF−α (57). CD200R is expressed in HLA-DR+CD14+ myeloid cells and CD11c+ dendritic cells (DCs), but poorly expressed in CD4+ and CD8+ T cells. Since the CD200-CD200R pathway downregulates anti-tumor immunity, also its blockade may be considered an interesting therapeutic option (57).
Among lymphocytes expanded from tumor biopsies, NKT cells could play a relevant role in anti-tumor immunity by enhancing both innate and adaptive immunity. In particular CD1d-restricted iNKT (type 1 NKT) cells (58–60), characterized by Vα24-Jα18 usage, may be more abundant in LR- than HR-NB and have been reported to be predictive of good OS (61). Alpha-Galactose Ceramide (α-GalCer) stimulation induces the proliferative response of type 1 NKT cells of patients resulting in an enhancement of cytotoxicity against NB cells (40). Activated type 1 NKT (and NK) cells inhibit TAMs and MDSCs, kill tumor cells downregulating MHC antigens and secrete chemokines to recruit additional immune effectors (62).
γδT and NKT cells have also been identified in the TME of NB patients. Notably, the γδT cells (mainly those producing IL-17) seem to favor rather than impair tumor cell proliferation and migration (46).
An inverse correlation between NKT cell- and MYCN-mRNA is reported in primary NB tumors (30). Type 1 NKT cells migrate toward NB cells through CCL2 which is preferentially expressed by MYCN-non amplified (NA) tumors since active MYCN downregulates CCL2 (30). In agreement, type 1 NKT (and NK) cells are decreased in MYCN-amplified (A) tumors showing an inverse correlation between MYCN and NKG2D/DNAM-1 ligand expression (36, 63).
The mechanism of impaired activity of iNKT cells in NB has been recently suggested: α‐Galcer‐pulsed DCs co‐cultured with supernatants of NB cell lines are unable to stimulate iNKT cells to release IFN-γ (which is reversed by IL‐12). CD40 expression and IL‐12 release by NB supernatant-treated DCs are increased with exogenous IFN‐γ, indicating that a new type of tolerogenic DCs are active in NB TME and impair the anti-tumor effect of type 1 NKT cells (64).
Treg cells are able to inhibit the activity of T effector cells and DCs and are usually correlated with tumor progression and poor survival even though, sometimes, they can regulate TME inflammation favoring anti-tumor response. The limitation with Treg cells in NB concerns their fine detection: in humans (not in mice) Foxp3 and CD25 molecules are co-expressed in both Treg- and activated T effector cells even though this latter subset expresses Foxp3 and CD25 at low and transient level (65, 66). Since it has been demonstrated that tumor-infiltrating CD4+ T effector cells can be activated by TME signals (24), the presence of the two cell types is not an exception. This problem can explain contradictory results on the proportion and activity of TME Treg cells and their correlation with prognosis in NB. For instance, some authors report no difference of Treg cells in PB from a cohort of LR and HR patients (67), while others have shown that circulating Treg cells were higher in NB patients than in healthy children (68). Another study showed a high density of CD25+T cells in the nests and Foxp3+T cells in the septa both being associated with better survival (25). A similar problem concerns reports using scRNA-seq analysis which exclusively evaluated Foxp3 and CD25 RNA expression: in primary tumor Foxp+CD25+T cells were less expressed in HR patients and did not correlate with OS (23), whereas in BM metastases they clearly presented exhausted profile suggesting a T effector phenotype (38). Lastly and importantly, Stigliani and coworkers showed that high Foxp3 expression of tumor biopsies predicting a better EFS and OS, was strictly correlated with perforin (PRF1) mRNA, definitively suggesting that Foxp3 is more an indicator of activation of T effector -rather than Treg cells (61). Indeed, few studies analyze accurately Treg cell phenotype (co-expression of Foxp3, CD25high, CD127low, CD39, CD73, PD1, LAG-3, Tim3 etc.) in NB TME or evaluate their suppressive function (69, 70). One of these rare reports indicates that CD4+Foxp3+CD25highCD127- Treg and CD4+CD45RO+CD49b+LAG3+ T regulatory-1 (Tr1) cells in bone marrow (BM) and blood of NB patients at a lower frequency than in healthy subjects (28). However, MYCN amplification correlates with a higher number of BM Treg- and circulating Tr1 cells (35), even though these cells do not associate with known NB prognostic factors (28). In conclusion, the information on the presence and activity of true Treg cells in the primary and metastatic NB and the evidence of their suppressive function inside TME are currently scarce. As a consequence, studies on Treg cell repertoire in NB are completely missing.
By contrast, since in mice Foxp3 is associated exclusively to Treg cells, Treg cell depletion (Foxp3-DTR mice) in para-orthopic NB murine model, resulted in 85% reduction of tumor volume and weight and increased amounts of cytokines (IFN-γ, TNF−α, IL-4, IL-6, and IL-10) in splenocytes as compared to control mice. By contrast, tumor growth was not affected in B regulatory cell-deficient- (μMT and CD19cre) mice (71), confirming that the ablation of true Treg cells to impairing tumor growth must be considered an option for NB therapy (72).
Table 1 summarizes the distinctive characteristics of NB subgroups according to different classifications.
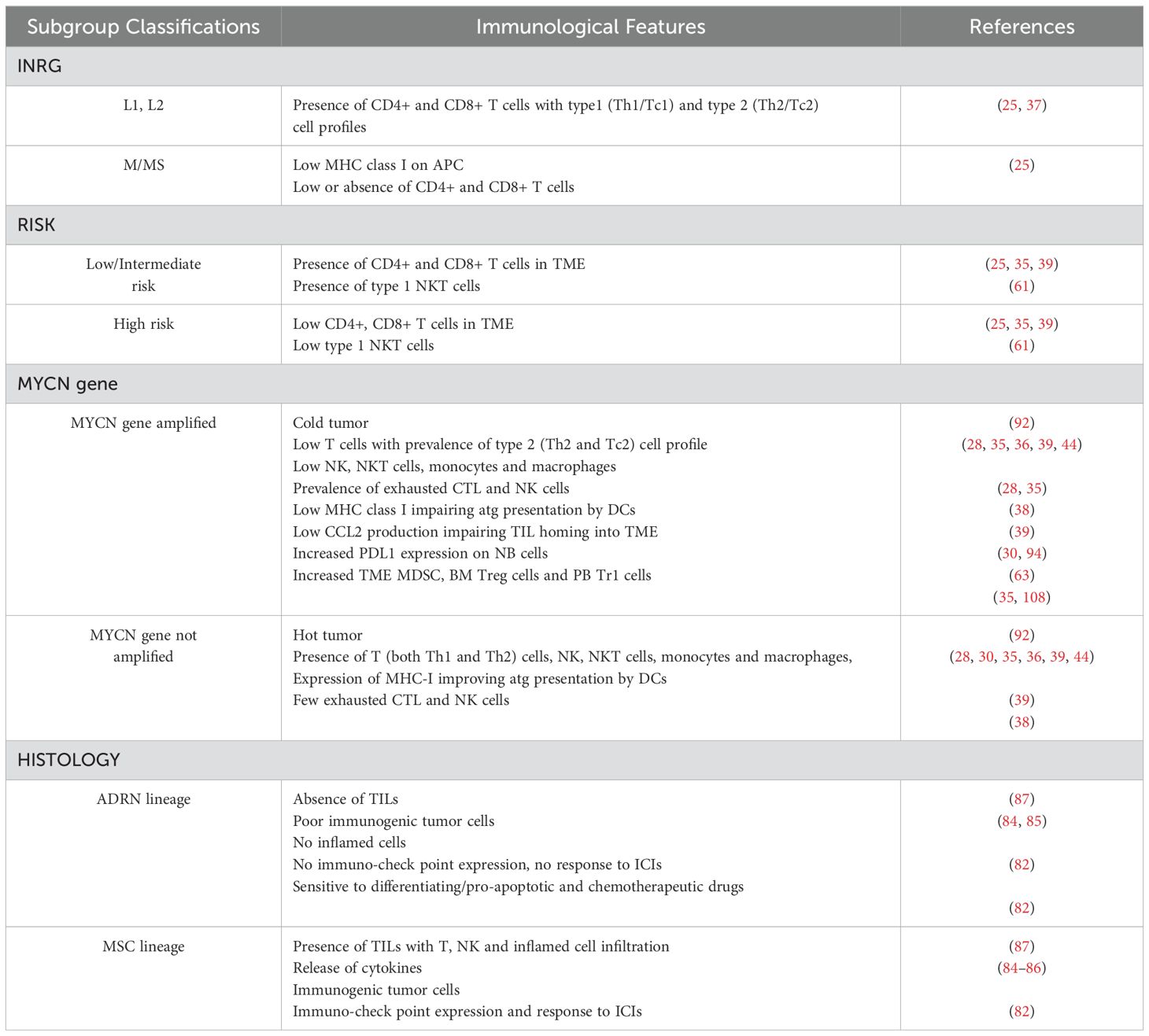
Table 1. Immunological features of subgroups of different NB classifications.
