
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 07 March 2025
Sec. Primary Immunodeficiencies
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1541899
This article is part of the Research Topic Decoding Syndromic Immunodeficiencies: Bridging Genetics and Immune Dysfunctions View all 3 articles
 Gabriel Emmanuel Arce-Estrada1
Gabriel Emmanuel Arce-Estrada1 Miguel Rodríguez-Morales2,3Selma Cecilia Scheffler-Mendoza4Marimar Sáez-de-Ocariz5
Miguel Rodríguez-Morales2,3Selma Cecilia Scheffler-Mendoza4Marimar Sáez-de-Ocariz5 Laura Berrón-Ruiz1
Laura Berrón-Ruiz1 Sara Elva Espinosa-Padilla1*
Sara Elva Espinosa-Padilla1* Francisco Alberto Contreras-Verduzco1*
Francisco Alberto Contreras-Verduzco1*Objective: To describe a novel IKBKB variant linked to immune dysregulation and disseminated tuberculosis, alongside a review of pathogenic variants to outline their phenotypic spectrum.
Material and methods: Observational case report and literature review.
Results: A five-month-old girl from an endogamous Mexican population developed symptoms suggestive of Kawasaki disease which progressed to hemophagocytic syndrome. Mycobacterium bovis was found in her skin, blood, and bone marrow. She had received the Bacillus Calmette-Guérin (BCG) vaccine on the second day of life. Genetic testing revealed a homozygous pathogenic variant (PV) in the IKBKB gene (c.1705G>T, p.Glu569*). Both parents were heterozygous. Fourteen publications were found, encompassing 33 patients with 14 different PV, including the case described in this work.
Discussion: Hypogammaglobulinemia, candidiasis and mycobacterial infections were common in most cases identified. Our case is unique in presenting with Kawasaki disease, hemophagocytic syndrome, and mycobacteria from skin, blood, and bone marrow.
Conclusions: We identified a novel homozygous PV in the IKBKB gene, highlighting new clinical manifestations.
● An infant girl received the BCG vaccine at birth.
● Kawasaki disease and hemophagocytic syndrome were the initial manifestations.
● Mycobacterium bovis was isolated from her skin, blood, and bone marrow.
● A homozygous PV in the IKBKB gene was found: c.1705G>T (p.Glu569*).
● 33 cases with 14 PV of the gene were found in a literature review.
The gene IKBKB encodes the protein IKKβ, which activates members of the nuclear factor kappa B (NF-κB) transcription factor family through the classical (or canonical) pathway. This activation occurs via the phosphorylation of IκB inhibitors (1, 2). In non-activated cells, NF-kB dimers are associated with molecules of the IkB protein family, which inhibit NF-κB binding to deoxyribonucleic acid (DNA) (1). PV in the IKBKB gene can result in severe or milder forms of combined immunodeficiency (3). This condition may be inherited in both autosomal dominant (immunodeficiency type 15A, Online Mendelian Inheritance in Man (OMIM) #618204) or autosomal recessive (immunodeficiency type 15B, OMIM #615592) patterns.
We present a previously unreported PV of a 15B immunodeficiency and its associated phenotypic spectrum.
A literature search was conducted for the development of this review in the Human Mutation Database, MEDLINE, EMBASE and LILACS databases using the terms “IKBKB gene” OR “IKBKB”, with emphasis on clinical cases, case series and reviews.
We describe a five-month-old girl from an endogamous Mexican population who was admitted to our hospital for persistent high fever over a week, malaise and a leukocytosis (29.0 x103/µL) in a complete blood cell test. She had received the BCG vaccine on the second day of life. Her weight and height were adequate for her age and she had no history of infections.
The patient was initially diagnosed with incomplete Kawasaki disease, this diagnosis was supported by the presence of high and prolonged fever (over a week), strawberry tongue, angular cheilitis, perineal erythema, BCG vaccination site erythema and induration, anemia, leukocytosis and sterile pyuria, with elevated inflammatory markers such as C reactive protein and pro-brain natriuretic peptide. Echocardiographic findings of pericardial effusion further strengthened this diagnosis and she was treated with intravenous gamma globulin. Also, she presented abdominal pain and elevated serum levels of procalcitonin, D-dimer and ferritin. Imaging studies revealed findings consistent with splenic abscesses and a nodule in the right lung. (Figures 1A–C).
