 Ying Zhang
Ying Zhang Haotian Wang
Haotian Wang Zhumei Zhan2
Zhumei Zhan2 Lin Gan
Lin Gan Out Bai
Out Bai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 07 April 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1529239
This article is part of the Research TopicPrecision Immunotherapy and Novel Target Discovery in Hematological MalignancyView all 11 articles
Histone deacetylases (HDACs) are a class of epigenetic regulators that play pivotal roles in key biological processes such as cell proliferation, differentiation, metabolism, and immune regulation. Based on this, HDAC inhibitors (HDACis), as novel epigenetic-targeted therapeutic agents, have demonstrated significant antitumor potential by inducing cell cycle arrest, activating apoptosis, and modulating the immune microenvironment. Current research is focused on developing highly selective HDAC isoform inhibitors and combination therapy strategies tailored to molecular subtypes, aiming to overcome off-target effects and resistance issues associated with traditional broad-spectrum inhibitors. This review systematically elaborates on the multidimensional regulatory networks of HDACs in tumor malignancy and assesses the clinical translation progress of next-generation HDACis and their prospects in precision medicine, providing a theoretical framework and strategic reference for the development of epigenetic-targeted antitumor drugs.
In eukaryotic cells, DNA is packaged into chromatin, with the nucleosome serving as its fundamental unit. Each nucleosome consists of approximately 148 base pairs of DNA wrapped around an octamer of core histone proteins, comprising two copies each of H2A, H2B, H3, and H4 (1). This packaging typically creates a repressive environment for gene expression. Therefore, transcriptional activation often requires chromatin modifications. Histone deacetylases (HDACs) and histone acetyltransferases (HATs) are two enzymes that regulate chromatin structure and function by adding and removing acetyl groups on lysine residues of core nucleosomal histones, respectively. The acetylation of histones H3 and H4 neutralizes the positive charge of lysine residues, leading to chromatin relaxation and enhanced accessibility for transcriptional activation. In contrast, histone deacetylation promotes chromatin condensation, thereby suppressing gene transcription (2, 3). Consequently, the dynamic equilibrium between acetylation and deacetylation levels plays a pivotal role in regulating physiological processes, cellular fate determination, and the pathogenesis of diseases. Growing evidence suggests that members of the HDAC family exhibit extensive functional heterogeneity during tumorigenesis and cancer progression. These enzymes contribute to oncogenic activation, inactivation of tumor suppressor pathways, metabolic adaptation, and alterations in the immune microenvironment, collectively driving malignant progression (4). Given the tumor-promoting properties of HDACs, targeted inhibition of HDAC activity has emerged as a critical therapeutic strategy in oncology. Although first-generation pan-HDAC inhibitors (HDACis), such as Vorinostat and Romidepsin, have been clinically approved for treating specific lymphomas, their off-target effects and drug resistance issues have limited broader clinical applications (5). Current research focuses on developing subtype-selective HDACis, exploring biomarker-guided precision dosing regimens, and designing combination therapeutic strategies integrating HDACis with chemotherapy, immune checkpoint inhibitors, or targeted agents to overcome current therapeutic efficacy limitations. This review systematically elucidates the pathological regulatory networks of HDACs in malignancies, dissects the mechanisms of action and pharmacological properties of HDACis, summarizes advancements in preclinical and clinical studies, and discusses future directions to optimize epigenetic-targeted therapies, thereby providing a scientific foundation for refining antitumor strategies.
The enzymatic capacity to remove acetyl groups from histones was first documented in 1969 through biochemical characterization of calf thymus extracts (6), though initial purification efforts using conventional chromatographic techniques proved unsuccessful. The field underwent transformative advancement in 1996 with the molecular cloning of HDAC1, the first bona fide histone deacetylase identified (7). Subsequent genomic analyses revealed 18 human HDACs categorized into four distinct classes based on sequence homology, subcellular localization, and enzymatic cofactor requirements. Notably, Classes I, II, and IV enzymes (HDAC1-11) are zinc-dependent hydrolases, whereas Class III sirtuins (SIRT1-7) utilize NAD+ as an essential cofactor for their catalytic activity (Figure 1). This phylogenetic classification reflects both conserved catalytic domains and divergent regulatory mechanisms across evolutionary lineages.
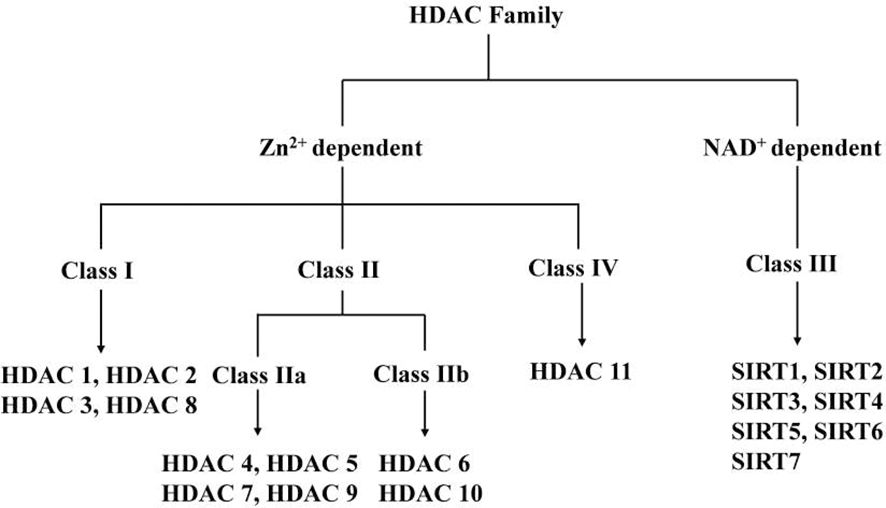
Figure 1. HDAC Family.
The Class I HDACs (HDAC1, 2, 3 and 8) are most closely related to the yeast (Saccharomyces cerevisiae) transcriptional regulator Rpd3 (8). It has now been widely stated in the literature that Class I HDACs are located in the nucleus and are widely expressed. More thorough findings reveal that HDAC3 expression is restricted to certain tissues, and HDAC1, HDAC2, HDAC3, and HDAC8 can localize in the cytoplasm or specialized cellular organelles (9).
The Class II HDACs have homology to yeast HDAC1, and are further subdivided into two subclasses, IIa and IIb, based on sequence homology and domain organization. The Class IIa HDACs, HDAC4, -5, -7, and -9, contain a highly conserved C-terminal deacetylase catalytic domain (*420 amino acids) homologous to yHda1 and share an N-terminal domain (*450–600 amino acids) with no similarity to HDACs in other classes. The N-terminal domain mediates interactions with the myocyte enhancer factor 2 transcription factor family, transcriptional corepressor C-terminal binding protein, and others. Class IIa HDACs shuttle between the cytoplasm and nucleus and their expression is tissue-specific. The Class IIb HDACs, HDAC6 and -10 are characterized by the presence of two catalytic HDAC domains arranged in tandem. Class IIb HDACs are predominantly cytoplasmic and expressed in a limited number of cell types (10, 11).
Class III HDACs are distinct from Class I and II and are homologous of the yeast silent information regulator 2 (Sir2). Sir2 enzymes (or sirtuins) are NAD (+)-dependent deacetylases that regulate gene silencing, aging, and energy metabolism. Earlier experiments showed that overexpression of Sir2 in yeast induced global deacetylation of histones, suggesting that Sir2 is an HDAC (12). Later, in a study by Frye et al. (13) the bacterial homolog of Sir2, cobB, was found to have ribosyltransferase activity, leading to experiments showing that Sir2 can also transfer adenosine-ribose (ADP-ribose) from nicotinamide adenine nucleotide (NAD). Subsequently, it was confirmed that Sir2 is an NAD-dependent HDAC and that the ADP-ribosylation of acetylated lysine residues is an intermediate state of the Sir2-catalyzed enzymatic reaction (14). Only Class III enzymes uses NAD as a cofactor. Therefore, they are referred to as NAD-dependent HDACs. Each of the seven mammalian sirtuin proteins (called SIRT1-SIRT7) has a different subcellular localization. SIRT1, SIRT6 and SIRT7 are localized in the nucleus, while SIRT2 is mainly cellular membrane and SIRT3, SIRT4 and SIRT5 seem to be present only in mitochondria. Although much is known about SIRT1, relatively little is known about the other Sirt family proteins (15). What is interesting is that some sirtuins have been recently shown to have additional enzymatic activities in addition to deacetylation, such as SIRT3 showing additional norylase activity, SIRT4 showing ADP-ribosyltransferase activity, SIRT5 with desmalonylase, desuccinylase and deglutaminase activities, and SIRT6 showing deacetylase and demyristoylase activities, which were further elaborated in the study by Chen et al (16).
It cannot be ignored that the role of some current sirtuins in tumorigenesis is still controversial. In a study by Heltweg et al. (17) it was demonstrated that SIRT1 is expressed at higher levels in cancer cells and promotes tumorigenesis through deacetylation of lysine 382 in Burkitt’s lymphoma cells. However, in a mouse model of cancer constructed by Firestein et al. (18) increased SIRT1 expression inhibited cell proliferation and tumor formation.
HDAC11 was first reported in 2002 only and is the smallest of the HDAC isoforms, with its catalytic domain accounting for > 80% of the protein sequence. It is mainly localized in the nucleus and plays an important role in immune regulation as a transcriptional regulator (19). HDAC11 is predominantly expressed in smooth muscle, heart, kidney and brain tissues (20), and is likely to be preferentially expressed in the gallbladder (21).
The location and biological functions of HDACs are detailed in Table 1.
