 David Kavanagh
David Kavanagh Jonathan Barratt2
Jonathan Barratt2 Anna Schubart
Anna Schubart- 1National Renal Complement Therapeutics Centre, Newcastle University, Newcastle-upon-Tyne, United Kingdom
- 2Department of Cardiovascular Sciences, University of Leicester, The John Walls Renal Unit, University Hospitals of Leicester NHS Trust, Leicester, United Kingdom
- 3Department of Immunology, Novartis BioMedical Research, Basel, Switzerland
- 4Novartis Pharma AG, Basel, Switzerland
- 5Service of Nephrology and Hypertension, Centre Hospitalier Universitaire Vaudois, UNIL, Lausanne, Switzerland
The complement system, consisting of three initiating pathways—classical, lectin and alternative, is an important part of innate immunity. Dysregulation of the complement system is implicated in the pathogenesis of several autoimmune and inflammatory diseases. Therapeutic inhibition of the complement system has been recognized as a viable approach to drug development and has been successful with the approval of a small number of complement inhibitors for diseases such as paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, neuromyelitis optica, myasthenia gravis and geographic atrophy. More recently, therapies selectively targeting the alternative pathway (AP), which drives the amplification of the complement responses, are being evaluated for these complement-mediated diseases. Complement Factor B, a serine protease, is a unique component of the AP that is essential for the catalytic activity of AP C3 convertase and AP C5 convertase. Inhibition of Factor B blocks the activity of the alternative pathway and the amplification loop, and subsequent generation of the membrane attack complex downstream; however, it has no effect on the initial activation mediated by the classical and lectin complement pathways. Therefore, Factor B is an attractive target for diseases in which the AP is overactivated. In this review, we provide an overview of Factor B and its critical role in the AP, discuss the benefit-risk of Factor B inhibition as a targeted therapeutic strategy, and describe the various Factor B inhibitors that are approved and/or in clinical development.
1 Introduction
The complement system is a central player in innate immunity, which serves as the first line of defense against invading pathogens and altered host cells, modulates adaptive immunity, and contributes to the maintenance of tissue homeostasis (1, 2). Its activities are mediated by sequentially activated serine proteases generating effector molecules that trigger inflammatory responses, enable opsonization, and promote phagocytosis and lysis of pathogens (1, 3–5). The balance between the activation and regulation of the complement system ensures that infectious pathogens and damaged cells are destroyed or removed, while leaving healthy host cells intact (1, 4). However, failure of regulatory mechanisms can lead to aberrant activation of the complement system, which underlies the pathogenesis of several inflammatory and autoimmune disorders and forms the rationale for the development of targeted complement therapies (1, 3, 4, 6, 7). In this review, we outline the role of Factor B (FB), a key protease of the alternative pathway (AP) of the complement system in health and disease, as an attractive therapeutic target for a number of complement-mediated diseases.
1.1 Overview of the complement system
Composed of more than 50 plasma and cell surface-bound proteins and regulators, the complement system can be activated through three distinct interconnected pathways: the classical pathway (CP), the lectin pathway (LP), and the AP (1, 4) (Figure 1).
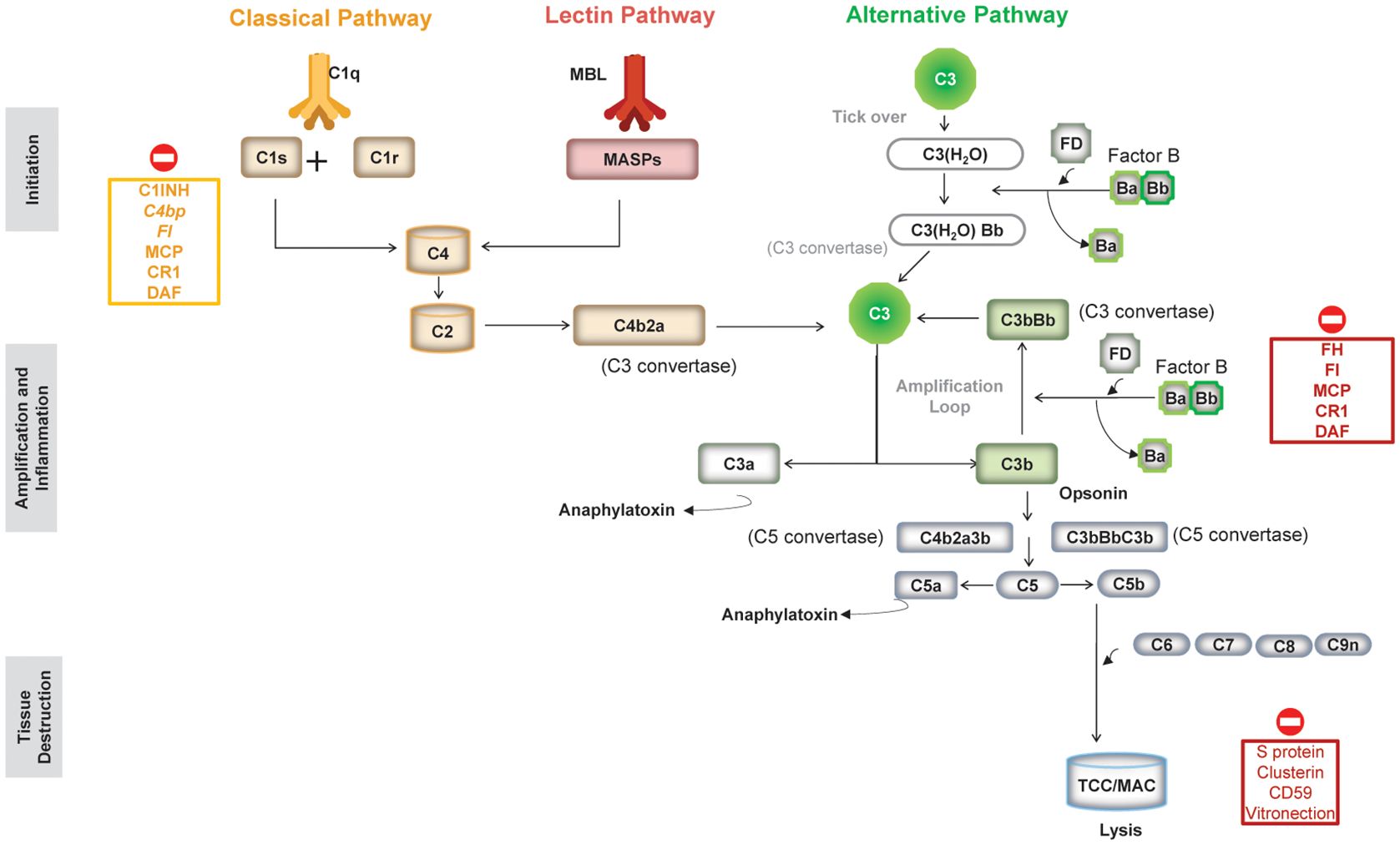
Figure 1. An overview of the three pathways of the complement system and complement regulators. (Figure adapted from Morgan BP et al. Front Cell Neurosci. 2021;14:600656 (https://doi.org/10.3389/fncel.2020.600656). Copyright 2021 Morgan, Gommerman and Ramaglia, CC BY (101). C1-INH, serum protein C1 inhibitor; C4Bp, complement inhibitor C4b binding protein; CR1, complement receptor type 1; DAF, decay accelerating factor (CD55); FI, factor I; FH, factor H; MAC, membrane attack complex; MBL, mannose-binding lectin; MCP, membrane cofactor protein (CD46); TCC, terminal complement complex.
The CP is activated by antigen–antibody complexes and the LP by conserved carbohydrate moieties present on pathogen surfaces, whereas the AP, evolutionarily the oldest, is constitutively active at low levels through a process of slow, spontaneous hydrolysis of C3 called “tick-over” (1). All three pathways converge at the cleavage of the central complement protein C3 by C3 convertases into C3a, an anaphylatoxin, and C3b, which is an opsonin that also triggers further activation of the AP acting as an amplification loop (1). Downstream of C3 convertase activation, the three pathways share the same sequence of events: C5 convertase formation, culminating in the assembly of the membrane attack complex (MAC), thus mediating cell lysis (Figure 1) (1).
1.1.1 AP and amplification loop
The AP is driven by two serine proteases that control activation (Factor D [FD]) and activity (Factor B [FB]) of the pathway. These two factors have distinct characteristics. The mannose-binding lectin-associated serine protease-3 (MASP-3), as a pro-FD activator, also plays an important role in AP activation.
FB is mainly produced in hepatocytes, found in high levels in serum (2-3 µM), and has a low clearance rate (plasma half-life 30-70 hours) (3). Factor D is largely produced in adipocytes, its latent proenzyme form (Pro-FD) is continuously converted into its active form (the predominant form that circulates in plasma) by MASP-3 (8, 9). FD is present in the serum at 2 µg/mL and has a rapid clearance rate (plasma half-life ~1 hours) (3, 10). While FD has long been considered rate-limiting for AP activation, it was recently shown that low levels of FD (<1% of serum levels) were sufficient for normal AP activation (11). Since FD is cleared through the kidneys, its levels can increase in renally impaired patients by 10-fold (10).
As shown in Figure 1, the AP is continuously active through spontaneous hydrolysis of a labile thioester bond in C3 [to form C3(H2O)] that induces a conformational change to expose the binding site for FB. The FB in this complex [C3(H2O)FB] is then cleaved by FD to form C3(H2O)Bb that acts as the initial fluid-phase C3 convertase of the AP to cleave C3 into C3a and C3b (1, 3).
