 Kai-long He1
Kai-long He1 Yan-dong Xie
Yan-dong Xie Chao-hui Jing
Chao-hui Jing
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 February 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1526786
This article is part of the Research TopicNeuroinflammation: Mechanisms and Therapeutic InterventionsView all 8 articles
Intracerebral hemorrhage (ICH), a specific subtype within the spectrum of stroke disorders, is characterized by its high mortality and significant risk of long-term disability. The initiation and progression of neuroinflammation play a central and critical role in the pathophysiology of ICH. The NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, a protein complex involved in initiating inflammation, is the central focus of this article. Microglia and astrocytes play critical roles in the inflammatory damage process associated with neuroinflammation. The NLRP3 inflammasome is expressed within both types of glial cells, and its activation drives these cells toward a pro-inflammatory phenotype, which exacerbates inflammatory damage in the brain. However, the regulatory relationship between these two cell types remains to be explored. Targeting NLRP3 inflammasomes in microglia or astrocytes may provide an effective approach to mitigate neuroinflammation following ICH. This article first provides an overview of the composition and activation mechanisms of the NLRP3 inflammasome. Subsequently, it summarizes recent research findings on novel signaling pathways that regulate NLRP3 inflammasome activity. Finally, we reviewed recent progress in NLRP3 inflammasome inhibitors, highlighting the clinical translation potential of certain candidates. These inhibitors hold promise as innovative strategies for managing inflammation following ICH.
Intracerebral hemorrhage(ICH) results from the rupture of blood vessels and is a severe subtype of stroke, characterized by the infiltration of blood into the brain tissue (1, 2). In all stroke cases, ICH accounts for approximately 10% to 15%, and it is associated with a high incidence and mortality rate (3). Primary brain injury triggered by ICH mainly refers to the space-occupying effect of the hematoma that forms after the rupture of cerebral blood vessels. This effect leads to a marked increase in intracranial pressure, which subsequently results in a gradual deterioration of neurological function (4). Secondary brain injury following ICH is mediated by blood and its metabolic byproducts, with the pathological processes encompassing inflammatory responses, mitochondrial dysfunction, oxidative stress, neuronal apoptosis, and disruption of the blood-brain barrier (BBB) (5, 6). Hematoma evacuation surgery is the primary treatment for ICH in neurosurgery. While this procedure can reduce morbidity associated with the surgery, it does not mitigate secondary brain injury resulting from the ICH (3, 7). Neuroinflammation is a primary driver of secondary brain injury following ICH (8). Thus, inhibiting neuroinflammation is a crucial strategy for improving the prognosis of ICH (9).
Neuroinflammation is defined as the robust host defense response immediately initiated when blood enters the brain parenchyma (6). After ICH, a cascade of immune cell activations initiates neuroinflammation. Among these immune responses, microglia play a particularly pivotal role, acting as central drivers that exacerbate inflammatory injury (9). The activation of microglia and other immune cells results in the release of pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, myeloperoxidase (MPO), and other cytotoxic substances (10). The sustained activation of the inflammatory cascade is maintained by pro-inflammatory cytokines, a process that leads to widespread neuronal loss, disruption of the BBB, the formation of cerebral edema, and ultimately, neurological dysfunction (6, 11, 12). Neuroinflammation increases BBB permeability, leading to edema that exacerbates the space-occupying effect, which in turn induces secondary ischemia. This ischemia accelerates cellular death and inflicts additional inflammatory damage on the surrounding brain tissue (13).
Among the different inflammasomes identified, the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome has been the subject of the most extensive research (14, 15). Growing evidence indicates that NLRP3 inflammasome activation after ICH amplifies the inflammatory cascade. Suppressing NLRP3 inflammasome activity has been shown to inhibit the maturation of pro-inflammatory cytokines, thereby promoting neurological recovery and improving outcomes for ICH patients (16, 17). Consequently, targeting NLRP3 inflammasome-mediated neuroinflammation presents a compelling therapeutic strategy for minimizing inflammatory damage and enhancing recovery outcomes after ICH. This review examines the pathways by which the NLRP3 inflammasome contributes to neuroinflammation following ICH and evaluates potential therapeutic approaches to counteract its impact.
