 Parker S. Woods
Parker S. Woods Gökhan M. Mutlu*
Gökhan M. Mutlu*- Department of Medicine, Section of Pulmonary and Critical Care Medicine, The University of Chicago, Chicago, IL, United States
Immunometabolism has emerged as a key area of focus in immunology and has the potential to lead to new treatments for immune-related diseases. It is well-established that glycolytic metabolism is essential for adaptation to hypoxia and for macrophage inflammatory function. Macrophages have been shown to upregulate their glycolytic metabolism in response to pathogens and pathogen-associated molecular patterns such as LPS. As a direct link to the external environment, the lungs’ distinctive nutrient composition and multiple macrophage subtypes provide a unique opportunity to study macrophage metabolism. This review aims to highlight how the steady-state airway and severely inflamed airway offer divergent environments for macrophage glycolytic metabolism. We describe the differences in glycolytic metabolism between tissue-resident alveolar macrophages, and other lung macrophages at steady-state and during inflammation/injury. We also provide an overview of experimental guidelines on how to assess metabolism at the cellular level using Seahorse-based bioenergetic analysis including a review of pharmacologic agents used to inhibit or activate glycolysis.
1 Introduction
It is well-established that glycolytic metabolism is essential for adaptation to hypoxia and for macrophage inflammatory function. Macrophages have been shown to upregulate their glycolytic metabolism in response to pathogens and pathogen-associated molecular patterns such as LPS. As a direct link to the external environment, the lungs’ distinctive nutrient composition and multiple macrophage subtypes provide a unique opportunity to study macrophage metabolism. This review aims to highlight how the steady-state airway and severely inflamed airway offer divergent environments for macrophage glycolytic metabolism. We describe how glycolytic metabolism in tissue resident alveolar macrophages (TR-AMs), interstitial macrophages (IMs), and monocyte-derived alveolar macrophages (Mo-AMs) relates to airway homeostasis and disease pathogenesis. We also provide an overview of experimental guidelines on how to assess metabolism at the cellular level using Seahorse-based bioenergetic analysis including a review of pharmacologic agents used to inhibit or activate glycolysis.
2 Airway microenvironment
The defining role of the lungs is to conduct gas exchange, and in doing so, oxygen is extracted from the external environment and into the bloodstream to drive metabolic processes at the cellular level throughout the body. This direct interface with the external environment necessitates cellular adaptation to the local microenvironment that is truly unique in relation to other specialized barrier sites. Under steady-state conditions, alveoli have one of the highest oxygen concentrations (PO2 ~100 mmHg at sea level, 13.2% O2) compared to other organs and compartments within the human body (1). Therefore, cells in the alveoli including TR-AMs are exposed to higher concentrations of O2 than most other cells in the body. In addition, luminal airway glucose concentrations are one-tenth of those observed in the blood, making the airway one of lowest glucose environments encountered by cells (2, 3). In contrast, the adjacent lung interstitium receives roughly 5.2% O2 (40 mm Hg), and maintains glucose levels more comparable to those in blood (4). (Table 1) The high oxygen environment in the alveolar lumen is a direct consequence of the basic anatomy and physiology of the respiratory system. When coupled with the low glucose environment of the airway, high oxygen does not only deter growth of some pathogens but also shapes the metabolic preferences of specialized airway cells, particularly TR-AMs, which lack direct access to interstitial nutrients. For instance, epithelial cells in the airway express high levels of apical glucose transporters, which are thought to maintain low glucose concentrations in the airway for two key reasons (5). Rapid removal of glucose from the airway restricts nutrient availability to greatly limit opportunities for pathogen growth (2, 6). In addition, sodium-dependent glucose cotransporters in alveolar type II (ATII) cells serve as an important regulator of the airway surface fluid that is essential for normal respiratory mechanics (7). These cotransporters simultaneously encourage fluid reabsorption and glucose removal from the airway as a means to balance airway fluid and provide energy to fuel epithelial cell processes (5, 8, 9). ATII cells also contribute to the unique composition of the airway through the production of pulmonary surfactant, a complex mixture of proteins and lipids that principally serve to reduce alveolar surface tension and prevent lung collapse. Surfactant proteins also have several immunoregulatory properties, and the lipid-rich portion likely serves as an energy for cells within the airway, particularly TR-AMs, which have high fatty acid oxidation (FAO) gene expression (10, 11).
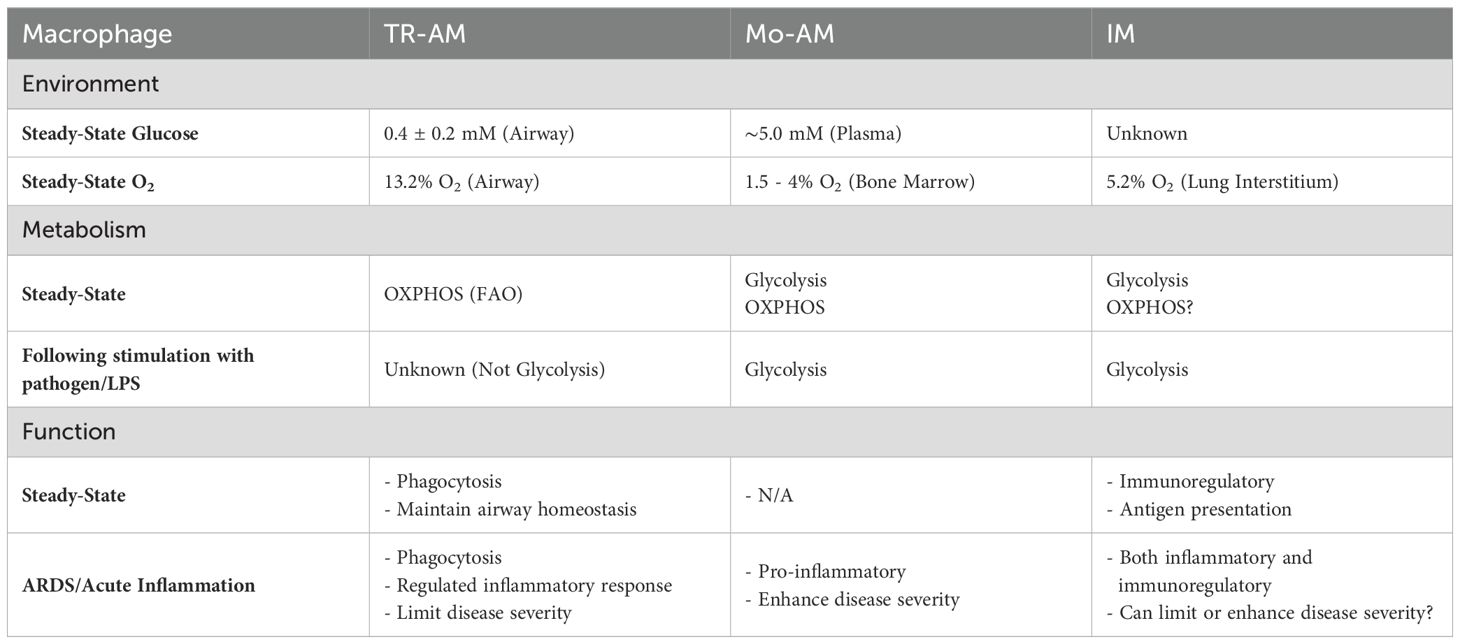
Table 1. Comparison of environmental and metabolic preferences amongst lung macrophage populations.
While the steady-state airway is marked by high oxygen levels, low glucose concentrations, and a lipid-rich environment, acute inflammation can significantly alter this composition. Viral pathogens, including influenza A and SARS-CoV-2, can cause severe lung injury and lead to acute respiratory distress syndrome (ARDS), which can lead to significant alterations to the airway microenvironment. ARDS is characterized by increased alveolar/endothelial permeability leading to alveolar fluid accumulation (12). Fluid accumulation results in increased airway glucose levels and impaired gas exchange creating local regions of high glucose and low oxygen, a near complete opposite environment compared to the steady-state lung (2, 13, 14). Moreover, ARDS dramatically reduces both the lipid and protein fractions of pulmonary surfactant within the airway to greatly alter homeostatic nutrient availability (15–18). Infiltrating immune cells also contribute to these alterations by sequestering oxygen and nutrients from resident cells (13).