The presence of different immunoregulatory subsets within the TME (mesenchymal stem cells -MSCs-, cancer-associated fibroblasts – CAFs-, TAMs-, tumor-associated neutrophils -TANs- and myeloid-derived suppressor cells -MDSCs-) has been correlated with unfavorable prognosis (73), conditioning the T cell infiltrate and contributing to orient the function of other TME cells. A full understanding of tumor–host interactions and T cell plasticity in “cold” tumors (as NB) will be crucial to enhance the ability of IT to attack efficiently tumor cells. Recent studies have confirmed the reduced CD4+ and CD8+ T cell infiltration in HR NB (especially in stage 4 tumors), while the higher (mainly CD4+) T cells have been associated with better OS (25, 35, 39). Some studies reported a survival benefit related to T cell infiltration also in stage 4 NB (39, 61). In this respect, Th1 cells modulate TAMs towards a pro-inflammatory antitumor phenotype (M1) while Th2 cells orient to an immunoregulatory phenotype (M2) favoring tumor progression. Whereas T cell density has a clear predictive value, the prognostic value of CD4+ or CD8+ T cells yielded conflicting results. Some reports indicated a beneficial prognostic value for CD8+ T cells (37) while CD4+ T cell prevalence occurred in tumors with unfavorable prognosis. Others highlighted that the prevalence of CD4+ over CD8+ T cells is associated with a better prognosis (25, 35). Both conditions can likely lead to favorable outcomes: the former emphasizes the role of cytotoxic cells while CD4+T cell infiltration may be crucial for the onset of an effective anti-tumor adaptive immunity. In addition, it is well known that some T effector cell subsets (such as Th2 and Th17) or Treg cells are highly plastic generating numerous transitory populations that are capable of heterogeneous cytokine release and variable cross-talk with other TME cells. As described, molecules such as the cytokine IL-12, can modulate in vitro Th2 or Th17 cells toward a protective Th1-oriented profile (74, 75). Treg cells also exert functional plasticity that allows them to partially acquire Th transcriptional programs referred to as Th1-, Th2-, Th17- and T follicular- Tfh-like Treg cells (76). Drugs interacting with TCR signaling, co-stimulation, and metabolic and epigenetic pathways can be used as a valid option to reprogram Treg cells toward potentially antitumor T cells. Unleashing Th programs in Treg cells, however, is not without risk, since it may upregulate auto-inflammatory and/or autoimmune mechanisms (76, 77).
The cellular and soluble signals deriving from tumor/stromal/innate cells play the major role in modifying T cell function and generating the diffuse immunosuppression of TME in NB which can explain the poor efficacy of cancer vaccines and of ICI- or CAR-T-mediated IT. Heterogenicity and immunosuppressive properties of NB cells (mainly regarding different phenotypes and MYCN gene amplification) and other TME components (as CAFs-, TAMs, TANs and MDSCs) and their released factors influencing the function of T effector cells will be analyzed here below.
Recently, van Groningen identified two different cell types, named respectively adrenergic (ADRN) and MSC lineages within NB tumors, which correlated with the absence or presence of TILs (78–80). Specific transcriptional circuits determine the phenotype of the two subsets: while ADRN cells express markers of sympathoadrenal differentiation, MSCs are undifferentiated and similar to neural crest progenitors (78). These cellular types can spontaneously switch each other under epigenetic signals. This tumor plasticity has a major impact on cancer pathogenesis and is a potential suitable target for IT (81, 82). ScRNA-seq analysis of NB samples has also shown a subset of “transitional cells” expressing genes of both ADRN and MSC lineages (83). Each subset is characterized by distinct immune gene expression, the MSC type having the capacity of an anti-tumor immune response and ADRN lineage being less immunogenic (84, 85). MSC lineage activation promotes T cell infiltration by secreting inflammatory cytokines. These tumors are efficiently killed by CTLs and NK cells and respond to ICIs, further highlighting the role of tumor cell lineage in modulating immune response (86).
The two ADRN and MSC lineages influence the activation of innate and adaptive immune pathways (87). MSC lineage, upregulating MHC class I and promoting CTL infiltration, also favors the expression of IC ligands and exhaustion markers on T and NK cells resembling blood MSCs from healthy donors which display a clearcut suppressive activity in vitro (88). The presence of both immune activation and suppression markers suggests that patients with MSC-type tumors may benefit from therapy with ICIs, whereas ADRN-type tumors are more sensitive to differentiating and chemotherapeutic agents. The high expression of GD2 and anaplastic lymphoma kinase (ALK) on the two lineages can be exploited through the use of anti-GD2 antibodies plus ALK inhibitors (ALKi) (82). Notably, relapsed NB with MSC features exhibit increased immunogenicity, including elevated expression of IC ligands, suggesting that ICI may be more effective at relapse (84).
TME signals tend to force NB cells towards the ADRN-type profile (89), whereas an MSC phenotype can be acquired prevalently during therapy. Several inhibitory molecules secreted in TME compromise the anti-tumor T cell response, stimulating cancer progression, MSC-ADRN type transition and reducing the efficacy of CAR-T/NK cell therapy. Recently, another subset of tumoral neuroblasts has been identified. These so-called mature NB-MSCs display features of BM-MSCs with suppressive activity towards NK cells (90). Initially identified in cell lines (90), this subset has been recently studied on primary NB cultures (91). Our recent data indicate that the primary cultures: i. interfere with NK (and CD8+T) cell activation and function through cell-to-cell contact, ii. express high levels of GD2 independently of their phenotype, differently from NB cell lines where the MSC-ADRN-type transition associated with GD2 loss and resistance to anti-GD2 mAb treatment, iii. are resistant to NK cell killing mediated by anti-GD2 mAbs through an ADCC mechanism, iv. include undifferentiated MSCs which, in contact with NK cells, cause an immune editing phenomenon by converting these cells into mature MSCs, increasing GD2 expression (91). These data highlight the relevance of the “transitional cells” that acquire the immunoregulatory properties typical of the NB-MSCs deeply interfering with the cytotoxic activity of NK and, likely, CTLs and NKT cells.
The MYCN oncogene amplification is currently considered one of the major prognostic values in NB: the TIL composition of NB differs between MYCN-A and MYCN-NA tumors, the former being “cold” and the latter “hot” tumor (92). Computational studies revealed the inverse correlation between MYCN amplification and leukocyte infiltration. CD8+ and CD4+ T, NK, NKT, B cells, macrophages, and monocytes, are lower in MYCN-A- than in MYCN-NA tumors (35, 36, 39, 44). ScRNA-seq of lymphocytes infiltrating untreated NB, validated by tissue microarray and TCR sequencing on NB TME, indicated that MYCN-NA tumors had significantly higher cytotoxic TIL signatures than MYCN-A tumors (28). MYCN silencing in NB cell lines enhances IFN-γ activity, promotes Th1-recruiting chemokines (CXCL9 and CXCL10), increases T cell infiltration (39) and expression of ligands (MICA, ULBPs and PVR) of NK cell activating receptors leading to increased cytotoxic activity (63). MYCN amplification impacts anti-tumor response, downregulating MHC class I antigens in NB cells and impairing TA presentation by APC leading to the decrease of anti-tumor CD8+ T cells (39). The absence of MHC class I in NB cells is responsible for the ability of MYCN-A tumors to escape the type 1 immune response (93). MYCN amplification inhibits CCL2 release which recruits TILs (30, 94). Lastly, MYCN activation correlates with PD-L1 expression by NB cells, favoring an immunosuppressive environment in a “hot” MYCN-NA tumor (63).
Activated Th2 cells have been detected in the TME of MYCN-A NB and positively correlated with the expression of the three hub genes (ZNF695, CHEK1 and C15ORF42) proposed as potential prognostic markers and IT targets (39). Some authors established the immune gene expression profile (as ADAM22, GAL, KLHL13, and TWISTT1) in HR NB and proposed an ultra-HR NB group with the worst prognosis (95). Finally, in univariate Cox regression analysis performed in the GSE49710 dataset, two risk scores negatively associated with the presence of DCs or CD8+ T cells and, positively, with Th2 cells have been established (96).
Two different clusters of CAFs have been identified in NB TME: the CAF-S1 and CAF-S4 each expressing different signatures of upregulated genes (97). Human CAF-S1 cluster strongly expresses CXCL12 and CCL2 chemokine genes (98); through CXCL12-CXCR4 interaction and CCL2 release CAFs may recruit monocytes into TME, thus favouring tumor progression, angiogenesis, metastasis, and low survival. In addition, CAFs induce monocyte differentiation into MDSCs through the IL-6/STAT3 axis (98). Likewise, signaling of the CCL2/CCR2 axis displays remarkable effects on myeloid cell survival and function and also directly enhances Treg cell suppressive activity (99). CAFs also attract Treg-cells and decrease CD8+ T cell infiltration into TME by secreting TGF-β and IL-6, likely interfering with anti-tumor immunity (100). DeClerck has isolated from primary NB tumors a cell subset of αFAP- and FSP-1-expressing CAFs sharing phenotypic and functional characteristics with BM-derived MSCs. These CAF-MSCs are classified into two different subsets on the basis of the GD2 expression. In vivo, the presence of the GD-2-neg subset associates with that of M2 TAMs and a type 2 immune response (101), while the GD-2-pos subset (expressed by 50% of MSCs) resulted refractory to anti-GD2 treatment (102). NB CAF-MSCs exhibit powerful immunoregulatory functions on T (103) and NK cells through the release of immunosuppressive molecules such as PGE2 and IDO-1 (104) that can be also conveyed by exosomes (105).