FB can also bind to C3b, generated from CP/LP or AP C3 convertases, to form the AP C3 convertase C3bBb (5, 12). Since C3b is both a result of the AP C3 convertase activity and a component of the convertase itself, this creates a positive feedback loop that amplifies the initial complement response rapidly, called amplification loop (1, 3, 5). With continued activation of the AP C3 convertase and increasing amounts of C3b in the vicinity of the C3bBb convertase, binding of an additional molecule of C3b to the C3 convertase switches the specificity of the convertase from cleaving C3 to C5 (into C5a and C5b) to form the AP C5 convertase (C3BbC3b), culminating in the formation of the MAC.
The amplification loop enables a rapid scale-up of the initial complement response, not only of the AP but also of those initiated by the CP/LP. Indeed, the amplification loop of the AP may contribute up to 80% of the initial complement response of the CP and LP (13, 14). However, the extent of the contribution by the AP is likely to depend on the strength of the trigger, and amplification may be redundant in the presence of strong LP/CP activation for example after vaccination against encapsulated bacteria, which can result in antibody titers that trigger bacterial clearance in vitro in the absence of AP activity (15–19). Not surprisingly, given its potential for rapidly producing copious amounts of C3b, a “runaway” activation of the amplification loop can result in uncontrolled inflammation and mediate the pathogenesis of several complement-mediated diseases (1, 3, 5).
1.2 AP in inflammatory and autoimmune diseases
The AP, and in particular the amplification loop is tightly regulated as its uncontrolled activation can induce excessive inflammation and tissue damage. The dysregulation of the AP is implicated in the pathogenesis of several distinct diseases (7, 20, 21). Here, we discuss the prototypical complement-mediated diseases involving the AP.
The pathogenesis of some diseases, such as atypical hemolytic uremic syndrome (aHUS), C3 glomerulopathy (C3G), but also paroxysmal nocturnal hemoglobinuria (PNH), and age-related macular degeneration (AMD), is a direct result of dysregulation of the AP, either due to pathogenic variants in genes encoding AP components and/or their regulators or due to acquired factors such as autoantibodies against components or regulators of the AP (Supplementary Table S1). In others, complement activation occurs downstream of disease-specific triggers, such as autoantibodies and immune complexes, which are the key drivers of pathophysiology and tissue injury in these diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis, immunoglobulin A nephropathy [IgAN], anti-neutrophil cytoplasmic autoantibody-associated vasculitis [AAV], and lupus nephritis [LN]) (Supplementary Table S1). Irrespective of the initial trigger, as noted earlier, the AP and its amplification loop may play a central role in turning up the magnitude of any initial complement response and, thus, play a key role in exacerbating tissue injury in all these complement-mediated diseases (6, 21).
Although most known complement-mediated diseases are rare or ultra-rare, and many progress slowly, collectively they impose a large burden, with a substantial proportion of patients continuing to remain at risk of disease progression and/or disease relapse despite optimal treatment with current standard of care (SoC). In many diseases, where the complement system mediates or contributes to the pathophysiology of the disease, there are no approved therapies that specifically target the complement pathway (Supplementary Table S1). The current management of these rare kidney diseases involving complement dysregulation relies on the use of broad, non-specific immunosuppressive and anti-inflammatory therapies such as corticosteroids, mycophenolate acids, calcineurin inhibitors, and cyclophosphamide. All of these therapies individually have a relatively high toxicity level, limiting their benefit-risk ratio; most are used in combination. A few regimens, which can be nephrotoxic, are associated with poor tolerability and several serious adverse events, including increased risk of serious infections. Moreover, they impose an additional burden on patients and their quality of life (QoL) (22). Furthermore, a majority of these drugs are not approved for most of the diseases/indications and are used off-label, as a rescue therapy, based on limited evidence (22).
PNH and aHUS, and to some extent AAV, have been success stories where complement inhibition has demonstrated efficacy and has emerged as the novel SoC (23–26).The C5 inhibitors eculizumab and ravulizumab (approved for PNH, aHUS, myasthenia gravis, and neuromyelitis optica spectrum disorder) are monoclonal antibodies that block the cleavage of the terminal complement protein C5 to C5a and C5b through C5 convertase, thereby preventing the formation of MAC, regardless of the initial pathway. While anti-C5 therapies can potently control intravascular hemolysis and the associated risk of thrombosis, the most severe complication of PNH, they exposed an additional pathomechanism (e.g. persistent anemia caused by extravascular hemolysis) necessitating more upstream complement-targeting therapies that are safe and well tolerated. Pegcetacoplan, a pegylated cyclic peptide inhibiting C3 cleavage prevents both intra-and extravascular hemolysis and is administered twice weekly by subcutaneous injection. Iptacopan, the first oral FB inhibitor that specifically targets the AP with high potency even during many complement amplifying conditions, has recently received FDA approval for treatment of PNH and IgAN (27–29). Iptacopan improved hemoglobin to near-normal levels and led to blood transfusion avoidance in the majority of treatment-naïve PNH patients and in PNH patients who had residual anemia despite anti-C5 therapy (27, 28).
2 Factor B
FB is an abundant plasma protein with circulating concentrations ranging from 0.2 to 0.4 mg/mL. FB is unique to the AP and provides the active protease site of the AP C3 convertase that is responsible for AP activity. The gene encoding FB (CFB) resides within the major histocompatibility complex class III region gene cluster on chromosome 6 (6p21.33), in proximity to its CP/LP homologue C2 (30). Like most other members of the complement system, FB is primarily, but not exclusively, synthesized in the liver. Low FB expression is observed in other cell types including renal tubular cells, monocytes, adipocytes, dendritic cells, and fibroblasts, and tissue-specific expression may be significantly upregulated during inflammation (30–33). Various proinflammatory cytokines (IL-1β, IL-6, TNF-α and IFN-γ) and endotoxins can induce the expression of FB (3, 30, 34, 35). FB is an acute phase reactant, with an average increase of ~50% in plasma soon after infection (30, 34, 35).
FB is a single-chain glycoprotein of 764 amino acids (~93kDa) consisting of two subunits: the non-catalytic N-terminal Ba subunit (~33 kDa; residues 26-259) linked to the C-terminal catalytic Bb subunit (~60 kDa, residues 260-764) via a 45–amino acid linker peptide encompassing the scissile bond (Arg 259-Lys260) that is cleaved by FD (Figures 2A, B) (30). The N-terminal Ba subunit consists of three complement control protein (CCP) modules or short consensus repeat domains of ~60 amino acids each (Figure 2B) that are common to several proteins of the complement system. The Bb subunit consists of a von Willebrand factor type A domain along with the MIDAS (metal-ion dependent adhesion site) motif and the serine protease domain (3, 30).
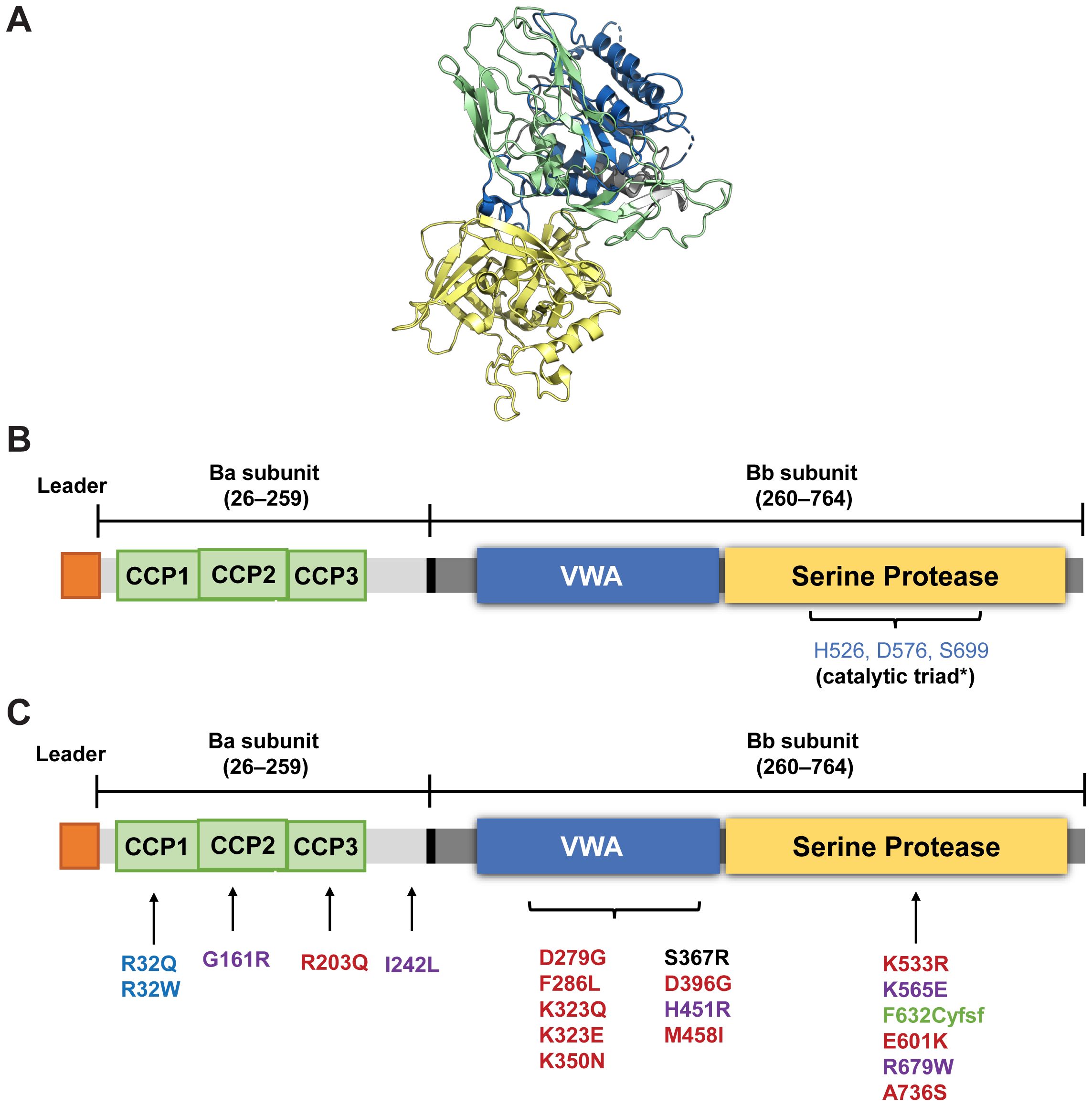
Figure 2. Structure of FB (A) and a schematic representation of its domains (B, C) Location of various polymorphic variants in FB reported in aHUS (red), C3G (black); MPGN (purple), and deficiency of FB (green). Slow-fast polymorphism variants R32Q (fast A) associated with reduced risk of age-related macular degeneration) and R32W (fast B) are in blue. *Iptacopan binds to the active site of FB containing the catalytic triad (HIS 526, Ser 699 and Asp576) in the serine protease domain of the Bb subunit (numbering according to UniProt (P00751). (Image A from RCSB PDB of 2OK5 created using Pymol: an open-source molecular visualization system. Figure B and C adapted with permission from Laskowski J, Thurman JM. Chapter 14 - Factor B. The Complement FactsBook: Academic Press; 2018. p. 135-46. Copyright 2018 by Elsevier) (30). CCP, complement control protein module; VWA, von Willebrand factor type A domain.