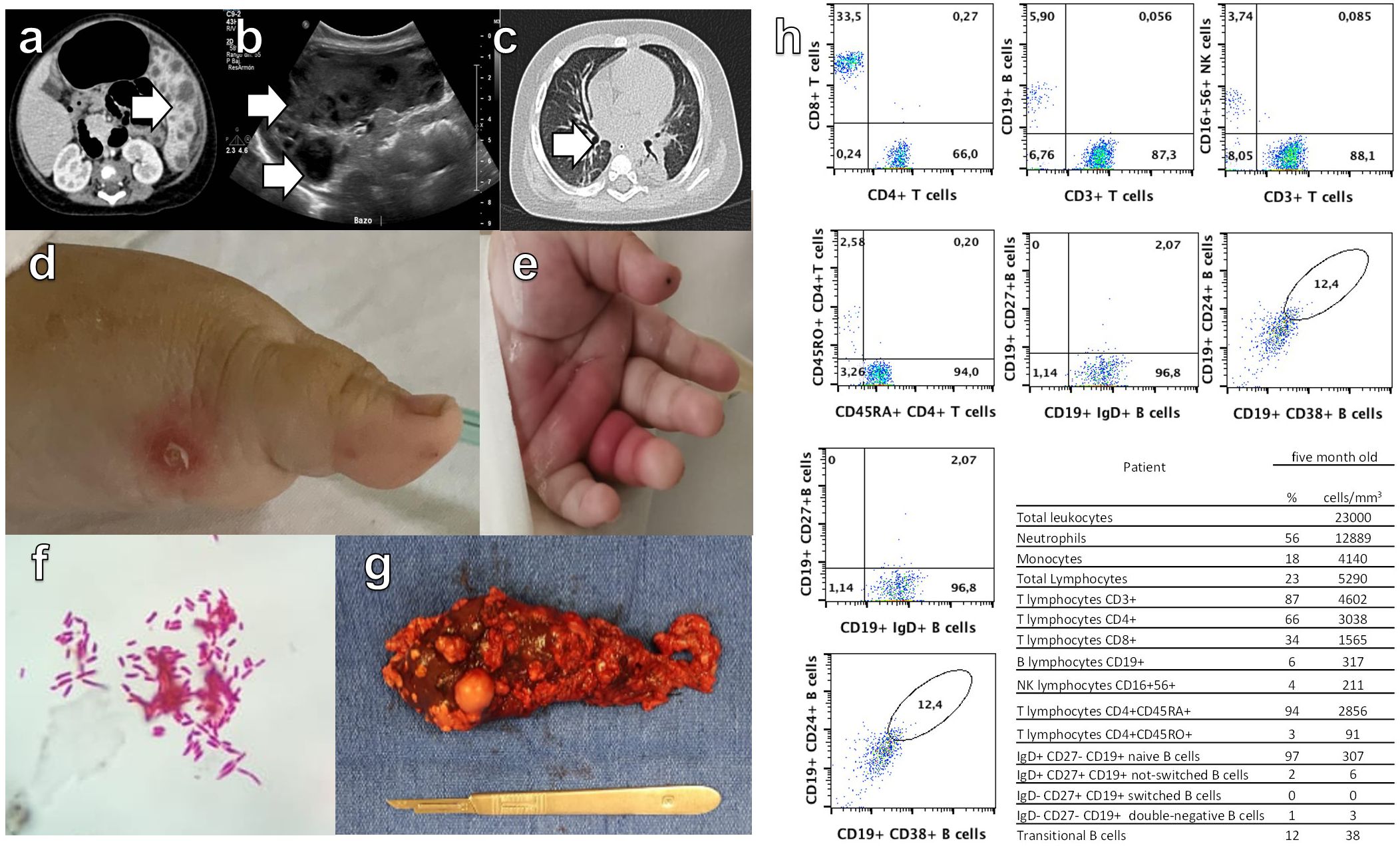
Figure 1. Abdominal computed tomography (CT) scan (a) and ultrasound (b), with white arrows indicating multiple abscesses. Image (c) shows a pulmonary nodule detected by CT scan. Images (d, e) display acral skin nodules. In (f), Mycobacterium bovis identification in blood culture is shown. Image (g) presents the macroscopic visualization of the spleen removed by surgery. Flow cytometry (h) reveals low levels of double-negative T cells, as well as decreased memory B cells without isotype switching and an absence of isotype-switched memory B cells.