Taken together, this information contextualizes the unique environment of the airway. At steady state, one would expect the high oxygen, low glucose, and lipid-rich airway environment to promote oxidative phosphorylation (OXPHOS) in resident airway cells. Under robust inflammation, the high glucose, low oxygen environment, and reduced lipid content would necessitate metabolic adaptation to anaerobic glycolysis to support resident airway cell effector function. These environmental features of the airway are critical in dictating resident cell effector function, and thus influence macrophage metabolism within the lung.
3 Tissue resident alveolar macrophages
TR-AMs serve as immune sentinels of the airway, where they must balance their maintenance roles of engulfing apoptotic cells, clearing cellular debris, and mitigating responses to benign antigens while also recognizing and mounting appropriate responses to pathogenic microorganisms (19). This often puts them at odds with traditional macrophage classification based on proinflammatory and anti-inflammatory polarization states (20). However, in general, TR-AMs are viewed to be less inflammatory than their monocytic counterparts and critical in regulating inflammation during ARDS and tissue repair following lung injury (21, 22). As mentioned in the previous section, the steady-state airway is an oxygen- and lipid-rich and glucose-limited environment. TR-AMs have evolved to become highly-specialized within this environment compared to their IM or Mo-AM counterparts. TR-AMs arise from fetal yolk monocytes and populate the airways during the first days of life (23, 24). Upon entry into the airway, TR-AMs undergo post-natal maturation where glycolysis is downregulated. Multiple groups have shown that preventing this downregulation of glycolysis through the deletion of Von Hippel-Lindau Protein (VHL), a key part of an E3 ubiquitin ligase complex responsible for targeting hypoxia-inducible factor 1-alpha (HIF-1α) for proteasomal degradation, produces immature TR-AMs (25, 26). VHL deletion leads to deficiencies in OXPHOS, lipid-handling, and self-renewal suggesting that downregulation of glycolysis is essential for normal TR-AM function at steady-state. It is thought that the airway environment provides cues for TR-AMs to downregulate HIF-1α target genes. This seems to be the case given that peritoneal macrophages and various macrophage precursors can be transferred into the airway and assume TR-AM phenotypical characteristics (27, 28).
The defining metabolic feature of TR-AMs is robust OXPHOS capacity. Deletion of mitochondrial transcription factor A (Tfam) reduces ETC complex expression and greatly reduces mitochondrial respiration across several immune cell subsets (29–31). When Tfam was deleted in macrophage populations across all cellular compartments in mice, TR-AMs experienced the largest decline in numbers (32). Tfam deletion resulted in impaired maturation, defective lipid handling, and apoptosis in TR-AMs, highlighting the importance of OXPHOS in TR-AM steady-state function (32, 33). We have previously shown that high dose ETC inhibition decreases TR-AM viability and that low dose ETC inhibition greatly reduces inflammatory capacity (34, 35). Collectively, these findings highlight the dominant role of OXPHOS in TR-AM steady-state metabolism.
Severe airway inflammation observed in ARDS leads to airway hypoxia and glucose influx. Bronchoscopic oxygen measurements in patients with cystic fibrosis harboring bacterial infections have recorded oxygen levels below 1%, and pimonidazole has been used to verify the presence of severely hypoxic alveoli during influenza infection in mice (36, 37). These conditions necessitate TR-AMs, which rely heavily on OXPHOS for maintenance function, to alter their metabolism in order to survive and aid in the resolution of severe inflammation. We and others have found that TR-AMs do not utilize glycolysis for proinflammatory processes in vitro (35, 38, 39). Moreover, we have shown that TR-AMs have comparatively low glycolytic protein expression and glycolysis alone cannot sustain normal TR-AM function in the presence of ETC inhibitors (34). This suggests that TR-AMs suffer from a glycolytic deficiency and cannot survive on glycolysis-derived energy production alone. However, when TR-AMs are exposed to hypoxia or pharmacological pseudohypoxia (inhibitors of prolyl hydroxylases), TR-AMs can assume a glycolytic phenotype that enhances TR-AM survival in the presence of ETC inhibitors (35). This hypoxic, glycolytic reprogramming is dependent on HIF-1α. We have shown that HIF-1α is not required for steady-state function or for proinflammatory processes in TR-AMs, but is critical in promoting TR-AM survival during influenza-induced ARDS in mice (35, 40). Significant TR-AM loss occurs during virus-induced (i.e., influenza, SARS-CoV-2) ARDS in humans and mice, and the degree of this loss is associated with poor outcomes (41–46). The exact mechanism of TR-AM depletion during ARDS remains unknown but evidence supports both cell-intrinsic and cell-extrinsic mechanisms of cell death. Gao et al. discovered that influenza infection decreases TFAM expression and causes mitochondrial damage in TR-AMs (33). After resolution of primary pneumonia in mice, surviving TR-AMs secrete higher levels of lactate in response to LPS (47). Mice treated intratracheally with HIF-1α activator (FG-4592) experienced reduced TR-AM loss, airway inflammation, and improved survival in a lethal model for influenza-induced ARDS (35). These findings suggest that TR-AM loss is linked to inadequate glycolytic adaptation under ARDS conditions that impair mitochondrial function (i.e. hypoxia and direct viral infection).
Given the well-established role of TR-AMs in ARDS, most of our knowledge on TR-AMs and their metabolism is based on data from clinical and experimental studies in ARDS. However, other lung diseases such as asthma or COPD may potentially affect or alter TR-AM metabolism and contribute to the pathogenesis of these chronic lung diseases. Others have suggested that glycolysis serves a role in TR-AM effector function outside of hypoxic adaptation. TR-AM glycolysis has been reported to be essential for interferon-driven cytokine production, IL-4 responsiveness, and allergen-induced inflammation (48–50). However, these studies rely heavily on 2-deoxy-D-glucose (2-DG) to inhibit glycolysis. 2-DG has extensive off-target effects, which are covered later in this review (See “Experimental Guidelines” section). Thus, while these studies are of value they should be interpreted with caution until genetic or other pharmacological approaches can confirm the results. Alveolar macrophages from smokers and patients with chronic obstructive pulmonary disease (COPD) exhibited impaired mitochondrial metabolism and increased glycolysis (51). Moreover, both monocyte-derived macrophages generated from blood and unsorted alveolar macrophages obtained via bronchoalveolar lavage from patients with COPD and treated in vitro exhibited alterations in mitochondrial reactive oxygen species generation (52, 53). However, one should be cautious about interpreting the results from these studies since alveolar macrophages are not sorted and the evaluation of metabolic profiles was performed on the whole alveolar macrophage population, which contains both TR-AMs and Mo-AMs; latter play a key driver role in the pathogenesis of COPD (54).Therefore, there is need for further studies in which alveolar macrophages are sorted to separate TR-AMs from Mo-AMs and other lung macrophages to determine whether COPD, asthma or other chronic lung diseases alter the metabolism of TR-AMs and other macrophages.
We have found that TR-AM metabolism and function is unperturbed by inhibiting glycolysis (34). We also observed no metabolic or functional changes when HIF-1α is deleted in TR-AMs, and that unlike other macrophage populations, LPS treatment does not result in HIF-1α translocation to the nucleus (40). In contrast, Zhu and colleagues have shown that in vitro treatment with Poly (I:C), a synthetic viral immunostimulant, or influenza infection induces HIF-1α in TR-AMs, but we were unable to reproduce these results (55, 56). In our hands, only hypoxia or pharmacological inhibition of prolyl hydroxylases induce TR-AM HIF-1α (34, 35, 40). Zhu and colleagues utilize GM-CSF in their TR-AM culture system. GM-CSF is critical in regulating TR-AM surfactant recycling in vivo, but it induces proliferation of TR-AMs in culture. Within the alveolus, TR-AMs form direct interactions with alveolar epithelial cells through extracellular receptors, like TGFBR and CD200R, that prevent aberrant proliferation at physiological concentrations of GM-CSF (19, 57). This could suggest that the observed, non-hypoxic HIF-1α induction in TR-AMs is associated with proliferation as opposed to inflammatory effector function. This will require new investigations examining the role of GM-CSF on HIF-1α induction in TR-AMs.