A complex immunosuppressive network of potential TANs, MDSCs and TAMs has been frequently detected in human NB TME.
TANs are often found at the tumor site and usually promote the growth and spread of tumor cells (106) since suppress anti-tumor CTL and NK responses. Through scRNA-seq analysis two TAN subsets, named Neutrophil 1 and Neutrophil 2, expressing different gene signatures have been detected in the metastatic BM microenvironment (38). Since Neutrophil 2 express CXCR1 and CXCR2 they are attracted by chemokines produced by tumor cells and infiltrate the TME (107). In addition, this subset producing matrix metalloproteinases (MMP) 9 and 25 favors angiogenesis and neuroblast dissemination. Expression analysis of TAN development indicates that they are modified by tumor-released signals into immunosuppressive cells within the metastatic BM microenvironment (38).
In addition, MDSCs result highly positive for both the neutrophil (PMN)- and monocyte (Mo)-derived MDSC gene signature. Interestingly, the proportions of MDSCs and non-classical monocytes are higher in MYCN-A- compared with MYCN-NA tumors. Notably, a higher proportion of MDSCs has been observed in relapsed- compared to at-diagnosis tumors (108). In a murine model of NB, MDSCs in vitro suppressed CD8+ and CD4+ T cell proliferation whereas the same fractions from wild-type spleens had no effect. The two tumoral MDSC subsets exhibit immunosuppressive activity on T cells inducing also an exhausted state in vivo (98). Of note, PMN-MDSCs have been found in the blood also in different human tumors (including NB) and their number inversely correlates with disease stages and may represent an useful prognostic marker (108, 109). Importantly, it has been observed that PMN-MDSCs impair the anti-tumor efficacy of GD2-CAR T and NK cells in patients with NB (108, 109).
Different clusters of TAMs expressing CD68 and apolipoprotein E (APOE) have been observed in NB TME. They express a M2 signature favouring pro-tumoral activity; their number correlate with clinical stage, MYCN amplification, BM metastasis, histological type and risk classification. TAMs express surface toll-like receptors (TLRs) and are activated by damage- and pathogen- associated molecular patterns (DAMPs and PAMPs) derived from tumor and/or other apoptotic cells of the TME. In turn, activated TAMs secrete a large number of molecules that influence tumor, stromal and immune cells. TAMs settled near the CAF area, suggesting their close interaction within the TME (97, 110), which induces TAM polarization towards the M2/pro-tumour profile secreting CXCL1 and CXCL2 and favouring further CAFs recruitment and activation. This interaction has been associated with NB progression, chemo-resistance, MYCN gene amplification, and immunosuppression including Treg cells (110).
The interactions of monocytes with NB cells expressing MSC lineage induce the release of pro-tumorigenic- (TGF-β1, MCP-1, IL-6, IL-8, and IL-4) but not of anti-tumorigenic- (TNF-α, IL-12) cytokines/chemokines (111). Among the pro-tumor immunosuppressive molecules, TGF-β, migration inhibitory factor -MIF-, soluble GD2 and secreted galectin-1 have been found (35, 37, 112–114) to impair CTL and/or NK cell function (115–117). Soluble galectin-1 and MIF induce T cell apoptosis and inhibit T cell proliferation and DC maturation (37, 118, 119). High tumoral MIF is associated with low infiltration of CTLs, NKT cells, B cells and DC, and a poor prognosis in stage 4 tumors even irrespective to MYCN amplification (120). Tumor TGF−β is associated with shorter EFS (121); it may be produced under MSC-NB cell-monocyte interaction, inducing an amplifying loop, where TGF-β stimulates the production of IL-6 and sIL-6Rα, protecting TAMs from spontaneous death and, in turn, promoting further TGF-β production (111). Soluble GD2 can be shed by tumor cells and may be detected even at >50-fold increased concentrations in plasma particularly in patients with stage 3/4 (122, 123). In addition, it inhibits T cell proliferation (122), favoring tumor immune evasion.
Moreover, NB cells produce arginase-2, which reduces the levels of arginine (an amino acid essential for lymphocyte cell cycle) suppressing T cell proliferation in vitro and in vivo. High arginase-2 and low arginine are detected in patients with NB, both locally and systemically, and correlate with poor survival (124).
Other regulatory molecules active in NB are soluble MICA, B7-H6, HLA-G, IL-10 and high Mobility Group Box 1 (HMGB1). Soluble MICA and B7-H6 (ligands for TCR of CTLs and activating receptors of NK cells) are released from NB cells. Elevated soluble MICA downregulates its receptor (NKG2D) on NK cells, thus inhibiting the killing of tumor cells (125). Likewise, the soluble B7-H6 levels inversely correlate with the expression of activating receptor NKp30, with the occurrence of BM metastases and chemoresistance (126). Soluble HLA-G is produced by monocytes in response to NB cell exposure, inhibits CTLs and NK cells, is detectable in serum and has a prognostic value for relapse (127). HMGB1 (an inflammatory molecule modulating chromatin) is overexpressed in 11% of NB patients, associated with an elevated risk of progression, relapse and disease-related death (128). IDO-1, which converts tryptophan into kynurenines, increases NB cell resistance to the immune cells and inhibits the efficacy of GD2-CAR T/NK cells by decreasing their IFN-γ production (129). All these molecules may favor the modulation/activation/exhaustion of T cells in TME and can be responsible for the frequent inefficacy of adoptive therapies.
Table 2 describes the new omics approaches used to characterize the NB-TME cells (130–132). Figure 2 summarizes the signals and amplifying circuits potentially able, at least in part, to modulate TME T cells in NB.
Table 2. Multi-Omics profiling for NB-TME characterization.

Figure 2. Immunosuppressive circuits between tumor cells and TAMs and their released signals in the TME of neuroblastoma reduce proliferation of T and NK cells improving their apoptosis and provide the recruitment and amplification of regulatory cells that favor tumor progression.
Novel NB therapies involving T cells include CAR-T cells, engineered (bi- and tri-specific Immune Cell Engagers -ICEs-) and nanobodies, nanoparticles carrying drugs or mAbs, aptamers and cancer vaccines and even micro-RNA and long-term-non-coding RNA with anti-tumor activity (133, 134). Some of them are undergoing clinical trials. (https://clinicaltrials.gov NCT06528496, NCT05990751). At present engineered CAR-T cells may result the best option to threat NB. Although many preclinical studies have demonstrated the promise of this strategy (135–138), many obstacles remain. The major problems include the need to use autologous T cells, the insufficient CAR-T-cell infiltration into TME, their expansion and persistence and, importantly a reduced function in a highly suppressive TME. In this context, the PMN-MDSCs, as mentioned above, have been detected not only in the TME but also in the blood, and their number inversely correlated with the response to anti-GD2 CAR-T cell treatment (108). To overcome these barriers CAR-T cells must be balanced against potential side effects causing toxicity. New-generation CAR cell types should be correctly chosen among T, NK, γδ T, NKT cells and macrophages: however, the most promising carriers are NK cells, which do not require autologous cells, but can be donor-derived. Their storage in large numbers may be immediately available and allows to design precise quick protocols and greatly reduce the costs. Evolution of CAR-T cells should include the engineered TCR recognizing TA specific of a given tumor (or some types of tumors) and secreting T cell-engager antibodies (CAR-T.BiTE; STAb-T cells) (139–141). Moreover, to overcome the poor infiltration capability (142), the resistance to inhibitory signals (135) or T cell exhaustion blockage, dual targeting or bispecific CARs are currently developed.
Since it is presently virtually impossible to overcome all these inhibitory mechanisms, it is important to define the prevalent inhibitors of a given tumor. The results obtained with anti-PD-1/PDL-1 therapy in some tumors suggest that this approach may be successful.
The Figure 3 summarizes the technologies to enhance CAR cell specificity and sensitivity for NB treatment.
