
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 January 2025
Sec. Immunological Tolerance and Regulation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1536020
This article is part of the Research TopicImmunometabolism in Immunological Tolerance and Regulation: Novel Mechanisms and Clinical InterventionsView all 6 articles
 Yuming Lu†‡Yifan Wang†‡Tiantian Ruan†‡Yihan Wang‡
Yuming Lu†‡Yifan Wang†‡Tiantian Ruan†‡Yihan Wang‡ Linling Ju‡Mengya Zhou‡Luyin Liu‡
Linling Ju‡Mengya Zhou‡Luyin Liu‡ Dengfu Yao‡
Dengfu Yao‡ Min Yao*‡
Min Yao*‡Immunometabolism is an emerging field that explores the intricate interplay between immune cells and metabolism. Regulatory T cells (Tregs), which maintain immune homeostasis in immunometabolism, play crucial regulatory roles. The activation, differentiation, and function of Tregs are influenced by various metabolic pathways, such as the Mammalian targets of rapamycin (mTOR) pathway and glycolysis. Correspondingly, activated Tregs can reciprocally impact these metabolic pathways. Tregs also possess robust adaptive capabilities, thus enabling them to adapt to various microenvironments, including the tumor microenvironment (TME). The complex mechanisms of Tregs in metabolic diseases are intriguing, particularly in conditions like MASLD, where Tregs are significantly upregulated and contribute to fibrosis, while in diabetes, systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA), they show downregulation and reduced anti-inflammatory capacity. These phenomena suggest that the differentiation and function of Tregs are influenced by the metabolic environment, and imbalances in either can lead to the development of metabolic diseases. Thus, moderate differentiation and inhibitory capacity of Tregs are critical for maintaining immune system balance. Given the unique immunoregulatory abilities of Tregs, the development of targeted therapeutic drugs may position them as novel targets in immunotherapy. This could contribute to restoring immune system balance, resolving metabolic dysregulation, and fostering innovation and progress in immunotherapy.
Metabolism, the fundamental process that organisms utilize to sustain life, can be broadly categorized into synthetic metabolism and breakdown metabolism (1). Synthetic metabolism involves the conversion of external nutrients into the organism’s own components, whereas breakdown metabolism refers to the transformation of a portion of the organism into metabolic byproducts, which are subsequently expelled from the body. The equilibrium between these processes is essential for maintaining life, preserving internal environmental stability, and ensuring continuous self-renewal.
The “Warburg effect” describes the metabolic preference of tumor cells, which differs from that of normal cells, which rely on oxidative phosphorylation (OXPHOS) for energy. Even under conditions of ample oxygen, tumor cells opt for the use of glycolytic metabolism instead of OXPHOS to generate energy. Via this metabolic reprogramming, tumor cells gain a growth advantage by utilizing lactate produced from glycolysis. This phenomenon aids in immune evasion and facilitates the proliferation and metastasis of tumor cells (2). This “aerobic glycolysis” phenomenon was later rediscovered in leukocytes, thus gaining increasing attention and evolving into the expansive field of immunometabolism in immunological research. Today, immunometabolism has become a hot research topic, with numerous studies reporting close connections between immune responses and metabolic pathways, such as the impact of various inflammatory factors, including tumor necrosis factor-alpha (TNF-α), on glucose metabolism (3). This series of studies on immunometabolism confirms that the differentiation and function of immune cells are influenced by various microenvironments within the body, thus leading to metabolic reprogramming and altering metabolic pathways in different environments (2, 3).
Regulatory T cells (Tregs), which are a special subset of CD4+ T cells, play a vital role in controlling the body’s immune response. Tregs can be classified into natural Tregs (nTregs) derived from thymic development, peripherally induced Tregs (pTregs) generated from initial CD4+ T cells in peripheral organs and tissues, and induced Tregs (iTregs) differentiated after external stimulation (4). Tregs from different sources can recognize not only shared antigens but also source-specific antigens, which is reflected in the heterogeneity of T-cell receptors (TCRs) (5). The heterogeneity of TCRs primarily arises from the random recombination of V (variable), D (diversity), and J (joining) gene segments during T-cell development in the thymus. This genetic recombination creates extensive diversity, enabling each T-cell to possess a unique TCR. This process grants T-cells broad antigen recognition capabilities, allowing them to respond to a wide variety of antigens, including both self and foreign antigens. nTregs predominantly recognize self-antigens, contributing to immune tolerance and preventing the development of autoimmune diseases. pTregs primarily recognize harmless foreign antigens, such as microbial and dietary antigens, maintaining peripheral immune homeostasis and preventing unnecessary immune responses to innocuous antigens. iTregs, on the other hand, are generated from conventional T-cells under conditions of infection or inflammation. They are capable of recognizing pathogen-derived antigens and modulating immune responses to prevent excessive inflammation and immune-mediated damage.
The key transcription factor for Tregs is Foxp3, which plays a crucial role in maintaining immune homeostasis (6). Sustained TCR signaling can maintain Foxp3 expression, thus promoting the inhibitory function of Tregs (7). The anti-inflammatory inhibitory capacity of Tregs dates back to the 1970s (8). Treg cells act as metabolic sensors, detecting both local and systemic metabolic signals such as fluctuations in nutrients and changes in peripheral energy sensors. Upon receiving these signals, Treg cells adjust their intracellular metabolism and anti-inflammatory functions to achieve immune tolerance. In different tissues, Treg cells regulate signaling pathways such as mTOR and AMP-activated protein kinase (AMPK) according to nutrient availability, which influences their proliferation and suppressive capabilities. For instance, in adipose tissue, Treg cells adapt to the lipid-rich environment, suppress inflammation, and maintain tissue homeostasis through mechanisms involving FoxP3 and the metabolism of prostaglandin E2, thus regulating local immune responses and preventing excessive inflammation. In the gut, Treg cells are influenced by signals from microbial fermentation products and gut hormones, adjusting their metabolism to maintain mucosal tolerance and epithelial barrier integrity, thereby preventing excessive immune responses that could damage intestinal tissues (9).
This study elucidates the connections between Tregs and relevant metabolic pathways, as well as different metabolic processes. The adaptability of Tregs in diverse microenvironments is also explored. From the perspectives of disease pathogenesis, adaptability, and drug therapy, this article describes the phenotypic differences, dynamic changes, and functional alterations of Tregs in various metabolic diseases. These findings highlight the bidirectional regulatory role of Tregs in disease progression. Finally, the article addresses the topic from a drug therapy standpoint, thus indicating the current efficacy and prospects of therapeutic drugs for different diseases. This study will reveal the diversity of Tregs that are involved in immuno- metabolism, thus emphasizing the critical importance of Tregs stability in immune metabolism.
The development, differentiation, and functional maintenance of Tregs are intricately regulated by key signaling pathways and metabolic processes. Their activation and proliferative capacity primarily depend on pathways such as the mTOR pathway and glycolysis, whereas their functionality relies heavily on lipid metabolism and other metabolic pathways. Herein, we provide a summary of the crucial signaling pathways and various metabolic processes that play regulatory roles in the differentiation, function, and homeostasis of Tregs (Figure 1).
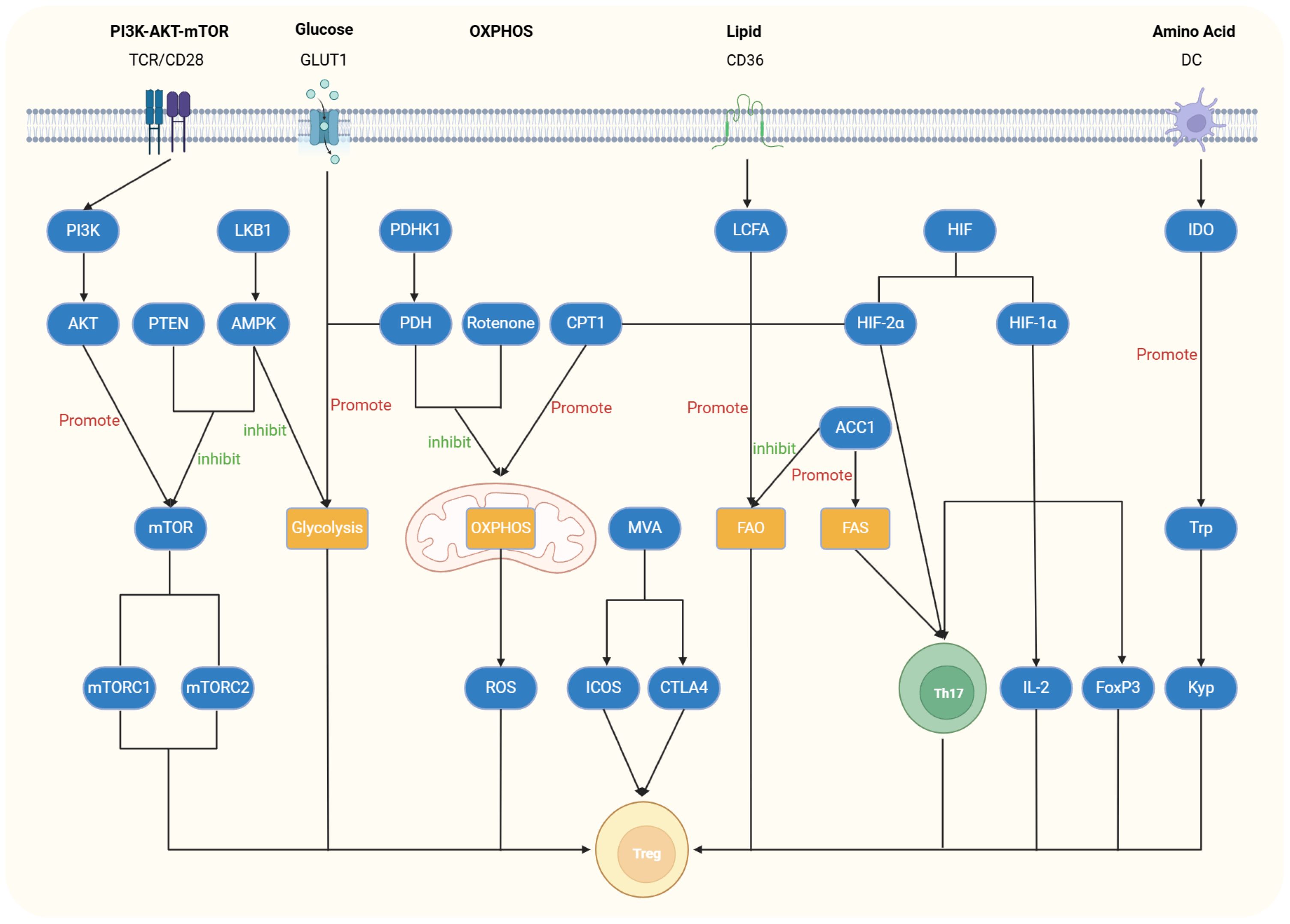
Figure 1. Regulatory role of relevant signaling or metabolic pathways in Tregs. The PI3K-AKT-mTOR pathway is activated by stimulation of T-cell receptors (TCRs) and CD28. The upstream activation of PI3K triggers downstream activation of AKT and Mammalian targets of rapamycin (mTOR), leading to reduced Foxp3 expression and inhibition of Treg cell differentiation and function. Activation of liver kinase B1 (LKB1), AMP-activated protein kinase (AMPK), or Phosphatase and Tensin Homolog (PTEN) can inhibit this pathway and restore Treg differentiation and function. In the glycolytic pathway, glucose transporter 1 (GLUT1) plays a crucial role, and its deficiency inhibits this pathway while enhancing the proliferative capacity of Treg cells. Pyruvate dehydrogenase kinase 1 (PDHK1), as an inhibitor of Pyruvate dehydrogenase (PDH), promotes glycolysis, thereby reducing Treg differentiation and function. Similarly, LKB1, which inhibits the mTOR pathway, also exerts an inhibitory effect in the glycolytic pathway. In the oxidative phosphorylation (OXPHOS) pathway, Carnitine palmitoyltransferase 1 (CPT1) enhances this process, activating acetyl-CoA and increasing reactive oxygen species (ROS) production, which promotes Treg differentiation. Conversely, PDHK1 and mitochondrial electron transport chain inhibitors such as rotenone inhibit OXPHOS, thereby suppressing Treg differentiation and function. In lipid metabolism, Treg cells absorb Long-chain fatty acids (LCFAs) through CD36, promoting the fatty acid oxidation (FAO) pathway and inducing their differentiation. CPT1 and hypoxia-inducible factor-2α (HIF-2α) also participate in the FAO pathway, promoting Treg differentiation. Acetyl-coenzyme A carboxylase 1 (ACC1) not only promotes the fatty acid synthesis (FAS) pathway and Th17 cell differentiation but also inhibits FAO and Treg differentiation. In amino acid metabolism, Indoleamine 2,3-dioxygenase (IDO) converts tryptophan into kynurenine, thereby promoting the proliferation, differentiation, and function of Treg cells.
