
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 February 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1530849
 Yuju Cai1,2†
Yuju Cai1,2† Luorui Shang1†
Luorui Shang1† Fangyuan Zhou1Mengqi Zhang1
Fangyuan Zhou1Mengqi Zhang1 Jinxiao Li1Shuhan Wang1Qifeng Lin1Jianghua Huang1
Jinxiao Li1Shuhan Wang1Qifeng Lin1Jianghua Huang1 Shenglan Yang1*
Shenglan Yang1*Acute lung injury(ALI)/acute respiratory distress syndrome(ARDS) is a severe clinical syndrome characterized by high morbidity and mortality, primarily due to lung injury. However, the pathogenesis of ALI/ARDS remains a complex issue. In recent years, the role of macrophage pyroptosis in lung injury has garnered extensive attention worldwide. This paper reviews the mechanism of macrophage pyroptosis, discusses its role in ALI/ARDS, and introduces several drugs and intervening measures that can regulate macrophage pyroptosis to influence the progression of ALI/ARDS. By doing so, we aim to enhance the understanding of the mechanism of macrophage pyroptosis in ALI/ARDS and provide novel insights for its treatment.
ALI/ARDS is a common respiratory disease, which is an acute hypoxic respiratory insufficiency caused directly or indirectly by various intrapulmonary or extrapulmonary factors, with progressive hypoxemia and respiratory distress as the main clinical feature (1, 2). About 3 million people suffer from ALI/ARDS around the world every year, and the mortality is as high as 35%-46% (3). The risk factors for ALI/ARDS include direct and indirect lung injuries. The direct factors include infectious pneumonia, aspiration of stomach contents and severe trauma. On the contrary, indirect factors arise from a systemic inflammatory response triggered outside the lungs, such as sepsis from non-pulmonary infections, non-thoracic trauma, pancreatitis, severe burns, blood product transfusion, and reperfusion edema following lung transplantation or thrombectomy (4). Currently, no effective treatment exists to reduce mortality or improve the prognosis of patients with ALI/ARDS. Therefore, exploring the pathogenesis of ALI/ARDS is crucial for developing effective treatments.
The pulmonary innate immune system serves as the first line of defense against external stimuli by recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Innate immune cells located in the lung epithelium play an essential role by producing pro-inflammatory factors to eliminate pathogens and releasing anti-inflammatory factors to maintain lung homeostasis (5). Pulmonary macrophage is a critical cell of the pneumonic innate immune system, expressing pattern recognition receptors(PRRs) to recognize PAMPs and DAMPs (6). Once the PAMPs or DAMPs are recognized by PRRs, macrophages will initiate a series of immune responses, including inflammasome activation and release of inflammatory cytokines. These processes, in addition to removing pathogens, also lead to damage and pyroptosis of macrophages. On the one hand, activated inflammasomes recruit and activate inflammatory caspases and subsequently lead to activation and release of pro-inflammatory factors like IL-1β and IL-18, which further aggravates the inflammatory response (7). On the other hand, activated caspases can also cleave and activate gasdermin protein, contributing to cell membrane perforation and cell swelling, eventually inducing cell pyroptosis (8). Generally, macrophage pyroptosis contributes to the occurrence and development of ALI/ARDS. However, the precise regulatory mechanisms of macrophage pyroptosis remain incompletely understood and involve a range of signaling pathways and other regulatory networks (9, 10). The regulation of macrophage pyroptosis may represent a novel therapeutic direction for ALI/ARDS. In this review, we summarize the known mechanisms of macrophage pyroptosis and its impact on ALI/ARDS.
Ashbaugh, with his co-workers, first put forward the term ALI/ARDS in 1967 (11). In 1994, American-European Consensus Conference(AECC)proposed the clinical definition of ALI/ARDS: rapid onset of respiratory failure(ALI is diagnosed when 200 mmHg<PaO2/FiO2<300 mmHg and the criteria for ARDS are met when PaO2/FiO2< 200mmHg); chest radiography showing bilateral diffuse pulmonary infiltration; absence of increased pulmonary artery wedge pressure; and no clinical manifestation of left atrial hypertension (12, 13). Subsequently, the ARDS was classified into the following three categories based on the degree of hypoxemia according to the later Berlin definition: mild ARDS(PaO2/FiO2 = 200-300mmHg); moderate ARDS(PaO2/FiO2 = 100~200mmHg); and severe ARDS(PaO2/FiO2<100mmHg) (14). Although the background of onset is different between ALI and ARDS, the clinical symptoms, pathophysiological features and specific targets for pharmacological interventions are similar, which contributes to the fact that ALI and ARDS are usually studied as a whole (15). The pathological characteristics of ALI/ARDS include depressed pulmonary compliance, increased pulmonary vascular permeability and alveolar and interstitial pulmonary edema (Figure 1) (16, 17), while the main clinical symptoms are refractory hypoxemia and progressive exacerbation of hypoxic respiratory failure (1).
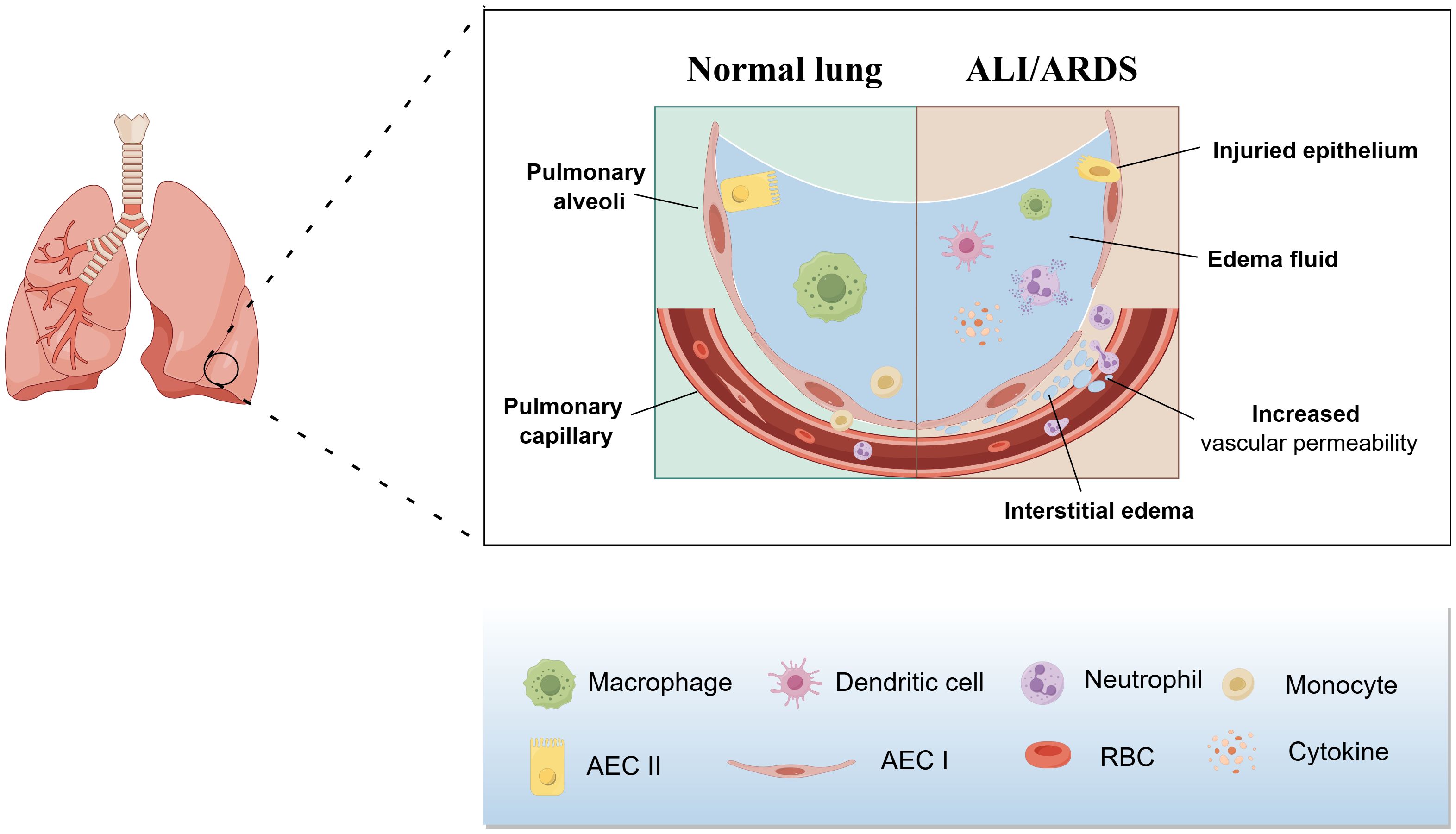
Figure 1. Pathological features of ALI/ARDS. The pathologic features of ALI/ARDS include diffuse necrosis of alveolar capillary endothelial cells and epithelial cells, increased permeability of the pulmonary capillary endothelial and alveolar epithelial barriers, accumulation of protein-rich edema fluid, extensive pulmonary hemorrhage, and thrombosis in the alveolar hyaloid membrane and capillaries.
ALI/ARDS is characterized by life-threatening lung injury, in which immune mechanisms play an essential role. In response to lung damage, the immune system initiates a series of inflammatory responses to remove pathogens and protect lung tissue. Pulmonary immune cells, such as dendritic cells, natural killer cells, macrophages, and neutrophils, maintain lung-tissue homeostasis (18). However, abnormal immune cell function can lead to continuous or excessive inflammatory reactions and subsequent lung tissue damage. Macrophages and neutrophils are additional immune cells that contribute to the pathogenesis of ALI/ARDS (18). Although macrophages play a key role in lung defense, their excessive activation aggravates lung damage. Various activation and death modes of macrophages, including polarization, apoptosis, autophagy, and pyroptosis et al., have different impacts on ALI/ARDS. Among them, pyroptosis is a newly discovered form of programmed cell death, depending on caspase-triggered cleavage of gasdermin proteins following recognition of the ligand in the cytoplasm (19). In ALI/ARDS caused by various factors, pulmonary damage is always accompanied by macrophage pyroptosis (20–22), suggesting that macrophage pyroptosis plays an essential role in ALI/ARDS.
At present, supportive treatment is the main therapeutic method for ALI/ARDS mainly including lung protective mechanical ventilation and fluid management therapy, supplemented by glucocorticoids, surfactants, and extracorporeal membrane oxygenation (23). Although supportive treatment can improve patients’ symptoms to some extent, it cannot significantly improve prognosis, instead the mortality remains high, and even patients in recovery may face long-term cognitive impairment and impaired quality of life (24). In addition, long-term or excessive mechanical ventilation may also lead to ventilator-induced lung injury(VILI)and even pulmonary fibrosis (25, 26). Therefore, it is of great significance to figure out the pathogenesis so that we can find much safer and more efficient treatment measures for ALI/ARDS.
Pyroptosis is defined as gasdermin-mediated programmed cell death and closely related to a variety of diseases (27). Pyroptosis was first identified by Zychlinsky et al. when they first discovered suicide events in macrophages infected with the Gram-negative pathogen Shigella flexneri in 1992 (28). In the initial study, it was found that there were some similar characteristics between apoptosis and such a cell death way, such as caspase independence, DNA damage and nuclear condensation et al., so it had been called apoptosis for a long time. The subsequent studies showed it was different from apoptosis, especially in the orderliness of DNA fragments and the integrity of cell nucleus (29). In 2001, D’souza et al. first proposed the term of pyroptosis to describe this kind of programmed proinflammatory cell death, thereby distinguishing pyroptosis from apoptosis (30).
