 Francesco Verona1†
Francesco Verona1† Sebastiano Di Bella1†
Sebastiano Di Bella1† Roberto Schirano2
Roberto Schirano2 Camilla Manfredi2
Camilla Manfredi2 Francesca Angeloro3
Francesca Angeloro3 Giulia Bozzari1
Giulia Bozzari1 Matilde Todaro3,4
Matilde Todaro3,4 Giuseppe Giannini2,5
Giuseppe Giannini2,5 Giorgio Stassi1*‡
Giorgio Stassi1*‡ Veronica Veschi2*‡
Veronica Veschi2*‡- 1Department of Precision Medicine in Medical, Surgical and Critical Care, University of Palermo, Palermo, Italy
- 2Department of Molecular Medicine, University La Sapienza, Rome, Italy
- 3Department of Health Promotion Sciences, Internal Medicine and Medical Specialties (PROMISE), University of Palermo, Palermo, Italy
- 4Azienda Ospedaliera Universitaria Policlinico “Paolo Giaccone” (AOUP), Palermo, Italy
- 5Istituto Pasteur, Fondazione Cenci-Bolognetti, Sapienza University of Rome, Rome, Italy
Cancer stem cells (CSCs) are a small subset within the tumor mass significantly contributing to cancer progression through dysregulation of various oncogenic pathways, driving tumor growth, chemoresistance and metastasis formation. The aggressive behavior of CSCs is guided by several intracellular signaling pathways such as WNT, NF-kappa-B, NOTCH, Hedgehog, JAK-STAT, PI3K/AKT1/MTOR, TGF/SMAD, PPAR and MAPK kinases, as well as extracellular vesicles such as exosomes, and extracellular signaling molecules such as cytokines, chemokines, pro-angiogenetic and growth factors, which finely regulate CSC phenotype. In this scenario, tumor microenvironment (TME) is a key player in the establishment of a permissive tumor niche, where CSCs engage in intricate communications with diverse immune cells. The “oncogenic” immune cells are mainly represented by B and T lymphocytes, NK cells, and dendritic cells. Among immune cells, macrophages exhibit a more plastic and adaptable phenotype due to their different subpopulations, which are characterized by both immunosuppressive and inflammatory phenotypes. Specifically, tumor-associated macrophages (TAMs) create an immunosuppressive milieu through the production of a plethora of paracrine factors (IL-6, IL-12, TNF-alpha, TGF-beta, CCL1, CCL18) promoting the acquisition by CSCs of a stem-like, invasive and metastatic phenotype. TAMs have demonstrated the ability to communicate with CSCs via direct ligand/receptor (such as CD90/CD11b, LSECtin/BTN3A3, EPHA4/Ephrin) interaction. On the other hand, CSCs exhibited their capacity to influence immune cells, creating a favorable microenvironment for cancer progression. Interestingly, the bidirectional influence of CSCs and TME leads to an epigenetic reprogramming which sustains malignant transformation. Nowadays, the integration of biological and computational data obtained by cutting-edge technologies (single-cell RNA sequencing, spatial transcriptomics, trajectory analysis) has significantly improved the comprehension of the biunivocal multicellular dialogue, providing a comprehensive view of the heterogeneity and dynamics of CSCs, and uncovering alternative mechanisms of immune evasion and therapeutic resistance. Moreover, the combination of biology and computational data will lead to the development of innovative target therapies dampening CSC-TME interaction. Here, we aim to elucidate the most recent insights on CSCs biology and their complex interactions with TME immune cells, specifically TAMs, tracing an exhaustive scenario from the primary tumor to metastasis formation.
Cancer stem cells hallmarks and crosstalk with TAMs: an old story new
In this review we revised the literature period of the last twenty years using as main keywords the following: cancer stem cells, stemness, tumor-associated macrophages, metastasis, metastatic niche, hallmark, proliferation, immune evasion, neo-angiogenesis, epithelial-mesenchymal transition, crosstalk, pathways, chemoresistance, therapy resistance, target therapy, preclinical model, clinical model, clinical trial, immunotherapy, stemness, self-renewal, invasion, tumorigenicity, oncogenic pathways, metastasis-associated macrophages, tumor microenvironment, scRNA-seq, spatial transcriptomic, trajectory analysis, stromal cells, extracellular matrix and immune cells.
Cancer stem cells (CSCs) are a small subpopulation within tumor bulk sharing features of normal stem cells, such as self-renewal and plasticity (1). Accordingly, the CSC model introduced the concept of the capability of CSCs to recapitulate the intertumoral heterogeneity, differentiating into various cancer cell phenotypes and, in parallel, guaranteeing their population maintenance (2). Due to their genetic flexibility, CSCs can be involved in different biological aspects such as tumor initiation, proliferation, invasion, migration, and chemoresistance (1). All these pro-tumoral traits underlined the critical role of CSCs in cancer progression and made CSCs a potential target for innovative therapeutic approaches (3). Tumor microenvironment (TME) provides an essential environmental niche necessary for cancer development (4). Among the immune cells that have a central role in orchestrating TME, tumor-associated macrophages (TAMs) represent a plastic immune cell population that drives multiple interactions within the TME, leading the spatiotemporal evolution from primary tumor to metastasis (5). TAMs can establish with CSCs an intricate complex communication in fueling different aspects of cancer progression: i) direct ligand-receptor interaction: TAMs expressing colony-stimulating factor (CSF1) receptor anchors CSC-derived CSF1, in the promotion of TAM survival and activation (6); ii) indirect interaction: TAMs release chemokines like chemokine (C–C motif) ligand 2 (CCL2), interleukin-6 (IL-6), interleukin-12 (IL-12), tumor necrosis factor-alpha (TNF-alpha), transforming growth factor-beta-1 (TGFB1) (7); TAMs release exosomes containing microRNAs and proteins that regulate CSC behavior by enhancing stemness and chemoresistance; conversely, CSC-derived exosomes can polarize TAM toward a tumor-promoting M2 phenotype (8, 9).
Overall, the interaction between CSCs and the surrounding environmental cells is a complex and ever-evolving process. CSCs arise in “ecological” niches in the TME. These niches, establishing intense trafficking of factors, promote a stem-like and chemoresistant phenotype in the CSCs (10). In this scenario, emerging bioinformatics technologies, such as trajectory analysis and spatial transcriptomics, shed light on unresolved biological complexities. Particularly, these tools enable a deeper investigation of the crosstalk between CSCs and TAMs dissecting unrevealed aspects of their communication. Comprehending the intricate symbiotic relationships between CSCs and TAMs could provide valuable insights to identify an efficacious innovative therapeutic approach. An overview of CSC hallmarks and how these characteristics critically contribute to the complex interplay between CSCs and TME components is illustrated in Figure 1.
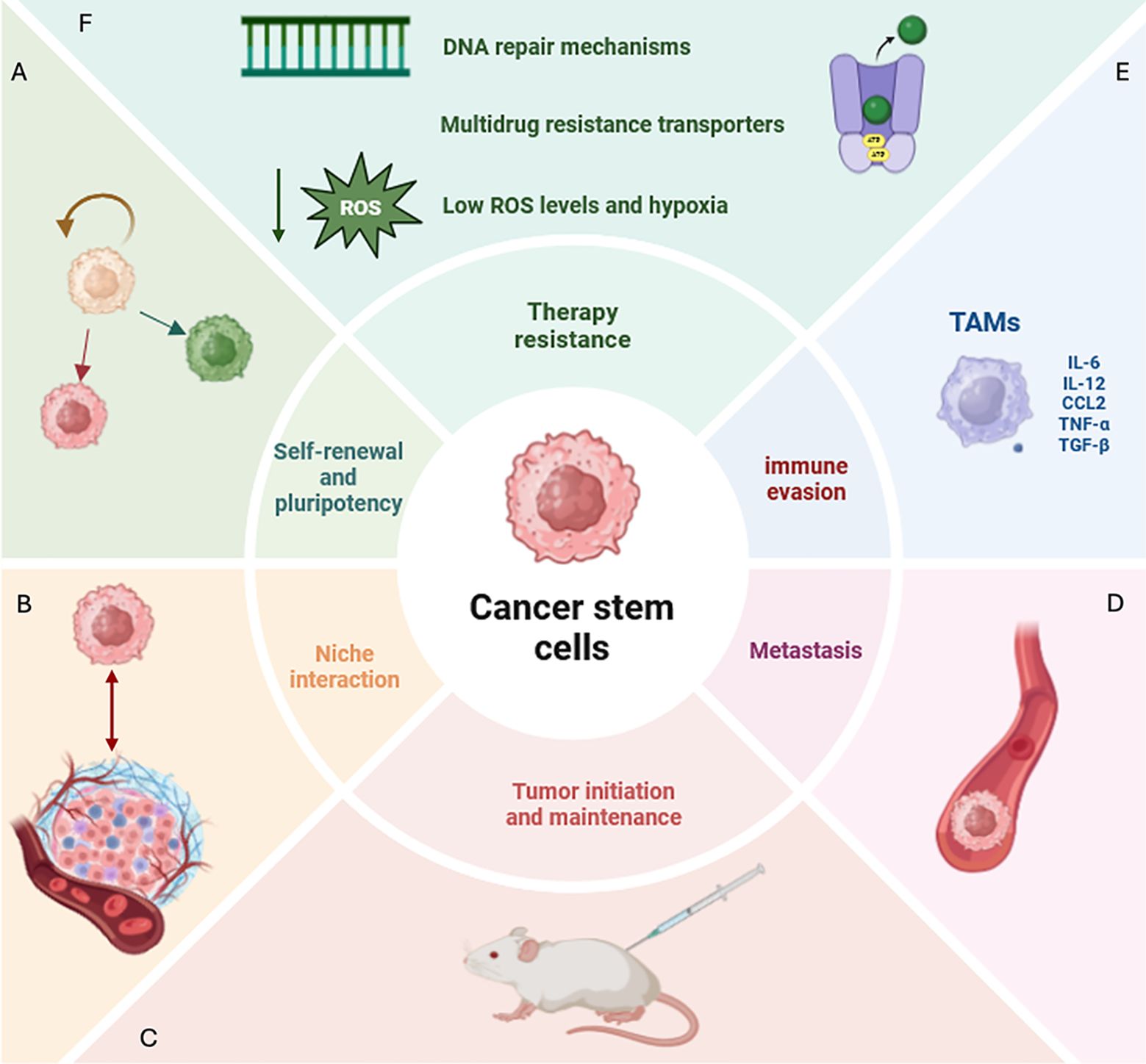
Figure 1. Defining CSC features and hallmarks. (A) CSCs (cancer stem cells) display the ability of self-renewal and pluripotency, disrupting tissue homeostasis and generating diverse lineages within the tumor. (B) CSCs create a niche in the tumor microenvironment (TME) with which they interact and that proliferates independently of the surrounding tissue. (C) CSCs show the ability to initiate tumor growth in immunocompromised mice. (D) CSCs represent the most aggressive tumor subpopulation able to spread and form metastases even at distant sites. (E) Among immune cells that create an immunosuppressive milieu in CSC-associated TME, in this review we will focus on tumor-associated macrophages (TAMs) which play a critical role. TAMs are macrophages characterized by both immunosuppressive and inflammatory phenotypes. Specifically, they produce a plethora of paracrine factors (IL-6, IL-12, TNF-alpha, TGFB1, CCL2) inducing the acquisition of a stem-like, invasive and metastatic phenotype in CSCs. (F) Several mechanisms contributing to therapy resistance in CSCs have been identified, including efficient DNA repair machinery, multidrug resistance transporters, low levels of reactive oxygen species (ROS) and hypoxia. CSCs, cancer stem cell; TME, tumor microenvironment; TAMs, tumor-associated macrophages; IL-6, interleukin-6; IL-12, interleukin-12; TNF-alpha, tumor necrosis factor-alpha, TGFB1, transforming growth factor-beta-1, CCL2:C-C Motif Chemokine Ligand 2; ROS, reactive oxygen species.
From normal to CSCs endowed with tumor-initiation and metastatic potential
In normal adult tissues stem cells are undifferentiated cells that reside in a proper niche, where they are protected and can exert their functions. Stem cells show the ability of self-renewal and differentiation in adult cell tissue, maintaining tissue homeostasis. Stem cell niche can be identified in several tissues such as the crypts of the intestine, the bone marrow, the liver or lung tissues (11). After tissue injury, the niche transmits activation signals such as adhesion molecules, matrix proteins, oxygen, growth factors or cytokines to the stem cells for tissue regeneration. These signals are factors that allow cell-cell interactions between stem cells and neighboring differentiated cells (12).
In normal tissues stem cells remain in an undifferentiated state throughout adult life. Stem cells reach a first stage by becoming transient amplifying cells and highly proliferative cells then they asymmetrically divide and finally reach the last stage of differentiated cells, that leads them to build up and support tissues (13). In both stem and differentiated cells, the potential accumulation of intracellular pathways mutations can lead to a tumor-type phenotype (14). When a critical mutation threshold is reached, cells become CSCs, changing to a more aggressive behavior (15). CSCs, as normal stem cells, have the ability of self-renewal and differentiation, while they create a niche that proliferates independently of the surrounding tissue. These characteristics contribute to tumor initiation, growth and maintenance (13). Among the most deregulated intracellular pathways, wingless-related integration site (WNT)/beta-catenin, NOTCH and Sonic Hedgehog emerge, as they promote self-renewal and tissue morphogenesis (16). In addition, cellular growth, migration, differentiation and epithelial-mesenchymal transition (EMT) are regulated by phosphatidylinositol 3-kinase/AKT serine/threonine kinase 1/Phosphatase and tensin homolog (PI3K/AKT1/PTEN) axis, one of the majors signaling pathways in CSCs (17). TGF, SMAD, peroxisome proliferator-activated receptor (PPAR), mitogen-activated protein kinases (MAPK) and Janus kinase/signal transducers and activators of transcription (JAK-STAT) are often deregulated in CSCs (18). CSCs are not only involved in the process of tumor initiation, growth and maintenance, but also in metastasis (19). CSCs represent the most aggressive tumor subpopulation able to spread and form metastases even at distant sites. One of the key requisites for successful metastasis formation is stemness. Indeed, depletion of various stemness markers such as cluster of differentiation 44 (CD44) in breast CSCs (20) or octamer-binding transcription factor 4 (OCT4) and SRY-Box Transcription Factor 2 (SOX2) in colon CSCs, prevented tumor metastasis and tumor growth (21). Beyond stemness markers, several studies have been focused on the identification of cell-surface markers specifically expressed in the subpopulation of CSCs endowed with metastatic potential such as CD44v6, a CD44 variant isoform, in colon CSCs (22). A broad and extensive description of CSCs hallmarks and the methodologies used to characterize the CSC state is reported in (23). In the next paragraphs, we will briefly introduce how CSCs evade the immune system and resist conventional therapies.
CSCs and immune evasion
Immunosurveillance is a set of immune-system related processes aimed at controlling the development of normal cells and detecting cancer cells. The innate and adaptive cells of the immune system respond to stress conditions, caused by tumor development, mainly by upregulating natural killer (NK) activator ligands and stimulating a more specific T lymphocyte response against cancer cells (24). NK cells are innate immune cells that recognize cells lacking major histocompatibility class I complex (MHC-I) and exert potent cytolytic activity releasing perforin and granzyme against transformed cells (24). NK cells mediate the tumor killing also triggering apoptotic pathways in tumor cells through the production of TNF-alpha or via direct cell–cell contact through activation of the Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and FAS ligand (FASL) pathways (24). Otherwise, T cells are the main component of the adaptive immunity that orchestrate a protective effector immune response, indeed, a high level of T cell infiltration in tumors is associated with a favorable prognosis in cancer patients (24). CD8+ T and CD4+ T helper 1 cells are the most prominent anti-tumor T cells, instead, through the exocytosis of perforin and granzyme containing granules, the former, and secretion of high amounts of proinflammatory cytokines, such as interleukin-2 (IL-2), TNF-alpha, and interferon-gamma (IFNG), the latter, promote T cell priming activation, cytotoxic T lymphocytes (CTL) cytotoxicity, but also, the anti-tumoral activity of macrophages and NK cells (24).
T and NK cells destructive effect on cancer cells is regulated even by TAMs, by increasing the number of active NKs, upregulating inhibitory T cell receptors programmed cell death protein 1 (PD-1) and Cytotoxic T lymphocyte associated protein 4 (CTLA-4), releasing factors such as TRAIL and inducing apoptosis in cancer cells (25, 26). However, during inflammation, TAMs can directly inhibit the proliferation of CD8+ T cell lymphocytes by regulating their metabolism or recruiting regulatory T cells (Tregs) (27). TAMs can also inhibit dendritic cell (DC) maturation and the secretion of IL-12 by DCs (28). TAMs and Tregs boost an immune-tolerant TME by secretion of molecules such as interleukin-10 (IL-10), TGFB1, and prostaglandins (28). Indeed, poor prognosis and reduced overall survival in oncological patients is correlated with high-grade TAMs (28). Tumor cells can evade the immune system by using different strategies like losing surface antigens that prevent recognition by cytotoxic T cells or downregulating cell surface NK activators, becoming invisible to detection by NK cells (28). However, the immune system can self-contribute to tumor development and progression, orchestrating an immunosuppressive inflammatory TME (24). This process is called “cancer immunoediting” and proceeds through three phases: elimination, equilibrium and escape (29). During the first phase the cytotoxic immune cells such as NK and CD8+ T cells kill transformed cells, although rare tumor subclones can survive (30, 31). These tumor subclones may enter the second phase where their growth is limited and stalled over time (30, 31). The steady pressure from the adaptive immune system and the genetic instability of cancer cells can make tumor subclones escaping immunosurveillance (30, 31). Cancer cells start proliferating unconditionally and adopt many features to escape from the immune system like downregulation of the antigen presentation machinery or inducting inhibitory immune checkpoint molecules (32). Moreover, cancer cells remodel the vasculature and extracellular matrix and supports cancer progression as well as therapy resistance (30, 31). This process can entail decreased IFN-gamma secretion by T cells, loss of antigen presentation and epigenetic changes (33).
Within the tumor CSCs control the immune system and regulate the composition of TME through the release of cytokines, chemokines, growth factors, metabolites and hormones playing an immunomodulatory role (34). CSCs develop different immunosuppressive strategies that promote tumor maintenance and growth. Downregulation of MHC-I complexes and activation of immune molecules such as cluster of differentiation 80 (CD80), human leukocyte antigen (HLA) and major Histocompatibility Complex Class I chain-related protein A/B (MICA/MICB), renders CSCs more resistant to cytotoxic effects exerted by CTL (35). Of note, the degree of tumor progression in the CSC niche has been attributed to a reduced CD8+ T cell infiltration and to an increase in TAMs (35). Moreover, CSCs interact through human leukocyte antigen G (HLA-G) with killer cell immunoglobulin like receptor, two Ig domains and long cytoplasmic tail 4 (KIR2DL4) and killer cell lectin like receptor C1 (KLRC1) to suppress NK activity (34). CSCs further drive recruitment and polarization Treg cells by secretion of factors like Chemokine (C-C motif) ligand 1 (CCL1), IL-2, interleukin-8 (IL-8), IL-10 and Transforming growth factor-beta-1 (TGFB1) (34). Moreover, Tregs produce TGFB1 and interleukin-17 (IL-17) to promote CSCs properties toward tumor progression and invasion (34). CSCs immune evasion properties are influenced by humoral factors: TGFB1, a cytokine that induces immune suppression, EMT and stemness; IL-6, secreted by TAMs, that induces and maintains CSCs, signal transducer and activator of transcription 3 (STAT3), a transcription factor required for the maintenance of pluripotency in stem cells or Chemokine (C-C motif) ligand 20 (CCL20) and its receptor that recruits Tregs to promote tumor progression enhanced by immune evasion (34). CTLA-4 and PD-1/programmed death-ligand 1 (PD-L1) represent two of the major immune checkpoints (34). Immunosuppressive myeloid cells, including macrophages and monocytic myeloid-derived suppressor cells (MDSCs) represent an additional layer of regulation of T cell activity and partially depend on secretion of factors like CSF1, CCL2, Chemokine (C-C motif) ligand 5 (CCL5), TGFB1 and prostaglandin E2 (PGE2), by CSCs (34). Collectively, all these interactions reshape the tumor microenvironment and create a habitat where immune cells support and are suppressed by CSCs (34).
Therapy resistance in CSCs
Conventional therapies developed for cancer treatment are based on the following approaches such as chemotherapy, radiation therapy and surgical excision (36, 37). Chemotherapy is the most widely used and effective treatment for cancer; however, cancer cells as well as CSCs often elaborate simultaneous resistance to many drugs, even if they are structurally and functionally quite different (36, 37). This phenomenon is called multidrug resistance (MDR) or multifactorial pleiotropic drug resistance (36, 37). Many in vivo and in vitro studies demonstrated that administering chemotherapeutic drugs led to an enrichment in CSCs (36, 37). Drug resistance is caused by regular administration of chemotherapy drugs, that are dose- or time-dependent. The multiple mechanisms underlying MDR can be listed as follows: increased drug efflux and reduced drug uptake, efficient DNA repair mechanisms, reduced presence of reactive oxygen species (ROS), apoptosis evasion, hypoxia, vasculogenic mimicry (VM) activation, increased autophagy and decreased ferroptosis (38).
Mechanisms responsible for therapy resistance in CSCs are summarized in Figure 2.