The T cell receptor (TCR) can activate the PI3K-AKT-mTOR signaling pathway, in which phosphoinositide 3-kinase (PI3K) is activated by the TCR and subsequently activates downstream AKT. Phosphorylated AKT can inhibit the expression of Foxp3, thereby suppressing the differentiation and function of Tregs (10). PI3K, which is positioned upstream of mTOR, contributes to the differentiation and function of Tregs when weakened. mTOR, which is a noncanonical serine/threonine protein kinase, exists in two complexes (mTORC1 and mTORC2) (11). mTOR plays crucial regulatory roles in the activation, proliferation, and function of Tregs (12). Specifically, mTOR regulates the availability of nutrients, particularly glucose and lipid metabolism, influencing the differentiation and function of Tregs. mTORC1 senses the cellular nutrient status (e.g., amino acids, glucose) to regulate Treg metabolism, promoting their proliferation and immune suppressive function. In glucose metabolism, mTORC1 promotes glycolysis, which is crucial for Treg activation and function. Simultaneously, mTORC1 also influences lipid metabolism, regulating the synthesis and breakdown of fatty acids, thus supporting Treg proliferation and maintenance.
Studies have indicated that Tregs lacking Phosphatase and Tensin Homolog (PTEN) enhance mTOR and impair Tregs differentiation and function, thereby promoting the development of various metabolic diseases (13). The serine-threonine kinase liver kinase B1 (LKB1) activates AMPK, thus inhibiting glucose transporter 1 (GLUT1) (This membrane protein is responsible for transporting glucose from the extracellular space into the cell. It belongs to the glucose transporter (GLUT) family and is widely expressed in various tissues, especially in cell types that require a continuous supply of glucose, such as the brain, red blood cells, and embryonic tissues) and mTOR and promoting Tregs differentiation and function (14, 15). Tregs lacking LKB1 exhibit compromised anti-inflammatory functions (16).
Compared with those in helper CD4+ T cells, GLUT1 expression levels are lower in Tregs. Therefore, GLUT1 deficiency or the use of glycolysis-inhibiting drugs such as 2-DG (2-deoxy-D-glucose) can increase the proliferation and viability of Tregs (17).
There is considerable controversy regarding the impact of hypoxia on Tregs. Some studies suggest that hypoxia-inducible factor-1α (HIF-1α) is upregulated during inflammation, thus increasing the expression of IL-2 and Foxp3 and thereby promoting the activation and proliferation of Tregs (18, 19). However, many other studies have reported that HIF-1α promotes the generation of Th17 cells and that its binding to Foxp3 leads to its degradation, thereby inhibiting the differentiation and function of Tregs (20–22).
Additionally, hypoxia-inducible factor-2α (HIF-2α) has been reported to influence the differentiation and function of Tregs, thus exhibiting a bidirectional regulatory effect. HIF-2α can restore the inhibitory function of Tregs by inhibiting HIF-1α, which reduces the differentiation of Th17 cells. It can also indirectly enhance glycolysis by inhibiting fatty acid oxidation (FAO), thereby suppressing the differentiation and function of Tregs (23). Furthermore, the coactivator of the Nedd4 family of E3 ubiquitin ligases (Ndfip1) maintains the stability of Tregs by limiting mTORC1 signaling and glycolysis. Although the number of Tregs lacking Ndfip1 remains unchanged, their phenotype undergoes significant alterations. These cells exhibited a highly proliferative state, elevated IL-4 levels, and a tendency to lose Foxp3 expression. This metabolic adaptation might be linked to the onset of inflammation (24).
FOXP3 can upregulate the activity of all the mitochondrial respiratory complexes, thus promoting OXPHOS and FAO (25). Unlike other T cells, Tregs can maintain their differentiation status and activity through the OXPHOS pathway. Consequently, Tregs can utilize OXPHOS to compensate for losses incurred by other impaired metabolic pathways, thus ensuring the continued maintenance of their differentiation and function. However, excessive enhancement of OXPHOS in Tregs may increase mitochondrial reactive oxygen species (ROS) production, thus leading to disruptions in cellular redox homeostasis and oxidative stress (26).
Carnitine palmitoyltransferase 1 (CPT1) enhances OXPHOS, activates acetyl- coenzyme A, and increases the number of Tregs (27). Conversely, mitochondrial electron transport chain inhibitors such as rotenone can inhibit mitochondrial OXPHOS, thus impairing the suppressive ability of Tregs (28). Pyruvate dehydrogenase (PDH) converts pyruvate to acetyl-coenzyme A, thus promoting mitochondrial OXPHOS. Pyruvate dehydrogenase kinase 1 (PDHK1), which is highly expressed in Th17 cells, inhibits PDH, which promotes aerobic glycolysis while inhibiting OXPHOS, thereby reducing the differentiation and function of Tregs. The inhibition of PDHK1 can weaken Th17 cells, enhance Tregs, and reduce the occurrence and development of inflammation (29).
Tregs absorb long-chain fatty acids through CD36, thus enabling their stable participation in the FAO pathway (30). Tregs can regulate the activity of CPT1, which leads to an increase in FAO. Inhibitors of CPT1 suppress Tregs differentiation, thus impacting FAO (31). Additionally, the upregulation of fatty acid synthesis (FAS) promotes the differentiation of Th17 cells, which downregulates FAO. Due to the fact that FAO is essential for Tregs differentiation, lipid metabolism to some extent determines the differentiation fate of CD4+ T cells. The inhibition of both FAS and FAO results in an imbalance between Th17 cells and Tregs (32). In a highly glycolytic environment, Th17 cells promote de novo fatty acid synthesis via acetyl-coenzyme A carboxylase 1 (ACC1). The inhibition of ACC1 not only suppresses glycolytic metabolism and de novo fatty acid synthesis but also reduces Th17 cell differentiation while increasing Tregs production (33). Methylhydroxybutyrate is crucial for the proliferation and inhibitory function of Tregs. The inhibition of the synthesis of methylhydroxybutyrate reduces the expression of inducible costimulatory molecule (ICOS) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4), thus weakening the inhibitory capacity of Tregs and enhancing inflammation (34). Moreover, the basic leucine zipper ATF-like transcription factor (BATF) can inhibit triglyceride synthesis. Tregs lacking BATF exhibit a significant increase in triglyceride expression, which severely affects the differentiation and function of Tregs (35).
Indoleamine 2,3-dioxygenase (IDO) in dendritic cells (DCs) and tumor cells converts tryptophan into kynurenine, thus promoting the proliferation and differentiation of Tregs (36). The absence of glutamine can inhibit mTOR, thus leading to biased differentiation of Th1 cells toward Tregs (37). Studies have shown that the small G proteins Rag and Rheb maintain mTOR activity by regulating amino acid metabolism, thereby inhibiting the differentiation and function of Tregs (38).
The self-adaptive ability of Tregs allows them to survive in various microenvironments, such as the gastrointestinal tract, liver, and adipose tissue. They undergo metabolic reprogramming based on the specific environment, thus correspondingly altering their phenotype and function. The roles played by Treg cells in different microenvironments can significantly vary. They can play protective anti-inflammatory roles; however, they can also act as pathogenic proinflammatory agents. The generation of these subtypes is often driven by the need to adapt to the specific microenvironment in which Treg cells find themselves (39). In this context, we discuss the adaptability of Tregs in different microenvironments and the resulting changes in their phenotype and function (Figure 2).
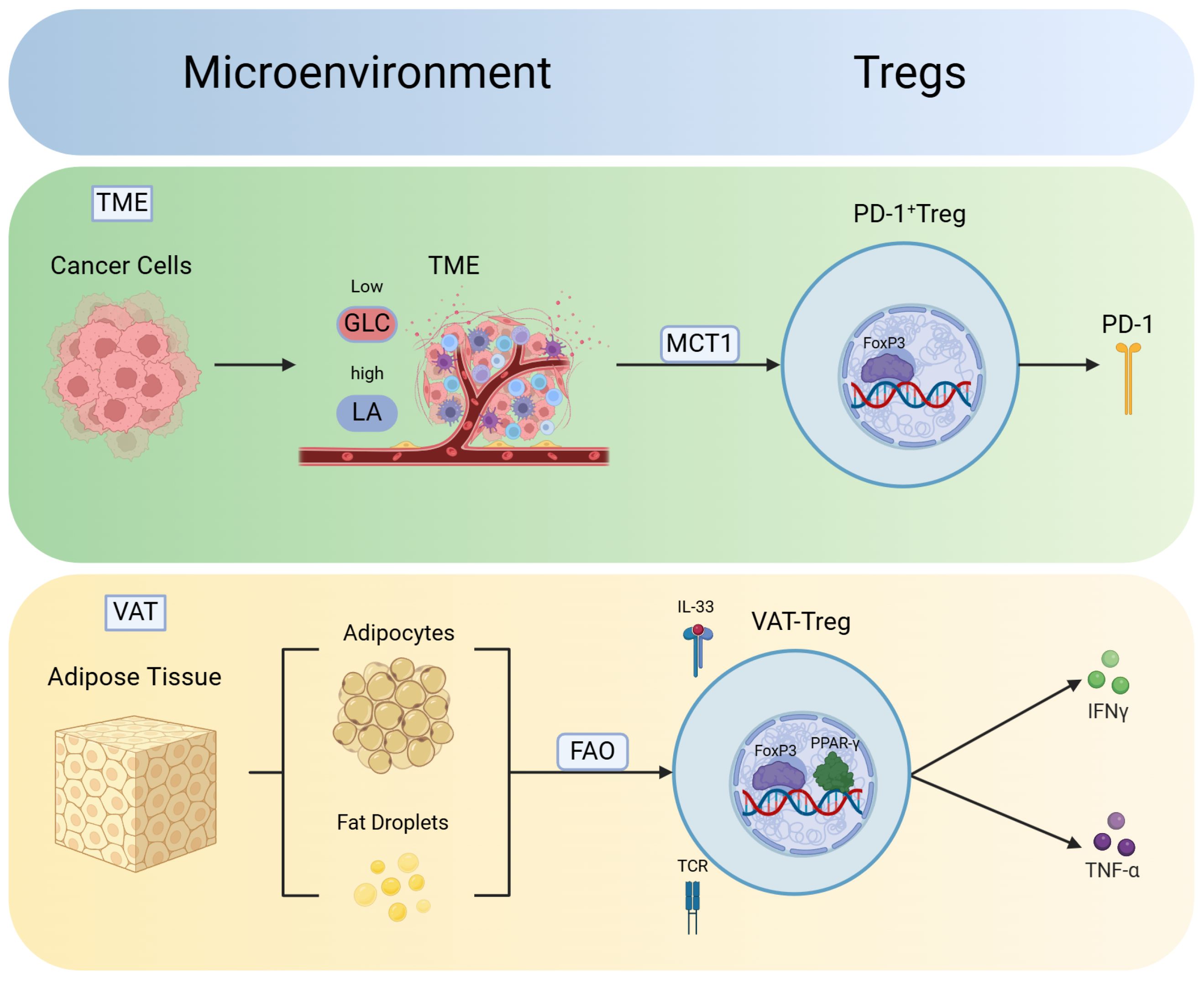
Figure 2. Phenotype and function of Tregs vary in different microenvironments. Tregs exhibit robust adaptability in different microenvironments, thus leading to the development of unique phenotypes. These findings indicate the plasticity of Tregs in response to various environmental conditions. Moreover, Tregs in different microenvironments acquire distinct functions to adapt to changes in the environment and phenotype. In the tumor microenvironment (TME), Tregs absorb lactate (LA) via monocarboxylate transporter 1 (MCT1), thus promoting the generation of PD-1 and differentiation into PD-1+ Tregs. In the visceral adipose tissue (VAT), Tregs utilize FAO pathways to mitigate lipotoxicity. T-cell receptors (TCRs) signaling and IL-33 contribute to the expression of Peroxisome proliferator-activated receptor gamma (PPARγ), thus facilitating the differentiation of VAT-Tregs and the release of inflammatory factors such as IFN-γ and TNF-α.
Tumor cells compete for nutrient resources from effector T cells, thus converting them into energy and auxiliary factors and creating a tumor-infiltrating microenvironment (TME) (40, 41). Cancer cells in the TME primarily produce lactate via glycolysis, which aids in immune escape. Various cell types, including Tregs, exist in the TME, which consists of tumor cells, immune cells, and fibroblasts, among other cell types (42). The low-glucose, high-lactate TME created by tumor cells not only is harmless to the normal survival of Tregs but also promotes their proliferation and differentiation (25). Tregs take up lactate (LA) through monocarboxylate transporter 1 (MCT1) and increases the expression of PD-1, thus activating the PD-1+ Treg subset. This subset significantly inhibits the function of effector T cells, such as CD8+ T cells, thus rendering anti-PD-1 therapy ineffective (43).