Subsequently, her clinical course evolved with features consistent with hemophagocytic lymphohistiocytosis (HLH). The patient fulfilled more than five diagnostic criteria, including fever, splenomegaly, neutropenia, thrombocytopenia, hypertriglyceridemia, hypofibrinogenemia, hyperferritinemia and confirmed hemophagocytosis in 2 bone marrow biopsies.
She was treated with intravenous steroids, cyclosporine and gamma globulin. The bone marrow biopsy also identified bacilliform microorganisms, so an inborn error of immunity was suspected. Immunoglobulin levels were not measured prior to the administration of intravenous gamma globulin. However, post-infusion levels were reported as follows: IgG 968 mg/dL (normal range: 215-704 mg/dL), IgA 5 mg/dL (normal range: 8.1-68 mg/dL), IgM 25 mg/dL (normal range: 35-102 mg/dL), and IgE 0.2 KUA/L.
She developed acral skin nodules with superinfection of some (Figures 1D, E) and the microscopic analysis revealed bacillary structures embedded in the cytoplasm of macrophages. Mycobacterium bovis was isolated from blood culture, bone marrow and the acral skin nodules biopsy (Figure 1F). Human Immunodeficiency Virus and leishmaniasis were excluded and the patient was treated for tuberculosis with isoniazid, rifampin, pyrazinamide, and ethambutol. There was no improvement in the febrile pattern until the spleen was surgically removed (Figure 1G). Histopathological examination revealed multiple splenic abscesses associated with bacillary colony infection with abundant macrophages.
Once the fever has subsided, flow cytometry was performed showing low levels of double negative T cells as well as low memory B cells without isotype switching and no isotype-switched memory B cells (Figure 1H).
Continuing with the diagnostic protocol and after informed consent, a massive parallel sequencing with the most current version of Invitae´s (Invitae Corp. San Francisco California, U.S.) primary immunodeficiencies panel available (407 genes) was requested. It reported a homozygous pathogenic variant in the IKBKB (NM_001556.2) gene: c.1705G>T (p.Glu569*).The extension study showed that parents are heterozygous. She received an haploidentical hematopoietic stem cell transplant (HSCT) with conditioning based on antithymocyte globulin, fludarabine and busulfan. She died ten days later because of septic shock. The timeline is shown in Figure 2.
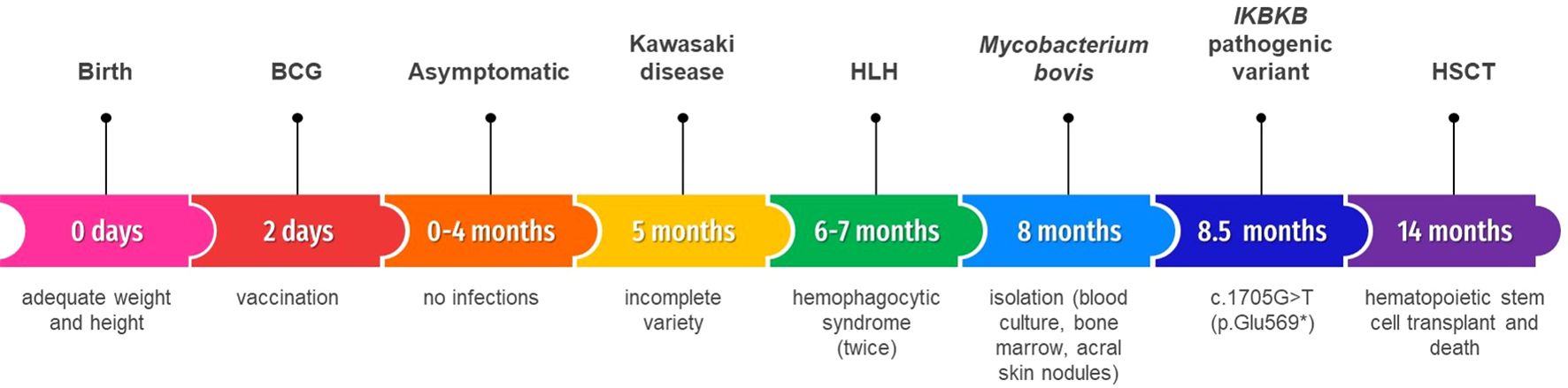
Figure 2. Timeline.