FB circulates in the plasma in a latent zymogen form. To prevent indiscriminate activation of FB, the Ba subunit folds back onto the Bb subunit, thus preventing binding of C3b [or C3(H2O)] with the MIDAS motif and preventing FD from accessing the scissile bond in unbound FB (36). Initial Mg2+-independent binding of C3b or [C3(H2O)] with the CCP domain of Ba subunits results in a conformational change, exposing the MIDAS motif. This allows the binding and stabilization of interaction between C3b [or C3(H2O)] and Bb subunit in an Mg2+-dependent manner and brings in another conformational change in FB that exposes the scissile bond in the linker region for cleavage by FD (Figure 3) (3, 30, 37). The FD–C3bB interaction induces a conformational change in FD, displacing its self-inhibitory loop (5, 38). This co-factor–dependent and substrate-induced proteolytic step is one of the regulatory mechanisms that restricts the uncontrolled activation and amplification of the AP on C3b-opsonized surfaces (38). FD cleaves the linker peptide at the scissile bond, generating the Ba fragment that dissociates from the complex, while Bb with its serine protease domain remains associated with C3b to become the catalytically active component of the newly formed AP C3 convertase (Figure 3) (3, 5).
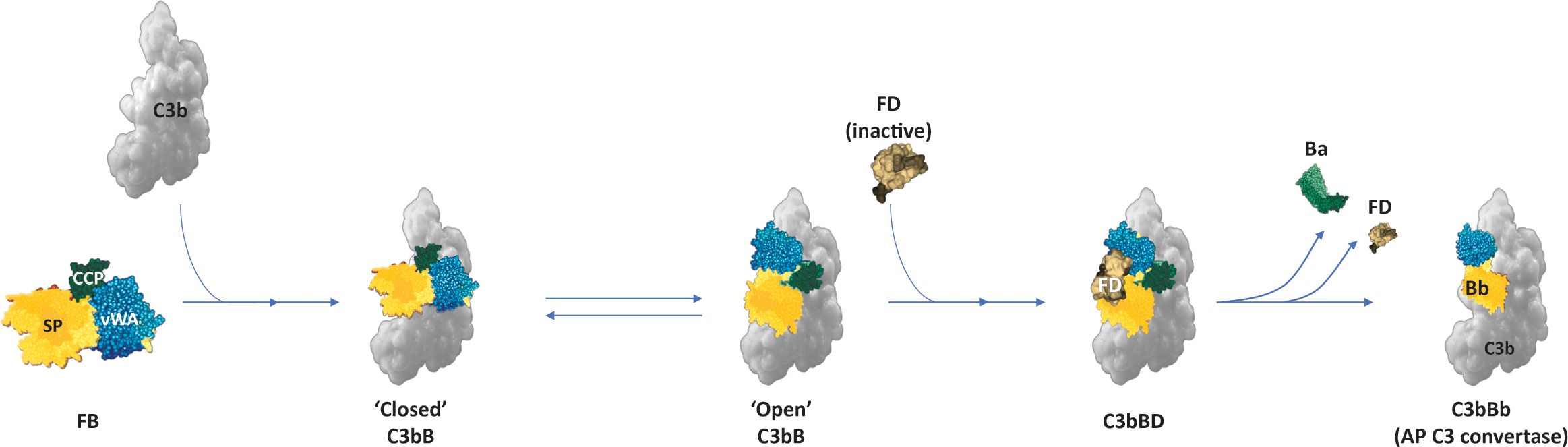
Figure 3. AP C3 convertase (C3bBb) formation. Binding of FB to C3b results in conformational changes in FB, with the serine protease domain dynamically oscillating between a “closed” and “open” form that allows the binding of FD and cleavage of the scissile bond to liberate the Ba fragment (1, 30). (Figure adapted from Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262, doi: 10.3389/fimmu.2015.00262. Copyright 2015 Merle, Church, Fremeaux-Bacchi and Roumenina. CC BY 4.0) (1). AP, alternative pathway; FB, Factor B; FD, Factor D.
For spontaneous AP activation started by C3 hydrolysis [C3(H2O)], an additional FD-independent mechanism may exist (39). With the formation of convertase, the serine protease domain in Bb re-orients and becomes catalytically active. The C3 convertase cleaves C3 at Arg726-Ser727 in the C3 α chain; the C5 convertase cleaves C5 at homologous Arg74-Leu75 in the C5 α chain (30).
C3 and C5 are the only known natural substrates of the convertases (C3bBb and C3bBbC3b, respectively). Both the initial AP fluid-phase [C3(H2O)Bb] and the C3bBb C3 convertases alone have very short half-lives (~90 seconds for C3bBb) and rapidly dissociate (3). The Bb fragment has only marginal catalytic activity outside of the C3bBb complex, and once Bb dissociates from the convertase complex, it cannot reassociate with C3b (3, 5).
The stability of the AP convertases can be affected by its interactome. Interaction with properdin can increase the stability of the AP C3 convertase (C3bBb) up to 10-fold on target cell surfaces as it prevents C3 convertase dissociation by Factor H (FH) and cleavage by Factor (FI), thus enhancing complement amplification. Negative regulators, such as FH, FI, membrane cofactor protein (MCP), complement receptor 1 (CR1), and decay accelerating factor (DAF), down-regulate the AP by facilitating AP convertase dissociation (1, 3, 5, 30, 40–43).
FH is the master regulator of the AP; it blocks the amplification loop both in the fluid phase and on surfaces by competitively inhibiting the interaction of FB with C3b, thus preventing the formation of new C3 convertase (1, 40, 41, 44). FH also controls the AP by promoting accelerated dissociation of the AP C3 convertases to C3b and Bb (decay-accelerating activity) and by acting as a cofactor for FI, which cleaves and deactivates C3b to iC3b on cell surfaces. DAF, MCP, and CR1 are membrane-bound proteins and contribute to the regulation of all three complement pathways (1, 40, 41).
2.1 FB deficiency and variants
FB deficiency is very rare, with only three reported cases (45–47). As with FD deficiency, a deficiency of FB is associated with an increased risk of encapsulated bacterial infections, including meningococcal infections.
Three major allelic variants of CFB (differing at nucleotide position 32 [amino acid 7] of the Ba domain), collectively referred to as the slow-fast polymorphism (based on electrophoretic mobility), have been described as “slow” A, “fast” A, and “fast” B. These variants have been shown to have an effect on complement activation. Compared to the common allele, the “slow” variant (32R) of CFB, the “fast” A variant (32Q) and “fast” B variant (32W) are less efficient in AP C3 convertase (C3bBb) formation. The “fast” A variant displays a disease-protective effect against AMD (30, 48). Several rare variants of FB have been associated with complement-mediated diseases such as aHUS, C3G (rarely), MPGN, and AMD (mainly gain-of-function, [GoF] variants), with some variants (loss-of-function [LoF] variants) conferring protection against developing AMD (Figure 2C) (30, 49–53).
Similarly, mice deficient in CFB develop normally but lack AP activity and are more susceptible to bacterial infections (54, 55). Deletion of CFB (−/−) has been shown to rescue the renal phenotype in CFH KO mice (that exhibit a C3G-like pathology due to uncontrolled AP activation) and leads to a less severe renal disease and immune complex deposition in LN disease models, highlighting the critical role of FB in driving the AP/amplification loop and in the pathophysiology of complement-mediated diseases (55–57).
3 Targeting FB in complement-mediated diseases
With increased knowledge of complement biology and its key role in the pathogenesis of several diseases, the complement system, and in particular the AP, has emerged as an innovative therapeutic target. Inhibition of the AP may be achieved either by preventing the formation of the new C3bBb complexes or by inhibiting the catalytic activity of the convertase (3, 5). Specific inhibition of the AP prevents formation of C3a and opsonization by C3b, halting the subsequent inflammation and phagocytosis of the opsonized cells (Figure 4), which are not impacted by distal inhibition of the complement pathway (e.g., at the level of C5). Several novel pharmacological agents targeting diverse aspects of the complement system are at various stages of clinical evaluation. Among potential targets that can specifically inhibit the AP and the amplification loop, FB, owing to its biology, has emerged as a promising pharmacological drug target (Table 1). FB is one of the upstream, proximal components unique to the AP and is essential for the activation of the pathway; it is an integral component that drives the amplification loop. FB has a relatively high plasma concentration; however, the plasma half-life of FB ranges from 30 to 70 hours, and it has fractional metabolic rates of 1.6%/h, allowing steady-state and stable inhibition (3). In comparison, FD inhibition might be more challenging than initially thought due to its rapid turn-over rate and self-inhibitory loop that may hinder the access of low-molecular-weight (LMW) inhibitors to the active site. Even a small amount of residual functional FD (1%-2%) is sufficient for restoring partial AP activity (11). This necessitates almost complete saturation of FD for complete and sustained inhibition (5, 6). While plasma FD levels are normally very low, they strongly increase in renally impaired individuals (10). Importantly, AP blockade (with FB and FD) inhibitors would also prevent amplification of the CP and LP (6). The anticipated effects of FB inhibitors on the pharmacodynamics of biomarkers of complement activation are summarized in Figure 4.
