
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 February 2025
Sec. T Cell Biology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1539265
 Bo Huang1
Bo Huang1 Suilan Zheng1
Suilan Zheng1 Kasireddy Sudarshan1
Kasireddy Sudarshan1 Ramesh Mukkamala1
Ramesh Mukkamala1 Madduri Srinivasarao1
Madduri Srinivasarao1 Tushar Sardesai1
Tushar Sardesai1 Xiaofei Yang1
Xiaofei Yang1 Haiyan Chu1,2
Haiyan Chu1,2 Philip S. Low1*
Philip S. Low1*CAR T cells therapies have demonstrated success in treating hematologic malignancies, but have proven less effective in eradicating solid tumors. While suppressive immune cells may contribute to reduced CAR T cell efficacies in malignant masses, cancer-associated fibroblasts (CAFs) are also believed to facilitate tumor survival by secreting growth factors, immunosuppressive cytokines, and extracellular matrix components that inhibit drug and immune cell filtration and facilitate metastasis. In an effort to eliminate both CAFs and cancer cells simultaneously, we have employed a universal CAR T cell that can attack both cell types when supplemented with appropriate bispecific adapters. We show here that tumor regression is indeed enhanced when CAR T cells are directed to concurrently kill both cancer cells and CAFs. We further demonstrate that simultaneous targeting of both cell types enhances CAR T cell proliferation, activation, tumor infiltration, and tumor distribution relative to targeting only a single cell type. Because all of these benefits are achieved in both cold and hot tumors without significant toxicity, we conclude that use of a universal CAR T cell in combination with multiple bispecific adapters can provide a safe, potent, cost-effective, and scalable alternative to the treatment of solid tumors with conventional CAR T cells.
Although chimeric antigen receptor (CAR) T cell therapies have proven effective in treating liquid tumors (1–4), their potencies in eradicating solid tumors have been less impressive (5–8). Reasons for this reduced potency have included poor CAR T cell penetration into tumor masses (5, 9), exhaustion of CAR T cells induced by chronic antigen exposure (10–12), selection for cancer cells that lack the targeted antigen (13–15), and/or an immunosuppressive tumor microenvironment generated by tumor stromal cells (16–18). Cancer-associated fibroblasts (CAFs) are believed to contribute to this CAR T cell inactivation by releasing immunosuppressive cytokines such as IL-10 and TGF-β (19), secreting tumor-stimulating growth factors including fibroblast growth factors, vascular endothelial growth factors, and hepatocyte growth factor (19–22), and depositing extracellular matrix components such as collagen and fibronectin that can create an impermeable barrier to entry of immune cells and therapeutic agents, and also facilitate cancer metastasis (19, 21, 23). Not surprisingly, CAF contents in solid tumors correlate inversely with overall survival (19, 24, 25).
Strategies to reduce the number of CAFs in solid tumors have exploited the upregulation of fibroblast activation protein (FAP), a cell surface serine protease, primarily on CAFs in solid tumors (26, 27). In these approaches, the over-expressed FAP has been targeted with cytotoxic drugs (28–31), radioligand therapies (27, 32–35), CAR T cell therapies (36–43), and/or bispecific T cell engagers (44–46). While each of these therapies may enhance tumor regression, many have suffered from insufficient potency or dose-limiting toxicity that has prevented their adoption in the clinic (42, 43). Although infusion of a second CAR T cell directed against CAFs has recently yielded encouraging results (36–38), this approach has required the engineering and expansion of a second CAR T cell preparation from the same patient that may render widespread adoption more difficult (47–49).
As a remedy to this problem, we have explored the use of a universal CAR T cell in which the CAR is comprised of a single chain variable fragment (scFv) that binds fluorescein rather than a tumor antigen (50–52). In this approach, the anti-fluorescein CAR T cell is induced to engage the cancer cell by injection of a bispecific adapter comprised of fluorescein linked to a low molecular weight tumor-targeting ligand that can bridge between the CAR T cell and cancer cell (Figure 1A). Formation of this bridge triggers killing of the cancer cell by the CAR T cell along with the subsequent proliferation of the CAR T cell. While the flexibility of this approach has enabled the use of two different bispecific adapters to force engagement of a universal CAR T cell with two antigenically orthogonal cancer cells in the same tumor mass (52), the approach has never been explored for its ability to enable simultaneously killing of both cancer cells and stromal cells. In the study below, we explore the ability of this universal CAR T cell to concurrently eliminate cancer cells and CAFs from the same tumor mass.
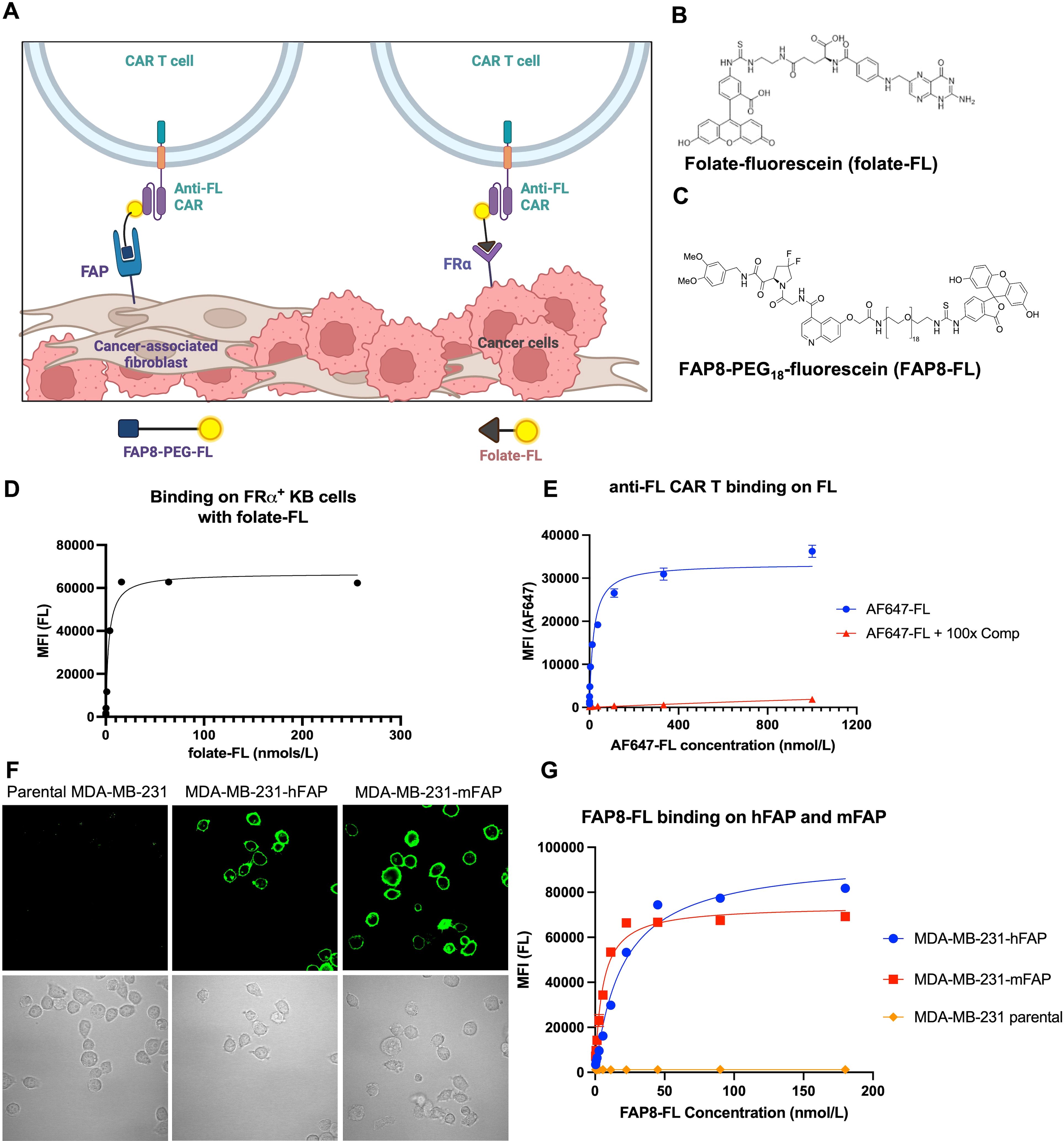
Figure 1. Development of a FAP8-FL bispecific adapter. (A) Sketch depicting the use of the same universal CAR T cell to engage both cancer cells and CAFs in a solid tumor. Created with BioRender.com. (B) Structure of folate-FL. (C) Structure of FAP8-FL. (D) Binding affinity of folate-FL for FRα-expressing KB cancer cells. (E) Binding affinity of AlexaFluor647-fluorescein conjugate to anti-FL CAR T cells in the absence (AF647-FL) or presence (AF647-FL + comp) of 100-fold molar excess of fluorescein. (F) Binding of FAP8-FL to unmodified MDA-MB-231 cells (left panel) or MDA-MB-231 cells transduced with human FAP (middle panel) or murine FAP (right panel), and (G) binding analysis of FAP8-FL to these same cells determined by flow cytometry. Mean ± SD, n=3.