We retrieved 14 publications documenting 33 patients (including the present case), of whom 14 were females, 10 were males and 9 without gender report. Age ranges at onset of symptoms were 1-11 months in most patients but 3 cases reported symptoms onset at 20, 27 and 52 years of age. Fourteen patients developed a mycobacterial infection (4 M. bovis, 1 M. avium, 1 M. tuberculosis and the rest did not specify the species of mycobacteria). Among them, 14 distinct PV in the IKBKB gene were found. Twenty six cases (including ours) exhibited autosomal recessive inheritance, while the remaining were autosomal dominant. The findings from this review are depicted in Table 1 and Figure 3.
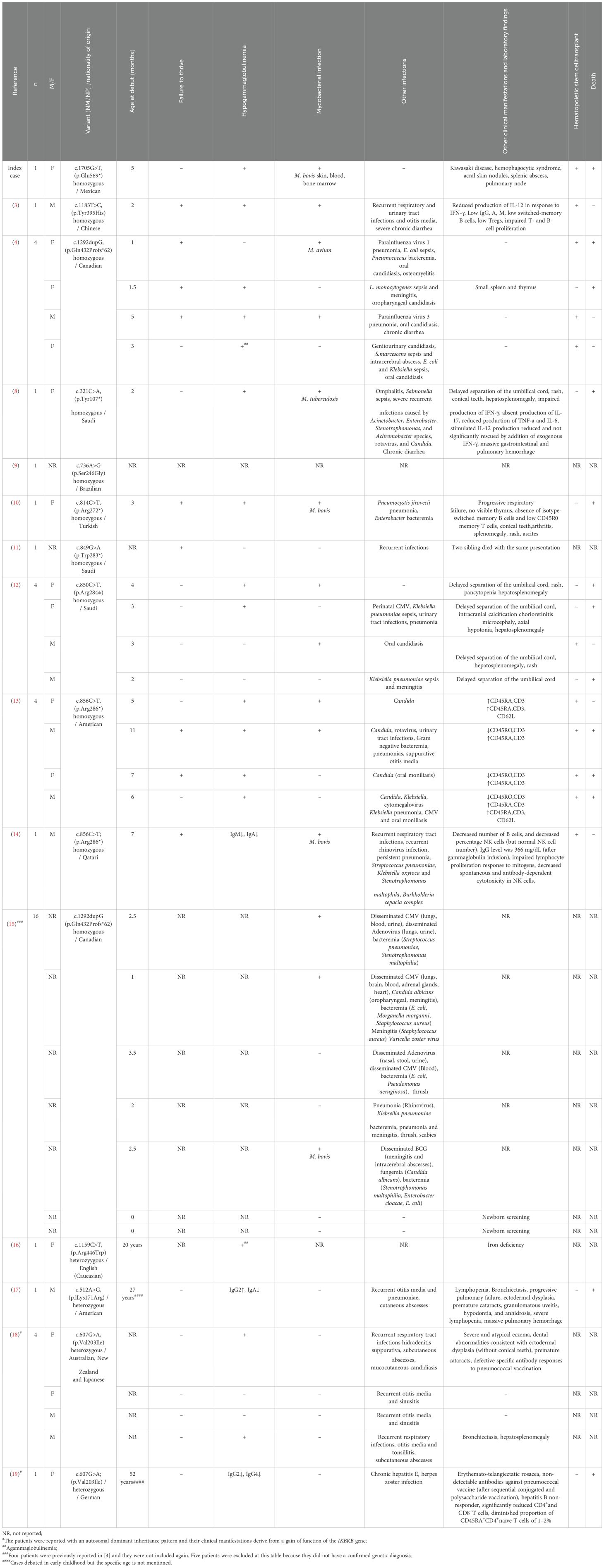
Table 1. Variants in the IKBKB gene are associated with disease.