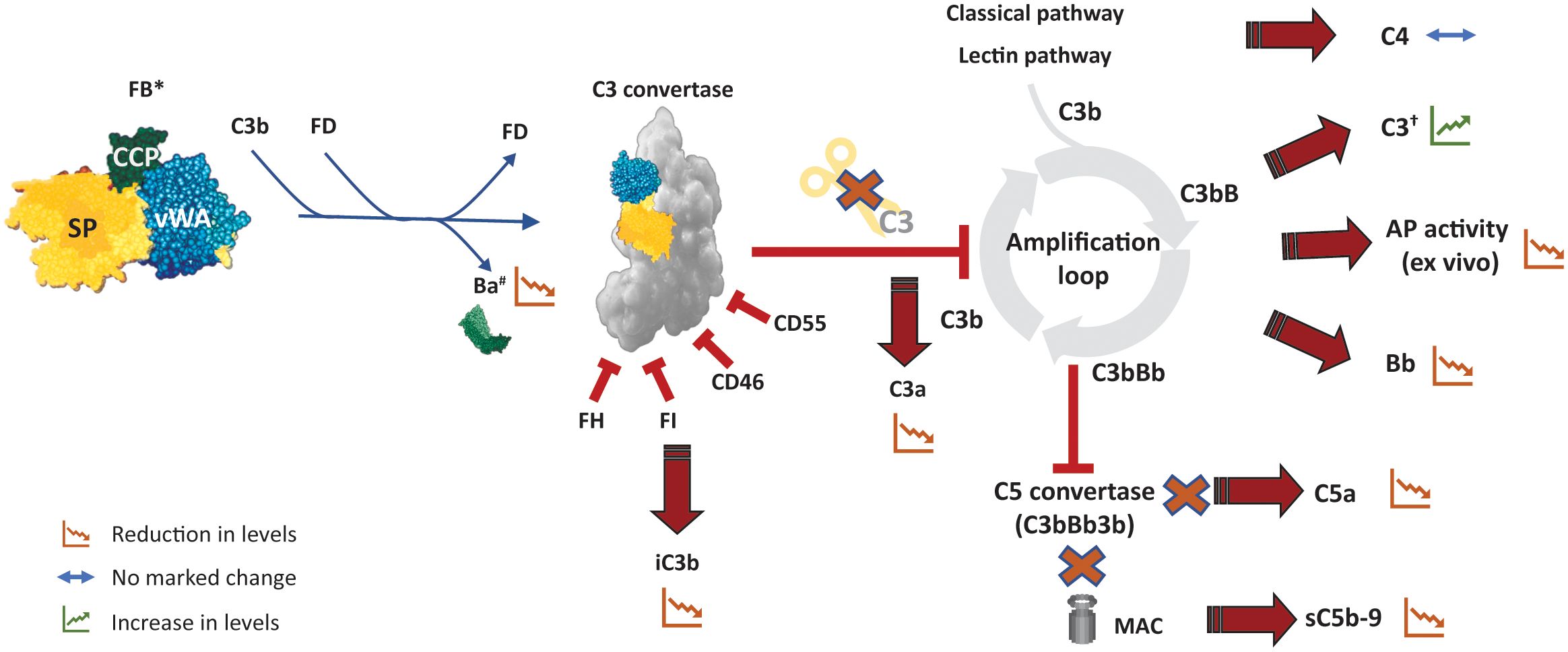
Figure 4. Potential pharmacodynamic effects of FB inhibition on complement activation. The effect of an inhibitor that inhibits catalytic activity of FB (and hence of C3/C5 convertase) is depicted here. Inhibition of FB abolishes cleavage of C3 by the AP C3 convertase and eventually inhibits the amplification loop as well as the downstream formation of AP C5 convertase. *An ASO (or siRNA) targeting FB would lead to reduction in the levels of circulating FB and its fragments in addition to the other effects described here. †Particularly likely to be evident in conditions that result in C3 hypocomplementemia such as C3G. AP, alternative pathway, ASO, antisense oligonucleotide; C3G, C3 glomerulopathy; DAF, decay accelerating factor; FB, factor B; FD, factor D; FH, factor H; MCP, membrane cofactor protein; sC5b-9, soluble C5b-9; siRNA, small interfering RNA.
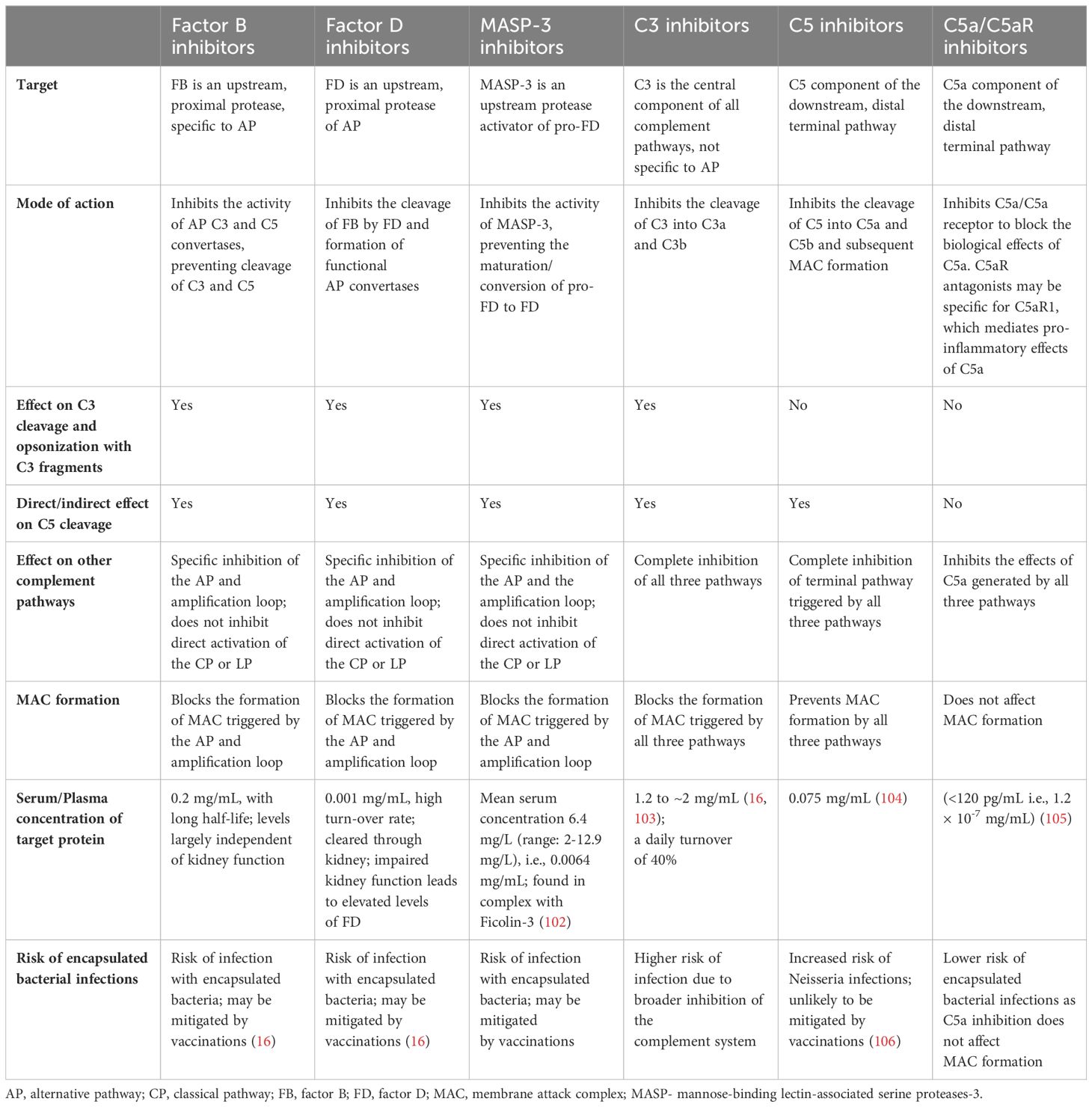
Table 1. A comparison of the various classes of complement inhibitors impacting the AP and terminal complement pathways.
4 FB inhibitors currently in clinical development
Development of selective FB inhibitors was considered challenging primarily because high drug levels would be needed for efficient inhibition of the high serum levels of FB. Nevertheless, multiple compounds targeting FB are currently in various stages of development. These range from highly selective small-molecule inhibitors to antisense oligonucleotides and antibodies; the efficacy and safety of a few of these are being evaluated in ongoing clinical trials.
4.1 Iptacopan (Fabhalta®)
Iptacopan (Novartis Pharma AG) is an oral, proximal complement inhibitor that specifically binds FB and inhibits AP activity. Iptacopan was approved by the US FDA as the first oral monotherapy for adults with PNH in December 2023 and is the first complement inhibitor to receive accelerated approval for reduction of proteinuria in adults with primary IgAN who are at risk of disease progression (i.e. urine protein to creatinine ratio ≥1.5 g/g) (27, 29, 58). It is also being evaluated as a potential therapy in several other complement-mediated diseases such as C3G, aHUS, IC-MPGN, LN, and AMD (Table 2). Iptacopan binds to the active site of FB present in the Bb subunit (both to the native FB and the Bb fragment attached to C3b) with high affinity (KD value 0.0079 ± 0.0019 μM) and is highly selective over all proteases tested, including FD and CP/LP activity (Figure 5) (3, 59).