For engagement of the cancer cells, we exploit folate receptor alpha (FRα) that is over-expressed on ~40% of human cancers (53) and induce cancer cell engagement with the CAR T cells by injection of a folate-fluorescein (folate-FL) bispecific adapter. For killing of the CAFs we employ an analogous FAP8-fluorescein (FAP8-FL) bispecific adapter that similarly bridges between CAFs and the CAR T cells. Importantly, concurrent administration of both bispecific adapters is found to significantly enhance tumor regression relative to injection of either adapter alone, i.e. demonstrating that the universal CAR T cell has the ability to kill both cells types concurrently and that elimination of CAFs enhances CAR T cell killing of cancer cells.
In order to evaluate the ability of a single anti-fluorescein (anti-FL) CAR T cell to kill both cancer cells and cancer-associated fibroblasts (CAFs) in the same tumor mass, we required two different bispecific adapters that would mediate engagement of our universal CAR T cell with each cell type. For CAR T cell killing of the folate receptor (FRα) over-expressing cancer cells, we employed the folate-fluorescein (folate-FL) bispecific adapter shown in (Figure 1B; Supplementary Figure S1), (50, 51, 54). Binding of this adapter to both FRα on the cancer cells and the anti-FL CAR on the CAR T cells was found to occur with high affinity, i.e. dissociation constants of ~3 nM and ~20 nM, respectively (Figures 1D, E).
A similar bispecific adapter to tether the same anti-FL CAR T cells to CAFs was then synthesized by attaching a fibroblast activation protein (FAP) ligand [FAP8 (55)] to fluorescein via a 54 atom PEG18 spacer (Figure 1C). This extended spacer was selected because shorter spacers were unable to mediate bridging of the CAR T cell to a FAP-expressing cell, and significantly longer spacers were found to display reduced affinities. Binding of FAP8-fluorescein (FAP8-FL) to cells transduced to express either human or murine FAP displayed strong cell surface localization (Figure 1F) characterized by dissociation constants of 20 nM for human FAP and 5 nM for murine FAP (Figure 1G). Since no binding to cells lacking FAP could be detected, we concluded that binding of this bispecific adapter was FAP specific.
To determine whether FAP8-FL might mediate CAR T cell killing of FAP-expressing cells in vitro, we incubated several FAP-expressing cell lines with both our universal CAR T cell and our FAP-linked bispecific adapter (Supplementary Figures S2, S3), with the relative FAP expression of these cell lines determined by anti-FAP antibodies (Supplementary Figure S4). As shown in Figure 2, FAP8-FL readily induced anti-FL CAR T cell killing of a human fibroblast cell line (Figure 2A), a mouse fibroblast cell line (Figure 2B), and MDA-MB-231 cells transduced to express human FAP (Figure 2C). Moreover, as FAP8-FL concentration was increased, cell killing first increased and then decreased in the predictable bell-shaped manner (i.e. due to eventual saturation of both fluorescein and FAP binding sites with different bispecific adapters). Since maximum killing potency was observed at ~1 nM adapter and since killing was always accompanied by release of IFNγ, we conclude that FAP8-FL mediates CAR T cell activation and the consequent killing of FAP-expressing cells with high potency. Because FAP-negative (nontransduced) MDA-MB-231 cells were not killed upon addition of FAP8-FL (Figure 2D), the data further demonstrate that killing was FAP-specific.