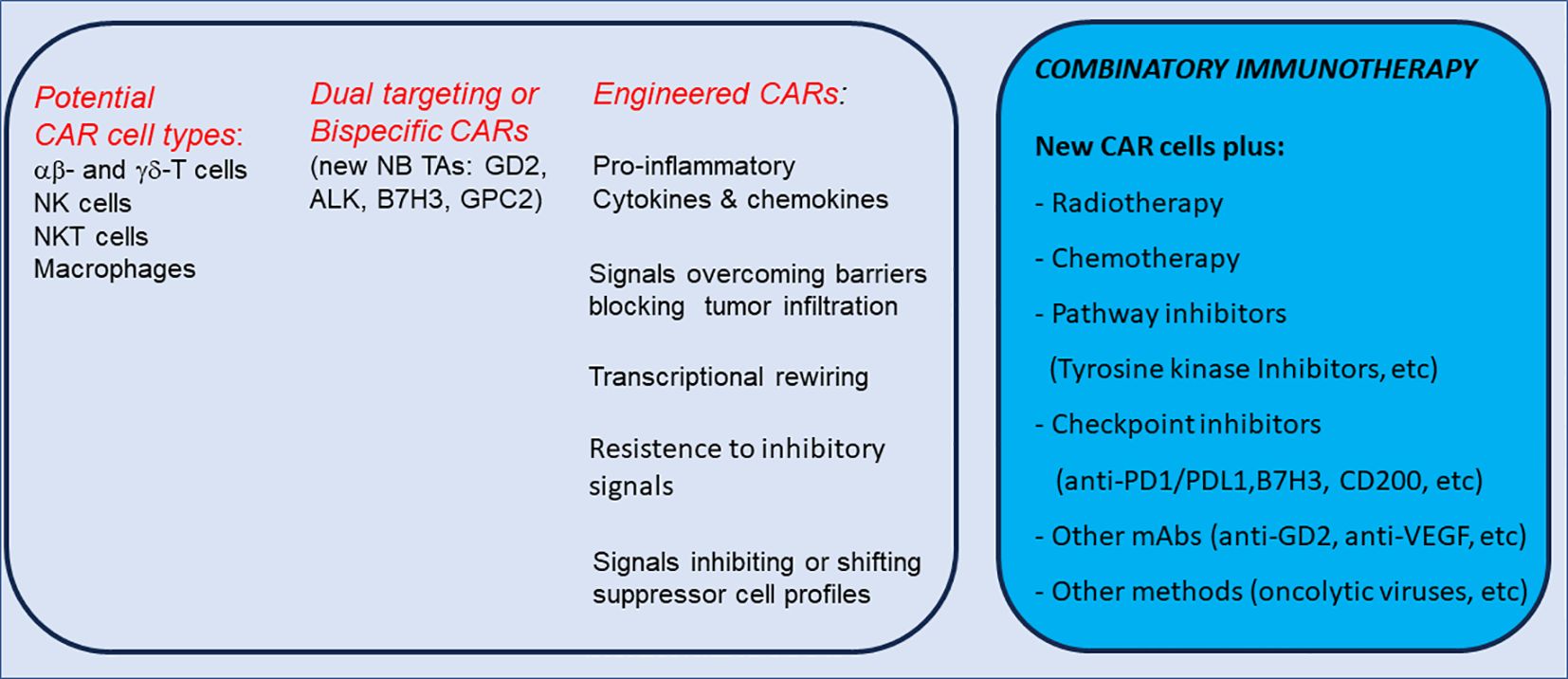
Figure 3. Cell types and novel technologies to enhance CAR cell specificity/sensitivity and potential combinatory immunotherapy for NB treatment.
Most ICEs are trans-binding bispecific antibodies (bs-Abs) consisting of two linked single-chains binding both TAs and the other cell surface trigger molecules as the activating receptors of effector cells (CD3, CD28 or 4-1BB for T cells, CD16a, NKG2D for NK cells, and CD64 for cytotoxic/phagocytic cells). Multi-specific antibodies as bs-Abs, tri-specific antibodies – ts-Abs- and even tetra-specific antibodies are currently in use in clinical trials of adults (143, 144).
As mentioned, the low number of somatic mutations in NB limits the immune responses that is induced by ICIs (145, 146). Thus, strategies of IT for NB differ significantly from those successfully used for some adult solid tumors. To date, IT is limited to target GD2, a glycolipid expressed on the surface of NB cells, which results poorly present on normal tissues. Therefore, GD2 represents the best IT target in NB. Subsequently, it has been demonstrated that some developmental proteins have been found to be highly expressed in NB cells but not in healthy tissues, thus serving as unique and specific targets for IT. These targets have been identified and include GPC2, B7-H3, and ALK.As discussed above, the NB TME prevents the infiltration of endogenous immune cells since it contains suppressor cells as type 2 TAMs, MDSCs and Treg cells interfering with immunotherapeutic strategies. Therapies as radio- or chemo-therapy, or CD47 “don’t eat me” signal blockade on tumor cells focused to reverse suppressive microenvironment, may improve the antitumor efficacy of endogenous immune cells or of genetically modified CAR-T cells. Indeed, radiotherapy can influence the suppressive NB TME turning cold- into hot tumors essentially by spreading: i. DAMPs from irradiated cancer cells promoting DC processing and presentation of TAs to T cells ii. cytoplasmic dsDNA which are accumulated after radiation, activating the cGAS-STING and TLR9 pathways and leading to type I IFN release and DC maturation (147). On the other hand, several chemotherapeutic drugs used in NB treatment inhibit Treg cell proliferation and induce MDSC lysis in TME in addition to triggering an immunogenic cell death pathway, which stimulates robust innate and adaptive immune response by exposing DC to TAs and DAMPs (148–150).Thus, a chemo-IT approach combining cytotoxic agents with anti-GD2 mAbs has resulted successful for several NB patients and is currently considered the standard of care for children with relapsed or refractory disease (151–154).
Additional combinations of IT with traditional cytotoxic agents and radiotherapy, small-molecule inhibitors of oncogenic pathways, and IT should be carefully considered in the future. Since next-generation small molecules, as ALK-, aurora-A- and CDK9/2 inhibitors (155–157), are being integrated into treatment for appropriate patients, their ability to really synergize with combination IT regimens will require careful analyses in precisely defined pre-clinical- and clinical studies (135, 142, 158).
Figure 4 summarizes the main novel therapeutical approaches for NB.
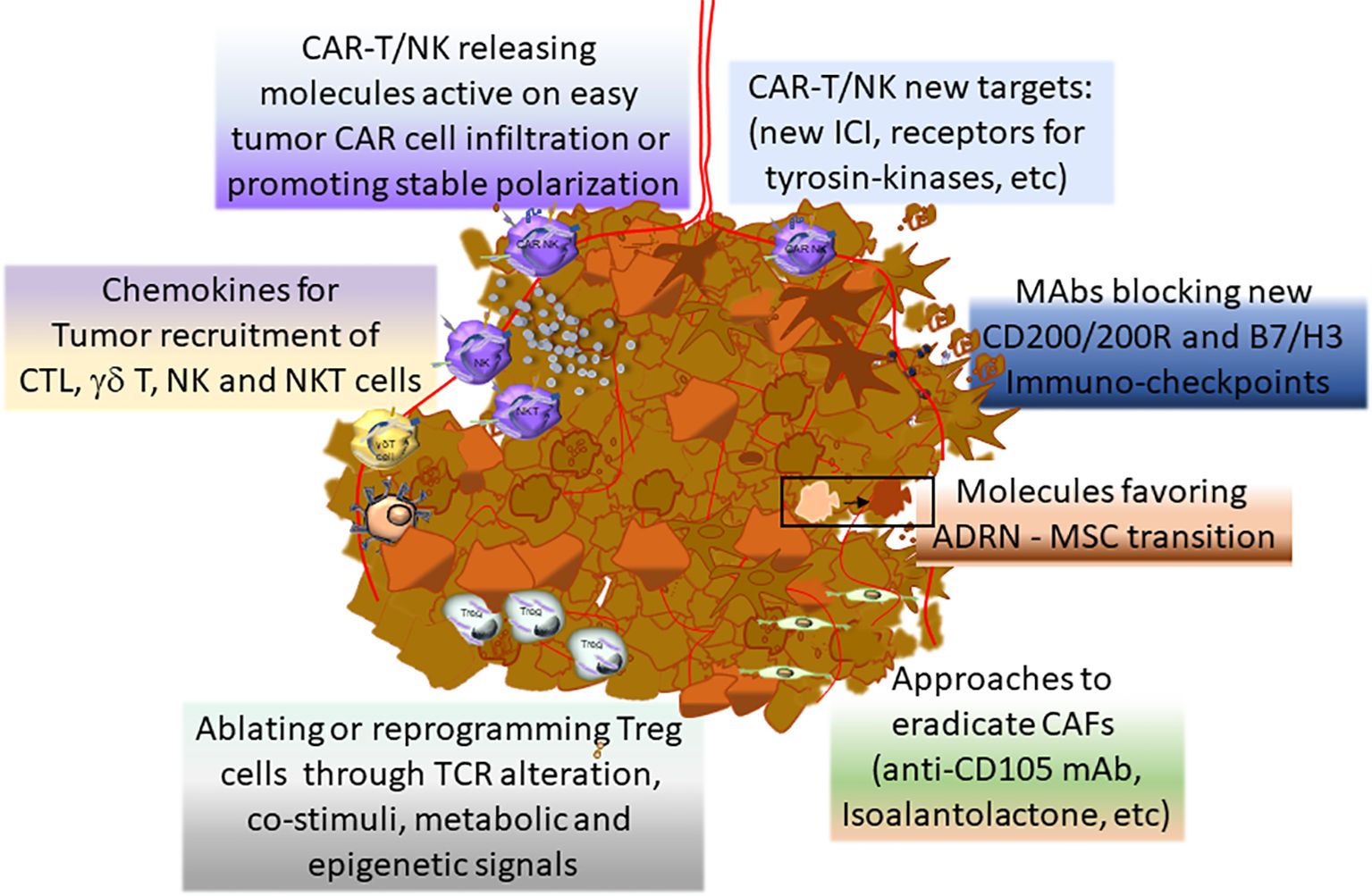
Figure 4. Novel therapeutical approaches to improve the efficacy of NB treatment.
The limited efficacy of current IT, CAR-T/NK therapy or cancer vaccines is the hallmark of the majority of solid pediatric tumors, including NB. While remaining potentially the most effective therapy, several problems on adoptive therapy of NB remain open, mostly those causing low efficacy of CAR-T cells. The ability of TCRs to target intracellular proteins (as oncoproteins) offers a valid opportunity to treat NB. This requires the need for CAR-T cells: i. to traffic easily to tumor sites, ii. to cross through tumor endothelial and stromal barriers iii. to recognize and kill efficiently cancer cells, iv. to survive and maintain their activity in a highly immunosuppressive environment.