The human gasdermin protein family consists of six proteins: gasdermin A–E and deafness autosomal recessive type 59 (DFNB59, also called Pejvakin). Except for DFNB59, all gasdermin proteins contain two conserved domains including the N-terminal pore-forming domain (PFD) and the C-terminal repressor domain (RD) connected by a peptide linker (31). Normally, the PFD and RD of gasdermin proteins interact to maintain oligomerization so that the cytotoxicity of PFD can be inhibited by RD (32, 33). After the host is stimulated, the activation of upstream molecules such as caspase-1 and caspase-11/4/5 will trigger the cleavage of gasdermin, separating the PFD from the RD (34). Then the PFD oligomer binds to cell membranes, contributing to damage of cell membranes and formation of perforation, which further leads to cell swelling and release of inflammatory factors such as IL-1β and IL-18 (32, 35) and meanwhile perturbs regulation of ions and water (36). The formation of perforation, cell cleavage and release of proinflammatory cytokine are significant characteristics of pyroptosis (37).
At present, there are three identified pathways of pyroptosis: canonical inflammasome pathway, non-canonical inflammasome pathway and inflammasome-independent pathway (Figure 2).
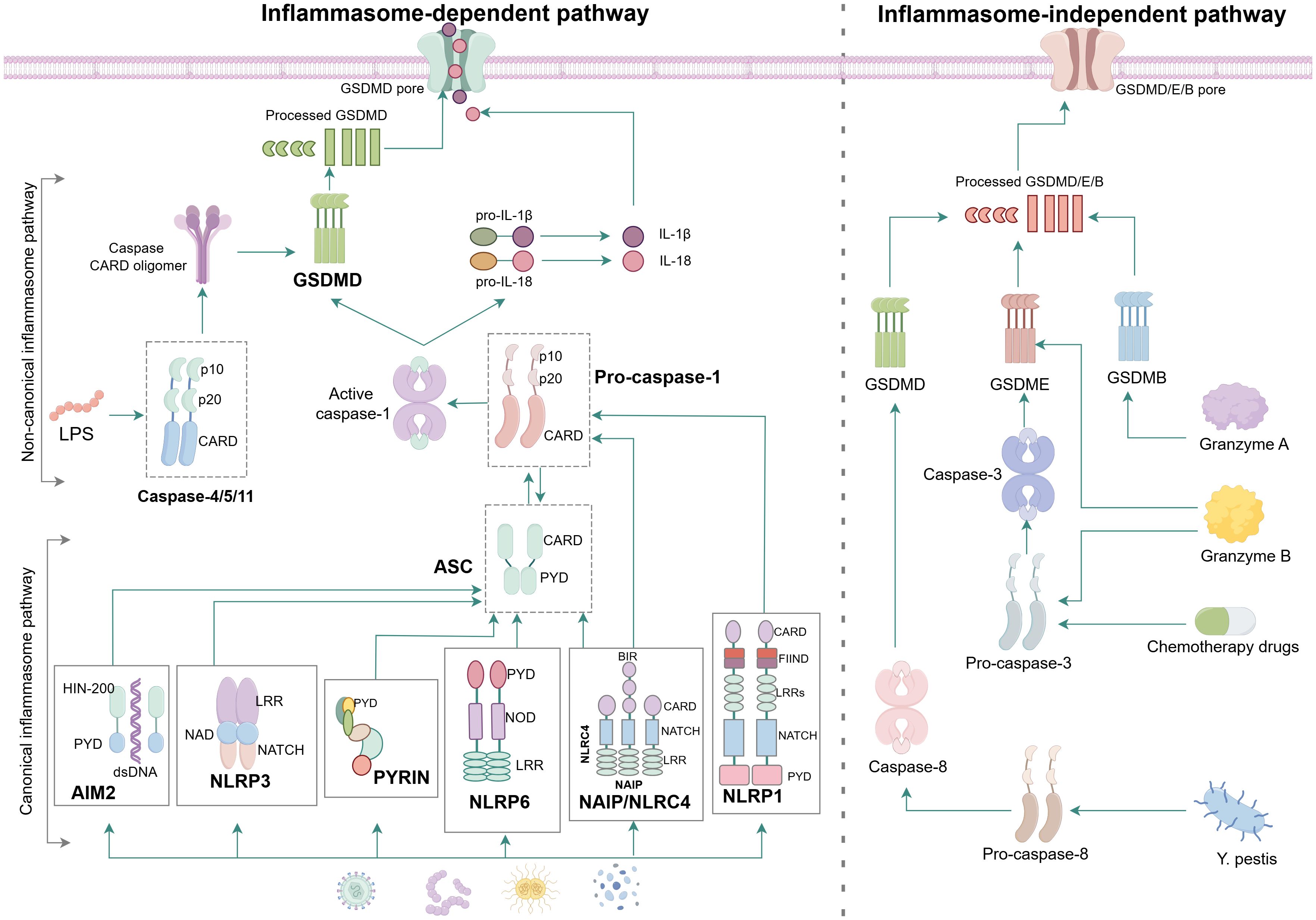
Figure 2. Molecular mechanism of pyroptosis. In the canonical inflammasome pathway, the majority of inflammasome complex is assembled from intracellular sensor protein, adapter proteins ASC and effector protein caspase. When PAMPs or DAMPs such as toxins, bacteria, viruses and dsDNA intrude, the NLRP1 and NAIP/NLRC4 can directly convert pro-caspase-1 into active caspase-1 because both of them have the CARD to connect with pro-caspase-1, while the AIM2, NLRP3, PYRIN and NLRP6 have to bind with ASC firstly before activating the pro-caspase-1 due to the lack of CARD. Active caspase-1 cleaves the GSDMD to causing damage to cell membrane and formation of perforations meanwhile cleaves pro-IL-1β and pro-IL-18 and results in the maturation of IL-1β and IL-18 that are subsequently released from the N-GSDMD pores. In the non-canonical inflammasome pathway, LPS directly binds to pro-caspase-4/5/11, resulting in activation of caspase-4/5/11, which cleaves GSDMD to trigger pyroptosis. In the granzyme-A/B-dependent pyroptosis pathway, GzmA and GzmB from NK cells and CD8 + T cells enter cancer cells via perforin and recognise GSDMB and GSDME, respectively, to induce pyroptosis. In addition, chemotherapeutic drugs and Y. pestis trigger pyroptosis through the caspase-3/GSDME or caspase-8/GSDMD cascades.
The canonical inflammasome pathway is mediated by assembly of inflammasomes. As a large cytoplasmic multiprotein complex, the inflammasome is assembled in response to pathogens, damage-associated stimulation, and other danger signals that disrupt cell homeostasis (38, 39). It is capable of regulating the activation of caspase (40). The majority of inflammasome complexes consist of sensor protein (also called pattern recognition receptor, PRR), adopter protein(an apoptosis-associated speck-like protein [ASC] containing caspase activation and recruitment domain [CARD]) and effector caspase (pro-caspase-1) (41). ASC is a bipartite molecule containing a pyrin domain(PYD) and a caspase activation and recruitment domain (CARD), enabling itself to bridge the sensor and effector pro-caspase-1 (42, 43). The structure of caspase also contains a CARD, allowing ASC to interact with it through CARD-CARD coaction to recruit pro-caspase-1 for inflammasome assembly.
Several inflammasomes have been identified up to now including NLRP1, NLRP3, NAIP/NLRC4, AIM2, PYRIN and CARD8 et al. It is fundamental and essential for assembly of inflammasome complex to produce spirochetes through CARD-CARD or PYD-PYD interactions, which will mediate oligomerization of homology and heterogeneity (44, 45). As the first identified key component of inflammasome, the human NLRP1 contains an N-terminal PYD and a C-terminal CARD (46). NLRP3 contains three conserved domains including the nucleotide binding and oligomerization (NACHT) domain in the middle, the leucine-rich repeat domain (LRR) in the C terminus, and the pyrin domain (PYD) in the N terminus (47). When activated and assembled, NACHT domain acts as a support structure for oligomerization of NLRP3 to recruit ASC and pro-caspase-1 to form multiprotein inflammasome complex (48, 49). NAIPs are cytoplasmic receptors for various bacterial protein ligands, leading to recruitment of the adaptor protein NLRC4 to form the NAIP-NLRC4 inflammasome (50, 51). NLRC4 containing CARD can serve as an adaptor in the downstream of NAIP to recruit CASP1, and NAIP, together with NLRC4 and CASP1, is sufficient to initiate pyroptosis although it is essential to recruit ASC adaptor to the complex for processing of IL-1 and IL-18 (41). As one of the members of PYHIN protein family, AIM2 contains a PYRIN domain(PYD) and a HIN-200 DNA-binding domain. AIM2 differs from other inflammasomes components due to the lack of NBD and LRR, but it can still coordinate the oligomerization of large inflammasome complexes by the way that AIM2 interacts with ASC via PYD (52), and the downstream pathway, including the processing of cytokines and pyroptosis, is similar to that of proteins containing NBD-LRR domains. The structure of PYRIN includes the N-terminal PYD, b30.2/SPRY domain, and central B box and coiled-coil domain, whose mode of action is similar to NLRP3. At present, NLRP12, NLRP6 and NLRP9b are also considered to be involved in the inflammasome complex, and NLRP6 is similar in structure and assembly to NLRP3. Overall, the assembly of NLRP3, NLRP6, AIM2 and PYRIN is strictly dependent on the adaptor protein ASC while NLRP1, NLRC4 and CARD8 can induce inflammasome assembly and the subsequent pyroptosis in an ASC-independent way because of the existence of CARD. However, participation of ASC is essential for NLRC4 to mediate the processing and release of cytokines.
In the non-canonical inflammasome pathway, caspase-11/4/5(caspase-11 is the mouse homologue of human caspase-4/5)can be activated by the way that the N-terminal CARD binds in direct to intracellular lipopolysaccharide(LPS) (53), and activated caspase-11/4/5 can also cleave gasdermin D(GSDMD) to N-GSDMD, oligomerizing it and transferring it to the cell membrane, ultimately forming membrane perforation (54). LPS is a major component of the outer membrane of Gram-negative bacteria and consists of three parts including the most conserved lipid moiety, a core oligosaccharide chain and a variable polysaccharide chain known as O-antigen (55). Although caspase-11/4/5 contains a CARD like caspase-1, their binding to LPS requires some specific charged residues. The predicted isoelectric point of CARD of caspase-11/4/5 is alkaline(>8) while that of caspase-1’s is about 6, which explains the binding between the CARD of caspase-11/4/5 and the acidic phosphate of the lipid A skeleton in LPS (53, 56). However, pro-IL-1β and pro-IL-18 can not be cut by caspase-11/4/5. In some cells, such as monocyte, GSDMD cleaved by caspase-11/4/5 generates efflux of K+, inducing assembly of NLRP3 inflammasome and then mediating maturement and secretion of IL-1β and IL-18 through NLRP3/caspase-1 pathway (27, 57, 58).
In addition to the above pathways, some studies have shown that with treatment of chemotherapeutic drugs the gasdermin-E(GSDME) when highly expressed can be characteristically cleaved and activated by caspase-3 to form N-GSDME termini, leading to pyroptosis of tumor cells (59, 60). Furthermore, PD-L1 transfers apoptosis mediated by TNF into pyroptosis in breast cancer cells and the main mechanism is that the p-STAT3 promotes nuclear translocation of PD-L1 as well as transcription of gasdermin-C(GSDMC) and then under the stimulation of TNF-α, caspase-8 specifically cleaves GSDMC to generate N-GSDMC, forming holes in cell membrane to induce pyroptosis (61).
Granzyme, a serine protease, is released from cytosolic granules of cytotoxic lymphocytes (CTLs) and natural killer cells (NK). Granzyme A from CTLs can cleave and activate gasdermin-B(GSDMB) and then release its pore-forming activity, triggering pyroptosis in target cells (62). Besides, CAR-T cell is able to rapidly activate caspase-3 in target cells by releasing granzyme B(GZMB), subsequently triggering caspase-3/GSDME-mediated pyroptosis (63).
Cell pyroptosis can trigger both immune protection, pathological inflammation and tissue damage (64). Overall, pyroptosis depends on the members of the gasdermin protein family to facilitate the formation of membrane pores, resulting in the release of pro-inflammatory mediators and cellular contents, which ultimately contributes to cell disruption. Under normal circumstances, these mediators play a vital role in activating and regulating the immune response, helping to control or eliminate invading pathogens. In ALI/ARDS, excessive release of cytokines and infiltration of immune cells can trigger a cytokine storm, leading to severe damage to lung tissue (5). In the case of ARDS, IL-1β has emerged as a potent pro-inflammatory factor found in the lungs of affected patients (65). Additionally, the cytokines released in pyroptosis promote the polarization of macrophages into M1 phenotype, aggravating the inflammatory response in the lungs (66).