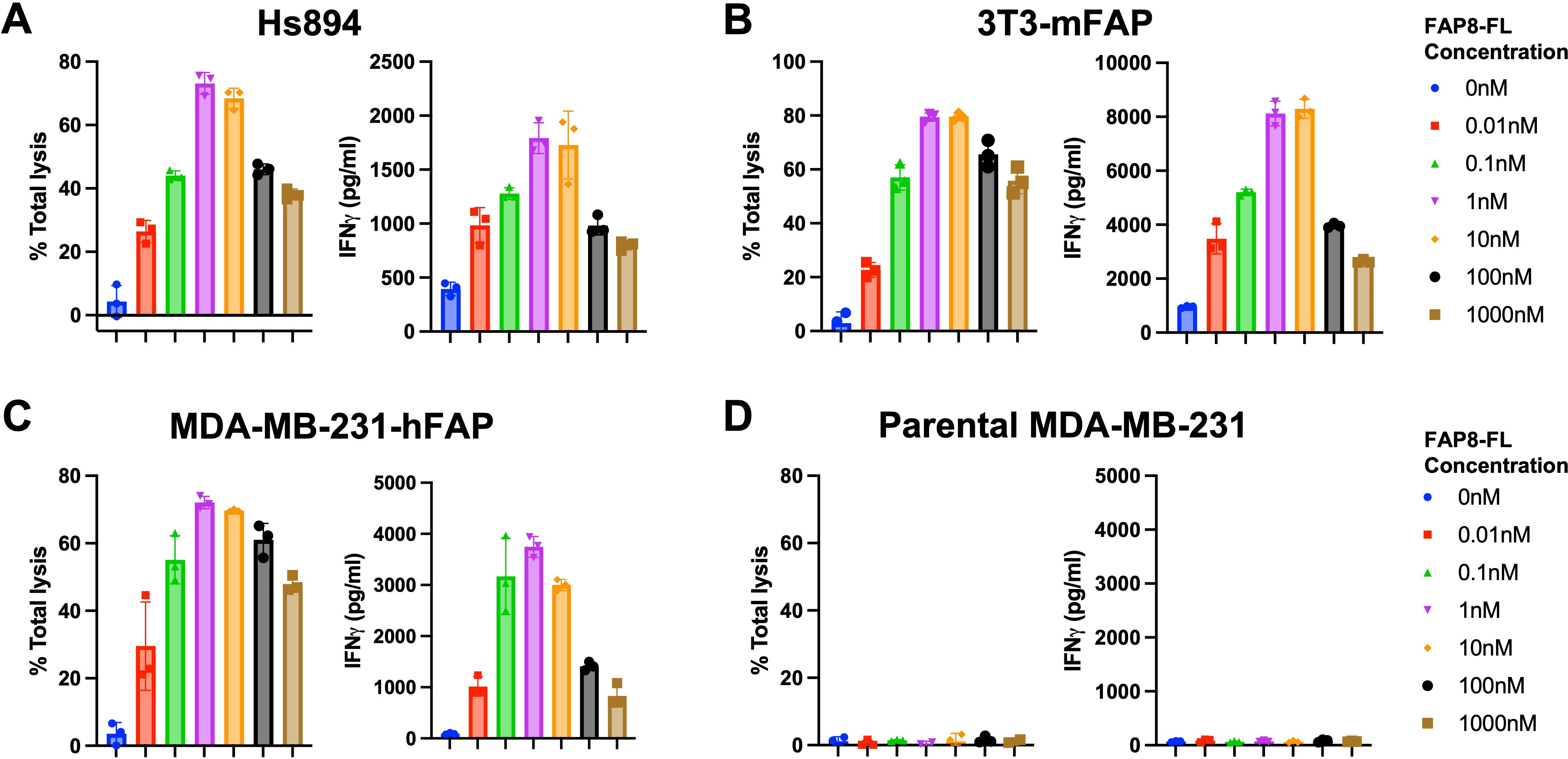
Figure 2. Evaluation of FAP8-FL mediated anti-FL CAR T cell engagement of different FAP-expressing cell lines. Percent cell lysis (left) and IFNγ release (right) were measured in a coculture killing assay in which the target cells were (A) Hs894 human CAFs, (B) 3T3 murine fibroblasts transduced with mouse FAP, (C) MDA-MB-231 cells transduced with human FAP, or (D) parental MDA-MB-231 with no FAP. Mean ± SD, n=3.
To evaluate whether the anti-FL CAR T cells might be capable of simultaneously eradicating both cancer cells and CAFs in the same tumor mass, we co-injected NSG mice with both unmodified (parental MDA-MB-231) cells that express no FAP and human Hs894 fibroblasts that naturally express FAP (Figure 3A) based on similar approach by others to build a physiologically relevant CAFs-containing human tumor in mice (36). After allowing the tumors to reach ~250 mm3, mice were injected intravenously with either CAR T cells alone or CAR T cells in combination FAP8-FL, folate-FL, or both adapters together. Folate-FL was dosed at once per week based on previously optimized dosing frequency (50), and FAP8-FL was dosed with at least one day gap to allow excess adapter excrete out of mouse bloodstream. As seen in Figure 3B, administration of FAP8-FL alone induced a small but statistically significant decrease in tumor growth, suggesting that elimination of fibroblasts may suppress tumor growth by killing the fibroblasts. Injection of folate-FL, in contrast, promoted a more profound reduction in tumor size, arguing that direct CAR T cell attack on the FRα-expressing MDA-MB-231 cells more effectively inhibited tumor growth. Importantly, concurrent treatment with both bispecific adapters induced nearly complete tumor eradication, demonstrating that diversion of some universal CAR T cells to engage FAP-expressing stromal cells (i.e. CAFs) not only did not hinder but actually augmented their abilities to inhibit tumor growth. Moreover, because tumor suppression could be achieved with no weight loss (Figure 3C), the data suggest that any FAP-expressing fibroblasts that might have been killed in healthy tissues must have occurred with little overt toxicity.
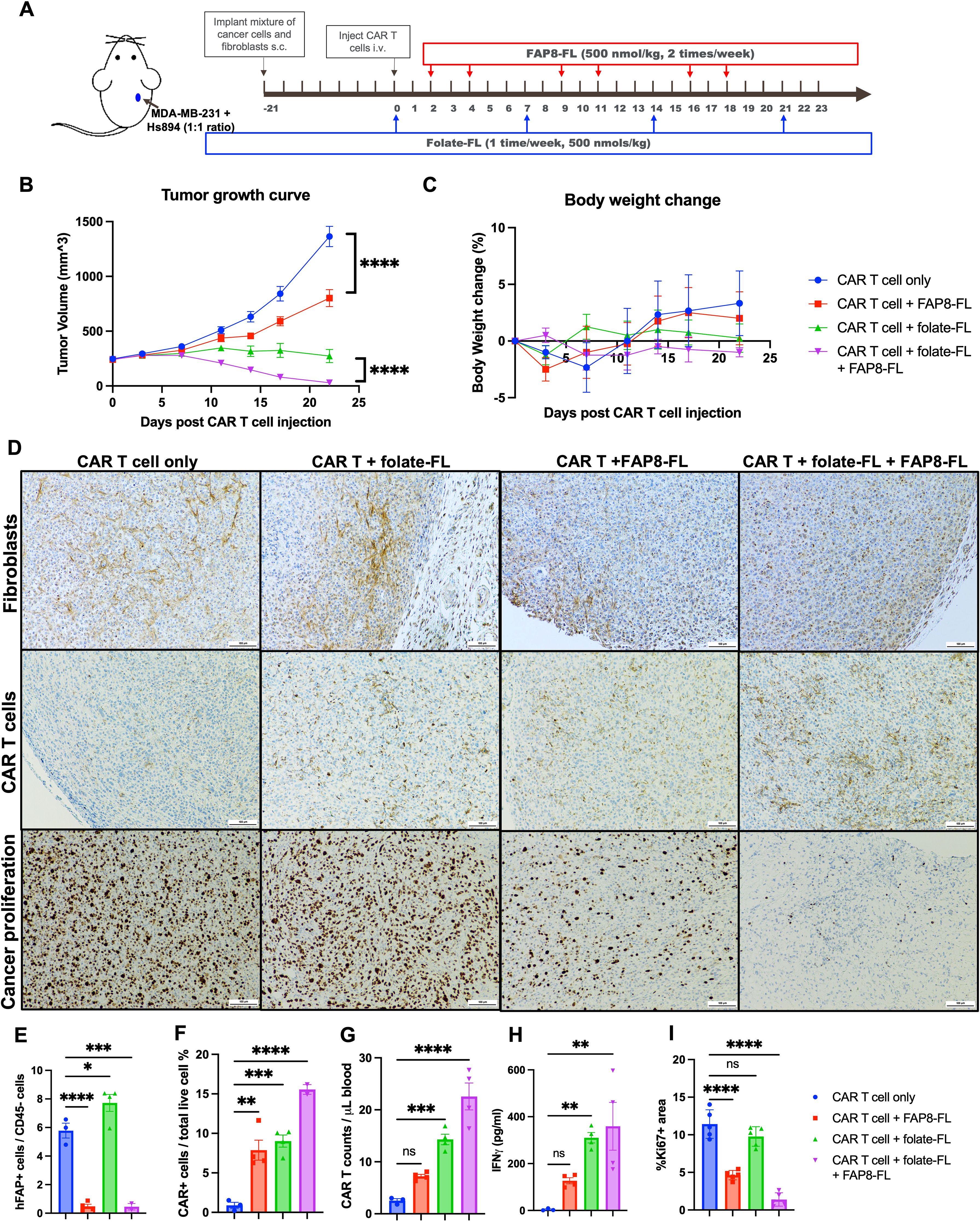
Figure 3. Evaluation of the ability of the same anti-FL CAR T cells to eradicate both cancer cells and CAFs in the same tumor mass and its impact on solid tumor. (A) Tumors were created by subcutaneous (s.c.) injection of a mixture of parental FRα+ MDA-MB-231 cells and Hs894 fibroblasts and allowed to grow to ~250 mm3 prior to injection of 3 million CAR T cells per mouse and dosing with bispecific adapters, as indicated. (B) Evaluation of tumor growth versus time in the different treatment groups. (C) Mouse body weights in the same study. (D) IHC staining of representative tumor sections obtained from the same treatment groups on day 23 for fibroblasts (murine FAP), human CAR T cells (human CD3), and cancer proliferation (Ki67). Tumors and peripheral blood samples collected from mice were quantitated for (E) residual Hs894 human FAP-expressing fibroblasts in the tumors (flow cytometry of digested tumors), (F) human CAR T cells in the same tumors (flow cytometry of digested tumors), (G) CAR T cells in the blood (flow cytometry), (H) IFNγ as a measure of CAR T cell activation (ELISA quantitation in mouse blood), and (I) Ki67 in the same tumors [ImageJ quantitation of stained sections (60) in panel (D)]. Mean ± SEM, n=3 in CAR T cell only group and n=4 in all treatment groups. Statistical significance between groups was determined using two-way ANOVA in (B) (****P < 0.0001) and using one-way ANOVA (*P < 0.05, **P<0.01, ***P < 0.001, ****P < 0.0001) in (E-I).
To obtain a more mechanistic understanding of the processes that might have contributed to the improved efficacy in the presence of both adapters, we analyzed the changes in CAR T cell and fibroblast abundances in all treatment groups by both immunohistochemistry of tumor sections and flow cytometry of isolated tumor cells (see Methods). As shown in Figure 3D, fibroblasts remained abundant in tumors from mice treated with either CAR T cells alone or CAR T cells plus folate-FL, but were largely eliminated from tumors in both cohorts treated with the FAP8-FL adapter. These data thus confirm that treatment with CAR T cells + FAP8-FL kills CAFs in solid tumors, and this conclusion is further supported by the flow cytometry data in Figure 3E; Supplementary Figure S5.