The NLRP3 inflammasome is pivotal in the development of numerous diseases, spanning neurodegenerative conditions such as multiple sclerosis, Alzheimer’s, and Parkinson’s diseases, as well as metabolic disorders like atherosclerosis, type 2 diabetes, and obesity (18). The NLRP3 inflammasome is a multiprotein complex typically composed of the NLRP3 sensor, the ASC (apoptosis-associated speck-like protein), and the pro-caspase-1 precursor (Figure 1) (19). NLRP3 is a complex multi-domain protein, consisting of three main components: the N-terminal pyrin domain (PYD), the central nucleotide-binding and oligomerization domain (NACHT), and the C-terminal leucine-rich repeat (LRR) region. It is one of the most widely studied members of the inflammasome family (20, 21). ASC, also known as PYCARD, is a protein containing an N-terminal PYD and a C-terminal caspase recruitment domain (CARD). It promotes the activation of caspase-1 (22). Pro-caspase-1 consists of two main domains: the CARD and the caspase activation domain (23).In the absence of immune activation signals, an autoinhibitory interaction between the NACHT domain and the LRR domain effectively prevents the binding of NLRP3 to ASC, thereby inhibiting the formation of the inflammasome (24). Upon detection of danger signals, the PYD domain of NLRP3 binds to ASC, which in turn recruits caspase-1 through its CARD domain, leading to the assembly of the NLRP3 inflammasome. This multiprotein complex activates pro-caspase-1 via autocatalytic cleavage, leading to the generation of active caspase-1. Active caspase-1 cleaves the precursor forms of the cytokines IL-1β and IL-18, producing their active forms. These activated cytokines play a crucial mediatory role in the inflammatory response (19, 24, 25).
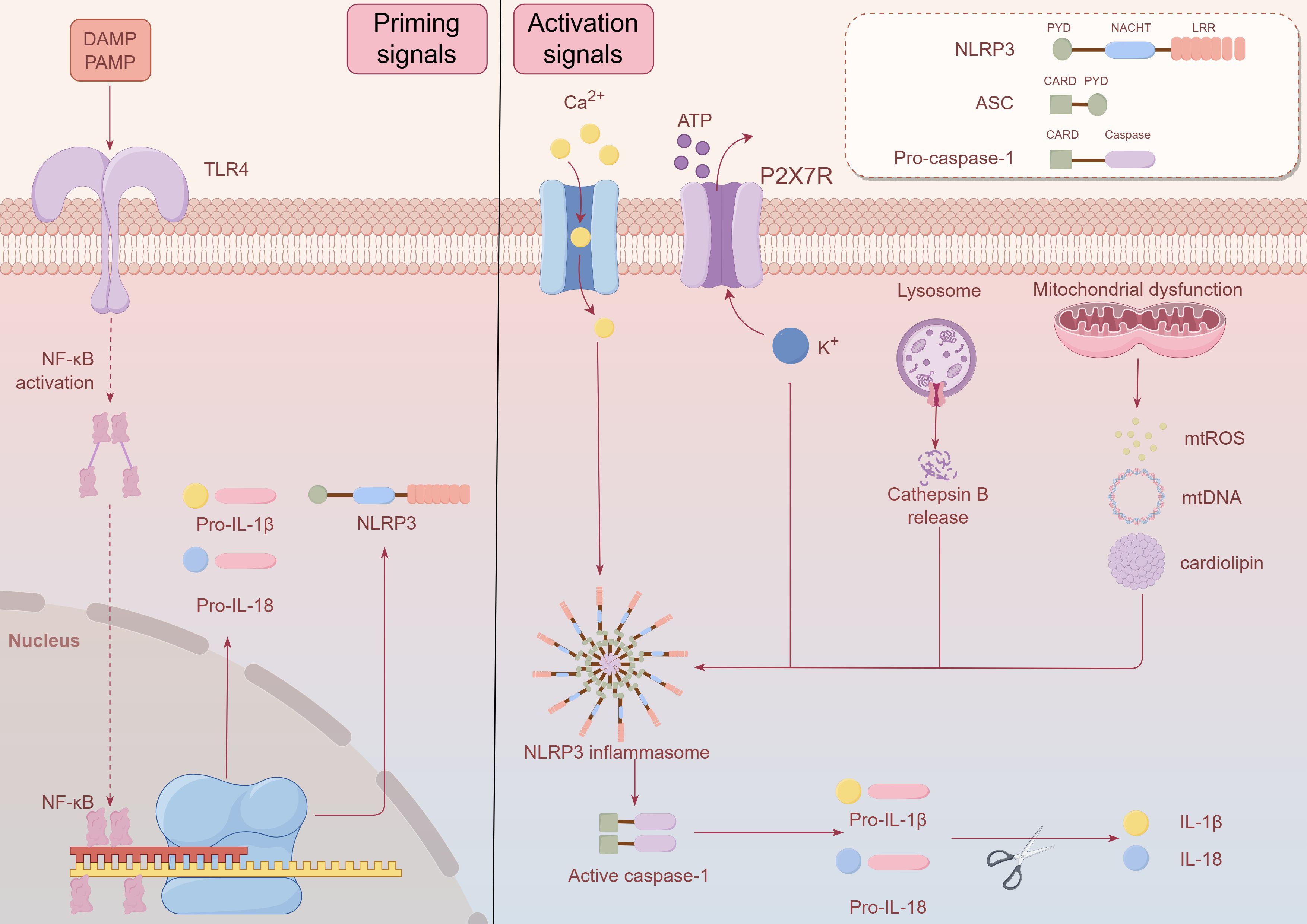