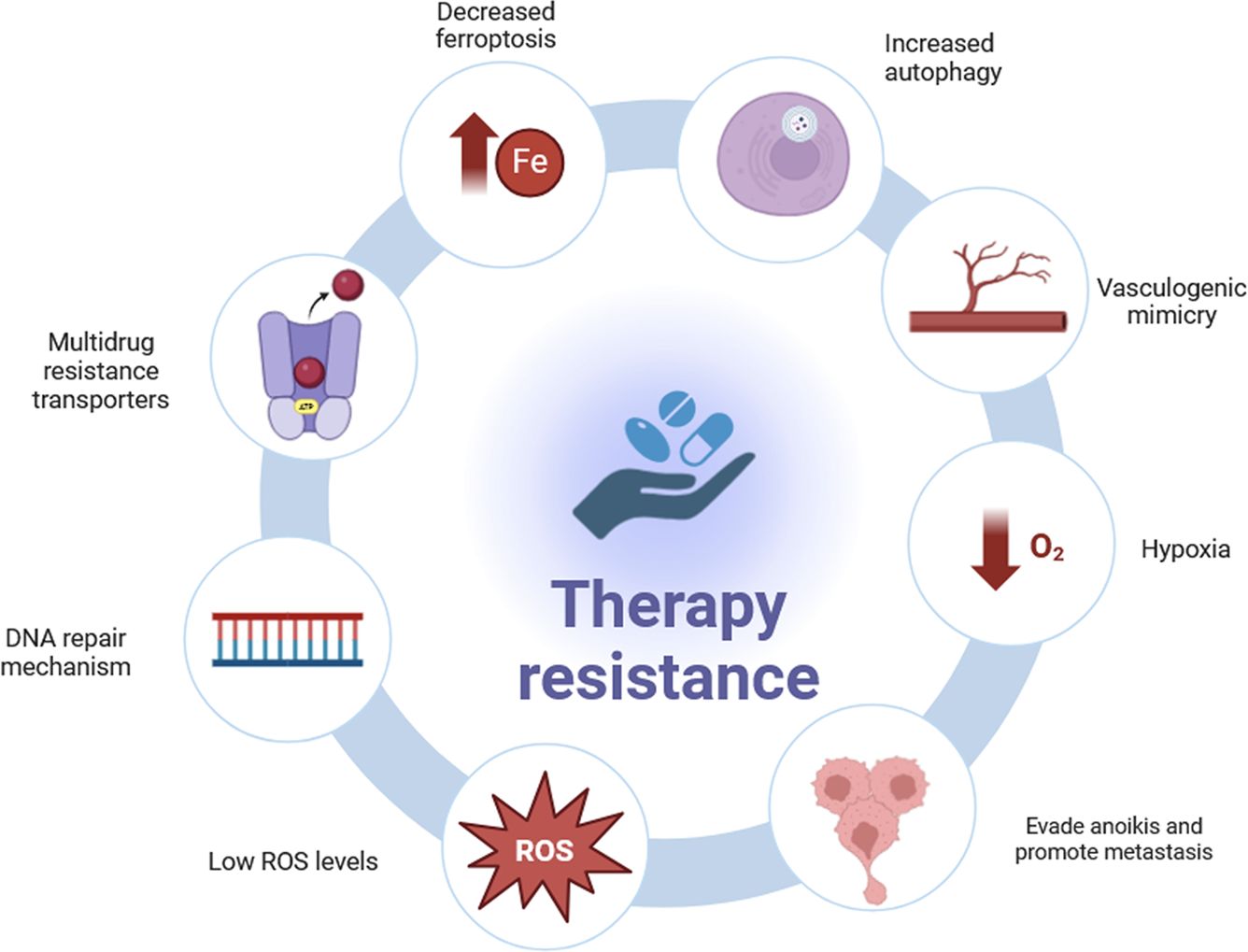
Figure 2. Mechanisms of therapy resistance in CSCs. Many CSCs strategies have been identified to resist to therapy: multidrug resistance transporters, efficient DNA repair mechanisms, lower Reactive Oxygen Species (ROS) levels, evading cell death or “anoikis” and promote metastasis, hypoxia, providing sufficient blood supply through vasculogenic mimicry (VM), increased autophagy and decreased ferroptosis.
Multidrug resistance transporters
Several studies demonstrated that many chemotherapeutic agents in clinical use are susceptible to ATP-binding cassette transporters-mediated efflux (ABC), such as microtubule-targeting, alkaloids, taxanes, topoisomerase inhibitors, DNA-damaging anthracyclines and tyrosine kinase inhibitors (38, 39). This subfamily of transporters is mainly localized in human tissues of the brain, lung, breast, kidneys, liver, ovaries, prostate, placenta and pancreas (40).
CSCs express higher levels of MDR transporters than cancer cells or healthy cells (41). ABCB1, ABCG2 and ABCB5 are overexpressed respectively in ovarian CSCs (41), breast CSCs (42) and malignant melanoma initiating cells (MMIC) (43). Inhibitors of the ABC transporters are currently used in clinical settings, although side-effects and high toxicity have been reported in patients (44).
DNA repair mechanisms
Efficient DNA repair mechanisms in CSCs are thought to be a major contributing factor in counteracting treatment-induced DNA damage (45). Efficient DNA damage repair system and the CSC long permanence in a quiescent G0 phase greatly reduce potential exogenous and endogenous DNA damage that could occur during DNA replication (45). Evidence demonstrates that DNA damage response (DDR) sensor proteins are upregulated in CSCs rather than tumor bulk cancer cells in monolayer cultures, thus conferring radio and chemotherapy resistance (46). Enhanced expression of DNA polymerase nu (POLN) contributes to chemoresistance in ovarian stem cells (47). Thus, cytotoxicity by chemotherapeutic drugs or radiotherapy-induced can be attenuated in CSCs based on an efficient DNA damage repair system (26).
ROS levels
CSCs show low intracellular levels of ROS, a group of highly reactive molecules, containing oxygen, that can promote DNA damage and influence the DDR machinery (48). Therefore, CSCs can dampen the entity of exogenous DNA damage induced by conventional therapy by expressing low levels of ROS, which production is mainly determined by the slow division rate of CSCs (48). Lower levels of ROS in CSCs result crucial in maintaining a stem cell-like phenotype, along with conferring resistance to radiation therapy and/or chemotherapy (49).
Anoikis
The ability of CSCs to metastasize and reach other organs should be reduced as cells undergo programmed cell death or apoptosis, where they lose contact with their extracellular matrix or neighboring cells (“anoikis”) (50). However, CSCs were reported to be anoikis resistant (50). Indeed, CSCs endowed with metastatic potential evade anoikis mechanism, therefore surviving and promoting the formation of metastatic lesions at a distant site (51). Notably, co-culturing CSCs with non-CSCs conferred anoikis resistance to non-stem cells in breast cancer (51). CSC-like cells protected non-stem cells from anoikis and promoted tumor growth (51).
Hypoxia
Oxygen is necessary for metabolism and cellular energy production. In many tumors, oxygen levels are usually between 0% and 2% compared to normal physiological levels that can reach up to 9% and therefore the high metabolic demand requires the activation of hypoxia-inducible factors (HIFs) (52, 53). HIFs are heterodimers consisting of two subunits a and b that can translocate in the nucleus and interact with specific sequences leading to activation or repression of gene expression (52, 53). There are three different genes encoding for HIF subunits: hypoxia inducible factor 1 subunit alpha (HIF1A), hypoxia inducible factor 2 subunit alpha (HIF2A), and hypoxia inducible factor 3 subunit alpha (HIF3A) (52, 53). All three heterodimerize with the hypoxia inducible factor 1 subunit beta (HIF1B) subunit and are subject to posttranslational regulation that is dependent on oxygen levels in the environment (52–54). HIF1A and HIF2A through the upregulation of regulators such as SOX2, Nanog homeobox (NANOG), OCT4, KLF Transcription Factor 4 (KLF4), and the transcription factor MYC proto-oncogene protein (MYC), have been shown to promote stemness and CSC phenotype (55). Upregulation of HIF-1 induces the expression of genes involved in angiogenesis, cell survival, and metabolism, conferring a selective advantage to CSCs (56). It has been demonstrated that breast cancer cells lines, MCF-7 and MDA-MB-231, display increased subpopulations of tumor cells with stem-like characteristics (56). Hypoxia is a hallmark of the CSCs environment that is essential for CSCs development, maintenance, tumor growth and resistance to therapy (57). Evidence suggests that the hypoxic niche in colon cancer protects CSCs from chemotherapy (58). Moreover, in ovarian cancer stem cell lines, SK-OV-3 and HO-8910, it has been demonstrated that chemotherapy treatment, under hypoxia conditions, induced CSC-like properties (59). The mechanisms through which hypoxia exerts its function are complex but can be summarized in shifting the metabolism toward aerobic glycolysis, reduced expression of pro-apoptotic factors, dysregulation of ROS and redox mechanisms, increasing genomic instability and aberrant cell cycling (48).
Vasculogenic mimicry
Vascularization plays an important role during carcinogenesis and metastasis. VM can provide sufficient blood supply for tumor growth, independently of endothelial cells (60, 61). VM is a process of blood vessel formation that cancer cells and CSCs employ to increase the blood supply of angiogenesis (60, 61). It is a mimicry process whereby malignant cells mimic the function of endothelial cells to form blood vessels by reshaping the extracellular matrix (60, 61). CSC VM has been observed in many tumors such as breast cancer and melanoma (62). Evidence shows that vasculogenic mimicry is mostly present at the early stages of tumor development when blood supply is most needed, as the tumor grows where the vessels created by endothelial cells are established (63). Studies also show that the early stage of CSC serves as tumor vasculogenic stem/progenitor cells that can differentiate into tumor vasculogenic endothelial cells (64, 65). New vessel formation, and particularly VM, makes the eradication of the tumor even more complex and unsuccessful, giving the tumor the ability to metastasize (64, 65).
Autophagy
Autophagy is a catabolic process that degrades and recycles cellular components and exhibits both protective and destructive roles in the TME under physiological stress conditions such as nutrient deprivation and hypoxia (66). The activation of autophagy may lead to an arrest of tumor development, but at the same time it can support CSC self-renewal and resistance to therapy (66). In CSCs, autophagy contributes to maintain self-renewal and proliferation properties, avoiding senescence (67). Evidence suggests that autophagy is involved in mechanisms that mediate resistance to therapy, in renal carcinoma and breast cancer (68, 69). Experiments carried out to inhibit autophagy have shown increasing sensitivity to radio- and chemotherapy in nasopharyngeal and breast CSCs, respectively (70, 71). In addition, the upregulation of signaling pathways mediating autophagy, such as SOX2- beta-catenin/BECLIN1, determines resistance to chemotherapy (72).
Ferroptosis
Iron is an essential cofactor for several metabolic reactions and contributes to the formation of ROS (73). Ferroptosis can be defined as a form of iron-catalyzed necrosis and occurs through the intracellular accumulation of ROS, induced by lipid peroxidation (74). Current studies demonstrate that during tumor development the levels of iron and its transporters increase in CSCs compared to cancer cells (74). Although iron accumulation promotes ferroptosis, CSCs maintain a balance that prevents toxic lipid peroxidation (75). Chemotherapeutic drugs generate ROS that can induce oxidative damage and apoptosis (75). However, CSC ability to control ferroptosis reduces the harmful effect of ROS species conferring chemotherapeutic resistance (75). Inducing high levels of ferroptosis is indeed currently used as an innovative approach to revert chemotherapy resistance, specifically in the CSC population (76).
TME as a key player in promoting CSC stemness and cancer development
Several studies have shown that various types of cells embedded in the TME contribute to maintain and sustain CSCs stemness properties. These findings prove that a crucial role in tumor progression is played by the specific TME surrounding tumor bulk cells and CSCs, which create the ideal conditions for tumor initiation. A detailed description of the key components present in the TME is reported in Figure 3.

Figure 3. Tumor microenvironment (TME) key components. TME is a highly complex player composed of cellular components and non-cellular components, where cancer stem cells (CSCs) engage in communications with diverse immune cells, playing a critical role in cancer progression. CSCs have the ability of self-renewal (in yellow) and differentiation (in dark pink) in adult cell tissue, disrupting tissue homeostasis. Cellular components include: heterogenous cancer cells, diverse immune cells (e.g., T lymphocytes, regulatory T cell or Treg, tumor-associated macrophages or TAMs and myeloid-derived suppressor cells or MDSCs), stromal cells (e.g. cancer‐associated fibroblasts or CAFs and mesenchymal stromal cells or MSCs) and endothelial cells. Noncellular components include extracellular matrix (ECM) molecules (e.g., collagen, fibronectin, laminin and hyaluronan) biochemical and biophysical cues. Immune cells largely determine TME secretome composed of IL-6, IFN-gamma, TNF-alpha, TGFB1, IL-12, CXCL12, CCL1, CCL18 and several others. TME, tumor microenvironment; CSCs, cancer stem cells; TAMs, tumor associated macrophages; MDSCs, myeloid-derived suppressor cells; NK, natural killer; Treg, regulatory T cell; CAFs, cancer-associated fibroblasts; MSCs, mesenchymal stromal cells; ECM, extracellular matrix; IL-6, interleukin-6; IFNG, interferon-gamma; TNF-alpha, tumor necrosis factor-alpha; TGFB1, transforming growth factor-beta-1; IL-12, interleukin-12; CXCL12, C-X-C motif chemokine ligand 12; CCL1, chemokine (C-C motif) ligand 1; CCL18, C-C motif chemokine ligand 18.
TME includes various host healthy cells which enfold the tumor, and by producing cytokines and hormones they can promote its growth and behavior (77). As the core of the TME, tumor cells exploit cellular and non-cellular components for their own advantage by the installation of a complex signaling network (78). The host healthy cells, like fibroblasts or immune cells, as well as the extracellular matrix, undergo a tumor-mediated reprogramming able to convert the host cells into tumor associated ones such as cancer-associated fibroblasts (CAFs) and TAMs. Following the conversion, the tumor-associated cells start to sustain and promote tumor growth in different ways. Hereinafter, an overview of the main cells which compose TME and their contribution to tumor progression will be provided.
Stromal cells and ECM
CAFs are highly heterogeneous stromal cells which represent the major modifiers of TME by the synthesis of soluble factors that promote tumor progression, stemness and angiogenesis in several cancers including prostate, gastric and non-small cell lung cancer (79–81). CAFs also contribute to tumor immune evasion both directly and indirectly. Different studies prove that CAFs are associated with T cells impairment, preventing their activation by secretion of C-X-C motif chemokine ligand 12 (CXCL12) and TGFB1 (82, 83). The primary role of CAFs is the establishment and apposition of the extracellular matrix (ECM) (84). ECM composes the scaffold for tissues and organs and facilitates cells crosstalk, both in healthy and malignant conditions. Jachetti et al. demonstrated that ECM proteins inhibit T cell proliferation and effector function (85). In addition, ECM can improve drug resistance by acting as a physical barrier. Besides, it has been shown that collagen, one of the most abundant proteins in ECM, can promotes stemness through the activation of an integrin/PI3K/AKT1/SNAIL signaling pathway (86).
Mesenchymal stromal cells (MSCs) are a substantial component of TME, recruited and re-educated by tumor cells in order to sustain tumorigenesis (87). Indeed, tumor associated-MSCs are crucial promoters of cancer hallmarks. It is shown that IL-6 produced by MSCs increases endothelin 1 (ET-1) expression in colorectal cancer (CRC) cells, resulting in the activation of AKT1 and ERK in endothelial cells which lead to tumor neo-angiogenesis enhancement (88). Several studies demonstrated that MSCs contribute also to tumor invasiveness and progression by regulating EMT regulators, like Twist, Snail and Zinc finger E-box binding homeobox 1 (ZEB1) (89–92). Finally, MSCs interact and suppress TME-embedded immune cells, either directly or through the release of factors like TGFB1, IL-2 and IL-10 (93) and, moreover, play a crucial role in enhancing stemness of cancer cells. Indeed, in physiological conditions, MSCs shape and support tissues and promote stemness features of the stem cell niches. Similarly, MSCs interact and promote CSC stemness in tumors via soluble factors (52).
Immune cells
Although immune cells should prevent and resolve tumor progression, they act as promoters of cancer development under the pressure of TME signalosome (94). MDSCs are regulators of immune homeostasis (95). Cancer cells exploit MDSCs activity to escape immune surveillance, indeed MDSCs are commonly present in TME for their capability to facilitate tumor progression by establishing immune-suppressive conditions in different ways (96). ROS, IL-10 and TGFB1 produced by MDSCs negatively regulates CD8+ T cells activity against cancer cells (96). Moreover, MDCSs up-regulate PD-L1 expression, resulting in suppression of the immune response against tumors (97). MDSCs also regulate indirectly the immune response exacerbating TME by factors essential for T lymphocytes functions, such as L-arginine, which is crucial for T cells proliferation and activity (98, 99). MDSCs promote CSC stemness by miRNAs able to trigger CSCs stemness program (100).
Tregs are spontaneously attracted by immunosuppressive cytokines produced by tumor and tumor-associated cells (101). As well as MDSCs, Tregs promote tumors immune evasion by releasing cytokines able to suppress the activation of the immune response effectors (102, 103). Recent evidence suggests that Tregs are important regulators of CSCs stemness. Indeed, in several cancers, Tregs promote stemness-related pathways (104), facilitate EMT (105) and angiogenesis (101).
Tumor cells and TME not only re-educate and exploit MDSCs and Tregs but also induce depletion of tumor killing activity exerted by immune response effectors cells, NK cells and lymphocytic cells (106, 107). Although in the early stages of tumorigenesis NK cells are lethal for tumor cells, they slowly exhausted their killing function under the pressure of TME factors (108). Indeed, TGF-beta produced by CSCs, MDSCs and Tregs, impairs NK cells cytotoxicity, inhibits the release of IFNG and reduces the expression levels of killer cell lectin like receptor K1 (KLRK1) receptor in several tumors (109–111). Also, TME hypoxia conditions inhibit NK cells by downregulating expression of NKp46, NKp30, NKp44, KLRK1, perforin (PRF1), and granzyme B (GZMB) (112). Finally, lactate produced by tumor cells leads to the acidification of TME which induces apoptosis of NK cells (113). The same conditions which inhibit NK cells affect also lymphocytic cells activity, the most potent immune weapons against tumor cells (114, 115). Besides, downregulation of MHC-I, along with the up-regulation of immune checkpoints, (i.e. PD-L1) allows tumor cells to ensure themselves immune evasion (116, 117). Among the immune cells present in the TME, a focus on TAMs and their hallmarks will be provided in the next paragraphs.
Recently, CSCs-TME interplay gained interest in cancer research as a potential therapeutic target against tumors. TME promotes a stem-like state in CSCs supporting their self-renewal, survival, and therapeutic resistance through different molecular mechanisms (118). CAFs, the most represented cells in TME, release cytokines like IL-6, able to sustain the expression of stemness-related genes like SOX2, NANOG and OCT4 in CSCs (119). On the other hand, CSCs drive TME immunosuppressive polarization and persistence (35). CSCs can regulate immune system activity through the release of immunosuppressive secretome (i.e. IL-10, TGFB1) showing a more efficient capability to recruit immune cells with pro-tumoral activity (Tregs, MDSCs and especially TAMs) which sustain CSCs stemness by releasing factors like platelet-derived growth factor (PDGF), IL-8, CXCL12 (120, 121).
The TAMS story
As tissue-resident immune cells, macrophages represent an anti-cancer first line of defense thanks to their capability to recognize and phagocyte malignant cells, but they are also the first allies of tumor initiation and development. After malignant transformation, TAMs are the result of the exploitation of macrophages plasticity (M1-M2 dichotomy), by cancer cells (122, 123). TAMs play a pivotal role in vascularization, inflammation, EMT and intravasation in different cancer models (124–127). This review aims to shed new light on the important role of macrophages in cancer development and the close link with TME modulation, the role of macrophages and monocytes, in relation with CSCs stemness and support.
TAMs Hallmarks
The hallmarks of cancer, initially introduced by Hanahan and Weinberg (128) mirror the complex and fundamental biological mechanisms that drive cancer cells to malignancy. In this context, TAMs have emerged as crucial players, within the TME, in cancer progression showing ability in tumor growth and metastasis processes (129). Particularly, TAMs originate from circulating monocytes, in the bloodstream, that migrate to tumor sites where they become macrophages (130). Macrophages are characterized by a peculiar plastic phenotype and can differentiate in wide spectrum of subclasses finely driven by super-enhancers activity (131, 132). Usually, they are classified as M1 or M2, which display pro-inflammatory and immunosuppressive phenotypes respectively. In cancer contexts, TAMs mainly display an M2-like state, which is correlated to oncogenic features such as cancer cell proliferation, immunosuppression, chemoresistance, angiogenesis and metastasis (133). Overall, the acquisition of an M2-like state is critical to create a microenvironment that supports both the survival and progression of cancer cells (134). Moreover, an enrichment of TAMs infiltration, in the context of TME is linked to a worse prognosis in several cancers (135–137).
The most significant TAMs hallmarks, which promote tumor progression are shown in Figure 4 and detailed below.