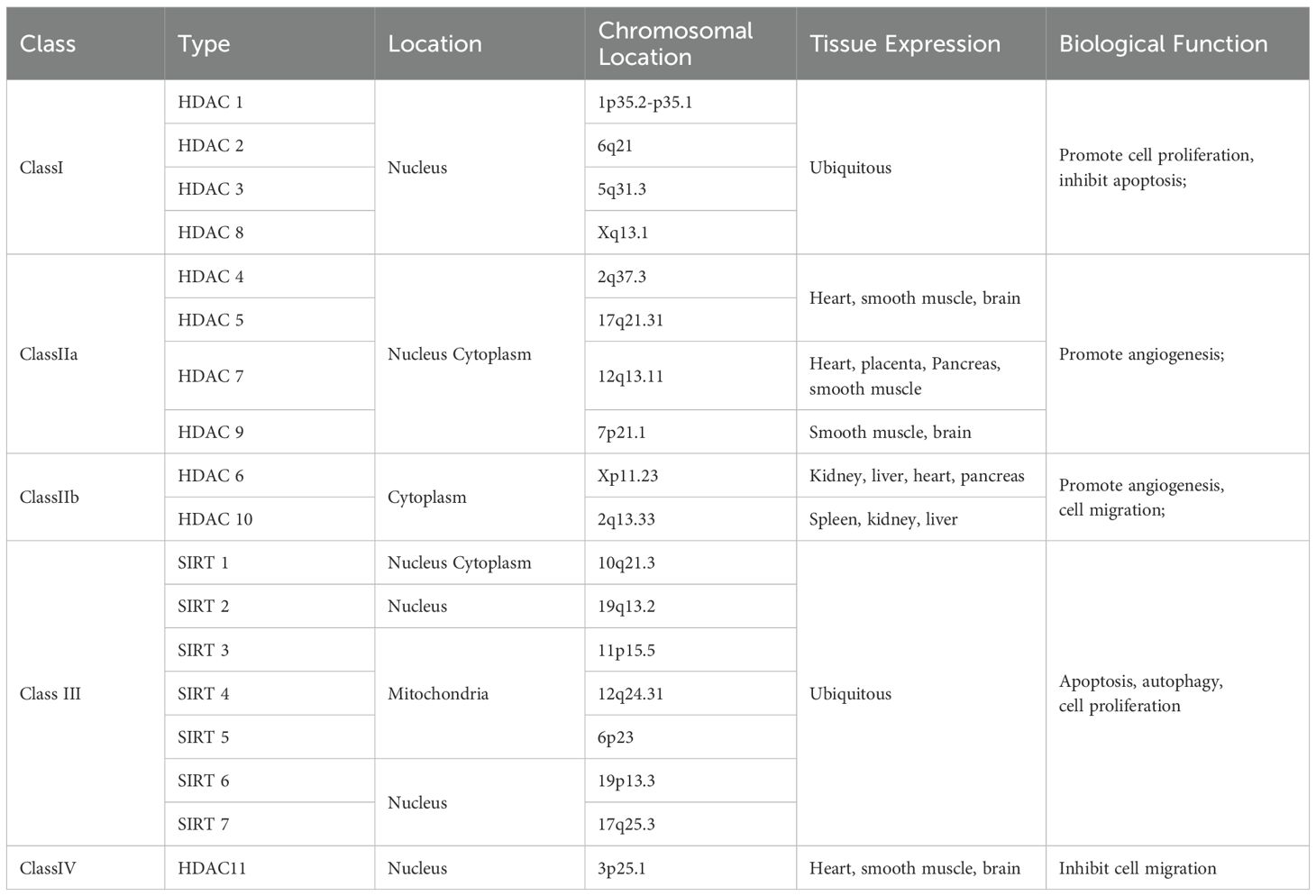
Table 1. Location and biological function of HDACs.
There are several main classes of HDAC inhibitors (HDACis), including those based on hydroxamic acid (e.g., suberoylanilide hydroxamic (SAHA), pyroxamide, trichostatin A (TSA), oxamflatin, cyclic hydroxamic acid-containing peptides (CHAP)s; LAQ824; BL1521); cyclic tetra/peptide (e.g., depsipeptide, trapoxin, apicidin, CHAPs); synthetic benzamide derivatives (e.g., MS-275 and CI-994); cyclic tetrapeptides (e.g., depsipeptide, trapoxin, apicidin, CHAPs); and short-chain fatty acids (e.g., sodium butyrate (SB), AN-9; phenylbutyrate (PB);phenylacetate (PA); valproic acid) (22).
The recent discovery of hydrazide-based HDACi further increases the diversity of HDACi (23, 24). Four HDACi are currently approved by the FDA (25–29) (Table 2). The first class of HDACi is hydroxyamide acid-based HDACi. Vorinostat is well-known as the first marketed HDACi with nanomolar affinity for HDACs. Initially, relevant studies suggested that this compound was able to inhibit all HDACs. However, further testing demonstrated that it could only inhibit HDAC 1, 2, 3, and 6 at reasonable concentrations (30). Currently, vorinostat is FDA-approved for the treatment of cutaneous T-cell lymphoma (CTCL). Amino-benzamide-based HDACi are the first inhibitors to selectively target Class I HDACs (31), and enzyme kinetic studies have shown that the aminobenzamide motif has a tight binding mechanism (slow-on/slow-off) and differs from classical fast on/fast off kinetics associated with hydroxamide acid-based HDACi (32–34). Entinostat is the first amino-benzamide-based HDACi to reach clinical trials (35). Recently, Wang et al (24) reported a novel HDACi family with a previously unutilized motif in HDACi.
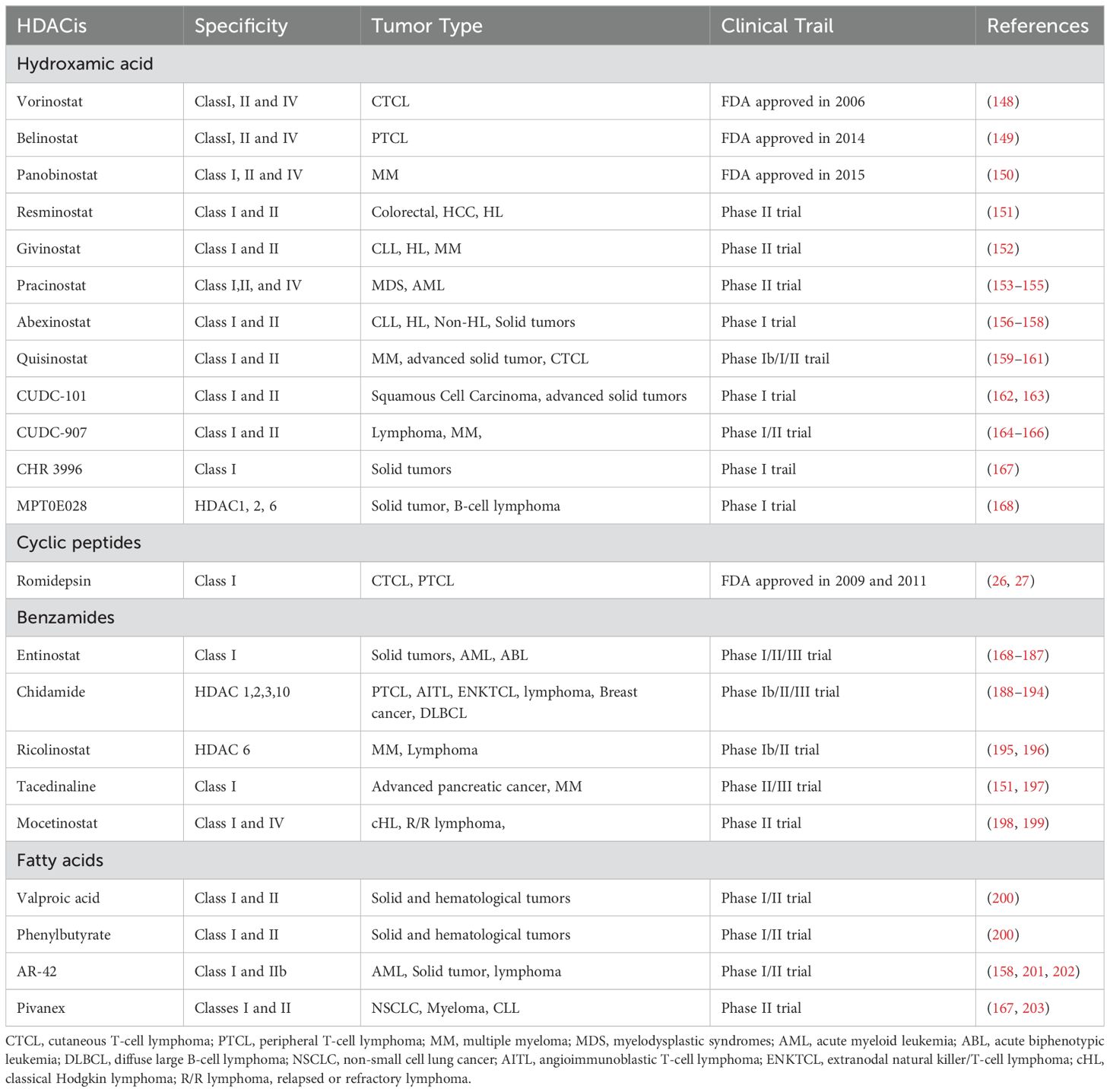
Table 2. HDACis currently under clinical investigations.
While the substrate specificity and biological functions of individual HDAC isoforms remain incompletely characterized, accumulating evidence has unequivocally established HDACs as central epigenetic orchestrators of oncogenic processes. Through dynamic deacetylation of both histone (e.g., H3K9, H4K16) and non-histone substrates (e.g., p53, STAT3, HIF-1α), HDACs mechanistically govern six hallmark cancer pathways: (1) cell cycle progression via CDK inhibitor silencing, (2) apoptosis resistance through Bcl-2 family modulation, (3) DNA damage tolerance by repair factor inactivation, (4) autophagic flux dysregulation, (5), VEGF-driven angiogenesis, and (6) EMT-mediated metastatic dissemination. In particular, recent studies have given great attention to the epigenetic regulation involved in HDAC. This functional pleiotropy positions HDACs as master regulators linking epigenetic plasticity to tumor microenvironment remodeling, but precise mechanistic mapping requires systematic subtype and context-specific interrogation (Figure 2).
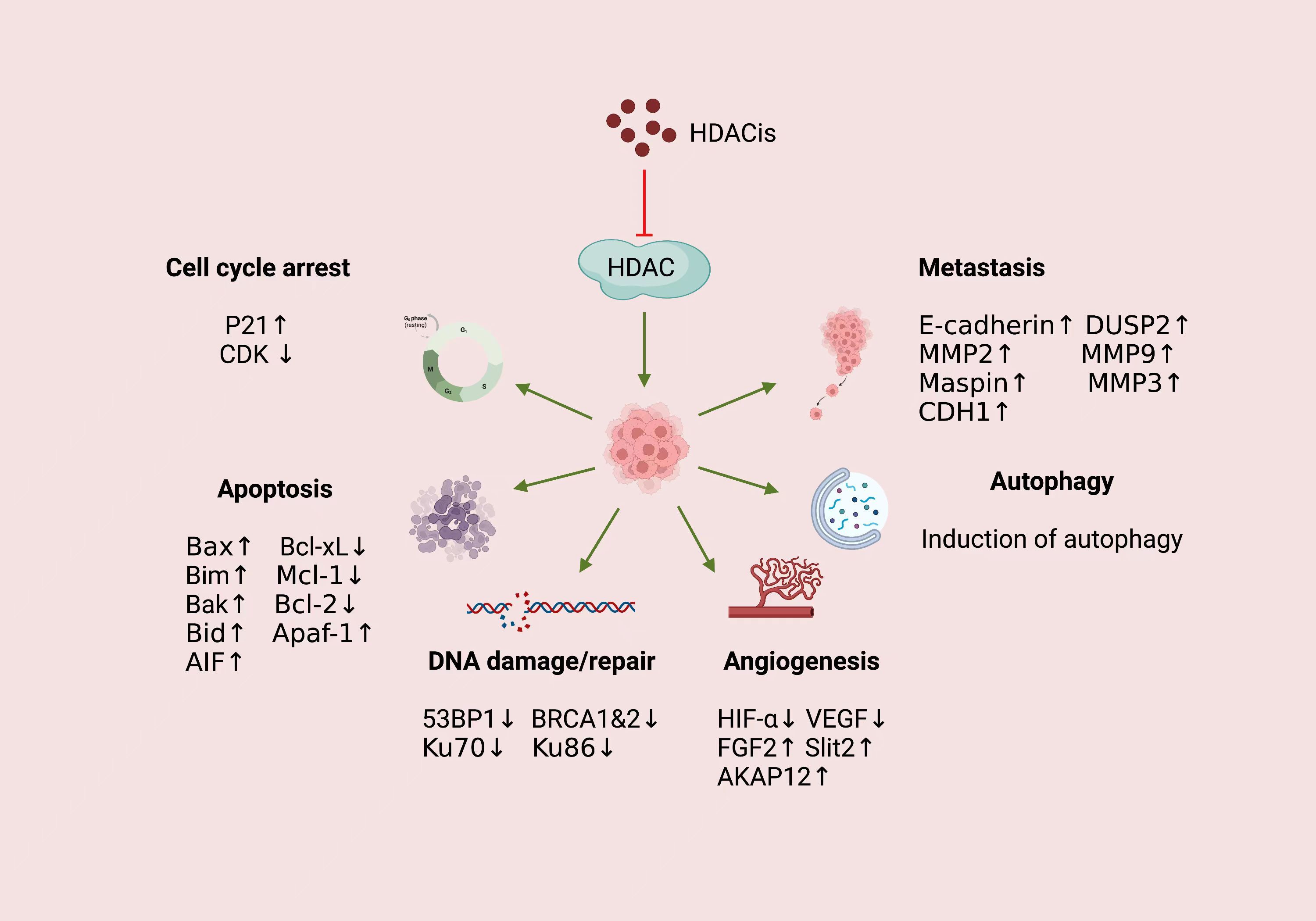
Figure 2. Mechanism of action of HDACis in cancer.