Macrophages are important components of the innate immune system and the activation of macrophage has been proven to be essential for immune defense, inflammatory response, tissue remodeling and homeostasis et al (67). Macrophages were first identified and named by immunologist Ellie Metchnikoff according to the characteristics of swallowing and killing bacteria in 1882 (13). At the very beginning, it was thought that macrophages originate from bone marrow-derived monocytes that are recruited to the tissue and differentiate into macrophages within tissue (68). Later research shows that the contribution of blood monocytes to the macrophage population homeostasis appears to be limited to a few specific tissues while a great number of tissue macrophages such as brain microglia, liver Kuffer cells, heart macrophages and alveolar macrophages originate from yolk sac or original macrophages existing in fetal livers and are maintained independently of blood monocytes (69, 70). Macrophages have the functions of phagocytosis, antigen presentation and immune defense and regulation (71, 72). In different environments and under various stimulations, macrophages will develop processes such as autophagy (73), polarization (67) and pyroptosis (74) to maintain tissue homeostasis. Based on their function and activation, macrophages are divided into two subtypes: classically activated M1 macrophages which produce mainly proinflammatory factors as part of the immune defense response and alternative activated M2 macrophages which mainly secrete anti-inflammatory factors to promote tissue repair (67, 75).
There are two subtypes of pulmonary macrophages including alveolar macrophages(AM) and pulmonary interstitial macrophages(IM) (76), and each type of macrophage contains tissue-resident and recruited macrophages, which are critical participants in innate immune and maintaining pulmonary homeostasis (Figure 3) (77). Acute inflammation and severe infection typically lead to loss of tissue-resident macrophages and recruitment of monocytes-derived macrophages, which become part of the pulmonary macrophage repertoire (77). The current study revealed that tissue-resident interstitial macrophages and monocyte-derived interstitial macrophages exhibit distinct profiles in omics analysis, but no significant differences were observed in their functional characteristics and properties (78).
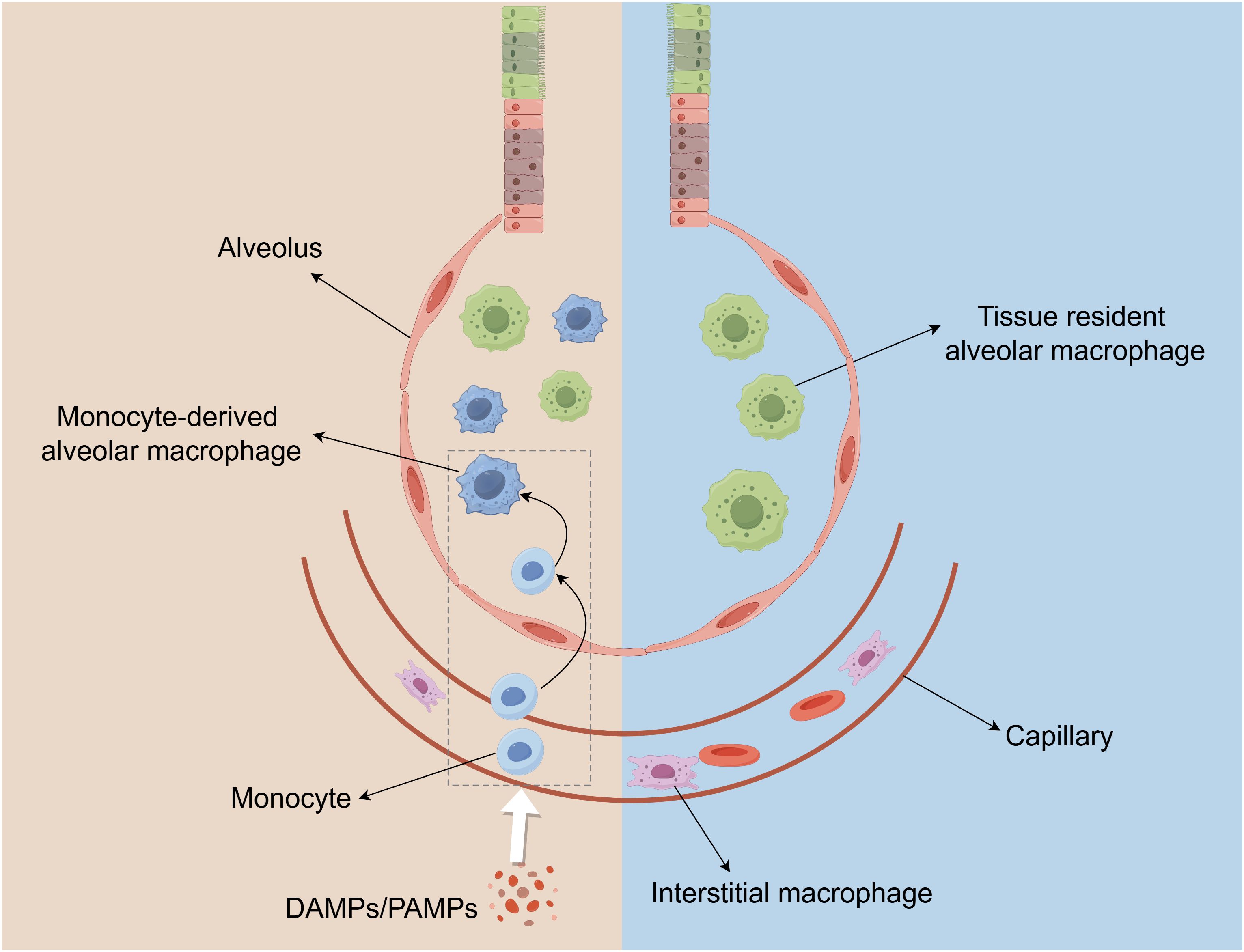
Figure 3. The distribution of macrophage populations in the lung. Macrophages in the lungs are composed of Alveolar macrophages (AMs) and interstitial macrophages (IMs). AMs are located in the alveoli, and IMs reside in the parenchyma between the microvascular endothelium and alveolar epithelium. When there is pulmonary inflammation and injury, circulating monocytes in capillaries are subsequently recruited to the lungs and converted into AM-like cells.
AM is an important cell line for catabolism of surface active material produced by alveolar type II epithelial cells and essential effector cells resisting external stimuli in lung, playing a critical role in the pathogenesis of pulmonary inflammation (79, 80). Generally, AM is able to continuously capture, swallow, conceal and neutralize vast inhaled pathogens and particles, which will not induce excessive inflammation triggered by influx of neutrophils (81). But when such a swallowing function is surpassed, AM will induce inflammation by producing chemokines and cytokines (such as type I IFNs, TNF-α, and IL-1β) that can recruit and activate neutrophils, monocytes and DCs (81). Tissue-resident alveolar macrophages(TR-AMs) and monocyte-derived alveolar macrophages(Mo-AMs) exhibit distinct characteristics during both homeostasis and disease states. TR-AMs play a crucial role in the clearance of dead alveolar cells and excess alveolar surfactants (82). In the absence of inflammation or tissue damage, TR-AMs maintain their cellular population through local proliferation, thereby minimizing the contribution of Mo-AMs (83). Upon the onset of inflammation, TR-AMs are rapidly activated, releasing a variety of cytokines and chemokines that modulate the inflammatory response and influence tissue integrity (84). Concurrently, the inflammatory process triggers the recruitment of monocytes to the lungs, where they differentiate into macrophages within the alveolar space, potentially exacerbating tissue damage through the release of pro-inflammatory mediators (85). Additionally, the time and cause of pyroptosis may vary among different macrophage populations. AMs emerged as the earliest cell type to initiate pyroptosis and act as pivotal regulators of cell communication (86). AMs usually die early in ALI/ARDS, and some pathogens can directly induce pyroptosis. In vitro experiments, the level of macrophage pyroptosis in RAW264.7 cells increased significantly within 24 hours after stimulation with LPS (87). The NETs and some cytokines can induce pyroptosis of monocyte-derived macrophages in the mid-stage (66, 88). In general, apoptosis is an important process in tissue homeostasis, which eliminates excessive cells in multicellular organisms, while pyroptosis usually leads to severe inflammatory response and tissue damage, aggravating disease progression. Although macrophage pyroptosis represents a distinct form of cell death compared to macrophage apoptosis, these two processes are not entirely independent. It has been identified that GSDME and GSDMD can induce the transition from apoptosis to pyroptosis. Caspase-3 and caspase-1, as the main performers of apoptosis and pyroptosis respectively, were also found to have crosstalk (89). However, there is uncertainty existing about whether apoptosis induced by pyroptosis protects cells from the pyroptosis-induced inflammatory response or further accelerates the inflammatory response of macrophages.
Pyroptosis is a double-edged sword. For one thing, it enhances adaptive immune response by releasing pro-inflammatory medium to gather the nearby immune cells around the pathogen-infected place and then destroy the replication niches (90). For another thing, excessive pyroptosis will aggravate multiple organ damage, circulatory failure and even death (91). Most of the time, macrophage pyroptosis significantly aggravates the progression of ALI/ARDS primarily through exacerbating pulmonary inflammation and tissue damage. Studies revealed that the mean caspase-1 levels, which are associated with pyroptosis, were significantly elevated in patients with ARDS compared to healthy individuals. It is noteworthy that the mean caspase-1 levels in nonsurviving patients were also significantly higher than those in survivors. Concurrently, the levels of inflammatory cytokines, including IL-1β, IL-18, and TNF-α, were significantly elevated and the extent of pulmonary injury was more severe in patients with ARDS compared to healthy controls (92, 93). In sepsis-induced acute lung injury, the incidence of macrophage pyroptosis were increased, followed by overexpression of pro-inflammatory mediators such as iNOS, IL-1β and TNF-α, and observable pulmonary tissue lesions (94).
Macrophages are capable of recognizing PAMPs and DAMPs, thereby initiating a cascade of responses during pyroptosis. Additionally, the impact of macrophage pyroptosis on ALI/ARDS is also modulated by protein post-translational modifications (95), mitochondrial function (96), exosome activity (97), neutrophil extracellular traps (98) and other various signaling pathways (99–101). The above factors can exert a positive or negative influences on the regulation of macrophage pyroptosis. Generally, modulating these factors to inhibit macrophage pyroptosis holds significant potential for mitigating ALI/ARDS.
Mitochondria are not only the major sites of cellular energy production through oxidative phosphorylation but also the participants in various cellular processes (96, 102). Studies have confirmed that mitochondria are involved in regulation of macrophage pyroptosis in ALI/ARDS.
Mitophagy plays a crucial role in mitochondrial quality control and cell survival by selectively eliminating excess or damaged mitochondria through the autophagic process (103). The present study indicates that mitophagy contributes to the mitigation of macrophage pyroptosis in ALI/ARDS. Defects in mitophagy result in the accumulation of mitochondrial reactive oxygen species (mtROS), leading to the overactivation of the NLRP3 inflammasome, and subsequently triggering Caspase-1-dependent pyroptosis (104). Dong et al. verified that the miR-138-5p promoter demethylation attenuates the pyroptosis of AMs in sepsis-associated ALI while increased mitophagy reduces cytoplasmic mtDNA levels, suppressing miR-138-5p promoter methylation, which suggested that enhanced mitophagy can reduce AM pyroptosis and alleviate sepsis-associated ALI (105).
Conversely, mitochondrial damage led to an increase of macrophage pyroptosis. Mitochondrial damage-associated molecular patterns (MTDs) are a type of damage-associated molecular patterns (DAMPs) that are released from mitochondrial rupture (106). MTDs have been reported to induce NLRP3 inflammasome activation, resulting in severe inflammatory response in alveolar macrophages (107). In sepsis-induced ALI, Z-DNA-binding protein 1 (ZBP1) deficiency in macrophages mitigates mitochondrial damage, consequently reducing macrophage pyroptosis mediated by NLRP3 inflammasome activation (108).