Table 2. Phase 2 and phase 3 clinical trials* of FB inhibitors.

Figure 5. (A) Chemical structure of iptacopan (Figure A reprinted from PubChem. https://pubchem.ncbi.nlm.nih.gov/compound/lnp023. In public domain) and (B) Crystal structure showing iptacopan (magenta) bound to the catalytic domain of human FB. The image on the right shows the indole moiety and the piperidine core of iptacopan binding into the S1 pocket and the S3 pocket, respectively; and the key hydrogen bonds. (Figure B reprinted from Schubart A et al. Immunol Rev. 2023; 313(1):339-357, doi: 10.1111/imr.v313. Copyrights 2022 Novartis Pharma AG. Immunological Reviews published by John Wiley & Sons Ltd. CC BY-NC-ND 4.0) (3).
Upon oral administration, iptacopan is rapidly absorbed, reaching peak plasma concentration in ~2 hours. In phase 2 studies, iptacopan dose-dependently inhibited AP activity, with a maximal sustained inhibition at a dose of 200 mg twice daily (60, 61). Similarly, iptacopan treatment also resulted in a reduction of Bb levels, which is reflective of the extent of the AP activation, as well as of urinary sC5b-9 levels, reflecting the inhibition of the AP and terminal complement pathway within the kidney (61). In patients with C3G and reduced C3 (<77 mg/dL), normalization of mean plasma C3 levels was observed following iptacopan treatment for 3 months in most patients (60). Taken together, these results highlight that treatment with iptacopan effectively blocks the AP.
Evidence from PNH clinical trials and those emerging from other phase 2 and phase 3 trials support the efficacy of iptacopan in various complement-mediated diseases. Initially, iptacopan was tested as an add-on therapy to eculizumab and was well-tolerated. It led to significant improvement in hemoglobin and reduction in lactate dehydrogenase (LDH) and the effect was maintained even when anti-C5 treatment was discontinued (62). Consistent with these findings, in another small phase 2 study in treatment-naïve patients with PNH, iptacopan monotherapy was well-tolerated and led to a significant reduction in serum LDH levels along with transfusion-free improvements in hemoglobin levels in most patients (62, 63). Corroborating these observations are the results from the pivotal phase 3 studies in APPLY-PNH and APPOINT-PNH (28). In APPLY-PNH, a significantly higher proportion of patients treated with iptacopan monotherapy achieved hemoglobin increases of ≥2 g/dL (82% vs 2%; p<0.0001) and hemoglobin levels of ≥12 g/dL (69% vs 2%; p <0.001) without the need for transfusions at 24-weeks compared to anti-C5 therapies; similar outcomes were achieved in treatment-naïve patients in the APPOINT study (28).
In a phase 2 study in patients with C3G, 12 months of iptacopan treatment resulted in a 45% reduction in proteinuria in patients with native kidneys and significantly reduced glomerular C3 deposition in patients with transplanted kidneys, with stabilization of kidney function in both cohorts (60). Similarly, in a phase 2 study in IgAN, iptacopan resulted in a dose-dependent reduction in proteinuria, with a clinically meaningful and statistically significant 23% reduction achieved at 3 months with iptacopan 200 mg twice daily compared to placebo (61). Iptacopan was generally safe and well tolerated in all the clinical studies, with no serious infections reported. Primary analysis results from the ongoing phase 3 studies in C3G and IgAN have corroborated the phase 2 findings. In the APPEAR-C3G study, patients treated with iptacopan achieved a 35.1% reduction in proteinuria (p=0.0014) at 6 months when compared to placebo on top of supportive care (64). Similarly, in the pre-specified interim analysis of APPLAUSE-IgAN study, iptacopan demonstrated superiority to placebo in reducing proteinuria in patients with IgAN, with a 38.3% (p<0.0001) reduction at 9 months when compared to placebo on top of supportive care (58). Based on these findings, iptacopan was granted accelerated approval by the US FDA for IgAN in August 2024 (29). Iptacopan is currently being evaluated in phase 3 studies for IgAN (APPLAUSE-IgAN), C3G (APPEAR-C3G), aHUS (APPELHUS), and IC-MPGN (APPARENT) (65–71) (Table 2). These promising data further strengthen the rationale that pharmacological inhibition of the AP, and of FB in particular, is an innovative therapeutic strategy.
4.2 IONIS-FB-LRx (RO743465/RG6299)
IONIS-FB-LRx (Ionis Pharmaceuticals/Roche) is an antisense oligonucleotide (ASO) that targets CFB mRNA in hepatocytes to prevent the synthesis of FB protein in the liver. It is a 2’-O-methoxyethyl (2’MOE) second-generation ASO (20 bp) conjugated to an N-acetylgalactosamine (GalNAc) ligand. As the liver is the main site of synthesis for FB, the GalNac moiety targets the ASO for uptake by hepatocytes (72). In a mouse model of LN, reduction of FB levels with ASO was associated with significant improvements in renal pathology, reduced glomerular C3 deposition and proteinuria, and improved survival (73). In a phase 1 study, administration of multiple, 20-mg doses of ASO for 36 days resulted in a 72% reduction in plasma FB levels and attenuation of AP activity by 62% (74).
In patients with IgAN, IONIS-FB-LRx administered every 4 weeks for 6 months resulted in a 44% reduction in proteinuria at week 29 (75). A phase 3 trial to evaluate the efficacy and safety of IONIS-FB-LRx in patients with IgAN has been initiated (Table 2) (76).
4.3 Compounds in early development phase
Several other compounds with diverse modes of action are being evaluated as potential FB inhibitors, including small molecule inhibitors such as the oral tablets MY008211A (in PNH) and HRS-5965 (in PNH and IgAN) (77, 78), a recombinant fragment of human FB (79), FB28.4.2 monoclonal antibody (mAb) targeting the Ba subunit to prevent interaction of C3b with FB (80, 81), mAbs (NM8074; SAR44380) targeting FB or its subunits, RNA interference therapies (ARO-CFB, ADX-038) targeting hepatic expression of FB (82, 83), and a cyclic hendecapeptide (complin) that prevents the cleavage of FB by FD and of C2 by activated C1s to inhibit the AP and CP/LP (84). Of these, the humanized mAb NM8074 (NovelMed) which binds to the Bb subunit (but not to the latent FB) to prevent the activity of C3/C5 convertases has entered phase 2 clinical development for C3G, aHUS, and PNH (Table 2) (85–89). Interim analysis from first-in-human studies of NM0874 in 40 healthy individuals suggests that NM8074 is safe and well tolerated across the doses tested (0.3-20 mg/kg), and specifically inhibits the AP but not the CP (90). Phase 2 trials have also been planned or are underway for HRS5965 (PNH and IgAN) and MY008211A (PNH) (77, 91–93). Phase 1 trial of SAR443809 has been completed, and phase 1/2a trial of ARO-CFB (in complement mediated kidney disease/IgAN) has been initiated (94–96).
5 Future perspectives and conclusions
In the last few decades, a vast body of data has demonstrated the role of complement and, in particular, the AP as key mediators of pathology in several diseases. An increased understanding of complement and its role in the pathogenesis of specific diseases, coupled with technological advances and a favorable regulatory framework for novel drugs in rare diseases, has spurred interest in the development of complement-targeted therapies that aim to halt or slow disease progression. Given the central role of the AP in amplifying the initial complement response, the AP and amplification loop have emerged as a key target, with FD, MASP-3, and FB being the prime candidates for inhibition (3, 6).
Several properties of FB make it an ideal therapeutic target for AP inhibition, and diverse approaches have been employed to target FB, each with its own strengths and limitations:
● Developing small-molecule inhibitors of FB was considered to be challenging due to the high serum concentration of FB. with iptacopan being the only FB inhibitor approved for PNH and IgAN (accelerated approval) and is in late-stage clinical development for several diseases (3, 5). A small-molecule inhibitor with good oral bioavailability and pharmacokinetic/pharmacodynamic properties has several advantages: a convenient route of administration for patients, potential for complete or near-complete therapeutic inhibition without disrupting the steady-state levels of the target protein, and owing to a reasonably short half-life, normal AP activity can be restored relatively quickly when needed (e.g., during infection) (3, 5, 6). In diseases such as aHUS, a rapid inhibition of the complement system with an oral inhibitor, as observed with iptacopan (maximal inhibition of the AP observed 2 hours post-dose), may be particularly relevant as an alternative therapeutic option vis-à-vis current intravenously administered SoC. On the other hand, a shorter half-life of the compound compared to an antibody requires good patient adherence to avoid incomplete complement inhibition after washout, in particular in acute indications.
● An antibody-mediated approach is perhaps less challenging: the convertase presents as a large substrate to a bulky antibody that needs to prevent access to C3b or FD (to prevent convertase formation) or to C3 (to inhibit its catalytic activity). This relative ease is reflected in several candidate antibodies that are being or have been explored as FB inhibitors. However, the bulky nature of antibodies necessitates an intravenous or subcutaneous mode of administration, Patients with aHUS report the need for chronic biweekly (or bimonthly) administration of anti-C5 antibodies as a significant burden (6, 26); estimated infusion duration is about 35 minutes to 1.5 hours (depending on body weight), and patients need to be under observation for 1 hour thereafter to monitor for infusion-related reactions (97, 98). In addition, the high plasma concentration of FB, as well as the increase in the half-life of targets of therapeutic antibodies, necessitate higher doses of antibodies to sustain FB inhibition.
● An ASO-based targeted approach to inhibit hepatic synthesis of FB is being evaluated/spearheaded by Ionis/Roche. However, similar to antibody-based approaches, ASOs would need chronic subcutaneous injections every month; moreover, ASO therapies may not be convenient in an acute setting since FB reduction occurs after a few days (74). In addition, whether inhibiting hepatic synthesis of FB with ASOs would have beneficial effects in instances where FB production occurs locally at the site of injury/inflammation remains to be elucidated.