As with our work and those of others, it is important to recognize the limitations of most of these studies. Current knowledge on TR-AM metabolism, as well as most macrophage populations, is largely derived from experiments in isolated cells cultured in artificial media. These conditions do not mimic the local nutrient composition or O2 concentration and fail to account for cell-to-cell interactions (i.e., TR-AMs with AT II cells), which may directly or indirectly influence macrophage metabolism. Therefore, there is need for further studies that model not only the metabolic/O2 environment, but also take neighboring cells into account. Studies in cell metabolism have revolutionized our understanding of tumorigenesis. Recent investigations have incorporated spatial metabolomics integrated with spatial transcriptomics and proteomics in tumors to better understand macrophage metabolism and function (58, 59). These new approaches are likely to improve our understanding of TR-AM metabolism and function and lead to new areas of research.
In summary, under steady-state conditions, TR-AMs do express glycolytic machinery albeit at a much lower level than their counterparts of monocytic origin (i.e., recruited macrophages). Only hypoxia-mediated HIF-1α induction allows TR-AMs to perform functional glycolysis, which is defined as sustained lactate production to support cellular function in the presence of ETC inhibition. This supports the notion that TR-AM glycolysis evolved to support cellular function under hypoxic conditions and demonstrates that HIF-1α induction promotes TR-AM survival during ARDS, a condition marked by TR-AM loss and mitochondrial dysfunction. Future studies utilizing spatial omics will strengthen our knowledge and clarify discrepancies within the field of TR-AM metabolism in a context-specific manner.
4 Interstitial macrophages
Interstitial macrophages are resident macrophages that reside with the lung interstitium. Much less is known about IMs than TR-AMs, but IMs have been classified into two or three distinct cell populations based on such characteristics as cell surface markers, gene signatures, developmental origin, and location within the interstitium (60–63). The exact origin of IMs has been controversial. Most studies find that IMs are of monocytic origin, while others suggest that certain populations arise from the developmental yolk-sac, like TR-AMs (61, 63). However, it is mostly thought that yolk sac IMs are slowly replaced by monocytes in adult mice. IMs have been described as having more specialized roles based on whether they localize near sympathetic nerve bundles or alongside blood vessels, but in general most IM populations have been described as being immunoregulatory in that they secreted high levels of IL-10 and are well-equipped for antigen presentation (60, 62, 63). Interestingly, lung IMs share many of the same cell surface markers and gene signatures as macrophages found in the interstitium of the heart, skin, and gut suggesting that the lung IM phenotype is dictated by the generalized interstitial environment as opposed the specialized lung compartment (61).
As mentioned earlier, the lung interstitium environment is replete with glucose, but has much lower oxygen levels than the airway. This environment would likely dictate a more glycolytic phenotype for IMs compared to TR-AMs, which would be aligned with their developmental origins as monocytes. Under inflammatory conditions, IM expansion occurs and IMs can be readily found in the airway. It is still debated whether IM expansion occurs as a result of monocyte trafficking or self-renewal but it is likely a result from a combination of both mechanisms. Regardless, investigators have found that IMs favor glycolysis as their primary means for energy production. IMs secret higher lactate, have higher glycolytic gene expression, and lower FAO gene expression than TR-AMs (64). Mice infected with Mycobacterium tuberculosis and treated with 2-DG experienced IM loss and had a higher bacterial burden. This study found that IMs, proficient in glycolysis, were responsible for reduced bacterial burden, while TR-AMs, proficient in FAO, promoted bacterial growth (64). Collectively, IMs are glycolytically-oriented, which is beneficial in limiting the growth of Mycobacterium tuberculosis. The study of IM effector function and metabolism is still in its infancy, which leaves much to explore within the field.
5 Monocyte-derived alveolar macrophages
Monocyte-derived alveolar macrophages (Mo-AMs) are of monocytic origin, and enter the airway during acute inflammation where they both simultaneously retain monocytic characteristics while also being shaped into macrophages by the local airway microenvironment. Monocytes arise from adult hematopoietic stems cells (HSCs) within the bone marrow where oxygen concentrations range from 1.5 to 4% (65). HSCs rely on glycolysis for cellular maintenance, but harbor mitochondria that largely remain inactive as a means to limit reactive oxygen species damage in the hypoxic bone marrow (66–68). HSCs give rise to monocytes though colony stimulating factor 1 (CSF-1), and these monocytes circulate in the blood with an average lifespan of 1-3 days. Glucose transporter-1 (GLUT-1)-dependent glucose metabolism maintains monocyte homeostasis and migratory capacity to the extent that serum glucose concentrations correlate with monocyte numbers (69, 70). Representative of monocyte-derived macrophages, murine bone marrow-derived macrophages (BMDMs) have been used extensively for in vitro immunometabolism studies. These studies have found that robust glycolysis is a critical mediator of proinflammatory responses in BMDMs (71–75).
Under diseased conditions, the affected tissue generates chemokine gradients that recruit monocytes to the origin of the inflammatory insult. In this case, both general macrophage differentiation pathways are activated as well as tissue-specific pathways that give rise to a more specialized recruited macrophage. For example, recruited macrophages existing in the tumor microenvironment share generalized macrophage characteristics with recruited Mo-AMs, but will also possess unique characters more aptly tailored to the specific tissue site (76, 77).
By the time Mo-AMs have constituted the airway in ARDS, the low glucose/high oxygen/lipid-rich environment of the steady-state lung has dramatically shifted. From a nutrient perspective, the inflamed airway more closely resembles the high glucose/low oxygen niche of the bone-marrow from which the Mo-AMs arise. These conditions, along with monocytic imprinting, favor a functional glycolytic phenotype to fuel cellular processes. In the case of viral infection, it is well accepted that Mo-AMs exacerbate lung injury (42, 46, 78–81). It may be that Mo-AMs are initially well-suited to limit viral replication or to later limit secondary bacterial infection upon recovery from viral infection, but their presence is associated with excessive inflammation and poor outcomes (82, 83). Our group has observed that Mo-AMs express high levels of glycolytic and proinflammatory genes and low levels of OXPHOS-related genes in a murine model of influenza-induced ARDS (34, 35). This Mo-AM gene expression pattern remains relatively stable across the acute phase of the infection time course, while TR-AMs, if they do not experience cell death, shift from an OXPHOS dominant gene profile to a glycolysis-dominant gene profile as ARDS progresses (35). Work by Li et al. confirms these findings where Mo-AMs have high glycolysis and low OXPHOS during influenza infection (81). Mo-AMs recovered from patients with severe SARS-CoV-2 infection exhibited high HIF-1α expression and enrichment of hypoxia genes while also exhibiting reduced enrichment of OXPHOS genes, suggesting that Mo-AM metabolism is conserved in viral-induced ARDS (56).
Following successful resolution of viral infection, both self-renewing TR-AMs and Mo-AMs reconstitute the alveolar niche in response to viral-induced TR-AM death. Several studies have found that these persisting Mo-AMs are significant contributors to pulmonary fibrosis (84–90). In murine models of bleomycin-induced pulmonary fibrosis, TR-AM numbers decrease during the inflammatory phase and are almost entirely replaced by Mo-AMs (85, 91). Misharin and colleagues demonstrated that murine lung injury with either influenza or bleomycin resulted in 50% of the alveolar niche being composed of Mo-AMs one year post injury (85). These long-lived Mo-AMs may contribute to collagen deposition through enhanced secretion of profibrotic factors, including TGF-β (90). Additionally, murine Mo-AMs remain glycolytic following injury and the development of fibrosis (86). Analysis of human lungs has also identified associations between Mo-AMs and profibrotic lesions, and airway macrophages from patients with idiopathic pulmonary fibrosis express higher levels of GLUT-1 compared to healthy controls (88, 92–94).