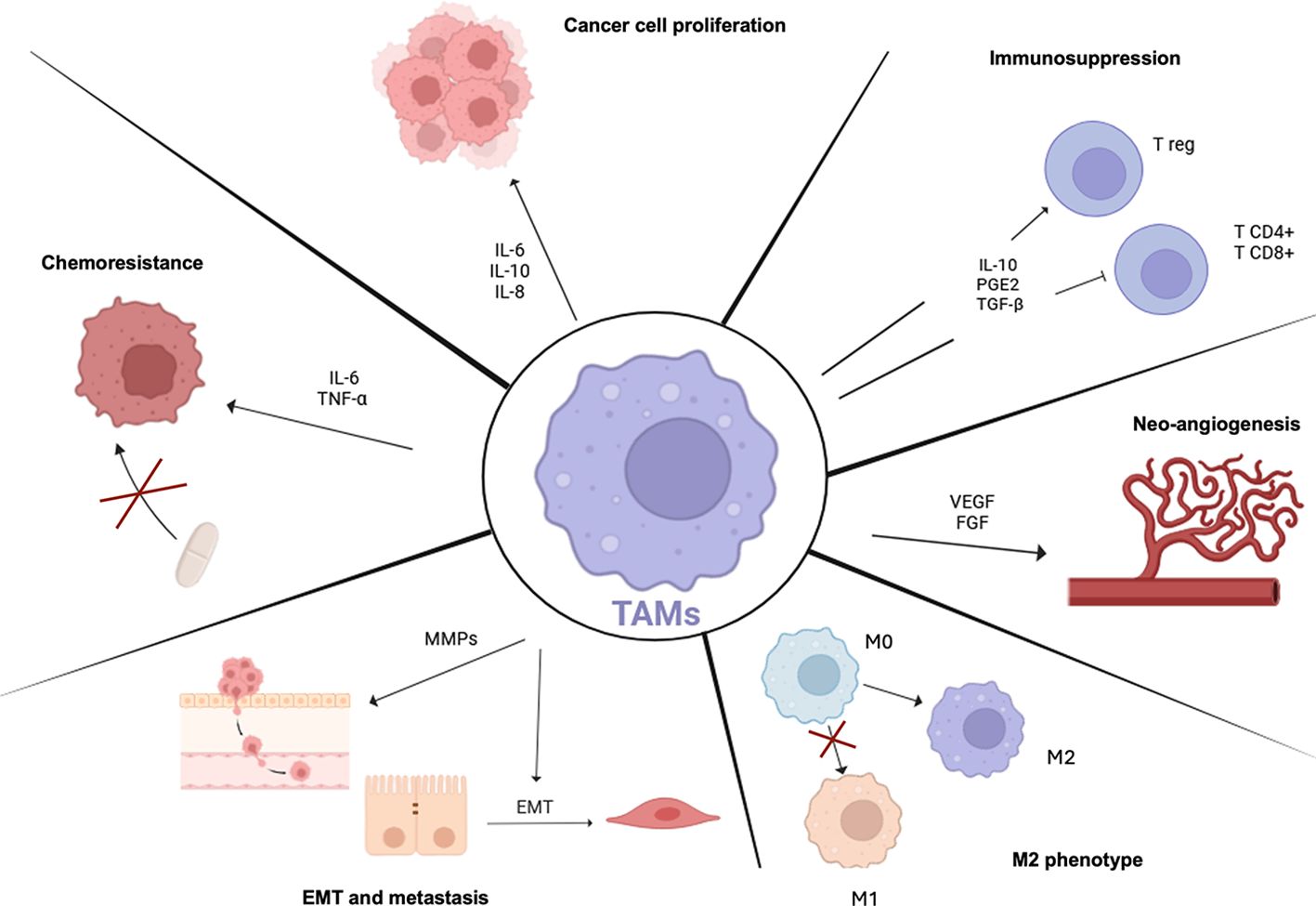
Figure 4. TAMs Hallmarks. Scheme showing TAMs properties in tumor progression. TAMs predominantly show an M2-like state which is mainly linked to pro-tumoral programs. TAMs are involved in many aspects of tumor cell biology such as T lymphocytes immunosuppression and increasing T reg recruitment, supporting tumor angiogenesis through pro-angiogenic factor production, inducing epithelial mesenchymal transition (EMT) and metastasis and promoting resistance to therapy activating pro-survival programs. TAMs, tumor-associated macrophages; IL-6, interleukin-6; IL-10, interleukin-10; IL-8, interleukin-8; Tregs, regulatory T cells; PGE2, prostaglandin E2; TGFB1, transforming growth factor-beta-1; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; EMT, epithelial-mesenchymal transition; MMPs, matrix metalloproteinases; TNF-alpha, tumor necrosis factor-alpha.
Cancer cell proliferation
A key hallmark of cancer is the ability to engage in an intricate communication with tumor cells and by activating proliferative signaling programs (138). TAMs positively support the cancer cell-cycle state by secreting various growth factors and cytokines. Among TAMs released factors, IL-6, IL-10 and IL-8 foster signaling pathways directly involved in stimulating cancer cell proliferation and tumor growth (139–142).
Immunosuppression
TAMs predominantly display an immunosuppressive M2 state in TME (130). M2 TAMs unbalance the immune surveillance role of T cells and favor the promotion of cancer cells escape from the immune system (130). More in detail, TAMs produce a plethora of molecules, such as TGFB1, IL-10, and PGE2 that act on T cells, disrupting the anti-tumoral role both CD4+ and CD8+ subtypes, and increasing the recruitment of Tregs, that enhance the pro-tumoral immune depletion (130, 143).
Chemoresistance
Chemoresistance is another cancer hallmark. TAMs play a crucial role in the acquisition of a cancer chemoresistant phenotype, through the secretion of inflammatory cytokines such as IL-6 and TNF-alpha which activate pro-survival programs (144, 145). Moreover, TAMs can enhance the efflux of chemotherapeutic drugs from cancer cells, reducing their therapeutic efficacy (146).
Neo-angiogenesis
TAMs promote neo-angiogenesis, vital for both tumor growth and metastasis. Accordingly, TAMs release pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), which drive the activation of new blood vessels formation signaling pathways (147, 148). The neo-angiogenesis not only is essential to feed cancer cells with nutrients and oxygen, but also it is critical for tumor mass growth and to guide metastatic spreading process (147).
EMT and metastasis
The pro-invasiveness and pro-metastatic role of TAMs is well-documented in literature (5, 149). Accordingly, TAMs can induce EMT in tumor cells toward a more mesenchymal phenotype, enhancing their more malignant invasive phenotype (149). Furthermore, TAMs can secrete matrix metalloproteinases through which they digest the extracellular matrix components, allowing cancer cells to reach the surrounding tissues (150).
In summary, TAMs significantly impact multiple hallmarks of cancer. Through their roles in sustaining proliferative signaling, immunosuppression, chemoresistance angiogenesis and metastasis, TAMs represent a critical player in all stages of cancer progression, from early to late ones.
CSCs and TAMs crosstalk and its impact on metastatic niche
One of the major drawbacks in counteracting cancer spread and resistance consists of the capacity of CSCs to migrate into secondary sites and avoid immune surveillance (2). Given their plastic behavior, their self-renewal capacity and treatment resistance, CSCs can foster metastasis formation from the primary tumor environment by disseminating into further districts, establishing the metastatic niche (151). In this context, a model that explains the architecture of the niche has been proposed by Lyden et al., in which the CSCs by migrating, reach a permissive and suitable microenvironment, the pre-metastatic niche, and by becoming disseminated tumor cells (DTCs), they can colonize and proliferate (152) through direct competition with normal stem cells for the niche occupation and establishment (153).
The metastatic niche characteristics vary depending on the specific components considered: the interacting cell types, ECM proteins, survival and self-renewal signals, but mostly the secondary site locations, that can either sustain and foster the metastatic niche, or set a hostile environment for the DTCs (154). The DTCs have to face several issues when colonizing a secondary site, including the lack of growth and extracellular matrix remodeling factors, that can hamper their survival and proliferation, thus adjusting into the new niche and metastasize (155). The disseminated cells will shape their surroundings to build a supportive metastatic niche and exploit the functions of both CSCs and metastatic stromal cells (2). However, studies analyzing human colorectal cancer samples displayed that metastatic occurrence arises from primary tumor cells, that are resistant to chemotherapy and might stay quiescent for a prolonged time (156).
Notwithstanding, little evidence investigating the genetic profiling of the tissue-derived and metastatic CSCs emerged, in the consideration of the metastasis signature mutations occurrence at the level of the primary tumor. This process can represent the first tool of selection in the CSCs population to direct a pool toward migration and extravasation into secondary sites (151). With this premises, the most accredited option relies on the fact that metastasis-driving alterations are present within the heterogeneous CSCs profile, and their expression selects the DTCs that will acquire a plastic and resistant profile (157).
Nevertheless, further evaluations need to be carried out, especially in the context of EMT pathways, and stem-like features involved in both the primary tumoral site and in the metastatic environment, highlighting similarities and differences among CSCs and DTCs (158).
Historically, the metastatic niche has been described as a cell-enriched environment constituted mostly by immune and stromal cells which secrete proteins and factors that sustain growth and self-renewal of CSCs, that consequentially stimulate the activation of angiogenic pathways aimed to the promotion of tumor invasion and metastatization (154, 159).
CSCs form the primary tumor can favor the diffusion of pro-tumorigenic and proangiogenic factors such as VEGF-A, TGFB1, TNF-alpha and lysyl oxidase (LOX) that induce the expression of S100A (a Ca2+ binding protein involved in endothelial remodeling) in the metastatic area (2). In the metastatic site the vasculature system boosts the recruitment of metastatic cancer stem cells (MetCSCs) by producing fibronectin and vascular endothelial cell adhesion molecule (VCAM). Consistently, it has been observed that the CCL2-CCR2 (C-C chemokine receptor type 2) axis promotes the establishment of inflammatory monocytes to the metastatic site, where they will transition into metastatic-associated macrophages (MAMs) and will enhance the extravasation and survival of metastasis-resident cancer cells (160).
Another important lead of metastasis formation is depicted by TAMs. Principally, TAMs promote tumor cell invasion and dissemination, and through their ability to release cytokines and factors that support growth and ECM-shaping (MMP-2, MMP-9), milk fat globule-EGF factor 8 (MFGE8), IL-6 are correlated with tumor progression and metastasis (161). TAMs derive from circulating Ly6C+CCR2+ inflammatory monocytes that are produced by hematopoietic stem cells (HSCs) in the bone marrow, that, when interacting with tumor tissue, are addressed toward a more cancerous-like profile (161, 162).
TAMs and CSCs crosstalk has been widely described in the last years, investigating whether their interaction may be direct or indirect, and which may be the effects on CSCs in the primary tumor, including chemoresistance, differentiation and proliferation (163). TAMs are essential in supporting metastasis establishment once CSCs migration has occurred. More specifically, studies conducted on lung and liver metastatic murine models, showed how inhibiting TAM recruitment in metastatic niches resulted in a reduced burden, indicating its paramount role in the onset and maintenance of metastasis by supporting both extravasation and intravasation in secondary sites of CSCs (164, 165).
One accredited metastasis hypothesis linking the role of TAMs in facilitating CSCs metastatization relies on the ability of metastatic cells to occupy niches in which are present CSCs (166). More in detail, it is thought that TAMs and CSCs derive from cell hybrids and set metastasis in further sites (166). The theory was proposed by John Pawelek in 2006, and he explained that myeloid and tumoral cells could perform a genomic hybridization (167). TAMs due to their migratory ability and the tissue-repair feature could transport the CSCs spheroids through either bloodstream or lymphatic circulation, and permit a favorable environment for metastatic initiation (168). Within the metastatic microenvironment, TAMs play a crucial role in shaping the behavior of CSCs, especially regarding tumor advancement and the colonization of cancer cells at secondary sites (169). A crucial aspect of TAMs is their role in promoting EMT, which is a vital process in the morphological alterations of cancer cells and contributes to the enhancement of their malignant traits (170). In triple-negative breast cancer, CCL2 secreted by TAMs activates AKT signaling pathways, resulting in heightened beta-catenin activity in CSCs (171). This pathway is essential for facilitating EMT and sustaining the properties of CSCs within the TME (172). In oral squamous cell carcinoma (OSCC), high levels of TAM-derived IL-6, promote EMT and enhance the expression of genes associated with stemness, via the IL-6/STAT3/thrombospondin 1 (THBS1) signaling pathway (173).
To sustain CSCs in pancreatic ductal adenocarcinoma (PDAC), TAMs utilize a critical mechanism involving the interferon-stimulated gene 15 (ISG15) signaling pathway (174). By releasing the ISG15, TAMs enhance the self-renewal, invasive potential and tumorigenic capabilities of CSCs (175). Among the several ways in which TAMs support CSCs behavior, the creation of an immunosuppressive microenvironment exerts a key function. Within the TME, TAMs predominantly display a M2 phenotype, which is known for its role in promoting immunosuppression (176). This phenotype fosters a protective microenvironment that shields CSCs from immune system attacks. By releasing immunosuppressive cytokines like IL-10 and TGFB1, TAMs effectively suppress the function of cytotoxic T cells and other immune cells, allowing CSCs to remain undetected and avoid destruction (177). TAMs influence the growth of CSCs through both direct contact and secretory mechanisms. In highly metastatic breast cancer, CSCs express hyaluronan synthase 2 (HAS2), which is crucial for creating a pro-metastatic microenvironment (177). This expression facilitates interactions between CSCs and TAMs, leading to the secretion of platelet-derived growth factor-B subunits (PDGFB) by TAMs (177).
PDGFB subsequently stimulates bone stromal cells to secrete fibroblast growth factors 7 and 9 (FGF7 and FGF9), which support CSC proliferation and survival (178). Moreover, in breast cancer, the EMT enhances the expression of cluster of differentiation 90 (CD90) and ephrin type-a receptor 4 (EPHA4), facilitating direct physical interactions between CSCs and TAMs through the binding with their respective receptors. When the EPHA4 receptor on carcinoma cells is activated, it triggers the sarcoma SRC proto-oncogene, non-receptor tyrosine kinase (SRC) and nuclear factor- kappa B (NF-kappa-B) signaling pathways. This activation leads to NF-kappa-B in CSCs induction of the secretion of various cytokines that help maintaining the stem cell state (179).
By preserving the stem-like properties of CSCs and boosting their migratory and invasive abilities, TAMs facilitate the detachment of CSCs from the primary tumor, enabling the formation of secondary tumors in distant organs (149).
Once malignant cells escape from the primary tumor, they intravasate and disseminate through the lymphatic and/or circulatory system, eventually establishing secondary tumors at distant sites. Research into lung metastasis reveals that when tumor cells reach their target location, they form micro-clots in conjunction with platelets, resulting in their entrapment within the blood vessels of the target tissue (180). Once arrested, the tumor cells secrete CCL2, which creates a gradient that attracts Ly6C monocytes (181). These recruited monocytes undergo differentiation into MAMs, which play a pivotal role in facilitating the extravasation of tumor cells by releasing VEGF, a factor known to enhance vascular permeability (182). Under the influence of CSF1, the primary lineage regulator for most macrophage populations, MAMs support the survival of tumor cells and contribute to their sustained growth through processes related to angiogenesis (183).
Recent studies conducted on CRC evidenced the interaction of CRC cells and TAM. Of note, a paramount interaction between CRC cells and M2 macrophages in the promotion of colorectal liver metastasis (CRLM) emerged (184). To date, CRLM is mediated by interactions between tumor cells and the TME in the liver and is considered one of the most common secondary liver cancers (185). Nevertheless, the mechanisms involved in the cancer cell-derived activation of M2 macrophages need further investigations in both CRC and CRLM. Notably, exosomes derived from tumors can polarize macrophages toward a M2 cellular profile, which in turn promotes metastasis (186, 187). Zhao et al, demonstrated that exosomes derived from CRC cells displayed a role in inducing M2 polarization through the secretion of microRNA-934 (miR-934) and the downregulation of PTEN expression, and activation of PI3K/AKT signaling cascade. Finally, miR-394 activated polarized M2 macrophages which promoted CRLM through C-X-C motif chemokine ligand 5 and 13 (CXCL5)/(CXCL13)/NF-kappa-B/p65/miR-394 positive feedback mechanism (188).
Another study (189) conducted on glioblastoma investigated the role of glioblastoma stem cells (GSCs) and TAMs in tumor progression and metastatic potential. The authors screened GSCs factors that could polarize macrophages, and they evaluated a potential group of proteins produced by GSCs with the ability of behaving as TAMs chemoattractant (189). Periostin (POSTN) emerged as a valuable factor expressed by the stem cells (189). It plays a role in the PI3K/AKT and WNT signaling pathways, which are involved in tumorigenesis (190, 191). In particular, evidence highlighted that CSCs profited from the POSTN-induced WNT augmented signaling, supporting a favorable metastatic colonization in breast cancer setting (192). Additionally, when silencing POSTN, TAM density was sensibly reduced, thus reinforcing the idea that GSCs can recruit TAMs and foster tumor growth by secreting POSTN. Consistently, GSCs established in the tumoral area, where they exploited the surrounding microenvironment by attracting TAMs from the peripheral circulation to set a more beneficial space for the reciprocal survival and growth of the resident populations and enhancing the metastatic CSCs potential. These observations need further investigation and open new scenarios regarding the involvement of TAMs, the role of CSCs, and their complex interplay in affecting the metastatic niche.
TAMs influence on tumor behavior, oncogenic pathways, immune inhibitory responses and therapy resistance
TAMs represent a cellular immune system subpopulation directly involved in the tumor formation and progression through the activation of several pro-tumoral signaling pathways within the CSCs, thus providing the creation of a tumor niche necessary for CSCs survival and expansion. Cellular matrix elements represent critical components for the tumor niche structure maintenance which help the direct crosstalk between CSCs and the surrounding cells, including TAMs. The intricate bi-directional communication between TAMs and CSCs is increasingly recognized as a critical factor in tumor biology. This interaction is underscored by a growing list of factors, ligands/receptors, shown in Table 1, derived from both TAMs and CSCs that are implicated in the mutual co-dependent maintenance of CSC stemness and the supportive actions of TAMs. The complex network of signaling molecules and pathways involved in this crosstalk not only influences tumor progression but also impacts therapeutic resistance, making it a focal point for cancer research.
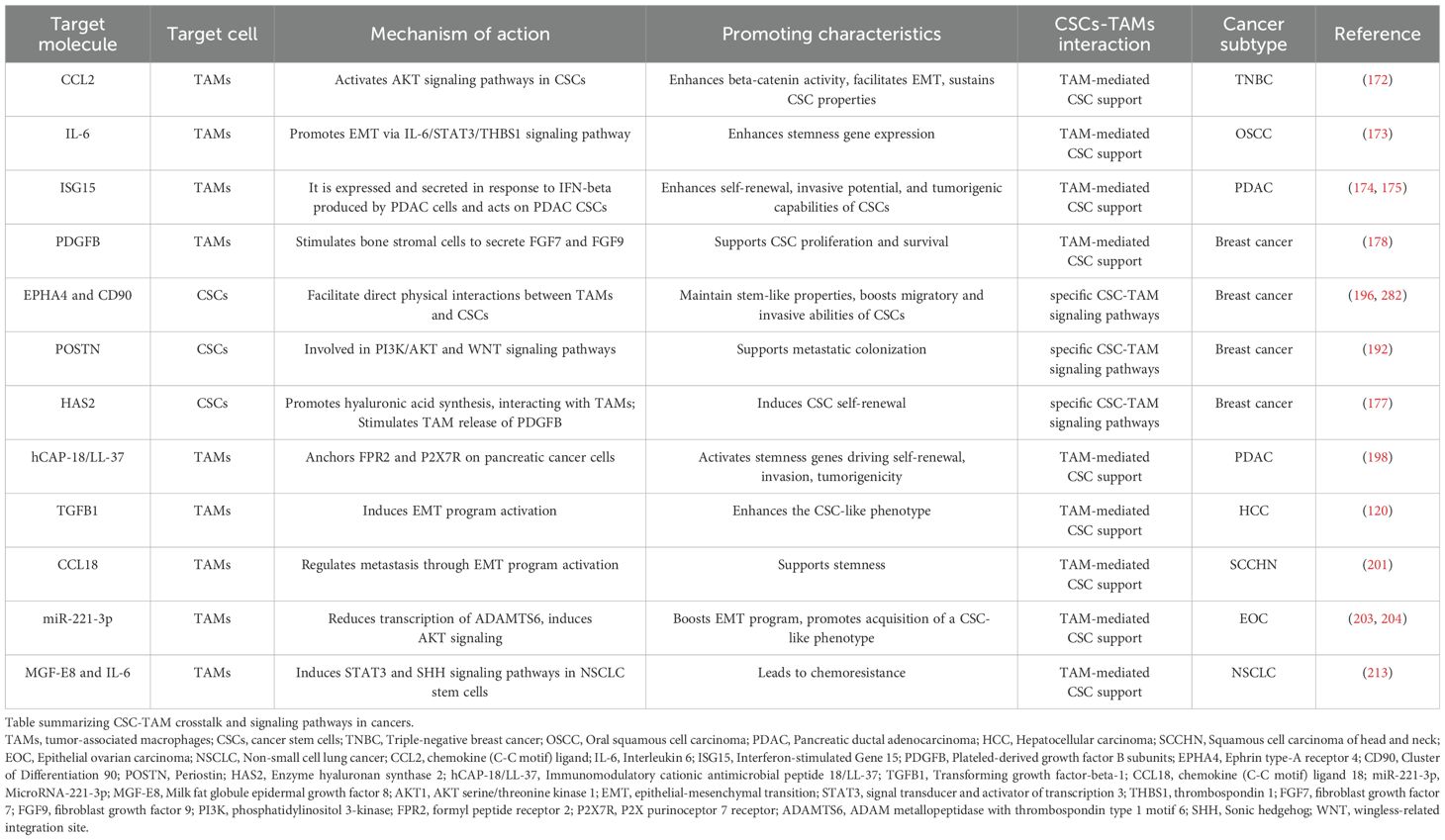
Table 1. CSCs-TAMs crosstalk and signaling pathways in cancers.