Most of the current complement-targeted therapies, including those targeting FB, have been or are currently being evaluated in prototypical complement-mediated kidney diseases where the AP drives pathogenesis and mediates tissue injury. In addition to these, FB inhibitors may also have a beneficial role in diseases where AP activation may be secondary to other triggers (3, 6). For example, in multiple autoimmune diseases, such as in LN where autoantibodies initially trigger the CP, the amplification loop is expected to subsequently contribute to the pathology.
Although most of the complement-mediated therapies in development or already in use have generally been safe, the increased risk of infection with encapsulated bacteria remains a safety concern for this class of drugs, given the important role the complement system plays in fighting invading pathogens. In general, the risk of severe infection, especially Neisseria infections, is expected to be higher with more distal complement inhibitors (such as anti-C5 therapies). Inhibition of C3 is also expected to be typically associated with the risk of a broader class of infections. Mitigation strategies such as vaccination against encapsulated bacteria and prophylactic antibiotics where needed are essential to reduce the risk of infections. Indeed, sera from vaccinated individuals treated with AP inhibitors retained higher bactericidal and opsonophagocytic activity against encapsulated bacteria compared to those treated with C5 or C3 inhibitors in vitro (16–18, 99). Such mitigation strategies have also been proposed as part of the design and conduct of clinical trials of complement-mediated therapies. Long-term, close safety monitoring for infections is nevertheless essential for patients being treated or considered for treatment with complement-targeted therapies (100).
In conclusion, AP blockade through FB inhibition is a promising addition to the armamentarium of potential therapies in complement-mediated kidney diseases. With the approval of iptacopan for treatment of adults with PNH and IgAN for patients at risk of progression with UPCR ≥1.5 g/g (accelerated approval), and potential approval for some of the other FB-inhibiting novel therapies in sight, patients with these rare complement-mediated diseases may soon have access to effective, targeted therapies that offer a better safety profile than conventional immunosuppression.
Author contributions
DK: Conceptualization, Supervision, Writing – review & editing. JB: Conceptualization, Supervision, Writing – review & editing. AS: Conceptualization, Supervision, Writing – review & editing. NW: Conceptualization, Supervision, Writing – review & editing. MM: Conceptualization, Supervision, Writing – review & editing. FF: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Medical writing and editorial support were provided by Nagabhushana Ananthamurthy, Shivani Vadapalli, and Rahul Lad of Novartis Healthcare Pvt. Ltd. in accordance with the Good Publication Practice (GPP-2022) guidelines. (https://www.acpjournals.org/doi/epdf/10.7326/M22-1460). The article processing fee was funded by Novartis Pharma AG.
Conflict of interest
AS is an employee of Novartis. MM and NW were employees of Novartis at the time of manuscript development. JB has received research funding and/or consultancy fees from Alexion, Alnylam, Arrowhead Pharmaceuticals, AstraZeneca, BioCryst, Kira Pharma, Novartis, Omeros, Roche/Genentech, Bio and Vifor. FF has received consultancy fees from Alexion, Apellis, Novartis, Roche, and Sobi. DK was academic founder of Gyroscope Therapeutics Ltd, now a Novartis Company, and has received consultancy income and equity from Gyroscope Therapeutics Ltd. DK has received honoraria for consultancy work from Alexion, Novartis, Sarepta Chemocentryx, Apellis and Idorsia. DK is an author of patent applications referencing recombinant complement factor I production and/or formation of the C3b/FH/FI trimolecular complex.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1537974/full#supplementary-material
References
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. (2015) 34:2735–57. doi: 10.15252/embj.201591881
3. Schubart A, Flohr S, Junt T, Eder J. Low-molecular weight inhibitors of the alternative complement pathway. Immunol Rev. (2023) 313:339–57. doi: 10.1111/imr.v313.1
4. Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol. (2019) 15:129–43. doi: 10.1038/s41581-018-0107-2
5. Dobo J, Kocsis A, Gal P. Be on target: strategies of targeting alternative and lectin pathway components in complement-mediated diseases. Front Immunol. (2018) 9:1851. doi: 10.3389/fimmu.2018.01851
6. Barratt J, Weitz I. Complement factor D as a strategic target for regulating the alternative complement pathway. Front Immunol. (2021) 12:712572. doi: 10.3389/fimmu.2021.712572
7. Lukawska E, Polcyn-Adamczak M, Niemir ZI. The role of the alternative pathway of complement activation in glomerular diseases. Clin Exp Med. (2018) 18:297–318. doi: 10.1007/s10238-018-0491-8
8. Dobo J, Szakacs D, Oroszlan G, Kortvely E, Kiss B, Boros E, et al. MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked. Sci Rep. (2016) 6:31877. doi: 10.1038/srep31877
9. Hayashi M, Machida T, Ishida Y, Ogata Y, Omori T, Takasumi M, et al. Cutting edge: role of MASP-3 in the physiological activation of factor D of the alternative complement pathway. J Immunol. (2019) 203:1411–6. doi: 10.4049/jimmunol.1900605
10. Pascual M, Steiger G, Estreicher J, Macon K, Volanakis JE, Schifferli JA. Metabolism of complement factor D in renal failure. Kidney Int. (1988) 34:529–36. doi: 10.1038/ki.1988.214
11. Wu X, Hutson I, Akk AM, Mascharak S, Pham CTN, Hourcade DE, et al. Contribution of adipose-derived factor D/adipsin to complement alternative pathway activation: lessons from lipodystrophy. J Immunol. (2018) 200:2786–97. doi: 10.4049/jimmunol.1701668
12. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. (2009) 104:115–49. doi: 10.1016/S0065-2776(08)04004-2
13. Harboe M, Garred P, Karlstrom E, Lindstad JK, Stahl GL, Mollnes TE. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol. (2009) 47:373–80. doi: 10.1016/j.molimm.2009.09.005
14. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. (2004) 138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x
15. de Boer EC, Thielen AJ, Langereis JD, Kamp A, Brouwer MC, Oskam N, et al. The contribution of the alternative pathway in complement activation on cell surfaces depends on the strength of classical pathway initiation. Clin Transl Immunol. (2023) 12:e1436. doi: 10.1002/cti2.1436
16. Ispasanie E, Muri L, Schubart A, Thorburn C, Zamurovic N, Holbro T, et al. Alternative complement pathway inhibition does not abrogate meningococcal killing by serum of vaccinated individuals. Front Immunol. (2021) 12:747594. doi: 10.3389/fimmu.2021.747594
17. Muri L, Ispasanie E, Schubart A, Thorburn C, Zamurovic N, Holbro T, et al. Alternative complement pathway inhibition abrogates pneumococcal opsonophagocytosis in vaccine-naive, but not in vaccinated individuals. Front Immunol. (2021) 12:732146. doi: 10.3389/fimmu.2021.732146
18. Muri L, Schubart A, Thorburn C, Zamurovic N, Holbro T, Kammuller M, et al. Inhibition of the different complement pathways has varying impacts on the serum bactericidal activity and opsonophagocytosis against Haemophilus influenzae type b. Front Immunol. (2022) 13:1020580. doi: 10.3389/fimmu.2022.1020580
19. Ispasanie E, Muri L, Schmid M, Schubart A, Thorburn C, Zamurovic N, et al. In vaccinated individuals serum bactericidal activity against B meningococci is abrogated by C5 inhibition but not by inhibition of the alternative complement pathway. Front Immunol. (2023) 14:1180833. doi: 10.3389/fimmu.2023.1180833
20. Zipfel PF, Wiech T, Grone HJ, Skerka C. Complement catalyzing glomerular diseases. Cell Tissue Res. (2021) 385:355–70. doi: 10.1007/s00441-021-03485-w
21. Poppelaars F, Thurman JM. Complement-mediated kidney diseases. Mol Immunol. (2020) 128:175–87. doi: 10.1016/j.molimm.2020.10.015
22. Kidney Disease: Improving Global Outcomes Glomerular Diseases Work G. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. (2021) 100:S1–S276. doi: 10.1016/j.kint.2021.05.021
23. Bodo I, Amine I, Boban A, Bumbea H, Kulagin A, Lukina E, et al. Complement inhibition in paroxysmal nocturnal hemoglobinuria (PNH): A systematic review and expert opinion from central Europe on special patient populations. Adv Ther. (2023) 40:2752–72. doi: 10.1007/s12325-023-02510-4
24. Prendecki M, McAdoo SP. Targeting complement in ANCA-associated vasculitis: insights from ADVOCATE. Nat Rev Nephrol. (2021) 17:439–40. doi: 10.1038/s41581-021-00417-3
25. Chen M, Jayne DRW, Zhao MH. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. (2017) 13:359–67. doi: 10.1038/nrneph.2017.37
26. Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. (2017) 390:681–96. doi: 10.1016/S0140-6736(17)30062-4
27. Food and Drug Administration. HIGHLIGHTS OF PRESCRIBING INFORMATION FABHALTA® (iptacopan) capsules, for oral use (2024). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/218276s001lbl.pdf (Accessed December 1, 2024).
28. Peffault de Latour R, Roth A, Kulasekararaj AG, Han B, Scheinberg P, Maciejewski JP, et al. Oral iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2024) 390:994–1008. doi: 10.1056/NEJMoa2308695
29. Novartis receives FDA accelerated approval for Fabhalta® (iptacopan), the first and only complement inhibitor for the reduction of proteinuria in primary IgA nephropathy (IgAN) (2024). Available online at: https://www.novartis.com/news/media-releases/novartis-receives-fda-accelerated-approval-fabhalta-iptacopan-first-and-only-complement-inhibitor-reduction-proteinuria-primary-iga-nephropathy-igan (Accessed August 9, 2024).