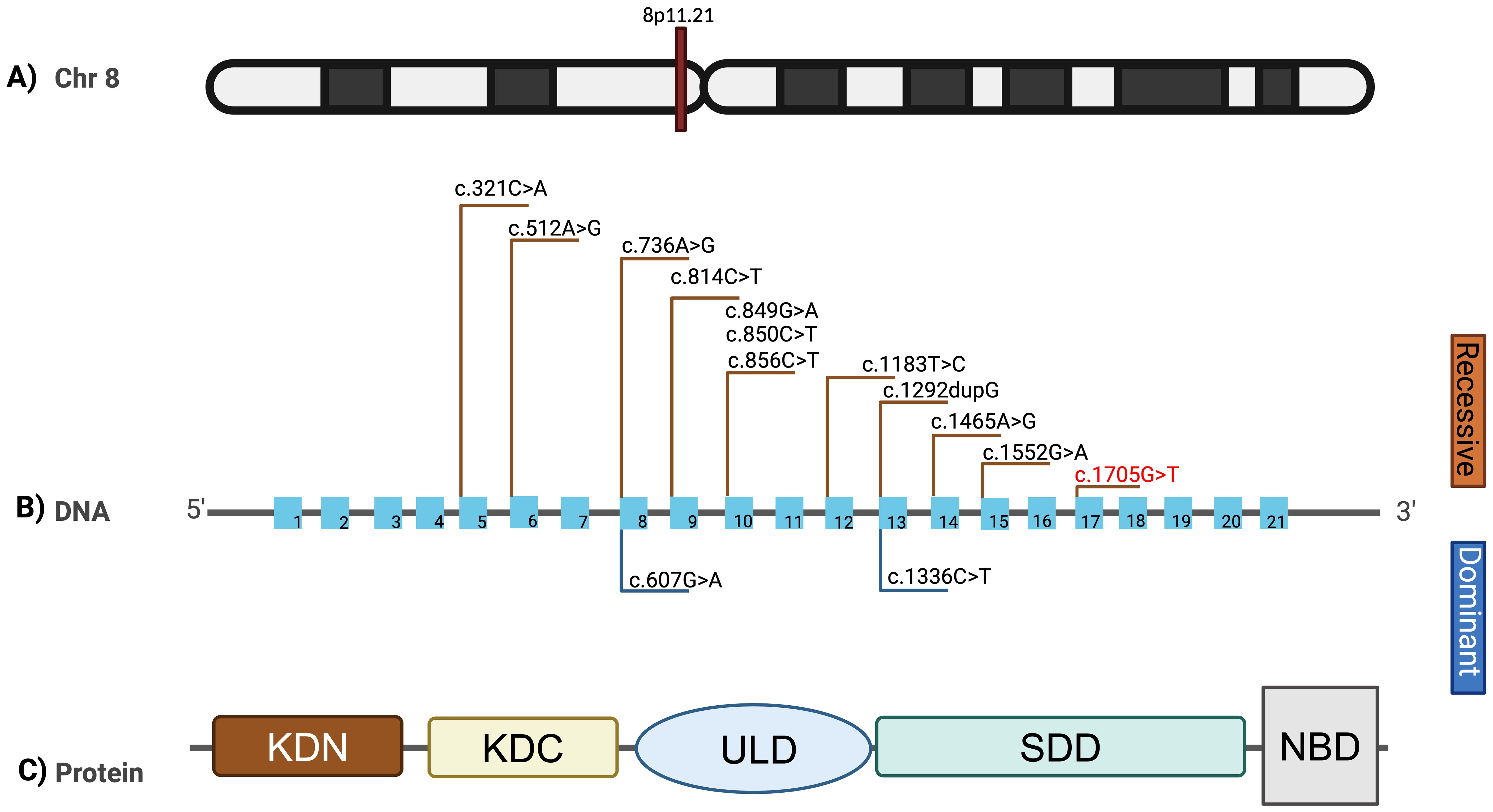
Figure 3. Mutational spectrum of the IKBKB gene. (A) Ideogram representing the 8p11.21 locus where the IKBKB gene is located. (B) Mutational spectrum of the variants reported in the IKBKB gene. The variants described with autosomal recessive inheritance are shown above, and the variant corresponding to this work is shown in red. The variants reported in a heterozygous state with an autosomal dominant inheritance pattern are shown below. (C) Structural representation of the domains of the IKBKB protein. N-terminal kinase domain (KDN), C-terminal kinase domain (KDC), ubiquitin-like domain (ULD), scaffold dimerization domain (SDD), and NEMO-binding domain (NBD). Created in BioRender. Contreras, (f) (2025) https://BioRendercom/x84m583.
The IKBKB gene encodes the IKKβ protein, a serine/threonine kinase that is a key component of the IκB kinase (IKK) complex, critical for activating the NF-κB signaling pathway. The IKKβ protein consists of several domains, each with specific functional roles:
● N-terminal kinase domain: Responsible for the phosphorylation of IκB proteins, leading to their degradation and subsequent activation of NF-κB.
● Helix-loop-helix domain: Involved in protein-protein interactions, particularly within the IKK complex.
● Leucine zipper domain: Facilitates dimerization and interaction with other components of the IKK complex.
● C-terminal NEMO-binding domain (NBD): Essential for binding to NEMO (IKKγ), stabilizing the IKK complex and enabling its activation.
In our patient, the identified variant according to Genome Reference Consortium Human Build 38 (GRCh38) (NM_001556.2) gene: c.1705G>T (p.Glu569*) results in a premature stop codon within the C-terminal region, truncating the protein and likely disrupting the NBD. This truncation would impair the assembly and function of the IKK complex, leading to defective NF-κB signaling and the observed severe immune dysregulation.