The precise coordination of cell cycle progression is fundamental to maintaining genomic stability and cellular homeostasis, governed by a tightly regulated network of cyclin-dependent kinases (CDKs), checkpoint proteins, and epigenetic modifiers. Among these regulators, histone deacetylases (HDACs) have emerged as critical players that bridge epigenetic dynamics with cell cycle control. By modulating the acetylation status of histones (e.g., H3K9, H4K16) and non-histone substrates (e.g., p53, E2F, Rb), HDACs exert spatiotemporal control over the transcription of cell cycle-related genes, DNA replication licensing, and mitotic fidelity.
The G1/S phase transition represents a critical checkpoint in the cell cycle, determining whether a cell commits to proliferation or exits the cycle into quiescence. Previous studies have confirmed that HDAC1 and HDAC2 bind to the promoter regions of the p21 and p57 genes, thereby inhibiting their expression and regulating the transition from the G1 phase to the S phase of the cell cycle (5). Fan et al. found that knockdown of HDAC5 led to a significant upregulation of p21 and a notable downregulation of cyclin D1 and CDK2/4/6, resulting in a strong G1-phase cell cycle arrest and inhibiting cell proliferation (36). Similarly, Qiu et al. (37) demonstrated that HDACi induce G1 arrest by upregulating p21. HDACi also reduce CDK activity by downregulating cyclins, leading to Rb dephosphorylation and inhibition of E2F activity, affecting G1 progression and G1/S transition (38). While HDACi-induced growth arrest is mainly linked to p21 induction, some evidence points to a p21-independent pathway. For instance, Trichostatin A (TSA) activates the p15Ink4b gene, inducing growth inhibition in colon cancer cells lacking p21 (39). Additionally, a study by Yamashita et al. (40) showed that TSA increased acetylation of histones H3 and H4 in hepatocellular carcinoma cells, causing G0/G1 phase arrest.
In addition to regulating the G1/S transition, HDACi have also been shown to interfere with the G2/M transition. HDAC1 knockdown in tumor cells disrupts G2/M progression and inhibits cell growth (41). Li et al. (42) demonstrated that HDAC10 controls G2/M transition by regulating cyclin A2 expression. Similarly, Kim et al. (43) showed that MHY218, a hydroxamic acid derivative, induces G2/M arrest in colon cancer cells through p21 upregulation, independent of p53. TSA further supports this by increasing p21 levels while decreasing Cyclin B1, Plk1, and Survivin, delaying G2/M progression (44). Interestingly, suberoylanilide hydroxamic acid (SAHA) causes a G1 phase block at low concentrations, but both G1 and G2/M blocks at higher concentrations (45).
Beyond transcriptional suppression of cell cycle genes at G1/S and G2/M checkpoints, HDACs may also regulate the cell cycle through transcription-independent mechanisms. For instance, the HDAC3-AKAP95/HA95 Aurora B pathway is essential for normal mitosis, as shown by Li et al (46). Additionally, LBH589, an HDAC inhibitor, prevents the degradation of Aurora A and B kinases by inhibiting HDAC3 and HDAC6, leading to G2/M arrest and apoptosis in renal cancer cells (47). HDAC3 also regulates the stability of cyclin A via acetylation, which is crucial for S phase progression and mitosis entry (48).
Overall, HDACi can block the cell cycle at the G1/S or G2/M phases, highlighting HDAC as a potential therapeutic target for abnormal cell growth in cancer.
The regulation of apoptosis—a genetically programmed cell death mechanism—is intricately governed by the dynamic interplay between pro-survival and pro-apoptotic signals, with HDACs emerging as pivotal modulators of this life-or-death equilibrium. HDACs exert dualistic control over apoptotic pathways through both epigenetic silencing of apoptosis-related genes and direct post-translational modification of key apoptotic executors. By deacetylating histones (e.g., H3K9, H4K16) at promoters of pro-apoptotic factors (BAX, PUMA, NOXA) and tumor suppressors (p53), HDACs enforce transcriptional repression under homeostatic conditions, favoring cell survival. Conversely, stress stimuli (e.g., DNA damage, oxidative stress) trigger HDAC inhibition or degradation, leading to chromatin relaxation and reactivation of apoptotic machinery. Apoptosis has been shown to be induced through two major signaling pathways, referred to as the endogenous and exogenous pathways, mediated by mitochondria and death receptors, respectively (49). This section Outlines the relationship between HDAC and apoptosis in terms of different apoptotic pathways, dissects their significance in the development and treatment of cancer, and evaluates strategies for selectively manipulating HDAC activity to restore apoptosis.
The extrinsic apoptotic pathway, initiated by extracellular death ligands (e.g., FasL, TRAIL) binding to transmembrane receptors (e.g., Fas, DR4/DR5), is tightly regulated by HDACs through both epigenetic and non-epigenetic mechanisms. HDACs modulate this pathway by altering the expression of death receptors, suppressing pro-apoptotic signaling, or directly modifying key apoptotic executors, thereby influencing cellular sensitivity to extrinsic death stimuli. The regulation of HDACi on TRAIL-induced apoptosis has been described in detail in the review of Fulda et al. (50), including the up-regulation of TRAIL receptor expression on the cell surface and the down-regulation of anti-apoptotic proteins (such as cFLIP) by HDACi, so that cancer cells are in a state of TRAIL-triggered apoptosis. Xia et al. (51) confirmed that downregulation of c-Jun expression in response to osmotic pressure was caused by transcriptional repression through caspase-7-dependent HDAC3 cleavage, which involved FAS ligand and MEK2-dependent caspase-8 activation. The downregulation of c-Jun promoted the osmotic stress-induced apoptosis.
The intrinsic (mitochondrial) apoptotic pathway, activated by intracellular stress signals such as DNA damage, oxidative stress, or metabolic imbalance, is critically regulated by HDACs through their dual roles in chromatin remodeling and direct modulation of mitochondrial apoptosis executors. HDACs orchestrate the balance between pro-survival and pro-apoptotic Bcl-2 family proteins, govern mitochondrial outer membrane permeabilization (MOMP), and fine-tune caspase activation, thereby determining cellular commitment to life or death. Activation of the intrinsic apoptotic pathway appears to be the major mechanism by which HDACi induces tumor cell death. In many cases, HDACi activates the intrinsic pathway by up-regulating some BH3-only pro-apoptotic Bcl-2 family genes including Bim, Bid and Bmf (52). Furthermore, recent studies have shown that HDACi can comprehensively alter the expression of pro-survival and pro-apoptotic Bcl-2 family genes, suggesting a pro-apoptotic biological response (53). Emerging evidence highlights the tumor-suppressive effects of HDAC2 depletion across cancer types. Jung et al. (54) demonstrated that HDAC2 silencing inhibits proliferation and induces apoptosis in human lung cancer cells via coordinated activation of p53 and Bax alongside Bcl-2 downregulation. Notably, Kim et al. (55) further revealed that targeted HDAC2 inactivation in gastric cancer cells restores pro-apoptotic activity of Bax, AIF, and Apaf-1 while suppressing Bcl-2, independent of p53 protein level alterations. This suggests HDAC2 ablation triggers caspase-dependent apoptosis through p53-independent mechanisms. However, the role of p53 in HDACi-mediated apoptosis remains context-dependent (56–59). Sonnemann et al. (60) observed that vorinostat, apicidin, and valproic acid exert antitumor effects largely independent of p53 status, whereas entinostat-induced cytotoxicity partially relies on p53 functionality. These findings collectively underscore the heterogeneous p53 dependency among HDACis, emphasizing the need for compound-specific mechanistic characterization. Notably, MHY218, an HDACi, significantly increased the Bax/Bcl-2 ratio in a concentration-dependent manner and activated caspases-3, -8, and -9, indicating that it induces apoptosis in colon cancer cells via both intrinsic and extrinsic pathways (43). It seems that the different effects of HDACi in the same cell type may be caused by the structural characteristics of the different HDACi.
The maintenance of genomic stability relies on a sophisticated network of DNA damage response (DDR) pathways that detect, signal, and repair lesions to prevent mutagenesis and cell death. HDACs, traditionally recognized as chromatin-modifying enzymes, have recently been implicated as critical regulators of DDR through their dual roles in modulating chromatin architecture and direct interactions with DNA repair machinery. DNA double strand breaks (DSBs) are lethal lesions detected by DNA damage signaling mechanisms, and most are repaired by non-homologous end-joining (NHEJ) or homologous recombination (HR) (61). NHEJ is a highly efficient process that joins DNA ends with minimal processing (62), while HR uses undamaged homologous sequences to ensure precise repair (63). Miller et al. (64) demonstrated that loss of HDAC1 or HDAC2 renders cells highly sensitive to DNA-damaging agents and exhibit persistent DNA damage signals, phenotypes that confirm defective DSBs repair, particularly through the NHEJ pathway. Specifically, depletion of HDAC9 or HDAC10 by RNA interference can specifically inhibit HR, leading to increased sensitivity to mitomycin C (65). Ataxia-telangiectasia mutated (ATM) is a master regulator of DNA damage response, promoting the activation of BRCA1, CHK2, and p53, which induces DNA repair response genes such as p21, GADD45A, and RRM2B. Previous studies by Thurn et al. (66) confirmed that selective silencing of HDAC1 and HDAC2 in vitro and in vivo is sufficient to modulate ATM activation, reduce GADD45A and RRM2B induction, and increase sensitivity to DNA strand breaks. Moreover, another study (67) found that silencing HDAC2 gene could alleviate the ATM/p53-mediated cell death pathway in osteosarcoma U2OS cells in response to adriamycin, confirming that HDAC2 is involved in the early molecular events of DNA damage response and is a coactivator of p53. HDAC6 has long been thought to play a unique role due to its cytosolic localization and ability to deacetylate non-histone proteins (68). The study by Yang et al. (69) confirmed that HDAC6 can regulate DDR-related genes by affecting Sp1 expression, eliminating DNA damage induced by MPT0B291, an HDAC6 inhibitor, providing evidence for MPT0B291 as a potential compound for glioblastoma (GBM) therapy. Strikingly, recent studies have revealed that HDAC6 functions as a valine sensor, which is retained in the nucleus upon valine deprivation, where it binds to and deacetylates ten-eleven translocation 2 (TET2). This process promotes DNA damage through thymine DNA glycosylase (TDG)-driven excision, thereby unveiling a novel therapeutic strategy for cancer treatment (70). In particular, in the latest study by Yao et al. (71), IHCH9033, a new selective class I HDACi, can selectively inhibit DNA repair in FLT3-ITD AML cells, leading to DNA damage accumulation and overcoming resistance to FLT3 inhibitors. Research on HDACi in DNA damage and repair is still developing, but it is known that therapeutic doses of HDACi not only induce DNA damage but also impair repair mechanisms, sensitizing cells to ionizing radiation (IR), topoisomerase inhibitors, and cisplatin (72–74).