30. Laskowski J, Thurman JM. Chapter 14- Factor B. In: Barnum S, Schein T, editors. The Complement FactsBook (Second Edition). Academic Press (2018). p. 135–46. doi: 10.1016/B978-0-12-810420-0.00014-6
31. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
32. Moreno-Navarrete JM, Martinez-Barricarte R, Catalan V, Sabater M, Gomez-Ambrosi J, Ortega FJ, et al. Complement factor H is expressed in adipose tissue in association with insulin resistance. Diabetes. (2010) 59:200–9. doi: 10.2337/db09-0700
33. Welch TR, Beischel LS, Frenzke M, Witte D. Regulated expression of complement factor B in the human kidney. Kidney Int. (1996) 50:521–5. doi: 10.1038/ki.1996.344
34. Vitale G, Mansueto S, Gambino G, Mocciaro C, Spinelli A, Rini GB, et al. The acute phase response in Sicilian patients with boutonneuse fever admitted to hospitals in Palermo, 1992-1997. J Infect. (2001) 42:33–9. doi: 10.1053/jinf.2000.0758
35. Volanakis JE. Transcriptional regulation of complement genes. Annu Rev Immunol. (1995) 13:277–305. doi: 10.1146/annurev.iy.13.040195.001425
36. Hourcade DE, Mitchell LM. Access to the complement factor B scissile bond is facilitated by association of factor B with C3b protein. J Biol Chem. (2011) 286:35725–32. doi: 10.1074/jbc.M111.263418
37. Janssen BJ, Gomes L, Koning RI, Svergun DI, Koster AJ, Fritzinger DC, et al. Insights into complement convertase formation based on the structure of the factor B-cobra venom factor complex. EMBO J. (2009) 28:2469–78. doi: 10.1038/emboj.2009.184
38. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. (2010) 330:1816–20. doi: 10.1126/science.1195821
39. Zhang Y, Keenan A, Dai DF, May KS, Anderson EE, Lindorfer MA, et al. C3(H2O) prevents rescue of complement-mediated C3 glomerulopathy in Cfh-/- Cfd-/- mice. JCI Insight. (2020) 5:e135758. doi: 10.1172/jci.insight.135758
40. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. (2008) 12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x
41. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. (2013) 33:479–92. doi: 10.1016/j.semnephrol.2013.08.001
42. Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. (2009) 10:728–33. doi: 10.1038/ni.1755
43. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A. (1976) 73:3268–72. doi: 10.1073/pnas.73.9.3268
44. Schmidt CQ, Herbert AP, Hocking HG, Uhrin D, Barlow PN. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol. (2008) 151:14–24. doi: 10.1111/j.1365-2249.2007.03553.x
45. Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. (2013) 369:1667–9. doi: 10.1056/NEJMc1306326
46. Gauthier A, Wagner E, Thibeault R, Lavoie A. A novel case of complement factor B deficiency. J Clin Immunol. (2021) 41:277–9. doi: 10.1007/s10875-020-00906-3
47. Dehoorne J, Haerynck F, Raes A, De Guchtenaere A, Joos R, Walle JV. Complement factor B deficiency associated with recurrent asceptic meningitis. Pediatr Rheumatol Online J. (2008) 6:266. doi: 10.1186/1546-0096-6-S1-P266
48. Montes T, Tortajada A, Morgan BP, Rodriguez de Cordoba S, Harris CL. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc Natl Acad Sci U S A. (2009) 106:4366–71. doi: 10.1073/pnas.0812584106
49. COMPLEMENT FACTOR B; CFB. Available online at: https://www.omim.org/entry/138470?search=cfb&highlight=cfb (Accessed April 30, 2024).
50. Aradottir SS, Kristoffersson AC, Roumenina LT, Bjerre A, Kashioulis P, Palsson R, et al. Factor D inhibition blocks complement activation induced by mutant factor B associated with atypical hemolytic uremic syndrome and membranoproliferative glomerulonephritis. Front Immunol. (2021) 12:690821. doi: 10.3389/fimmu.2021.690821
51. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol. (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
52. Imamura H, Konomoto T, Tanaka E, Hisano S, Yoshida Y, Fujimura Y, et al. Familial C3 glomerulonephritis associated with mutations in the gene for complement factor B. Nephrol Dial Transplant. (2015) 30:862–4. doi: 10.1093/ndt/gfv054
53. Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, et al. Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol. (2014) 25:2053–65. doi: 10.1681/ASN.2013070796
54. Tong HH, Li YX, Stahl GL, Thurman JM. Enhanced susceptibility to acute pneumococcal otitis media in mice deficient in complement C1qa, factor B, and factor B/C2. Infect Immun. (2010) 78:976–83. doi: 10.1128/IAI.01012-09
55. Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, et al. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U.S.A. (1997) 94:8720–5. doi: 10.1073/pnas.94.16.8720
56. Gibson BG, Cox TE, Marchbank KJ. Contribution of animal models to the mechanistic understanding of Alternative Pathway and Amplification Loop (AP/AL)-driven Complement-mediated Diseases. Immunol Rev. (2023) 313:194–216. doi: 10.1111/imr.v313.1
57. Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, et al. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. (2000) 164:786–94. doi: 10.4049/jimmunol.164.2.786
58. Perkovic V, Barratt J, Rovin B, Kashihara N, Maes B, Zhang H, et al. Alternative Complement Pathway Inhibition with Iptacopan in IgA Nephropathy. N Engl J Med. (2025) 392(6):531–43. doi: 10.1056/NEJMoa2410316
59. Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci. (2019) 116:7926–31. doi: 10.1073/pnas.1820892116
60. Wong E, Nester C, Cavero T, Karras A, Le Quintrec M, Lightstone L, et al. Efficacy and safety of iptacopan in patients with C3 glomerulopathy. Kidney Int Rep. (2023) 8:2754–64. doi: 10.1016/j.ekir.2023.09.017
61. Zhang H, Rizk DV, Perkovic V, Maes B, Kashihara N, Rovin B, et al. Results of a randomized double-blind placebo-controlled Phase 2 study propose iptacopan as an alternative complement pathway inhibitor for IgA nephropathy. Kidney Int. (2024) 105:189–99. doi: 10.1016/j.kint.2023.09.027
62. Risitano AM, Roth A, Soret J, Frieri C, de Fontbrune FS, Marano L, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol. (2021) 8:e344–e54. doi: 10.1016/S2352-3026(21)00028-4
63. Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study. Blood Adv. (2022) 6:4450–60. doi: 10.1182/bloodadvances.2022006960
64. Novartis presents latest Phase III Fabhalta® (iptacopan) data in C3 glomerulopathy (C3G) showing clinically meaningful and statistically significant 35.1% proteinuria reduction vs. placebo (2024). Available online at: https://www.novartis.com/news/media-releases/novartis-presents-latest-phase-iii-fabhalta-iptacopan-data-c3-glomerulopathy-c3g-showing-clinically-meaningful-and-statistically-significant-351-proteinuria-reduction-vs-placebo (Accessed June 1, 2024).
65. Study of Efficacy and Safety of Iptacopan in Participants With IC-MPGN (APPARENT). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05755386 (Accessed December 1, 2024).
66. Efficacy and Safety of Iptacopan (LNP023) in Adult Patients With Atypical Hemolytic Uremic Syndrome Naive to Complement Inhibitor Therapy (APPELHUS). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT04889430 (Accessed December 1, 2024).
67. Study of Efficacy and Safety of Iptacopan in Patients With C3 Glomerulopathy. (APPEAR-C3G). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT04817618 (Accessed December 1, 2024).
68. Study of Efficacy and Safety of LNP023 in Primary IgA Nephropathy Patients (APPLAUSE-IgAN). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT04578834 (Accessed December 1, 2024).
69. Bomback AS, Kavanagh D, Vivarelli M, Meier M, Wang Y, Webb NJA, et al. Alternative omplement pathway inhibition with iptacopan for the treatment of C3 glomerulopathy-study design of the APPEAR-C3G trial. Kidney Int Rep. (2022) 7:2150–9. doi: 10.1016/j.ekir.2022.07.004
70. Kavanagh D, Greenbaum LA, Bagga A, Karki RG, Chen CW, Vasudevan S, et al. Design and rationale of the APPELHUS phase 3 open-label study of factor B inhibitor iptacopan for atypical hemolytic uremic syndrome. Kidney Int Rep. (2023) 8:1332–41. doi: 10.1016/j.ekir.2023.04.029
71. Rizk DV, Rovin BH, Zhang H, Kashihara N, Maes B, Trimarchi H, et al. TaRgeting the Alternative Complement Pathway With Iptacopan to Treat IgA Nephropathy: Design and Rationale of the APPLAUSE-IgAN Study. Kidney Int Rep. (2023) 8:968–79. doi: 10.1016/j.ekir.2023.01.041
72. Dreismann AK, Hallam TM, Tam LC, Nguyen CV, Hughes JP, Ellis S, et al. Gene targeting as a therapeutic avenue in diseases mediated by the complement alternative pathway. Immunol Rev. (2023) 313:402–19. doi: 10.1111/imr.13149
73. Grossman TR, Hettrick LA, Johnson RB, Hung G, Peralta R, Watt A, et al. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology. (2016) 221:701–8. doi: 10.1016/j.imbio.2015.08.001
74. McCaleb M, Hughes S, Greary R, Monia B, Grossman T. Pharmacodynamic efficacy of IONIS-FB-LRX, a complement factor B antisense oligonucleotide, in a phase 1 clinical study. Mol Immunol. (2018) 102:187–8. doi: 10.1016/j.molimm.2018.06.154
75. Barbour S, Hladunewich MA, Irvine J, Makris A, Robson R, Tan S-J, et al. An Exploratory Trial of an Investigational RNA Therapeutic, IONIS-FB-LRx, for Treatment of IgA Nephropathy. ABSTRACT: SA-PO714. Orlando, Florida: American Society of Nephrology (ASN) Kidney Week (2022). Available at: https://www.asn-online.org/education/kidneyweek/archives/KW22Abstracts.pdf (Accessed April 30, 2024).