Additionally, the study conducted by Han et al. identified that the 18-kDa translocator protein (TSPO), a mitochondrial outer membrane protein, is a crucial mediator regulating NLRP3 inflammasome activation in macrophages during ALI/ARDS (109). They found that the expression of TSPO was rapidly upregulated in response to inflammatory stimulation and its deficiency resulted in enhanced activation of the NLRP3 inflammasome pathway in LPS-injured lung tissue (109).
Exosome is the nanoscale membrane-bound extracellular vesicle that plays crucial roles in intercellular communications by carrying bioactive molecules, such as proteins, RNAs, microRNAs (miRNAs) and DNAs, from one cell to the others (97). Crosstalk between exosome and inflammasome activation has been verified in many studies. As the upstream of inflammasome, exosome can either promote or suppress activation of inflammasome, subsequently affecting macrophage pyroptosis and ALI/ARDS. This discrepancy of exosome effects is likely affected by the type of the cells producing exosomes and interventions or conditions that induce cells to release the exosomes (97, 110).
Polymorphonuclear neutrophils (PMN) play an important role in ALI/ARDS, and exosomes derived from it are a new subcellular entity which is basic links between inflammation and tissue damage driven by PMN (111). The study shows that TNF-α-stimulated exosomes(TNF-Exo) derived from PMN is able to transfer the miR-30d-5p of miRNA family into macrophages and then activate the NF-κB signaling to up-regulate the expression of NLRP3 inflammation, which triggers pyroptosis in macrophages and therefore promotes sepsis-associated ALI (94). Another kind of exosome called Tenascin-C(TNC) induces macrophage pyroptosis by DNA damage response. The specific mechanism is that the DNA damage response could be induced by excessive TNC-produced ROS and activation of p38/ERK/NF-κB signaling to trigger macrophage pyroptosis to make ALI/ARDS severe (112). However, exosomes derived from mesenchymal stem cells(MSCs-Exo) can inhibit AM pyroptosis by down-regulation activated caspase-1, thereby alleviating LPS-induced ALI (113). Bone marrow mesenchymal stem cells (BMSCS) -derived exosomes can deliver miR-125b-5p and reduce macrophage pyroptosis by regulating STAT3 expression, subsequently improving sepsis-induced acute lung injury (114).
In conclusion, although exosomes from different cells have different impacts on macrophage pyroptosis, regulating exosomes can inhibit macrophage pyroptosis and reduce ALI/ARDS.
Neutrophils are short-lived granulocytes, serving as the primary line of defense against pathogens (115). Activated neutrophils release neutrophil extracellular traps (NETs) in response to various stimuli, which was identified as part of innate immune response, and this response can either be beneficial or pathological (98, 116).
In ALI/ARDS, NETs aggravate lung injury mainly by promoting macrophage pyroptosis (90, 93). The study proved that neutrophils undergo NETosis to produce a large amount of NETs. These NETs are engulfed by alveolar macrophages, leading to AIM2 inflammasome activation and caspase-1-dependent pyroptosis in LPS-priming alveolar macrophages. This, in turn, results in the release of a large amount of cytokines and more neutrophil infiltration, leading to ARDS through inflammatory storm development due to a vicious cycle (93). A study conducted by Liu et al. found that pretreatment of alpha-linolenic acid(ALA) might alleviate NETs-induced alveolar macrophage pyroptosis by mediating Pyrin inflammasome activation, which alleviated ALI/ARDS (117). They utilized Mefv (Pyrin)-/- mice to demonstrate that NETs induce macrophage pyroptosis depending on Pyrin inflammasome. In addition, The research found that IgG IC-induced formation of NETs stimulates pulmonary macrophage pyroptosis in ALI/ARDS, which could be inhibited by verbenalin (118).
Post-translational modifications (PTMs) refer to a series of covalent modifications of proteins following the translation of RNA, constituting a critical phase in protein biosynthesis (119). Throughout the life cycle, PTMs contribute to increasing complexity of the proteome, modulating subcellular localization of associated proteins, facilitating or inhibiting protein-protein interactions, and activating or inactivating target proteins (120). Recent reports have identified multiple PTMs in pyroptosis process. For example, (de)ubiquitylation, (de)phosphorylation, (de)SUMOylation, (de)acetylation, alkylation and citrullination are involved in the assembly and activation of inflammasomes (NLRP3, NLRC4, AIM2) (121–123), while ubiquitination, phosphorylation and ADP- ribosylation participate in caspase activation, which is essential for the cleavage of GSDMD (124). Additionally, several PTMs can directly modulate the cleavage of GSDMD, such as ubiquitylation, phosphorylation, palmitoylation and succinylation, while ubiquitylation exerts a negative regulatory effect on the release of mature IL-1β and IL-18 through the pores formed by GSDMD-N (125, 126).
The majority of PTMs are reversible. For instance, deubiquitinating enzymes (DUBs) can reverse ubiquitination by hydrolyzing the peptide or isopeptide bonds that link ubiquitin molecules to each other or to substrate proteins. Ubiquitination typically suppresses inflammasome activity, thereby inhibiting pyroptosis, while deubiquitination has the opposite effect (127, 128). SUMO-conjugating enzyme (UBC9) interacts with NLRP3 and promotes SUMO1 to catalyse SUMOylation at Lys204 in NLRP3, subsequently promoting NLRP3 activation and macrophage pyroptosis. Conversely, SUMO-specific protease 3 (SENP3) catalyses deSUMOylation of NLRP3, reducing ASC recruitment, ultimately suppressing NLRP3 inflammasome activation, as well as IL-1β cleavage and secretion (129). However, there are also some PTMs that are irreversible, such as citrullination (130). The current studies suggest that protein citrullination can promote macrophage pyroptosis through regulating activation of inflammasomes and caspase proteins, thereby aggravating ALI/ARDS (131, 132).
It has been demonstrated that Heat Shock Factor 1 (HSF1) mitigates sepsis-induced lung injury through the promotion of NLRP3 ubiquitination in macrophages (122). In human THP-1 cells and mouse bone marrow-derived macrophages lacking the DUB USP50, reduced activation of pyroptosis was observed, leading to decreased levels of IL-1β and IL-18. Further investigations revealed that USP50 interacts with ASC proteins and deubiquitinates K63-linked ASC polyubiquitination (133). Immunostaining of human macrophages indicates that the bacterial effector protein NLeA inhibits inflammasome activity through its binding to ubiquitinated NLRP3, thereby disrupting the deubiquitination process. This interaction prevents inflammasome assembly, caspase-1 activation, and subsequent IL-1β secretion (134). The peptidylarginine deiminase 2(PAD2) is a calcium-dependent enzyme that promotes the conversion of arginine to citrulline. It has been demonstrated that PAD2 knockout can reduce the mortality of Pseudomonas aeruginosa induced pneumonia mice by reducing the caspase-1-dependent inflammasome activation in macrophages (135). In sepsis-associated acute lung injury, PAD2 deficiency decreased caspase-11-dependent macrophage pyroptosis, increasing survival and organ functions following the onset of sepsis (132). Furthermore, the study found that inhibition of PAD2 leads to a reduction of ASC citrullination, suggesting that PAD2 may influence inflammasome assembly through the mediation of ASC citrullination, ultimately impacting macrophage pyroptosis (136). NLRC4 is a cytosolic member of the NOD-like receptor family that is expressed in innate immune cells. The phosphorylation of residue Ser 533 in NLRC4 had been identified to activate NLRC4 inflammasome activity and induce conformational changes that are essential for host immune responses (137).
The effect of PTMs on caspase protein and gasdermin protein is not as extensive as that on inflammasome. There is a study demonstrating that the E3 ubiquitin ligase, synoviolin (SYVN1), mediates the K27-linked polyubiquitination of GSDMD at K203 and K204, promoting GSDMD-induced pyroptosis (138). In addition, upon activation by inflammasome, palmitoyl transferases ZDHHC5/9 can induce S-palmitoylation at Cys191 (human)/Cys192 (mouse) on GSDMD, generating palmitoylated GSDMD-N which effectively triggers pyroptosis (139).
Nuclear factor-erythroid 2 related factor 2(Nrf2)is a ubiquitous master transcription factor that upregulates antioxidant response elements (AREs)-mediated expression of antioxidant enzymes and cytoprotective proteins (140). Activation of Nrf2 has been shown to be protective against lung injury (140), and increasing evidence also demonstrates the crosstalk between the Nrf2 and NLRP3 inflammasome axis at different levels (141). Liu et al. found that Nrf2 knockdown can substantially increase the mRNA level of NLRP3 (142). Further studies showed that upregulating of AMPK phosphorylation can promote expression of Nrf2 followed by inhibition of NLRP3 transcription, thereby suppressing pyroptosis in alveolar macrophages and ultimately alleviating ALI/ARDS (142). In the models of LPS-induced sepsis, melatonin inhibits NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis to reduce ALI/ARDS in vivo and in vitro (100). In addition, Nrf2 is able to inhibit activation of NLRP3 inflammasome by reducing intracellular ROS levels and avoiding inflammation (143). A study showed that chicoric acid alleviated NLRP3-mediated macrophage pyroptosis in the ALI model. This effect was achieved through ROS-induced mitochondrial damage by activating the Akt/Nrf2 pathway via PDPK1 ubiquitination (144). In conclusion, it is of great significance in reducing ALI/ARDS to inhibit activation of NLRP3 by up-regulating Nrf2 level.
Stimulator of interferon genes(STING)is a signaling molecule associated with the endoplasmic reticulum (ER) and is essential for controlling the transcription of numerous host defence genes, including type I interferons (IFNs) and pro-inflammatory cytokines, following the recognition of aberrant DNA species or cyclic dinucleotides (CDNs) in the cytosol of the cell (145, 146). STING signaling has now been shown to be essential for protecting the cell against a variety of pathogens (147). At present, growing studies have shown that STING involved in regulation of macrophage pyroptosis mainly by affecting NLRP3 inflammasome activation in ALI/ARDS. Peng et al. found that up-gratulation of STING signaling can promote NLRP3-mediated pyroptosis in macrophages and knockout of cGAS/STING could ameliorate NLRP3 activation and macrophage pyroptosis, ultimately improving SAP-ALI in mouse model (148). In LPS-induced ALI, LPS could activate STING in a cytosolic DNA-dependent manner and upregulate the expression of STING in a c-Myc-dependent manner, which cooperatively promote NLRP3-mediated macrophage pyroptosis following contributing to acute pulmonary damage (149). Furthermore, another study suggested that histone deacetylase 3 (HDAC3) activates cGAS/STING pathway through its histone deacetylation function, playing an essential role in mediating macrophage pyroptosis and ALI (101). The specific mechanism is that HDAC3 and H3K9Ac are recruited by LPS to the miR-4767 gene promoter, which repressed expression of miR-4767 to promote the expression of cGAS (101).
Nuclear factor kappa-B(NF-κB)is a Rel family transcription factor consisting of five members in mammalian cells, namely RelA (p65), RelB, Rel (c-Rel), NF-κB1 (p50/p105) and NF-κB2 (p52/p100) (150), which controls both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system (151). NF-κB is widely recognized as a key regulator of inflammation due to its pivotal role in governing diverse aspects of the inflammatory response, including evolution and resolution (152). NF-κB signaling is also involved in the progression of ALI/ARDS (153, 154), and NLRP3 inflammasome activation requires involvement of NF-κB (122). Triggered by PAMPs, TNF, and IL-1β, the mRNA expression of NLRP3 and pro-IL-1β is upregulated by activating NF-κB. In septic ALI, heat shock factor 1 (HSF1) inhibited the NF-κB signaling pathway by upregulating tumor necrosis factor receptor-associated factors 3(TRAF3)expression, thereby inhibiting the production of NLRP3 at the transcriptional level and ultimately inhibiting the activation of the NLRP3 inflammasome, which reduces macrophage pyroptosis and subsequently alleviates pulmonary damage (122). Sun et al. discovered that angiopoietin-like 4 (ANGPTL4) silencing could disrupt the activation of the NF-κB pathway to repress the pyroptosis and polarization of M1 macrophages, whereby suppressing the CLP-induced sepsis-related ALI (155). Other studies demonstrated that macrophage pyroptosis mediated by NLRP3/GSDMD signaling can be inhibited by suppressing the NF-κB activation (99, 156), suggesting that inhibiting pyroptosis of macrophages by targeting NF-κB pathway can reduce acute lung injury. In addition, S100A9 gene deficiency inhibits pyroptosis of macrophages through TLR4/MyD88/NF-κB pathway, alleviating LPS-induced acute lung injury (157), which suggests a potential therapeutic strategy for the treatment of ALI/ARDS.