Other groups have identified long-lived Mo-AMs to be beneficial. Aegerter et al. showed that Mo-AMs residing in the lung following influenza challenge secret more IL-6 than their TR-AM counterparts making them more effective in limiting secondary bacterial infection (82). Another group found that Mo-AMs constituted the airway following gamma herpesvirus infection and that these cells conferred protection against allergic asthma (95).
Collectively, glycolytically oriented Mo-AMs repopulate the airway following airway injury and inflammation. These cells have been associated with poor outcomes in ARDS and have identified in driving pulmonary fibrosis but may be beneficial in limiting secondary bacterial infection and allergic responses. More studies are needed to understand the appropriate balance of Mo-AMs and TR-AMs to not only prevent the development of chronic lung disease, but to also bolster the beneficial functions of Mo-AMs.
6 Experimental guidelines
Given the unique metabolic characteristics exhibited by TR-AMs compared to other macrophage populations within the body, it is worthwhile to discuss best experimental practices when assessing the glycolytic phenotype of lung macrophages. In this section, we will discuss the existing TR-AM culture systems including their metabolic implications and review experimental reagent selection for inhibiting or activating glycolysis and the interpretation of bioenergetics data. We will also provide information about the emerging new techniques that enable evaluation of metabolic profiles at single cell level. Ensuring that these best practices are followed will allow for the most accurate interpretation of macrophage metabolic phenotype and promote rigorous data reproducibility.
6.1 TR-AM culture systems
Sample collection is a major limitation to studying TR-AMs. A single mouse will yield approximately 300,000-500,000 TR-AMs via bronchoalveolar lavage, and obtaining human TR-AM specimens has several barriers making the endeavor impractical for routine use. Moreover, in vitro TR-AMs maintain high expression of many core TR-AM-specific genes, but genes related to the expression of key TR-AM functions, including surfactant handling and lipid metabolism, decrease with extended time in culture. This likely reflects the loss of cues from alveolar niche, and highlights the challenges of studying TR-AM metabolism in vitro (96). Granulocyte-macrophage colony-stimulating factor (GM-CSF), transforming growth factor β receptor (TGF-βR), and peroxisome proliferator-activated receptor gamma (PPARγ) have all been identified as critical regulators of TR-AM maturation and homeostasis (11, 23, 97). Gorki et al. established a system where isolated TR-AMs were cultured in the presence of GM-CSF, TGF-β, and rosiglitazone (PPARγ activator) (98). This combination yielded TR-AMs that could be expanded for months while maintaining functional and morphologic features found in freshly isolated TR-AMs. Another group took human monocytes and transformed them into “TR-AM-like cells” using bovine surfactant, GM-CSF, TGF-β, and IL-10 (99). Like the murine culture system, these human “TR-AM-like cells” exhibited a morphological, transcriptional, and metabolic profile more similar to freshly isolated human TR-AMs than to human monocyte-derived macrophages.
The full extent of how these different culture systems recreate functional TR-AM metabolism is debatable and necessitates further studies. In isolation, GM-CSF has been shown to be critical in regulating mitochondrial function in both TR-AMs and BMDMs (100, 101). GM-CSF has also been shown to enhance inflammation and glycolysis in murine BMDMs (102). TGF-β signaling regulates lipid metabolism in TR-AMs but was found to enhance glycolysis and reduce proinflammatory cytokine production in murine peritoneal macrophages (97, 103). Trying to recreate the lung niche through different cytokines and nutrients is an important endeavor. Providing TR-AMs with lipids through surfactant supplementation goes a step beyond adding lung specific cytokines and growth factors (99). However, to truly capture the alveolar niche, we may have to go a step further and consider co-culturing TR-AMs with alveolar epithelial cells. As mentioned before, TR-AMs form direct interactions with alveolar epithelial cells through extracellular receptors, like TGFBR and CD200R. Under these circumstances, TR-AMs do not proliferate at physiological concentrations of GM-CSF, and TGF-β serves as an inhibitor of macrophage activation. Thus, it is likely that GM-CSF and TGF-β may both keep TR-AMs more “AM-like” in culture, while also promoting a TR-AM phenotype that would not be observed within the context of the alveolar environment. Nevertheless, long-term TR-AM culture systems should be utilized with continual modifications being made in an attempt to make it easier and more physiologically relevant to study TR-AM function in vitro.
6.2 Agents used to inhibit or activate glycolysis
Inhibiting or activating glycolysis allows one to assess the full functional relevance of glucose catabolism in a given cell type. The most commonly used reagent for inhibiting glycolysis is 2-deoxy-D-glucose (2-DG), a stable glucose analog that is taken up by glucose transporters and retained in the cell following phosphorylation by hexokinase. 2-DG accumulation competitively inhibits the conversion of glucose to glucose-6-phoasphate leading to glycolytic arrest. While this is the desired outcome in assessing the effects of glycolysis inhibition on cellular function, 2-DG has been identified to have off several off target effects (104). Mainly, 2-DG inhibits N-linked glycosylation of proteins by also serving as a mannose mimic. N-linked glycosylation inhibition prevents proper protein folding and promotes protein retention in the endoplasmic reticulum (ER) leading to activation of the unfolded protein response and subsequent apoptosis (105). 2-DG-induced ER stress has been shown to inhibit both inflammatory capacity through COX-2 and fatty acid synthesis independently of glycolysis inhibition (106–108). Most importantly, commonly used 2-DG concentrations (10 mM) have been shown to inhibit oxidative phosphorylation independently of glycolysis in macrophages within one hour of treatment (109). These off target effects render 2-DG a less than optimal tool for studying the reliance of macrophages on glycolysis at steady-state or for effector functions. Alternatively, one could use oxamate, a competitive inhibitor of lactate dehydrogenase, that not only inhibits lactate production, but also inhibits overall glycolytic activity (110, 111). Manipulating culture media conditions to greatly reduce glucose availability via low glucose concentrations or by replacing glucose with galactose to greatly inhibit the rate of glycolysis, serve as valuable tools to study macrophages reliance on glycolysis without compromising cellular glycosylation (109). We have shown that overnight treatment of bone marrow-derived macrophages with 2-DG, but not oxamate or glucose-free media [non-dialyzed fetal bovine serum media supplementation provides roughly 0.4-0.8mM glucose (112)], induces cleaved caspase 3 expression, signifying a commitment to apoptosis (34, 113). Thus, we would suggest that 2-DG be avoided in studying the role of glycolysis in cellular effector function, and for 2-DG-based studies to be interpreted in such a way as to not overestimate the role of glycolysis in shaping metabolic phenotype.
Glycolysis can also be induced through the HIF-1α activation, which provides a method for assessing the full range of glycolytic phenotype in lung macrophages. This can be done through prolyl hydroxylase inhibition using hypoxia or pharmacological inhibitors. Under normal oxygen conditions, oxygen-dependent prolyl hydroxylases mark HIF-1α for proteasomal degradation. As oxygen levels decrease, prolyl hydroxylase activity becomes reduced so that HIF-1α accumulates, then enters the nucleus, and activates the hypoxic transcriptional response resulting in the upregulation of glycolytic enzyme expression (114). Hypoxia may alter macrophage inflammatory capacity independently of HIF-1α induction or changes in glycolysis so pharmacological inhibitors may offer a cleaner approach to studying how upregulation of glycolytic metabolism alters cellular function (35, 115). The most commonly used pharmacological prolyl hydroxylase inhibitor is dimethyloxalylglycine (DMOG), a synthetic analogue of α-ketoglutarate. This is a great tool for assessing HIF-1α induction and glycolytic phenotype, but it can also directly inhibit mitochondrial function and significantly reduce macrophage inflammatory capacity (34, 116–118). Thus, we suggest using second generation pharmacological prolyl hydroxylase inhibitors such as Roxadustat (FG-4592). We have found that Roxadustat can stabilize HIF-1α and significantly enhance glycolysis without significantly altering macrophage inflammatory capacity (35, 40). Roxadustat mildly suppress mitochondrial function in macrophages, but this effect is lost in HIF-1α knockout cells demonstrating that decreased respiration is linked to canonical HIF-1α activation (40). In contrast, decreases in cellular respiration following DMOG treatment precede HIF-1α activation and are independent of HIF-1α signaling, highlighting that DMOG has unspecified effects on cell metabolism (119).