76. A Study to Evaluate the Efficacy and Safety of RO7434656 in Participants With Primary Immunoglobulin A (IgA) Nephropathy at High Risk of Progression (IMAGINATION). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05797610 (Accessed December 1, 2024).
77. A Trial of HRS-5965 Tablets in Primary IgA Nephropathy. ClinicalTrials.gov. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT06137768 (Accessed December 1, 2024).
78. A Study of Single-dose MY008211A in Healthy Adults. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05642585 (Accessed December 1, 2024).
79. Sultan EY, Rizk DE, Kenawy HI, Hassan R. A small fragment of factor B as a potential inhibitor of complement alternative pathway activity. Immunobiology. (2021) 226:152106. doi: 10.1016/j.imbio.2021.152106
80. Subias M, Tortajada A, Gastoldi S, Galbusera M, Lopez-Perrote A, Lopez Lde J, et al. A novel antibody against human factor B that blocks formation of the C3bB proconvertase and inhibits complement activation in disease models. J Immunol. (2014) 193:5567–75. doi: 10.4049/jimmunol.1402013
81. Jager NM, van Zanden JE, Subias M, Leuvenink HGD, Daha MR, Rodriguez de Cordoba S, et al. Blocking complement factor B activation reduces renal injury and inflammation in a rat brain death model. Front Immunol. (2019) 10:2528. doi: 10.3389/fimmu.2019.02528
82. Arrowhead pharmaceuticals initiates phase 1/2a study of ARO-CFB for treatment of complement mediated kidney disease. (2024). Available online at: https://arrowheadpharma.com/news-press/arrowhead-pharmaceuticals-initiates-phase-1-2a-study-of-aro-cfb-for-treatment-of-complement-mediated-kidney-disease/.
83. Safety, Tolerability, PK and PD of ADX-038 in Healthy Participants and Paroxysmal Nocturnal Hemoglobinuria (PNH) Patients (PNH). ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/study/NCT05876312. (Accessed January 2025)
84. Kadam AP, Sahu A. Identification of Complin, a novel complement inhibitor that targets complement proteins factor B and C2. J Immunol. (2010) 184:7116–24. doi: 10.4049/jimmunol.1000200
85. Study of NM8074 in Patients With aHUS With Evidence of Ongoing Thrombotic Microangiopathy. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05684159 (Accessed December 1 2024).
86. Study of NM8074 in Adult C3 Glomerulopathy Patients. ClinicalTrials. Available at: https://clinicaltrials.gov/study/NCT05647811 (Accessed December 1, 2024).
87. Study of NM8074 in Soliris-Treated Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05731050 (Accessed December 1, 2024).
88. Study of Efficacy and Safety of NM8074 in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05646524 (Accessed December 1, 2024).
89. Study of NM8074 in Adult PNH Patients With Inadequate Response to Soliris. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05646563 (Accessed December 1, 2024).
90. NovelMed Announces Interim Positive Results from Its Phase I Clinical Trial of NM8074, a Bb Complement Alternative Pathway Blocker Antibody . Available online at: https://www.prnewswire.com/news-releases/novelmed-announces-interim-positive-results-from-its-phase-i-clinical-trial-of-nm8074-a-bb-complement-alternative-pathway-blocker-antibody-301485271.html?tc=eml_cleartime (Accessed December 1, 2024).
91. A Study of MY008211A in Adult Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH). ClinicalTrials.gov. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT06050226 (Accessed December 1, 2024).
92. Study of Safety and Efficacy of MY008211A in in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH). ClinicalTrials.gov. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT06134414 (Accessed December 1, 2024).
93. Proof of Concept Study to Assess the Efficacy, Safety of HRS-5965 in Patients With Paroxysmal Nocturnal Hemoglobinuria. ClinicalTrials.gov. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT06051357 (Accessed December 1, 2024).
94. Rajagopal V, Leksa NC, Gorham RD, Jindal S, Nair SV, Knockenhauer KE, et al. SAR443809: A selective inhibitor of the complement alternative pathway, targeting complement factor bb. Blood Adv. (2023) 7:4258–68. doi: 10.1182/bloodadvances.2022009028
95. A Study to Test if SAR443809 is Tolerated and Safe When Taken as a Single Dose in Healthy Adults. ClinicalTrials.gov. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT06326814 (Accessed December 1, 2024).
96. Study of ARO-CFB in Adult Healthy Volunteers and Patients With Complement-Mediated Kidney Disease. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT06209177 (Accessed December 1, 2024).
97. Food and Drug administration. SOLIRIS® (eculizumab) injection, for intravenous use. HIGHLIGHTS OF PRESCRIBING INFORMATION (2020). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125166s431lbl.pdf (Accessed December 1, 2024).
98. Food and Drug Administration. ULTOMIRIS® (ravulizumab-cwvz) injection, for intravenous use.HIGHLIGHTS OF PRESCRIBING INFORMATION (2022). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761108s023lbl.pdf (Accessed December 1, 2024).
99. Granoff DM, Kim H, Topaz N, MacNeil J, Wang X, McNamara LA. Differential effects of therapeutic complement inhibitors on serum bactericidal activity against non-groupable meningococcal isolates recovered from patients treated with eculizumab. Haematologica. (2019) 104:e340–e4. doi: 10.3324/haematol.2018.209692
100. Bomback AS, Appel GB, Gipson DS, Hladunewich MA, Lafayette R, Nester CM, et al. Improving clinical trials for anticomplement therapies in complement-mediated glomerulopathies: report of a scientific workshop sponsored by the national kidney foundation. Am J Kidney Dis. (2022) 79:570–81. doi: 10.1053/j.ajkd.2021.07.025
101. Morgan BP, Gommerman JL, Ramaglia V. An "Outside-in" and "Inside-out" Consideration of complement in the multiple sclerosis brain: lessons from development and neurodegenerative diseases. Front Cell Neurosci. (2020) 14:600656. doi: 10.3389/fncel.2020.600656
102. Skjoedt MO, Palarasah Y, Munthe-Fog L, Jie Ma Y, Weiss G, Skjodt K, et al. MBL-associated serine protease-3 circulates in high serum concentrations predominantly in complex with Ficolin-3 and regulates Ficolin-3 mediated complement activation. Immunobiology. (2010) 215:921–31. doi: 10.1016/j.imbio.2009.10.006
103. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
104. Struijf EM, De la O Becerra KI, Ruyken M, de Haas CJC, van Oosterom F, Siere DY et al. Inhibition of cleavage of human complement component C5 and the R885H C5 variant by two distinct high affinity anti-C5 nanobodies. J Biol Chem. (2023) 299:104956. doi: 10.1016/j.jbc.2023.104956
105. Cyprian FS, Suleman M, Abdelhafez I, Doudin A, Masud Danjuma M, Mir FA, et al. Complement C5a and Clinical Markers as Predictors of COVID-19 Disease Severity and Mortality in a Multi-Ethnic Population. Front Immunol. (2021) 12:707159. doi: 10.3389/fimmu.2021.707159
106. Brocklebank V, Walsh PR, Smith-Jackson K, Hallam TM, Marchbank KJ, Wilson V, et al. Atypical hemolytic uremic syndrome in the era of terminal complement inhibition: an observational cohort study. Blood (2023) 142:1371–86. doi: 10.1182/blood.2022018833
107. Efficacy and Safety of Switching From Anti-C5 Antibody Treatment to Iptacopan Treatment in Study Participants With Atypical Hemolytic Uremic Syndrome (aHUS). ClinicalTrials.gov. Available at: https://www.clinicaltrials.gov/study/NCT05935215 (Accessed December 1, 2024).
108. Study of Efficacy and Safety of LNP023 in Participants With Active Lupus Nephritis Class III-IV, +/- V. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05268289 (Accessed December 1, 2024).
109. A Masked, Placebo-controlled Study to Assess Iptacopan in Age-related Macular Degeneration. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT05230537 (Accessed December 1, 2024).
110. A Phase III Study to Investigate Efficacy, Safety and Tolerability of Iptacopan Compared With Placebo in Participants Aged 18 to 75 Years With gMG. ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT06517758 (Accessed December 1 2024).
111. A Study to Evaluate the Effectiveness and Safety of IONIS-FB-LRx, an Antisense Inhibitor of Complement Factor B, in Adult Participants With Primary IgA Nephropathy. Clinical Trials.gov. Available at: https://www.clinicaltrials.gov/ct2/show/NCT04014335 (Accessed December 1, 2024).
112. GOLDEN STUDY: A Study to Assess Safety and Efficacy of Multiple Doses of IONIS-FB-LRx in Participants With Geographic Atrophy Secondary to Age-Related Macular Degeneration (AMD). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT03815825 (Accessed December 1, 2024).
113. Safety and Efficacy of IONIS-FB-Lrx in up to 120 Patients 55 and Older With Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/study/NCT03446144 (Accessed December 1, 2024).
Keywords: complement, alternative pathway, factor B, therapeutic target, inhibitors, pharmacological treatment
Citation: Kavanagh D, Barratt J, Schubart A, Webb NJA, Meier M and Fakhouri F (2025) Factor B as a therapeutic target for the treatment of complement-mediated diseases. Front. Immunol. 16:1537974. doi: 10.3389/fimmu.2025.1537974
Received: 02 December 2024; Accepted: 13 January 2025;
Published: 14 February 2025.
Edited by:
Marcin Okrój, Intercollegiate Faculty of Biotechnology of University of Gdańsk and Medical University of Gdańsk, PolandReviewed by:
Peter F. Zipfel, Leibniz Institute for Natural Product Research and Infection Biology, GermanyMargarita López-Trascasa, Autonomous University of Madrid, Spain
Copyright © 2025 Kavanagh, Barratt, Schubart, Webb, Meier and Fakhouri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Kavanagh, ZGF2aWQua2F2YW5hZ2hAbmV3Y2FzdGxlLmFjLnVr