The difficulty of lymphocyte infiltration and of their sufficient expansion in vitro for a possible adoptive treatment (159, 160) are due to some evasion strategies of NB, as expression of IC, induction of different immunosuppressive myeloid and stromal cells and secretion of immunoregulatory molecules leading to poor TIL infiltration and deficient/inadequate anti-tumor reactivity (20). The last Pediatric Strategy Forum of the Consortium of European Society for Pediatric Oncology and Innovative Therapies for Children with Cancer in Europe, established that the lack of knowledge on the immunological landscape of SPT is responsible for the failure of IT assayed so far (161). For instance, the eradication of CAFs with anti-CD105 mAbs-mediated ADCC or the use of drugs such as iso-alantolactone able to induce aging and functional inactivation of CAFs (162), could lead to the loss of M2 macrophages and MDSCs and, therefore to efficient CTL recovery. Finally, the development of CAR-T cells engineered to secrete proinflammatory cytokines (such as IL-12, IL-15 and IL-18), to produce molecules engaging tumor cells by not engineered T cells (162–166), as well as the development of strategies to overcome cell exhaustion could also be required (140, 167, 168).
EMa: Writing – original draft, Writing – review & editing. NL: Writing – review & editing. EMu: Writing – review & editing. FM: Writing – review & editing. NT: Writing – review & editing. PV: Writing – review & editing. BA: Writing – review & editing. LM: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Associazione Italiana per la Ricerca sul Cancro (project no. 5x1000 2018 Id 21147 to LM and Investigator Grant ID 27065 to PV) and by the Italian Ministry of Health with “Current Research funds”.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
2. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
3. Stolk D, van der Vliet HJ, de Gruijl TD, van Kooyk Y, Exley MA. Positive & Negative roles of innate effector cells in controlling cancer progression. Front Immunol. (2018) 9:1990. doi: 10.3389/fimmu.2018.01990
4. Taube JM, Galon J, Sholl LM, Rodig SJ, Cottrell TR, Giraldo NA, et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod Pathol. (2018) 31:214–34. doi: 10.1038/modpathol.2017.156
5. Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. (2015) 368:7–13. doi: 10.1016/j.canlet.2015.07.039
6. Jacquelot N, Pitt JM, Enot DP, Roberti MP, Duong CPM, Rusakiewicz S, et al. Immune biomarkers for prognosis and prediction of responses to immune checkpoint blockade in cutaneous melanoma. Oncoimmunology. (2017) 6:e1299303. doi: 10.1080/2162402X.2017.1299303
7. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. (2017) 8:16073. doi: 10.1038/ncomms16073
8. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. (2017) 168:487–502.e15. doi: 10.1016/j.cell.2016.12.022
9. Kadoki M, Patil A, Thaiss CC, Brooks DJ, Pandey S, Deep D, et al. Organism-level analysis of vaccination reveals networks of protection across tissues. Cell. (2017) 171:398–413.e21. doi: 10.1016/j.cell.2017.08.024
10. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. doi: 10.1126/science.1129139
11. Munari E, Marconi M, Querzoli G, Lunardi G, Bertoglio P, Ciompi F, et al. Impact of PD-L1 and PD-1 expression on the prognostic significance of CD8(+) tumor-infiltrating lymphocytes in non-small cell lung cancer. Front Immunol. (2021) 12:680973. doi: 10.3389/fimmu.2021.680973
12. Casey DL, Cheung NV. Immunotherapy of pediatric solid tumors: treatments at a crossroads, with an emphasis on antibodies. Cancer Immunol Res. (2020) 8:161–6. doi: 10.1158/2326-6066.CIR-19-0692
13. Le TP, Thai TH. The state of cellular adoptive immunotherapy for neuroblastoma and other pediatric solid tumors. Front Immunol. (2017) 8:1640. doi: 10.3389/fimmu.2017.01640
14. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
16. Ora I, Eggert A. Progress in treatment and risk stratification of neuroblastoma: impact on future clinical and basic research. Semin Cancer Biol. (2011) 21:217–28. doi: 10.1016/j.semcancer.2011.07.002
17. Davidoff AM. Neuroblastoma. Semin Pediatr Surg. (2012) 21:2–14. doi: 10.1053/j.sempedsurg.2011.10.009
18. Katzenstein HM, Bowman LC, Brodeur GM, Thorner PS, Joshi VV, Smith EI, et al. Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D(S) neuroblastoma: the pediatric oncology group experience–a pediatric oncology group study. J Clin Oncol. (1998) 16:2007–17. doi: 10.1200/JCO.1998.16.6.2007
19. Wolfl M, Jungbluth AA, Garrido F, Cabrera T, Meyen-Southard S, Spitz R, et al. Expression of MHC class I, MHC class II, and cancer germline antigens in neuroblastoma. Cancer Immunol Immunother. (2005) 54:400–6. doi: 10.1007/s00262-004-0603-z
20. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Morandi F, Bocca P, et al. Mechanisms of immune evasion of human neuroblastoma. Cancer Lett. (2005) 228:155–61. doi: 10.1016/j.canlet.2004.11.064
21. Wienke J, Dierselhuis MP, Tytgat GAM, Kunkele A, Nierkens S, Molenaar JJ. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur J Cancer. (2021) 144:123–50. doi: 10.1016/j.ejca.2020.11.014
22. Wienke J, Visser LL, Kholosy WM, Keller KM, Barisa M, Poon E, et al. Integrative analysis of neuroblastoma by single-cell RNA sequencing identifies the NECTIN2-TIGIT axis as a target for immunotherapy. Cancer Cell. (2024) 42:283–300 e8. doi: 10.1016/j.ccell.2023.12.008
23. Verhoeven BM, Mei S, Olsen TK, Gustafsson K, Valind A, Lindstrom A, et al. The immune cell atlas of human neuroblastoma. Cell Rep Med. (2022) 3:100657. doi: 10.1016/j.xcrm.2022.100657
24. Carlson LM, De Geer A, Sveinbjornsson B, Orrego A, Martinsson T, Kogner P, et al. The microenvironment of human neuroblastoma supports the activation of tumor-associated T lymphocytes. Oncoimmunology. (2013) 2:e23618. doi: 10.4161/onci.23618
25. Mina M, Boldrini R, Citti A, Romania P, D’Alicandro V, De Ioris M, et al. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology. (2015) 4:e1019981. doi: 10.1080/2162402X.2015.1019981
26. Apps JR, Hasan F, Campus O, Behjati S, Jacques TS, JS N, et al. The immune environment of paediatric solid Malignancies: evidence from an immunohistochemical study of clinical cases. Fetal Pediatr Pathol. (2013) 32:298–307. doi: 10.3109/15513815.2012.754527
27. Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, Santi M, et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. (2017) 123:3807–15. doi: 10.1002/cncr.v123.19
28. Morandi F, Pozzi S, Barco S, Cangemi G, Amoroso L, Carlini B, et al. CD4(+)CD25(hi)CD127(-) Treg and CD4(+)CD45R0(+)CD49b(+)LAG3(+) Tr1 cells in bone marrow and peripheral blood samples from children with neuroblastoma. Oncoimmunology. (2016) 5:e1249553. doi: 10.1080/2162402X.2016.1249553
29. Jin W, Zhang Y, Liu Z, Che Z, Gao M, Peng H. Exploration of the molecular characteristics of the tumor-immune interaction and the development of an individualized immune prognostic signature for neuroblastoma. J Cell Physiol. (2021) 236:294–308. doi: 10.1002/jcp.v236.1
30. Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med. (2004) 199:1213–21. doi: 10.1084/jem.20031462
31. Facchetti P, Prigione I, Ghiotto F, Tasso P, Garaventa A, Pistoia V. Functional and molecular characterization of tumour-infiltrating lymphocytes and clones thereof from a major-histocompatibility-complex-negative human tumour: neuroblastoma. Cancer Immunol Immunother. (1996) 42:170–8. doi: 10.1007/s002620050267
32. Coughlin CM, Fleming MD, Carroll RG, Pawel BR, Hogarty MD, Shan X, et al. Immunosurveillance and survivin-specific T-cell immunity in children with high-risk neuroblastoma. J Clin Oncol. (2006) 24:5725–34. doi: 10.1200/JCO.2005.05.3314
33. Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med. (2020) 26:792–802. doi: 10.1038/s41591-020-0844-1
34. Camisaschi C, Renne SL, Beretta V, Rini F, Spagnuolo RD, Tuccitto A, et al. Immune landscape and in vivo immunogenicity of NY-ESO-1 tumor antigen in advanced neuroblastoma patients. BMC Cancer. (2018) 18:983. doi: 10.1186/s12885-018-4910-8
35. Zhang P, Wu X, Basu M, Dong C, Zheng P, Liu Y, et al. MYCN amplification is associated with repressed cellular immunity in neuroblastoma: an in silico immunological analysis of TARGET database. Front Immunol. (2017) 8:1473. doi: 10.3389/fimmu.2017.01473
36. Zhong X, Zhang Y, Wang L, Zhang H, Liu H, Liu Y. Cellular components in tumor microenvironment of neuroblastoma and the prognostic value. PeerJ. (2019) 7:e8017. doi: 10.7717/peerj.8017