IKBKB deficiency is a rare immunophenotype characterized by severe combined immunodeficiency (SCID), usually evident in neonates with persistent respiratory or gastrointestinal infections caused by viruses, bacteria, or fungi, often associated with prolonged diarrhea and failure to thrive (3). In contrast, our patient did not present with the typical somatometric involvement seen in neonates. Instead, she debuted later, in infancy, with normal weight and height for her age, initially exhibiting incomplete Kawasaki disease, which later progressed to HLH. SCID was suspected since she came from an endogamous community and Mycobacterium bovis was isolated from skin, bone marrow and blood. Also, she had low levels of IgA and IgM and probably IgG, and the flow cytometry reported only that both double-negative T cells and memory B cells were decreased without isotype switching. This is similar to what has already been described in PV of the IKBKB gene, since these patients may present normal B-cell and T-cell counts and very low levels of immunoglobulins, as well as a severe defect in immune-cell activation that affects both innate and adaptive immune pathways (4). IKBKB deficiency induces abnormal IKKβ protein degradation, leading to impaired NF-kB signaling and immune function. In the absence of stimulatory signals the majority of inactive NF-κB is bound to IkBα and remains in the cell cytosol (3). The BCR and CD40 trigger the canonical NF-kB pathway via activation of the IkB kinase (IKK) complex, which comprises IKKα, IKKβ and NEMO (also known as IKKγ). IKK activation leads to the phosphorylation of IkBα and the subsequent release of the active p50–p65 NF-kB heterodimer, which then translocates to the nucleus to regulate gene transcription. So, impairment of the canonical NF-kB pathway leads to abnormal B cell activation and the patients with hypomorphic variants in IKBKB exhibit variable hypogammaglobulinemia (2).