Mammalian sirtuins (SIRT 1-7), which belong to NAD+ -dependent class III HDACs, have different modes of action, targets and subcellular compartments, and play unique roles in DNA damage and repair. SIRT1, a NAD+-dependent deacetylase, has been demonstrated to directly interact with and deacetylate multiple core DNA repair factors, including Ku70 (15) (critical for non-homologous end joining), apurinic/apyrimidinic endonuclease-1 (APE1) (base excision repair effector) (75), NBS1 (nibrin/p95, a central component of the MRE11-RAD50-NBS1 [MRN] complex for DNA damage sensing) (76), and xeroderma pigmentosum group A (XPA) (key mediator of nucleotide excision repair) (77). This post-translational modification modulates their enzymatic activities, subcellular localization, and recruitment to DNA lesion sites, thereby fine-tuning repair fidelity across distinct DNA damage response pathways. The relationship between SIRT1 and DNA damage response has been elaborated in recent reviews, including the interaction with different proteins in the major DNA repair mechanisms and DDR pathway, recruiting them to DNA damage foci or activating proteins involved in DNA repair through deacetylation (78). Previous studies have found that SIRT5 is highly expressed in colorectal cancer (CRC), and knockdown of SIRT5 impairs the production of ribo-5-phosphate, which is required for nucleotide synthesis, resulting in continuous and irreparable DNA damage. This results in CRC cell cycle arrest and significant apoptosis, indicating that SIRT5 can be a promising target for CRC therapy (79). SIRT6 plays a critical role in DNA repair by enhancing resistance to DNA damage and maintaining genomic stability, particularly through base excision repair (BER), as demonstrated by Mostoslavsky et al. (80). Moreover, SIRT6-deficient mice exhibit decreased chromatin-associated levels of SNF2H in specific tissues, a finding that correlates with elevated DNA damage (81). In addition, research by Mao et al. (82) has shown that SIRT6 interacts with poly(ADP-ribose) polymerase 1 (PARP1) and catalyzes its mono-ADP-ribosylation at lysine 521, thereby enhancing PARP1 activity and facilitating the repair of DSBs under oxidative stress conditions. Members of the SIRT family play multifaceted roles in DNA damage repair, exerting effects that are equally as important as those of classical HDACs.
The interplay between HDACs and autophagy represents a pivotal nexus in cellular adaptation to metabolic stress, genomic instability, and environmental challenges. Emerging evidence underscores HDACs as dual regulators of autophagy, operating through both epigenetic and non-epigenetic mechanisms to fine-tune autophagic flux. By deacetylating histones (e.g., H3K9, H4K16) at promoters of autophagy-related genes (ATG5, LC3, BECN1), HDACs suppress basal autophagy under nutrient-replete conditions, while their inhibition or stress-induced degradation triggers transcriptional activation of autophagic machinery (83–85). The effects of autophagy on cancer development and the interaction between autophagy and HDAC have been described in detail in the latest review by Koeneke et al (86).
Early work by Moresi et al. (87) demonstrated that the simultaneous deletion of HDAC1 and HDAC2 in mice inhibited autophagosome formation. Similarly, Kang et al. (88) found that knockdown of HDAC4 induced autophagy, increasing LC3-II, Beclin-1, and ATG7 levels. Not only that, the researchers found that LC3-II increased over time when HDAC5 was depleted in breast cancer cells, and this effect was enhanced with the use of lysosomal inhibitors, suggesting that HDAC5 downregulation increased autophagic flux (89). HDAC6 is involved in the regulation of autophagy at multiple levels, including post-translational modification (PTM) of transcription factors involved in autophagy-related transcription (90, 91), formation of aggregates routinely cleared through the autophagic pathway (92, 93), and transport and degradation of autophagosomes (94). Liu et al. (95) observed that the level of acetylated phosphotidylethanolamine (PE)-conjugated LC3B (LC3B-II) was increased in cells treated with tubacin, a specific inhibitor of HDAC6, under normal medium. However, tubacin only partially inhibited serum starvation-induced deacetylation of LC3B-II, suggesting that HDAC6 is not the only deacetylase acting on LC3B-III during serum starvation-induced autophagy. Therefore, HDAC6 depletion impairs serum starvation-induced autophagy. The post-transcriptional modification (PTM) of autophagy-related transcription factors, such as transcription factor EB (TFEB) and forkhead box 1 (FOXO1), notably affects their activity and thus regulates the autophagy-lysosome pathway (96), and current studies have reported that HDAC6 deacetylates TFEB and FOXO1 to reduce their activity and inhibit autophagy (91, 97, 98). In addition, in a study (99) on breast cancer, it was also found that HDAC6 knockdown resulted in reduced LC3B protein and reduced autophagy. Another work (100) showed that productive autophagy with efficient autophagosome-lysosome fusion is dependent on HDAC10 and that depletion of HDAC10 enhances the sensitivity of tumor cells to chemotherapeutic agents such as doxorubicin.
Sirtuins on autophagy in cancer has been detailed in a recent review by Aventaggiato et al. (101) and will not be repeated here.
Hypoxia is a common characteristic of many solid tumors, where tumor cells in hypoxic regions adapt to low oxygen conditions by activating several survival pathways. Among these, the activation of the hypoxia-inducible factor-1 (HIF-1) transcription factor is the most well-known mechanism employed by hypoxic cells in this hostile microenvironment. Notably, there is a strong correlation between HIF-1 and tumor angiogenesis (102). As a result, drugs that inhibit HIF-1 expression hold significant potential as antitumor agents. HIF-1 is a heterodimeric transcription factor, consisting of two subunits: HIF-1α (or its analogs HIF-2α and HIF-3α) and HIF-1β. Histone deacetylase inhibitors (HDACIs) have been shown to significantly reduce HIF-1α expression and are currently being evaluated in clinical trials, partly due to their potent anti-angiogenic effects. However, the precise mechanism by which HDACIs function as HIF-1α inhibitors remains unclear.
The research of Yoo et al. (103) demonstrated that the expression of metastasis-associated protein 1 (MTA1) is strongly induced under hypoxic conditions, and MTA1 promotes the deacetylation of HIF-1α by upregulating histone deacetylase 1 (HDAC1). In a separate study by Geng et al. (104), it was shown that acetylation of the HIF-1α protein increased with HDAC4 shRNA and decreased with HDAC4 overexpression. In contrast, HDAC5 and 6 promote the maturation and stabilization of HIF-1α by deacetylating its chaperones HSP70 and HSP90 (105, 106). Lim et al. (107) observed that SIRT1-mediated deacetylation of HIF-1α at Lys674 inhibits HIF-1α activity by preventing the recruitment of p300. Under hypoxic conditions, SIRT1 inhibition creates a positive feedback loop that maintains high levels of HIF-1 activity.
Vascular endothelial growth factor (VEGF) is a key angiogenic factor that promotes angiogenesis in various pathological conditions, including inflammation, ischemic diseases, and cancer. In a report by Ray et al. (108) involving breast cancer, it was found that Kruppel-like factor-4 (KLF-4) recruits HDAC2 and HDAC3 at the VEGF promoter and represses their transcription, and that upregulation of VEGF in cancer is associated with loss of KLF-4-HDAC-mediated transcriptional repression. In another research (109), it was confirmed that silencing of HDAC5 increased the expression of fibroblast growth factor 2 (FGF2) and angiogenesis directing factors including Slit2. Kaluza et al. (110) found that HDAC6 interacts with and deacetylates the actin remodeling protein cortactin in endothelial cells (ECs), thereby regulating endothelial cell migration and germination. However, in another study, it was found that knockdown of HDAC6 significantly upregulated the expression of HIF-1α and VEGFA in vivo and in vitro and promoted HIF-1α-mediated hepatocellular carcinoma (HCC) angiogenesis (111). Turtoi et al. (112) were the first to show that HDAC7 epigenetically targets the AKAP12 tumor/angiogenesis suppressor gene. Not surprisingly, HDAC regulates angiogenesis through a variety of pro- and anti-angiogenic factors.
Epithelial-mesenchymal transition (EMT) is a critical process in cancer cell invasion and metastasis, with recent studies highlighting the role of histone deacetylases (HDACs) in regulating EMT across various cancers. EMT is marked by the loss of the epithelial cell marker E-cadherin (CDH1), and several transcriptional repressors of CDH1 have been identified, including Snail, Slug, Twist, ZEB1, and ZEB2. The breakdown of the basement membrane (BM) barrier allows cancer cells to directly invade the surrounding stromal region, a process driven by active proteolysis, primarily through the action of matrix metalloproteinases (MMPs) (113).
The recruitment of HDACs to the CDH1 promoter has been shown to be regulated by the transcription factor ZEB1, as first demonstrated by Aghdassi et al. in their study on pancreatic cancer (114). In research on pancreatic cancer (115), the genetic inactivation of E-cadherin (encoded by the CDH1 gene) was found to induce EMT and promote metastasis in vivo. The silencing of E-cadherin was mediated by a transcriptional repressor complex involving Snail, HDAC1, and HDAC2. Additionally, this Snail/HDAC1/HDAC2 complex is essential for EZH2-mediated repression of CDH1 in nasopharyngeal carcinoma cells (116). A previous study (117) confirmed that class I HDAC inhibitors enhance the acetylation of Y-box binding protein 1 (YB-1) and increase oxidative stress, thereby blocking sarcoma metastasis. More recently, it was shown that DNTTIP1 represses DUSP2 gene expression by recruiting HDAC1 to its promoter, maintaining the deacetylated state of histone H3K27 (118), and downregulation of DUSP2 leads to abnormal activation of ERK signaling and elevated MMP2 levels, which promote metastasis in nasopharyngeal carcinoma (NPC). In a research by Ma et al. (119), the cytoplasmic expression of HDAC3 was found to be upregulated in brain metastases from breast cancer, while its nuclear expression was conversely downregulated. This suggests that HDAC3 plays a key role in the development and progression of brain metastases in breast cancer patients, though the study did not explore the underlying mechanisms in detail. Additionally, heat shock protein 90 (HSP90), another non-histone substrate of HDAC6, is primarily responsible for promoting protein maturation and maintaining protein structure (120), which is crucial for the stability and function of proteins involved in tumor metastasis (121). Recent studies (122) have shown that targeted inhibition of HDAC6 increases the acetylation of HSP90, which weakens the binding between HSP90 and ATP, thereby reducing the interaction between chaperone proteins and oncogenes. Additionally, cytoplasmic linker protein 170 (CLIP-170), a microtubule-binding protein, regulates cell motility by modulating microtubule dynamics. In a study by Li et al. (123) HDAC6 was found to interact with CLIP-170 and the two proteins acted together to stimulate the migration of pancreatic cancer cells. Additionally, HDAC6, a novel estrogen-regulated gene, was found to have increased expression in estrogen receptor-positive breast cancer MCF-7 cells, as reported by Saji et al (124). Elevated HDAC6 expression enhanced cell motility by promoting its binding to α-tubulin and increasing microtubule (MT) activity. This finding was further confirmed in a separate study on neuroblastoma (125). HDAC8 is highly expressed in breast cancer compared to other types of cancers. In a study (126) triple-negative breast cancer (TNBC), HDAC8 was found to promote TNBC cell migration by regulating Hippo-YAP signaling. Additionally, HDAC8 was shown to drive breast cancer cell dissemination through the AKT/GSK-3β/Snail signaling pathway (127). Not only that, HDAC8 cooperates with the SMAD3/4 complex to inhibit SIRT7 and promote cell survival and migration (128). HDAC8 promotes cancer metastasis by suppressing maspin expression in prostate cancer (129). HDAC8 also promotes glioma migration by regulating the acetylation levels of α-tubulin (130). Additionally, the long non-coding RNA (lncRNA) ID2-AS1, which downregulates inhibitor of DNA binding 2 (ID2), enhanced ID2 transcription by blocking HDAC8 occupancy at the ID2 enhancer region, thereby inhibiting hepatocellular carcinoma (HCC) invasion and migration both in vitro and in vivo (131). In a study by Song et al. (132) on cervical cancer, it was confirmed that HDAC10 suppresses the expression of MMP2 and MMP9, genes known to be critical for cancer cell invasion and metastasis. Furthermore, HDAC11 was found to inhibit the migration and invasion of colorectal cancer cells by downregulating MMP3 expression (133).