In breast cancer stem cells (BCSCs) the overexpression of the HAS2 is implicated in the new synthesis of hyaluronic acid, a major polysaccharide component of the ECM which drives the physical interaction to TAMs, via CD44 receptor expressed on their surface. The hyaluronic acid/CD44 interaction stimulates TAMs to release the growth factor PDGFB, which induces CSC self-renewal (178, 193). In addition, TAMs/CSCs in vitro co-culture confirmed the oncogenic role of hyaluronic acid-expressing CSCs/CD44-TAMs interaction in the activation of different signaling pathways such as PI3K–Eukaryotic Translation Initiation Factor 4E Binding Protein 1 (EIF4EBP1)–SOX2, implicated in CSCs pool maintenance (194, 195). Interestingly, it has been found that BCSCs cooperate directly with TAMs through cluster of differentiation 11b (CD11b) and CD90 binding. This anchoring stimulates EPHA4 receptor-mediated induction of both the NF-kappa-B and SRC signaling pathways ensuring CSCs pool stemness state (196). Similarly, in a triple negative breast cancer (TNBC) model, the butyrophilin subfamily member A3 (BTN3A3) receptor enhances cancer stemness markers (i.e. NANOG, OCT4, SOX2) via juxtacrine interaction with its ligand, liver and lymph node sinusoidal endothelial cell C-type lectin (LSECtin), a transmembrane protein expressed on TAMs surface (197). Furthermore, CSCs engage a juxtacrine signaling pathway with the TAMs via GPI-anchored protein CD90/CD11b. Specifically, CSCs express the membrane GPI-anchored protein CD90 and EPHA4. Mechanistically, CD90 creates a bridge to bind the integrin CD11b on TAM surface, whereas the receptor EPHA4 interacts with its ligand, Ephrin, expressed by TAMs, inducing the expression of both SRC and NF-kappa-B driving tumor progression and metastatic dissemination (196). In pancreatic cancer, immunomodulatory cationic antimicrobial peptide 18/LL-37 (hCAP-18/LL-37) on TAM, anchors the formyl peptide receptor 2 (FPR2) and the P2X purinoceptor 7 receptor (P2X7R) expressed on pancreatic cancer cells, which lead to the activation of stemness genes (i.e. KLF4, SOX2, OCT3/4 and NANOG) driving CSC self-renewal, invasion, tumorigenicity (198).
Different studies showed that an indirect paracrine interaction between TAMs and CSCs, driven by a plethora of inflammatory molecules including cytokines, chemokines, growth factors, was also crucial in the determination of CSCs fate and behavior. Particularly, IL-6 is one of the most representative pro-inflammatory cytokines in the context of TME. It is critically upregulated in many tumors, underlying the strong correlation between inflammatory stimuli and tumor progression by affecting multiple cancer signaling pathways (199). IL-6 derived from TAMs induces the proliferation of CD44+ Human Hepatocellular Carcinoma Stem Cells (HHCSCs) via STAT3 pathway induction (139). In addition, TAMs secrete high levels of IL-6 increasing stemness markers (i.e. SOX2, OCT3/4 and NANOG) and consequently CSCs expansion in breast cancer cells via STAT3 pathway supporting tumor cells migration and angiogenesis (125).
Paracrine communication mechanisms between TAM and CSCs are driven by several molecules. TAMs can enhance the CSC-like phenotype via TGFB1, which induces EMT program activation in a hepatocellular carcinoma (HCC) (120). Similarly, TAMs induce stemness, EMT and chemoresistance in HCC by realizing TNF-alpha via the WNT/β-catenin axis (200). It has been discovered that TAMs can produce Chemokine (C-C motif) ligand 18 (CCL18). In squamous cell carcinoma of the head and neck model (SCCHN), CCL18 produced by TAMs regulates metastasis through the activation of EMT program and cancer stemness (201). TAMs releasing CCL2 is correlated with worse prognosis in breast cancer. Particularly, TAM-produced CCL2 in the context of breast cancer microenvironment activates AKT/beta-catenin signaling resulting in EMT and CSC properties in TNBC (172).
Exosomes derived from TAMs have shown unrevealed aspects about the role of TAMs in the support of cancer progression. Specifically, it has been found that annexin A3 (ANXA3)-loaded exosomes derived from TAMs impaired ferroptosis process in laryngeal cancer cells supporting lymphatic metastasis. More in detail, ANXA3 in exosomes regulates negatively the ubiquitination of activating transcription factor 2 (ATF2), a transcription factor that induces ChaC Glutathione Specific Gamma-Glutamylcyclotransferase 1 (CHAC1) expression, thus blocking ferroptosis in lung squamous cell carcinoma (LSCC) cells (202). Moreover, CD163+ TAMs release exosomes that are absorbed by epithelial ovarian cancer cells (EOCCs) (203, 204).
During the tumor progression, TAMs can create an immunosuppressive TME facilitating the immune escape of CSCs. The creation of an immunosuppressive milieu depends on a fine balance between the inhibition of pro-inflammatory immune cells and the activation of immunosuppressive TAMs-dependent counterparts. Accordingly, TAMs promote the upregulation of cluster of differentiation 47 (CD47) ligand on different cancers stem cells (including pancreatic, HCC and leukemia), which interacts to signal-regulatory protein alpha (SIRPA) on immune cells inhibiting phagocytic process (205–207). Parallelly, TAMs can also inhibit the adaptive immune system. Particularly, TAMs boost both inhibitor immune checkpoints expression PD-1 and its ligand PD-L1 in T cells and CSCs, respectively (208). The concomitant expression of PD-L1 and PD-1 impedes the cytotoxicity in T-cells (208).
Overall, some evidence showed how TAM-derived factors and TAM-CSCs physical interactions drive the activation of a great number of pathways in CSCs that are responsible of the maintenance of stemness in different cancer histotypes. These stemness-related hallmark pathways include Sonic hedgehog (SHH), STAT3, NOTCH, PI3K/AKT, WNT/beta-catenin, and NANOG (18). Particularly, TAMs induce STAT3 pathway regulating the expression of stemness genes, via NF-kappa-B activation, in CSCs in different malignancies including breast cancer, liver cancer, prostate cancer, pancreatic cancer and colon cancer (139, 209–213). TAMs activate WNT/beta-catenin and SHH pathways, in CSCs, by leading transcriptional activation of stemness related genes in liver cancer, prostate cancer and lymphoma after secreting TNF-alpha, CCL5, pleiotrophin respectively (200, 211, 214). Furthermore, TAMs support cancer stemness through the direct activation of SHH pathway or through the induction of stemness-related alternative pathways (196, 213, 215–217). Specifically, TAMs sustain stemness via direct activation of SHH pathway in colon cancer (213), meanwhile SHH alternative signaling pathways are TAM-induced in pancreatic cancer (TGFB1/SMAD2/SMAD3/NANOG pathway) (215), in liver cancer (via the NOTCH pathway) (216), breast cancer (via the SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase (SRC) pathway) (196), and in glioma via extracellular regulated kinase 1/2 (ERK1/2) pathway (217).
Innovative studies indicate that the complex communication between CSCs and TAMs has a critical pivotal role in the acquisition of a chemoresistant phenotype refractory to anticancer therapies. In OSCC TAMs influence positively the formation of CSC-like cells, via the induction of stemness markers of the SOX2, OCT4, and NANOG genes, leading to a strong reduction of the percentage of apoptosis in OSCC, supporting cell migration and chemoresistance to vincristine (218). Similarly, TAMs release Pleiotrophin (PTN), which interacts with the protein tyrosine phosphatase receptor type Z1 (PTPRZ1) receptor on the surface of CSCs, in OSCC model. The ligand/receptor interaction activates the FYN proto-oncogene (FYN)-AKT pathway, sustaining both the expression of stemness characteristics in CSCs and chemoresistance in tumor cells (219). Furthermore, MFGE8 in cooperation with IL-6, from TAMs induces both STAT3 and SHH signaling pathways in non-small cell lung cancer stem cells (NSCLCCSCs) leading to chemoresistance (213). Despite a growing body of research that has elucidated various molecular mechanisms underlying the interactions between TAMs and CSCs, significant gaps in our understanding remain. The intricate crosstalk between these two cellular populations is a complex phenomenon that has not yet been fully characterized.
An overview of the most significant mechanisms of indirect and direct interaction between TAMs and CSCs are shown in Figure 5.
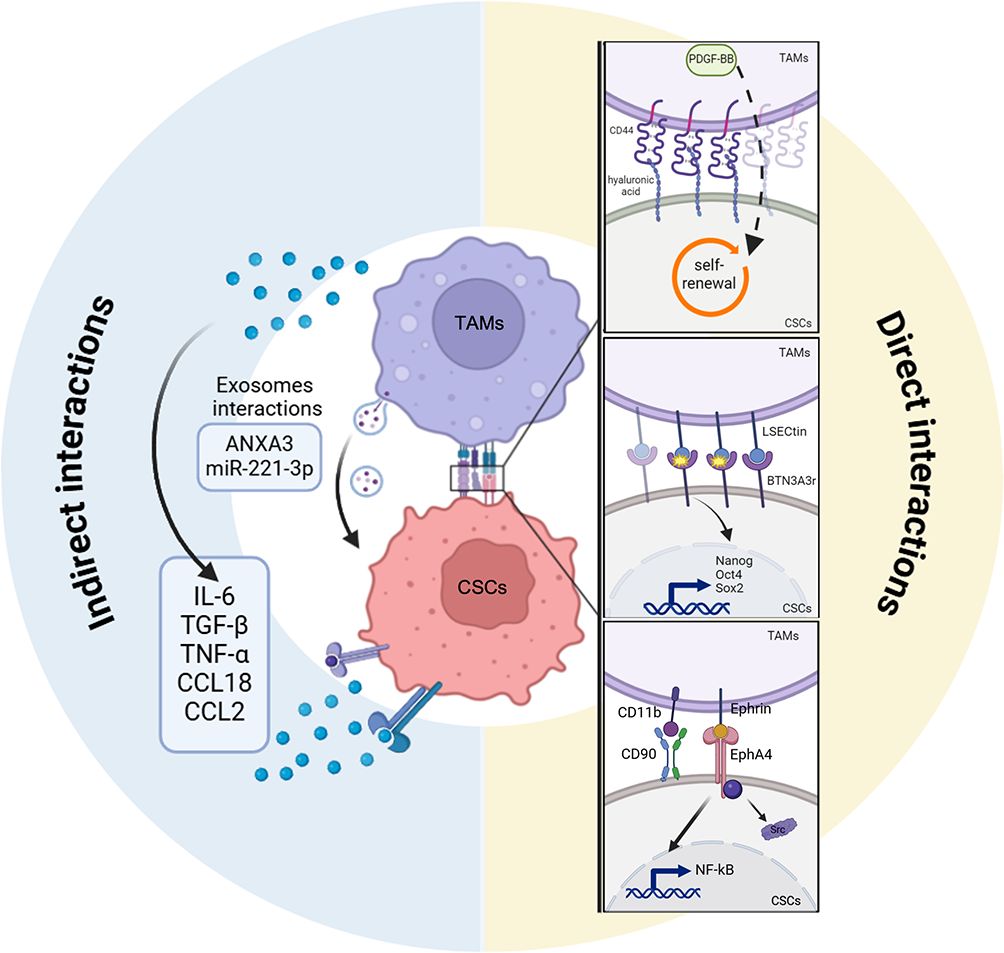
Figure 5. Indirect and direct interactions between CSCs and TAMs. Scheme showing the indirect (left) and the direct (right) mechanisms of crosstalk between tumor associated macrophages (TAMs) and cancer stem cells (CSCs). CSCs directly regulate TAMs activity to improve their own stemness conditions through different ligand/receptor interactions (hylaronic acids/CD44, BTN3A3r/LSECtin, CD11b/CD90, Ephrin/EPHA4. TAMs secretome including cytokines (IL-6, TGF- β, TNF-alpha, CCL18, CCL2) or exosomes cargo (ANXA3, microRNA-221-3p or miR-221-3p) promotes, indirectly, CSCs stem-like state by activating CSCs stemness programs. TAMs, tumor-associated macrophages; CSCs, cancer stem cells; CD44, cluster of differentiation 44; BTN3A3, butyrophilin subfamily member A3; LSECtin, liver and lymph node sinusoidal endothelial cell C-type lectin; OCT4, octamer-binding transcription factor 4; SOX2, SRY-Box transcription factor 2; CD90, cluster of differentiation 90; CD11b, cluster of differentiation 11b; EPHA4, ephrin type-a receptor 4; Src SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase; NF-kappa-B, nuclear factor-kappa B; IL-6, interleukin 6; TGFB1, transforming growth factor-beta-1; TNF-alpha, tumor necrosis factor-alpha; CCL18, chemokine (C-C motif) ligand 18; CCL2, chemokine (C-C motif) ligand 2; ANXA3, annexin A3.
Therapeutic strategies targeting the interactions between CSCS and TAMS to improve cancer treatment outcomes
The innovative targeting of the crosstalk between TAMs and CSCs represents a promising frontier in cancer therapy, although several strategies have been already developed to specifically target the CSC subpopulation including differentiative agents, chimeric antigen receptor T cell (CAR-T) therapy, natural compounds and epigenetic inhibitors (56, 220–225). This interaction is crucial as TAMs can enhance the stemness and survival of CSCs, contributing to tumor progression and resistance to conventional treatments (163). Nowadays the aim is to disrupt this communication, for the development of more effective therapeutic strategies that could potentially improve cancer patient prognosis. A therapeutic strategy could be represented by the disruption of CSC-TAM communication centers by blocking soluble factors that reciprocally support each cell type.
IL-6 is an important regulator in paracrine communication between TAMs and CSCs (125, 139). The IL-6 downstream pathway can be unpaired by both anti-IL-6, interleukin-6 receptor (IL-6R) antibodies and by STAT3 inhibitor pathway. Inhibitors against TGF-beta pathway are crucial to target CSCs (226, 227). Additionally, it has been discovered that IL-6 inhibition can impair MFGE8 functionality, which sustains CSC phenotype and cancer chemoresistance (213). Notably, the anti-IL-6R, tocilizumab, has been approved by the FDA for treating rheumatoid arthritis (228). It is currently in phase II clinical study for the treatment of unresectable late-stage melanoma in combination with the anti PD-1 and anti CTLA-4 immune checkpoint inhibitors nivolumab and ipilimumab (NCT03999749) (229). IL-8 is another important TAM-secreted regulator in cancer stemness (230). Reparixin is an anti-IL-8 receptor (IL-8R), known as CXCR1, that reduces CSC population in breast cancer setting (231). Phase I clinical trial study NCT02001974 showed that Reparixin provides a synergistic effect in combination with paclitaxel (231). The inhibition of the glioblastoma multiforme (GBM) CSC-released POSTN has shown a significant reduction in TAMs recruitment in pre-clinical glioblastoma model xenografts (189). In addition, Huang et al. demonstrated that TAMs-secreted CCL5 inhibition could impair stemness and metastasis formation in in pre-clinical prostate model xenografts (211). An alternative targeting strategy is to re-educate the biological role of TAMs toward an anti-tumor phenotype. Specifically, it has been demonstrated that dasatinib inhibitors, directed against SRC, drive the reprogramming from TAMs to M1 anti-tumor macrophages affecting the SRC/cluster of differentiation 155 (CD155)/macrophage inhibitory factor (MIF) signaling (232). This leads to downregulation of stemness markers, NOTCH1 and beta-catenin in cisplatin-resistant lung cancer cells (232).
The reactivation of phagocytic activity in anti-tumoral macrophages toward dead tumor cells represents a really important resource for obtaining cancer cells antigens to boost T cell-mediated immune responses. Accordingly, macrophage phagocytosis can be restored via anti- CD47 administration in immunodeficient pre-clinical xenograft models (233–236). Particularly, anti-CD47 antibodies are currently being designed in clinical trials (NCT02216409, NCT02367196) to overcome the phagocytosis-driven CD47+ TAMs/SIRPA+ CSCs inhibition with promising results (235, 237).
Interestingly, pre-clinical models showed a strong synergism between anti-CD47 and chemotherapies (i.e. paclitaxel, cyclophosphamide) in triggering T cell responses in immunogenic colon and lymphoma tumors (238). ALX148, a CD47 blocking protein, displayed high efficacy in combination with anti-PD-1, anti-human epidermal growth factor 2 (HER-2), anti-vascular endothelial growth factor receptor 2 (VEGFR-2) and anti-CD20 antibodies (known as pembrolizumab, trastuzumab, ramucirumab, rituximab respectively) and conventional chemotherapy (Paclitaxel, fluorouracil, cisplatin) in patients with malignant solid tumor and Non-Hodgkin Lymphoma (NCT03013218) (239).
Humanized IgG4 antibody (Hu5F9-G4), an anti-CD47 antibody, showed combinatorial effect with chemotherapy azacitidine in leukemia stem cells (NCT03248479) (240).
Zoledronic acid represents a double effects drug affecting both TAMs in liver cancer infiltration and decreasing tumor growth in CSCs-derived cervical cancer (241, 242). Zoledronic acid has been chosen for phase III clinical trials aiming at the prevention of bone metastasis in late-stage lung cancer patients (NCT02622607).
Of note, another innovative target is represented by myeloid-epithelial-reproductive tyrosine kinase (MERTK), a tyrosine kinase receptor discovered both in TAMs and several malignancies. MERTK, on TAMs surface, binds to the “eat-me” signal presented on apoptotic cells, activating a biological process known as “efferocytosis”. It drives the shift of macrophages to the pro-tumoral immunosuppressive M2 phenotype (243). MERTK is also overexpressed in cancer cells and is directly correlated to CSC maintenance in glioblastoma multiforme (244). The block of the MERTK signaling pathway represents a promising therapeutic strategy able to have a bidirectional effect both on TAMs and CSCs. Additionally, the administration of the agonist anti-CD40 regulates the activation of the TAM receptor CD40. Anti-CD40 mimics the homonymous ligand physiologically produced by T cells, and it leads to the reprogram of TAMs into anti-cancer macrophages with the establishment of immune surveillance (179, 245, 246). Accordingly, NG-350A, an adenoviral vector encoding for an anti-CD40 monoclonal antibody directed against tumor cells has been used to remodel the immunosuppressive TME. Interestingly, an ongoing phase I trial is investigating its systemic intravenous infusion alone or as a combinatorial treatment with pembrolizumab (NCT05165433) or chemoradiotherapy/radiotherapy (NCT06459869) in patients with advanced epithelial tumors, in particular locally advanced rectal cancer (LARC) (247). Lastly, TAMs reprogramming involves different specific biological sensors for ectopic nucleic acids such as (stimulator of interferon response cGAMP interactor (STING) and some members of toll-like receptors family (TLRs), such as TLR3, TLR7 and TLR8. The design of several synthetic compounds, which regulate these receptors on TAMs endosomal compartments, induces the activation of NF-kappa-B signaling and the consequent release of several immunostimulatory cytokines, including type I interferon (IFN-1), the master regulator of anti-cancer immunity (248–250).
As discussed above, TAMs can create an immunosuppressive TME to facilitate CSCs spreading and progression. Accordingly, the specific TAMs Inhibitor of DNA Binding 1 (ID1) + subpopulation can interact with STAT1 to localize it in the cytoplasm and inhibiting its nuclear translocation for Plasminogen activator inhibitor 2 (SERPINB2) and CCL4 transcription (251). These two factors are responsible for cancer stemness inhibition and CD8+ T cell recruitment (251). Shang et al. demonstrated that ML323 administration reduced ID1 affecting CSCs and increasing CD8+ T cells infiltration (251). In addiction ML323 treatment showed a synergistic effect with both anti-CTLA-4 antibody and 5-fluorouracil (5-FU) alone and in combination, in a colon cancer preclinical model (251). Despite the efforts made in researching therapeutic treatments to address the complex communication between TAMs and CSCs, much remains unresolved and requires further investigation and studies. The main preclinical models and clinical trials targeting CSCs-TAMs axis are summarized in Tables 2, 3, respectively.