Comparison of CAR T cell abundances in the different treatment groups then revealed that administration of CAR T cells plus FAP8-FL increased CAR T cell proliferation and infiltration relative to cohorts not treated with the FAP8-FL adapter. Thus, CAR T cell numbers in the tumors (Figure 3F; Supplementary Figure S6) and peripheral blood (Figure 3G; Supplementary Figure S7) were both increased in mice treated with CAR T cells + FAP8-FL relative to CAR T cells alone. CAR T cells were also elevated in mice treated with CAR T cells + FAP8-FL + folate-FL relative to mice treated with CAR T cells + folate-FL alone. Taken together these data establish that co-administration of FAP8-FL adapter with folate-FL adapter significantly augments infiltration of the CAR T cells into tumor masses. The data also argue that any competition between the FAP8-FL and folate-FL for binding to the anti-FL CAR must be minimal, since tumor eradication (Figure 3A), CAR T cell infiltration (Figure 3F), and IFNγ release (Figure 3H) all improved by concurrent administration of the two adapters. In fact, the data on IFNγ release may be an underestimate of the true impact of dual adapter administration, since the tumors had already been eradicated in the two mice with low IFNγ levels, suggesting that their IFNγ levels may have already been returning to normal when their peripheral blood samples were collected.
To assess the impact of CAF elimination on cancer cell proliferation, we next quantitated Ki67 staining of multiple tumor sections from the same treatment groups using ImageJ analysis. As shown in Figure 3I, injection of FAP8-FL adapter invariably reduced cancer cell proliferation relative to similar treatment groups lacking FAP8-FL. Although already reported by others (38, 40), these data confirm that CAFs can promote cancer cell proliferation, presumably by their release of growth factors (19–22), and that elimination of CAFs thereby suppresses tumor growth.
Next, because both endogenous immune cells and exogenous CAR T cells are frequently excluded from immunologically “cold” tumors, at least in part due to the “barriers” created by accumulation of CAFs near the tumor periphery (19, 38, 40), the question arose whether a universal CAR T cell in combination with both a CAF- and cancer cell-killing bispecific adapter might more effectively penetrate and destroy these resistant tumors than a CAR T cell capable of killing only one tumor cell type. Although MDA-MB-231 tumors have been shown to be readily eradicated by our universal CAR T cells in combination with folate-FL adapters, similarly treated KB tumors have been found to be extremely resistant (12). To obtain an initial indication of whether this difference in CAR T cell responsiveness might have derived from a difference in CAF content/distribution, we first stained both KB and MDA-MB-231 tumors for CAR T cells and CAFs to evaluate their distributions and abundances when treated with CAR T cells plus folate-FL adapter. As shown in Supplementary Figure 8, CAR T cells are highly enriched and uniformly distributed in the MDA-MB-231 tumors, but few in number and concentrated at the periphery in KB tumors. Moreover, consistent with previous observations of immunologically cold tumors (12, 38, 40), CAFs are more abundant and peripherally confined in KB tumors (Supplementary Figure S8), but less numerous and more evenly distributed in MDA-MB-231 tumors. Based on suggestions by others that this barrier-like distribution of fibroblasts restricting CAR T cell penetration defines an immunologically cold tumor (19, 21, 38), we elected to test the ability of our universal CAR T cells to concurrently eradicate both cancer cells and CAFs in these cold KB tumors (Figure 4A). As shown in Figure 4B, neither the CAR T cells plus folate-FL (targeting the cancer cells) nor CAR T cells plus FAP8-FL (targeting the CAFs) alone exerted an impact on KB tumor growth, i.e. consistent with their immunologically cold nature. However, the combination of CAR T cells with both bispecific adapters promoted an ~50% suppression of tumor growth. The fact that co-administration of both adapters also induced the largest IFNγ release (Figure 4D) and proliferation of CAR T cells (Figures 4E, F) further emphasized the benefit that the two bispecific adapters provided in treating the cold KB tumors. Because body weights throughout the study (Figure 4C) increased, regardless of the adapter combination used, we surmise that all combinations analyzed were also largely nontoxic.