Visceral adipose tissue (VAT) serves as the primary site for energy storage in organisms, whereby it regulates biological activities through the intake of nutrients. VAT-Tregs can adapt to the lipid-rich environment and utilize fatty acids via pathways such as FAO. This adaptation allows them to avoid lipid toxicity while maintaining homeostasis in the VAT environment. However, in obese individuals, the production of type I interferons (IFN-α) secreted by plasmacytoid dendritic cells (pDCs) is increased, which directly inhibits the accumulation of VAT-Tregs, leading to their dysfunction and impaired adaptability in the lipid-rich environment. The quantity of these cells decreases with increases in indicators such as body mass index, thus leading to elevated levels of inflammatory factors such as IFN-γ and TNF-α (44, 45). Peroxisome proliferator-activated receptor gamma (PPARγ) is a key transcription factor that drives the expression of VAT-Tregs. The specific loss of PPARγ significantly reduces the quantity of VAT-Tregs (46). Studies have indicated that TCR signal transduction and IL-33 can increase the number of VAT-Tregs and increase PPARγ expression, thereby reducing inflammation in obese individuals (47).
The central nervous system (CNS) has long been considered an immune-privileged site, but recent evidence indicates that immune cells, including Tregs, can infiltrate the CNS under both physiological and pathological conditions (48). Tregs play a critical role in regulating neuroinflammation, particularly in diseases such as multiple sclerosis (MS) (49). In the CNS microenvironment, Tregs primarily regulate immune responses by inhibiting the activation of microglia and effector T cells. Additionally, they maintain the integrity of the blood-brain barrier, preventing immune cell infiltration and reducing inflammation (50). CNS-resident Tregs undergo metabolic reprogramming via glycolysis and fatty acid oxidation (FAO) to adapt to the nutrient-scarce environment. Moreover, CNS-resident Tregs respond to signals such as IL-10 and TGF-β, regulating neuroinflammatory responses and promoting tissue repair through interactions with glial cells (51).
In the respiratory system, Tregs play a crucial role in maintaining immune homeostasis and preventing excessive immune responses to inhaled antigens, such as allergens, pathogens, and pollutants. In the pulmonary microenvironment, Tregs control inflammation by regulating the activity of alveolar macrophages, dendritic cells, and other effector immune cells (52). They are particularly important in chronic respiratory diseases, where Tregs prevent excessive immune responses and tissue damage (53). Pulmonary Tregs are regulated by signals such as IL-10 and TGF-β, and they undergo metabolic adaptation to efficiently function in the hypoxic and inflammatory environment of the lungs. Furthermore, in cases of acute respiratory infections, such as influenza or COVID-19 viral infections, Tregs help attenuate excessive immune responses, thereby protecting lung tissue from damage caused by inflammation (54).
The development of metabolic diseases is significantly influenced by disruptions in Treg metabolism. Metabolic changes, such as altered nutrient availability and energy metabolism, can impact Treg differentiation, function, and stability. For example, shifts in fatty acid oxidation, glycolysis, and mitochondrial function can impair Treg activity, leading to an imbalance in immune regulation. This dysfunction of Tregs may result in either excessive or insufficient immune tolerance, promoting the imbalance of Th17/Treg cells and triggering inflammation, which contributes to the progression of metabolic diseases (55) (Figure 3, Table 1).
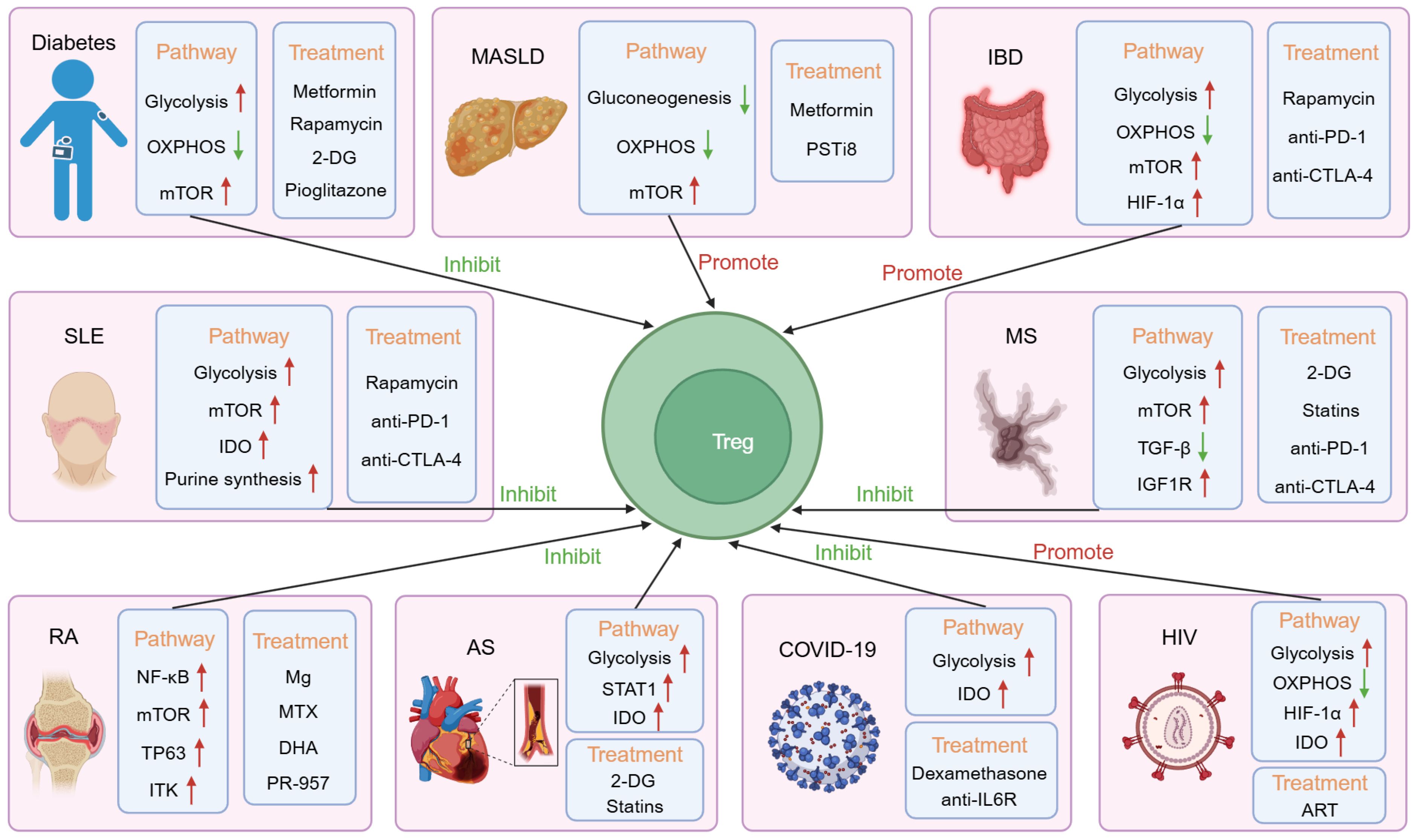
Figure 3. The mechanisms, adaptability, and therapeutic implications of Treg immunemetabolism in various diseases. In diabetes, both glycolysis and the mammalian targets of rapamycin (mTOR) pathway are upregulated, while the oxidative phosphorylation (OXPHOS) pathway is downregulated, which has an inhibitory effect on Tregs. The use of metformin, rapamycin, 2-deoxy-D-glucose (2-DG), and pioglitazone can alleviate the condition. In Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD), the mTOR pathway is upregulated, while gluconeogenesis and the oxidative phosphorylation pathway are downregulated, promoting Treg function. Metformin and pancreatic suppressor (PST) inhibitor (PSTi8) can alleviate the condition. In Inflammatory Bowel Disease (IBD), glycolysis, mTOR, and the hypoxia-inducible factor-1α (HIF-1α) pathway are upregulated, while the oxidative phosphorylation pathway is downregulated, promoting Treg function. Rapamycin, anti-PD-1, and anti-CTLA-4 can alleviate the condition. In Systemic Lupus Erythematosus (SLE), glycolysis, mTOR, Indoleamine 2,3-dioxygenase (IDO), and purine synthesis pathways are upregulated, inhibiting Treg function. Rapamycin, anti-PD-1, and anti-CTLA-4 can alleviate the condition. In Rheumatoid arthritis (RA), the NF-κB, mTOR, TP63, and interleukin-2-inducible T cell kinase (ITK) pathways are upregulated, inhibiting Treg function. Mg, Methotrexate (MTX), dihydroartemisinin (DHA), and PR-957 can alleviate the condition. In Multiple Sclerosis (MS), glycolysis, mTOR, and Insulin-like Growth Factor 1 Receptor (IGF1R) pathways are upregulated, while the TGF-β pathway is downregulated, inhibiting Treg function. 2-DG, statins, anti-PD-1, and anti-CTLA-4 can alleviate the condition. In Arteriosclerosis (AS), glycolysis, STAT1, and IDO pathways are upregulated, inhibiting Treg function. 2-DG and statins can alleviate the condition. In COVID-19, glycolysis and IDO pathways are upregulated, inhibiting Treg function. Dexamethasone and anti-IL-6R can alleviate the condition. In HIV, glycolysis, HIF-1α, and IDO pathways are upregulated, while the oxidative phosphorylation pathway is downregulated, promoting Treg function. Antiretroviral therapy (ART) can alleviate the condition.
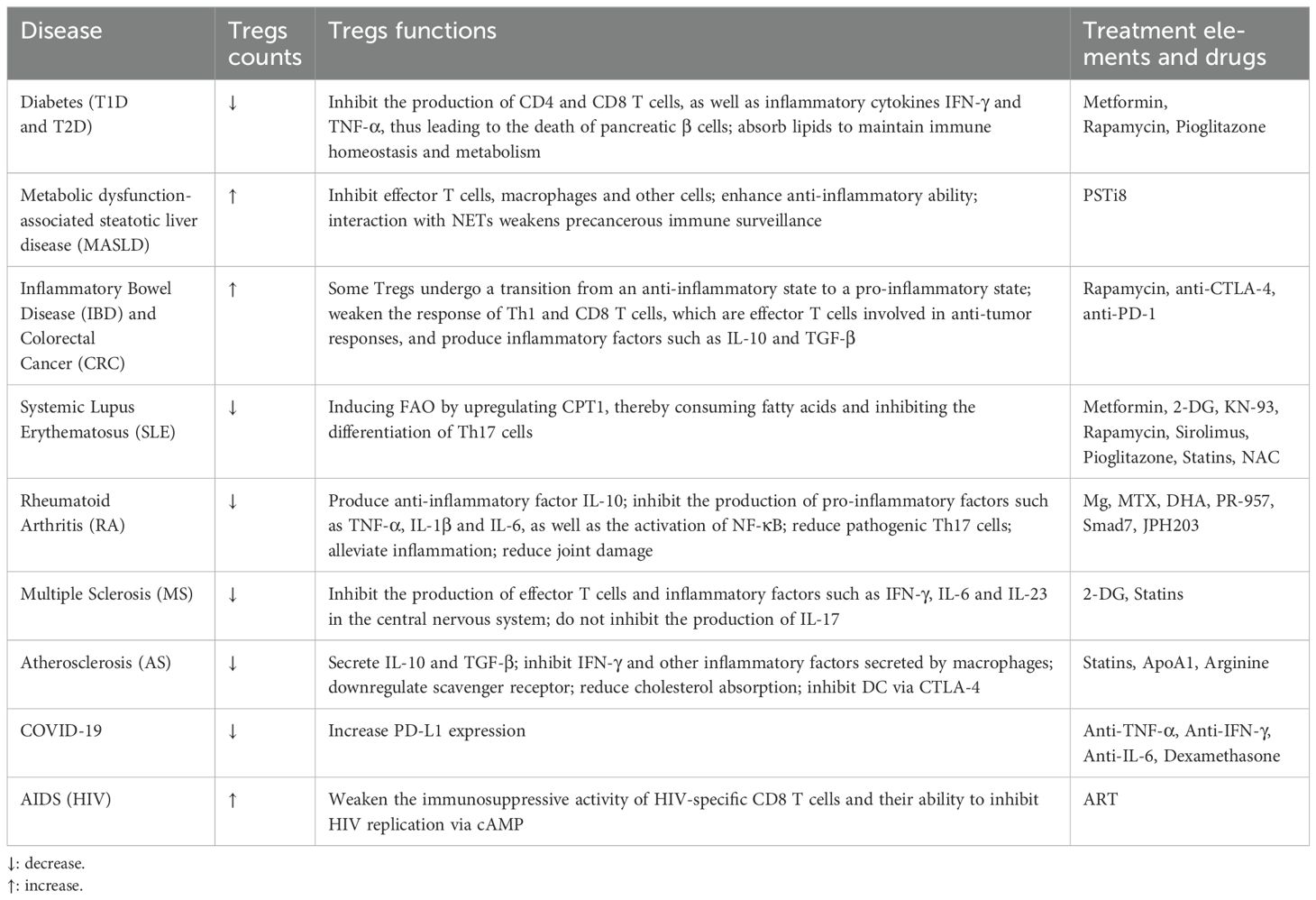
Table 1. Differentiation, functions, and therapeutic approaches of tregs in various metabolic diseases.