In prostate cancer, SIRT1 was found to promote cell migration in vitro and metastasis in vivo by synergistically inhibiting CDH1 transcription alongside ZEB1 (134). Furthermore, a study by Eades et al. (135) on breast cancer showed that SIRT1 overexpression was associated with reduced levels of miR-200a, which normally acts as a negative regulator of SIRT1 and suppresses EMT. A similar mechanism was identified in oral cancer, where research by Chen et al. (136) demonstrated the key role of the SIRT1/Smad4/MMP7 pathway in the EMT process.
HDACs, beyond their canonical roles in chromatin remodeling and transcriptional repression, have emerged as pivotal regulators of immune cell differentiation, activation, and functional polarization. By dynamically modulating the acetylation status of both histones (e.g., H3K9, H4K16) and non-histone immune-related proteins (e.g., STATs, NF-κB, Foxp3), HDACs fine-tune the expression of cytokines, chemokines, and immune checkpoint molecules, thereby shaping innate and adaptive immune responses. Early studies by Xiao et al. (137) have confirmed that HDAC5 can reduce immune responses and the de novo expansion of T regulatory (Treg) cells, highlighting the importance of HDAC5 in antitumor immune responses. Recent research has also found that HDAC5 regulates PD-L1 expression by directly interacting with NF-κB p65. Therefore, silencing or inhibiting HDAC5 in pancreatic ductal adenocarcinoma can sensitize tumors to immune checkpoint blockade therapy (138). Foxp3 is a key transcription factor of Treg cells, and its expression and activity level are determined by post-translational modification. In the transplantation environment, regulating the acetylation or deacetylation of key lysine residues of Foxp3 can promote its stability and function, thus regulating the generation and activity of Tregs. The role of HDACs in regulating Treg function has been described in detail in the latest review by Wang et al (139). In particular, the latest research found that targeting HDAC3 could enhance CXCL12 secretion through the ATF3 dependent pathway, thereby stimulating the recruitment and activation of NK cells and inhibiting the progression of T-cell lymphoma (140). Regarding HDACi, ACY241, a selective inhibitor of HDAC6, has been shown to significantly reduce the frequency of CD138+MM cells, CD4+CD25+FoxP3+ regulatory T cells, and HLA-DRLow/-CD11b+CD33+ myeloid-derived suppressor cells. Reduced immune checkpoint PD1/PD-L1 expression in CD8+T cells and bone marrow cells from myeloma patients. ACY241 increases the expression of B7 (CD80, CD86) and MHC (class I, class II) in tumor and dendritic cells. ACY241 also enhanced the antitumor activity of antigen-specific cytotoxic T lymphocytes (CTL) in a dose - and time-dependent manner, as indicated by increased production of perforin/CD107a, IFN-γ/IL-2/TNF-α, and antigen-specific central memory CD8+ T cells (141).
Stem cells, with their unparalleled capacity for self-renewal and lineage-specific differentiation, serve as the cornerstone of tissue homeostasis and regenerative medicine. HDACs, long recognized as epigenetic modifiers, have recently emerged as master regulators of stem cell fate, dynamically balancing pluripotency maintenance and differentiation through both chromatin-dependent and -independent mechanisms. In the latest study (142), inhibition of histone deacetylase activity in LSD1-HDAC1/2 corepressor complex by sodium butyrate (NaB) increased the number of 2C-like cells in mouse embryonic hepatocytes and directly reprogrammed embryonic hepatocytes into trophoblast stem cells. Paradoxically, dysregulated HDAC activity drives pathological stemness in cancer stem cells (CSCs), where aberrant deacetylation stabilizes oncogenic transcription programs (e.g., Wnt/β-catenin, Notch) and confers therapy resistance (143). This duality underscores the context-dependent roles of HDAC isoforms, positioning them as both guardians of regenerative capacity and accomplices in malignant transformation. The latest study (144) found that the combination of HDACi CS055 and chiglitazar can synergistically act on leukemia cell lines and leukemia stem cell-like cells in patient samples. Chiglitazar enhanced the inhibitory effect of CS055 on HDAC3 by down-regulating the expression of SLC7A11, an inhibitor of ferroptosis, and induced ferroptosis in leukemia stem cell-like cells. HDAC profoundly affects the pluripotency, differentiation and therapeutic application of stem cells by regulating chromatin dynamics and signaling pathways, which still needs to be further explored in the future.
HDAC and HDACi have shown extraordinary potential in the treatment of cancer, and widely regulate the key processes of tumorigenesis. However, they still face many challenges. Firstly, the efficacy of HDACi as a single agent in solid tumors is insufficient, which may be partially related to tumor heterogeneity, microenvironment hypoxia and poor drug permeability (145, 146). Moreover, most of the existing HDACis are broad-spectrum inhibitors, lack of subtype selectivity, and have hematologic and cardiac-related toxicity (147). In addition, predictive markers for efficacy of HDACi have not been clearly defined, leading to a dilemma in patient stratification. In summary, HDAC and HDACi are the new candidates for epigenetic regulation, and their future development needs to break through the bottleneck of solid tumor efficacy, improve subtype selectivity, and further identify HDAC-related markers to guide clinical diagnosis and treatment. Combined treatment strategies (such as HDACi + immunotherapy or targeted drugs) and precision medicine models (individualized treatment based on molecular subtypes) are the directions we need to actively explore in the future to finally achieve the “precision endurance” of cancer treatment.
YZ: Writing – original draft. HW: Writing – review & editing. ZZ: Writing – review & editing. LG: Writing – review & editing. OB: Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Department of Finance of Jilin Province (JLSWSRCZX2023-8).
We thank all participants for their contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. (1997) 389:251–260. doi: 10.1038/38444
2. Loidl P. Towards an understanding of the biological function of histone acetylation. FEBS Lett. (1988) 227:91–5. doi: 10.1016/0014-5793(88)80874-3
3. Mizzen CA, Allis CD. Linking histone acetylation to transcriptional regulation. Cell Mol Life Sci. (1998) 54:6–20. doi: 10.1007/s000180050121
4. Narasumani M, Harrison PM. Discerning evolutionary trends in post-translational modification and the effect of intrinsic disorder: Analysis of methylation, acetylation and ubiquitination sites in human proteins. PloS Comput Biol. (2018) 14:e1006349. doi: 10.1371/journal.pcbi.1006349
5. Shah RR. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. (2019) 42:235–45. doi: 10.1007/s40264-018-0773-9
6. Inoue A, Fujimoto D. Enzymatic deacetylation of histone. Biochem Biophys Res Commun. (1969) 36:146–50. doi: 10.1016/0006-291x(69)90661-5
7. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. (1996) 272:408–11. doi: 10.1126/science.272.5260.408
8. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. (2003) 370:737–49. doi: 10.1042/BJ20021321
9. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. (2014) 6:a018713. doi: 10.1101/cshperspect.a018713
10. Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. (2001) 79:243–52.
11. Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class II histone deacetylases. Biochem Cell Biol. (2001) 79:337–48.