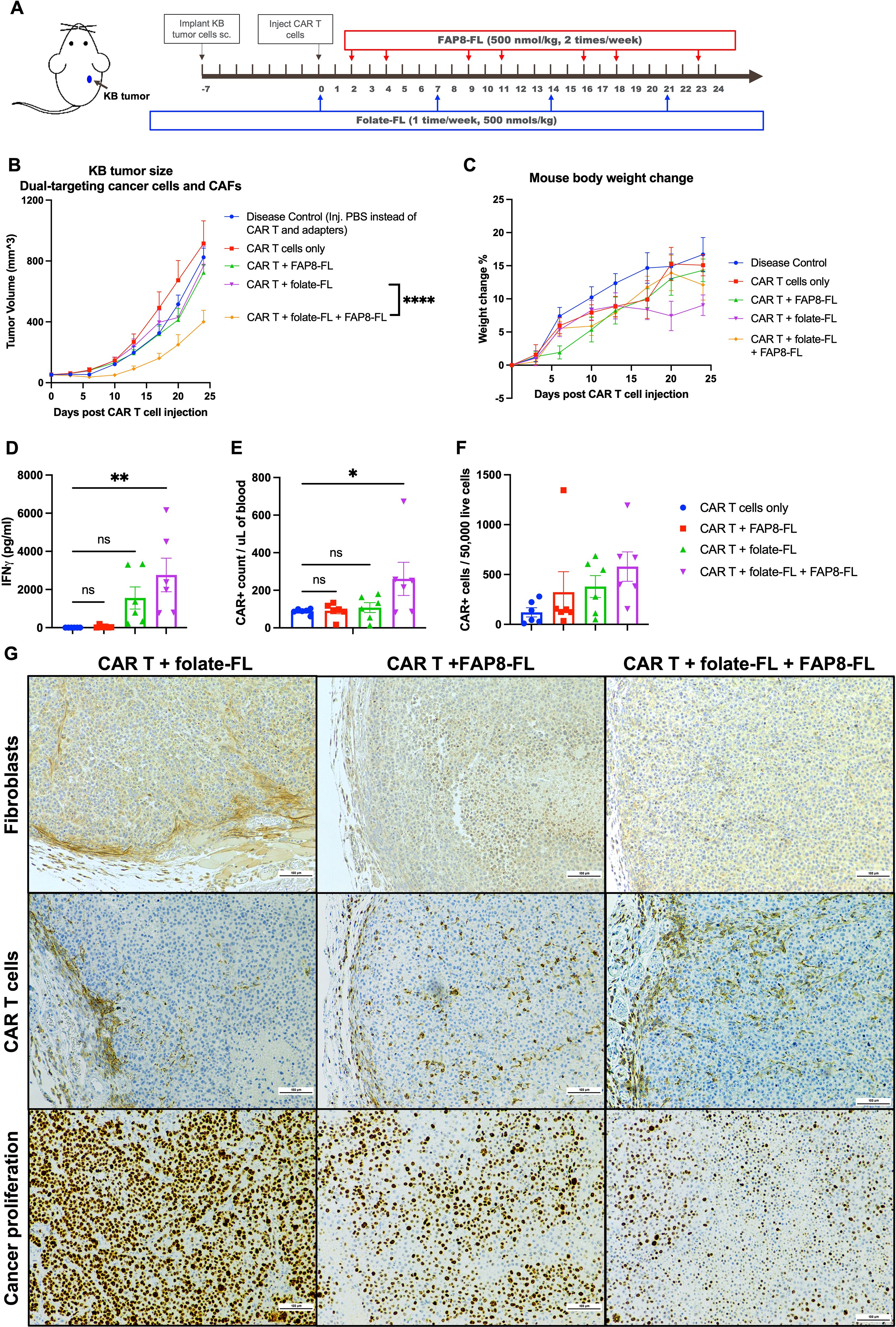
Figure 4. Evaluation of the abilities of the same anti-FL CAR T cells to target both cancer cells and CAFs in the same immunologically cold KB tumors. (A) KB tumors were implanted by s.c. injection of cultured KB cells into NSG mice and allowed to grow to ~50 mm3 prior to injection of 8 million CAR T cells per mouse and dosing with bispecific adapters, as indicated. (B) Evaluation of tumor growth versus time. (C) Evaluation of body weight changes versus time. (D) Blood CAR T cell counts were quantitated by flow cytometry, and (E) peripheral blood IFNγ concentrations were determined by ELISA at the study endpoint as a measure of T cell activation. (F) CAR T cell infiltration into the tumors was assessed by flow cytometry of dissociated tumors. (G) Distributions of fibroblasts (stained with anti-mouse FAP antibody), CAR T cells (anti-human CD3 antibody), and proliferating cancer cells (Ki67 antibody) in tumor sections were determined by IHC staining. Mean ± SEM, n=6, statistical significance between groups (B) was determined using two-way ANOVA (****P < 0.0001), and between groups (D, E) was determined using one-way ANOVA (*P < 0.05, **P<0.01).
To determine whether the peripherally confined distribution of CAR T cells and CAFs characteristic of cold KB tumors may have been at least partially disrupted by the above therapy, we next stained sections from all of the tumors for CAFs and CAR T cells (Figure 4G). As anticipated, CAR T cells were largely limited to tumor boundaries in the untreated specimens, but encouragingly distributed throughout the tumors in mice treated with either FAP8-FL alone or FAP8-FL + folate-FL. Since CAFs were largely absent in both of these latter treatment cohorts, it could be concluded that elimination of CAFs can permit greater penetration of CAR T cells into solid KB tumors, i.e. perhaps by removing the physical barriers created by the CAFs. However, the fact that CAR T cell infiltration is even more improved by simultaneous targeting of both tumor cells and CAFs further suggests that the KB cells may also contribute to the confinement of CAR T cells to the tumor periphery.
Finally, since CAFs secrete growth factors that promote tumor growth (19–22), we decided to assess the impact of FAP8-FL-directed CAR T cell treatment on cancer cell proliferation. As shown in the lower panel of Figure 4G, a remarkable reduction in the cell proliferation marker (Ki67) was seen upon targeting of the CAFs alone and this was further suppressed when both bispecific adapters were administered, i.e. confirming that CAFs indeed contribute to cancer cell proliferation and that dual targeting of both CAFs and cancer cells is more effective than targeting either cell type alone.
The above studies establish a therapeutic benefit associated with employing the same CAR T cell to concurrently attack both cancer cells and CAFs. Not only was an immunologically hot tumor more efficiently destroyed in the presence of both bispecific adapters, but an immunologically cold tumor was also successfully suppressed under conditions where neither adapter alone achieved a response. Because tasking the universal CAR T cell with two concurrent killing assignments did not detract from its ability to perform either task independently, we conclude that the CAR T cell still had untapped capacity that could be successful redirected to another cell type. Whether administration of still other bispecific adapters for elimination of other unwanted cells (e.g regulatory T cells, myeloid derived suppressor cells, tumor-associated macrophages, etc.) could have further enhanced therapeutic outcomes was not explored, but this possibility should not be considered unreasonable, since ~100,000 anti-fluorescein CARs are expressed per CAR T cell and only a small number of bridges between a CAR T cell and its target cell are required to induce killing (56). While production of multiple classical CAR T cells dedicated to different tumor cell types could have conceivably achieved the same objectives, their production would have required additional resources, and their activation and proliferation would have only been stimulated by independent engagement of each CAR T cell’s specific target (i.e. not by either CAFs and cancer cells as seen in Figure 3F). Thus, use of a universal CAR T cell in combination with multiple adapters can be argued to offer a safe, cost-effective and scalable alternative to the production of multiple CAR T cells. Moreover, if administration of still additional bispecific adapters would allow elimination of still other unwanted stromal cells or perhaps even cancer cells that have downregulated their targeted antigens (52), even more advantages associated with use of a universal CAR T cell would accrue. A potential obstacle to translation of the above CAR T cell technology into humans, however, should also be mentioned. The CAR T cells employed here were first prepared using human T cells and then tested in NSG immunocompromised mice. Although all CAR T cell therapies approved for use in humans to date have followed a similar development path, it will still be important to validate the above results in immune competent animals to assure that immunogenicity issues do arise when universal CAR T cell strategy is translated into humans.