6.3 Seahorse-based assessment of glycolysis
Seahorse (Agilent) metabolic flux assays have enabled users to assess the overall metabolic fitness of a given cell type through the measurement of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Mitochondrial oxygen consumption can be determined as the total OCR minus the OCR that remains after exposure to specific electron transport chain (ETC) inhibitors (120, 121). Thus, the OCR of a given cell type serves as a direct readout for the mitochondrial electron transport rate. The ECAR is routinely used as a direct surrogate in the measurement for glycolytic metabolism, but the ECAR requires a more nuanced analysis to determine whether acidification is derived from glycolysis. The conversion of one glucose molecule to two lactate molecules results in the net release of two protons that acidify the media, however, other metabolic reactions can also contribute to the ECAR. CO2 generated via the TCA cycle is readily converted to carbonic acid which can contribute media acidification (122). In fact, the complete oxidation of one glucose molecule by the TCA cycle yields six protons that can contribute to media acidification. This suggests that the ECAR alone can be an unreliable measure of cellular glycolysis, and we have found this to be the case in studying TR-AMs.
The standard Seahorse assay for assessing glycolytic metabolism is the glycolysis stress test. While this test is essential in measuring macrophages responsiveness to glucose, it often fails to assess the cells’ overall reliance on glycolysis for essential metabolic activity. For this reason, we would also suggest a thorough interrogation of the ECAR during a Mitochondrial Stress Test to get a complete understanding of the metabolic phenotype (Figure 1). One can determine cell’s reliance on glycolysis by assessing the ECAR changes in the presence of mitochondrial inhibitors. If a cell is truly glycolytic, then the ECAR rate should increase, and remain at a sustained, higher rate in the presence of oligomycin. The ECAR will also increase following the decoupler (FCCP), and this increase is a result of CO2-derived carbonic acid from the TCA cycle. Lastly, the ECAR of a cell that exhibits true glycolytic capabilities will remain relatively unresponsive to complex I and III inhibitors (Rotenone and Antimycin) suggesting that the majority of acidification is derived by glycolysis. This is quite visible in BMDMs (Figure 1B). In the case of TR-AMs, a higher, sustained ECAR is only observed following FCCP injection, and then the ECAR immediately returns to below baseline rates following Rotenone/Antimycin injection demonstrating that the contribution to the ECAR is entirely mitochondria-derived carbonic acid (Figure 1D). The spike in TR-AM ECAR in response to oligomycin, and its subsequent and immediate collapse is harder to parse out, but it does suggest that TR-AMs cannot sustain metabolic activity through anaerobic glycolysis. Others have documented the spike and crash in TR-AM ECAR following oligomycin injection (50, 81, 123).

Figure 1. Analysis of reciprocal ECAR measurements during mitochondrial stress test to assess glycolytic capabilities in response to mitochondrial inhibition. (A, B) BMDMs and (C-D) TR-AMs were treated sequentially with oligomycin (ATP synthase inhibitor), FCCP (uncoupler) and Rotenone/Antimycin A (complex I and III inhibitor, respectively). Red shading denotes glycolytic contribution to ECAR. Blue shading denotes Mitochondrial (Mito) contribution to ECAR. Purple shading denotes unknown contribution to ECAR.
Seahorse is a valuable tool in evaluating cellular metabolism ex vivo but is more limited in assessing cellular metabolism of rare and heterogeneous cell populations. New approaches like single-cell energetic metabolism by profiling translation inhibition (SCENITH) aim to circumvent these limitations (124). SCENITH relies on the knowledge that a substantial amount of the total energy produced through metabolic pathways is immediately consumed by protein synthesis machinery so that ATP production and protein synthesis are tightly coupled (125, 126). Measuring protein synthesis (puromycin incorporation) via fluorescence-activated cell sorting (FACS) in the presence of metabolic inhibitors can be used for metabolic profiling (124). More importantly, this methodology could provide single cell resolution of energy metabolism from heterogenous blood and tumor samples. This method is not without caveats in that the processing of tissue and FACS can lead to alterations in cellular metabolism (127). Likewise, selecting metabolic inhibitors with off-target effects could lead to inaccurate data interpretation. This is why the best approach for understanding cellular metabolism is to utilize multiple technologies. An “integrated toolbox to profile macrophage immunometabolism” that utilizes functional assays, Seahorse analysis, metabolic dyes, metabolomics, and SCENITH has been proposed to more conclusively determine the metabolic characteristics of a cell (128). Each methodology alone has its limitations, which can be counteracted through a multifaceted approach. We hope that this experimental guideline section can provide researchers with the basic tools for generating the most accurate and rigorous data in lung macrophage metabolism.
7 Conclusion
Despite recent advances in our understanding of pulmonary macrophage metabolism, key questions remain. Gene expression data suggest that TR-AMs utilize FAO at steady-state, but no one has mechanistically identified their preferred metabolic fuel through functional assays. Moreover, outside of developmental and environmental cues, we do not fully understand why TR-AMs cannot sustain cellular processes through glycolysis. Gaining a better understanding of the dynamics of TR-AMs, Mo-AMs, and IMs in disease inflammation and resolution may provide clues to some of these questions and also aid in the development of therapeutics to reduce mortality from ARDS and pulmonary fibrosis.
Author contributions
PW: Writing – original draft, Writing – review & editing, Investigation. GM: Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the Department of Defense grant HT9425-24-1-0138 (GM), and NIH R01ES015024 (GM). The funders did not have any role in the writing or submission of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Carreau A, El-Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. (2011) 15:1239–53. doi: 10.1111/j.1582-4934.2011.01258.x
2. Baker EH, Baines DL. Airway glucose homeostasis: A new target in the prevention and treatment of pulmonary infection. Chest. (2018) 153:507–14. doi: 10.1016/j.chest.2017.05.031
3. Baker EH, Clark N, Brennan AL, Fisher DA, Gyi KM, Hodson ME, et al. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol. (2007) 102:1969–75. doi: 10.1152/japplphysiol.01425.2006
4. Le Q-T, Chen E, Salim A, Cao H, Kong CS, Whyte R, et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non–small cell lung cancers. Clin Cancer Res. (2006) 12:1507–14. doi: 10.1158/1078-0432.Ccr-05-2049
5. Garnett JP, Baker EH, Baines DL. Sweet talk - insights into the nature and importance of glucose transport in lung epithelium. Eur Respir J. (2012), erj00526–2012 40:1269–76. doi: 10.1183/09031936.00052612
6. Gill SK, Hui K, Farne H, Garnett JP, Baines DL, Moore LSP, et al. Increased airway glucose increases airway bacterial load in hyperglycaemia. Sci Rep. (2016) 6:27636. doi: 10.1038/srep27636
7. Kemp PJ, Boyd CA. Pathways for glucose transport in type ii pneumocytes freshly isolated from adult Guinea pig lung. Am J Physiology-Lung Cell Mol Physiol. (1992) 263:L612–L6. doi: 10.1152/ajplung.1992.263.5.L612