12. Shankar S, Srivastava RK. Histone deacetylase inhibitors: Mechanisms and clinical significance in cancer - HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. (2008) 615:261–98. doi: 10.1007/978-1-4020-6554-5_13
13. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. (1999) 260:273–9. doi: 10.1006/bbrc.1999.0897
14. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. (2000) 403:795–800. doi: 10.1038/35001622
15. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. (2004) 305:390–2. doi: 10.1126/science.1099196
16. Chen B, Zang WW, Wang J, Huang YJ, He YH, Yan LL, et al. The chemical biology of sirtuins. Chem Soc Rev. (2015) 44:5246–64. doi: 10.1039/c4cs00373j
17. Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li HZ, Goehle S, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. (2006) 66:4368–77. doi: 10.1158/0008-5472.Can-05-3617
18. Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, et al. The SIRT1 Deacetylase Suppresses Intestinal Tumorigenesis and Colon Cancer Growth. PloS One. (2008) 3:e2020. doi: 10.1371/journal.pone.0002020
19. Yanginlar C, Logie C. HDAC11 is a regulator of diverse immune functions. Biochim Et Biophys Acta-Gene Regul Mechanisms. (2018) 1861:54–9. doi: 10.1016/j.bbagrm.2017.12.002
20. Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. (2002) 277:25748–55. doi: 10.1074/jbc.M111871200
21. Boltz TA, Khuri S, Wuchty S. Promoter conservation in HDACs points to functional implications. BMC Genomics. (2019) 20:613. doi: 10.1186/s12864-019-5973-x
22. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discovery. (2014) 13:673–91. doi: 10.1038/nrd4360
23. McClure JJ, Zhang C, Inks ES, Peterson YK, Li J, Chou CJ. Development of allosteric hydrazide-containing class I histone deacetylase inhibitors for use in acute myeloid leukemia. J Med Chem. (2016) 59:9942–59. doi: 10.1021/acs.jmedchem.6b01385
24. Wang YF, Stowe RL, Pinello CE, Tian GM, Madoux F, Li DW, et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem Biol. (2015) 22:273–84. doi: 10.1016/j.chembiol.2014.12.015
25. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. (2007) 25:3109–15. doi: 10.1200/JCO.2006.10.2434
26. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. (2010) 28:4485–91. doi: 10.1200/JCO.2010.28.9066
27. Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. (2012) 30:631–6. doi: 10.1200/Jco.2011.37.4223
28. O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. (2015) 33:2492–U21. doi: 10.1200/Jco.2014.59.2782
29. San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. (2014) 15:1195–206. doi: 10.1016/S1470-2045(14)70440-1
30. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. (2010) 6:238–43. doi: 10.1038/nchembio.313
31. Suzuki T, Ando T, Tsuchiya K, Fukazawa N, Saito A, Mariko Y, et al. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem. (1999) 42:3001–3. doi: 10.1021/jm980565u
32. Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. (2008) 283:35402–9. doi: 10.1074/jbc.M807045200
33. Beconi M, Aziz O, Matthews K, Moumne L, O’Connell C, Yates D, et al. Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PloS One. (2012) 7:e44498. doi: 10.1371/journal.pone.0044498
34. Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J Biol Chem. (2013) 288:26926–43. doi: 10.1074/jbc.M113.490706
35. Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. (2005) 23:3912–22. doi: 10.1200/JCO.2005.02.188
36. Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF, et al. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumor Biol. (2014) 35:11523–32. doi: 10.1007/s13277-014-2358-2
37. Qiu L, Burgess A, Fairlie DP, Leonard H, Parsons PG, Gabrielli BG. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol Biol Cell. (2000) 11:2069–+. doi: 10.1091/mbc.11.6.2069
38. Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci United States America. (2005) 102:16090–5. doi: 10.1073/pnas.0505585102
39. Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15(INK4b) in HDAC inhibitor-induced growth arrest. FEBS Letters. (2003) 554:347–50. doi: 10.1016/S0014-5793(03)01186-4
40. Yamashita Y, Shimada M, Harimoto N, Rikimaru T, Shirabe K, Tanaka S, et al. Histone, deacetylase inhibitor trichostatin a induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. (2003) 103:572–6. doi: 10.1002/ijc.10699
41. Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. (2007) 27:4784–95. doi: 10.1128/MCB.00494-07
42. Li Y, Peng L, Seto E. Histone deacetylase 10 regulates the cell cycle G2/M phase transition via a novel let-7-HMGA2-cyclin A2 pathway. Mol Cell Biol. (2015) 35:3547–65. doi: 10.1128/MCB.00400-15
43. Kim MK, Kang YJ, Kim DH, Hossain MA, Jang JY, Lee SH, et al. A novel hydroxamic acid derivative, MHY218, induces apoptosis and cell cycle arrest through downregulation of NF-kappa B in HCT116 human colon cancer cells. Int J Oncol. (2014) 44:256–64. doi: 10.3892/ijo.2013.2163
44. Noh EL, Lim DS, Jeong GJ, Lee JS. An HDAC inhibitor, trichostatin A, induces a delay at G(2)/M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner. Biochem Biophys Res Commun. (2009) 378:326–31. doi: 10.1016/j.bbrc.2008.11.057
45. Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21(WAF1) expression and gene-associated histone acetylation. Proc Natl Acad Sci United States America. (2000) 97:10014–9. doi: 10.1073/pnas.180316197
46. Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. (2006) 20:2566–79. doi: 10.1101/gad.1455006
47. Cha TL, Chuang MJ, Wu ST. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. (2009) 15:1843. doi: 10.1158/1078-0432.CCR-08-1918
48. Vidal-Laliena M, Gallastegui E, Mateo F, Martinez-Balbas M, Pujol MJ, Bachs O. Histone deacetylase 3 regulates cyclin A stability. J Biol Chem. (2013) 288:21096–104. doi: 10.1074/jbc.M113.458323
49. Watanabe K, Okamoto K, Yonehara S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. (2005) 12:10–8. doi: 10.1038/sj.cdd.4401507
50. Fulda S. Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Exp Cell Res. (2012) 318:1208–12. doi: 10.1016/j.yexcr.2012.02.005
51. Xia Y, Wang J, Liu TJ, Yung WK, Hunter T, Lu Z. c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol Cell. (2007) 25:219–32. doi: 10.1016/j.molcel.2007.01.005
52. Matthews GM, Newbold A, Johnstone RW. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res. (2012) 116:165–97. doi: 10.1016/B978-0-12-394387-3.00005-7
53. Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk Res. (2008) 32:1382–92. doi: 10.1016/j.leukres.2008.02.018
54. Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Xie HJ, et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J Cell Biochem. (2012) 113:2167–77. doi: 10.1002/jcb.24090
55. Kim JK, Noh JH, Eun JW, Jung KH, Bae HJ, Shen Q, et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Mol Cancer Res. (2013) 11:62–73. doi: 10.1158/1541-7786.MCR-12-0332
56. Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. (2009) 114:380–93. doi: 10.1182/blood-2008-10-182758
57. Bajbouj K, Mawrin C, Hartig R, Schulze-Luehrmann J, Wilisch-Neumann A, Roessner A, et al. P53-dependent antiproliferative and pro-apoptotic effects of trichostatin A (TSA) in glioblastoma cells. J Neuro-Oncology. (2012) 107:503–16. doi: 10.1007/s11060-011-0791-2
58. Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. (1999) 18:7016–25. doi: 10.1038/sj.onc.1203176
59. Meng J, Zhang HH, Zhou CX, Li C, Zhang F, Mei QB. The histone deacetylase inhibitor trichostatin A induces cell cycle arrest and apoptosis in colorectal cancer cells via p53-dependent and -independent pathways. Oncol Rep. (2012) 28:384–8. doi: 10.3892/or.2012.1793
60. Sonnemann J, Marx C, Becker S, Wittig S, Palani CD, Kramer OH, et al. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br J Cancer. (2014) 110:656–67. doi: 10.1038/bjc.2013.742
61. Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. (2010) 17:11–6. doi: 10.1038/nsmb.1710
62. Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. (2008) 283:1–5. doi: 10.1074/jbc.R700039200
63. Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. (2004) 38:233–71. doi: 10.1146/annurev.genet.38.072902.091500
64. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. (2010) 17:1144–51. doi: 10.1038/nsmb.1899
65. Kotian S, Liyanarachchi S, Zelent A, Parvin JD. Histone deacetylases 9 and 10 are required for homologous recombination. J Biol Chem. (2011) 286:7722–6. doi: 10.1074/jbc.C110.194233
66. Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. (2013) 12:2078–87. doi: 10.1158/1535-7163.Mct-12-1242
67. Sun D, Yu M, Li Y, Xing H, Gao Y, Huang Z, et al. Histone deacetylase 2 is involved in DNA damage-mediated cell death of human osteosarcoma cells through stimulation of the ATM/p53 pathway. FEBS Open Bio. (2019) 9:478–89. doi: 10.1002/2211-5463.12585
68. Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, et al. Histone deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes (Basel). (2020) 11. doi: 10.3390/genes11050556
69. Yang WB, Wu AC, Hsu TI, Liou JP, Lo WL, Chang KY, et al. Histone deacetylase 6 acts upstream of DNA damage response activation to support the survival of glioblastoma cells. Cell Death Dis. (2021) 12:884. doi: 10.1038/s41419-021-04182-w
70. Jin J, Meng T, Yu Y, Wu S, Jiao CC, Song S, et al. Human HDAC6 senses valine abundancy to regulate DNA damage. Nature. (2025) 637:215–23. doi: 10.1038/s41586-024-08248-5
71. Yao M, Yan W, Wang Y, Zhao Y, Xu X, Chen Y, et al. IHCH9033, a novel class I HDAC inhibitor, synergizes with FLT3 inhibitor and rescues quizartinib resistance in FLT3-ITD AML via enhancing DNA damage response. Exp Hematol Oncol. (2025) 14:15. doi: 10.1186/s40164-025-00605-y
72. Baschnagel A, Russo A, Burgan WE, Carter D, Beam K, Palmieri D, et al. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther. (2009) 8:1589–95. doi: 10.1158/1535-7163.Mct-09-0038
73. Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem. (2004) 92:223–37. doi: 10.1002/jcb.20045
74. Suzuki M, Endo M, Shinohara F, Echigo S, Rikiishi H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemotherapy Pharmacol. (2009) 64:1115–22. doi: 10.1007/s00280-009-0969-x
75. Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, et al. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. (2010) 38:832–45. doi: 10.1093/nar/gkp1039
76. Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the nijmegen breakage syndrome protein. Mol Cell. (2007) 27:149–62. doi: 10.1016/j.molcel.2007.05.029
77. Fan W, Luo JY. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. (2010) 39:247–58. doi: 10.1016/j.molcel.2010.07.006
78. Alves-Fernandes DK, Jasiulionis MG. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20133153
79. Wang HL, Chen Y, Wang YQ, Tao EW, Tan J, Liu QQ, et al. Sirtuin5 protects colorectal cancer from DNA damage by keeping nucleotide availability. Nat Commun. (2022) 13:6121. doi: 10.1038/s41467-022-33903-8
80. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. (2006) 124:315–29. doi: 10.1016/j.cell.2005.11.044
81. Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. (2013) 51:454–68. doi: 10.1016/j.molcel.2013.06.018
82. Mao ZY, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. (2011) 332:1443–6. doi: 10.1126/science.1202723
83. Kawano T, Akiyama M, Agawa-Ohta M, Mikami-Terao Y, Iwase S, Yanagisawa T, et al. Histone deacetylase inhibitors valproic acid and depsipeptide sensitize retinoblastoma cells to radiotherapy by increasing H2AX phosphorylation and p53 acetylation-phosphorylation. Int J Oncol. (2010) 37:787–95. doi: 10.3892/ijo_00000728
84. Hsu YF, Sheu JR, Hsiao G, Lin CH, Chang TH, Chiu PT, et al. p53 in trichostatin A induced C6 glioma cell death. Biochim Biophys Acta. (2011) 1810:504–13. doi: 10.1016/j.bbagen.2011.02.006