37. Soldati R, Berger E, Zenclussen AC, Jorch G, Lode HN, Salatino M, et al. Neuroblastoma triggers an immunoevasive program involving galectin-1-dependent modulation of T cell and dendritic cell compartments. Int J Cancer. (2012) 131:1131–41. doi: 10.1002/ijc.26498
38. Mei S, Alchahin AM, Embaie BT, Gavriliuc IM, Verhoeven BM, Zhao T, et al. Single-cell analyses of metastatic bone marrow in human neuroblastoma reveals microenvironmental remodeling and metastatic signature. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.173337
39. Layer JP, Kronmuller MT, Quast T, van den Boorn-Konijnenberg D, Effern M, Hinze D, et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology. (2017) 6:e1320626. doi: 10.1080/2162402X.2017.1320626
40. Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, PeRussia B, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol. (2001) 167:3114–22. doi: 10.4049/jimmunol.167.6.3114
41. Kroesen M, Lindau D, Hoogerbrugge P, Adema GJ. Immunocombination therapy for high-risk neuroblastoma. Immunotherapy. (2012) 4:163–74. doi: 10.2217/imt.11.169
42. Seeger RC. Immunology and immunotherapy of neuroblastoma. Semin Cancer Biol. (2011) 21:229–37. doi: 10.1016/j.semcancer.2011.09.012
43. Melaiu O, Mina M, Chierici M, Boldrini R, Jurman G, Romania P, et al. PD-L1 is a therapeutic target of the bromodomain inhibitor JQ1 and, combined with HLA class I, a promising prognostic biomarker in neuroblastoma. Clin Cancer Res. (2017) 23:4462–72. doi: 10.1158/1078-0432.CCR-16-2601
44. Wei JS, Kuznetsov IB, Zhang S, Song YK, Asgharzadeh S, Sindiri S, et al. Clinically relevant cytotoxic immune cell signatures and clonal expansion of T-cell receptors in high-risk MYCN-not-amplified human neuroblastoma. Clin Cancer Res. (2018) 24:5673–84. doi: 10.1158/1078-0432.CCR-18-0599
45. Sarkar AK, Burlingame SM, Zang YQ, Dulai V, Hicks MJ, Strother DR, et al. Major histocompatibility complex-restricted lysis of neuroblastoma cells by autologous cytotoxic T lymphocytes. J Immunother. (2001) 24:305–11. doi: 10.1097/00002371-200107000-00006
46. Olle Hurtado M, Wolbert J, Fisher J, Flutter B, Stafford S, Barton J, et al. Tumor infiltrating lymphocytes expanded from pediatric neuroblastoma display heterogeneity of phenotype and function. PloS One. (2019) 14:e0216373. doi: 10.1371/journal.pone.0216373
47. Spel L, Boelens JJ, van der Steen DM, Blokland NJ, van Noesel MM, Molenaar JJ, et al. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget. (2015) 6:35770–81. doi: 10.18632/oncotarget.v6i34
48. Maris JM. Recent advances in neuroblastoma. N Engl J Med. (2010) 362:2202–11. doi: 10.1056/NEJMra0804577
49. De Geer A, Carlson LM, Kogner P, Levitskaya J. Soluble factors released by activated cytotoxic T lymphocytes interfere with death receptor pathways in neuroblastoma. Cancer Immunol Immunother. (2008) 57:731–43. doi: 10.1007/s00262-007-0412-2
50. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol. (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
51. Aoki T, Hino M, Koh K, Kyushiki M, Kishimoto H, Arakawa Y, et al. Low frequency of programmed death ligand 1 expression in pediatric cancers. Pediatr Blood Cancer. (2016) 3:1461–4. doi: 10.1002/pbc.26018
52. Inoue S, Takeuchi Y, Horiuchi Y, Murakami T, Odaka A. CD69 on tumor-infiltrating cells correlates with neuroblastoma suppression by simultaneous PD-1 and PD-L1 blockade. J Surg Res. (2023) 289:190–201. doi: 10.1016/j.jss.2023.03.042
53. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
54. Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. (1997) 94:8099–103. doi: 10.1073/pnas.94.15.8099
55. Gregorio A, Corrias MV, Castriconi R, Dondero A, Mosconi M, Gambini C, et al. Small round blue cell tumours: diagnostic and prognostic usefulness of the expression of B7-H3 surface molecule. Histopathology. (2008) 53:73–80. doi: 10.1111/j.1365-2559.2008.03070.x
56. Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci U S A. (2004) 101:12640–5. doi: 10.1073/pnas.0405025101
57. Xin C, Zhu J, Gu S, Yin M, Ma J, Pan C, et al. CD200 is overexpressed in neuroblastoma and regulates tumor immune microenvironment. Cancer Immunol Immunother. (2020) 69:2333–43. doi: 10.1007/s00262-020-02589-6
58. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. (2016) 17:1025–36. doi: 10.1038/ni.3518
59. Hishiki T, Mise N, Harada K, Ihara F, Takami M, Saito T, et al. Frequency and proliferative response of circulating invariant natural killer T cells in pediatric patients with Malignant solid tumors. Pediatr Surg Int. (2018) 34:169–76. doi: 10.1007/s00383-017-4185-1
60. McNerney KO, Karageorgos SA, Hogarty MD, Bassiri H. Enhancing neuroblastoma immunotherapies by engaging iNKT and NK cells. Front Immunol. (2020) 11:873. doi: 10.3389/fimmu.2020.00873
61. Stigliani S, Croce M, Morandi F, Scaruffi P, Rigo V, Carlini B, et al. Expression of FOXP3, CD14, and ARG1 in neuroblastoma tumor tissue from high-risk patients predicts event-free and overall survival. BioMed Res Int. (2015) 2015:347867. doi: 10.1155/2015/347867
62. HaDuong JH, Blavier L, Baniwal SK, Frenkel B, Malvar J, Punj V, et al. Interaction between bone marrow stromal cells and neuroblastoma cells leads to a VEGFA-mediated osteoblastogenesis. Int J Cancer. (2015) 137:797–809. doi: 10.1002/ijc.29465
63. Brandetti E, Veneziani I, Melaiu O, Pezzolo A, Castellano A, Boldrini R, et al. MYCN is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. Oncoimmunology. (2017) 6:e1316439. doi: 10.1080/2162402X.2017.1316439
64. Harada K, Ihara F, Takami M, Kamata T, Mise N, Yoshizawa H, et al. Soluble factors derived from neuroblastoma cell lines suppress dendritic cell differentiation and activation. Cancer Sci. (2019) 110:888–902. doi: 10.1111/cas.2019.110.issue-3
65. Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. Human FOXP3 and cancer. Oncogene. (2010) 29:4121–9. doi: 10.1038/onc.2010.174
66. deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. (2012) 18:3022–9. doi: 10.1158/1078-0432.CCR-11-3216
67. Gowda M, Godder K, Kmieciak M, Worschech A, Ascierto ML, Wang E, et al. Distinct signatures of the immune responses in low risk versus high risk neuroblastoma. J Transl Med. (2011) 170. doi: 10.1186/1479-5876-9-170
68. Tilak T, Sherawat S, Agarwala S, Gupta R, Vishnubhatla S, Bakhshi S. Circulating T-regulatory cells in neuroblastoma: a pilot prospective study. Pediatr Hematol Oncol. (2014) 31:717–22. doi: 10.3109/08880018.2014.886002
69. Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. (2011) 23:424–30. doi: 10.1016/j.smim.2011.10.002
70. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
71. Weissenborn C, von Lenthe S, Hinz N, Langwisch S, Busse M, Schumacher A, et al. Depletion of Foxp3+ regulatory T cells but not the absence of CD19+IL-10+ regulatory B cells hinders tumor growth in a para-orthotopic neuroblastoma mouse model. Int J Cancer. (2022) 151:2031–42. doi: 10.1002/ijc.34262
72. Chen J, Sun M, Chen C, Jiang B, Fang Y. Identification of hub genes and their correlation with infiltration of immune cells in MYCN positive neuroblastoma based on WGCNA and LASSO algorithm. Front Immunol. (2022) 13:1016683. doi: 10.3389/fimmu.2022.1016683
73. Hashimoto O, Yoshida M, Koma Y, Yanai T, Hasegawa D, Kosaka Y, et al. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J Pathol. (2016) 240:211–23. doi: 10.1002/path.2016.240.issue-2
74. Hegazy AN, Peine M, Helmstetter C, Panse I, Frohlich A, Bergthaler A, et al. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. (2010) 32:116–28. doi: 10.1016/j.immuni.2009.12.004
75. Veneziani I, Landolina N, Ricci B, Rossi O, Moretta L, Maggi E. How the immune system responds to allergy immunotherapy. Biomedicines. (2022) 10:2825. doi: 10.3390/biomedicines10112825
76. Kang JH, Zappasodi R. Modulating Treg stability to improve cancer immunotherapy. Trends Cancer. (2023) 9:911–27. doi: 10.1016/j.trecan.2023.07.015
77. Malviya V, Yshii L, Junius S, Garg AD, Humblet-Baron S, Schlenner SM. Regulatory T-cell stability and functional plasticity in health and disease. Immunol Cell Biol. (2023) 101:112–29. doi: 10.1111/imcb.v101.2
78. van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. (2017) 49:1261–6. doi: 10.1038/ng.3899
79. Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. (2017) 49:1408–13. doi: 10.1038/ng.3921
80. Upton K, Modi A, Patel K, Kendsersky NM, Conkrite KL, Sussman RT, et al. Epigenomic profiling of neuroblastoma cell lines. Sci Data. (2020) 7:116. doi: 10.1038/s41597-020-0458-y
81. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
82. D’Amico S, Tempora P, Gragera P, Krol K, Melaiu O, De Ioris MA, et al. Two bullets in the gun: combining immunotherapy with chemotherapy to defeat neuroblastoma by targeting adrenergic-mesenchymal plasticity. Front Immunol. (2023) 14:1268645. doi: 10.3389/fimmu.2023.1268645
83. Yuan X, Seneviratne JA, Du S, Xu Y, Chen Y, Jin Q, et al. Single-cell profiling of peripheral neuroblastic tumors identifies an aggressive transitional state that bridges an adrenergic-mesenchymal trajectory. Cell Rep. (2022) 41:111455. doi: 10.1016/j.celrep.2022.111455
84. Sengupta S, Das S, Crespo AC, Cornel AM, Patel AG, Mahadevan NR, et al. Mesenchymal and adrenergic cell lineage states in neuroblastoma possess distinct immunogenic phenotypes. Nat Cancer. (2022) 3:1228–46. doi: 10.1038/s43018-022-00427-5
85. Kaufman ME, Vayani OR, Moore K, Chlenski A, Wu T, Lee SM, et al. Characterizing relationships between T-cell inflammation and outcomes in patients with high-risk neuroblastoma according to mesenchymal and adrenergic signatures. Cancer Res Commun. (2024) 4:2255–66. doi: 10.1158/2767-9764.CRC-24-0214
86. Wolpaw AJ, Grossmann LD, Dessau JL, Dong MM, Aaron BJ, Brafford PA, et al. Epigenetic state determines inflammatory sensing in neuroblastoma. Proc Natl Acad Sci U.S.A. (2022) 119:e2102358119. doi: 10.1073/pnas.2102358119
87. van Wezel EM, van Zogchel LMJ, van Wijk J, Timmerman I, Vo NK, Zappeij-Kannegieter L, et al. Mesenchymal neuroblastoma cells are undetected by current mRNA marker panels: the development of a specific neuroblastoma mesenchymal minimal residual disease panel. JCO Precis Oncol. (2019) 3:PO.18.00413. doi: 10.1200/PO.18.00413
88. Liotta F, Angeli R, Cosmi L, Fili L, Manuelli C, Frosali F, et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells. (2008) 26:279–89. doi: 10.1634/stemcells.2007-0454
89. Thirant C, Peltier A, Durand S, Kramdi A, Louis-Brennetot C, Pierre-Eugene C, et al. Reversible transitions between noradrenergic and mesenchymal tumor identities define cell plasticity in neuroblastoma. Nat Commun. (2023) 14:2575. doi: 10.1038/s41467-023-38239-5
90. Canzonetta C, Pelosi A, Di Matteo S, Veneziani I, Tumino N, Vacca P, et al. Identification of neuroblastoma cell lines with uncommon TAZ(+)/mesenchymal stromal cell phenotype with strong suppressive activity on natural killer cells. J Immunother Cancer. (2021) 9:e001313. doi: 10.1136/jitc-2020-001313
91. Di Matteo S, Bilotta MT, Pelosi A, Haas D, Theinert T, Weber G, et al. Transition to a mesenchymal state in neuroblastoma may be characterized by a high expression of GD2 and by the acquisition of immune escape from NK cells. Front Immunol. (2024) 15:1382931. doi: 10.3389/fimmu.2024.1382931
92. Lenardo M, Rustgi AK, Schievella AR, Bernards R. Suppression of MHC class I gene expression by N-myc through enhancer inactivation. EMBO J. (1989) 8:3351–5. doi: 10.1002/j.1460-2075.1989.tb08497.x
93. Kushner BH. Neuroblastoma: a disease requiring a multitude of imaging studies. J Nucl Med. (2004) 45:1172–88.
94. Song L, Ara T, Wu HW, Woo CW, Reynolds CP, Seeger RC, et al. Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest. (2007) 117:2702–12. doi: 10.1172/JCI30751
95. Liu Z, Grant CN, Sun L, Miller BA, Spiegelman VS, Wang HG. Expression patterns of immune genes reveal heterogeneous subtypes of high-risk neuroblastoma. Cancers. (2020) 12:1739. doi: 10.3390/cancers12071739
96. Zhong X, Tao Y, Chang J, Zhang Y, Zhang H, Wang L, et al. Prognostic signature of immune genes and immune-related lncRNAs in neuroblastoma: A study based on GEO and TARGET datasets. Front Oncol. (2021) 11:631546. doi: 10.3389/fonc.2021.631546
97. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun. (2020) 11:404. doi: 10.1038/s41467-019-14134-w
98. Costa A, Thirant C, Kramdi A, Pierre-Eugene C, Louis-Brennetot C, Blanchard O, et al. Single-cell transcriptomics reveals shared immunosuppressive landscapes of mouse and human neuroblastoma. J Immunother Cancer. (2022) 10:e004807. doi: 10.1136/jitc-2022-004807
99. Fei L, Ren X, Yu H, Zhan Y. Targeting the CCL2/CCR2 axis in cancer immunotherapy: one stone, three birds? Front Immunol. (2021) 12:771210. doi: 10.3389/fimmu.2021.771210
100. Li Y, Xiang S, Pan W, Wang J, Zhan H, Liu S. Targeting tumor immunosuppressive microenvironment for pancreatic cancer immunotherapy: Current research and future perspective. Front Oncol. (2023) 13:1166860. doi: 10.3389/fonc.2023.1166860
101. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. (2017) 77:5142–57. doi: 10.1158/0008-5472.CAN-16-2586
102. Wu HW, Sheard MA, Malvar J, Fernandez GE, DeClerck YA, Blavier L, et al. Anti-CD105 antibody eliminates tumor microenvironment cells and enhances anti-GD2 antibody immunotherapy of neuroblastoma with activated natural killer cells. Clin Cancer Res. (2019) 25:4761–74. doi: 10.1158/1078-0432.CCR-18-3358
103. Pelizzo G, Veschi V, Mantelli M, Croce S, Di Benedetto V, D’Angelo P, et al. Microenvironment in neuroblastoma: isolation and characterization of tumor-derived mesenchymal stromal cells. BMC Cancer. (2018) 18:1176. doi: 10.1186/s12885-018-5082-2
104. Di Matteo S, Avanzini MA, Pelizzo G, Calcaterra V, Croce S, Spaggiari GM, et al. Neuroblastoma tumor-associated mesenchymal stromal cells regulate the cytolytic functions of NK cells. Cancers. (2022) 15:19. doi: 10.3390/cancers15010019
105. Jahangiri L, Ishola T. Exosomes in neuroblastoma biology, diagnosis, and treatment. Life (Basel). (2022) 12:1714. doi: 10.3390/life12111714
106. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol. (2019) 9:1146. doi: 10.3389/fonc.2019.01146
107. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
108. Tumino N, Weber G, Besi F, Del Bufalo F, Bertaina V, Paci P, et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol. (2021) 14:191. doi: 10.1186/s13045-021-01193-0
109. Tumino N, Besi F, Martini S, Di Pace AL, Munari E, Quatrini L, et al. Polymorphonuclear myeloid-derived suppressor cells are abundant in peripheral blood of cancer patients and suppress natural killer cell anti-tumor activity. Front Immunol. (2021) 12:803014. doi: 10.3389/fimmu.2021.803014
110. Navarro G, Gomez-Autet M, Morales P, Rebassa JB, Llinas Del Torrent C, Jagerovic N, et al. Homodimerization of CB(2) cannabinoid receptor triggered by a bivalent ligand enhances cellular signaling. Pharmacol Res. (2024) 208:107363. doi: 10.1016/j.phrs.2024.107363
111. Louault K, Porras T, Lee MH, Muthugounder S, Kennedy RJ, Blavier L, et al. Fibroblasts and macrophages cooperate to create a pro-tumorigenic and immune resistant environment via activation of TGF-beta/IL-6 pathway in neuroblastoma. Oncoimmunology. (2022) 11:2146860. doi: 10.1080/2162402X.2022.2146860
112. Cousin JM, Cloninger MJ. The role of galectin-1 in cancer progression, and synthetic multivalent systems for the study of galectin-1. Int J Mol Sci. (2016) 17:1566. doi: 10.3390/ijms17091566
113. Van Woensel M, Mathivet T, Wauthoz N, Rosiere R, Garg AD, Agostinis P, et al. Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci Rep. (2017) 7:1217. doi: 10.1038/s41598-017-01279-1
114. Buchel G, Schulte JH, Harrison L, Batzke K, Schuller U, Hansen W, et al. Immune response modulation by Galectin-1 in a transgenic model of neuroblastoma. Oncoimmunology. (2016) 5:e1131378. doi: 10.1080/2162402X.2015.1131378
115. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
116. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
117. Bin Q, Johnson BD, Schauer DW, Casper JT, Orentas RJ. Production of macrophage migration inhibitory factor by human and murine neuroblastoma. Tumour Biol. (2002) 23:123–9. doi: 10.1159/000064028
118. Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. (1995) 378:736–9. doi: 10.1038/378736a0
119. Yan X, Orentas RJ, Johnson BD. Tumor-derived macrophage migration inhibitory factor (MIF) inhibits T lymphocyte activation. Cytokine. (2006) 33:188–98. doi: 10.1016/j.cyto.2006.01.006
120. Cavalli E, Mazzon E, Mammana S, Basile MS, Lombardo SD, Mangano K, et al. Overexpression of macrophage migration inhibitory factor and its homologue D-dopachrome tautomerase as negative prognostic factor in neuroblastoma. Brain Sci. (2019) 9:284. doi: 10.3390/brainsci9100284
121. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
122. Li R, Villacreses N, Ladisch S. Human tumor gangliosides inhibit murine immune responses. vivo. Cancer Res. (1995) 55:211–4.