8. Ridsdale R, Post M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type ii cells. Am J Physiology-Lung Cell Mol Physiol. (2004) 287:L743–L51. doi: 10.1152/ajplung.00146.2004
9. Wang Z, Wei D, Bin E, Li J, Jiang K, Lv T, et al. Enhanced glycolysis-mediated energy production in alveolar stem cells is required for alveolar regeneration. Cell Stem Cell. (2023) 30:1028–42.e7. doi: 10.1016/j.stem.2023.07.007
10. Baker AD, Malur A, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. Pparγ Regulates the expression of cholesterol metabolism genes in alveolar macrophages. Biochem Biophys Res Commun. (2010) 393:682–7. doi: 10.1016/j.bbrc.2010.02.056
11. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor ppar-Γ by the cytokine gm-csf is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. (2014) 15:1026–37. doi: 10.1038/ni.3005
12. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. (2000) 342:1334–49. doi: 10.1056/nejm200005043421806
13. Campbell Eric L, Bruyninckx Walter J, Kelly Caleb J, Glover Louise E, McNamee Eóin N, Bowers Brittelle E, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. (2014) 40:66–77. doi: 10.1016/j.immuni.2013.11.020
14. Fröhlich S, Boylan J, McLoughlin P. Hypoxia-induced inflammation in the lung. Am J Respir Cell Mol Biol. (2013) 48:271–9. doi: 10.1165/rcmb.2012-0137TR
15. Cui L, Zheng D, Lee YH, Chan TK, Kumar Y, Ho WE, et al. Metabolomics investigation reveals metabolite mediators associated with acute lung injury and repair in a murine model of influenza pneumonia. Sci Rep. (2016) 6:26076. doi: 10.1038/srep26076
16. Hofer CC, Woods PS, Davis IC. Infection of mice with influenza a/wsn/33 (H1n1) virus alters alveolar type ii cell phenotype. Am J Physiology-Lung Cell Mol Physiol. (2015) 308:L628–L38. doi: 10.1152/ajplung.00373.2014
17. Schousboe P, Ronit A, Nielsen HB, Benfield T, Wiese L, Scoutaris N, et al. Reduced levels of pulmonary surfactant in covid-19 ards. Sci Rep. (2022) 12:4040. doi: 10.1038/s41598-022-07944-4
18. Woods PS, Doolittle LM, Rosas LE, Joseph LM, Calomeni EP, Davis IC. Lethal H1n1 influenza a virus infection alters the murine alveolar type ii cell surfactant lipidome. Am J Physiology-Lung Cell Mol Physiol. (2016) 311:L1160–L9. doi: 10.1152/ajplung.00339.2016
19. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
20. Murray Peter J, Allen Judith E, Biswas Subhra K, Fisher Edward A, Gilroy Derek W, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
21. Morimoto K, Amano H, Sonoda F, Baba M, Senba M, Yoshimine H, et al. Alveolar macrophages that phagocytose apoptotic neutrophils produce hepatocyte growth factor during bacterial pneumonia in mice. Am J Respir Cell Mol Biol. (2001) 24:608–15. doi: 10.1165/ajrcmb.24.5.4292
22. Yoon Y-S, Lee Y-J, Choi Y-H, Park YM, Kang JL. Macrophages programmed by apoptotic cells inhibit epithelial-mesenchymal transition in lung alveolar epithelial cells via pge2, pgd2, and hgf. Sci Rep. (2016) 6:20992. doi: 10.1038/srep20992
23. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via gm-csf. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
24. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
25. Izquierdo HM, Brandi P, Gómez M-J, Conde-Garrosa R, Priego E, Enamorado M, et al. Von hippel-lindau protein is required for optimal alveolar macrophage terminal differentiation, self-renewal, and function. Cell Rep. (2018) 24:1738–46. doi: 10.1016/j.celrep.2018.07.034
26. Zhang W, Li Q, Li D, Li J, Aki D, Liu Y-C. The E3 ligase vhl controls alveolar macrophage function via metabolic–epigenetic regulation. J Exp Med. (2018) 215:3180–93. doi: 10.1084/jem.20181211
27. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
28. van de Laar L, Saelens W, De Prijck S, Martens L, Scott Charlotte L, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
29. Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. (2015) 22:485–98. doi: 10.1016/j.cmet.2015.07.020
30. Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science. (2020) 368:1371–6. doi: 10.1126/science.aax0860
31. Larsson N-G, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor a is necessary for mtdna maintance and embryogenesis in mice. Nat Genet. (1998) 18:231–6. doi: 10.1038/ng0398-231
32. Wculek SK, Heras-Murillo I, Mastrangelo A, Mañanes D, Galán M, Miguel V, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. (2023) 56:516–30.e9. doi: 10.1016/j.immuni.2023.01.011
33. Gao X, Zhu B, Wu Y, Li C, Zhou X, Tang J, et al. Tfam-dependent mitochondrial metabolism is required for alveolar macrophage maintenance and homeostasis. J Immunol. (2022) 208:1456–66. doi: 10.4049/jimmunol.2100741
34. Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O’Leary EM, et al. Tissue-resident alveolar macrophages do not rely on glycolysis for lps-induced inflammation. Am J Respir Cell Mol Biol. (2020) 62:243–55. doi: 10.1165/rcmb.2019-0244OC
35. Woods PS, Kimmig LM, Sun KA, Meliton AY, Shamaa OR, Tian Y, et al. Hif-1α Induces glycolytic reprograming in tissue-resident alveolar macrophages to promote cell survival during acute lung injury. eLife. (2022) 11:e77457. doi: 10.7554/eLife.77457
36. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway pseudomonas infections of cystic fibrosis patients. J Clin Invest. (2002) 109:317–25. doi: 10.1172/jci13870
37. Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol. (2017) 19:904–14. doi: 10.1038/ncb3580
38. Pereverzeva L, van Linge CCA, Schuurman AR, Klarenbeek AM, Ramirez Moral I, Otto NA, et al. Human alveolar macrophages do not rely on glucose metabolism upon activation by lipopolysaccharide. Biochim Biophys Acta (BBA) - Mol Basis Dis. (2022) 1868:166488. doi: 10.1016/j.bbadis.2022.166488
39. Russo S, Kwiatkowski M, Wolters JC, Gerding A, Hermans J, Govorukhina N, et al. Effects of lysine deacetylase inhibitor treatment on lps responses of alveolar-like macrophages. J Leukocyte Biol. (2023) 115:435–49. doi: 10.1093/jleuko/qiad121
40. Woods PS, Cetin-Atalay R, Meliton AY, Sun KA, Shamaa OR, Shin KWD, et al. Hif-1α Regulates mitochondrial function in bone marrow-derived macrophages, but not in tissue-resident alveolar macrophages. bioRxiv. (2024). doi: 10.1101/2024.10.14.618294
41. Bosteels C, Van Damme KFA, De Leeuw E, Declercq J, Maes B, Bosteels V, et al. Loss of gm-csf-dependent instruction of alveolar macrophages in covid-19 provides a rationale for inhaled gm-csf treatment. Cell Rep Med. (2022) 3:100833. doi: 10.1016/j.xcrm.2022.100833
42. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with covid-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
43. Nelson B, Zhou X, White M, Hartshorn K, Takahashi K, Kinane TB, et al. Recombinant human mannose-binding lectin dampens human alveolar macrophage inflammatory responses to influenza a virus in vitro. J Leukocyte Biol. (2014) 95:715–22. doi: 10.1189/jlb.0313161
44. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PloS Pathog. (2014) 10:e1004053. doi: 10.1371/journal.ppat.1004053
45. Wendisch D, Dietrich O, Mari T, von Stillfried S, Ibarra IL, Mittermaier M, et al. Sars-cov-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. (2021) 184:6243–61.e27. doi: 10.1016/j.cell.2021.11.033
46. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. Circuits between infected macrophages and T cells in sars-cov-2 pneumonia. Nature. (2021) 590:635–41. doi: 10.1038/s41586-020-03148-w
47. Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol. (2020) 21:636–48. doi: 10.1038/s41590-020-0673-x
48. Albers GJ, Michalaki C, Ogger PP, Lloyd AF, Causton B, Walker SA, et al. Airway macrophage glycolysis controls lung homeostasis and responses to aeroallergen. Mucosal Immunol. (2024) 18:121–34. doi: 10.1016/j.mucimm.2024.10.002
49. Cox DJ, Connolly SA, Maoldomhnaigh CÓ, Brugman AAI, Thomas OS, Duffin E, et al. Human airway macrophages are metabolically reprogrammed by Ifn-Γ resulting in glycolysis dependent functional plasticity. eLife (2024) 13:RP98449. doi: 10.7554/elife.98449.3