Figure 1. Schematic diagram of NLRP3 inflammasome composition and activation after ICH. The NLRP3 inflammasome is a multiprotein complex typically composed of the NLRP3 sensor, the ASC (apoptosis-associated speck-like protein), and the pro-caspase-1 precursor. NLRP3 is a complex multi-domain protein, consisting of three main components: the N-terminal pyrin domain (PYD), the central nucleotide-binding and oligomerization domain (NACHT), and the C-terminal leucine-rich repeat (LRR) region. ASC is a protein containing an N-terminal PYD and a C-terminal caspase recruitment domain (CARD).Pro-caspase-1 consists of two main domains: the CARD and the caspase activation domain. Upon detection of danger signals, the PYD domain of NLRP3 binds to ASC, which in turn recruits caspase-1 through its CARD domain, leading to the assembly of the NLRP3 inflammasome. In the priming signals, Toll-like receptors (TLRs) are capable of recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which in turn trigger the nuclear factor kappa B (NF-κB) signaling pathway activation. The activation of NF-κB promotes its translocation to the nucleus, where it binds to DNA, thereby enhancing the transcriptional expression of NLRP3, pro-IL-1β, and pro-IL-18. During the Activation Signals, the NLRP3 inflammasome activation can be initiated by various stimuli, including purinergic P2X7 receptor (P2X7R)-mediated potassium efflux, intracellular calcium influx, reactive oxygen species (ROS) production caused by mitochondrial stress, the secretion of mitochondrial DNA or cardiolipin from mitochondria, and the release of cathepsin B following lysosomal rupture. Activation of the NLRP3 inflammasome promotes the autocatalytic maturation of pro-caspase-1 into active caspase-1, which then cleaves the precursors of IL-1β and IL-18 into their biologically active mature forms. This figure was created by Figdraw.