123. Floutsis G, Ulsh L, Ladisch S. Immunosuppressive activity of human neuroblastoma tumor gangliosides. Int J Cancer. (1989) 43:6–9. doi: 10.1002/ijc.2910430103
124. Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. (2015) 75:3043–53. doi: 10.1158/0008-5472.CAN-14-3443
125. Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. (2004) 6:558–68. doi: 10.1593/neo.04316
126. Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vely F, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med. (2015) 7:283ra55. doi: 10.1126/scitranslmed.aaa2327
127. Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, et al. Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer Res. (2007) 67:6433–41. doi: 10.1158/0008-5472.CAN-06-4588
128. Vanichapol T, Chiangjong W, Panachan J, Anurathapan U, Chutipongtanate S, Hongeng S. Secretory high-mobility group box 1 protein affects regulatory T cell differentiation in neuroblastoma microenvironment. In Vitro. J Oncol. (2018) 2018:7946021. doi: 10.1155/2018/7946021
129. Caforio M, Sorino C, Caruana I, Weber G, Camera A, Cifaldi L, et al. GD2 redirected CAR T and activated NK-cell-mediated secretion of IFNgamma overcomes MYCN-dependent IDO1 inhibition, contributing to neuroblastoma cell immune escape. J Immunother Cancer. (2021) 9:e001502. doi: 10.1136/jitc-2020-001502
130. Wang X, Fan D, Yang Y, Gimple RC, Zhou S. Integrative multi-omics approaches to explore immune cell functions: Challenges and opportunities. iScience. (2023) 26:106359. doi: 10.1016/j.isci.2023.106359
131. Ma A, Xin G, Ma Q. The use of single-cell multi-omics in immuno-oncology. Nat Commun. (2022) 13:2728. doi: 10.1038/s41467-022-30549-4
132. Ali SS, Li Q, Agrawal PB. Implementation of multi-omics in diagnosis of pediatric rare diseases. Pediatr Res. (2024). doi: 10.1038/s41390-024-03728-w
133. Qin X, Ning W, Liu H, Liu X, Luo W, Xia N. Stepping forward: T-cell redirecting bispecific antibodies in cancer therapy. Acta Pharm Sin B. (2024) 14:2361–77. doi: 10.1016/j.apsb.2024.03.027
134. Chehelgerdi M, Chehelgerdi M, Allela OQB, Pecho RDC, Jayasankar N, Rao DP, et al. Progressing nanotechnology to improve targeted cancer treatment: overcoming hurdles in its clinical implementation. Mol Cancer. (2023) 22:169. doi: 10.1186/s12943-023-01865-0
135. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discovery. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
136. Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. (2019) 35:221–37.e8. doi: 10.1016/j.ccell.2019.01.002
137. Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. (2022) 40:53–69 e9. doi: 10.1016/j.ccell.2021.12.005
138. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci Transl Med. (2020) 12:eabd6169. doi: 10.1126/scitranslmed.abd6169
139. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
140. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
141. Diez-Alonso L, Falgas A, Arroyo-Rodenas J, Romencin PA, Martinez A, Gomez-Rosel M, et al. Engineered T cells secreting anti-BCMA T cell engagers control multiple myeloma and promote immune memory in vivo. Sci Transl Med. (2024) 16:eadg7962. doi: 10.1126/scitranslmed.adg7962
142. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
143. Fuca G, Spagnoletti A, Ambrosini M, de Braud F, Di Nicola M. Immune cell engagers in solid tumors: promises and challenges of the next generation immunotherapy. ESMO Open. (2021) 6:100046. doi: 10.1016/j.esmoop.2020.100046
144. Tapia-Galisteo A, Alvarez-Vallina L, Sanz L. Bi- and trispecific immune cell engagers for immunotherapy of hematological Malignancies. J Hematol Oncol. (2023) 16:83. doi: 10.1186/s13045-023-01482-w
145. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
146. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
147. Guo S, Yao Y, Tang Y, Xin Z, Wu D, Ni C, et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct Target Ther. (2023) 8:205. doi: 10.1038/s41392-023-01462-z
148. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
149. Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. (2022) 23:487–500. doi: 10.1038/s41590-022-01132-2
150. Behranvand N, Nasri F, Zolfaghari Emameh R, Khani P, Hosseini A, Garssen J, et al. Chemotherapy: a double-edged sword in cancer treatment. Cancer Immunol Immunother. (2022) 71:507–26. doi: 10.1007/s00262-021-03013-3
151. Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. (2017) 18:946–57. doi: 10.1016/S1470-2045(17)30355-8
152. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. (2017) 23:6441–9. doi: 10.1158/1078-0432.CCR-17-0379
153. Mody R, Yu AL, Naranjo A, Zhang FF, London WB, Shulkin BL, et al. Irinotecan, temozolomide, and dinutuximab with GM-CSF in children with refractory or relapsed neuroblastoma: A report from the children’s oncology group. J Clin Oncol. (2020) 38:2160–9. doi: 10.1200/JCO.20.00203
154. Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, et al. A phase II trial of hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin Cancer Res. (2019) 25:6320–8. doi: 10.1158/1078-0432.CCR-19-1452
155. Poon E, Liang T, Jamin Y, Walz S, Kwok C, Hakkert A, et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J Clin Invest. (2020) 130:5875–92. doi: 10.1172/JCI134132
156. Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin Cancer Res. (2017) 23:2856–68. doi: 10.1158/1078-0432.CCR-16-1114
157. Roeschert I, Poon E, Henssen AG, Garcia HD, Gatti M, Giansanti C, et al. Combined inhibition of Aurora-A and ATR kinase results in regression of MYCN-amplified neuroblastoma. Nat Cancer. (2021) 2:312–26.
158. Anderson J, Majzner RG, Sondel PM. Immunotherapy of neuroblastoma: facts and hopes. Clin Cancer Res. (2022) 28:3196–206. doi: 10.1158/1078-0432.CCR-21-1356
159. Rivoltini L, Arienti F, Orazi A, Cefalo G, Gasparini M, Gambacorti-Passerini C, et al. Phenotypic and functional analysis of lymphocytes infiltrating paediatric tumours, with a characterization of the tumour phenotype. Cancer Immunol Immunother. (1992) 34:241–51. doi: 10.1007/BF01741792
160. Nanno M, Seki H, Mathioudakis G, Suzuki R, Itoh K, Ioannides CG, et al. Gamma/delta T cell antigen receptors expressed on tumor-infiltrating lymphocytes from patients with solid tumors. Eur J Immunol. (1992) 22:679–87. doi: 10.1002/eji.1830220310
161. Pearson ADJ, Rossig C, Lesa G, Diede SJ, Weiner S, Anderson J, et al. ACCELERATE and European Medicines Agency Paediatric Strategy Forum for medicinal product development of checkpoint inhibitors for use in combination therapy in paediatric patients. Eur J Cancer. (2020) 127:52–66. doi: 10.1016/j.ejca.2019.12.029
162. Huang H, Li P, Ye X, Zhang F, Lin Q, Wu K, et al. Isoalantolactone increases the sensitivity of prostate cancer cells to cisplatin treatment by inducing oxidative stress. Front Cell Dev Biol. (2021) 9:632779. doi: 10.3389/fcell.2021.632779
163. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
164. Morgan MA, Schambach A. Engineering CAR-T cells for improved function against solid tumors. Front Immunol. (2018) 9:2493. doi: 10.3389/fimmu.2018.02493
165. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. (2020) 21:914–26. doi: 10.1038/s41590-020-0676-7
166. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. (2015) 23:769–78. doi: 10.1038/mt.2015.4
167. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
Keywords: immunosuppression, immunotherapy of tumors, solid pediatric tumors, T effector cells, T regulatory cells, tumor infiltrating lymphocytes
Citation: Maggi E, Landolina N, Munari E, Mariotti FR, Tumino N, Vacca P, Azzarone B and Moretta L (2025) T cells in the microenvironment of solid pediatric tumors: the case of neuroblastoma. Front. Immunol. 16:1544137. doi: 10.3389/fimmu.2025.1544137
Received: 12 December 2024; Accepted: 12 February 2025;
Published: 28 February 2025.
Edited by:
Silvano Sozzani, Sapienza University of Rome, ItalyReviewed by:
Giovanni Bernardini, Sapienza University of Rome, ItalyCopyright © 2025 Maggi, Landolina, Munari, Mariotti, Tumino, Vacca, Azzarone and Moretta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Moretta, bG9yZW56by5tb3JldHRhQG9wYmcubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.