Although multiple distinct subtypes of fibroblasts have been described in the literature, only the myofibroblast subtype has been shown to express FAP (57, 58). Because induction of this FAP is primarily stimulated by TGFβ, which is in turn released in response to tissue damage or stress (e.g. within a wound or tumor), FAP-expressing fibroblasts are primarily observed in traumatized and malignant tissues, where they are thought to stimulate proliferation and regeneration (19–22). Since their contributions to tissue growth may be important to wound healing, the question naturally arises whether a patient recovering from tissue trauma might be retarded in this recovery by treatment with a FAP-targeted CAR T cell therapy. While such risks have not yet been investigated, we suspect that CAR T cell treatment of a cancer may in some cases have to be delayed to allow adequate healing of the concomitant wound.
While the universal CAR T cell approach may come with some advantages, it may also introduce complexities into the clinical application of the therapies. Thus, as noted in Figure 2, the concentration dependence of bispecific adapter-mediated killing follows a bell-shaped curve, where target cell engagement/killing first increases and then decreases with increasing adapter concentration. While the plateau of maximum potency is generally seen to extend over an adapter concentration range of two orders of magnitude, the ideal concentration may still not be the same for all patients. And although this complexity may at first appear undesirable, closer scrutiny should reveal that it could also become advantageous, since it would permit sensitive control of the therapy for each patient. Thus, simply lowering the bispecific adapter concentration has been shown to prevent a cytokine storm (50), and decreasing adapter dosing frequency has been similarly documented to avoid CAR T cell exhaustion (10–12, 50). When considered together with the user’s ability to independently adjust the composition of adapters to address each tumor’s cancer and stromal cell composition, these complexities could become tools for personalization. Thus, with the expanding data sets expected to emerge from treatment of increasingly more patients, artificial intelligence methods should enable an informed selection of an optimal adapter composition, concentration, and dosing frequency for each patient, thereby allowing each patient to be treated with a highly personalized therapy.
Hs894 cells (Cat# CRL-7631_FL), 3T3 cells (Cat# CRL-1658), and MDA-MB-231 cells (Cat# HTB-26), KB cells (Cat# CRL-3596) were purchased from ATCC. Hs894 and 3T3 cells were cultured in DMEM medium (Gibco, Cat# 11995073), while MDA-MB-231 and KB cells were cultured in folate-free RPMI 1640 medium (Gibco, Cat# 27016021), all containing 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin in 5% CO2 at 37°C.
Human peripheral blood mononuclear cells (PBMCs) were isolated from fresh peripheral blood samples obtained from healthy donors using Ficoll (GE Healthcare Lifesciences) density gradient centrifugation. CD3+ T cells were then isolated with an EasySep Human T-Cell Isolation Kit (STEMCELL Technologies) and cultured in TexMACS medium (Miltenyi Biotec) supplemented with 1% penicillin-streptomycin, 5% human serum (Valley Biomedical), and human IL-2 (100 IU/mL, Miltenyi Biotec). T cells were count and maintained at 0.5 × 10^6 cells/mL. Human anti-fluorescein CAR T cells were generated using a lentiviral vector protocol as previously described (12, 50–52).
Six- to eight-week-old female NSG mice were purchased from Jackson Laboratory (Stain# 005557) and used for all live animal studies. Mice were housed in accordance with protocols approved by Purdue University Animal Care and Use Committee. Water and folate-deficient chow (Envigo, Cat#TD.00434) were freely available.
Mice were subcutaneously injected with MDA-MB-231 + Hs894 cell mixtures (3 million cells each/mouse), or KB cells (1 million cells/mouse), and tumor sizes were measured every other day. Mice were sacrificed when control tumors reached ~1500mm3 for MDA-MB-231 + Hs894 tumors, and 900 mm3 for KB tumors. Part of each tumor was then dissociated with tumor dissociation kit (Miltenyi, Cat# 130-095-929) and erythrocytes were depleted using RBC lysis buffer. After two washes with cold PBS, the resulting single cell suspensions were analyzed by flow cytometry for the desired phenotypic markers (Supplementary Table S1). The remaining portion of each tumor was fixed in 4% formalin, followed by storage in 70% ethanol. Subsequent paraffin and immunohistochemistry staining was performed by the Purdue Histology Research Laboratory.
For analysis of CAR T cells from the peripheral blood of treated mice, peripheral blood was collected by cardiac puncture prior to washing 3x in PBS. Erythrocytes in the resulting cell suspensions were then lysed using RBC lysis buffer and residual cells were washed 2x in PBS. The washed PBMCs were then stained with the desired antibodies and analyzed by flow cytometry (Supplementary Table S1) for quantitation of CAR T cell numbers in the blood.
Single cell suspensions obtained as described from murine blood or tumor samples were first stained with Zombie Violet (BioLegend, Cat#423114) and then washed 2x with PBS prior to incubation with anti-mouse TruStain FcX™ (BioLegend, Cat#101319) and anti-human TruStain FcX™ (BioLegend, Cat# 422301), respectively, to block nonspecific Fc domain binding. Cells were then stained with the desired antibodies listed in Supplementary Table S1 and washed 2x with PBS. After washing, cells were resuspended in FACS buffer and examined using an Attune NxT flow cytometer prior to analysis of the data using Attune Cytometric Software and FlowJo V10.
Human IFNγ was quantified using an ELISA MAX™ Deluxe Set (BioLegend, Cat# 430116) kit following manufacturer’s protocol.
Hs894, 3T3-mFAP, MDA-MB-231-hFAP and parental MDA-MB-231 cells were stained with viability dye (Sartorius, #4839) and seeded onto 96 wells plates (~8000 cells/well for MDA-MB-231 cells and 3T3-mFAP cells, ~5000 cells/well for Hs894 cells), after which anti-FL CAR T cells were then added at a 1:3 effector to target cell (E:T) ratio for MDA-MB-231 cells, and 1:2 E:T ratio for both Hs894 and 3T3-mFAP fibroblasts. To initiate CAR T cell killing, increasing concentrations (0 nM-1000nM) of FAP8-FL bispecific adaptor were added and the incubation was continued for 48 hours. Target cell lysis was then quantitated by dislodging the surviving cells with 0.25% trypsin, washing them twice in complete cell culture media, and counting surviving cells by flow cytometry. Percent target cell lysis was determined by comparing treatment wells to control wells with target cells alone (18, 59).