The activation of the NLRP3 inflammasome can be triggered by various stimuli, including viruses, bacteria, fungi, pore-forming toxins, crystal substances (such as urate crystals, silica, asbestos, and alum), and extracellular ATP. These factors activate the NLRP3 inflammasome through different mechanisms, initiating a cascade of intracellular reactions (26, 27). The NLRP3 inflammasome activation can be initiated by various stimuli, including purinergic P2X7 receptor (P2X7R)-mediated potassium efflux, intracellular calcium influx, reactive oxygen species (ROS) production caused by mitochondrial stress, the secretion of mitochondrial DNA or cardiolipin from mitochondria, and the release of cathepsin B following lysosomal rupture (25, 27). The activation of the inflammasome is a process involving two stages: the initial priming signals and the subsequent activation signals (Figure 1). In the priming signals, Toll-like receptors (TLRs) are capable of recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which in turn trigger the nuclear factor kappa B (NF-κB) signaling pathway activation. The activation of NF-κB promotes its translocation to the nucleus, where it binds to DNA, thereby enhancing the transcriptional expression of NLRP3, pro-IL-1β, and pro-IL-18 (24, 28). Various factors, including TNF, IL-1, high-mobility group box 1 (HMGB1), interferon (IFN), transforming growth factor-β (TGF-β), and lipopolysaccharide (LPS), can promote the priming signals of NLRP3 inflammasome activation. These molecules play crucial roles in triggering the NF-κB signaling pathway, which leads to the upregulation of key inflammasome components and preparing the system for subsequent activation (29, 30). During the activation signals, NLRP3 undergoes oligomerization, forming a complex consisting of NLRP3, ASC, and pro-caspase-1. The formation of this complex promotes the autocatalytic maturation of pro-caspase-1 into active caspase-1, which then cleaves the precursors of IL-1β and IL-18 into their biologically active mature forms (31–33). Recent studies have established an important link between the NLRP3 inflammasome and the inflammatory response triggered in ICH (34). After ICH, various upstream signals can activate the NLRP3 inflammasome, with heme, released during hemoglobin degradation, playing a crucial role (35). Following ICH, the NLRP3 inflammasome activation exacerbates both the degree of cerebral edema and the neuroinflammatory response (36).
The complex immune response is a critical factor in the development of neuroinflammation after intracerebral hemorrhage, involving various immune cells, among which microglia and astrocytes play pivotal roles. The NLRP3 inflammasome is expressed in both microglia and astrocytes, and numerous studies have demonstrated that the activation of the NLRP3 inflammasome in these cells can alleviate neuroinflammation under specific pathological conditions. In the following section, we focus on neuroinflammation mediated by microglia and astrocytes, as well as the potential relationship between the NLRP3 inflammasome expressed in these cells and neuroinflammation after intracerebral hemorrhage.
Microglia play a critical immune regulatory role in the central nervous system (CNS), which is vital for both the physiological and pathological processes of neuroinflammation (37). The pathological characteristics of neuroinflammation are usually marked by an increase in pro-inflammatory cytokine expression, enhanced activation of microglia, infiltration of peripheral immune cells like leukocytes into neural tissue, and subsequent structural and functional damage to the nervous system (38). Microglia-mediated neuroinflammation is crucial in the inflammatory injury process. Upon activation, microglia transition from their branched morphology to an amoeboid shape, enhancing their phagocytic activity and reactivity (9, 39). M1-like microglia are characterized by a pro-inflammatory and neurotoxic phenotype, marked by the secretion of TNF-α, IL-1β, IL-12, and inducible nitric oxide synthase (INOS). These factors contribute to the exacerbation of neuroinflammation and the progression of tissue damage (40–42). M2 microglia secrete anti-inflammatory factors, suppress immune responses, and promote tissue repair and neuroregeneration. These functions are crucial for alleviating inflammation and aiding recovery after brain injury. They stimulate the release of anti-inflammatory cytokines, such as IL-4, IL-10, IL-13, and transforming growth factor-β (TGF-β), and are characterized by markers like arginase-1 (Arg-1) and CD206, which contribute to the repair process (40, 42). Simultaneously, M2 microglia facilitate axonal regeneration and vascular remodeling by secreting remodeling factors such as vascular endothelial growth factor (VEGF), brain-derived neurotrophic factor (BDNF), which play crucial roles in tissue repair and neurovascular recovery (43). Furthermore, during the remodeling phase following a stroke, microglia and infiltrating macrophages function as essential phagocytic cell populations, collaborating to clear dead cells and tissue debris, thus facilitating the repair and reconstruction of damaged neural tissues (44). ICH may benefit from the exploration of cell therapies that target microglial phenotype switching, potentially improving patient outcomes. (There seems to be controversy regarding the M1/M2 classification of microglia, with some scholars arguing that such a classification is overly simplistic (45). However, this classification is widely used and still plays a role in understanding the functions of microglia in brain hemorrhage).