50. Svedberg FR, Brown SL, Krauss MZ, Campbell L, Sharpe C, Clausen M, et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat Immunol. (2019) 20:571–80. doi: 10.1038/s41590-019-0352-y
51. O’Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV, et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2020) 201:735–9. doi: 10.1164/rccm.201908-1683LE
52. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in copd macrophages. Eur Respir J. (2019) 54:1802244. doi: 10.1183/13993003.02244-2018
53. Bewley MA, Preston JA, Mohasin M, Marriott HM, Budd RC, Swales J, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med. (2017) 196:845–55. doi: 10.1164/rccm.201608-1714OC
54. Wohnhaas CT, Baßler K, Watson CK, Shen Y, Leparc GG, Tilp C, et al. Monocyte-derived alveolar macrophages are key drivers of smoke-induced lung inflammation and tissue remodeling. Front Immunol. (2024) 15:1325090. doi: 10.3389/fimmu.2024.1325090
55. Zhu B, Wei X, Narasimhan H, Qian W, Zhang R, Cheon IS, et al. Inhibition of the mitochondrial pyruvate carrier simultaneously mitigates hyperinflammation and hyperglycemia in covid-19. Sci Immunol. (2023) 8:eadf0348. doi: 10.1126/sciimmunol.adf0348
56. Zhu B, Wu Y, Huang S, Zhang R, Son YM, Li C, et al. Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity. (2021) 54:1200–18.e9. doi: 10.1016/j.immuni.2021.04.001
57. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for cd200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. (2008) 9:1074–83. doi: 10.1038/ni.1637
58. Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci. (2017) 114:2934–9. doi: 10.1073/pnas.1700600114
59. Ma R-Y, Black A, Qian B-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. (2022) 43:546–63. doi: 10.1016/j.it.2022.04.008
60. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. (2019) 363:eaau0964. doi: 10.1126/science.aau0964
61. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol. (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
62. Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun. (2019) 10:3964. doi: 10.1038/s41467-019-11843-0
63. Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol. (2020) 5:eaax8756. doi: 10.1126/sciimmunol.aax8756
64. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
65. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. (2014) 508:269–73. doi: 10.1038/nature13034
66. Mantel C, Messina-Graham S, Broxmeyer HE. Upregulation of nascent mitochondrial biogenesis in mouse hematopoietic stem cells parallels upregulation of cd34 and loss of pluripotency: A potential strategy for reducing oxidative risk in stem cells. Cell Cycle. (2010) 9:2008–17. doi: 10.4161/cc.9.10.11733
67. Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. (2010) 7:380–90. doi: 10.1016/j.stem.2010.07.011
68. Xu X, Duan S, Yi F, Ocampo A, Liu G-H, Izpisua Belmonte Juan C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. (2013) 18:325–32. doi: 10.1016/j.cmet.2013.06.005
69. Gallerand A, Dolfi B, Stunault MI, Caillot Z, Castiglione A, Strazzulla A, et al. Glucose metabolism controls monocyte homeostasis and migration but has no impact on atherosclerosis development in mice. Nat Commun. (2024) 15:9027. doi: 10.1038/s41467-024-53267-5
70. Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell. (2019) 178:1102–14.e17. doi: 10.1016/j.cell.2019.07.050
71. Corcoran SE, O’Neill LA. Hif1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/jci84431
72. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. Hif-1alpha is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/s0092-8674(03)00154-5
73. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (Glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
74. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of il-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
75. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates hif-1α Activity and il-1β Induction and is a critical determinant of the warburg effect in lps-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
76. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S, et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. (2022) 7:eabf7777. doi: 10.1126/sciimmunol.abf7777
77. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. (2021) 54:1883–900.e5. doi: 10.1016/j.immuni.2021.07.007
78. Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of ccr5 and ccr2 deficiency in the pulmonary inflammatory response to influenza a virus. Am J Pathol. (2000) 156:1951–9. doi: 10.1016/S0002-9440(10)65068-7
79. Gurczynski SJ, Nathani N, Warheit-Niemi HI, Hult EM, Podsiad A, Deng J, et al. Ccr2 mediates increased susceptibility to post-H1n1 bacterial pneumonia by limiting dendritic cell induction of il-17. Mucosal Immunol. (2019) 12:518–30. doi: 10.1038/s41385-018-0106-4
80. Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD. Ccr2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol. (2011) 186:508–15. doi: 10.4049/jimmunol.1001002
81. Li F, Piattini F, Pohlmeier L, Feng Q, Rehrauer H, Kopf M. Monocyte-derived alveolar macrophages autonomously determine severe outcome of respiratory viral infection. Sci Immunol. (2022) 7:eabj5761. doi: 10.1126/sciimmunol.abj5761
82. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. (2020) 21:145–57. doi: 10.1038/s41590-019-0568-x
83. Hoeve MA, Nash AA, Jackson D, Randall RE, Dransfield I. Influenza virus a infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PloS One. (2012) 7:e29443. doi: 10.1371/journal.pone.0029443
84. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. (2008) 14:633–40. doi: 10.1038/nm1770
85. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
86. Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S, et al. Metabolic characterization and rna profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiology-Lung Cell Mol Physiol. (2017) 313:L834–L44. doi: 10.1152/ajplung.00235.2017
87. McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest. (2021) 131:e140299. doi: 10.1172/jci140299
88. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. (2019) 20:163–72. doi: 10.1038/s41590-018-0276-y
89. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-csf/M-csfr signalling in monocyte-derived alveolar macrophages. Eur Respir J. (2020) 55:1900646. doi: 10.1183/13993003.00646-2019
90. Zhang N, Yang K, Bai J, Yi J, Gao C, Zhao J, et al. Myeloid-specific blockade of notch signaling alleviates murine pulmonary fibrosis through regulating monocyte-derived ly6c(Lo) mhcii(Hi) alveolar macrophages recruitment and tgf-B Secretion. FASEB J. (2020) 34:11168–84. doi: 10.1096/fj.201903086RR
91. McCowan J, Fercoq F, Kirkwood PM, T’Jonck W, Hegarty LM, Mawer CM, et al. The transcription factor egr2 is indispensable for tissue-specific imprinting of alveolar macrophages in health and tissue repair. Sci Immunol. (2021) 6:eabj2132. doi: 10.1126/sciimmunol.abj2132
92. Brody SL, Gunsten SP, Luehmann HP, Sultan DH, Hoelscher M, Heo GS, et al. Chemokine receptor 2-targeted molecular imaging in pulmonary fibrosis. A clinical trial. Am J Respir Crit Care Med. (2021) 203:78–89. doi: 10.1164/rccm.202004-1132OC
93. El-Chemaly S, Malide D, Yao J, Nathan SD, Rosas IO, Gahl WA, et al. Glucose transporter-1 distribution in fibrotic lung disease: association with [¹⁸f]-2-fluoro-2-deoxyglucose-pet scan uptake, inflammation, and neovascularization. Chest. (2013) 143:1685–91. doi: 10.1378/chest.12-1359
94. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36. doi: 10.1164/rccm.201712-2410OC
95. Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. (2017) 18:1310–20. doi: 10.1038/ni.3857