To evaluate the binding affinity of folate-FL and FAP8-FL for the anti-FL scFv on the CAR T cell and its tumor antigen on the cancer cell, folate-FL and FAP8-FL were dissolved in PBS. We quantitated folate-FL binding to the CAR T cells by analyzing displacement of fluorescein-AlexaFluor647 (FL-AF647) from the anti-FL CAR T cells by FAP8-FL. For this purpose, CAR T cells were incubated with various concentrations of FL-AF647 for 1 hour at room temperature, followed by washing 3 times with PBS + 2% FBS and measurement of cell-associated AF647 mean fluorescence intensity by flow cytometry (52). To confirm the specificity of bispecific adapter binding to anti-FL CAR T cells, CAR T cells were also incubated with FL-AF647 in the presence of 100x excess competitive ligands for 1 hour at room temperature. For analysis of the binding affinity of folate-FL and FAP8-FL for its tumor antigen on the cancer cells, increasing concentrations of each of bispecific adapters were incubated with either KB cells (for folate-FL) or MDA-MB-231-hFAP cells (for FAP8-FL) for 1 hour at room temperature. After incubation, cancer cells were washed 3x with PBS + 2% FBS and analyzed by flow cytometry (50, 52). The GraphPad Prism version 10 software was used to analyze binding affinity.
To qualitatively determine the binding of FAP8-FL to human FAP and murine FAP qualitatively under confocal microscopy, MDA-MB-231-hFAP, MDA-MB-231-mFAP, and parental MDA-MB-231 cells were pre-seeded in confocal chambers (Thermo Scientific, Cat# 155360) using complete RPMI at a density of 0.1 million cells per well. The cells were incubated with 100 nM of FAP8-FL at room temperature for 1 hour, followed by three washes with PBS + 2% FBS, and then processed for confocal microscopy measuring fluorescein signals.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Purdue University Human Research Protection Program/Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by Purdue University Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
BH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. SZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing, Software. KS: Formal analysis, Investigation, Methodology, Validation, Writing – review & editing, Software. RM: Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. MS: Formal analysis, Investigation, Project administration, Resources, Writing – review & editing. TS: Data curation, Formal analysis, Investigation, Validation, Writing – review & editing. XY: Data curation, Investigation, Methodology, Writing – review & editing. HC: Formal analysis, Project administration, Resources, Supervision, Writing – review & editing. PL: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from Umoja Biopharma. The funders were not involved in the collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
We thank Umoja Biopharma for providing the MDA-MB-231-hFAP cells, the lentivirus for generating anti-fluorescein CAR T cells, and the antibody for detecting anti-FL CAR. We also thank Chemical Genomics Facility (CGF) at Purdue Institute for Drug Discovery for their support in flow cytometry, and Purdue Histology Research Laboratory for their help in immunohistochemistry staining. We acknowledge Brooklyn Jolie Stocksdale, Gabriel Paul Bachman, and Emily Ann Raine for help with the studies.
Author HC was employed by company Umoja Biopharma, author MS was a consultant of company Umoja Biopharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1539265/full#supplementary-material
1. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
3. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
4. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
5. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:282. doi: 10.1038/s41419-018-0278-6
6. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
7. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
8. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. (2016) 59:468–79. doi: 10.1007/s11427-016-5023-8
9. Xia A-L, Wang X-C, Lu Y-J, Lu X-J, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. (2017) 8:90521. doi: 10.18632/oncotarget.19361
10. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. (2021) 372:eaba1786. doi: 10.1126/science.aba1786
11. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. (2022) 19:775–90. doi: 10.1038/s41571-022-00689-z
12. Luo Q, Napoleon JV, Liu X, Zhang B, Zheng S, Low PS. Targeted rejuvenation of exhausted chimeric antigen receptor T cells regresses refractory solid tumors. Mol Cancer Res. (2022) 20:823–33. doi: 10.1158/1541-7786.MCR-21-0711
13. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr., Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
14. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
15. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
16. Liu Z, Zhou Z, Dang Q, Xu H, Lv J, Li H, et al. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. (2022) 12:6273. doi: 10.7150/thno.76854
17. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
18. Luo W, Napoleon JV, Zhang F, Lee YG, Wang B, Putt KS, et al. Repolarization of tumor-infiltrating myeloid cells for augmentation of CAR T cell therapies. Front Immunol. (2022) 13:816761. doi: 10.3389/fimmu.2022.816761
19. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol cancer. (2021) 20:1–30. doi: 10.1186/s12943-021-01428-1
20. Suh J, Kim DH, Lee YH, Jang JH, Surh YJ. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol Carcinogenesis. (2020) 59:1028–40. doi: 10.1002/mc.23233
21. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers. (2015) 7:2443–58. doi: 10.3390/cancers7040902
22. Suzuki E, Yamazaki S, Naito T, Hashimoto H, Okubo S, Udagawa H, et al. Secretion of high amounts of hepatocyte growth factor is a characteristic feature of cancer-associated fibroblasts with EGFR-TKI resistance-promoting phenotype: A study of 18 cases of cancer-associated fibroblasts. Pathol Int. (2019) 69:472–80. doi: 10.1111/pin.12838
23. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. (2017) 216:3799–816. doi: 10.1083/jcb.201704053
24. Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. (2014) 25:61–8. doi: 10.1016/j.semcancer.2014.02.006
25. Knops AM, South A, Rodeck U, Martinez-Outschoorn U, Harshyne LA, Johnson J, et al. Cancer-associated fibroblast density, prognostic characteristics, and recurrence in head and neck squamous cell carcinoma: A meta-analysis. Front Oncol. (2020) 10:565306. doi: 10.3389/fonc.2020.565306
26. Scanlan MJ, Raj B, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, et al. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc Natl Acad Sci. (1994) 91:5657–61. doi: 10.1073/pnas.91.12.5657
27. Lindeman SD, Mukkamala R, Horner A, Tudi P, Booth OC, Huff R, et al. Fibroblast activation protein–targeted radioligand therapy for treatment of solid tumors. J Nucl Med. (2023) 64:759–66. doi: 10.2967/jnumed.122.264494
28. Millul J, Bassi G, Mock J, Elsayed A, Pellegrino C, Zana A, et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc Natl Acad Sci. (2021) 118:e2101852118. doi: 10.1073/pnas.2101852118