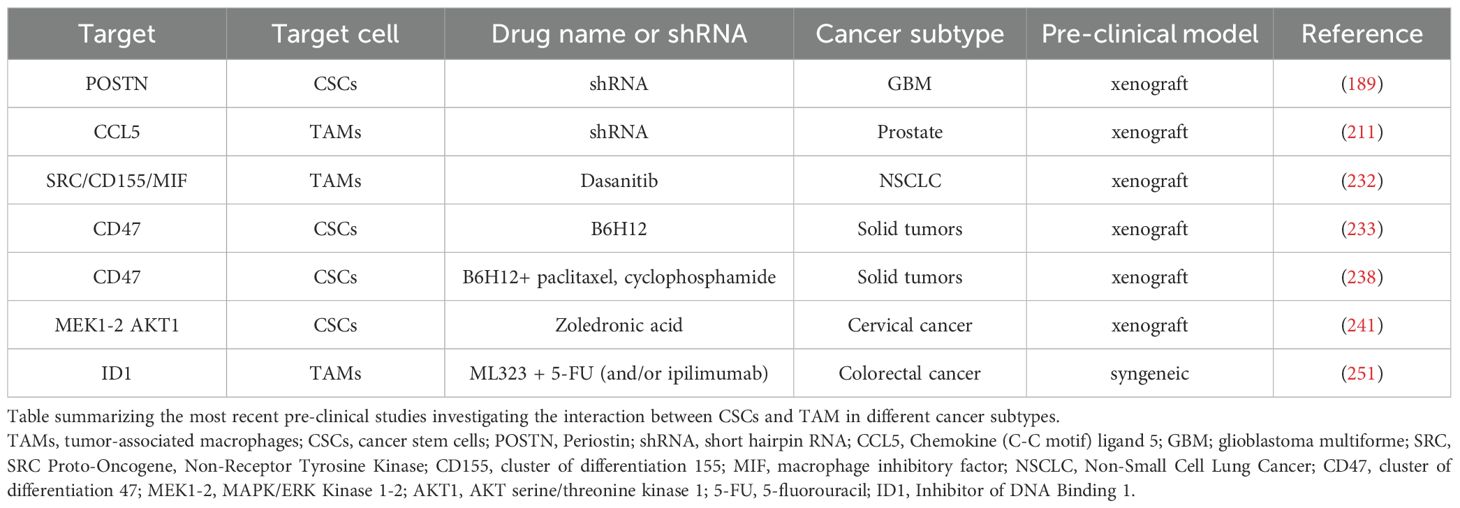
Table 2. Preclinical studies targeting CSCs-TAMs interactions.
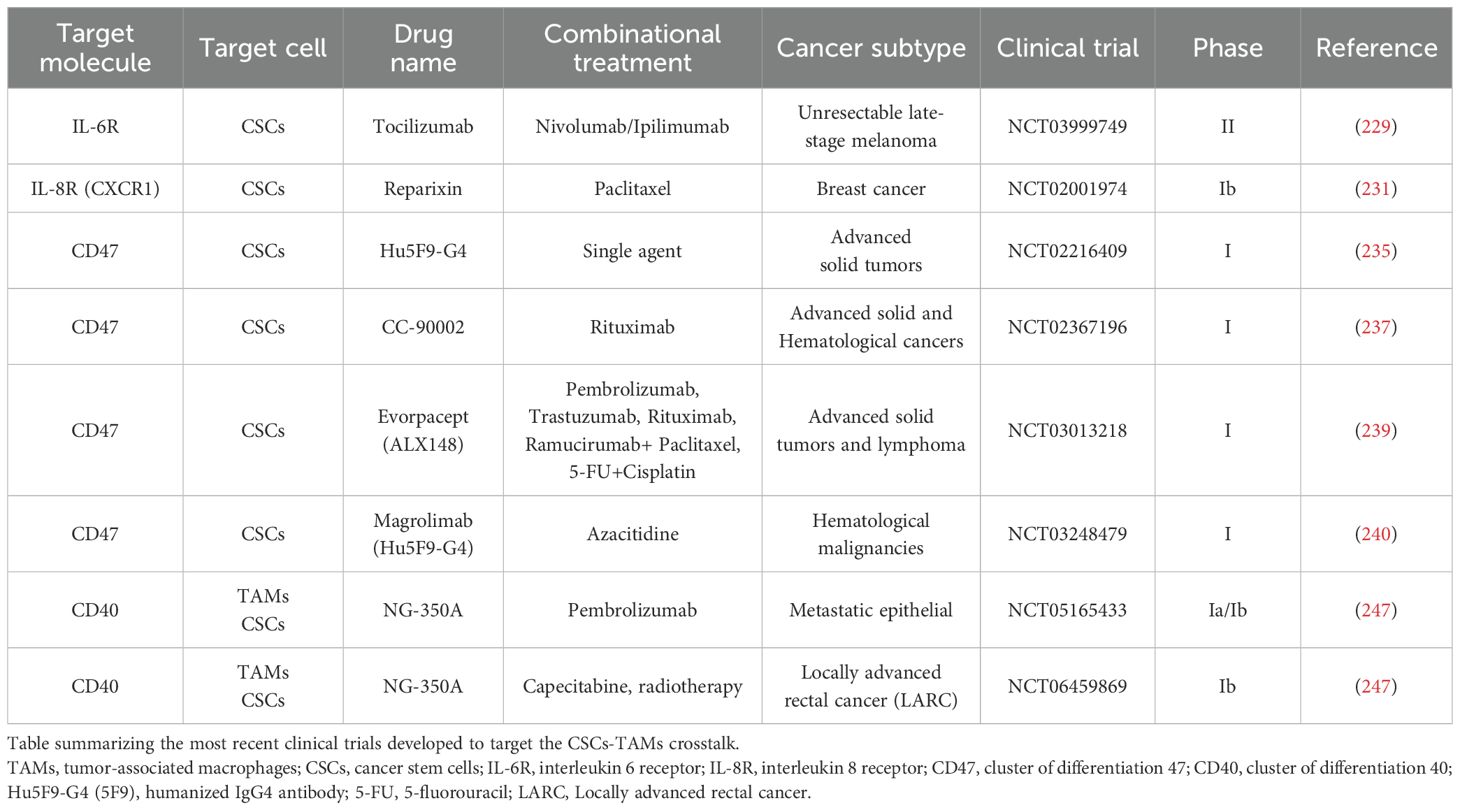
Table 3. Clinical trials targeting CSCs-TAMs interactions.
Advanced bioinformatics techniques based on single-cell and spatial transcriptomics focused on dissecting the role of TAMS
In the next paragraphs we summarize recent discoveries, enabled by advanced bioinformatics techniques including single-cell RNA-sequencing (scRNA-seq), spatial transcriptomics and trajectories analyses, to study TAMs in cancer progression (252). These techniques have provided crucial insights into the interactions in the TME, highlighting the pivotal role of TAMs in promoting cancer progression, influencing tumor growth, metastasis, and modulating therapeutic responses (252). TAMs exhibit functional plasticity, adopting pro- or anti-tumorigenic roles depending on environmental cues (252). The integration of scRNA-seq and spatial transcriptomics, has facilitated the dissection of TAM trajectories, signaling pathways, and their interactions with other TME components, including CSCs the dissection of TAM trajectories, signaling pathways, and their interactions with other TME components, including CSCs (252). Recent advances have revealed the dynamic interplay between TAMs and CSCs (253). The plasticity of TAMs, influenced by factors such as cytokines, chemokines, and direct cellular interactions, plays a key role in tumor dynamics (252). This review focuses on the transformative impact of bioinformatics in understanding TAM trajectories and signaling within the TME, with an emphasis on their potential for novel therapeutic interventions (252). These bioinformatics techniques are great tools for analyzing all kinds of cells, but in this review, we will focus on applications and studies for the role of TAMs. An overview regarding the bioinformatic tools to specifically study TAMs is reported in Table 4.
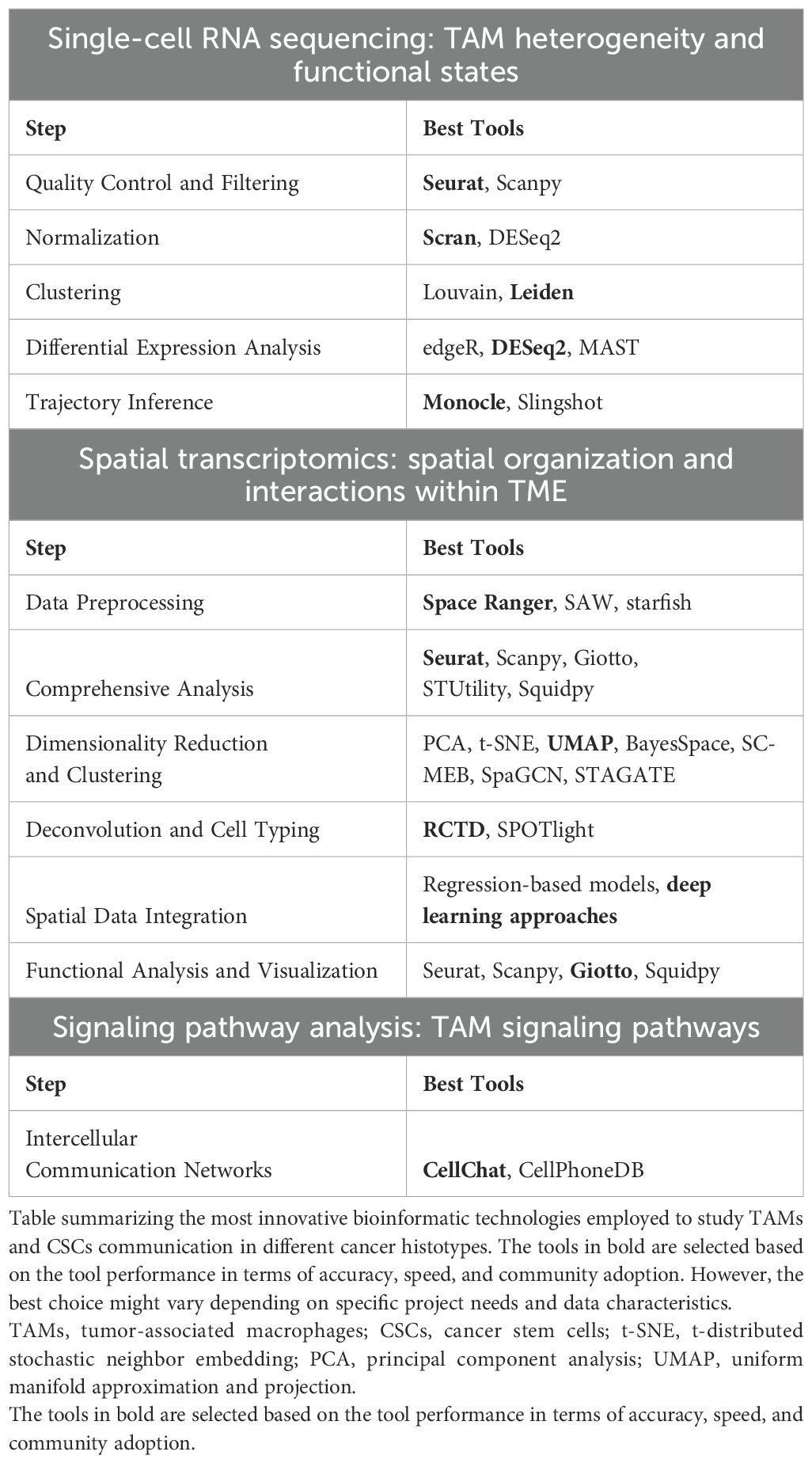
Table 4. Bioinformatic tools to study TAMs.
Single cell RNA-seq
scRNA-seq has emerged as a pivotal tool for dissecting the heterogeneity of TAMs within the TME. This technology allows the analysis of gene expression at the resolution of individual cells, providing unprecedented insights into the distinct subpopulations of TAMs and their functional states. Numerous studies have highlighted significant variations in the transcriptional profiles of TAMs across different tumor types, underscoring their role in modulating the immunosuppressive landscape of the TME (133). The application of scRNA-seq in cancer research has revealed the coexistence of TAMs with pro-tumor (M2-like) and anti-tumor (M1-like) phenotypes within tumors. This duality emphasizes the functional plasticity of TAMs in cancer progression. Recent advancements have utilized scRNA-seq to trace the developmental trajectories of TAMs, identifying key signaling pathways that regulate their polarization and function. Valdes-Mora et al. demonstrated the utility of high-throughput scRNA-seq for analyzing thousands of tumor cells, including TAMs, revealing transcriptional programs associated with different TAM states, further elucidating their roles within the TME (254).
To fully leverage scRNA-seq for TAM characterization, several bioinformatics methodologies are employed (252):
Quality Control and Filtering: Tools such as Seurat (255) and Scanpy (https://scanpy.readthedocs.io/en/stable/) are commonly used to filter low-quality cells based on the number of detected genes and mitochondrial content.
Normalization: Normalization of scRNA-seq data is essential for accurate downstream analysis. Techniques like Scran or DESeq2 (256) provide effective approaches for normalization.
Clustering: To identify distinct cell populations, clustering algorithms like Louvain or Leiden are employed, allowing for robust community detection in high-dimensional datasets (257).
Differential Expression Analysis: To uncover differences in gene expression across TAM subpopulations, tools such as edgeR (258), DESeq2 (256), or MAST (259) are frequently used, depending on the analysis framework.
scRNA-seq has provided groundbreaking insights into the transcriptional diversity and functional heterogeneity of TAMs across various cancer types (260). This approach has enabled the identification of distinct TAM subtypes, each contributing differently to tumor immunity and progression. Specifically, studies in breast cancer have delineated M1-like and M2-like TAM populations, revealing their unique roles in promoting or inhibiting tumor growth (261, 262). This emerging knowledge is crucial for the development of targeted therapies aimed at reprogramming TAMs to a more anti-tumor state, offering new avenues for therapeutic intervention in cancer. scRNA-seq represents a transformative approach in TAM research, providing high-resolution profiling of individual TAMs and enabling the identification of diverse subpopulations based on their gene expression profiles. By combining scRNA-seq with advanced bioinformatics tools, researchers can uncover the full spectrum of TAM heterogeneity and its implications for cancer progression and therapy. This method excels at revealing transcriptional heterogeneity and elucidating the cellular and molecular mechanisms underlying TAM function within the TME. However, it requires tissue dissociation, which disrupts the spatial organization of the tumor microenvironment and leads to a loss of spatial information. This limitation prevents a direct understanding of TAM interactions within their native tissue context, which is crucial for fully characterizing TAM functionality in relation to the TME.
Spatial transcriptomics for TAM trajectories
While scRNA-seq has provided significant insights into the heterogeneity of TAMs, it lacks spatial resolution, which is crucial for understanding their interactions within the TME. Spatial transcriptomics bridges this gap by integrating gene expression data with spatial information, allowing for the precise mapping of TAM distribution and organization within tumor tissues. This spatial context is essential for capturing the complexity of TAM interactions with other cell types and their influence on tumor progression. Spatial transcriptomics has been extensively applied to study the spatial dynamics of TAMs across various cancer types. It has been demonstrated that TAMs located within the tumor stroma and at invasive tumor margins exhibit distinct gene expression profiles and functional states, which play a pivotal role in driving tumor progression and metastasis (263). By integrating spatial transcriptomics with scRNA-seq data, researchers can gain a more comprehensive understanding of TAM trajectories and their interactions with other cells in the TME. Recent developments in bioinformatics have facilitated the analysis of spatial transcriptomics data, from preprocessing to functional interpretation.
These advancements include:
Data Preprocessing: The initial steps of spatial transcriptomics analysis involve generating a gene expression matrix along with spatial coordinates. Tools such as Space Ranger (10X Genomics), SAW (Stereo-seq), and starfish (ISS/ISH) are widely used for data preprocessing, depending on the platform and methodology employed (264).
Comprehensive Analysis Tools: Seurat (255) and Scanpy (https://scanpy.readthedocs.io/en/stable/) are versatile tools frequently used for both scRNA-seq and spatial transcriptomics analysis. These platforms offer functionalities for filtering, normalization, and various downstream analyses. For more specialized spatial transcriptomics tasks, Giotto (265), STUtility (266), and Squidpy (267) provide extended capabilities, including advanced spatial analyses (264).
Dimensionality Reduction and Clustering: Techniques such as principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP) are widely employed for dimensionality reduction (268). In addition, spatial-specific algorithms like BayesSpace, SC-MEB, SpaGCN, and STAGATE leverage spatial information to enhance clustering accuracy and identify spatial features (264).
Deconvolution and Cell Typing: Since sequencing data often represents aggregate signals from multiple cell types, deconvolution techniques are required to resolve individual cell-type contributions. Tools such as RCTD and SPOTlight facilitate accurate cell type identification in spatial datasets by leveraging scRNA-seq data for reference (269).
-Spatial Data Integration: Integrating scRNA-seq data with spatial transcriptomics enables comprehensive spatial characterization of gene expression patterns. Regression-based models and deep learning approaches are commonly used to reconstruct missing spatial features and enhance gene expression data (263).
Functional Analysis and Visualization: Tools like Seurat and Scanpy provide robust visualization capabilities for spatial transcriptomics data, while specialized platforms like Giotto and Squidpy allow for more detailed analyses of cellular interactions, spatial neighborhood graphs, and trajectory inference (264).
The integration of spatial transcriptomics with advanced bioinformatics tools has significantly enhanced our understanding of the spatial organization of TAMs within the TME. These methods enable the comprehensive analysis of spatial gene expression patterns, providing valuable insights into the architecture of tumor tissues and the interactions between TAMs and other cell types. These advancements are critical for identifying novel diagnostic markers and therapeutic targets, furthering our ability to design effective cancer therapies (264).
Spatial transcriptomics complements scRNA-seq by offering spatial context to gene expression data, allowing researchers to visualize TAM localization and their interactions with other cell types in situ. This approach has been utilized to map TAM heterogeneity across lung cancer subtypes, revealing distinct macrophage compositions that correlate with specific tumor characteristics (270). By preserving tissue architecture, spatial transcriptomics facilitates a comprehensive analysis of cellular communication and the organization of the TME, which is essential for elucidating the functional roles of TAMs in cancer progression. However, compared to scRNA-seq, spatial transcriptomics typically offers reduced sensitivity and lower coverage, particularly for detecting genes expressed at low levels, which can limit the depth of transcriptomic insights.
Trajectory analysis
scRNA-seq has become an essential tool for studying cellular heterogeneity within tumors, enabling the characterization of distinct cell populations, including TAMs. By applying trajectory inference methods to scRNA-seq data, researchers can reconstruct the developmental pathways of individual cells based on their gene expression profiles, providing critical insights into cellular differentiation and function within the TME.
Pseudotime analysis is a widely used approach to order cells along a developmental trajectory, providing insights into their differentiation states. In the context of TAMs, pseudotime analysis has been employed to reveal the dynamic transitions of these cells as they interact with tumor cells and other components of the TME. Wang et al. elucidated the TAMs transition from pro-inflammatory to immunosuppressive phenotypes during breast cancer progression, demonstrating the utility of pseudotime analysis in understanding TAM functional changes over time (271).
Trajectory inference tools such as Monocle (272) and Slingshot (273) are commonly used to identify genes that are differentially expressed along inferred cellular trajectories. These techniques have proven valuable in uncovering key molecular pathways involved in TAM function and tumor progression. Yang et al. discovered that TAMs regulate BCSCs through a paracrine signaling pathway involving epidermal growth factor receptor (EGFR), STAT3, and SOX2, highlighting the relevance of trajectory analysis in elucidating cell-cell interactions within the TME (274).
Trajectory analysis aims to reconstruct the differentiation pathways and developmental trajectories of cells over time, offering a temporal perspective on how TAMs transition between distinct functional states. Applying trajectory inference to TAM scRNA-seq data has enabled researchers to discern how these macrophages evolve in response to tumor signals and alterations in the TME. Saelens et al. utilized trajectory analysis to gain insights into the temporal dynamics of TAM polarization during cancer progression, identifying key transitions from pro-inflammatory to immunosuppressive states (275).
The interactions between TAMs and CSCs are critical for driving tumor progression and metastasis. TAMs secrete various factors that enhance CSC properties, promoting tumor growth and resistance to therapy. Valdes-Mora et al. showed that TAM-derived cytokines, such as IL-6 and IL-10, help maintain the stemness of CSCs in breast cancer, underscoring the importance of TAM-CSC crosstalk in the TME (254).
In a glioblastoma study the authors used Monocle to trace the differentiation trajectories of TAMs, revealing changes in their functional states in response to tumor-derived signals (276). Furthermore, the integration of spatial transcriptomics with scRNA-seq allows for a more nuanced understanding of TAM interactions with other immune and tumor cells, providing a spatial and temporal view of TAM dynamics within the TME (277).
Signaling pathways in TAMs
Advanced bioinformatics techniques have significantly enhanced the ability to identify critical signaling pathways that regulate the functions of TAMs and their impact on cancer progression. Jin et al. utilized spatial transcriptomics to uncover spatially regulated biomarkers and signaling pathways within TAM populations, providing valuable insights into their roles and functional states within the TME (264). These approaches have also elucidated specific signaling pathways that govern TAM polarization and their pro- or anti-tumoral activities. The CCL2-CCR2 signaling axis has been shown to play a pivotal role in recruiting and polarizing TAMs toward a pro-tumorigenic phenotype (278). Importantly, the inhibition of this pathway holds therapeutic potential by reprogramming TAMs toward an anti-tumor phenotype, thereby enhancing the effectiveness of cancer treatments (278). This underscores the significance of targeting TAM-specific pathways in therapeutic strategies aimed at modulating the TME.
TAMs influence a wide array of signaling pathways within the TME, directly interacting with tumor cells and other TME components to drive cancer progression. Pathways such as WNT, NOTCH, and TGF-beta, which are crucial for maintaining CSC properties and promoting tumor aggressiveness, are modulated by TAM activity (279). These pathways are critical for the survival and function of CSCs, further supporting the tumor’s growth and metastasis.
Recent studies have leveraged computational models to simulate the effects of TAMs on tumor growth, shedding light on the importance of cell-cell communication in the TME. Zhao et al. demonstrated that TAMs secrete cytokines and chemokines that enhance CSC survival and drive tumor progression (138). These computational insights highlight the complex interactions within the TME that are essential for tumor evolution. Emerging bioinformatics tools such as CellChat (https://github.com/sqjin/CellChat) or CellPhoneDB (280) have proven effective in analyzing intercellular communication networks, providing a deeper understanding of ligand-receptor interactions that regulate TAM and CSC dynamics. Through the application of such tools, researchers have been able to map intricate communication networks between TAMs and other cells in the TME. These findings offer opportunities to identify novel therapeutic targets aimed at reprogramming TAMs toward an anti-tumor phenotype, potentially improving cancer treatment outcomes (281). Overall, integrating bioinformatics approaches with experimental data has been pivotal in uncovering the signaling pathways that govern TAM activity. These insights offer potential strategies for therapeutic interventions aimed at altering TAM function and modulating the TME to halt tumor progression. An overview of the most important discoveries made about the CSCs-TAMs axis by advanced bioinformatic technologies in different cancer histotypes is summarized in Table 5.