Diabetes is a chronic metabolic disorder characterized by elevated blood sugar levels; in severe cases, it can lead to a range of complications, such as cardiovascular diseases and kidney disorders. It can be classified into type 1 diabetes (T1D) and type 2 diabetes (T2D) (56, 57). T1D is a T cell-mediated chronic metabolic disease characterized by impaired pancreatic beta-cell function (56, 58). Various factors, including genetic and environmental factors, contribute to the development of T1D (59). CD4+ and CD8+ T cells release inflammatory cytokines such as TNF-α and IFN-γ, thus causing the death of pancreatic beta cells and triggering T1D (60). iTregs play a crucial role in immune tolerance by suppressing the production of inflammatory cytokines, such as IFN-γ, by CD4+ and CD8+ T cells, and by blocking the mTOR signaling pathway. This helps alleviate inflammation and reduce the severity of T1D (61).
Compared with T1D, T2D accounts for the majority of diabetes cases. T2D is a chronic inflammatory disorder caused by metabolic disturbances in glucose and lipid metabolism and is characterized by insulin resistance and dysfunction of pancreatic beta cells (57, 62). T2D patients exhibit an increased Th1/Treg and Th17/Treg ratio, characterized by a significant elevation in Th1 and Th17 levels and a notable reduction in Treg levels. As a result, there is an increase in pro-inflammatory factors such as IL-6 and IL-17, leading to the formation of a pro-inflammatory environment, while anti-inflammatory factors such as IL-10 are suppressed (63). Tregs are crucial for T2D patients and are capable of absorbing lipids and maintaining immune homeostasis and metabolism. However, T2D patients exhibit a significant reduction in pTregs in adipose tissue, which may be a crucial factor in the development of this disease (44, 64).
The first-line diabetes treatment known as metformin inhibits Th1 and Th17 cell differentiation while enhancing Treg cell proliferation and differentiation, which slows disease progression (65). The mTOR inhibitor rapamycin, combined with IL-2 therapy, can suppress Th17 cells, increase Treg cells, and restore beta-cell function, thus achieving therapeutic effects (66). The glycolysis inhibitor 2-DG balances the Th17/Treg ratio by reducing glycolysis and promoting OXPHOS, thereby increasing the granularity of pancreatic β-cells (67). Additionally, PPARγ-driven lipid metabolism regulates the phenotype of adipose tissue pTregs. The use of synthetic PPARγ agonists such as pioglitazone effectively enhances the expression of lipid metabolism genes, thus allowing pTregs to utilize lipid metabolism for energy and thereby resisting inflammation and reducing the progression of diabetes (44, 68).
Non-alcoholic fatty liver disease (NAFLD), has been renamed as MASLD, refers to a clinical and pathological syndrome characterized by excessive fat deposition in the liver, with the exclusion of alcohol-related factors. It encompasses a spectrum of conditions ranging from simple hepatic steatosis (SFL) and metabolic dysfunction- associated steatohepatitis (MASH) to liver cirrhosis (LC) and hepatocellular carcinoma (HCC). This progression involves liver fat degeneration, inflammation, hepatocellular damage, and liver fibrosis (69, 70).
The role of Tregs in MASLD is currently a subject of considerable debate. Some studies have suggested that iTregs reduce liver inflammation by suppressing effector T cells, neutrophils, and macrophages (71, 72). It has been reported that a pancreatic suppressor (PST) inhibitor (PSTi8) can treat MASH in mice fed a methionine-choline-deficient (MCD) diet (73). PST regulates glucose and lipid metabolism in the liver and adipose tissue, thus promoting fat degeneration and inflammation. It acts against insulin while altering mitochondrial function (74). PSTi8 enhances Tregs differentiation and strengthens their suppressive ability. Moreover, PSTi8 alleviates hepatic fat degeneration and fibrosis in MCD-fed mice, reduces liver damage markers such as ALT, LDL, and TC, and ameliorates mitochondrial dysfunction. Research has demonstrated that PSTi8 alleviates MASH-related inflammation and fat degeneration by enhancing the anti-inflammatory ability of iTregs. However, at times, the suppressive capabilities of Tregs can be exploited by other diseases and cells, thus leading to unintended consequences. For example, the interaction between Tregs and neutrophil extracellular traps (NETs) may promote the transition from MASH to HCC.
Neutrophils, which are abundant in MASH patients, release chromatin-related traps known as NETs (75). NETs can upregulate the expression of genes related to OXPHOS, increase mitochondrial respiration, and enhance the activity of transcription factors associated with Treg differentiation, thereby increasing the number and function of Tregs. In liver cancer tissues, the increased number of Tregs can suppress immune surveillance through multiple mechanisms, such as inhibiting the function and proliferation of effector T cells (Teffs) and reducing the cytotoxic activity of immune cells against tumor cells. Through Toll-like receptor 4 (TLR4), NETs induce metabolic reprogramming and promote the initial differentiation of CD4+ T cells into Tregs via the mitochondrial OXPHOS pathway. This effect weakens immune surveillance in the precancerous stage, thus creating an immune-suppressive environment that is conducive to cancer cell survival and growth (76). Additionally, the dual-regulatory protein of Tregs known as amphiregulin (AREG) can activate hepatic stellate cells (HSCs) via the EGFR signaling pathway, thereby promoting liver fibrosis and insulin resistance in MASH (77).
Inflammatory bowel disease (IBD), which is comprised of Crohn’s disease (CD) and ulcerative colitis (UC), is characterized by chronic inflammation in the gastrointestinal tract, which often leads to tissue damage (78). Prolonged IBD can increase the risk of colorectal cancer (CRC) development (79). In general, Tregs play a role in anti-inflammatory responses and the maintenance of immune homeostasis. However, research suggests that in IBD, the upregulation of IL-21 disrupts multiple metabolic pathways, including oxidative phosphorylation (OXPHOS) and glycolysis, thus leading to disturbances in the structure of the mitochondria-endoplasmic reticulum (ER) network (80). This upregulation enhances the activity of glycogen synthase kinase 3β (GSK3β), thus inhibiting the voltage-dependent anion channel (VDAC) in the mitochondria and causing some Tregs to undergo a transition from an anti-inflammatory state to a proinflammatory state. Research has also confirmed that the inhibition of IL-21 or GSK3β can alleviate the symptoms of IBD (81). Additionally, the increase in glycolysis in IBD contributes to the differentiation of Th17 cells, which leads to an imbalance between proinflammatory Th17 cells and anti-inflammatory iTregs, thus further exacerbating inflammation (82).
Numerous studies have indicated that inhibition of the mechanistic target of rapamycin (mTOR) can reduce the differentiation of Th17 cells while increasing the population of anti-inflammatory iTregs (83, 84). These findings suggest that the targeting of mTOR may be a potential therapeutic approach to ameliorate inflammation caused by IBD (83, 84). Moreover, HIF-1α plays a crucial role in controlling the differentiation of Tregs in IBD, and its regulation is vital for improving inflammation (20, 85). Therefore, the targeting of key metabolic hubs (such as mTOR and HIF-1α) to restore the function of anti-inflammatory iTregs could be beneficial for targeted immuno- metabolic therapy in IBD (86).
Furthermore, previous studies have indicated that when IBD progresses to CRC, the number and function of Tregs change, thus resulting in characteristics similar to those of Th17 cells (87). In CRC, tumor cells rely on glycolysis, glutaminolysis, and FAS for metabolic reprogramming. Tregs adapt to these changes in metabolic pathways and enhance their differentiation and inhibitory functions in the TME. This correspondingly weakens the antitumor immune response of Th1 and CD8+ T cells, thus promoting the immune escape of tumor cells and contributing to the development of CRC. In CRC, Tregs also express coinhibitory receptors such as LAG-3 and TIM-3 (88). LAG-3+ Treg cells increase the production of IL-10 and TGF-β, as well as the expression of CTLA-4. Currently, therapeutic approaches targeting tumor-associated Treg cells, such as anti-CTLA-4 and anti-PD-1 treatments, are crucial for alleviating and suppressing CRC. The combination of anti-CTLA-4 monoclonal antibodies with anti-PD-1 monoclonal antibodies holds promise for achieving enhanced therapeutic effects (89, 90).
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by chronic inflammation and excessive activation of immune cells such as B cells and T cells (91). The activation and differentiation of immune cells in SLE patients are closely linked to alterations in metabolic pathways. Metabolic abnormalities can significantly impact immune cell function, thereby exacerbating autoimmune responses and inflammation. For example, in SLE patients, T cells and B cells undergo metabolic reprogramming, which alters their energy metabolism and functional characteristics. This may promote the release of inflammatory cytokines and cause tissue damage. Tregs play a crucial role in these metabolic changes. nTregs suppress inflammation by maintaining immune tolerance, but their function is also affected by metabolic abnormalities, which may lead to immune dysregulation, further exacerbating the pathogenesis of SLE (92, 93).
In SLE mice, oral administration of metformin can increase nTregs, reduce Th17 cells, inhibit mTOR and signal transducer and activator of transcription 3 (STAT3) signaling, as well as decrease the severity of inflammation. Furthermore, the addition of 2-DG can restore T cell metabolism and alleviate inflammation in SLE patients (94, 95). The overexpression of calcium/calmodulin-dependent protein kinase IV (CaMK4) can increase mTOR activity, thus promoting Th17 cell differentiation (96). The CaMK4 inhibitor KN-93 can increase the number of Tregs in diseased mice and decrease the number of inflammatory immune cells, such as Th17 cells (97). Similarly, rapamycin and sirolimus, as mTOR inhibitors, can increase the number of Tregs by inhibiting mTOR, thus reducing pathogenic T-cell subgroups (including Th17 cells) and decreasing proinflammatory cytokines, among others (98, 99). Additionally, the insulin sensitizer pioglitazone can increase the proportion of Tregs by inhibiting mTOR and activating AMPK in SLE patients (100).
Acetyl-CoA carboxylase 1 (ACC1) in lipid metabolism is an important regulator of Th1 and Th17 cells, thus promoting their differentiation via metabolic reprogramming (33, 101). Conversely, Tregs induce FAO by upregulating CPT I, thus inhibiting the differentiation of Th17 cells by consuming fatty acids. Statin drugs may also help in regulating the balance between Tregs and Th17 cells, thus contributing to the treatment of SLE patients (102). In amino acid metabolism, the expression of IDO can limit the secretion of inflammatory cytokines, promote the generation of Treg cells, and regulate the Th17/Treg balance (103). Additionally, mycophenolic acid can inhibit purine synthesis, suppress CD4+ T cell proliferation, and alter its phenotype, as well as activate the CD70 signaling pathway. Consequently, mycophenolic acid can reduce the production of the inflammatory cytokine IL-17 and inflammatory factors IFN-γ and TNF-α, decrease cellular oxygen consumption and glycolytic activity, and enhance the activity and function of Tregs (104).
Previous studies have revealed changes in the mitochondrial membrane potential and increased ROS in the lymphocytes of SLE patients (105). Treatment with N-acetylcysteine (NAC) can alleviate this phenomenon by reducing mTOR activity, thus increasing T cell mitochondrial membrane potential and cell apoptosis, as well as inducing the expression of nTregs in SLE patients (106). The above studies suggest that SLE is not only a typical autoimmune disease but also closely associated with immune metabolic abnormalities. Changes in immune metabolism play a crucial role in immune dysregulation, inflammatory responses, and the clinical manifestations of SLE.
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by joint destruction and synovial inflammation, with severe cases potentially leading to disability (107). The development of RA is closely linked to changes in immune metabolism, particularly the metabolic reprogramming of Tregs, which plays a crucial role in the immune imbalance seen in RA. Alterations in metabolic pathways not only affect the quantity and function of nTregs but also have the potential to modulate the metabolic environment, thereby influencing the activity of immune cells and exacerbating inflammation and joint damage in RA (108).
Studies have shown that nutritional elements such as magnesium (Mg) play an important role in regulating immune metabolism in RA. Magnesium can reduce the production of pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6, and exert anti-inflammatory effects by inhibiting NF-κB activation (109). A high-magnesium diet alters the metabolic environment of the gut microbiota, promotes the differentiation of Tregs, and increases the production of anti-inflammatory cytokine IL-10. This, in turn, reduces the differentiation of pathogenic Th17 cells, alleviates inflammation, and lessens the clinical symptoms of RA (110, 111). TP63 is highly expressed in Th17 cells; however, its expression is lower in nTregs because of its inhibition of Foxp3 (112). Methotrexate (MTX), which is a folate antagonist, is a standard first-line treatment for RA. It inhibits TP63 and Th17 cells while restoring the inhibitory function of nTregs, thereby improving RA (113).
TCR activation induces interleukin-2-inducible T cell kinase (ITK), which regulates the development of CD4+ T cell subtypes (114). Elevated levels of ITK have been observed in RA patients (115). The inhibitor of ITK known as dihydroartemisinin (DHA) acts by inhibiting the PI3K-Akt-mTOR signaling pathway. This mechanism results in a reduction in Th17 cells and an increase in nTregs, thus restoring the balance between the two cell types. As a result, the progression of RA is impeded (115, 116). Low-molecular-mass polypeptide (LMP7) is an immuno- proteasome subunit that can amplify inflammatory T cells and cytokines. It is highly expressed in patients with RA (117). Research indicates that the LMP7 inhibitor PR-957 can reduce the differentiation of Th1 and Th17 cells and promote the production of nTregs, thus alleviating RA (118). Smad7 is a key negative regulator of the TGF-β signaling pathway; specifically, it inhibits the TGF-β and NF-κB pathways, limits RA development, and maintains the balance between Th17 cells and Tregs (119). DNA methylation is a chemical modification that alters gene expression and is capable of changing chromosome structure and DNA stability, among other effects (120). Research has shown that DNA methylation of Smad7 in patients with RA can decrease its expression levels, thus leading to joint inflammation and damage and promoting Th17 cell differentiation while reducing nTregs differentiation (121).