85. Feng L, Pan M, Sun J, Lu H, Shen Q, Zhang S, et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J Mol Med (Berl). (2013) 91:49–58. doi: 10.1007/s00109-012-0932-x
86. Koeneke E, Witt O, Oehme I. HDAC family members intertwined in the regulation of autophagy: A druggable vulnerability in aggressive tumor entities. Cells. (2015) 4:135–68. doi: 10.3390/cells4020135
87. Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci United States America. (2012) 109:1649–54. doi: 10.1073/pnas.1121159109
88. Kang ZH, Wang CY, Zhang WL, Zhang JT, Yuan CH, Zhao PW, et al. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PloS One. (2014) 9:e98894. doi: 10.1371/journal.pone.0098894
89. Peixoto P, Castronovo V, Matheus N, Polese C, Peulen O, Gonzalez A, et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differentiation. (2012) 19:1239–52. doi: 10.1038/cdd.2012.3
90. Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, et al. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front Pharmacol. (2018) 9:34. doi: 10.3389/fphar.2018.00034
91. Zhang LQ, Zhang ZG, Li CB, Zhu TT, Gao J, Zhou H, et al. S100A11 promotes liver steatosis via FOXO1-mediated autophagy and lipogenesis. Cell Mol Gastroenter. (2021) 11:697–724. doi: 10.1016/j.jcmgh.2020.10.006
92. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. (2003) 115:727–38. doi: 10.1016/S0092-8674(03)00939-5
93. Su M, Shi JJ, Yang YP, Li J, Zhang YL, Chen J, et al. HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP plus -induced stress. J Neurochemistry. (2011) 117:112–20. doi: 10.1111/j.1471-4159.2011.07180.x
94. Li Z, Liu S, Fu T, Peng Y, Zhang J. Microtubule destabilization caused by silicate via HDAC6 activation contributes to autophagic dysfunction in bone mesenchymal stem cells. Stem Cell Res Ther. (2019) 10:351. doi: 10.1186/s13287-019-1441-4
95. Liu KP, Zhou D, Ouyang DY, Xu LH, Wang Y, Wang LX, et al. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem Biophys Res Commun. (2013) 441:970–5. doi: 10.1016/j.bbrc.2013.11.007
96. Zhu L, Duan W, Wu GJ, Zhang D, Wang L, Chen D, et al. Protective effect of hydrogen sulfide on endothelial cells through Sirt1-FoxO1-mediated autophagy. Annals Translational Med. (2020) 8:1586. doi: 10.21037/atm-20-3647
97. Zhang JB, Ng SK, Wang JG, Zhou J, Tan SH, Yang ND, et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. (2015) 11:629–42. doi: 10.1080/15548627.2015.1023981
98. Zhang JB, Wang JG, Zhou ZH, Park JE, Wang LM, Wu S, et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy. (2018) 14:1043–59. doi: 10.1080/15548627.2018.1447290
99. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res. (2009) 69:4415–23. doi: 10.1158/0008-5472.Can-08-2839
100. Oehme I, Linke JP, Bock BC, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci United States America. (2013) 110:E2592–E601. doi: 10.1073/pnas.1300113110
101. Aventaggiato M, Vernucci E, Barreca F, Russo MA, Tafani M. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol Ther. (2021) 221:107748. doi: 10.1016/j.pharmthera.2020.107748
102. Masoud GN, Li W. HIF-1 alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
103. Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. (2006) 25:1231–41. doi: 10.1038/sj.emboj.7601025
104. Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue CH, et al. HDAC4 protein regulates HIF1 alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem. (2011) 286:38095–102. doi: 10.1074/jbc.M111.257055
105. Kong XG, Lin Z, Liang DM, Fath D, Sang NL, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1 alpha. Mol Cell Biol. (2006) 26:2019–28. doi: 10.1128/Mcb.26.6.2019-2028.2006
106. Chen SY, Yin CQ, Lao TT, Liang DM, He D, Wang CG, et al. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1 alpha and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle. (2015) 14:2520–36. doi: 10.1080/15384101.2015.1055426
107. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. (2010) 38:864–78. doi: 10.1016/j.molcel.2010.05.023
108. Ray A, Alalem M, Ray BK. Loss of epigenetic kruppel-like factor 4 histone deacetylase (KLF-4-HDAC)-mediated transcriptional suppression is crucial in increasing vascular endothelial growth factor (VEGF) expression in breast cancer. J Biol Chem. (2013) 288:27232–42. doi: 10.1074/jbc.M113.481184
109. Urbich C, Rossig L, Kaluza D, Potente M, Boeckel JN, Knau A, et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. (2009) 113:5669–79. doi: 10.1182/blood-2009-01-196485
110. Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. (2011) 30:4142–56. doi: 10.1038/emboj.2011.298
111. Lv Z, Weng XY, Du CL, Zhang C, Xiao H, Cai XL, et al. Downregulation of HDAC6 promotes angiogenesis in hepatocellular carcinoma cells and predicts poor prognosis in liver transplantation patients. Mol Carcinogenesis. (2016) 55:1024–33. doi: 10.1002/mc.22345
112. Turtoi A, Mottet D, Matheus N, Dumont B, Peixoto P, Hennequiere V, et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis. (2012) 15:543–54. doi: 10.1007/s10456-012-9279-8
113. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
114. Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. (2012) 61:439–48. doi: 10.1136/gutjnl-2011-300060
115. von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. (2009) 137:361–71. doi: 10.1053/j.gastro.2009.04.004
116. Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ, Liu YH, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene. (2012) 31:583–94. doi: 10.1038/onc.2011.254
117. El-Naggar AM, Somasekharan SP, Wang YM, Cheng HW, Negri GL, Pan M, et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. (2019) 20:e48375. doi: 10.15252/embr.201948375
118. Ding S, Gao Y, Lv D, Tao Y, Liu S, Chen C, et al. DNTTIP1 promotes nasopharyngeal carcinoma metastasis via recruiting HDAC1 to DUSP2 promoter and activating ERK signaling pathway. Ebiomedicine. (2022) 81:104100. doi: 10.1016/j.ebiom.2022.104100
119. Ma L, Qi L, Li S, Yin Q, Liu J, Wang J, et al. Aberrant HDAC3 expression correlates with brain metastasis in breast cancer patients. Thorac Cancer. (2020) 11:2493–505. doi: 10.1111/1759-7714.13561
120. Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. (2003) 425:407–10. doi: 10.1038/nature01913
121. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A, et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br J Haematol. (2015) 168:811–9. doi: 10.1111/bjh.13222
122. Dallavalle S, Pisano C, Zunino F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem Pharmacol. (2012) 84:756–65. doi: 10.1016/j.bcp.2012.06.014
123. Li DW, Sun XD, Zhang LL, Yan B, Xie SB, Liu RM, et al. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein Cell. (2014) 5:214–23. doi: 10.1007/s13238-013-0010-3
124. Saji S, Kawakami M, Hayashi S, Yoshida N, Hirose M, Horiguchi S, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. (2005) 24:4531–9. doi: 10.1038/sj.onc.1208646
125. Zhang LL, Liu NN, Xie SB, He XF, Tala, Zhou J, et al. HDAC6 regulates neuroblastoma cell migration and may play a role in the invasion process. Cancer Biol Ther. (2014) 15:1561–70. doi: 10.4161/15384047.2014.956632
126. An PP, Li JX, Lu LL, Wu YM, Ling YY, Du J, et al. Histone deacetylase 8 triggers the migration of triple negative breast cancer cells via regulation of YAP signals. Eur J Pharmacol. (2019) 845:16–23. doi: 10.1016/j.ejphar.2018.12.030
127. An PP, Chen F, Li ZH, Ling YY, Peng YX, Zhang HS, et al. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3 beta/Snail signals. Oncogene. (2020) 39:4956–69. doi: 10.1038/s41388-020-1337-x
128. Tang XL, Li G, Su FT, Cai YL, Shi L, Meng Y, et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. (2020) 48:2912–23. doi: 10.1093/nar/gkaa039
129. Shankar E, Pandey M, Verma S, Abbas A, Candamo M, Kanwal R, et al. Role of class I histone deacetylases in the regulation of maspin expression in prostate cancer. Mol Carcinogenesis. (2020) 59:955–66. doi: 10.1002/mc.23214
130. Mormino A, Cocozza G, Fontemaggi G, Valente S, Esposito V, Santoro A, et al. Histone-deacetylase 8 drives the immune response and the growth of glioma. Glia. (2021) 69:2682–98. doi: 10.1002/glia.24065
131. Zhou YQ, Huan L, Wu YJ, Bao CY, Chen B, Wang L, et al. LncRNA ID2-AS1 suppresses tumor metastasis by activating the HDAC8/ID2 pathway in hepatocellular carcinoma. Cancer Letters. (2020) 469:399–409. doi: 10.1016/j.canlet.2019.11.007
132. Song CL, Zhu SC, Wu CY, Kang JH. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J Biol Chem. (2013) 288:28021–33. doi: 10.1074/jbc.M113.498758
133. Wen YQ, Zhang XM, Li XY, Tian L, Shen SR, Ma J, et al. Histone deacetylase (HDAC) 11 inhibits matrix metalloproteinase (MMP) 3 expression to suppress colorectal cancer metastasis. J Cancer. (2022) 13:1923–32. doi: 10.7150/jca.66914
134. Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, et al. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. (2012) 31:4619–29. doi: 10.1038/onc.2011.612
135. Eades G, Yao Y, Yang MH, Zhang YS, Chumsri S, Zhou Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J Biol Chem. (2011) 286:25992–6002. doi: 10.1074/jbc.M111.229401
136. Chen IC, Chiang WF, Huang HH, Chen PF, Shen YY, Chiang HC. Role of SIRT1 in regulation of epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis. Mol Cancer. (2014) 13:254. doi: 10.1186/1476-4598-13-254
137. Xiao H, Jiao J, Wang L, O’Brien S, Newick K, Wang LC, et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int J Cancer. (2016) 138:2477–86. doi: 10.1002/ijc.29979
138. Zhou Y, Jin X, Yu H, Qin G, Pan P, Zhao J, et al. HDAC5 modulates PD-L1 expression and cancer immunity via p65 deacetylation in pancreatic cancer. Theranostics. (2022) 12:2080–94. doi: 10.7150/thno.69444
139. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am J Transplant. (2018) 18:1596–603. doi: 10.1111/ajt.14749
140. Zhu J, Wang F, Wang L, Dai B, Xu G, Zhao L, et al. HDAC inhibition increases CXCL12 secretion to recruit natural killer cells in peripheral T-cell lymphoma. Cancer Res. (2024) 84:2450–67. doi: 10.1158/0008-5472.CAN-23-3250
141. Bae J, Hideshima T, Tai YT, Song Y, Richardson P, Raje N, et al. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia. (2018) 32:1932–47. doi: 10.1038/s41375-018-0062-8
142. Huang B, Peng X, Zhai X, Hu J, Chen J, Yang S, et al. Inhibition of HDAC activity directly reprograms murine embryonic stem cells to trophoblast stem cells. Dev Cell. (2024) 59:2101–17 e8. doi: 10.1016/j.devcel.2024.05.009
143. Debeb BG, Lacerda L, Xu W, Larson R, Solley T, Atkinson R, et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells. (2012) 30:2366–77. doi: 10.1002/stem.1219
144. Zhou H, Qin D, Xie C, Zhou J, Jia S, Zhou Z, et al. Combinations of HDAC inhibitor and PPAR agonist induce ferroptosis of leukemic stem cell-like cells in acute myeloid leukemia. Clin Cancer Res. (2024) 30:5430–44. doi: 10.1158/1078-0432.CCR-24-0796
145. Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. (2012) 4:505–24. doi: 10.4155/fmc.12.3
146. O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic Malignancies. J Clin Oncol. (2006) 24:166–73. doi: 10.1200/JCO.2005.01.9679
147. Li W, Fu Y, Wang W. A real-world pharmacovigilance study investigating the toxicities of histone deacetylase inhibitors. Ann Hematol. (2024) 103:3207–17. doi: 10.1007/s00277-024-05691-2
148. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. (2007) 12:1247–52. doi: 10.1634/theoncologist.12-10-1247
149. Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. (2015) 21:2666–70. doi: 10.1158/1078-0432.CCR-14-3119
150. Panobinostat approved for multiple myeloma. Cancer Discovery. (2015) 5:OF4. doi: 10.1158/2159-8290.CD-NB2015-040