Apart from the above regulatory modes, there are many other signaling pathways that can affect macrophage pyroptosis in ALI/ARDS. A body of evidence confirmed that p38 mitogen-activated protein kinase (MAPK) signaling pathway participates in the progression of ALI/ARDS (158, 159). Li et al. demonstrated that macrophage cell death could be skewed from proinflammatory pyroptosis towards non−inflammatory apoptosis through blockage of p38 MAPK signaling pathway, which may contribute to amelioration of acute lung injury and excessive inflammation in mice of ALI induced by LPS (160).
Tumor necrosis factor receptor-associated factor 3 (TRAF3) is the member of TRAF family, playing a crucial role in regulating both immune and inflammatory response. The study showed the TRAF3/ULK1/NLRP3 axis promoted the development of ALI in mice by inducing alveolar macrophage pyroptosis. The vitro cell experiments verified that TRAF3 can downregulate ULK1 through ubiquitination and activate the NLRP3 inflammasome to induce alveolar macrophage pyroptosis (161).
Basic helix-loop-helix family member e40 (Bhlhe40), belonging to the subfamily of transcription factors, is considered as an important regulator of inflammation and immunity (162). A study showed the expression of Bhlhe40 significantly increased in AMs treated with LPS. Meanwhile, Bhlhe40−/− mice exhibited decreased macrophages pyroptosis and inflammation by inhibiting signaling pathway mediated by caspase-1 and caspase-11, and they were resistant to LPS-induced ALI (163).
Natural products derived from plants play an important role in the treatment of ALI/ARDS. Many studies show that natural active ingredients from Chinese herbs can regulate pyroptosis in macrophages, alleviating ALI/ARDS.
Alpha-linolenic acid (ALA) is a plant-based omega-3 fatty acid. Studies show that ALA reduces ALI/ARDS by inhibiting the activation of macrophage pyroptosis driven by Pyrin inflammasome (117). Emodin, a natural active ingredient extracted from the Chinese herb Radix et Rhizoma Rhei, alleviates LPS-induced ALI by inhibiting the NLRP3 inflammasome-dependent pyroptosis signaling pathway of macrophages in vitro and in vivo (164). Arctiin (ARC) is a lignan glycoside isolated from Fructus arctii that exerts strong anti-inflammatory and antioxidant effects (165, 166). Studies demonstrated that a nanoparticle (NP)-based delivery system, ARC@DPBNP, could be applied to reduce LPS-induced acute lung injury through attenuating pyroptosis in alveolar macrophages (167). Sinensetin (SIN) is a polymethoxylated flavonoid, which is proven to improve LPS-induced ALI by inhibiting Txnip/NLRP3/Caspase-1/GSDMD signaling-mediated macrophage pyroptosis. Dehydroandrographolide(Deh), as one of main components of Andrographis paniculata (Burm.f.) Wall, can weaken ROS production in mitochondria to suppress NLRP3-mediated pyroptosis in macrophages, thereby alleviating ALI/ARDS (168). Shao et al. found that Britannin extracted from Inula japonica Thunb. is an innate inhibitor that effectively targeted NLRP3, suppressing activation of NLRP3 inflammasome in an NF-κB-independent manner and inhibiting assembly of the NLRP3 inflammasome by directly binding to the NACHT domain of NLRP3 (169), which suggests that Britannin may be an effective drug inhibiting macrophages to reduce ALI/ARDS.
Buformin (BF), a number of the biguanide family, was originally used clinically as a hypoglycemic agent in the treatment of type 2 diabetes (170). Studies conducted by Liu et al. demonstrated that BF can reduce sepsis-induced lung injury by inhibiting NLRP3-mediated macrophages pyroptosis in an AMPK-dependent manner in vivo and in vitro (142). Sacubitril/valsartan (SV) is an angiotensin receptor-neprilysin inhibitor used for treating heart failure in the clinical settings (171). The study showed that SV treatment effectively alleviated sepsis-induced lung injury in caecal ligation and puncture (CLP) mice (172). Further study found that the SV could inhibit GSDMD-mediated macrophage pyroptosis through the caspase-1-dependent signaling pathway, contributing to the resolution of the inflammatory response and lung injury in sepsis (172). As an N-methyl-D-aspartic acid receptor (NMDAR) antagonist, memantine suppresses macrophage pyroptosis through inhibiting NLRP3 inflammasome (173). In cardiopulmonary bypass (CPB)–induced ALI, hydromorphone (Hyd) alleviated NLRP3 inflammasome-mediated pyroptosis via upregulating the Nrf2/HO-1 pathway, which may be achieved by AMs (174).
Apart from medications, there are alternative interventions that can mitigate ALI/ARDS by suppressing macrophage pyroptosis. More and more studies have confirmed it is a promising strategy for ALI/ARDS to regulate exosomes. Liu et al. demonstrated that mesenchymal stem cells-derived exosomes(MSCs-Exo) can alleviate acute lung injury by inhibiting alveolar macrophage pyroptosis (113). The specific mechanism is MSCs-Exo inhibited J774A.1 cell pyroptosis by inhibiting the activation of caspase-1 (113). In addition, exosomes from bone marrow-derived mesenchymal stem cells, serving as carriers for delivering miR-125b-5p, can downregulate STAT3, thereby inhibiting macrophage pyroptosis and alleviating sepsis-associated ALI (114). The above studies suggest that the regulation of mesenchymal stem cell-derived exosomes can effectively inhibit macrophage pyroptosis and alleviate ALI/ARDS.
In LPS-induced ALI/ARDS, researchers found that NETs directly promoted alveolar macrophage pyroptosis through NET DNA-mediated activation of the AIM2 inflammasome, suggesting that NETs and the AIM2 sensor may be crucial therapeutic targets for the regulation of alveolar macrophage inflammasome-mediated immunopathology in ARDS (93). Melatonin (Mel, N-acetyl-5-methoxytryptamine) is a neurosecretory hormone, as well as a potential modulator of Nrf2 signaling based on its abilities to scavenge ROS and inflammatory cytokines (175). The study proved that melatonin significantly inhibits LPS-induced pyroptosis, attributed to its regulation of NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis (100). Irisin, a hormone−like myokine, can attenuate ALI by inhibiting the HSP90/NLRP3/caspase−1/GSDMD signaling pathway, reducing the pyroptosis of macrophages (176). The following section discusses the treatments and mechanisms related to macrophage pyroptosis in ALI/ARDS (Table 1).
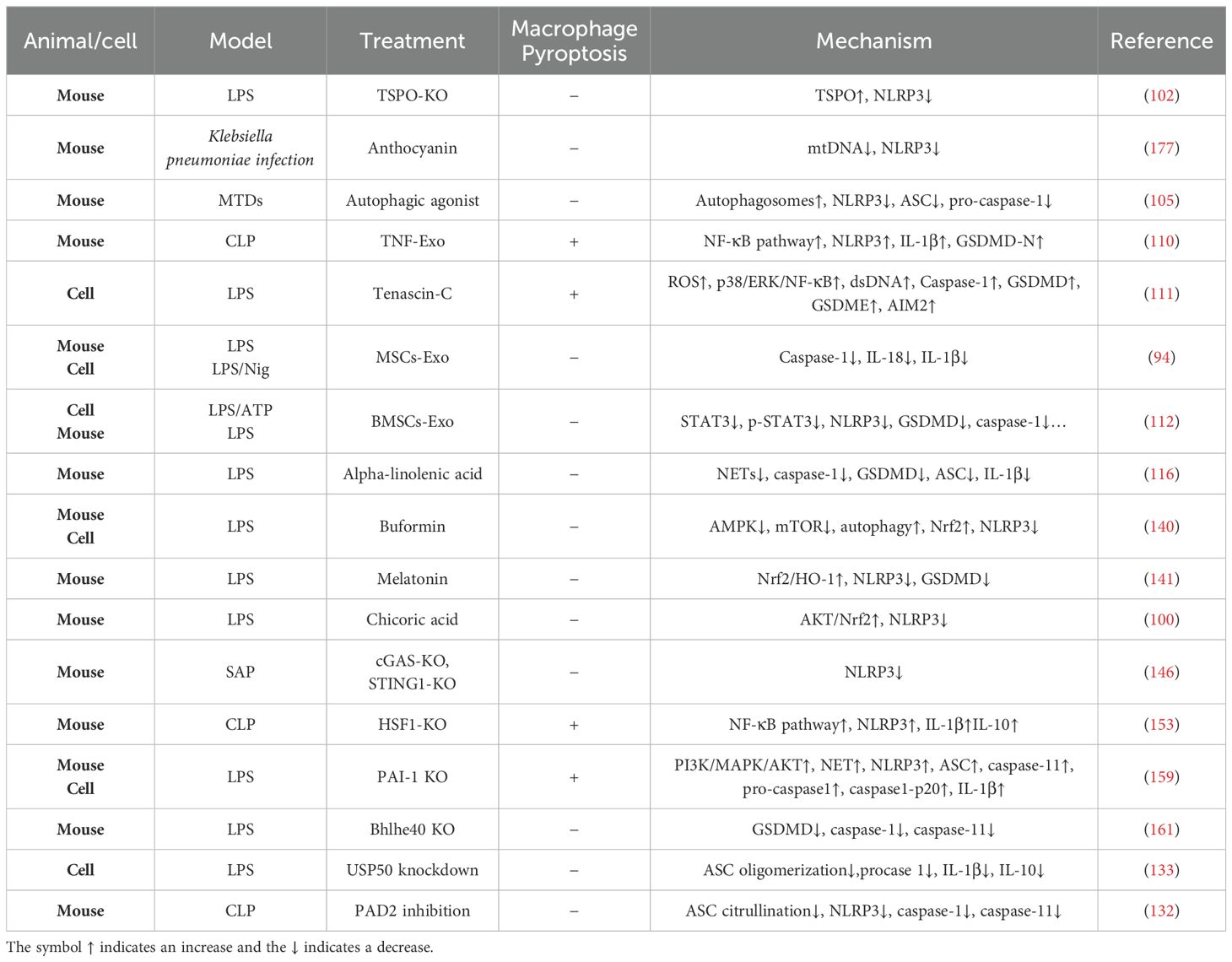
Table 1. The treatment and mechanisms of macrophage pyroptosis in ALI/ARDS.
Undoubtedly, the therapeutic intervention of targeted macrophage pyroptosis is still challenging to some extent. Although the mechanism of macrophage pyroptosis in ALI/ARDS have been increasingly investigated in recent studies, the specific mechanisms of macrophage pyroptosis remain unclear. Whether additional inflammasome complexes or gasdermin proteins revolves in cell pyroptosis remains an open question. At the same time, because of complexity of drugs’ action mechanism, their absorption and distribution in the body are diverse. Therefore, how to select an optimal delivery method for inhibitors to maximize their suppression of macrophage pyroptosis presents a significant challenge. The advent of nanotechnology has opened new avenues for the development of novel therapeutic strategies for the treatment of ARDS/ALI that can utilize targeting macrophage pyroptosis pathways (178). However, its clinical transformation still faces a series of challenges (179).