29. Fabre M, Ferrer C, Domínguez-Hormaetxe S, Bockorny B, Murias L, Seifert O, et al. OMTX705, a novel FAP-targeting ADC demonstrates activity in chemotherapy and pembrolizumab-resistant solid tumor models. Clin Cancer Res. (2020) 26:3420–30. doi: 10.1158/1078-0432.CCR-19-2238
30. Roy J, Hettiarachchi SU, Kaake M, Mukkamala R, Low PS. Design and validation of fibroblast activation protein alpha targeted imaging and therapeutic agents. Theranostics. (2020) 10:5778. doi: 10.7150/thno.41409
31. Bocci M, Zana A, Principi L, Lucaroni L, Prati L, Gilardoni E, et al. In vivo activation of FAP-cleavable small molecule-drug conjugates for the targeted delivery of camptothecins and tubulin poisons to the tumor microenvironment. J Control Release. (2024) 367:779–90. doi: 10.1016/j.jconrel.2024.02.014
32. Zboralski D, Hoehne A, Bredenbeck A, Schumann A, Nguyen M, Schneider E, et al. Preclinical evaluation of FAP-2286 for fibroblast activation protein targeted radionuclide imaging and therapy. Eur J Nucl Med Mol imaging. (2022) 49:3651–67. doi: 10.1007/s00259-022-05842-5
33. Syed M, Flechsig P, Liermann J, Windisch P, Staudinger F, Akbaba S, et al. Fibroblast activation protein inhibitor (FAPI) PET for diagnostics and advanced targeted radiotherapy in head and neck cancers. Eur J Nucl Med Mol imaging. (2020) 47:2836–45. doi: 10.1007/s00259-020-04859-y
34. Baum RP, Schuchardt C, Singh A, Chantadisai M, Robiller FC, Zhang J, et al. Feasibility, biodistribution, and preliminary dosimetry in peptide-targeted radionuclide therapy of diverse adenocarcinomas using (177)Lu-FAP-2286: first-in-humans results. J Nucl Med. (2022) 63:415–23. doi: 10.2967/jnumed.120.259192
35. Privé BM, Boussihmad MA, Timmermans B, van Gemert WA, Peters SMB, Derks YHW, et al. Fibroblast activation protein-targeted radionuclide therapy: background, opportunities, and challenges of first (pre)clinical studies. Eur J Nucl Med Mol Imaging. (2023) 50:1906–18. doi: 10.1007/s00259-023-06144-0
36. Das S, Valton J, Duchateau P, Poirot L. Stromal depletion by TALEN-edited universal hypoimmunogenic FAP-CAR T cells enables infiltration and anti-tumor cytotoxicity of tumor antigen-targeted CAR-T immunotherapy. Front Immunol. (2023) 14:1172681. doi: 10.3389/fimmu.2023.1172681
37. Liu Y, Sun Y, Wang P, Li S, Dong Y, Zhou M, et al. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J Transl Med. (2023) 21:255. doi: 10.1186/s12967-023-04080-z
38. Xiao Z, Todd L, Huang L, Noguera-Ortega E, Lu Z, Huang L, et al. Desmoplastic stroma restricts T cell extravasation and mediates immune exclusion and immunosuppression in solid tumors. Nat Commun. (2023) 14:5110. doi: 10.1038/s41467-023-40850-5
39. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
40. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. (2015) 75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041
41. Zhou L, Li Y, Zheng D, Zheng Y, Cui Y, Qin L, et al. Bispecific CAR-T cells targeting FAP and GPC3 have the potential to treat hepatocellular carcinoma. Mol Ther Oncol. (2024) 32(2):200817. doi: 10.1016/j.omton.2024.200817
42. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. (2013) 210:1137–51. doi: 10.1084/jem.20122344
43. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. (2013) 210:1125–35. doi: 10.1084/jem.20130110
44. de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. (2019) 7:19. doi: 10.1186/s40425-019-0505-4
45. Ding Z, Sun S, Wang X, Yang X, Shi W, Huang X, et al. Nanobody-based trispecific T cell engager (Nb-TriTE) enhances therapeutic efficacy by overcoming tumor-mediated immunosuppression. J Hematol Oncol. (2023) 16:115. doi: 10.1186/s13045-023-01507-4
46. Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. (2018) 78:6852–65. doi: 10.1158/0008-5472.CAN-18-1750
47. Fiorenza S, Ritchie DS, Ramsey SD, Turtle CJ, Roth JA. Value and affordability of CAR T-cell therapy in the United States. Bone Marrow Transplant. (2020) 55:1706–15. doi: 10.1038/s41409-020-0956-8
48. Harkins RA, Patel SP, Flowers CR. Cost burden of diffuse large B-cell lymphoma. Expert Rev Pharmacoecon Outcomes Res. (2019) 19:645–61. doi: 10.1080/14737167.2019.1680288
49. Tkacz J, Garcia J, Gitlin M, McMorrow D, Snyder S, Bonafede M, et al. The economic burden to payers of patients with diffuse large B-cell lymphoma during the treatment period by line of therapy. Leuk Lymphoma. (2020) 61:1601–9. doi: 10.1080/10428194.2020.1734592
50. Lee YG, Chu H, Lu Y, Leamon CP, Srinivasarao M, Putt KS, et al. Regulation of CAR T cell-mediated cytokine release syndrome-like toxicity using low molecular weight adapters. Nat Commun. (2019) 10:2681. doi: 10.1038/s41467-019-10565-7
51. Lu YJ, Chu H, Wheeler LW, Nelson M, Westrick E, Matthaei JF, et al. Preclinical evaluation of bispecific adaptor molecule controlled folate receptor CAR-T cell therapy with special focus on pediatric Malignancies. Front Oncol. (2019) 9:151. doi: 10.3389/fonc.2019.00151
52. Lee YG, Marks I, Srinivasarao M, Kanduluru AK, Mahalingam SM, Liu X, et al. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. (2019) 79:387–96. doi: 10.1158/0008-5472.CAN-18-1834
53. Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res. (2008) 41:120–9. doi: 10.1021/ar7000815
54. van Dam GM, Themelis G, Crane LMA, Harlaar NJ, Pleijhuis RG, Kelder W, et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting: first in-human results. Nat Med. (2011) 17:1315–9. doi: 10.1038/nm.2472
55. Mukkamala R, Carlson DJ, Miller NK, Lindeman SD, Bowen ER, Tudi P, et al. Design of a fibroblast activation protein–targeted radiopharmaceutical therapy with high tumor–to–healthy-tissue ratios. J Nucl Med. (2024) 65(8):1257–63. doi: 10.2967/jnumed.124.267756
56. Watanabe K, Kuramitsu S, Posey AD Jr., June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
57. Cords L, Tietscher S, Anzeneder T, Langwieder C, Rees M, de Souza N, et al. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat Commun. (2023) 14:4294. doi: 10.1038/s41467-023-39762-1
58. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
59. Wu X, Zhang Y, Li Y, Schmidt-Wolf IGH. Improvements in flow cytometry-based cytotoxicity assay. Cytometry A. (2021) 99:680–8. doi: 10.1002/cyto.a.24242
Keywords: universal CAR T cells, tumor microenvironment, cancer-associated fibroblasts (CAFs), solid tumors, fibroblast activation protein (FAP)
Citation: Huang B, Zheng S, Sudarshan K, Mukkamala R, Srinivasarao M, Sardesai T, Yang X, Chu H and Low PS (2025) Use of a universal targeting CAR T cell to simultaneously kill cancer cells and cancer-associated fibroblasts. Front. Immunol. 16:1539265. doi: 10.3389/fimmu.2025.1539265
Received: 04 December 2024; Accepted: 30 January 2025;
Published: 17 February 2025.
Edited by:
Dean Tantin, The University of Utah, United StatesReviewed by:
John Scholler, University of Pennsylvania, United StatesCopyright © 2025 Huang, Zheng, Sudarshan, Mukkamala, Srinivasarao, Sardesai, Yang, Chu and Low. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip S. Low, cGxvd0BwdXJkdWUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.