The overexpression of Smad7 can inhibit the activation of NF-κB, block IL-1R and TNFR signaling, reduce proinflammatory cytokine levels and anti- apoptotic signal activity, thereby restoring the Th17/Treg balance and effectively alleviating RA to some extent (122). CD4+ T cells in the synovium of RA patients express L-type amino acid transporter 1 (LAT1), which is a potential diagnostic and therapeutic target for emerging diseases such as cancer (123, 124). The loss of LAT1 can inhibit Th17 cell differentiation, reduce the number of proinflammatory cells such as IFN-γ+ CD4+ T cells and TNF-α+ CD4+ T cells, maintain the quantity and suppressive capacity of nTregs, and significantly alleviate arthritis (123). LAT1 loss also downregulates AKT expression, thus inhibiting the PI3K-AKT-mTOR pathway, which is crucial for maintaining the Th17/Treg balance. Research has demonstrated that the LAT1 inhibitor JPH203 successfully alleviates arthritis severity in individuals with RA (125).
Multiple sclerosis (MS) is an autoimmune disease characterized by immune cell infiltration and abnormal immune responses in the central nervous system (CNS) (126). Currently, the experimental autoimmune encephalomyelitis (EAE) animal model is predominantly used in MS research (127). Th17 cells and Tregs are key participants in coordinating the immune response in MS, and their balance largely determines the autoimmune response in the central nervous system and the course of the disease. In MS patients, pro-inflammatory cytokines such as IL-1β, IL-6, TGF-β, and IL-23 are elevated. These cytokines not only affect the differentiation of nTregs but also influence their function through metabolic pathways such as glycolysis and lipid metabolism.
In metabolic regulation, Tregs rely on fatty acid oxidation (FAO) to maintain their suppressive function, while Th17 cells depend more on glycolysis. Therefore, changes in metabolic states, such as alterations in fatty acid and glucose metabolism, can directly affect Tregs’ functionality and promote the generation of pathogenic Th17 cells (128, 129). IL-23 is a crucial driving factor for the pathogenic subset of Th17 cells, thus leading to the development of EAE in mice, whereas nonpathogenic Th17 cells do not contribute to EAE development (130). nTregs in EAE mice are severely depleted and functionally impaired (131). The inhibitory capacity of nTregs in EAE is limited; although they can suppress the production of effector T cells and IFN-γ in the central nervous system, they cannot inhibit the production of IL-17 (132). Currently, the use of 2-DG to inhibit glycolysis, the loss of ACC1 and LDHA, and the use of statin drugs are essential pathways to reduce the differentiation of Th1 and Th17 cells, promote nTregs activation, and improve EAE (20, 33, 133). CTLA-4 can inhibit T cell activation and expansion, thus preventing the occurrence and development of inflammation (134).
In EAE mice, the lack of CTLA-4 in nTregs affects their inhibitory function, and supplementation with IL-10 compensates for the partial loss of CTLA-4, which partially restores the inhibitory function of nTregs (135). Treg-specific inducible PD-1 deficiency can also increase the levels of nTregs in the central nervous system and alleviate the severity of EAE (136). Additionally, the immunoreceptor tyrosine-based inhibitory motif (TIGIT) enhances the ability of Tregs to inhibit Th1 and Th17 cells by promoting the production of IL-10 (137). TGF-β signal transduction affects the differentiation of Tregs and Th17 cells by inducing Smad3 phosphorylation to induce Foxp3 and inhibit IL-6 and IL-23, thereby suppressing the differentiation of Th17 cells and reducing the severity of EAE (138).
An imbalance of Th17/Treg cells in MS also leads to changes in lipid profiles. Unsaturated fatty acids such as oleic acid induce the expression of IL-10 and adiponectin to activate AMPK and inhibit the production of IL-2 and IFN-γ (139). The PPAR family of lipid receptors regulates the differentiation of Th17 cells and Tregs by modulating IL-2 (140). Long-chain fatty acids (LCFAs) and short-chain fatty acids (SCFAs) have opposing effects on T cell differentiation. Specifically, LCFAs promote the differentiation of Th17 cells, thus contributing to the production of IFN-γ and IL-17, whereas SCFAs induce the proliferation of Tregs, thus increasing the expression of FOXP3 (141). Insulin-like growth factor (IGF) is a key regulatory factor in the balance of Th17/Treg cells, primarily by activating AKT and mTOR to suppress the developmental program of early-stage nTregs (142). IGF1R signal transduction promotes glycolysis and activates the PI3K-AKT-mTOR pathway, thereby promoting the differentiation of pathogenic Th17 cells and limiting the activity of nTregs. Therefore, the lack of IGF1R in EAE can reduce pathogenic Th17 cells, thereby alleviating the severity of EAE.
In terms of amino acid metabolism, glutamine uptake increases during Th17 cell activation, thus enhancing the differentiation of Th17 cells and making them pathogenic. The activation of mTOR promotes their pathogenicity during EAE (143). When the glutamine supply is insufficient, initial CD4+ T cells begin to differentiate into pTregs, which results in a corresponding decrease in the production of Th17 cells, thereby mitigating the severity of EAE (144). Additionally, prolonged inflammation in MS leads to changes in the phenotype and function of nTregs, thus resulting in the production of new pathogenic Treg subsets (Th1-like Tregs). This type of Treg, via the activation of the PI3K-AKT-mTOR pathway, secretes IFN-γ and can also play a role in promoting disease (145).
Arteriosclerosis (AS) is a chronic inflammatory disease characterized by the formation of atherosclerotic plaques in arterial walls, thus leading to reduced blood flow or direct blockage; moreover, it is a major cause of cardiovascular diseases (146). During AS, the imbalance between pathogenic Th1 and Th17 cells and protective Th2 and pTregs is a major factor in the occurrence and development of the disease (147). Research indicates that IFNγ secreted by Th1 cells can promote the deterioration of atherosclerotic plaques through various mechanisms, including the promotion of the recruitment of white blood cells and the formation of foam cells, as well as the inhibition of the proliferation of vascular smooth muscle cells and the production of collagen; in contrast, pTregs can effectively prevent the occurrence and development of AS in mice (148). Therefore, maintaining the balance between Th1 cells and pTregs helps to alleviate the progression of arteriosclerosis.
AS leads to a decrease in the number and impaired function of pTregs, as well as a decrease in IL-10 and TGF-β (149). Studies have shown that IL-10 secreted by pTregs can reduce the expression of the inflammatory factor IFN-γ, thus inhibiting AS; furthermore, the overexpression of TGF-β can also have the same effect (150). The microenvironment under AS promotes glycolysis, and Tregs are well suited to this low-glucose, high-lactate environment. However, although this environment promotes glycolysis, the expression of Foxp3, the key transcription factor of Tregs, is affected, leading to impaired proliferation and function of Tregs that should otherwise adapt to this environment, thereby contributing to pathogenesis (25).
Hypoxia is a key driving factor for the formation of atherosclerotic plaques; however, its regulation of Tregs is controversial (151). Some studies have suggested that hypoxia can increase the expression of Foxp3, thus promoting the differentiation and inhibitory functions of Tregs; however, other studies have indicated that hypoxia induces the ubiquitination and degradation of Foxp3 and, in the absence of HIF-1α, contributes to the differentiation of Tregs and the inhibition of the activation of Th17 cells (152). Therefore, the mechanism by which Tregs act on AS in a hypoxic environment still requires further exploration. As self-antigens in AS are presented to T cells by major histocompatibility complex class II (MHC II) molecules on antigen-presenting cells (APCs) and DCs are the predominant APCs, DCs play an indispensable role in atherosclerotic lesions. pTregs can inhibit DCs via coinhibitory molecules such as CTLA-4, thus disrupting T cell activation and reducing AS (153).
Macrophages can secrete proinflammatory cytokines, accelerating the formation of atherosclerotic plaques. Elevated plasma cholesterol levels also lead to AS, affecting the differentiation and function of pTregs. The accumulation of cholesterol alters the phenotype of pTregs, transforming them into proinflammatory Th1-like Tregs. These Tregs secrete the proinflammatory cytokine IFN-γ, activating the IFN-γ/IL-27-mediated STAT1 signaling pathway. As a result, Tregs, which originally have immunosuppressive functions, partially lose their suppressive activity, rendering them ineffective in controlling arterial inflammation and thereby accelerating the progression of atherosclerosis (154). pTregs can inhibit the secretion of inflammatory factors by macrophages, downregulate scavenger receptors on macrophages, reduce cholesterol absorption, and inhibit the formation of foam cells, which are required for AS development (155). As hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, statins can induce the accumulation of pTregs in atherosclerotic plaques and have a protective effect (156). In addition, the use of apolipoprotein A1 (ApoA1) can increase the expression of the cholesterol efflux transporter ABCA1 in pTregs, restore cholesterol levels, prevent the transformation of pTregs into follicular helper T (Tfh) cells, and thereby improve AS (157).
It has been reported that disturbances in amino acid metabolism also affect the development of AS (158). Some studies have suggested that supplementation with high arginine can activate CD4+ T cells and alleviate atherosclerotic plaques via the inhibitory ability of pTregs (159). Furthermore, IDO can induce the differentiation of pTregs and reduce the extent of AS lesions (160).
At the end of 2019, cases of pneumonia of unknown origin first appeared in Wuhan, China, and later led to a widespread outbreak. This sudden pneumonia was later diagnosed as coronavirus disease 2019 (COVID-19) caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (161). To date, more than one hundred million people have been affected by COVID-19, with a significantly higher probability of infection for elderly individuals, individuals with comorbidities, and cancer patients (162). The onset of COVID-19 is accompanied by inflammation and metabolic disturbances (163). COVID-19 infection can activate Th17 cells and suppress Tregs, thus disrupting the balance of Th17/Treg cells and leading to mitochondrial dysfunction (164). Compared to healthy individuals, COVID-19 patients show increased levels of inflammatory factors IL-6 and TNF-α due to the activation of Th17 cells, along with elevated expression of PD-L1 in CD8+ T cells and iTregs, accompanied by disturbances in tryptophan, amino acid, and glucose-lipid metabolism (165).
Research on the immunometabolism of Tregs in COVID-19 is relatively limited; therefore, we have summarized some of the existing studies. Several metabolic pathways pose a risk for increased severity of COVID-19. Studies have shown that patients with metabolic diseases such as diabetes and obesity have an increased mortality rate, which is associated with increased glycolysis and mitochondrial dysfunction in the immune cells of COVID-19 patients: Elevated blood glucose levels directly promote SARS-CoV-2 replication and pro-inflammatory cytokine expression. Glycolytic flux is essential for viral replication, and SARS- CoV-2-induced mtROS production stabilizes HIF-1α, thereby influencing glycolysis and cytokine expression (166). In patients with COVID-19 and diabetes, the levels of pro-inflammatory cytokines such as IL-6 are significantly elevated (167). Furthermore, COVID-19 alters tryptophan metabolism, in which IFN-γ-regulated IDO catalyzes the conversion of tryptophan to kynurenine, thus leading to reduced tryptophan and weakened antitumor capabilities (165). Changes in these metabolic pathways involve the regulation of Th17 cells and Tregs. Currently, treatments targeting TNF-α and IFN-γ are effective at reducing mortality in COVID-19-infected mice (168).
HIV/AIDS is a chronic disease characterized by immune activation and chronic inflammation. HIV infection can lead to immune metabolic imbalance, with CD4+ T cells being its primary target (169). The proliferation, differentiation, maintenance, and migration of iTregs to inflammatory sites require glycolysis. Therefore, in HIV patients, an environment with high glycolysis is often observed, thus leading to an increased number of iTregs (170). During HIV infection, HIV replication directly or by activating innate immune cells induces an increase in the number of iTregs in peripheral blood and mucosal tissues. At the same time, Th17 cells are rapidly depleted, disrupting the Th17/Treg balance. This imbalance leads to systemic immune dysfunction and triggers chronic inflammation (171).
HIV is often co-infected with HBV, and HIV or HIV gp120 can bind to co-receptors such as CCR5 and CXCR4, activating the AKT and ERK signaling pathways, which upregulate HIF-1α in hepatocytes and hepatic stellate cells. This upregulation increases HBV-induced TGF-β1 and pro-fibrotic gene expression, promoting liver fibrosis. Additionally, TGF-β1 can upregulate HIF-1α expression via the SMAD signaling pathway, forming a positive feedback loop that exacerbates the progression of liver fibrosis (172). Under hypoxic conditions, HIF-1α promotes glycolysis and de novo fatty acid synthesis while inhibiting OXPHOS and carnitine palmitoyl- transferase 1A (CPT1A), thereby reducing FAO. HIF-1α promotes Th17 cell differentiation, but its impact on the increase or decrease of Tregs remains controversial (173).