96. Subramanian S, Busch CJ-L, Molawi K, Geirsdottir L, Maurizio J, Vargas Aguilar S, et al. Long-term culture-expanded alveolar macrophages restore their full epigenetic identity after transfer in vivo. Nat Immunol. (2022) 23:458–68. doi: 10.1038/s41590-022-01146-w
97. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine tgf-B Promotes the development and homeostasis of alveolar macrophages. Immunity. (2017) 47:903–12.e4. doi: 10.1016/j.immuni.2017.10.007
98. Gorki A-D, Symmank D, Zahalka S, Lakovits K, Hladik A, Langer B, et al. Murine ex vivo cultured alveolar macrophages provide a novel tool to study tissue-resident macrophage behavior and function. Am J Respir Cell Mol Biol. (2022) 66:64–75. doi: 10.1165/rcmb.2021-0190OC
99. Pahari S, Arnett E, Simper J, Azad A, Guerrero-Arguero I, Ye C, et al. A new tractable method for generating human alveolar macrophage-like cells in vitro to study lung inflammatory processes and diseases. mBio. (2023) 14:e00834–23. doi: 10.1128/mbio.00834-23
100. Staitieh BS, Auld SC, Ahmed M, Fan X, Smirnova N, Yeligar SM. Granulocyte macrophage-colony stimulating factor reverses hiv protein-induced mitochondrial derangements in alveolar macrophages. AIDS Res Hum Retroviruses. (2021) 37:224–32. doi: 10.1089/aid.2020.0176
101. Wessendarp M, Watanabe-Chailland M, Liu S, Stankiewicz T, Ma Y, Kasam RK, et al. Role of gm-csf in regulating metabolism and mitochondrial functions critical to macrophage proliferation. Mitochondrion. (2022) 62:85–101. doi: 10.1016/j.mito.2021.10.009
102. Na YR, Gu GJ, Jung D, Kim YW, Na J, Woo JS, et al. Gm-csf induces inflammatory macrophages by regulating glycolysis and lipid metabolism. J Immunol. (2016) 197:4101–9. doi: 10.4049/jimmunol.1600745
103. Gauthier T, Yao C, Dowdy T, Jin W, Lim YJ, Patiño LC, et al. Tgf-B Uncouples glycolysis and inflammation in macrophages and controls survival during sepsis. Sci Signal. (2023) 16:eade0385. doi: 10.1126/scisignal.ade0385
104. Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, et al. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci U.S.A. (2008) 105:17807–11. doi: 10.1073/pnas.0803090105
105. Kurtoglu M, Maher JC, Lampidis TJ. Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D -glucose in hypoxic and normoxic tumor cells. Antioxidants Redox Signaling. (2007) 9:1383–90. doi: 10.1089/ars.2007.1714
106. Yu S-M, Kim S-J. Endoplasmic reticulum stress (Er-stress) by 2-deoxy-D-glucose (2dg) reduces cyclooxygenase-2 (Cox-2) expression and N-glycosylation and induces a loss of cox-2 activity via a src kinase-dependent pathway in rabbit articular chondrocytes. Exp Mol Med. (2010) 42:777–86. doi: 10.3858/emm.2010.42.11.079
107. Harada N, Yoshikatsu A, Yamamoto H, Nakaya Y. 2-deoxy-D-glucose downregulates fatty acid synthase gene expression via an endoplasmic reticulum stress-dependent pathway in hela cells. Cell Biochem Biophys. (2024) 82:2285–96. doi: 10.1007/s12013-024-01339-0
108. Andresen L, Skovbakke SL, Persson G, Hagemann-Jensen M, Hansen KA, Jensen H, et al. 2-deoxy D-glucose prevents cell surface expression of nkg2d ligands through inhibition of N-linked glycosylation. J Immunol. (2012) 188:1847–55. doi: 10.4049/jimmunol.1004085
109. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–75.e4. doi: 10.1016/j.cmet.2018.08.012
110. Coe EL, Strunk RC. The effect of oxamate on glycolysis in intact ascites tumor cells I. Kinetic evidence for a dual glycolytic system. Biochim Biophys Acta (BBA) - Gen Subj. (1970) 208:189–202. doi: 10.1016/0304-4165(70)90237-0
111. Moreno-Sánchez R, Marín-Hernández Á, Del-Mazo-Monsalvo I, Saavedra E, Rodríguez-Enríquez S. Assessment of the low inhibitory specificity of oxamate, aminooxyacetate and dichloroacetate on cancer energy metabolism. Biochim Biophys Acta (BBA) - Gen Subj. (2017) 1861:3221–36. doi: 10.1016/j.bbagen.2016.08.006
112. Crooks DR, Fan TW, Linehan WM. Metabolic labeling of cultured mammalian cells for stable isotope-resolved metabolomics: practical aspects of tissue culture and sample extraction. Methods Mol Biol. (2019) 1928:1–27. doi: 10.1007/978-1-4939-9027-6_1
113. Sun X-M, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis*. J Biol Chem. (1999) 274:5053–60. doi: 10.1074/jbc.274.8.5053
114. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
115. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U.S.A. (1998) 95:11715–20. doi: 10.1073/pnas.95.20.11715
116. Hams E, Saunders SP, Cummins EP, O’Connor A, Tambuwala MT, Gallagher WM, et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and il-10 production by B1 cells. Shock. (2011) 36:295–302. doi: 10.1097/SHK.0b013e318225ad7e
117. Hirai K, Furusho H, Hirota K, Sasaki H. Activation of hypoxia-inducible factor 1 attenuates periapical inflammation and bone loss. Int J Oral Sci. (2018) 10:12. doi: 10.1038/s41368-018-0015-0
118. Palaniyandi S, Kumari R, Venniyil Radhakrishnan S, Strattan E, Hakim N, Munker R, et al. The prolyl hydroxylase inhibitor dimethyl oxalyl glycine decreases early gastrointestinal gvhd in experimental allogeneic hematopoietic cell transplantation. Transplantation. (2020) 104:2507–15. doi: 10.1097/tp.0000000000003383
119. Zhdanov AV, Okkelman IA, Collins FWJ, Melgar S, Papkovsky DB. A novel effect of dmog on cell metabolism: direct inhibition of mitochondrial function precedes hif target gene expression. Biochim Biophys Acta (BBA) - Bioenergetics. (2015) 1847:1254–66. doi: 10.1016/j.bbabio.2015.06.016
120. Brand MD. The proton leak across the mitochondrial inner membrane. Biochim Biophys Acta (BBA) - Bioenergetics. (1990) 1018:128–33. doi: 10.1016/0005-2728(90)90232-S
121. Hütter E, Renner K, Jansen-Dürr P, Gnaiger E. Biphasic oxygen kinetics of cellular respiration and linear oxygen dependence of antimycin a inhibited oxygen consumption. Mol Biol Rep. (2002) 29:83–7. doi: 10.1023/A:1020322922732
122. Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta (BBA) - Bioenergetics. (2015) 1847:171–81. doi: 10.1016/j.bbabio.2014.10.005
123. Guo Z, Yang H, Zhang JR, Zeng W, Hu X. Leptin receptor signaling sustains metabolic fitness of alveolar macrophages to attenuate pulmonary inflammation. Sci Adv. (2022) 8:eabo3064. doi: 10.1126/sciadv.abo3064
124. Argüello RJ, Combes AJ, Char R, Gigan J-P, Baaziz AI, Bousiquot E, et al. Scenith: A flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab. (2020) 32:1063–75.e7. doi: 10.1016/j.cmet.2020.11.007
125. Buttgereit F, Brand MD. A hierarchy of atp-consuming processes in mammalian cells. Biochem J. (1995) 312:163–7. doi: 10.1042/bj3120163
126. Lindqvist LM, Tandoc K, Topisirovic I, Furic L. Cross-talk between protein synthesis, energy metabolism and autophagy in cancer. Curr Opin Genet Dev. (2018) 48:104–11. doi: 10.1016/j.gde.2017.11.003
127. Llufrio EM, Wang L, Naser FJ, Patti GJ. Sorting cells alters their redox state and cellular metabolome. Redox Biol. (2018) 16:381–7. doi: 10.1016/j.redox.2018.03.004
Keywords: alveolar macrophage, monocyte-derived alveolar macrophage, interstitial macrophage, bone marrow-derived macrophage, metabolism, glycolysis, bioenergetics, cytokines
Citation: Woods PS and Mutlu GM (2025) Differences in glycolytic metabolism between tissue-resident alveolar macrophages and recruited lung macrophages. Front. Immunol. 16:1535796. doi: 10.3389/fimmu.2025.1535796
Received: 27 November 2024; Accepted: 13 February 2025;
Published: 28 February 2025.
Edited by:
Hao Wu, University of Würzburg, GermanyReviewed by:
Peter Sporn, Northwestern University, United StatesFotios Karagiannis, University Hospital Bonn, Germany
Li Yu, First Affiliated Hospital of Southern University of Science and Technology, China
Somenath Banerjee, University of Illinois Chicago, United States
Łukasz Gałgański, Adam Mickiewicz University, Poland
Copyright © 2025 Woods and Mutlu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gökhan M. Mutlu, Z211dGx1QHVjaGljYWdvLmVkdQ==