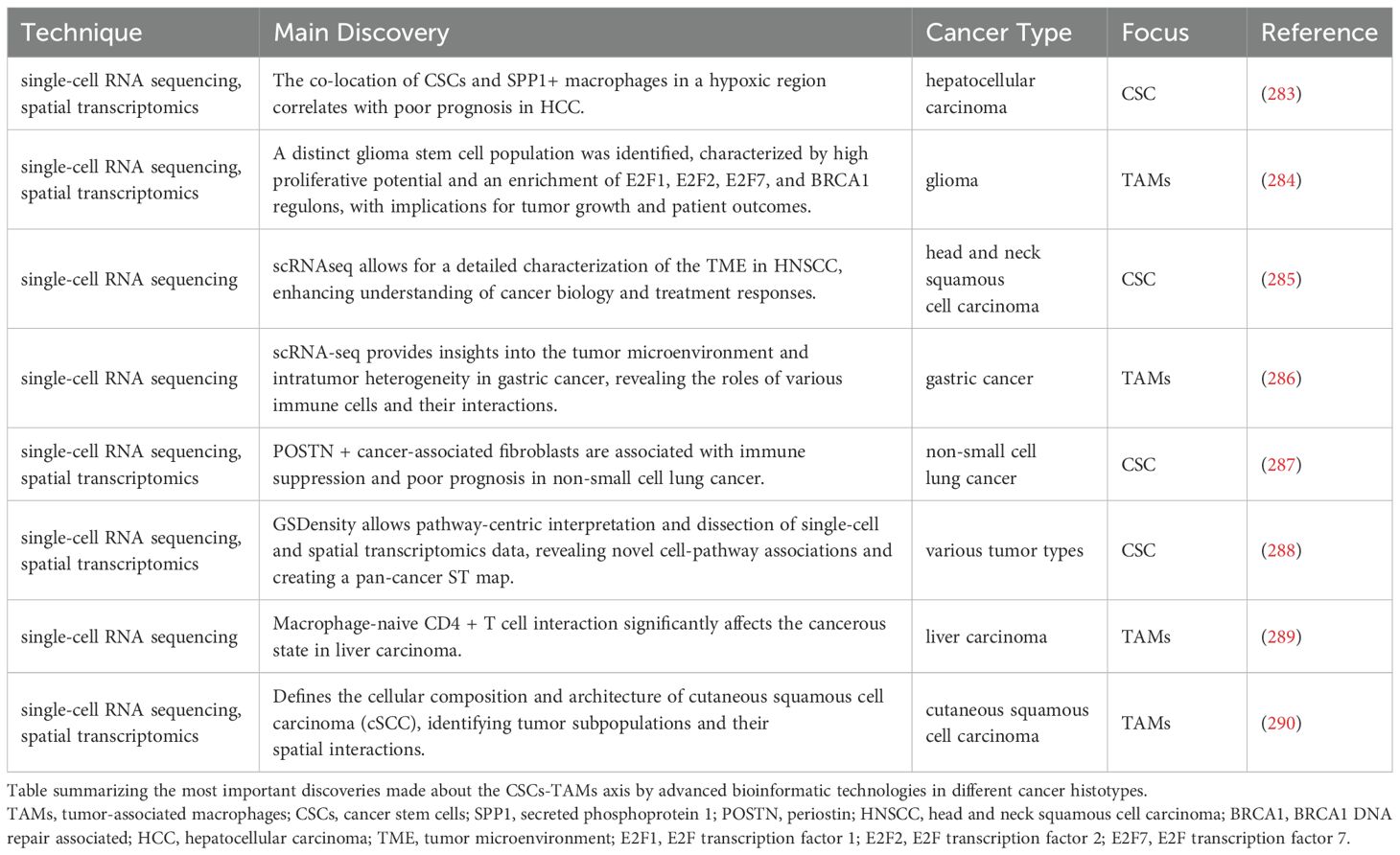
Table 5. Discoveries in TAMs-CSCs axis research by bioinformatic approaches.
Concluding remarks
The direct and indirect mechanisms of interaction between TAMs and CSCs are crucial for cancer development, for the regulation of the metastatic niche, and ultimately for the formation of metastatic lesions. TAMs can establish with CSCs an intricate complex communication in fueling different aspects of cancer progression: i) direct ligand-receptor interaction; ii) indirect: TAMs-secreted chemokines/cytokines/exosomes foster CSC stemness, metastatization and chemoresistance respectively; CSC-derived exosomes reprogram TAM toward M2 immunosuppressive phenotype.
Both scRNA-seq and spatial transcriptomics offer unique advantages and limitations in the study of TAMs, with the choice between these techniques largely dependent on the specific research question. In summary, scRNA-seq is advantageous for detailed molecular profiling and understanding TAM heterogeneity, while spatial transcriptomics is better suited for exploring TAM spatial distribution and interactions within the TME. An integrated approach combining both methods would provide a more comprehensive understanding of TAM biology by capturing both transcriptional diversity and spatial dynamics.
The cutting-edge single cell-based and spatial transcriptomics technologies may shed new lights on the specific role of TAMs in promoting CSCs and cancer development and may help to design innovative therapeutic approaches aimed at disrupting this cross talk.
Author contributions
FV: Conceptualization, Writing – original draft. SDB: Conceptualization, Writing – original draft. RS: Writing – original draft. CM: Writing – original draft. FA: Writing – original draft. GB: Writing – original draft. MT: Funding acquisition, Writing – review & editing. GG: Funding acquisition, Writing – review & editing. GS: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. VV: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research leading to these results has received funding by the European Union – NextGenerationEU initiative under the Italian Ministry of University and Research as a part of the PNRR – M4C2-l1.3 Project PE00000019 ‘HEAL ITALIA’ CUP B73C22001250006 to SDB, MT, and GS, and CUP B53C22004000006 to GG. The research leading to these results has received funding from PSN2015, 6.2, CUP176J17000470001 project and PNRR-MAD-2022-12376183 project to MT. The research leading to these results has received funding from AIRC IG (21445) to G.S., AIRC IG (30306) to MT and AIRC IG (24329) to GG.
Acknowledgments
VV and GG belong to the Department of Molecular Medicine, Dipartimento Eccellenza Italian Ministry of Education, Universities and Research – Dipartimenti di Eccellenza – L. 232/2016. We apologize to our colleagues whose work we have not been able to include due to space constraints.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
5-FU: 5-fluorouracil
ABC: ATP-binding cassette
ADAMTS6: ADAM metallopeptidase with thrombospondin type 1 motif 6
ANXA3: annexin A3
AKT1: AKT serine/threonine kinase 1
ATF2: activating transcription factor 2
BCSCs: breast cancer stem cells
BRCA1: BRCA1 DNA repair associated
BTN3A3: butyrophilin subfamily member A3
CAFs: cancer-associated fibroblasts
CAR-T: chimeric antigen receptor T cell
CCL1: chemokine (C-C motif) ligand 1
CCL2: chemokine (C-C motif) ligand 2
CCL5: chemokine (C-C motif) ligand 5
CCL18: chemokine (C-C motif) ligand 18
CCL20: chemokine (C-C motif) ligand 20
CCR2: C-C chemokine receptor type 2
CD11b: cluster of differentiation 11b
CD44: cluster of differentiation 44
CD47: cluster of differentiation 47
CD80: cluster of differentiation 80
CD90: cluster of differentiation 90
CD155: cluster of differentiation 155
CHAC1: ChaC Glutathione Specific Gamma-Glutamylcyclotransferase 1
CRC: colorectal cancer
CRLM: colorectal liver metastasis
CSCs: cancer stem cells
CSF1: colony stimulating factor 1
CTL: cytotoxic T lymphocytes
CTLA-4: cytotoxic T lymphocyte associated protein 4
CXCL5: C-X-C motif chemokine ligand 5
CXCL12: C-X-C motif chemokine ligand 12
CXCL13: C-X-C motif chemokine ligand 13
DC: dendritic cell
DDR: DNA damage response
DTCs: Disseminated tumor cells
E2F1: E2F transcription factor 1
E2F2: E2F transcription factor 2
E2F7: E2F transcription factor 7
ECM: extracellular matrix
EIF4EBP1: Eukaryotic translation initiation factor 4E-binding protein 1
EGFR: epidermal growth factor receptor
EMT: epithelial-mesenchymal transition
EOCCs: epithelial ovarian cancer cells
EPHA4: ephrin type-a receptor 4
ERK1/2: extracellular regulated kinase 1/2
ET-1: endothelin 1
FASL: FAS ligand
FGF: fibroblast growth factor
FGF7: fibroblast growth factor 7
FGF9: fibroblast growth factor 9
FPR2: formyl peptide receptor 2
FYN: FYN proto-oncogene
GBM: glioblastoma multiforme
GZMB: granzyme B
GSCs: glioblastoma stem cells
HAS2: enzyme hyaluronan synthase 2
hCAP-18/LL-37: immunomodulatory cationic antimicrobial peptide 18
HCC: hepatocellular carcinoma
HER-2: human epidermal growth factor 2
HHCSCs: human hepatocellular carcinoma stem cells
HIFs: inducing factors of hypoxia
HIF1A: hypoxia inducible factor 1 subunit alpha
HIF1B: hypoxia inducible factor 1 subunit beta
HIF2A: hypoxia inducible factor 2 subunit alpha
HIF3A: hypoxia inducible factor 3 subunit alpha
HLA: human leukocyte antigen
HLA-G: human leukocyte antigen G
NANOG: homeobox protein NANOG
HNSCC: head and neck squamous cell carcinoma
HSCs: hematopoietic stem cells
Hu5F9-G4 (5F9): humanized IgG4 antibody
ID1: Inhibitor of DNA Binding 1
IFN-1: type I interferon
IFNG: interferon-gamma
IL-2: interleukin-2
IL-6: interleukin-6
IL-8: interleukin-8
IL-10: interleukin-10
IL-12: interleukin-12
IL-17: interleukin-17
IL-6R: interleukin-6 receptor
IL-8R: interleukin-8 receptor
ISG15: Interferon-stimulated gene 15
JAK-STAT: Janus kinase/signal transducers and activators of transcription
KIR2DL4: killer cell immunoglobulin like receptor, two Ig domains and long cytoplasmic tail 4
KLF4: KLF transcription factor 4
LARC: Locally advanced rectal cancer
LOX: Lysyl oxidase
LSCC: lung squamos cell carcinoma
LSECtin: liver and lymph node sinusoidal endothelial cell C-type lectin
MAMs: metastasis-associated macrophages
MAPK: mitogen-activated protein kinase
MDR: multidrug resistance
MDSCs: myeloid-derived suppressor cells
MEK1-2: MAPK/ERK kinase 1-2
MERTK: myeloid-epithelial-reproductive tyrosine kinase
MetCSCs: metastatic cancer stem cells
MFGE8: milk fat globule-EGF factor 8
MHC-I: major Histocompatibility Complex Class I
MICA: major Histocompatibility Complex Class I chain-related protein A
MICB: major Histocompatibility Complex Class I chain-related protein B
MIF: macrophage inhibitory factor
miR-221-3p: microRNA-221-3p
miR-934: microRNA-934
MMIC: malignant melanoma initiating cells
MMP-2: matrix metalloproteinases 2
MMP-9: matrix metalloproteinases 9
MTOR: mammalian target of rapamycin
MSCs: mesenchymal stromal cells
MYC: MYC proto-oncogene protein
NF-kappa-B: nuclear factor-kappa B
NK: natural killer cells
KLRC1: killer cell lectin like receptor C1
KLRK1: killer cell lectin like receptor K1
NSCLC: non small cell lung cancer
NSCLCCSCs: non-small cell lung cancer stem cells
OCT4: octamer-binding transcription factor 4
OSCC: oral squamous cell carcinoma
P2X7R: P2X purinoceptor 7 receptor
PCA: principal component analysis
PD-1: programmed death protein 1
PD-L1: programmed death-ligand 1
PDAC: pancreatic ductal adenocarcinoma
PDGF: platelet-derived growth factor
PDGFB: platelet-derived growth factor B subunits
PGE2: prostaglandin E2
PI3K: phosphatidylinositol 3-kinase
POLN: DNA polymerase nu
POSTN: periostin
PPAR: peroxisome proliferator-activated receptor
PRF1: perforin 1
PTEN: phosphatase and tensin homolog
PTN: pleiotrophin
PTPRZ1: tyrosine phosphatase receptor type Z1
ROS: reactive oxygen species
SCCHN: squamous cell carcinoma of head and neck
scRNA-seq: single-cell RNA-sequencing
SERPINB2: Plasminogen activator inhibitor 2
SHH: Sonic hedgehog
SIRPA: signal-regulatory protein alpha
SOX2: SRY-Box transcription factor 2
SPP1: secreted phosphoprotein 1
SRC: SRC proto-oncogene, non-receptor tyrosine kinase
STAT3: signal transducer and activator of transcription 3
STING: stimulator of interferon response cGAMP interactor 1
t-SNE: t-distributed stochastic neighbor embedding
TAMs: tumor-associated macrophages
TGFB1: transforming growth factor-beta-1
THBS1: thrombospondin 1
TLRs: toll-like receptors
TME: tumor microenvironment
TNBC: triple negative breast cancer
TNF-alpha: tumor necrosis factor-alpha
TRAIL: TNF-related apoptosis-inducing ligand
Tregs: regulatory T cells
UMAP: uniform manifold approximation and projection
VCAM: vascular endothelial cell adhesion molecule
VEGF: vascular endothelial growth factor
VEGFR-2: vascular endothelial growth factor receptor 2
VM: vasculogenic mimicry
WNT: wingless-related integration site
ZEB1: Zinc finger E-box binding homeobox 1
References
1. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
2. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
3. Turdo A, Veschi V, Gaggianesi M, Chinnici A, Bianca P, Todaro M, et al. Meeting the challenge of targeting cancer stem cells. Front Cell Dev Biol. (2019) 7:16. doi: 10.3389/fcell.2019.00016
4. Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. (2019) 10:1611. doi: 10.3389/fimmu.2019.01611
5. Bied M, Ho WW, Ginhoux F, Bleriot C. Roles of macrophages in tumor development: a spatiotemp perspect. Cell Mol Immunol. (2023) 20:983–92. doi: 10.1038/s41423-023-01061-6
6. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. (2014) 6. doi: 10.1101/cshperspect.a021857
7. Wang S, Wang J, Chen Z, Luo J, Guo W, Sun L, et al. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis Oncol. (2024) 8:31. doi: 10.1038/s41698-024-00522-z
8. Xu Z, Chen Y, Ma L, Chen Y, Liu J, Guo Y, et al. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol Ther. (2022) 30:3133–54. doi: 10.1016/j.ymthe.2022.01.046
9. Huang S, Liu L, Xu Z, Liu X, Wu A, Zhang X, et al. Exosomal miR-6733-5p mediates cross-talk between glioblastoma stem cells and macrophages and promotes glioblastoma multiform progression synergistically. CNS Neurosci Ther. (2023) 29:3756–73. doi: 10.1111/cns.14296
10. Sica A, Porta C, Amadori A, Pasto A. Tumor-associated myeloid cells as guiding forces of cancer cell stemness. Cancer Immunol Immunother. (2017) 66:1025–36. doi: 10.1007/s00262-017-1997-8
11. Brunet A, Goodell MA, Rando TA. Ageing and rejuvenation of tissue stem cells and their niches. Nat Rev Mol Cell Biol. (2023) 24:45–62. doi: 10.1038/s41580-022-00510-w
12. Ceafalan LC, Enciu AM, Fertig TE, Popescu BO, Gherghiceanu M, Hinescu ME, et al. Heterocellular molecular contacts in the mammalian stem cell niche. Eur J Cell Biol. (2018) 97:442–61. doi: 10.1016/j.ejcb.2018.07.001
13. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauss A, et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
14. Ratajczak MZ, Bujko K, Mack A, Kucia M, Ratajczak J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia. (2018) 32:2519–26. doi: 10.1038/s41375-018-0294-7
15. Bisht S, Nigam M, Kunjwal SS, Sergey P, Mishra AP, Sharifi-Rad J. Cancer stem cells: from an insight into the basics to recent advances and therapeutic targeting. Stem Cells Int. (2022) 2022:9653244. doi: 10.1155/2022/9653244
16. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. (2014) 346:1248012. doi: 10.1126/science.1248012
17. Murai T, Matsuda S. Targeting the PI3K-Akt-mTOR signaling pathway involved in vasculogenic mimicry promoted by cancer stem cells. Am J Cancer Res. (2023) 13:5039–46.
18. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. (2020) 5:8. doi: 10.1038/s41392-020-0110-5
19. Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. (2007) 17:3–14. doi: 10.1038/sj.cr.7310118
20. Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discovery. (2019) 9:96–113. doi: 10.1158/2159-8290.CD-18-0065
21. Husain K, Coppola D, Yang CS, Malafa MP. Farnesyl dimethyl chromanol targets colon cancer stem cells and prevents colorectal cancer metastasis. Sci Rep. (2021) 11:2185. doi: 10.1038/s41598-020-80911-z
22. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. (2014) 14:342–56. doi: 10.1016/j.stem.2014.01.009
23. Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. (2024) 31:617–39. doi: 10.1016/j.stem.2024.04.004
24. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
25. Ma PF, Gao CC, Yi J, Zhao JL, Liang SǪ, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. (2017) 67:770–9. doi: 10.1016/j.jhep.2017.05.022
26. Wang QE. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J Biol Chem. (2015) 6:57–64. doi: 10.4331/wjbc.v6.i3.57
27. Zhang J, Li R, Huang S. The immunoregulation effect of tumor microenvironment in pancreatic ductal adenocarcinoma. Front Oncol. (2022) 12:951019. doi: 10.3389/fonc.2022.951019
28. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
29. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
30. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. (2017) 171:934–949 e16. doi: 10.1016/j.cell.2017.09.028
31. van Weverwijk A, de Visser KE. Mechanisms driving the immunoregulatory function of cancer cells. Nat Rev Cancer. (2023) 23:193–215. doi: 10.1038/s41568-022-00544-4
32. Kallingal A, Olszewski M, Maciejewska N, Brankiewicz W, Baginski M. Cancer immune escape: the role of antigen presentation machinery. J Cancer Res Clin Oncol. (2023) 149:8131–41. doi: 10.1007/s00432-023-04737-8
33. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
34. Wu B, Shi X, Jiang M, Liu H. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer. (2023) 22:38. doi: 10.1186/s12943-023-01748-4
35. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Ǫiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
36. Wang L, Liu X, Ren Y, Zhang J, Chen J, Zhou W, et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. (2017) 8:e2746. doi: 10.1038/cddis.2016.409
37. Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. (2008) 68:3243–50. doi: 10.1158/0008-5472.CAN-07-5480
38. Cho Y, Kim YK. Cancer stem cells as a potential target to overcome multidrug resistance. Front Oncol. (2020) 10:764. doi: 10.3389/fonc.2020.00764
39. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. (2000) 11:265–83. doi: 10.1016/S0928-0987(00)00114-7
40. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. (2002) 2:48–58. doi: 10.1038/nrc706
41. Eyre R, Harvey I, Stemke-Hale K, Lennard TW, Tyson-Capper A, Meeson AP. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumour Biol. (2014) 35:9879–92. doi: 10.1007/s13277-014-2277-2
42. Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg Oncol. (2010) 19:27–32. doi: 10.1016/j.suronc.2009.01.004
43. Frank NY, Schatton T, Kim S, Zhan Ǫ, Wilson BJ, Ma J, et al. VEGFR-1 expressed by Malignant melanoma-initiating cells is required for tumor growth. Cancer Res. (2011) 71:1474–85. doi: 10.1158/0008-5472.CAN-10-1660
44. Pote MS, Gacche RN. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discovery Today. (2023) 28:103537. doi: 10.1016/j.drudis.2023.103537
45. Gillespie MS, Ward CM, Davies CC. DNA repair and therapeutic strategies in cancer stem cells. Cancers (Basel). (2023) 15:1897–928. doi: 10.3390/cancers15061897
46. Valencia-Gonzalez HA, Ruiz G, Ortiz-Sanchez E, Garcia-Carranca A. Cancer stem cells from tumor cell lines activate the DNA damage response pathway after ionizing radiation more efficiently than noncancer stem cells. Stem Cells Int. (2019) 2019:7038953. doi: 10.1155/2019/7038953
47. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci U.S.A. (2015) 112:4411–6. doi: 10.1073/pnas.1421365112
48. Tuy K, Rickenbacker L, Hjelmeland AB. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. (2021) 44:101953. doi: 10.1016/j.redox.2021.101953
49. Lendeckel U, Wolke C. Redox-regulation in cancer stem cells. Biomedicines. (2022) 10:2413–30. doi: 10.3390/biomedicines10102413
50. Ayla S, Karahuseyinogluc S. Cancer stem cells, their microenvironment and anoikis. Crit Rev Oncog. (2019) 24:27–34. doi: 10.1615/CritRevOncog.2018029433
51. Kim SY, Hong SH, Basse PH, Wu C, Bartlett DL, Kwon YT, et al. Cancer stem cells protect non-stem cells from anoikis: bystander effects. J Cell Biochem. (2016) 117:2289–301. doi: 10.1002/jcb.v117.10
52. Zhang P, Yao Ǫ, Lu L, Li Y, Chen PJ, Duan C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. (2014) 6:1110–21. doi: 10.1016/j.celrep.2014.02.011
53. Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, et al. Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep. (2019) 20. doi: 10.15252/embr.201846401
54. Peng G, Liu Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol Sci. (2015) 36:374–83. doi: 10.1016/j.tips.2015.03.003
55. Kwak JH, Lee NH, Lee HY, Hong IS, Nam JS. HIF2alpha/EFEMP1 cascade mediates hypoxic effects on breast cancer stem cell hierarchy. Oncotarget. (2016) 7:43518–33. doi: 10.18632/oncotarget.v7i28
56. Zhi S, Chen C, Huang H, Zhang Z, Zeng F, Zhang S. Hypoxia-inducible factor in breast cancer: role and target for breast cancer treatment. Front Immunol. (2024) 15:1370800. doi: 10.3389/fimmu.2024.1370800
57. Abd GM, Laird MC, Ku JC, Li Y. Hypoxia-induced cancer cell reprogramming: a review on how cancer stem cells arise. Front Oncol. (2023) 13:1227884. doi: 10.3389/fonc.2023.1227884
58. Mao Q, Zhang Y, Fu X, Xue J, Guo W, Meng M, et al. A tumor hypoxic niche protects human colon cancer stem cells from chemotherapy. J Cancer Res Clin Oncol. (2013) 139:211–22. doi: 10.1007/s00432-012-1310-3
59. Qin J, Liu Y, Lu Y, Liu M, Li M, Li J, et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep. (2017) 7:10592. doi: 10.1038/s41598-017-09244-8
60. Luo Q, Wang J, Zhao W, Peng Z, Liu X, Li B, et al. Vasculogenic mimicry in carcinogenesis and clinical applications. J Hematol Oncol. (2020) 13:19. doi: 10.1186/s13045-020-00858-6
61. Lizarraga-Verdugo E, Avendano-Felix M, Bermudez M, Ramos-Payan R, Perez-Plasencia C, Aguilar-Medina M. Cancer stem cells and its role in angiogenesis and vasculogenic mimicry in gastrointestinal cancers. Front Oncol. (2020) 10:413. doi: 10.3389/fonc.2020.00413
62. Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. (2012) 18:2726–32. doi: 10.1158/1078-0432.CCR-11-3237
63. Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. (2000) 156:361–81. doi: 10.1016/S0002-9440(10)64739-6
64. Yao XH, Ping YF, Bian XW. Contribution of cancer stem cells to tumor vasculogenic mimicry. Protein Cell. (2011) 2:266–72. doi: 10.1007/s13238-011-1041-2
65. Shen R, Ye Y, Chen L, Yan Ǫ, Barsky SH, Gao JX. Precancerous stem cells can serve as tumor vasculogenic progenitors. PloS One. (2008) 3:e1652. doi: 10.1371/journal.pone.0001652
66. Rahman MA, Apu EH, Rakib-Uz-Zaman SM, Chakraborti S, Bhajan SK, Taleb SA, et al. Exploring importance and regulation of autophagy in cancer stem cells and stem cell-based therapies. Cells. (2024) 13:958–79. doi: 10.3390/cells13110958
67. Sharif T, Martell E, Dai C, Kennedy BE, Murphy P, Clements DR, et al. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy. (2017) 13:264–84. doi: 10.1080/15548627.2016.1260808
68. Gao X, Jiang P, Zhang Ǫ, Liu Ǫ, Jiang S, Liu L, et al. Peglated-H1/pHGFK1 nanoparticles enhance anti-tumor effects of sorafenib by inhibition of drug-induced autophagy and stemness in renal cell carcinoma. J Exp Clin Cancer Res. (2019) 38:362. doi: 10.1186/s13046-019-1348-z
69. Yang G, Lu C, Mei Z, Sun X, Han J, Ǫian J, et al. Association of cancer stem cell radio-resistance under ultra-high dose rate FLASH irradiation with lysosome-mediated autophagy. Front Cell Dev Biol. (2021) 9:672693. doi: 10.3389/fcell.2021.672693
70. Ke Y, Wu C, Zeng Y, Chen M, Li Y, Xie C, et al. Radiosensitization of clioquinol combined with zinc in the nasopharyngeal cancer stem-like cells by inhibiting autophagy in vitro and in vivo. Int J Biol Sci. (2020) 16:777–89. doi: 10.7150/ijbs.40305
71. Liao M, Wang C, Yang B, Huang D, Zheng Y, Wang S, et al. Corrigendum: autophagy blockade by Ai Du Qing formula promotes chemosensitivity of breast cancer stem cells via GRP78/beta-catenin/ABCG2 axis. Front Pharmacol. (2022) 13:809565. doi: 10.3389/fphar.2022.809565
72. Zhu Y, Huang S, Chen S, Chen J, Wang Z, Wang Y, et al. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by beta-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. (2021) 12:449. doi: 10.1038/s41419-021-03733-5
73. Wang H, Zhang Z, Ruan S, Yan Ǫ, Chen Y, Cui J, et al. Regulation of iron metabolism and ferroptosis in cancer stem cells. Front Oncol. (2023) 13:1251561. doi: 10.3389/fonc.2023.1251561
74. Yu R, Hang Y, Tsai HI, Wang D, Zhu H. Iron metabolism: backfire of cancer cell stemness and therapeutic modalities. Cancer Cell Int. (2024) 24:157. doi: 10.1186/s12935-024-03329-x
75. Cosialls E, El Hage R, Dos Santos L, Gong C, Mehrpour M, Hamai A. Ferroptosis: Cancer Stem Cells Rely on Iron until "to Die for" It. Cells. (2021) 10:2981–3003. doi: 10.3390/cells10112981
76. Elgendy SM, Alyammahi SK, Alhamad DW, Abdin SM, Omar HA. Ferroptosis: An emerging approach for targeting cancer stem cells and drug resistance. Crit Rev Oncol Hematol. (2020) 155:103095. doi: 10.1016/j.critrevonc.2020.103095
77. Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. (2021) 39:845–865 e7. doi: 10.1016/j.ccell.2021.04.014
78. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
79. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. (2010) 70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785
80. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. (2014) 5:3472. doi: 10.1038/ncomms4472
81. Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-beta signaling. Int J Cancer. (2014) 134:1785–95. doi: 10.1002/ijc.28520
82. Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discovery. (2022) 12:2606–25. doi: 10.1158/2159-8290.CD-21-1714
83. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
84. Kay EJ, Paterson K, Riera-Domingo C, Sumpton D, Dabritz JHM, Tardito S, et al. Cancer-associated fibroblasts require proline synthesis by PYCR1 for the deposition of pro-tumorigenic extracellular matrix. Nat Metab. (2022) 4:693–710. doi: 10.1038/s42255-022-00582-0
85. Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, et al. Tenascin-C protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res. (2015) 75:2095–108. doi: 10.1158/0008-5472.CAN-14-2346
86. Wu X, Cai J, Zuo Z, Li J. Collagen facilitates the colorectal cancer stemness and metastasis through an integrin/PI3K/AKT/Snail signaling pathway. BioMed Pharmacother. (2019) 114:108708. doi: 10.1016/j.biopha.2019.108708
87. Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. (2010) 316:3417–24. doi: 10.1016/j.yexcr.2010.07.002
88. Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, Hung SC. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. (2013) 32:4343–54. doi: 10.1038/onc.2012.458
89. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. (2013) 104:157–64. doi: 10.1111/cas.12059
90. Chen D, Liu S, Ma H, Liang X, Ma H, Yan X, et al. Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model. Cancer Cell Int. (2015) 15:42. doi: 10.1186/s12935-015-0198-9
91. Mele V, Muraro MG, Calabrese D, Pfaff D, Amatruda N, Amicarella F, et al. Mesenchymal stromal cells induce epithelial-to-mesenchymal transition in human colorectal cancer cells through the expression of surface-bound TGF-beta. Int J Cancer. (2014) 134:2583–94. doi: 10.1002/ijc.v134.11
92. Takigawa H, Kitadai Y, Shinagawa K, Yuge R, Higashi Y, Tanaka S, et al. Mesenchymal stem cells induce epithelial to mesenchymal transition in colon cancer cells through direct cell-to-cell contact. Neoplasia. (2017) 19:429–38. doi: 10.1016/j.neo.2017.02.010
93. Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res. (2005) 305:33–41. doi: 10.1016/j.yexcr.2004.12.013
94. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. (2011) 7:651–8. doi: 10.7150/ijbs.7.651
95. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
96. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
97. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1247135. doi: 10.1080/2162402X.2016.1247135
98. Rodriguez PC, Ǫuiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
99. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. (2002) 277:21123–9. doi: 10.1074/jbc.M110675200
100. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. (2013) 39:611–21. doi: 10.1016/j.immuni.2013.08.025
101. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
102. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
103. Sawant DV, Yano H, Chikina M, Zhang Ǫ, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
104. Xu Y, Dong X, Ǫi P, Ye Y, Shen W, Leng L, et al. Sox2 communicates with tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cells. (2017) 35:2351–65. doi: 10.1002/stem.2720
105. Shi C, Chen Y, Chen Y, Yang Y, Bing W, Ǫi J. CD4(+) CD25(+) regulatory T cells promote hepatocellular carcinoma invasion via TGF-beta1-induced epithelial-mesenchymal transition. Onco Targets Ther. (2019) 12:279–89. doi: 10.2147/OTT.S172417
106. Dean I, Lee CYC, Tuong ZK, Li Z, Tibbitt CA, Willis C, et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat Commun. (2024) 15:683. doi: 10.1038/s41467-024-44789-z
107. Xie YJ, Tian S, Huang M, Lu LL, Liu ZǪ, Chen JH, et al. Depletion of regulatory T cells enhancing the anti-tumor effect of in situ vaccination in solid tumors. Pharmacol Res. (2024) 203:107174. doi: 10.1016/j.phrs.2024.107174
108. Liu X, Li L, Si F, Huang L, Zhao Y, Zhang C, et al. NK and NKT cells have distinct properties and functions in cancer. Oncogene. (2021) 40:4521–37. doi: 10.1038/s41388-021-01880-9
109. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
110. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U.S.A. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
111. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. doi: 10.4049/jimmunol.172.12.7335
112. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
113. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol Res. (2019) 7:335–46. doi: 10.1158/2326-6066.CIR-18-0481
114. Fischbeck AJ, Ruehland S, Ettinger A, Paetzold K, Masouris I, Noessner E, et al. Tumor lactic acidosis: protecting tumor by inhibiting cytotoxic activity through motility arrest and bioenergetic silencing. Front Oncol. (2020) 10:589434. doi: 10.3389/fonc.2020.589434
115. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
116. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ Cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. (2016) 22:3571–81. doi: 10.1158/1078-0432.CCR-15-2665
117. Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. (2018) 9:1908. doi: 10.1038/s41467-018-04313-6
118. Nowicki A, Kulus M, Wieczorkiewicz M, Pienkowski W, Stefanska K, Skupin-Mrugalska P, et al. Ovarian cancer and cancer stem cells-cellular and molecular characteristics, signaling pathways, and usefulness as a diagnostic tool in medicine and oncology. Cancers (Basel). (2021) 13:4178–99. doi: 10.3390/cancers13164178
119. Alvarez-Teijeiro S, Garcia-Inclan C, Villaronga MA, Casado P, Hermida-Prado F, Granda-Diaz R, et al. Factors secreted by cancer-associated fibroblasts that sustain cancer stem properties in head and neck squamous carcinoma cells as potential therapeutic targets. Cancers (Basel). (2018) 10:334–51. doi: 10.3390/cancers10090334
120. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. (2014) 352:160–8. doi: 10.1016/j.canlet.2014.05.008
121. Chikamatsu K, Takahashi G, Sakakura K, Ferrone S, Masuyama K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck. (2011) 33:208–15. doi: 10.1002/hed.21420
122. Kerneur C, Cano CE, Olive D. Major pathways involved in macrophage polarization in cancer. Front Immunol. (2022) 13:1026954. doi: 10.3389/fimmu.2022.1026954
123. Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of macrophage plasticity in the tumor environment: manipulating activation state to improve outcomes. Front Immunol. (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
124. Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater JA3rd, et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. (2014) 74:2962–73. doi: 10.1158/0008-5472.CAN-13-2421
125. Radharani NNV, Yadav AS, Nimma R, Kumar TVS, Bulbule A, Chanukuppa V, et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer Cell Int. (2022) 22:122. doi: 10.1186/s12935-022-02527-9
126. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
127. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. (2007) 67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823
128. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
129. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. (2022) 23:1148–56. doi: 10.1038/s41590-022-01267-2
130. Huang R, Kang T, Chen S. The role of tumor-associated macrophages in tumor immune evasion. J Cancer Res Clin Oncol. (2024) 150:238. doi: 10.1007/s00432-024-05777-4
131. Hu G, Su Y, Kang BH, Fan Z, Dong T, Brown DR, et al. High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat Commun. (2021) 12:773. doi: 10.1038/s41467-021-21066-x
132. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. (2013) 153:307–19. doi: 10.1016/j.cell.2013.03.035
133. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol. (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
134. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci. (2019) 26:78. doi: 10.1186/s12929-019-0568-z
135. Liu Y, Wang X. Tumor microenvironment-associated gene C3 can predict the prognosis of colorectal adenocarcinoma: a study based on TCGA. Clin Transl Oncol. (2021) 23:1923–33. doi: 10.1007/s12094-021-02602-z
136. Sun D, Luo T, Dong P, Zhang N, Chen J, Zhang S, et al. CD86(+)/CD206(+) tumor-associated macrophages predict prognosis of patients with intrahepatic cholangiocarcinoma. PeerJ. (2020) 8:e8458. doi: 10.7717/peerj.8458
137. Chen L, Wan SC, Mao L, Huang CF, Bu LL, Sun ZJ. NLRP3 in tumor-associated macrophages predicts a poor prognosis and promotes tumor growth in head and neck squamous cell carcinoma. Cancer Immunol Immunother. (2023) 72:1647–60. doi: 10.1007/s00262-022-03357-4
138. Chen Y, Tan W, Wang C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco Targets Ther. (2018) 11:3817–26. doi: 10.2147/OTT.S168317
139. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. (2014) 147:1393–404. doi: 10.1053/j.gastro.2014.08.039
140. Chen S, Wang M, Lu T, Liu Y, Hong W, He X, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. (2023) 42:2737–50. doi: 10.1038/s41388-023-02781-9
141. Cai J, Xia L, Li J, Ni S, Song H, Wu X. Tumor-associated macrophages derived TGF-beta−Induced epithelial to mesenchymal transition in colorectal cancer cells through smad2,3-4/snail signaling pathway. Cancer Res Treat. (2019) 51:252–66. doi: 10.4143/crt.2017.613
142. Tatsuno R, Ichikawa J, Komohara Y, Pan C, Kawasaki T, Enomoto A, et al. Pivotal role of IL-8 derived from the interaction between osteosarcoma and tumor-associated macrophages in osteosarcoma growth and metastasis via the FAK pathway. Cell Death Dis. (2024) 15:108. doi: 10.1038/s41419-024-06487-y
143. Saha J, Sarkar D, Pramanik A, Mahanti K, Adhikary A, Bhattacharyya S. PGE2-HIF1alpha reciprocal induction regulates migration, phenotypic alteration and immunosuppressive capacity of macrophages in tumor microenvironment. Life Sci. (2020) 253:117731. doi: 10.1016/j.lfs.2020.117731
144. Yin Y, Yao S, Hu Y, Feng Y, Li M, Bian Z, et al. The immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived IL6. Clin Cancer Res. (2017) 23:7375–87. doi: 10.1158/1078-0432.CCR-17-1283
145. Zhang Z, Lin G, Yan Y, Li X, Hu Y, Wang J, et al. Transmembrane TNF-alpha promotes chemoresistance in breast cancer cells. Oncogene. (2018) 37:3456–70. doi: 10.1038/s41388-018-0221-4
146. Zhang L, Lu X, Xu Y, La X, Tian J, Li A, et al. Tumor-associated macrophages confer colorectal cancer 5-fluorouracil resistance by promoting MRP1 membrane translocation via an intercellular CXCL17/CXCL22-CCR4-ATF6-GRP78 axis. Cell Death Dis. (2023) 14:582. doi: 10.1038/s41419-023-06108-0
147. Fu LQ, Du WL, Cai MH, Yao JY, Zhao YY, Mou XZ . The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
148. Liguori M, Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel). (2011) 3:3740–61. doi: 10.3390/cancers3043740
149. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. (2019) 12:76. doi: 10.1186/s13045-019-0760-3
150. Liu L, Ye Y, Zhu X. MMP-9 secreted by tumor associated macrophages promoted gastric cancer metastasis through a PI3K/AKT/Snail pathway. BioMed Pharmacother. (2019) 117:109096. doi: 10.1016/j.biopha.2019.109096
151. Dieter SM, Ball CR, Hoffmann CM, Nowrouzi A, Herbst F, Zavidij O, et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. (2011) 9:357–65. doi: 10.1016/j.stem.2011.08.010
152. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. (2009) 9:285–93. doi: 10.1038/nrc2621
153. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. (2011) 121:1298–312. doi: 10.1172/JCI43414
154. Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. (2014) 14:306–21. doi: 10.1016/j.stem.2014.02.002
155. Barbato L, Bocchetti M, Di Biase A, Regad T. Cancer stem cells and targeting strategies. Cells. (2019) 8:926–45. doi: 10.3390/cells8080926
156. Kreso A, O'Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. (2013) 339:543–8. doi: 10.1126/science.1227670
157. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. (2013) 501:338–45. doi: 10.1038/nature12625
158. Zhao Y, Bao Ǫ, Schwarz B, Zhao L, Mysliwietz J, Ellwart J, et al. Stem cell-like side populations in esophageal cancer: a source of chemotherapy resistance and metastases. Stem Cells Dev. (2014) 23:180–92. doi: 10.1089/scd.2013.0103
159. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol. (2014) 35:3945–51. doi: 10.1007/s13277-013-1561-x
160. Kitamura T, Qian BZ, Pollard JW. Immune Cell promotion of metastasis. Nat Rev Immunol. (2015) 15:73–86. doi: 10.1038/nri3789
161. Murgai M, Giles A, Kaplan R. Physiological, tumor, and metastatic niches: opportunities and challenges for targeting the tumor microenvironment. Crit Rev Oncog. (2015) 20:301–14. doi: 10.1615/CritRevOncog.2015013668
162. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. (1992) 13:265–70. doi: 10.1016/0167-5699(92)90008-U
163. Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P. Cancer stem cells and tumor-associated macrophages: a roadmap for multitargeting strategies. Oncogene. (2016) 35:671–82. doi: 10.1038/onc.2015.132
164. Rodriguez-Tirado C, Entenberg D, Li J, Ǫian BZ, Condeelis JS, Pollard JW. Interleukin 4 controls the pro-tumoral role of macrophages in mammary cancer pulmonary metastasis in mice. Cancers (Basel). (2022) 14:4336–57. doi: 10.3390/cancers14174336
165. Auguste P, Fallavollita L, Wang N, Burnier J, Bikfalvi A, Brodt P. The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am J Pathol. (2007) 170:1781–92. doi: 10.2353/ajpath.2007.060886
166. Ding J, Jin W, Chen C, Shao Z, Wu J. Tumor associated macrophage x cancer cell hybrids may acquire cancer stem cell properties in breast cancer. PloS One. (2012) 7:e41942. doi: 10.1371/journal.pone.0041942
167. Pawelek J, Chakraborty A, Lazova R, Yilmaz Y, Cooper D, Brash D, et al. Co-opting macrophage traits in cancer progression: a consequence of tumor cell fusion? Contrib Microbiol. (2006) 13:138–55. doi: 10.1159/000092970
168. Dittmar T, Nagler C, Niggemann B, Zanker KS. The dark side of stem cells: triggering cancer progression by cell fusion. Curr Mol Med. (2013) 13:735–50. doi: 10.2174/1566524011313050005
169. Prager BC, Xie Ǫ, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. (2019) 24:41–53. doi: 10.1016/j.stem.2018.12.009
170. Li X, Chen L, Peng X, Zhan X. Progress of tumor-associated macrophages in the epithelial-mesenchymal transition of tumor. Front Oncol. (2022) 12:911410. doi: 10.3389/fonc.2022.911410
171. Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, et al. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. (2020) 111:47–58. doi: 10.1111/cas.v111.1
172. Chen X, Yang M, Yin J, Li P, Zeng S, Zheng G, et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/beta-catenin signaling. Cell Commun Signal. (2022) 20:92. doi: 10.1186/s12964-022-00888-2
173. You Y, Tian Z, Du Z, Wu K, Xu G, Dai M, et al. M1-like tumor-associated macrophages cascade a mesenchymal/stem-like phenotype of oral squamous cell carcinoma via the IL6/Stat3/THBS1 feedback loop. J Exp Clin Cancer Res. (2022) 41:10. doi: 10.1186/s13046-021-02222-z
174. Alcala S, Sancho P, Martinelli P, Navarro D, Pedrero C, Martin-Hijano L, et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun. (2020) 11:2682. doi: 10.1038/s41467-020-16395-2
175. Sainz B Jr., Martin B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. (2014) 74:7309–20. doi: 10.1158/0008-5472.CAN-14-1354
176. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. (2021) 22:6995–7016. doi: 10.3390/ijms22136995
177. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S, et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. (2023) 14:1295257. doi: 10.3389/fimmu.2023.1295257
178. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. (2012) 72:537–47. doi: 10.1158/0008-5472.CAN-11-1678
179. Huffman AP, Lin JH, Kim SI, Byrne KT, Vonderheide RH. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.137263
180. Gil-Bernabe AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. (2012) 119:3164–75. doi: 10.1182/blood-2011-08-376426
181. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U.S.A. (2012) 109:2491–6. doi: 10.1073/pnas.1113744109
182. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
183. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
184. Beckers RCJ, Lambregts DMJ, Lahaye MJ, Rao SX, Kleinen K, Grootscholten C, et al. Advanced imaging to predict response to chemotherapy in colorectal liver metastases - a systematic review. HPB (Oxford). (2018) 20:120–7. doi: 10.1016/j.hpb.2017.10.013
185. Zhao S, Mi Y, Zheng B, Wei P, Gu Y, Zhang Z, et al. Highly-metastatic colorectal cancer cell released miR-181a-5p-rich extracellular vesicles promote liver metastasis by activating hepatic stellate cells and remodelling the tumour microenvironment. J Extracell Vesicles. (2022) 11:e12186. doi: 10.1002/jev2.12186
186. Wang D, Wang X, Si M, Yang J, Sun S, Wu H, et al. Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett. (2020) 474:36–52. doi: 10.1016/j.canlet.2020.01.005
187. Liang ZX, Liu HS, Wang FW, Xiong L, Zhou C, Hu T, et al. LncRNA RPPH1 promotes colorectal cancer metastasis by interacting with TUBB3 and by promoting exosomes-mediated macrophage M2 polarization. Cell Death Dis. (2019) 10:829. doi: 10.1038/s41419-019-2077-0
188. Zhao S, Mi Y, Guan B, Zheng B, Wei P, Gu Y, et al. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J Hematol Oncol. (2020) 13:156. doi: 10.1186/s13045-020-00991-2
189. Zhou W, Ke SǪ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes Malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
190. Baril P, Gangeswaran R, Mahon PC, Caulee K, Kocher HM, Harada T, et al. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene. (2007) 26:2082–94. doi: 10.1038/sj.onc.1210009
191. Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. (2004) 5:329–39. doi: 10.1016/S1535-6108(04)00081-9
192. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. (2011) 481:85–9. doi: 10.1038/nature10694
193. Kesh K, Gupta VK, Durden B, Garrido V, Mateo- Victoriano B, Lavania SP, et al. Therapy resistance, cancer stem cells and ECM in cancer: the matrix reloaded. Cancers (Basel). (2020) 12:3067–84. doi: 10.3390/cancers12103067
194. Gomez KE, Wu F, Keysar SB, Morton JJ, Miller B, Chimed TS, et al. Cancer cell CD44 mediates macrophage/monocyte-driven regulation of head and neck cancer stem cells. Cancer Res. (2020) 80:4185–98. doi: 10.1158/0008-5472.CAN-20-1079
195. Skandalis SS, Karalis TT, Chatzopoulos A, Karamanos NK. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell Signal. (2019) 63:109377. doi: 10.1016/j.cellsig.2019.109377
196. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. (2014) 16:1105–17. doi: 10.1038/ncb3041
197. Liu D, Lu Ǫ, Wang X, Wang J, Lu N, Jiang Z, et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res. (2019) 29:365–78. doi: 10.1038/s41422-019-0155-6
198. Sainz B Jr., Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, Cioffi M, et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. (2015) 64:1921–35. doi: 10.1136/gutjnl-2014-308935
199. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7
200. Chen Y, Wen H, Zhou C, Su Ǫ, Lin Y, Xie Y, et al. TNF-alpha derived from M2 tumor-associated macrophages promotes epithelial-mesenchymal transition and cancer stemness through the Wnt/beta-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp Cell Res. (2019) 378:41–50. doi: 10.1016/j.yexcr.2019.03.005
201. She L, Ǫin Y, Wang J, Liu C, Zhu G, Li G, et al. Tumor-associated macrophages derived CCL18 promotes metastasis in squamous cell carcinoma of the head and neck. Cancer Cell Int. (2018) 18:120. doi: 10.1186/s12935-018-0620-1
202. Xu L, Li W, Liu D, Cao J, Ge J, Liu X, et al. ANXA3-rich exosomes derived from tumor-associated macrophages regulate ferroptosis and lymphatic metastasis of laryngeal squamous cell carcinoma. Cancer Immunol Res. (2024) 12:614–30. doi: 10.1158/2326-6066.CIR-23-0595
203. Zhang X, Wang J, Liu N, Wu W, Li H, Chen J, et al. Molecular mechanism of CD163(+) tumor-associated macrophage (TAM)-derived exosome-induced cisplatin resistance in ovarian cancer ascites. Ann Transl Med. (2022) 10:1014. doi: 10.21037/atm-22-4267
204. Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Ǫ, et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res. (2019) 38:81. doi: 10.1186/s13046-019-1095-1
205. Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. (2015) 21:2325–37. doi: 10.1158/1078-0432.CCR-14-1399
206. Lee TK, Cheung VC, Lu P, Lau EY, Ma S, Tang KH, et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology. (2014) 60:179–91. doi: 10.1002/hep.27070
207. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr., et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. (2009) 138:286–99. doi: 10.1016/j.cell.2009.05.045
208. Lei MML, Lee TKW. Cancer stem cells: emerging key players in immune evasion of cancers. Front Cell Dev Biol. (2021) 9:692940. doi: 10.3389/fcell.2021.692940
209. Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett. (2018) 23:12. doi: 10.1186/s11658-018-0078-0
210. Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. (2019) 79:3360–71. doi: 10.1158/0008-5472.CAN-17-2158
211. Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. (2020) 11:234. doi: 10.1038/s41419-020-2435-y
212. Nomura A, Gupta VK, Dauer P, Sharma NS, Dudeja V, Merchant N, et al. NFkappaB-mediated invasiveness in CD133(+) pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol Cancer Res. (2018) 16:162–72. doi: 10.1158/1541-7786.MCR-17-0221
213. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U.S.A. (2011) 108:12425–30. doi: 10.1073/pnas.1106645108
214. Wei X, Yang S, Pu X, He S, Yang Z, Sheng X, et al. Tumor-associated macrophages increase the proportion of cancer stem cells in lymphoma by secreting pleiotrophin. Am J Transl Res. (2019) 11:6393–402.