HIV infection results in lower GLUT1 levels in Tregs than in Th17 cells. In contrast, HIV infection increases the number of pathogenic Th17 cells, which are more glycolytic than nonpathogenic Th17 cells. Moreover, via PI3K/AKT-mediated inhibition of mTOR, TGF-β1 promotes iTreg differentiation, reduces the expression of GLUT1 and HK2 in iTregs, inhibits glycolysis, and promotes FAO (174). Although iTregs play a protective role by suppressing immune activation, studies have indicated that iTregs primarily trigger specific immunity in HIV, which is harmful. This effect could be observed through the imbalance between Th17 cells and Tregs (171).
In terms of lipid metabolism, HIV patients exhibit decreased expression of PPAR-γ and pro-lipid genes, which are necessary for lipid synthesis and storage. This suggests that FAO is weakened during HIV infection (175) and that weakened FAO may reciprocally inhibit iTregs production (176). The purine pathway involves various high-energy phosphorylated compounds, such as ATP (177), and it functions via the ectonucleotidases CD39 and CD73, thus generating immunosuppressive adenosine, which binds to G protein-coupled purinergic receptor 1 (P1) (178). P1-adenosine signaling in effector T cells promotes iTregs differentiation and ensures their inhibitory activity (179). An increased proportion of CD39 Tregs is a marker of HIV progression. This can weaken the immunosuppressive activity of HIV-specific CD8+ T cells and the ability of CD8+ T cells to inhibit HIV replication via cAMP (180). During HIV infection, an increased proportion of iTregs is associated with Tregs IDO induction and activity, as well as CTLA-4 expression. CTLA-4 expressed by iTregs can interact with B7 expressed by DCs, thus promoting IDO expression, which leads to an increase in the proportion of iTregs and a decrease in the proportion of Th17 cells (181).
Currently, antiretroviral therapy (ART) for HIV has transformed it from an almost fatal disease to a manageable chronic condition; however, the complete eradication of HIV has not been achieved (182). A consensus has not been reached concerning the impact of ART on the balance between Tregs and Th17 cells. Some studies have suggested that ART can decrease the proportion of iTregs in patients, whereas others have argued that ART cannot restore the proportion of iTregs to normal levels in treated patients. Similarly, there is controversy regarding changes in Th17 cells (183). The main reason for these disparate results may involve differences in ART duration, methods, and Th17/Treg identification methods (184).
Immunometabolism has become a popular research area in recent years. Crosstalk between immune cells and metabolism is a dynamic process involving the activation, inflammation, and resolution of immune cells. This entire process involves numerous metabolic mechanisms, which can be summarized as changes in the phenotype, differentiation, and function of immune cells under the influence of the microenvironment, thus leading to the reprogramming of various metabolic pathways. Therefore, mutual coordination between the functions of immune cells and metabolic activities is crucial for maintaining overall immune metabolic environment homeostasis.
As negative regulators of immune metabolism, Tregs play a role in immune tolerance and in maintaining immune homeostasis. Tregs participate in various metabolic pathways, including glycolysis, lipid metabolism, and amino acid metabolism; moreover, they play different roles in these pathways. The mTOR pathway and glycolysis support Treg development, proliferation, and migration, while OXPHOS enhances their suppressive abilities.
Tregs play dual roles in different metabolic diseases, and the maintenance of the normal differentiation and function of Tregs is the key to maintaining immune metabolic balance. The absorption and discontinuation of nutrients and elements, as well as targeted drug therapy, are important methods for regulating the differentiation and function of Tregs and for restoring metabolic pathways. Different hormones and drugs can have markedly different effects on the proliferative capacity and inhibitory function of Tregs. Insulin and leptin can inhibit the proliferation and function of Tregs, whereas glucocorticoids, rapamycin, metformin, and pioglitazone can maintain and promote the differentiation and function of Tregs. Therefore, Treg-based immuno- therapy contributes to a deeper understanding of Treg differentiation, migration, and function, as well as innovation and optimization of treatment methods.
With the advancement of single-cell sequencing technology in recent years, the related subgroups and cytokines of Tregs will be further revealed. According to various single-cell transcriptomic analyses, Tregs can adapt to different pathological environments and establish entirely new transcriptional characteristics.
In summary, Tregs are a unique immune-tolerant regulatory cell type that plays an indispensable role in maintaining immune system balance and acts as a crucial component of immune metabolism. Therefore, a better understanding of their mechanism of action through the key signaling pathways and metabolic pathways in which they participate is essential for developing Treg cell-based therapeutic drugs and for providing new perspectives for the development of the field of immunometabolism.
MY: Funding acquisition, Writing – original draft, Writing – review & editing. YL: Conceptualization, Visualization, Writing – original draft. YFW: Conceptualization, Writing – original draft. TR: Visualization, Writing – original draft. YHW: Conceptualization, Writing – original draft. LJ: Visualization, Data curation, Writing – original draft. MZ: Conceptualization, Data curation, Writing – original draft. LL: Visualization, Data curation, Writing – original draft. DY: Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The National Natural Science Foundation of China, No. 32470985 and No. 81873915 The Key Plan of Nantong S&T Development, No. MS12020021 The Scientific Research Program of Nantong, No. JC12022091.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hashimoto H, McCallion O, Kempkes RWM, Hester J, Issa F. Distinct metabolic pathways mediate regulatory T cell differentiation and function. Immunol Lett. (2020) 223:53–61. doi: 10.1016/j.imlet.2020.04.011
2. Warburg O, Wind F, Negelein E. THE METABOLISM OF TUMORS IN THE BODY. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
3. Liu J, Ibi D, Taniguchi K, Lee J, Herrema H, Akosman B, et al. Inflammation improves glucose homeostasis through IKKβ-XBP1s interaction. Cell. (2016) 167:1052–66.e18. doi: 10.1016/j.cell.2016.10.015
4. Shi H, Chi H. Metabolic control of treg cell stability, plasticity, and tissue-specific heterogeneity. Front Immunol. (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
5. Yang K. Regulation of treg cell metabolism and function in non-lymphoid tissues. Front Immunol. (2022) 13:909705. doi: 10.3389/fimmu.2022.909705
6. Sumida TS, Cheru NT, Hafler DA. The regulation and differentiation of regulatory T cells and their dysfunction in autoimmune diseases. Nat Rev Immunol. (2024) 24:503–17. doi: 10.1038/s41577-024-00994-x
7. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
8. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. (1970) 18:723–37.
9. de Candia P, Procaccini C, Russo C, Lepore MT, Matarese G. Regulatory T cells as metabolic sensors. Immunity. (2022) 55:1981–92. doi: 10.1016/j.immuni.2022.10.006
10. Pompura SL, Dominguez-Villar M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J Leukoc Biol. (2018) 103:1065–76. doi: 10.1002/JLB.2MIR0817-349R
11. Battaglioni S, Benjamin D, Wälchli M, Maier T, Hall MN. mTOR substrate phosphorylation in growth control. Cell. (2022) 185:1814–36. doi: 10.1016/j.cell.2022.04.013
12. Chapman NM, Chi H. mTOR signaling, Tregs and immune modulation. Immunotherapy. (2014) 6:1295–311. doi: 10.2217/imt.14.84
13. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. (2015) 16:178–87. doi: 10.1038/ni.3076
14. Pokhrel RH, Acharya S, Ahn JH, Gu Y, Pandit M, Kim JO, et al. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol Cancer. (2021) 20:133. doi: 10.1186/s12943-021-01420-9
15. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. (2009) 9:563–75. doi: 10.1038/nrc2676
16. Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, et al. Homeostatic control of metabolic and functional fitness of T(reg) cells by LKB1 signalling. Nature. (2017) 548:602–6. doi: 10.1038/nature23665
17. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
18. Groneberg M, Hoenow S, Marggraff C, Fehling H, Metwally NG, Hansen C, et al. HIF-1α modulates sex-specific Th17/Treg responses during hepatic amoebiasis. J Hepatol. (2022) 76:160–73. doi: 10.1016/j.jhep.2021.09.020
19. Luo J, Wang H, Chen J, Wei X, Feng J, Zhang Y, et al. The application of drugs and nano-therapies targeting immune cells in hypoxic inflammation. Int J nanomedicine. (2024) 19:3441–59. doi: 10.2147/IJN.S456533
20. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
21. Zhou Y, Kang L, Yin G, Yang L, Chen B, Liu B, et al. Adenosine A2B receptor activation regulates the balance between T helper 17 cells and regulatory T cells, and inhibits regulatory T cells exhaustion in experimental autoimmune myositis. J cachexia sarcopenia muscle. (2024) 15:2460–75. doi: 10.1002/jcsm.13581
22. Wang Y, Li M, Zha A. mTOR promotes an inflammatory response through the HIF1 signaling pathway in ulcerative colitis. Int Immunopharmacol. (2024) 134:112217. doi: 10.1016/j.intimp.2024.112217
23. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai AC, et al. HIF-2α is indispensable for regulatory T cell function. Nat Commun. (2020) 11:5005. doi: 10.1038/s41467-020-18731-y
24. Layman AAK, Deng G, O'Leary CE, Tadros S, Thomas RM, Dybas JM, et al. Ndfip1 restricts mTORC1 signalling and glycolysis in regulatory T cells to prevent autoinflammatory disease. Nat Commun. (2017) 8:15677. doi: 10.1038/ncomms15677
25. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
26. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. (2020) 32:591–604.e7. doi: 10.1016/j.cmet.2020.07.001
27. Hurrell BP, Helou DG, Howard E, Painter JD, Shafiei-Jahani P, Sharpe AH, et al. PD-L2 controls peripherally induced regulatory T cells by maintaining metabolic activity and Foxp3 stability. Nat Commun. (2022) 13:5118. doi: 10.1038/s41467-022-32899-5
28. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2015) 29:2315–26. doi: 10.1096/fj.14-268409
29. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. (2015) 125:194–207. doi: 10.1172/JCI76012
30. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. (2020) 21:298–308. doi: 10.1038/s41590-019-0589-5
31. Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature. (2021) 591:306–11. doi: 10.1038/s41586-021-03235-6
32. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
33. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
34. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
35. Xu C, Fu Y, Liu S, Trittipo J, Lu X, Qi R, et al. BATF regulates T regulatory cell functional specification and fitness of triglyceride metabolism in restraining allergic responses. J Immunol (Baltimore Md: 1950). (2021) 206:2088–100. doi: 10.4049/jimmunol.2001184
36. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. (2017) 8:15207. doi: 10.1038/ncomms15207
37. Metzler B, Gfeller P, Guinet E. Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human T cells with high FOXP3 expression and regulatory properties. J Immunol (Baltimore Md: 1950). (2016) 196:3618–30. doi: 10.4049/jimmunol.1501756
38. Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, et al. Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity. (2019) 51:1012–27.e7. doi: 10.1016/j.immuni.2019.10.001
39. Omenetti S, Pizarro TT. The treg/th17 axis: A dynamic balance regulated by the gut microbiome. Front Immunol. (2015) 6:639. doi: 10.3389/fimmu.2015.00639
40. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
41. Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. J Exp Clin Cancer research: CR. (2019) 38:403. doi: 10.1186/s13046-019-1409-3
42. Cadenas-De Miguel S, Lucianer G, Elia I. The metabolic cross-talk between cancer and T cells. Trends Biochem Sci. (2023) 48:597–609. doi: 10.1016/j.tibs.2023.03.004
43. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–18.e9. doi: 10.1016/j.ccell.2022.01.001
44. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
45. Li C, Wang G, Sivasami P, Ramirez RN, Zhang Y, Benoist C, et al. Interferon-α-producing plasmacytoid dendritic cells drive the loss of adipose tissue regulatory T cells during obesity. Cell Metab. (2021) 33:1610–23.e5. doi: 10.1016/j.cmet.2021.06.007