The NF-kB transcription factors are key regulators of inflammatory and immune responses, mediating cell activation, proliferation, survival, and effector functions. The ubiquitously expressed IKK complex links these transcription factors to immune receptors, including T-cell and B-cell receptors, Toll-like receptors, and inflammatory cytokine (TNF- and interleukin [IL]-1ß) receptors (4) and certain variants in the IKBKB have been linked to increased susceptibility to autoimmunity and autoinflammation (5, 6). Dysregulation of the NF-κB pathway led to uncontrolled inflammatory responses in our patient, manifesting in conditions such as Kawasaki disease and HLH. In this regards, upon binding to its receptor, IFN-γ activates both the STAT1 and NF-κB pathways, leading to macrophage activation and enhanced IL-12 production, which is essential for eliminating mycobacteria (7). Impairment of the IFN-γ/IL-12 axis predisposes individuals to mycobacterial infections, aligning with previous findings (3).
Our case exhibited hypogammaglobulinemia and a mycobacterial infection, similar to several other cases associated with IKBKB PV. However, the isolation of mycobacteria from the skin, blood, and bone marrow in another patient has not been previously reported. Additionally, manifestations of immune dysregulation such as Kawasaki disease and hemophagocytic syndrome have not been documented in connection with this condition.
This case provides valuable insights into the spectrum of alterations in the IKBKB gene, including the identification of a novel PV. It also highlights the challenging realities faced by patients undergoing HSCT. Consistent with reports by Cuvelier, et al. (19), our patient developed septic shock just ten days post-transplantation and unfortunately succumbed to it. These findings illustrate the considerable vulnerability of HSCT recipients to severe infections, despite HSCT remaining the only curative treatment currently available.
In conclusion, we describe a novel homozygous PV in the IKBKB gene in our patient, with each parent carrying an affected allele. This finding indicates an autosomal recessive inheritance pattern and expands the mutational spectrum of the gene. Additionally, it confirms the presence of immunodeficiency 15B in our population, revealing clinical manifestations that have not been described until now, such as Kawasaki disease, hemophagocytic syndrome and disseminated tuberculosis affecting skin, blood and bone marrow.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
GA-E: Writing – original draft, Writing – review & editing. MR-M: Writing – original draft, Writing – review & editing. SS-M: Writing – original draft, Writing – review & editing, Investigation. MS-d-O: Conceptualization, Data curation, Formal analysis, Supervision, Writing – original draft, Writing – review & editing. LB-R: Writing – original draft, Writing – review & editing, Data curation, Resources. SE-P: Writing – original draft, Writing – review & editing. FC-V: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Supervision, Validation.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. (2008) 19:157–65. doi: 10.1016/j.cytogfr.2008.01.006
2. Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. (2013) 13:519–33. doi: 10.1038/nri3466
3. Qin T, Jia Y, Liu Y, Dai R, Zhou L, Okada S, et al. A novel homozygous mutation destabilizes IKKβ and leads to human combined immunodeficiency. Front Immunol. (2021) 11:517544. doi: 10.3389/fimmu.2020.517544
4. Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med. (2013) 369:2504–14. doi: 10.1056/NEJMoa1309199
5. Sacco K, Kuehn HS, Kawai T, Alsaati N, Smith L, Davila B, et al. A heterozygous gain-of-function variant in IKBKB associated with autoimmunity and autoinflammation. J Clin Immunol. (2023) 43:512–20. doi: 10.1007/s10875-022-01395-2