At the same time, it has not been thoroughly studied whether the potential side effects of inhibitors on the liver, kidney and other organs will aggravate the multiple organs dysfunction of ALI/ARDS. There are few successful examples of macrophage pyroptosis interventions successfully applied in clinical practice, and the application of therapies that inhibit excessive macrophage pyroptosis in clinical practice is still facing severe challenges, while how to clarify the changes of macrophage pyroptosis markers in ALI/ARDS patients is also a key point of carrying out clinical interventions targeting the modulation of pulmonary macrophage pyroptosis. At present, there are few clinical studies related to the treatment of ALI/ARDS patients by modulating macrophage pyroptosis, and more preclinical studies are still needed to confirm the potential beneficial role of regulating macrophage pyroptosis in ALI/ARDS. Some studies have confirmed the role of markers related to pyroptosis in early identification and prognosis assessment of ARDS patients. For example, a study demonstrated that the mean levels of caspase-1, IL-1β and IL-18 in aspirates from ARDS patients were significantly elevated compared to those in healthy individuals (93). Modulation of macrophage pyroptosis-related indicators may become a potential therapeutic approach for ARDS patients, but future clinical studies are still needed to demonstrate the beneficial effects of interventions to modulate macrophage pyroptosis in ARDS patients.
As an acute diffuse lung injury occurring in a short period of time, ALI/ARDS has significantly severe clinical prognosis, and therefore its clinical treatment is full of challenges. Macrophage pyroptosis, as a special way of cell death, has attracted much attention in study of development and treatment of ALI/ARDS. Up to now, many studies have shown that macrophage pyroptosis could be affected by regulating mitochondrial function (107, 180), adjusting the generation of exosomes (181) or neutrophil extracellular traps (93), and controlling its upstream signaling transduction (100, 149, 182), thus influencing development and outcome of ALI/ARDS. The regulation of macrophage pyroptosis may become a therapeutic target for ALI/ARDS, thus providing more new research directions for the treatment of ALI/ARDS.
However, there are still some problems unsolved. Firstly, the current understanding of the inflammasomes and their role in pyroptosis is incomplete, with only a few inflammasomes and their mechanisms of action in pyroptosis being well understood. Secondly, most of the current research on macrophage pyroptosis is limited to preclinical studies, and the clinical studies that have been conducted also have problems such as small sample sizes. In clinical practice, there are very few cases where the actual reduction of lung injury through intervention in macrophage pyroptosis has been achieved, making it difficult to prove the clinical utility. Therefore, more and more in-depth basic and clinical trials are needed to investigate the specific role of macrophage pyroptosis in ALI/ARDS, with the aim of selecting more effective targets for the treatment of ALI/ARDS.
YC: Conceptualization, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. LS: Conceptualization, Formal analysis, Methodology, Writing – review & editing. FZ: Investigation, Methodology, Writing – review & editing. MZ: Data curation, Investigation, Writing – review & editing. JL: Conceptualization, Writing – review & editing. SW: Investigation, Methodology, Writing – review & editing. QL: Writing – review & editing. JH: Writing – review & editing. SY: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No: 82274314).
We would like to express our gratitude to people who helped. We would like to express our gratitude to people who helped. Figures were created using Figdraw (www.figdraw.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. (2017) 377:562–72. doi: 10.1056/NEJMra1608077
2. Rubenfeld GD. Epidemiology of acute lung injury. Crit Care Med. (2003) 31:S276–284. doi: 10.1097/01.Ccm.0000057904.62683.2b
3. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama. (2016) 315:788–800. doi: 10.1001/jama.2016.0291
4. Wick KD, Ware LB, Matthay MA. Acute respiratory distress syndrome. Bmj. (2024) 387:e076612. doi: 10.1136/bmj-2023-076612
5. Wang J, Peng X, Yuan N, Wang B, Chen S, Wang B, et al. Interplay between pulmonary epithelial stem cells and innate immune cells contribute to the repair and regeneration of ALI/ARDS. Transl Res. (2024) 272:111–25. doi: 10.1016/j.trsl.2024.05.012
6. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
7. Oh C, Spears TJ, Aachoui Y. Inflammasome-mediated pyroptosis in defense against pathogenic bacteria. Immunol Rev. (2024) 329:e13408. doi: 10.1111/imr.13408
8. Nadella V, Kanneganti TD. Inflammasomes and their role in PANoptosomes. Curr Opin Immunol. (2024) 91:102489. doi: 10.1016/j.coi.2024.102489
9. Makuch M, Stepanechko M, Bzowska M. The dance of macrophage death: the interplay between the inevitable and the microenvironment. Front Immunol. (2024) 15:1330461. doi: 10.3389/fimmu.2024.1330461
10. Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
11. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. (1967) 2:319–23. doi: 10.1016/s0140-6736(67)90168-7
12. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. (1994) 149:818–24. doi: 10.1164/ajrccm.149.3.7509706
13. Liu C, Xiao K, Xie L. Progress in preclinical studies of macrophage autophagy in the regulation of ALI/ARDS. Front Immunol. (2022) 13:922702. doi: 10.3389/fimmu.2022.922702
14. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. Jama. (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
15. Raghavendran K, Napolitano LM. Definition of ALI/ARDS. Crit Care Clin. (2011) 27:429–37. doi: 10.1016/j.ccc.2011.05.006
16. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. (2021) 398:622–37. doi: 10.1016/s0140-6736(21)00439-6
17. Swenson KE, Swenson ER. Pathophysiology of acute respiratory distress syndrome and COVID-19 lung injury. Crit Care Clin. (2021) 37:749–76. doi: 10.1016/j.ccc.2021.05.003
18. Zheng J, Li Y, Kong X, Guo J. Exploring immune-related pathogenesis in lung injury: Providing new insights Into ALI/ARDS. BioMed Pharmacother. (2024) 175:116773. doi: 10.1016/j.biopha.2024.116773
19. Tan S, Chen S. The mechanism and effect of autophagy, apoptosis, and pyroptosis on the progression of silicosis. Int J Mol Sci. (2021) 22:8110. doi: 10.3390/ijms22158110
20. Hsu CG, Chávez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. (2022) 29:1790–803. doi: 10.1038/s41418-022-00966-5
21. Hou L, Yang Z, Wang Z, Zhang X, Zhao Y, Yang H, et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab Invest. (2018) 98:1052–64. doi: 10.1038/s41374-018-0073-0
22. Pu Z, Wang W, Xie H, Wang W. Apolipoprotein C3 (ApoC3) facilitates NLRP3 mediated pyroptosis of macrophages through mitochondrial damage by accelerating of the interaction between SCIMP and SYK pathway in acute lung injury. Int Immunopharmacol. (2024) 128:111537. doi: 10.1016/j.intimp.2024.111537
23. Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. Jama. (2018) 319:698–710. doi: 10.1001/jama.2017.21907
24. Sasannejad C, Ely EW, Lahiri S. Long-term cognitive impairment after acute respiratory distress syndrome: a review of clinical impact and pathophysiological mechanisms. Crit Care. (2019) 23:352. doi: 10.1186/s13054-019-2626-z
25. Curley GF, Contreras M, Higgins B, O’Kane C, McAuley DF, O’Toole D, et al. Evolution of the inflammatory and fibroproliferative responses during resolution and repair after ventilator-induced lung injury in the rat. Anesthesiology. (2011) 115:1022–32. doi: 10.1097/ALN.0b013e31823422c9
26. Cabrera-Benitez NE, Laffey JG, Parotto M, Spieth PM, Villar J, Zhang H, et al. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: a significant contributor to poor outcome. Anesthesiology. (2014) 121:189–98. doi: 10.1097/aln.0000000000000264
27. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
28. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. (1992) 358:167–9. doi: 10.1038/358167a0
29. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. (2015) 265:130–42. doi: 10.1111/imr.12287
30. D’Souza CA, Heitman J. Dismantling the cryptococcus coat. Trends Microbiol. (2001) 9:112–3. doi: 10.1016/s0966-842x(00)01945-4
31. Zhu C, Xu S, Jiang R, Yu Y, Bian J, Zou Z. The gasdermin family: emerging therapeutic targets in diseases. Signal Transduct Target Ther. (2024) 9:87. doi: 10.1038/s41392-024-01801-8
32. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
33. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. (2019) 10:1689. doi: 10.1038/s41467-019-09397-2
34. Zhao H, Yang Y, Si X, Liu H, Wang H. The role of pyroptosis and autophagy in ischemia reperfusion injury. Biomolecules. (2022) 12:1010. doi: 10.3390/biom12071010
35. Zhang K-J, Wu Q, Jiang S-M, Ding L, Liu C-X, Xu M, et al. Pyroptosis: A new frontier in kidney diseases. Oxid Med Cell Longev. (2021) 2021:6686617. doi: 10.1155/2021/6686617
36. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity. (2018) 49:413–426.e415. doi: 10.1016/j.immuni.2018.07.006
37. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
38. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med. (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
39. Fu J, Schroder K, Wu H. Mechanistic insights from inflammasome structures. Nat Rev Immunol. (2024) 24:518–35. doi: 10.1038/s41577-024-00995-w
40. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
41. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
42. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
43. Franklin BS, Latz E, Schmidt FI. The intra- and extracellular functions of ASC specks. Immunol Rev. (2018) 281:74–87. doi: 10.1111/imr.12611
44. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
45. Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C, et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A. (2018) 115:10845–52. doi: 10.1073/pnas.1810524115
46. Calabrese L, Fiocco Z, Mellett M, Aoki R, Rubegni P, French LE, et al. Role of the NLRP1 inflammasome in skin cancer and inflammatory skin diseases. Br J Dermatol. (2024) 190:305–15. doi: 10.1093/bjd/ljad421
47. Yu L, Hong W, Lu S, Li Y, Guan Y, Weng X, et al. The NLRP3 inflammasome in non-alcoholic fatty liver disease and steatohepatitis: therapeutic targets and treatment. Front Pharmacol. (2022) 13:780496. doi: 10.3389/fphar.2022.780496
48. Mulay SR. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. (2019) 96:58–66. doi: 10.1016/j.kint.2019.01.014
49. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. (2004) 430:213–8. doi: 10.1038/nature02664
50. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. (2011) 477:592–5. doi: 10.1038/nature10394
51. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U.S.A. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
52. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U.S.A. (2010) 107:9771–6. doi: 10.1073/pnas.1003738107
53. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
54. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
55. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. (2002) 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414
56. Rathinam VAK, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol. (2019) 20:527–33. doi: 10.1038/s41590-019-0368-3
57. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. (2015) 45:2918–26. doi: 10.1002/eji.201545655
58. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
59. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. (2017) 547:99–103. doi: 10.1038/nature22393
60. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. (2017) 8:14128. doi: 10.1038/ncomms14128
61. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:128. doi: 10.1038/s41392-021-00507-5
62. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
63. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. (2020) 5:eaax7969. doi: 10.1126/sciimmunol.aax7969
64. Liu Y, Pan R, Ouyang Y, Gu W, Xiao T, Yang H, et al. Pyroptosis in health and disease: mechanisms, regulation and clinical perspective. Signal Transduct Target Ther. (2024) 9:245. doi: 10.1038/s41392-024-01958-2
65. Zhu W, Zhang Y, Wang Y. Immunotherapy strategies and prospects for acute lung injury: Focus on immune cells and cytokines. Front Pharmacol. (2022) 13:1103309. doi: 10.3389/fphar.2022.1103309
66. Ahmed NT, Kummarapurugu AB, Zheng S, Bulut G, Kang L, Batheja A, et al. Neutrophil elastase targets select proteins on human blood-monocyte-derived macrophage cell surfaces. Int J Mol Sci. (2024) 25:13038. doi: 10.3390/ijms252313038
67. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/jci59643
68. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. (1972) 46:845–52.
69. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
70. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
71. Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. (2019) 26:715–27. doi: 10.1038/s41418-019-0297-6
72. Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol. (2021) 18:579–87. doi: 10.1038/s41423-020-00541-3
73. Qiu P, Liu Y, Zhang J. Review: the role and mechanisms of macrophage autophagy in sepsis. Inflammation. (2019) 42:6–19. doi: 10.1007/s10753-018-0890-8
74. Xie D, Ouyang S. The role and mechanisms of macrophage polarization and hepatocyte pyroptosis in acute liver failure. Front Immunol. (2023) 14:1279264. doi: 10.3389/fimmu.2023.1279264
75. Chen X, Tang J, Shuai W, Meng J, Feng J, Han Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflammation Res. (2020) 69:883–95. doi: 10.1007/s00011-020-01378-2
76. Wang Z, Wang Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front Immunol. (2023) 14:1209438. doi: 10.3389/fimmu.2023.1209438
77. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. (2022) 55:1564–80. doi: 10.1016/j.immuni.2022.08.010
78. Shi T, Denney L, An H, Ho LP, Zheng Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J Leukoc Biol. (2021) 110:107–14. doi: 10.1002/jlb.3ru0720-418r
79. Martin WJ 2nd, Wu M, Pasula R. A novel approach to restore lung immunity during systemic immunosuppression. Trans Am Clin Climatol Assoc. (2005) 116:221–226; discussion 226-227.
80. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine TGF-β Promotes the development and homeostasis of alveolar macrophages. Immunity. (2017) 47:903–912.e904. doi: 10.1016/j.immuni.2017.10.007
81. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol. (2022) 15:223–34. doi: 10.1038/s41385-021-00480-w
82. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. (2017) 47:913–927.e916. doi: 10.1016/j.immuni.2017.10.006
83. Hetzel M, Ackermann M, Lachmann N. Beyond “Big eaters”: the versatile role of alveolar macrophages in health and disease. Int J Mol Sci. (2021) 22:3308. doi: 10.3390/ijms22073308
84. Pervizaj-Oruqaj L, Ferrero MR, Matt U, Herold S. The guardians of pulmonary harmony: alveolar macrophages orchestrating the symphony of lung inflammation and tissue homeostasis. Eur Respir Rev. (2024) 33:230263. doi: 10.1183/16000617.0263-2023
85. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol. (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
86. Zhang M, Lan H, Jiang M, Yang M, Chen H, Peng S, et al. NLRP3 inflammasome mediates pyroptosis of alveolar macrophages to induce radiation lung injury. J Hazard Mater. (2024) 484:136740. doi: 10.1016/j.jhazmat.2024.136740
87. Wu D, Pan P, Su X, Zhang L, Qin Q, Tan H, et al. Interferon regulatory factor-1 mediates alveolar macrophage pyroptosis during LPS-induced acute lung injury in mice. Shock. (2016) 46:329–38. doi: 10.1097/shk.0000000000000595
88. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. (2018) 9:597. doi: 10.1038/s41419-018-0538-5
89. Cong L, Liu X, Bai Y, Qin Q, Zhao L, Shi Y, et al. Melatonin alleviates pyroptosis by regulating the SIRT3/FOXO3α/ROS axis and interacting with apoptosis in Atherosclerosis progression. Biol Res. (2023) 56:62. doi: 10.1186/s40659-023-00479-6
90. Wu J, Lan Y, Wu J, Zhu K. Sepsis-induced acute lung injury is alleviated by small molecules from dietary plants via pyroptosis modulation. J Agric Food Chem. (2023) 71:12153–66. doi: 10.1021/acs.jafc.2c08926
91. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol. (2021) 6:eabj3859. doi: 10.1126/sciimmunol.abj3859
92. Wu DD, Pan PH, Liu B, Su XL, Zhang LM, Tan HY, et al. Inhibition of alveolar macrophage pyroptosis reduces lipopolysaccharide-induced acute lung injury in mice. Chin Med J (Engl). (2015) 128:2638–45. doi: 10.4103/0366-6999.166039
93. Li H, Li Y, Song C, Hu Y, Dai M, Liu B, et al. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis via AIM2 inflammasome activation in LPS-induced ALI/ARDS. J Inflammation Res. (2021) 14:4839–58. doi: 10.2147/jir.S321513
94. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care. (2021) 25:356. doi: 10.1186/s13054-021-03775-3
95. Zhou YR, Dang JJ, Yang QC, Sun ZJ. The regulation of pyroptosis by post-translational modifications: molecular mechanisms and therapeutic targets. EBioMedicine. (2024) 109:105420. doi: 10.1016/j.ebiom.2024.105420
96. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. (2011) 11:389–402. doi: 10.1038/nri2975
97. Noonin C, Thongboonkerd V. Exosome-inflammasome crosstalk and their roles in inflammatory responses. Theranostics. (2021) 11:4436–51. doi: 10.7150/thno.54004
98. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. (2012) 198:773–83. doi: 10.1083/jcb.201203170
99. Feng M, Wei S, Zhang S, Yang Y. Anti-inflammation and anti-pyroptosis activities of mangiferin via suppressing NF-κB/NLRP3/GSDMD signaling cascades. Int J Mol Sci. (2022) 23:10124. doi: 10.3390/ijms231710124
100. Kang JY, Xu MM, Sun Y, Ding ZX, Wei YY, Zhang DW, et al. Melatonin attenuates LPS-induced pyroptosis in acute lung injury by inhibiting NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis. Int Immunopharmacol. (2022) 109:108782. doi: 10.1016/j.intimp.2022.108782
101. Li N, Liu B, He R, Li G, Xiong R, Fu T, et al. HDAC3 promotes macrophage pyroptosis via regulating histone deacetylation in acute lung injury. iScience. (2023) 26:107158. doi: 10.1016/j.isci.2023.107158
102. Jusic A, Devaux Y. Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res Cardiol. (2020) 115:23. doi: 10.1007/s00395-020-0783-5
103. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. (2012) 111:1208–21. doi: 10.1161/circresaha.112.265819
104. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. (2018) 103:115–24. doi: 10.1016/j.molimm.2018.09.010
105. Dong G, Xu N, Wang M, Zhao Y, Jiang F, Bu H, et al. Anthocyanin extract from purple sweet potato exacerbate mitophagy to ameliorate pyroptosis in Klebsiella pneumoniae infection. Int J Mol Sci. (2021) 22:11422. doi: 10.3390/ijms222111422
106. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
107. Peng W, Peng F, Lou Y, Li Y, Zhao N, Shao Q, et al. Autophagy alleviates mitochondrial DAMP-induced acute lung injury by inhibiting NLRP3 inflammasome. Life Sci. (2021) 265:118833. doi: 10.1016/j.lfs.2020.118833
108. Gong T, Fu Y, Wang Q, Loughran PA, Li Y, Billiar TR, et al. Decoding the multiple functions of ZBP1 in the mechanism of sepsis-induced acute lung injury. Commun Biol. (2024) 7:1361. doi: 10.1038/s42003-024-07072-x
109. Han J, Zhang X, Cai M, Tian F, Xu Y, Chen H, et al. deficiency exacerbates acute lung injury via NLRP3 inflammasome-mediated pyroptosis. Chin Med J (Engl). (2024) 137:1592–602. doi: 10.1097/cm9.0000000000003105
110. Zhang J, Li S, Li L, Li M, Guo C, Yao J, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinf. (2015) 13:17–24. doi: 10.1016/j.gpb.2015.02.001
111. Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. (2019) 76:2031–42. doi: 10.1007/s00018-019-03060-1
112. Gong T, Zhang X, Liu X, Ye Y, Tian Z, Yin S, et al. Exosomal Tenascin-C primes macrophage pyroptosis amplifying aberrant inflammation during sepsis-induced acute lung injury. Transl Res. (2024) 270:66–80. doi: 10.1016/j.trsl.2024.04.001
113. Liu P, Yang S, Shao X, Li C, Wang Z, Dai H, et al. Mesenchymal stem cells-derived exosomes alleviate acute lung injury by inhibiting alveolar macrophage pyroptosis. Stem Cells Transl Med. (2024) 13:371–86. doi: 10.1093/stcltm/szad094
114. Tao Y, Xu X, Yang B, Zhao H, Li Y. Mitigation of sepsis-induced acute lung injury by BMSC-derived exosomal miR-125b-5p through STAT3-mediated suppression of macrophage pyroptosis. Int J Nanomed. (2023) 18:7095–113. doi: 10.2147/ijn.S441133
115. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
116. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
117. Liu C, Zhou Y, Tu Q, Yao L, Li J, Yang Z. Alpha-linolenic acid pretreatment alleviates NETs-induced alveolar macrophage pyroptosis by inhibiting pyrin inflammasome activation in a mouse model of sepsis-induced ALI/ARDS. Front Immunol. (2023) 14:1146612. doi: 10.3389/fimmu.2023.1146612
118. Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, et al. HMGB1-mediated neutrophil extracellular trap formation exacerbates intestinal ischemia/reperfusion-induced acute lung injury. J Immunol. (2022) 208:968–78. doi: 10.4049/jimmunol.2100593
119. Song L, Jiang W, Lin H, Yu J, Liu K, Zheng R. Post-translational modifications in sepsis-induced organ dysfunction: mechanisms and implications. Front Immunol. (2024) 15:1461051. doi: 10.3389/fimmu.2024.1461051
120. Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. (2005) 44:7342–72. doi: 10.1002/anie.200501023
121. Sun X, Guo C, Huang C, Lv N, Chen H, Huang H, et al. GSTP alleviates acute lung injury by S-glutathionylation of KEAP1 and subsequent activation of NRF2 pathway. Redox Biol. (2024) 71:103116. doi: 10.1016/j.redox.2024.103116
122. Shi X, Li T, Liu Y, Yin L, Xiao L, Fu L, et al. HSF1 protects sepsis-induced acute lung injury by inhibiting NLRP3 inflammasome activation. Front Immunol. (2022) 13:781003. doi: 10.3389/fimmu.2022.781003
123. Liu F, Gao C. Regulation of the inflammasome activation by ubiquitination machinery. Adv Exp Med Biol. (2024) 1466:123–34. doi: 10.1007/978-981-97-7288-9_9
124. Xia J, Jiang S, Dong S, Liao Y, Zhou Y. The role of post-translational modifications in regulation of NLRP3 inflammasome activation. Int J Mol Sci. (2023) 24:6126. doi: 10.3390/ijms24076126
125. Zhang N, Yang Y, Xu D. Emerging roles of palmitoylation in pyroptosis. Trends Cell Biol. (2024). doi: 10.1016/j.tcb.2024.10.005
126. Johnson AG, Mayer ML, Schaefer SL, McNamara-Bordewick NK, Hummer G, Kranzusch PJ. Structure and assembly of a bacterial gasdermin pore. Nature. (2024) 628:657–63. doi: 10.1038/s41586-024-07216-3
127. Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J Exp Med. (2020) 217:e20182091. doi: 10.1084/jem.20182091
128. Singh M, Kumari B, Yadav UCS. Regulation of oxidized LDL-induced inflammatory process through NLRP3 inflammasome activation by the deubiquitinating enzyme BRCC36. Inflammation Res. (2019) 68:999–1010. doi: 10.1007/s00011-019-01281-5
129. Sheng Z, Zhu J, Deng YN, Gao S, Liang S. SUMOylation modification-mediated cell death. Open Biol. (2021) 11:210050. doi: 10.1098/rsob.210050
130. Yu K, Proost P. Insights into peptidylarginine deiminase expression and citrullination pathways. Trends Cell Biol. (2022) 32:746–61. doi: 10.1016/j.tcb.2022.01.014
131. Yu X, Song Y, Dong T, Ouyang W, Shao L, Quan C, et al. Loss of PADI2 and PADI4 ameliorates sepsis-induced acute lung injury by suppressing NLRP3+ macrophages. JCI Insight. (2024) 9:e181686. doi: 10.1172/jci.insight.181686
132. Tian Y, Qu S, Alam HB, Williams AM, Wu Z, Deng Q, et al. Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. JCI Insight. (2020) 5:e138873. doi: 10.1172/jci.insight.138873
133. Lee JY, Seo D, You J, Chung S, Park JS, Lee JH, et al. The deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the ASC adaptor protein. FEBS Lett. (2017) 591:479–90. doi: 10.1002/1873-3468.12558
134. Yen H, Sugimoto N, Tobe T. Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PloS Pathog. (2015) 11:e1005121. doi: 10.1371/journal.ppat.1005121
135. Wu Z, Tian Y, Alam HB, Li P, Duan X, Williams AM, et al. Peptidylarginine deiminases 2 mediates caspase-1-associated lethality in Pseudomonas aeruginosa pneumonia-induced sepsis. J Infect Dis. (2021) 223:1093–102. doi: 10.1093/infdis/jiaa475