46. Elkins C, Li C. Cytokine and metabolic regulation of adipose tissue Tregs. Immunometabolism (Cobham Surrey). (2022) 4:e00013. doi: 10.1097/IN9.0000000000000013
47. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. (2015) 16:276–85. doi: 10.1038/ni.3085
48. Nakajima A, Yanagimura F, Saji E, Shimizu H, Toyoshima Y, Yanagawa K, et al. Stage-dependent immunity orchestrates AQP4 antibody-guided NMOSD pathology: a role for netting neutrophils with resident memory T cells in situ. Acta Neuropathol. (2024) 147:76. doi: 10.1007/s00401-024-02725-x
49. Shi C, Zhang J, Wang H, Chen C, Han M, Gao L, et al. Trojan horse nanocapsule enabled in situ modulation of the phenotypic conversion of th17 cells to treg cells for the treatment of multiple sclerosis in mice. Advanced materials (Deerfield Beach Fla). (2023) 35:e2210262. doi: 10.1002/adma.202210262
50. Kim MW, Kipnis J. Picking a (neuroimmune) fight against fragile regulation of addiction. Cell. (2023) 186:464–6. doi: 10.1016/j.cell.2023.01.003
51. Llorián-Salvador M, de la Fuente AG. Brain-specific regulatory T cell expansion limits cognitive decline. Trends Mol Med. (2023) 29:481–3. doi: 10.1016/j.molmed.2023.05.001
52. Zagorulya M, Yim L, Morgan DM, Edwards A, Torres-Mejia E, Momin N, et al. Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity. (2023) 56:386–405.e10. doi: 10.1016/j.immuni.2023.01.010
53. Mengistu DT, Curtis JL, Freeman CM. A model of dysregulated crosstalk between dendritic, natural killer, and regulatory T cells in chronic obstructive pulmonary disease. Trends Immunol. (2024) 45:428–41. doi: 10.1016/j.it.2024.04.010
54. Griffith JW, Faustino LD, Cottrell VI, Nepal K, Hariri LP, Chiu RS, et al. Regulatory T cell-derived IL-1Ra suppresses the innate response to respiratory viral infection. Nat Immunol. (2023) 24:2091–107. doi: 10.1038/s41590-023-01655-2
55. Qin Y, Gao C, Luo J. Metabolism characteristics of th17 and regulatory T cells in autoimmune diseases. Front Immunol. (2022) 13:828191. doi: 10.3389/fimmu.2022.828191
56. Norris JM, Johnson RK, Stene LC. Type 1 diabetes-early life origins and changing epidemiology. Lancet Diabetes endocrinology. (2020) 8:226–38. doi: 10.1016/S2213-8587(19)30412-7
57. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
58. Zhang M, Zhou Y, Xie Z, Luo S, Zhou Z, Huang J, et al. New developments in T cell immunometabolism and therapeutic implications for type 1 diabetes. Front Endocrinol (Lausanne). (2022) 13:914136. doi: 10.3389/fendo.2022.914136
59. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet (London England). (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
60. Burrack AL, Martinov T, Fife BT. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (Lausanne). (2017) 8:343. doi: 10.3389/fendo.2017.00343
61. Mahne AE, Klementowicz JE, Chou A, Nguyen V, Tang Q. Therapeutic regulatory T cells subvert effector T cell function in inflamed islets to halt autoimmune diabetes. J Immunol (Baltimore Md: 1950). (2015) 194:3147–55. doi: 10.4049/jimmunol.1402739
62. Hameed I, Masoodi SR, Mir SA, Nabi M, Ghazanfar K, Ganai BA. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J Diabetes. (2015) 6:598–612. doi: 10.4239/wjd.v6.i4.598
63. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
64. Lu J, Zhao J, Meng H, Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol. (2019) 10:1173. doi: 10.3389/fimmu.2019.01173
65. Putilin DA, Evchenko SY, Fedoniuk LY, Tokarskyy OS, Kamyshny OM, Migenko LM, et al. The influence of metformin to the transcriptional activity of the mTOR and FOX3 genes in parapancreatic adipose tissue of streptozotocin-induced diabetic rats. J Med Life. (2020) 13:50–5. doi: 10.25122/jml-2020-0029
66. Bolla AM, Gandolfi A, Borgonovo E, Laurenzi A, Caretto A, Molinari C, et al. Rapamycin plus vildagliptin to recover β-cell function in long-standing type 1 diabetes: A double-blind, randomized trial. J Clin Endocrinol Metab. (2021) 106:e507–e19. doi: 10.1210/clinem/dgaa791
67. Tanimine N, Germana SK, Fan M, Hippen K, Blazar BR, Markmann JF, et al. Differential effects of 2-deoxy-D-glucose on in vitro expanded human regulatory T cell subsets. PloS One. (2019) 14:e0217761. doi: 10.1371/journal.pone.0217761
68. Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc Natl Acad Sci United States America. (2015) 112:482–7. doi: 10.1073/pnas.1423486112
69. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
70. Rui L, Lin JD. Reprogramming of hepatic metabolism and microenvironment in nonalcoholic steatohepatitis. Annu Rev Nutr. (2022) 42:91–113. doi: 10.1146/annurev-nutr-062220-105200
71. Vaikunthanathan T, Landmann E, Correa DM, Romano M, Trevelin SC, Peng Q, et al. Dysregulated anti-oxidant signalling and compromised mitochondrial integrity negatively influence regulatory T cell function and viability in liver disease. EBioMedicine. (2023) 95:104778. doi: 10.1016/j.ebiom.2023.104778
72. Ramadori P, Kam S, Heikenwalder M. T cells: Friends and foes in NASH pathogenesis and hepatocarcinogenesis. Hepatology. (2022) 75:1038–49. doi: 10.1002/hep.32336
73. Goand UK, Patel I, Verma S, Yadav S, Maity D, Singh N, et al. Immunometabolic impact of pancreastatin inhibitor PSTi8 in MCD induced mouse model of oxidative stress and steatohepatitis. Cytokine. (2023) 171:156354. doi: 10.1016/j.cyto.2023.156354
74. González-Yanes C, Sánchez-Margalet V. Pancreastatin, a chromogranin-A-derived peptide, inhibits insulin-stimulated glycogen synthesis by activating GSK-3 in rat adipocytes. Biochem Biophys Res Commun. (2001) 289:282–7. doi: 10.1006/bbrc.2001.5967
75. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol hepatology. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
76. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. (2021) 75:1271–83. doi: 10.1016/j.jhep.2021.07.032
77. Savage TM, Fortson KT, de Los Santos-Alexis K, Oliveras-Alsina A, Rouanne M, Rae SS, et al. Amphiregulin from regulatory T cells promotes liver fibrosis and insulin resistance in non-alcoholic steatohepatitis. Immunity. (2024) 57:303–18.e6. doi: 10.1016/j.immuni.2024.01.009
78. Adolph TE, Meyer M, Schwärzler J, Mayr L, Grabherr F, Tilg H. The metabolic nature of inflammatory bowel diseases. Nat Rev Gastroenterol hepatology. (2022) 19:753–67. doi: 10.1038/s41575-022-00658-y
79. Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. (2013) 35:229–44. doi: 10.1007/s00281-012-0352-6
80. Chen L, Ruan G, Cheng Y, Yi A, Chen D, Wei Y. The role of Th17 cells in inflammatory bowel disease and the research progress. Front Immunol. (2022) 13:1055914. doi: 10.3389/fimmu.2022.1055914
81. Bamidele AO, Mishra SK, Piovezani Ramos G, Hirsova P, Klatt EE, Abdelrahman LM, et al. Interleukin 21 drives a hypermetabolic state and CD4(+) T-cell-associated pathogenicity in chronic intestinal inflammation. Gastroenterology. (2024) 166:826–41.e19. doi: 10.1053/j.gastro.2024.01.026
82. Lee H, Jeon JH, Kim ES. Mitochondrial dysfunctions in T cells: focus on inflammatory bowel disease. Front Immunol. (2023) 14:1219422. doi: 10.3389/fimmu.2023.1219422
83. Guan Y, Zhang L, Li X, Zhang X, Liu S, Gao N, et al. Repression of mammalian target of rapamycin complex 1 inhibits intestinal regeneration in acute inflammatory bowel disease models. J Immunol (Baltimore Md: 1950). (2015) 195:339–46. doi: 10.4049/jimmunol.1303356
84. Yin H, Li X, Zhang B, Liu T, Yuan B, Ni Q, et al. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology. (2013) 139:494–502. doi: 10.1111/imm.2013.139.issue-4
85. Higashiyama M, Hokari R, Hozumi H, Kurihara C, Ueda T, Watanabe C, et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J Leukoc Biol. (2012) 91:901–9. doi: 10.1189/jlb.1011518
86. Zaiatz Bittencourt V, Jones F, Doherty G, Ryan EJ. Targeting immune cell metabolism in the treatment of inflammatory bowel disease. Inflammation Bowel Dis. (2021) 27:1684–93. doi: 10.1093/ibd/izab024
87. Aristin Revilla S, Kranenburg O, Coffer PJ. Colorectal cancer-infiltrating regulatory T cells: functional heterogeneity, metabolic adaptation, and therapeutic targeting. Front Immunol. (2022) 13:903564. doi: 10.3389/fimmu.2022.903564
88. Szeponik L, Ahlmanner F, Sundström P, Rodin W, Gustavsson B, Bexe Lindskog E, et al. Intratumoral regulatory T cells from colon cancer patients comprise several activated effector populations. BMC Immunol. (2021) 22:58. doi: 10.1186/s12865-021-00449-1
89. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326-6066.CIR-13-0013
90. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. (2020) 21:1346–58. doi: 10.1038/s41590-020-0769-3
91. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
92. Saadh MJ, Kazemi K, Khorramdelazad H, Mousavi MJ, Noroozi N, Masoumi M, et al. Role of T cells in the pathogenesis of systemic lupus erythematous: Focus on immunometabolism dysfunctions. Int Immunopharmacol. (2023) 119:110246. doi: 10.1016/j.intimp.2023.110246
93. Ma L, Roach T, Morel L. Immunometabolic alterations in lupus: where do they come from and where do we go from there? Curr Opin Immunol. (2022) 78:102245. doi: 10.1016/j.coi.2022.102245
94. Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J Immunol (Baltimore Md: 1950). (2017) 198:2661–70. doi: 10.4049/jimmunol.1403088
95. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol (Baltimore Md: 1950). (2016) 196:80–90. doi: 10.4049/jimmunol.1501537
96. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. (2014) 124:2234–45. doi: 10.1172/JCI73411
97. Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispín JC, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3+ regulatory T cells in MRL/lpr mice. Autoimmunity. (2014) 47:445–50. doi: 10.3109/08916934.2014.915954
98. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol (Hoboken NJ). (2016) 68:2728–39. doi: 10.1002/art.v68.11
99. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (London England). (2018) 391:1186–96. doi: 10.1016/S0140-6736(18)30485-9
100. Zhao W, Berthier CC, Lewis EE, McCune WJ, Kretzler M, Kaplan MJ. The peroxisome-proliferator activated receptor-γ agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin Immunol (Orlando Fla). (2013) 149:119–32. doi: 10.1016/j.clim.2013.07.002
101. Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, et al. Obesity drives th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. (2015) 12:1042–55. doi: 10.1016/j.celrep.2015.07.014
102. Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest. (2015) 125:2220–7. doi: 10.1172/JCI78087
103. Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci United States America. (2012) 109:3909–14. doi: 10.1073/pnas.1117736109
104. He X, Smeets RL, Koenen HJ, Vink PM, Wagenaars J, Boots AM, et al. Mycophenolic acid-mediated suppression of human CD4+ T cells: more than mere guanine nucleotide deprivation. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2011) 11:439–49. doi: 10.1111/j.1600-6143.2010.03413.x
105. Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol (Orlando Fla). (2012) 144:200–13. doi: 10.1016/j.clim.2012.07.001
106. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis rheumatism. (2012) 64:2937–46. doi: 10.1002/art.34502
107. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (London England). (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
108. Zhang X, Olsen N, Zheng SG. The progress and prospect of regulatory T cells in autoimmune diseases. J Autoimmun. (2020) 111:102461. doi: 10.1016/j.jaut.2020.102461
109. Laragione T, Harris C, Azizgolshani N, Beeton C, Bongers G, Gulko PS. Magnesium increases numbers of Foxp3+ Treg cells and reduces arthritis severity and joint damage in an IL-10-dependent manner mediated by the intestinal microbiome. EBioMedicine. (2023) 92:104603. doi: 10.1016/j.ebiom.2023.104603
110. Kanjana K, Chevaisrakul P, Matangkasombut P, Paisooksantivatana K, Lumjiaktase P. Inhibitory activity of FOXP3+ regulatory T cells reveals high specificity for displaying immune tolerance in remission state rheumatoid arthritis. Sci Rep. (2020) 10:19789. doi: 10.1038/s41598-020-76168-1
111. Jacobs JP, Wu HJ, Benoist C, Mathis D. IL-17-producing T cells can augment autoantibody-induced arthritis. Proc Natl Acad Sci United States America. (2009) 106:21789–94. doi: 10.1073/pnas.0912152106
112. Suga K, Suto A, Tanaka S, Sugawara Y, Kageyama T, Ishikawa J, et al. TAp63, a methotrexate target in CD4+ T cells, suppresses Foxp3 expression and exacerbates autoimmune arthritis. JCI Insight. (2023) 8:e164778. doi: 10.1172/jci.insight.164778
113. Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol. (2020) 16:145–54. doi: 10.1038/s41584-020-0373-9
114. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med. (2014) 211:529–43. doi: 10.1084/jem.20131459