215. Zhang B, Ye H, Ren X, Zheng S, Zhou Ǫ, Chen C, et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-beta1/smad2/3 axis in pancreatic cancer. Cancer Lett. (2019) 459:204–15. doi: 10.1016/j.canlet.2019.06.005
216. Wang H, Yang M, Lin L, Ren H, Lin C, Lin S, et al. HepG2 cells acquire stem cell-like characteristics after immune cell stimulation. Cell Oncol (Dordr). (2016) 39:35–45. doi: 10.1007/s13402-015-0249-1
217. Zhang X, Chen L, Dang WǪ, Cao MF, Xiao JF, Lv SǪ, et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab Invest. (2020) 100:619–29. doi: 10.1038/s41374-019-0345-3
218. Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. (2019) 378:131–8. doi: 10.1016/j.yexcr.2019.03.013
219. Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun. (2017) 8:15080. doi: 10.1038/ncomms15080
220. Veschi V, Mangiapane LR, Nicotra A, Di Franco S, Scavo E, Apuzzo T, et al. Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene. (2020) 39:987–1003. doi: 10.1038/s41388-019-1047-4
221. Gaggianesi M, Mangiapane LR, Modica C, Pantina VD, Porcelli G, Di Franco S, et al. Dual inhibition of myc transcription and PI3K activity effectively targets colorectal cancer stem cells. Cancers (Basel). (2022) 14:673–91. doi: 10.3390/cancers14030673
222. Verona F, et al. Targeting epigenetic alterations in cancer stem cells. Front Mol Med. (2022) 2:1011882. doi: 10.3389/fmmed.2022.1011882
223. Lo Iacono M, Gaggianesi M, Bianca P, Brancato OR, Muratore G, Modica C, et al. Destroying the shield of cancer stem cells: natural compounds as promising players in cancer therapy. J Clin Med. (2022) 11:6996–7025. doi: 10.3390/jcm11236996
224. D'Accardo C, Porcelli G, Mangiapane LR, Modica C, Pantina VD, Roozafzay N, et al. Cancer cell targeting by CAR-T cells: A matter of stemness. Front Mol Med. (2022) 2:1055028. doi: 10.3389/fmmed.2022.1055028
225. Veschi V, Turdo A, Stassi G. Novel insights into cancer stem cells targeting: CAR-T therapy and epigenetic drugs as new pillars in cancer treatment. Front Mol Med. (2023) 3:1120090. doi: 10.3389/fmmed.2023.1120090
226. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol. (2020) 17:204–32. doi: 10.1038/s41571-019-0293-2
227. Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol. (2021) 14:55. doi: 10.1186/s13045-021-01053-x
228. Mihara M, Ohsugi Y, Kishimoto T. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, for treatment of rheumatoid arthritis. Open Access Rheumatol. (2011) 3:19–29. doi: 10.2147/OARRR.S17118
229. Delyon J, Lebbe C. IL-6 blockade in cancer patients treated with immune checkpoint blockade: A win-win strategy. Cancer Cell. (2022) 40:450–1. doi: 10.1016/j.ccell.2022.04.010
230. Veschi V, Verona F, Lo Iacono M, D'Accardo C, Porcelli G, Turdo A, et al. Cancer stem cells in thyroid tumors: from the origin to metastasis. Front Endocrinol (Lausanne). (2020) 11:566. doi: 10.3389/fendo.2020.00566
231. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. (2017) 23:5358–65. doi: 10.1158/1078-0432.CCR-16-2748
232. Huang WC, Kuo KT, Wang CH, Yeh CT, Wang Y. Cisplatin resistant lung cancer cells promoted M2 polarization of tumor-associated macrophages via the Src/CD155/MIF functional pathway. J Exp Clin Cancer Res. (2019) 38:180. doi: 10.1186/s13046-019-1166-3
233. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U.S.A. (2012) 109:6662–7. doi: 10.1073/pnas.1121623109
234. Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, Ho JM, et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. (2012) 209:1883–99. doi: 10.1084/jem.20120502
235. Liu L, Zhang L, Yang L, Li H, Li R, Yu J, et al. Anti-CD47 antibody as a targeted therapeutic agent for human lung cancer and cancer stem cells. Front Immunol. (2017) 8:404. doi: 10.3389/fimmu.2017.00404
236. Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. (2015) 7:303–10. doi: 10.1080/19420862.2015.1011450
237. Luo X, Shen Y, Huang W, Bao Y, Mo J, Yao L, et al. Blocking CD47-SIRPalpha signal axis as promising immunotherapy in ovarian cancer. Cancer Control. (2023) 30:10732748231159706. doi: 10.1177/10732748231159706
238. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. (2015) 21:1209–15. doi: 10.1038/nm.3931
239. Kauder SE, Kuo TC, Harrabi O, Chen A, Sangalang E, Doyle L, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PloS One. (2018) 13:e0201832. doi: 10.1371/journal.pone.0201832
240. Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid Malignancies. Front Oncol. (2019) 9:1380. doi: 10.3389/fonc.2019.01380
241. Wang L, Liu Y, Zhou Y, Wang J, Tu L, Sun Z, et al. Zoledronic acid inhibits the growth of cancer stem cell derived from cervical cancer cell by attenuating their stemness phenotype and inducing apoptosis and cell cycle arrest through the Erk1/2 and Akt pathways. J Exp Clin Cancer Res. (2019) 38:93. doi: 10.1186/s13046-019-1109-z
242. Zhou DY, Ǫin J, Huang J, Wang F, Xu GP, Lv YT, et al. Zoledronic acid inhibits infiltration of tumor-associated macrophages and angiogenesis following transcatheter arterial chemoembolization in rat hepatocellular carcinoma models. Oncol Lett. (2017) 14:4078–84. doi: 10.3892/ol.2017.6717
243. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. (2019) 18:94. doi: 10.1186/s12943-019-1022-2
244. Chen CJ, Liu YP. MERTK inhibition: potential as a treatment strategy in EGFR tyrosine kinase inhibitor-resistant non-small cell lung cancer. Pharm (Basel). (2021) 14:130–55. doi: 10.3390/ph14020130
245. Allavena P, Anfray C, Ummarino A, Andon FT. Therapeutic manipulation of tumor-associated macrophages: facts and hopes from a clinical and translational perspective. Clin Cancer Res. (2021) 27:3291–7. doi: 10.1158/1078-0432.CCR-20-1679
246. Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med. (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
247. Naing A, Khalil D, Rosen O, Camidge DR, Lillie T, Ji RR, et al. First-in-human clinical outcomes with NG-350A, an anti-CD40 expressing tumor-selective vector designed to remodel immunosuppressive tumor microenvironments. J Immunother Cancer. (2024) 12. doi: 10.1136/jitc-2024-010016
248. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
249. McWhirter SM, Jefferies CA. Nucleic acid sensors as therapeutic targets for human disease. Immunity. (2020) 53:78–97. doi: 10.1016/j.immuni.2020.04.004
250. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
251. Shang S, Yang C, Chen F, Xiang RS, Zhang H, Dai SY, et al. ID1 expressing macrophages support cancer cell stemness and limit CD8(+) T cell infiltration in colorectal cancer. Nat Commun. (2023) 14:7661. doi: 10.1038/s41467-023-43548-w
252. Heumos L, Schaar AC, Lance C, Litinetskaya A, Drost F, Zappia L, et al. Best practices for single-cell analysis across modalities. Nat Rev Genet. (2023) 24:550–72. doi: 10.1038/s41576-023-00586-w
253. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
254. Valdes-Mora F, Handler K, Law AMK, Salomon R, Oakes SR, Ormandy CJ, et al. Single-cell transcriptomics in cancer immunobiology: the future of precision oncology. Front Immunol. (2018) 9:2582. doi: 10.3389/fimmu.2018.02582
255. Hao Y, Stuart T, Kowalski MH, Choudhary S, Hoffman P, Hartman A, et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. (2024) 42:293–304. doi: 10.1038/s41587-023-01767-y
256. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
257. Blair AP, Hu RK, Farah EN, Chi NC, Pollard KS, Przytycki PF, et al. Cell Layers: uncovering clustering structure in unsupervised single-cell transcriptomic analysis. Bioinform Adv. (2022) 2:vbac051. doi: 10.1093/bioadv/vbac051
258. Geng Y, Feng J, Huang H, Wang Y, Yi X, Wei S, et al. Single-cell transcriptome analysis of tumor immune microenvironment characteristics in colorectal cancer liver metastasis. Ann Transl Med. (2022) 10:1170. doi: 10.21037/atm-22-5270
259. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. (2015) 16:278. doi: 10.1186/s13059-015-0844-5
260. Cao J, Spielmann M, Ǫiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. (2019) 566:496–502. doi: 10.1038/s41586-019-0969-x
261. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. (2018) 174:1293–1308 e36. doi: 10.1016/j.cell.2018.05.060
262. Wang X, Chen J, Jia G. From dichotomy to diversity: deciphering the multifaceted roles of tumor-associated macrophages in cancer progression and therapy. Cancer Biol Med. (2023) 21:132–8. doi: 10.20892/j.issn.2095-3941.2023.0370
263. Toninelli M, Rossetti G, Pagani M. Charting the tumor microenvironment with spatial profiling technologies. Trends Cancer. (2023) 9:1085–96. doi: 10.1016/j.trecan.2023.08.004
264. Jin Y, Zuo Y, Li G, Liu W, Pan Y, Fan T, et al. Advances in spatial transcriptomics and its applications in cancer research. Mol Cancer. (2024) 23:129. doi: 10.1186/s12943-024-02040-9
265. Dries R, Zhu Ǫ, Dong R, Eng CL, Li H, Liu K, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. (2021) 22:78. doi: 10.1186/s13059-021-02286-2
266. Bergenstrahle J, Larsson L, Lundeberg J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics. (2020) 21:482. doi: 10.1186/s12864-020-06832-3
267. Palla G, Spitzer H, Klein M, Fischer D, Schaar AC, Kuemmerle LB, et al. Squidpy: a scalable framework for spatial omics analysis. Nat Methods. (2022) 19:171–8. doi: 10.1038/s41592-021-01358-2
268. Dong X, Leary JR, Yang C, Brusko MA, Brusko TM, Bacher R. Data-driven selection of analysis decisions in single-cell RNA-seq trajectory inference. Brief Bioinform. (2024) 25. doi: 10.1093/bib/bbae216
269. Meylan M, Petitprez F, Becht E, Bougouin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. (2022) 55:527–541 e5. doi: 10.1016/j.immuni.2022.02.001
270. Su P, Li O, Ke K, Jiang Z, Wu J, Wang Y, et al. Targeting tumor−associated macrophages: Critical players in tumor progression and therapeutic strategies (Review). Int J Oncol. (2024) 64. doi: 10.3892/ijo.2024.5648
271. Wang L, Jin Z, Master RP, Maharjan CK, Carelock ME, Reccoppa TBA, et al. Breast cancer stem cells: signaling pathways, cellular interactions, and therapeutic implications. Cancers (Basel). (2022) 14:3287–314. doi: 10.3390/cancers14133287
272. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. (2014) 32:381–6. doi: 10.1038/nbt.2859
273. Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. (2018) 19:477. doi: 10.1186/s12864-018-4772-0
274. Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. (2013) 31:248–58. doi: 10.1002/stem.1281
275. Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. (2019) 37:547–54. doi: 10.1038/s41587-019-0071-9
276. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
277. Liu S, Iorgulescu JB, Li S, Borji M, Barrera- Lopez IA, Shanmugam V, et al. Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response. Immunity. (2022) 55:1940–1952 e5. doi: 10.1016/j.immuni.2022.09.002
278. Hao Q, Vadgama JV, Wang P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal. (2020) 18:82. doi: 10.1186/s12964-020-00589-8
279. Manni W, Min W. Signaling pathways in the regulation of cancer stem cells and associated targeted therapy. MedComm. (2020) 3:e176. doi: 10.1002/mco2.176
280. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. (2020) 15:1484–506. doi: 10.1038/s41596-020-0292-x
281. Li M, He L, Zhu J, Zhang P, Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. (2022) 12:85. doi: 10.1186/s13578-022-00823-5
282. Zhang Y, Ding X, Zhang X, Li Y, Xu R, Li HJ, et al. Unveiling the contribution of tumor-associated macrophages in driving epithelial-mesenchymal transition: a review of mechanisms and therapeutic Strategies. Front Pharmacol. (2024) 15:1404687. doi: 10.3389/fphar.2024.1404687
283. Fan G, Xie T, Li L, Tang L, Han X, Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. (2024) 8:75. doi: 10.1038/s41698-024-00564-3
284. Cao L, Lu X, Wang X, Wu H, Miao X. From single-cell to spatial transcriptomics: decoding the glioma stem cell niche and its clinical implications. Front Immunol. (2024) 15:1475235. doi: 10.3389/fimmu.2024.1475235
285. Conde-Lopez C, Marripati D, Elkabets M, Hess J, Kurth I. Unravelling the complexity of HNSCC using single-cell transcriptomics. Cancers (Basel). (2024) 16:3265–84. doi: 10.3390/cancers16193265
286. Xu J, Yu B, Wang F, Yang J. Single-cell RNA sequencing to map tumor heterogeneity in gastric carcinogenesis paving roads to individualized therapy. Cancer Immunol Immunother. (2024) 73:233. doi: 10.1007/s00262-024-03820-4
287. Chen C, Guo Ǫ, Liu Y, Hou Ǫ, Liao M, Guo Y, et al. Single-cell and spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin Transl Med. (2023) 13:e1515. doi: 10.1002/ctm2.v13.12
288. Liang Q, Huang Y, He S, Chen K. Pathway centric analysis for single-cell RNA-seq and spatial transcriptomics data with GSDensity. Nat Commun. (2023) 14:8416. doi: 10.1038/s41467-023-44206-x
289. Mo Z, Liu D, Chen Y, Luo J, Li W, Liu J, et al. Single-cell transcriptomics reveals the role of Macrophage-Naive CD4 + T cell interaction in the immunosuppressive microenvironment of primary liver carcinoma. J Transl Med. (2022) 20:466. doi: 10.1186/s12967-022-03675-2
Keywords: cancer stem cells, TAMs, single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, signaling pathway analysis, trajectory analysis
Citation: Verona F, Di Bella S, Schirano R, Manfredi C, Angeloro F, Bozzari G, Todaro M, Giannini G, Stassi G and Veschi V (2025) Cancer stem cells and tumor-associated macrophages as mates in tumor progression: mechanisms of crosstalk and advanced bioinformatic tools to dissect their phenotypes and interaction. Front. Immunol. 16:1529847. doi: 10.3389/fimmu.2025.1529847
Received: 17 November 2024; Accepted: 17 January 2025;
Published: 06 February 2025.
Edited by:
Chuanwen Fan, Linköping University, SwedenCopyright © 2025 Verona, Di Bella, Schirano, Manfredi, Angeloro, Bozzari, Todaro, Giannini, Stassi and Veschi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronica Veschi, dmVyb25pY2EudmVzY2hpQHVuaXJvbWExLml0; Giorgio Stassi, Z2lvcmdpby5zdGFzc2lAdW5pcGEuaXQ=
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share senior authorship