136. Wu Z, Li P, Tian Y, Ouyang W, Ho JW, Alam HB, et al. Peptidylarginine deiminase 2 in host immunity: current insights and perspectives. Front Immunol. (2021) 12:761946. doi: 10.3389/fimmu.2021.761946
137. Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature. (2012) 490:539–42. doi: 10.1038/nature11429
138. Shi Y, Yang Y, Xu W, Shi D, Xu W, Fu X, et al. E3 ubiquitin ligase SYVN1 is a key positive regulator for GSDMD-mediated pyroptosis. Cell Death Dis. (2022) 13:106. doi: 10.1038/s41419-022-04553-x
139. Balasubramanian A, Hsu AY, Ghimire L, Tahir M, Devant P, Fontana P, et al. The palmitoylation of gasdermin D directs its membrane translocation and pore formation during pyroptosis. Sci Immunol. (2024) 9:eadn1452. doi: 10.1126/sciimmunol.adn1452
140. Zhao H, Eguchi S, Alam A, Ma D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L155–L162. doi: 10.1152/ajplung.00449.2016
141. Zhang X, Ding M, Zhu P, Huang H, Zhuang Q, Shen J, et al. New insights into the Nrf-2/HO-1 signaling axis and its application in pediatric respiratory diseases. Oxid Med Cell Longev. (2019) 2019:3214196. doi: 10.1155/2019/3214196
142. Liu B, Wang Z, He R, Xiong R, Li G, Zhang L, et al. Buformin alleviates sepsis-induced acute lung injury via inhibiting NLRP3-mediated pyroptosis through an AMPK-dependent pathway. Clin Sci (Lond). (2022) 136:273–89. doi: 10.1042/cs20211156
143. Zhang H, Xue L, Li B, Tian H, Zhang Z, Tao S. Therapeutic potential of bixin in PM2.5 particles-induced lung injury in an Nrf2-dependent manner. Free Radic Biol Med. (2018) 126:166–76. doi: 10.1016/j.freeradbiomed.2018.08.015
144. Zhang W, Zhao M, Pu Z, Yin Q, Shui Y. Chicoric acid presented NLRP3-mediated pyroptosis through mitochondrial damage by PDPK1 ubiquitination in an acute lung injury model. Am J Chin Med. (2023) 51:1431–57. doi: 10.1142/s0192415x23500659
145. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
146. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
147. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
148. Peng Y, Yang Y, Li Y, Shi T, Xu N, Liu R, et al. Mitochondrial (mt)DNA-cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling promotes pyroptosis of macrophages via interferon regulatory factor (IRF)7/IRF3 activation to aggravate lung injury during severe acute pancreatitis. Cell Mol Biol Lett. (2024) 29:61. doi: 10.1186/s11658-024-00575-9
149. Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Transl Med. (2020) 10:e228. doi: 10.1002/ctm2.228
150. Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. (1996) 14:649–83. doi: 10.1146/annurev.immunol.14.1.649
151. Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. (2011) 21:223–44. doi: 10.1038/cr.2011.13
152. Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol. (2014) 15:239–47. doi: 10.1038/ni.2823
153. Millar MW, Fazal F, Rahman A. Therapeutic targeting of NF-κB in acute lung injury: A double-edged sword. Cells. (2022) 11:3317. doi: 10.3390/cells11203317
154. Peng J, Tang R, He J, Yu Q, Wang D, Qi D. S1PR3 inhibition protects against LPS-induced ARDS by inhibiting NF-κB and improving mitochondrial oxidative phosphorylation. J Transl Med. (2024) 22:535. doi: 10.1186/s12967-024-05220-9
155. Sun B, Bai L, Li Q, Sun Y, Li M, Wang J, et al. Knockdown of angiopoietin-like 4 suppresses sepsis-induced acute lung injury by blocking the NF-κB pathway activation and hindering macrophage M1 polarization and pyroptosis. Toxicol In Vitro. (2024) 94:105709. doi: 10.1016/j.tiv.2023.105709
156. Tao H, Li W, Zhang W, Yang C, Zhang C, Liang X, et al. Urolithin A suppresses RANKL-induced osteoclastogenesis and postmenopausal osteoporosis by, suppresses inflammation and downstream NF-κB activated pyroptosis pathways. Pharmacol Res. (2021) 174:105967. doi: 10.1016/j.phrs.2021.105967
157. Gong C, Ma J, Deng Y, Liu Q, Zhan Z, Gan H, et al. S100A9(-/-) alleviates LPS-induced acute lung injury by regulating M1 macrophage polarization and inhibiting pyroptosis via the TLR4/MyD88/NFκB signaling axis. BioMed Pharmacother. (2024) 172:116233. doi: 10.1016/j.biopha.2024.116233
158. Ma L, Zhao Y, Wang R, Chen T, Li W, Nan Y, et al. 3,5,4’-tri-O-acetylresveratrol attenuates lipopolysaccharide-induced acute respiratory distress syndrome via MAPK/SIRT1 pathway. Mediators Inflamm. (2015) 2015:143074. doi: 10.1155/2015/143074
159. Zhang D, Li X, Hu Y, Jiang H, Wu Y, Ding Y, et al. Tabersonine attenuates lipopolysaccharide-induced acute lung injury via suppressing TRAF6 ubiquitination. Biochem Pharmacol. (2018) 154:183–92. doi: 10.1016/j.bcp.2018.05.004
160. Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep. (2018) 18:4399–409. doi: 10.3892/mmr.2018.9427
161. Aji N, Wang L, Wang S, Pan T, Song J, Chen C, et al. PAI-1 deficiency promotes NET-mediated pyroptosis and ferroptosis during Pseudomonas aeruginosa-induced acute lung injury by regulating the PI3K/MAPK/AKT axis. Inflammation. (2024). doi: 10.1007/s10753-024-02102-6
162. Ow JR, Tan YH, Jin Y, Bahirvani AG, Taneja R. Stra13 and Sharp-1, the non-grouchy regulators of development and disease. Curr Top Dev Biol. (2014) 110:317–38. doi: 10.1016/b978-0-12-405943-6.00009-9
163. Hu X, Zou M, Zheng W, Zhu M, Hou Q, Gao H, et al. Bhlhe40 deficiency attenuates LPS-induced acute lung injury through preventing macrophage pyroptosis. Respir Res. (2024) 25:100. doi: 10.1186/s12931-024-02740-2
164. Liu Y, Shang L, Zhou J, Pan G, Zhou F, Yang S. Emodin attenuates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-dependent pyroptosis signaling pathway in vitro and in vivo. Inflammation. (2022) 45:753–67. doi: 10.1007/s10753-021-01581-1
165. Zhou B, Weng G, Huang Z, Liu T, Dai F. Arctiin prevents LPS-induced acute lung injury via inhibition of PI3K/AKT signaling pathway in mice. Inflammation. (2018) 41:2129–35. doi: 10.1007/s10753-018-0856-x
166. Jin X, Liu S, Chen S, Wang L, Cui Y, He J, et al. A systematic review on botany, ethnopharmacology, quality control, phytochemistry, pharmacology and toxicity of Arctium lappa L. fruit. J Ethnopharmacol. (2023) 308:116223. doi: 10.1016/j.jep.2023.116223
167. Gao F, Xiong D, Sun Z, Shao J, Wei D, Nie S. ARC@DPBNPs suppress LPS-induced acute lung injury via inhibiting macrophage pyroptosis and M1 polarization by ERK pathway in mice. Int Immunopharmacol. (2024) 131:111794. doi: 10.1016/j.intimp.2024.111794
168. Pu Z, Sui B, Wang X, Wang W, Li L, Xie H. The effects and mechanisms of the anti-COVID-19 traditional Chinese medicine, Dehydroandrographolide from Andrographis paniculata (Burm.f.) Wall, on acute lung injury by the inhibition of NLRP3-mediated pyroptosis. Phytomedicine. (2023) 114:154753. doi: 10.1016/j.phymed.2023.154753
169. Shao JJ, Li WF, Sun JF, Zhuang ZS, Min JL, Long XH, et al. Britannin as a novel NLRP3 inhibitor, suppresses inflammasome activation in macrophages and alleviates NLRP3-related diseases in mice. Acta Pharmacol Sin. (2024) 45:803–14. doi: 10.1038/s41401-023-01212-5
170. Ohara S, Komatsu R, Matsuyama T. Short-term effect of buformin, a biguanide, on insulin sensitivity, soluble fraction of tumor necrosis factor receptor and serum lipids in overweight patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. (2004) 66:133–8. doi: 10.1016/j.diabres.2004.03.007
171. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381:1609–20. doi: 10.1056/NEJMoa1908655
172. Wang J, Li J, Lou A, Lin Y, Xu Q, Cui W, et al. Sacubitril/valsartan alleviates sepsis-induced acute lung injury via inhibiting GSDMD-dependent macrophage pyroptosis in mice. FEBS J. (2023) 290:2180–98. doi: 10.1111/febs.16696
173. Ding H, Yang J, Chen L, Li Y, Jiang G, Fan J. Memantine alleviates acute lung injury via inhibiting macrophage pyroptosis. Shock. (2021) 56:1040–8. doi: 10.1097/shk.0000000000001790
174. Zhang J, Li J, An Z, Qi J. Hydromorphone mitigates cardiopulmonary bypass-induced acute lung injury by repressing pyroptosis of alveolar macrophages. Shock. (2023) 60:92–9. doi: 10.1097/shk.0000000000002138
175. Ahmadi Z, Ashrafizadeh M. Melatonin as a potential modulator of Nrf2. Fundam Clin Pharmacol. (2020) 34:11–9. doi: 10.1111/fcp.12498
176. Han Z, Ma J, Han Y, Yuan G, Jiao R, Meng A. Irisin attenuates acute lung injury by suppressing the pyroptosis of alveolar macrophages. Int J Mol Med. (2023) 51:32. doi: 10.3892/ijmm.2023.5235
177. Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. (2016) 49:529–35. doi: 10.5483/bmbrep.2016.49.10.115
178. He JX, Zhu CQ, Liang GF, Mao HB, Shen WY, Hu JB. Targeted-lung delivery of bardoxolone methyl using PECAM-1 antibody-conjugated nanostructure lipid carriers for the treatment of lung inflammation. BioMed Pharmacother. (2024) 178:116992. doi: 10.1016/j.biopha.2024.116992
179. Hofmann-Amtenbrink M, Grainger DW, Hofmann H. Nanoparticles in medicine: Current challenges facing inorganic nanoparticle toxicity assessments and standardizations. Nanomedicine. (2015) 11:1689–94. doi: 10.1016/j.nano.2015.05.005
180. Ding HG, Li Y, Li XS, Liu XQ, Wang KR, Wen MY, et al. Hypercapnia promotes microglial pyroptosis via inhibiting mitophagy in hypoxemic adult rats. CNS Neurosci Ther. (2020) 26:1134–46. doi: 10.1111/cns.13435
181. Zhang T, Lu L, Li M, Zhang D, Yu P, Zhang X, et al. Exosome from BMMSC attenuates cardiopulmonary bypass-induced acute lung injury via YAP/β-catenin pathway: downregulation of pyroptosis. Stem Cells. (2022) 40:1122–33. doi: 10.1093/stmcls/sxac063
Keywords: ALI (acute lung injury), ARDS (acute respiratory distress syndrome), pyroptosis, macrophage - cell, treatment
Citation: Cai Y, Shang L, Zhou F, Zhang M, Li J, Wang S, Lin Q, Huang J and Yang S (2025) Macrophage pyroptosis and its crucial role in ALI/ARDS. Front. Immunol. 16:1530849. doi: 10.3389/fimmu.2025.1530849
Received: 19 November 2024; Accepted: 27 January 2025;
Published: 14 February 2025.
Edited by:
Yongqing Li, University of Michigan, United StatesReviewed by:
Lixin Xie, People ‘s Liberation Army General Hospital, ChinaCopyright © 2025 Cai, Shang, Zhou, Zhang, Li, Wang, Lin, Huang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenglan Yang, eWFuZ3NoZW5nbGFuMDA1QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.