115. Chen Y, Liang R, Shi X, Shen R, Liu L, Liu Y, et al. Targeting kinase ITK treats autoimmune arthritis via orchestrating T cell differentiation and function. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 169:115886. doi: 10.1016/j.biopha.2023.115886
116. Eken A, Cansever M, Somekh I, Mizoguchi Y, Zietara N, Okus FZ, et al. Genetic deficiency and biochemical inhibition of ITK affect human th17, treg, and innate lymphoid cells. J Clin Immunol. (2019) 39:391–400. doi: 10.1007/s10875-019-00632-5
117. Jin J, Wang W, Li A, Wu J. LMP7 inhibits the activation of NLRP3 inflammasome through interaction with NLRP3. Biochem Biophys Res Commun. (2020) 531:152–9. doi: 10.1016/j.bbrc.2020.07.091
118. Liu Q, Shen J, Wang J, Xia J, Yin J, Cheng G, et al. PR-957 retards rheumatoid arthritis progression and inflammation by inhibiting LMP7-mediated CD4(+) T cell imbalance. Int Immunopharmacol. (2023) 124:110860. doi: 10.1016/j.intimp.2023.110860
119. Zhou G, Sun X, Qin Q, Lv J, Cai Y, Wang M, et al. Loss of smad7 promotes inflammation in rheumatoid arthritis. Front Immunol. (2018) 9:2537. doi: 10.3389/fimmu.2018.02537
120. Bogdanović O, Lister R. DNA methylation and the preservation of cell identity. Curr Opin Genet Dev. (2017) 46:9–14. doi: 10.1016/j.gde.2017.06.007
121. Hu Y, Xu B, He J, Shan H, Zhou G, Wang D, et al. Hypermethylation of Smad7 in CD4(+) T cells is associated with the disease activity of rheumatoid arthritis. Front Immunol. (2023) 14:1104881. doi: 10.3389/fimmu.2023.1104881
122. Chen SY, Shiau AL, Wu CL, Wang CR. Intraarticular overexpression of Smad7 ameliorates experimental arthritis. Sci Rep. (2016) 6:35163. doi: 10.1038/srep35163
123. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
124. Häfliger P, Charles RP. The L-type amino acid transporter LAT1-an emerging target in cancer. Int J Mol Sci. (2019) 20:2428. doi: 10.3390/ijms20102428
125. Ogbechi J, Wright HL, Balint S, Topping LM, Kristina Z, Huang YS, et al. LAT1 enables T cell activation under inflammatory conditions. J Autoimmun. (2023) 138:103031. doi: 10.1016/j.jaut.2023.103031
126. Spiljar M, Kuchroo VK. Metabolic regulation and function of T helper cells in neuroinflammation. Semin Immunopathol. (2022) 44:581–98. doi: 10.1007/s00281-022-00959-z
127. Mangiardi M, Crawford DK, Xia X, Du S, Simon-Freeman R, Voskuhl RR, et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol (Zurich Switzerland). (2011) 21:263–78. doi: 10.1111/j.1750-3639.2010.00444.x
128. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat Immunol. (2017) 18:74–85. doi: 10.1038/ni.3632
129. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor blimp-1 promotes pathogenicity of T helper 17 cells. Immunity. (2016) 44:131–42. doi: 10.1016/j.immuni.2015.11.009
130. Gyülvészi G, Haak S, Becher B. IL-23-driven encephalo-tropism and Th17 polarization during CNS-inflammation in vivo. Eur J Immunol. (2009) 39:1864–9. doi: 10.1002/eji.200939305
131. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
132. Lowther DE, Chong DL, Ascough S, Ettorre A, Ingram RJ, Boyton RJ, et al. Th1 not Th17 cells drive spontaneous MS-like disease despite a functional regulatory T cell response. Acta Neuropathol. (2013) 126:501–15. doi: 10.1007/s00401-013-1159-9
133. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. (2016) 354:481–4. doi: 10.1126/science.aaf6284
134. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
135. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med. (2015) 212:1603–21. doi: 10.1084/jem.20141030
136. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic cell PD-L1 limits autoimmunity and follicular T cell differentiation and function. J Immunol (Baltimore Md: 1950). (2018) 200:2592–602. doi: 10.4049/jimmunol.1701231
137. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
138. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol (Baltimore Md: 1950). (2008) 181:2277–84. doi: 10.4049/jimmunol.181.4.2277
139. Palomer X, Barroso E, Zarei M, Botteri G, Vázquez-Carrera M. PPARβ/δ and lipid metabolism in the heart. Biochim Biophys Acta. (2016) 1861:1569–78. doi: 10.1016/j.bbalip.2016.01.019
140. Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol (Baltimore Md: 1950). (2007) 178:4129–35. doi: 10.4049/jimmunol.178.7.4129
141. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. (2015) 43:817–29. doi: 10.1016/j.immuni.2015.09.007
142. DiToro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA, et al. Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity. (2020) 52:650–67.e10. doi: 10.1016/j.immuni.2020.03.013
143. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
144. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of T(H)17 and induced T(reg) cell balance by an epigenetic mechanism. Nature. (2017) 548:228–33. doi: 10.1038/nature23475
145. Kitz A, de Marcken M, Gautron AS, Mitrovic M, Hafler DA, Dominguez-Villar M. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep. (2019) 20:e48624. doi: 10.15252/embr.201948624
146. Baardman J, Lutgens E. Regulatory T cell metabolism in atherosclerosis. Metabolites. (2020) 10:279. doi: 10.3390/metabo10070279
147. Yang J, Chen Y, Li X, Qin H, Bao J, Wang C, et al. Complex interplay between metabolism and CD4(+) T-cell activation, differentiation, and function: a novel perspective for atherosclerosis immunotherapy. Cardiovasc Drugs Ther. (2024) 38:1033–46. doi: 10.1007/s10557-023-07466-9
148. Boshuizen MC, de Winther MP. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:1579–88. doi: 10.1161/ATVBAHA.115.305464
149. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
150. Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. (2002) 90:1064–71. doi: 10.1161/01.RES.0000018941.10726.FA
151. Hsu TS, Lai MZ. Hypoxia-inducible factor 1α plays a predominantly negative role in regulatory T cell functions. J Leukoc Biol. (2018) 104:911–8. doi: 10.1002/JLB.MR1217-481R
152. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
153. Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2016) 36:1141–51. doi: 10.1161/ATVBAHA.115.306848
154. Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, et al. Atherosclerosis-driven treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/tregs. Circ Res. (2016) 119:1190–203. doi: 10.1161/CIRCRESAHA.116.309764
155. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–77.e6. doi: 10.1016/j.immuni.2018.07.015
156. Meng X, Zhang K, Li J, Dong M, Yang J, An G, et al. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol Med (Cambridge Mass). (2012) 18:598–605. doi: 10.2119/molmed.2011.00471
157. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. (2018) 9:1095. doi: 10.1038/s41467-018-03493-5
158. Zaric BL, Radovanovic JN, Gluvic Z, Stewart AJ, Essack M, Motwalli O, et al. Atherosclerosis linked to aberrant amino acid metabolism and immunosuppressive amino acid catabolizing enzymes. Front Immunol. (2020) 11:551758. doi: 10.3389/fimmu.2020.551758
159. Nitz K, Lacy M, Bianchini M, Wichapong K, Kücükgöze IA, Bonfiglio CA, et al. The amino acid homoarginine inhibits atherogenesis by modulating T-cell function. Circ Res. (2022) 131:701–12. doi: 10.1161/CIRCRESAHA.122.321094
160. Yun TJ, Lee JS, Machmach K, Shim D, Choi J, Wi YJ, et al. Indoleamine 2,3-dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T cells. Cell Metab. (2016) 23:852–66. doi: 10.1016/j.cmet.2016.04.010
161. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. New Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
162. Sica A, Colombo MP, Trama A, Horn L, Garassino MC, Torri V. Immunometabolic status of COVID-19 cancer patients. Physiol Rev. (2020) 100:1839–50. doi: 10.1152/physrev.00018.2020
163. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. (2020) 26:1623–35. doi: 10.1038/s41591-020-1038-6
164. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun. (2020) 11:3434. doi: 10.1038/s41467-020-17292-4
165. Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. (2020) 5:e140327. doi: 10.1172/jci.insight.140327
166. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated glucose levels favor SARS-coV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. (2020) 32:437–46.e5. doi: 10.2139/ssrn.3606770
167. Aggarwal G, Lippi G, Lavie CJ, Henry BM, Sanchis-Gomar F. Diabetes mellitus association with coronavirus disease 2019 (COVID-19) severity and mortality: A pooled analysis. J Diabetes. (2020) 12:851–5. doi: 10.1111/1753-0407.13091
168. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers inflammatory cell death, tissue damage, and mortality in SARS-coV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–68.e17. doi: 10.1016/j.cell.2020.11.025
169. Teer E, Mukonowenzou NC, Essop MF. The role of immunometabolism in HIV-1 pathogenicity: links to immune cell responses. Viruses. (2022) 14:1813. doi: 10.3390/v14081813
170. Yero A, Bouassa RM, Ancuta P, Estaquier J, Jenabian MA. Immuno-metabolic control of the balance between Th17-polarized and regulatory T-cells during HIV infection. Cytokine Growth Factor Rev. (2023) 69:1–13. doi: 10.1016/j.cytogfr.2023.01.001
171. Chevalier MF, Petitjean G, Dunyach-Rémy C, Didier C, Girard PM, Manea ME, et al. The Th17/Treg ratio, IL-1RA and sCD14 levels in primary HIV infection predict the T-cell activation set point in the absence of systemic microbial translocation. PloS Pathog. (2013) 9:e1003453. doi: 10.1371/journal.ppat.1003453
172. Xu M, Warner C, Duan X, Cheng Z, Jeyarajan AJ, Li W, et al. HIV coinfection exacerbates HBV-induced liver fibrogenesis through a HIF-1α- and TGF-β1-dependent pathway. J Hepatol. (2024) 80:868–81. doi: 10.1016/j.jhep.2024.01.026
173. Tao JH, Barbi J, Pan F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. (2015) 309:C580–9. doi: 10.1152/ajpcell.00204.2015
174. Qiu R, Yu X, Wang L, Han Z, Yao C, Cui Y, et al. Inhibition of Glycolysis in Pathogenic T(H)17 Cells through Targeting a miR -21-Peli1-c-Rel Pathway Prevents Autoimmunity. J Immunol (Baltimore Md: 1950). (2020) 204:3160–70. doi: 10.4049/jimmunol.2000060
175. Koethe JR, Lagathu C, Lake JE, Domingo P, Calmy A, Falutz J, et al. HIV and antiretroviral therapy-related fat alterations. Nat Rev Dis primers. (2020) 6:48. doi: 10.1038/s41572-020-0181-1
176. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab. (2020) 31:422–37.e5. doi: 10.1016/j.cmet.2019.11.021
177. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. New Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
178. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
179. Jenabian MA, Seddiki N, Yatim A, Carriere M, Hulin A, Younas M, et al. Regulatory T cells negatively affect IL-2 production of effector T cells through CD39/adenosine pathway in HIV infection. PloS Pathog. (2013) 9:e1003319. doi: 10.1371/journal.ppat.1003319
180. Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol. (2017) 14:521–8. doi: 10.1038/cmi.2016.30
181. Hryniewicz A, Boasso A, Edghill-Smith Y, Vaccari M, Fuchs D, Venzon D, et al. CTLA-4 blockade decreases TGF-beta, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood. (2006) 108:3834–42. doi: 10.1182/blood-2006-04-010637
182. Deme P, Rubin LH, Yu D, Xu Y, Nakigozi G, Nakasujja N, et al. Immunometabolic reprogramming in response to HIV infection is not fully normalized by suppressive antiretroviral therapy. Viruses. (2022) 14:1313. doi: 10.3390/v14061313
183. Kleinman AJ, Sivanandham R, Pandrea I, Chougnet CA, Apetrei C. Regulatory T cells as potential targets for HIV cure research. Front Immunol. (2018) 9:734. doi: 10.3389/fimmu.2018.00734
Keywords: immunometabolism, Tregs, metabolic diseases, metabolic pathways, microenvironment, inflammation
Citation: Lu Y, Wang Y, Ruan T, Wang Y, Ju L, Zhou M, Liu L, Yao D and Yao M (2025) Immunometabolism of Tregs: mechanisms, adaptability, and therapeutic implications in diseases. Front. Immunol. 16:1536020. doi: 10.3389/fimmu.2025.1536020
Received: 28 November 2024; Accepted: 06 January 2025;
Published: 23 January 2025.
Edited by:
Yang Zhang, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Pei Xiong Liew, Augusta University, United StatesCopyright © 2025 Lu, Wang, Ruan, Wang, Ju, Zhou, Liu, Yao and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Yao, ZXJiZWlAbnR1LmVkdS5jbg==
†These authors have contributed equally to this work
‡ORCID: Yuming Lu, orcid.org/0009-0009-2792-2818
Yifan Wang, orcid.org/0009-0009-2384-7013
Tiantian Ruan, orcid.org/0009-0004-9895-0051
Yihan Wang, orcid.org/0009-0001-5719-5828
Linling Ju, orcid.org/0000-0002-9541-0533
Mengya Zhou, orcid.org/0009-0004-4798-047X
Luyin Liu, orcid.org/0009-0004-2294-2362
Dengfu Yao, orcid.org/0000-0002-3448-7756
Min Yao, orcid.org/0000-0002-5473-0186
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.