6. Sgrulletti M, Cifaldi C, Di Cesare S, Kroegler B, Del Duca E, Ferradini V, et al. Case Report: Crossing a rugged road in a primary immune regulatory disorder. Front Pediatr. (2023) 10:1055091. doi: 10.3389/fped.2022.1055091
7. Riazi S, Zeligs B, Yeager H, Peters SM, Benavides GA, Di Mita O, et al. Rapid diagnosis of Mycobacterium tuberculosis infection in children using interferon-gamma release assays (IGRAs). Allergy Asthma Proc. (2012) 33:217–26. doi: 10.2500/aap.2012.33.3574
8. Burns SO, Plagnol V, Gutierrez BM, Al Zahrani D, Curtis J, Gaspar M, et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKβ. J Allergy Clin Immunol. (2014) 134:215–8. doi: 10.1016/j.jaci.2013.12.1093
9. Al-Herz W, Chou J, Delmonte OM, Massaad MJ, Bainter W, Castagnoli R, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. (2019) 9:3146. doi: 10.3389/fimmu.2018.03146
10. Nielsen C, Jakobsen MA, Larsen MJ, Müller AC, Hansen S, Lillevang ST, et al. Immunodeficiency associated with a nonsense mutation of IKBKB. J Clin Immunol. (2014) 34:916–21. doi: 10.1007/s10875-014-0097-1
11. Al-Shamsi A, Hertecant JL, Souid AK, Al-Jasmi FA. Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet J Rare Dis. (2016) 11:94. doi: 10.1186/s13023-016-0474-3
12. Alsum Z, AlZahrani MS, Al-Mousa H, Alkhamis N, Alsalemi AA, Shamseldin HE, et al. Multiple family members with delayed cord separtion and combined immunodeficiency with novel mutation in IKBKB. Front Pediatr. (2020) 8:9. doi: 10.3389/fped.2020.00009
13. Mousallem T, Yang J, Urban TJ, Wang H, Adeli M, Parrott RE, et al. A nonsense mutation in IKBKB causes combined immunodeficiency. Blood. (2014) 124:2046–50. doi: 10.1182/blood-2014-04-571265
14. Taylor MG, Nicholas SK, Forbes Satter LR, Martinez C, Cameron LH. Plasma metagenomic sequencing expedites diagnosis of disseminated BCG in an infant with IKBKB mutation. Pediatr Infect Dis J. (2022) 41:430–5. doi: 10.1097/INF.0000000000003465
15. Cuvelier GDE, Rubin TS, Junker A, Sinha R, Rosenberg AM, Wall DA, et al. Clinical presentation, immunologic features, and hematopoietic stem cell transplant outcomes for IKBKB immune deficiency. Clin Immunol. (2019) 205:138–47. doi: 10.1016/j.clim.2018.10.019
16. van Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol. (2015) 160:301–14. doi: 10.1016/j.clim.2015.05.020
17. Abbott J, Ehler AC, Jayaraman D, Reynolds PR, Otsu K, Manka L, et al. Heterozygous IKKβ activation loop mutation results in a complex immunodeficiency syndrome. J Allergy Clin Immunol. (2021) 147:737–40. doi: 10.1016/j.jaci.2020.06.007
18. Cardinez C, Miraghazadeh B, Tanita K, da Silva E, Hoshino A, Okada S, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. (2018) 215:2715–24. doi: 10.1084/jem.20180639
Keywords: IKBKB, disseminated tuberculosis, splenic abscesses, acral skin nodules, hemophagocytic syndrome
Citation: Arce-Estrada GE, Rodríguez-Morales M, Scheffler-Mendoza SC, Sáez-de-Ocariz M, Berrón-Ruiz L, Espinosa-Padilla SE and Contreras-Verduzco FA (2025) Case Report: A novel IKBKB variant (c.1705G>T) is associated with immune dysregulation and disseminated tuberculosis. Front. Immunol. 16:1541899. doi: 10.3389/fimmu.2025.1541899
Received: 09 December 2024; Accepted: 14 February 2025;
Published: 07 March 2025.
Edited by:
Maria Cecilia Poli, Universidad del Desarrollo, ChileReviewed by:
Kunihiko Moriya, National Defense Medical College, JapanCopyright © 2025 Arce-Estrada, Rodríguez-Morales, Scheffler-Mendoza, Sáez-de-Ocariz, Berrón-Ruiz, Espinosa-Padilla and Contreras-Verduzco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Elva Espinosa-Padilla, c2FyYWVsdmFlc3Bpbm9AZ21haWwuY29t; Francisco Alberto Contreras-Verduzco, ZHIuZmFjdkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.