151. Mottamal M, Zheng SL, Huang TL, Wang GD. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. (2015) 20:3898–941. doi: 10.3390/molecules20033898
152. Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. (2010) 150:446–55. doi: 10.1111/j.1365-2141.2010.08266.x
153. Yalniz FF, Berdeja JG, Maris MB, Lyons RM, Reeves JA, Essell JH, et al. A phase II study of addition of pracinostat to a hypomethylating agent in patients with myelodysplastic syndromes who have not responded to previous hypomethylating agent therapy. Brit J Haematol. (2020) 188:404–12. doi: 10.1111/bjh.16173
154. Garcia-Manero G, Abaza Y, Takahashi K, Medeiros BC, Arellano M, Khaled SK, et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: results of a phase 2 study. Blood Adv. (2019) 3:508–18. doi: 10.1182/bloodadvances.2018027409
155. Garcia-Manero G, Montalban-Bravo G, Berdeja JG, Abaza Y, Jabbour E, Essell J, et al. Phase 2, randomized, double-blind study of pracinostat in combination with azacitidine in patients with untreated, higher-risk myelodysplastic syndromes. Cancer-Am Cancer Soc. (2017) 123:994–1002. doi: 10.1002/cncr.30533
156. Ribrag V, Kim WS, Bouabdallah R, Lim ST, Coiffier B, Illes A, et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica. (2017) 102:903–9. doi: 10.3324/haematol.2016.154377
157. Evens AM, Balasubramanian S, Vose JM, Harb W, Gordon LI, Langdon R, et al. A phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma. Clin Cancer Res. (2016) 22:1059–66. doi: 10.1158/1078-0432.Ccr-15-0624
158. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. (2014) 124:30–9. doi: 10.1172/Jci69738
159. Moreau P, Facon T, Touzeau C, Benboubker L, Delain M, Badamo-Dotzis J, et al. Quisinostat, bortezomib, and dexamethasone combination therapy for relapsed multiple myeloma. Leukemia Lymphoma. (2016) 57:1546–59. doi: 10.3109/10428194.2015.1117611
160. Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A, et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. (2013) 19:4262–72. doi: 10.1158/1078-0432.Ccr-13-0312
161. Child F, Ortiz-Romero PL, Alvarez R, Bagot M, Stadler R, Weichenthal M, et al. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Seezary syndrome. Brit J Dermatol. (2016) 175:80–8. doi: 10.1111/bjd.14427
162. Galloway TJ, Wirth LJ, Colevas AD, Gilbert J, Bauman JE, Saba NF, et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin Cancer Res. (2015) 21:1566–73. doi: 10.1158/1078-0432.Ccr-14-2820
163. Shimizu T, LoRusso PM, Papadopoulos KP, Patnaik A, Beeram M, Smith LS, et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. (2014) 20:5032–40. doi: 10.1158/1078-0432.Ccr-14-0570
164. Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. (2017) 102:1923–30. doi: 10.3324/haematol.2017.172882
165. Landsburg DJ, Barta SK, Ramchandren R, Batlevi C, Iyer S, Kelly K, et al. Fimepinostat (CUDC-907) in patients with relapsed/refractory diffuse large B cell and high-grade B-cell lymphoma: report of a phase 2 trial and exploratory biomarker analyses. Brit J Haematol. (2021) 195:201–9. doi: 10.1111/bjh.17730
166. Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. (2016) 17:622–31. doi: 10.1016/S1470-2045(15)00584-7
167. Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M, et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res. (2012) 18:2687–94. doi: 10.1158/1078-0432.Ccr-11-3165
168. Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Brit J Cancer. (2012) 106:77–84. doi: 10.1038/bjc.2011.527
169. Ny L, Jespersen H, Karlsson J, Alsen S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. (2021) 12:5155. doi: 10.1038/s41467-021-25332-w
170. Connolly RM, Zhao FM, Miller KD, Lee MJ, Piekarz RL, Smith KL, et al. E2112: randomized phase III trial of endocrine therapy plus entinostat or placebo in hormone receptor-positive advanced breast cancer. J Clin Oncol. (2021) 39:3171–+. doi: 10.1200/Jco.21.00944
171. Hellmann MD, Janne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin Cancer Res. (2021) 27:1019–28. doi: 10.1158/1078-0432.Ccr-20-3305
172. Torres ETR, Rafie C, Wang CG, Lim D, Brufsky A, LoRusso P, et al. Phase I study of entinostat and nivolumab with or without ipilimumab in advanced solid tumors (ETCTN-9844). Clin Cancer Res. (2021) 27:5828–37. doi: 10.1158/1078-0432.Ccr-20-5017
173. Yeruva SLH, Zhao F, Miller KD, Tevaarwerk AJ, Wagner LI, Gray RJ, et al. E2112: randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. Breast Cancer. (2018) 4:1. doi: 10.1038/s41523-017-0053-3
174. Lin JQ, Elkon J, Ricart B, Palmer E, Zevallos-Delgado C, NooNepalle S, et al. Phase I study of entinostat in combination with enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer. Oncologist. (2021) 26:E2136–E42. doi: 10.1002/onco.13957
175. Karasic TB, Brown TJ, Schneider C, Teitelbaum UR, Reiss KA, Mitchell TC, et al. Phase I trial of regorafenib, hydroxychloroquine, and entinostat in metastatic colorectal cancer. Oncologist. (2022) 27:716–+. doi: 10.1093/oncolo/oyac078
176. Masuda N, Tamura K, Yasojima H, Shimomura A, Sawaki M, Lee MJ, et al. Phase 1 trial of entinostat as monotherapy and combined with exemestane in Japanese patients with hormone receptor-positive advanced breast cancer. BMC Cancer. (2021) 21:1269. doi: 10.1186/s12885-021-08973-4
177. Carraway HE, Sawalha Y, Gojo I, Lee MJ, Lee S, Tomita Y, et al. Phase 1 study of the histone deacetylase inhibitor entinostat plus clofarabine for poor-risk Philadelphia chromosome-negative (newly diagnosed older adults or adults with relapsed refractory disease) acute lymphoblastic leukemia or biphenotypic leukemia. Leuk Res. (2021) 110:106707. doi: 10.1016/j.leukres.2021.106707
178. Tomita Y, Lee MJ, Lee S, Tomita S, Chumsri S, Cruickshank S, et al. The interplay of epigenetic therapy and immunity in locally recurrent or metastatic estrogen receptor-positive breast cancer: Correlative analysis of ENCORE 301, a randomized, placebo-controlled phase II trial of exemestane with or without entinostat. Oncoimmunology. (2016) 5:e1219008. doi: 10.1080/2162402X.2016.1219008
179. Bukowinski A, Chang B, Reid JM, Liu X, Minard CG, Trepel JB, et al. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr Blood Cancer. (2021) 68:e28892. doi: 10.1002/pbc.28892
180. Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, et al. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: A multicenter, single-arm, phase I/II trial (NCI-CTEP7870). Clin Cancer Res. (2017) 23:7199–208. doi: 10.1158/1078-0432.CCR-17-1178
181. Lim B, Murthy RK, Lee J, Jackson SA, Lwase T, Davis DW, et al. A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Brit J Cancer. (2019) 120:1105–12. doi: 10.1038/s41416-019-0473-y
182. van Tilburg CM, Witt R, Heiss M, Pajtler KW, Plass C, Poschke I, et al. INFORM2 NivEnt: The first trial of the INFORM2 biomarker driven phase I/II trial series: the combination of nivolumab and entinostat in children and adolescents with refractory high-risk Malignancies. BMC Cancer. (2020) 20:523. doi: 10.1186/s12885-020-07008-8
183. Jespersen H, Olofsson Bagge R, Ullenhag G, Carneiro A, Helgadottir H, Ljuslinder I, et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study): protocol for a multicenter phase II open label study. BMC Cancer. (2019) 19:415. doi: 10.1186/s12885-019-5623-3
184. Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. (2013) 31:2128–+. doi: 10.1200/Jco.2012.43.7251
185. Connolly RM, Li HL, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, et al. Combination epigenetic therapy in advanced breast cancer with 5-azacitidine and entinostat: A phase II national cancer institute/stand up to cancer study. Clin Cancer Res. (2017) 23:2691–701. doi: 10.1158/1078-0432.Ccr-16-1729
186. Ngamphaiboon N, Dy GK, Ma WW, Zhao YJ, Reungwetwattana T, DePaolo D, et al. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Invest New Drug. (2015) 33:225–32. doi: 10.1007/s10637-014-0174-6
187. Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. (2012) 30:2248–55. doi: 10.1200/Jco.2011.38.9411
188. Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. (2015) 26:1766–71. doi: 10.1093/annonc/mdv237
189. Wang YW, Zhang MZ, Song W, Cai QQ, Zhang LL, Sun XH, et al. Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: A multicenter phase II trial. Am J Hematol. (2022) 97:623–9. doi: 10.1002/ajh.26499
190. Chai Y, Chen B, Qi F, Fang H, Qi SN, Guo RY, et al. First-line chemoradiation with or without chidamide (Tucidinostat) in patients with intermediate- and high-risk early-stage extranodal nasal-type natural killer/T-cell lymphoma: A randomized phase 2 study in China. Int J Radiat Oncol. (2022) 113:833–44. doi: 10.1016/j.ijrobp.2022.04.001
191. Zhang W, Su LP, Liu LH, Gao YH, Wang QS, Su H, et al. The combination of chidamide with the CHOEP regimen in previously untreated patients with peripheral T-cell lymphoma: a prospective, multicenter, single arm, phase 1b/2 study. Cancer Biol Med. (2021) 18:841–8. doi: 10.20892/j.issn.2095-3941.2020.0413
192. Chen X, Wang H, Sun X, Su L, Liu F, Zhao K, et al. Safety of chidamide plus rituximab in elderly patients with relapsed or refractory B-cell lymphoma in China: a multicenter, single-arm, phase II study. Ann Transl Med. (2021) 9:1769. doi: 10.21037/atm-21-6019
193. Dong M, Ning ZQ, Xing PY, Xu JL, Cao HX, Dou GF, et al. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemoth Pharm. (2012) 69:1413–22. doi: 10.1007/s00280-012-1847-5
194. Zhang MC, Fang Y, Wang L, Cheng S, Fu D, He Y, et al. Clinical efficacy and molecular biomarkers in a phase II study of tucidinostat plus R-CHOP in elderly patients with newly diagnosed diffuse large B-cell lymphoma. Clin Epigenet. (2020) 12:160. doi: 10.1186/s13148-020-00948-9
195. Yee AJ, Bensinger WI, Supko JRG, Voorhees PM, Berdeja JG, Richardson PG, et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. (2016) 17:1569–78. doi: 10.1016/S1470-2045(16)30375-8
196. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18071414
197. Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. (2006) 17:1096–102. doi: 10.1093/annonc/mdl081
198. Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S, et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. (2011) 12:1222–8. doi: 10.1016/S1470-2045(11)70265-0
199. Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. (2017) 178:434–41. doi: 10.1111/bjh.14698
200. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discovery. (2006) 5:769–84. doi: 10.1038/nrd2133
201. Collier KA, Valencia H, Newton H, Hade EM, Sborov DW, Cavaliere R, et al. A phase 1 trial of the histone deacetylase inhibitor AR-42 in patients with neurofibromatosis type 2-associated tumors and advanced solid Malignancies. Cancer Chemoth Pharm. (2021) 87:599–611. doi: 10.1007/s00280-020-04229-3
202. Sborov DW, Canella A, Hade EM, Mo XK, Khountham S, Wang J, et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leukemia Lymphoma. (2017) 58:2310–8. doi: 10.1080/10428194.2017.1298751
Keywords: histone acetyltransferases, histone deacetylases, cancer, HDACi, lymphoma
Citation: Zhang Y, Wang H, Zhan Z, Gan L and Bai O (2025) Mechanisms of HDACs in cancer development. Front. Immunol. 16:1529239. doi: 10.3389/fimmu.2025.1529239
Received: 16 November 2024; Accepted: 17 March 2025;
Published: 07 April 2025.
Edited by:
Yu Pan, Macau University of Science and Technology, Macao SAR, ChinaReviewed by:
Ayad A. Al-Hamashi, University of Baghdad, IraqCopyright © 2025 Zhang, Wang, Zhan, Gan and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Out Bai, YmFpb3VAamx1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.