The NLRP3 inflammasome is crucial in microglia-mediated neuroinflammation after ICH and is predominantly expressed in microglia (34). Studies have shown that pre-treatment with epigallocatechin-3-gallate (EGCG) can reduce the levels of NLRP3 inflammasome in microglia following ICH and promote the transition of microglia from M1 phenotype to M2 phenotype (46). Xia et al. experimentally demonstrated that activation of sirtuin 1 (SIRT1), a sirtuin family member, suppresses NLRP3 inflammasome signaling in microglia during subarachnoid hemorrhage (SAH). Additionally, it facilitates microglia from M1 phenotype to M2 phenotype, which could be beneficial in mitigating neuroinflammation (47). Based on the hypothesis that both SAH and ICH are classified as hemorrhagic strokes, we speculate that the activation of SIRT1 may exhibit similar therapeutic effects in the context of ICH. Although neither of the two referenced studies directly addresses the connection between the NLRP3 inflammasome and microglial polarization towards M1 or M2 phenotypes, It is reasonable to propose that the NLRP3 inflammasome may play a role in regulating this phenotypic transition in microglia. However, due to the limited amount of literature on this topic, this area remains underexplored and represents one of our future research directions.
Astrocytes, the most prevalent glial cells in the CNS, are essential for supporting neurons and maintaining brain homeostasis. In response to pathological conditions, astrocytes become activated, transforming into reactive astrocytes. This process involves cell hypertrophy and an increase in the release of neurotoxic factors, playing a critical role in the development of inflammation and neuronal damage within the central CNS (48, 49). Activated microglia possess the capacity to stimulate astrocyte activation, as well as modulate astrocyte proliferation and differentiation (34). Astrocytes can be categorized into A1 and A2 phenotypes. Much like microglia, astrocytes are capable of secreting mediators, either promoting inflammation or performing immunoregulatory functions, depending on their specific polarized phenotype (50, 51). A1 astrocytes play a pro-inflammatory role by secreting neurotoxins that induce neuronal death and the apoptosis of mature oligodendrocytes, while inhibiting processes essential for neuronal survival and synaptogenesis. In contrast, A2 astrocytes exhibit anti-inflammatory activities, upregulating anti-inflammatory genes to support neuronal survival and growth. They also contribute to glial scar formation, aid in clearing cellular debris, and promote the repair of the BBB, thus facilitating tissue recovery and regeneration following CNS injury (50, 52). (Similarly, the A1/A2 classification of astrocytes is also debated internationally, and a new consensus on the classification of astrocytes has yet to be reached (53). We hold a similar view on the A1/A2 classification of astrocytes as we do for the M1/M2 classification of microglia.) ICH triggers astrocyte activation, regulating the release of pro-inflammatory factors. Research indicates a significant increase in reactive astrocytes surrounding the hematoma within 12 to 48 hours post-ICH, suggesting their involvement in the early inflammatory response and potential contribution to both neuroprotection and neurotoxicity in the acute phase of injury (54, 55). After ICH, oxidative stress triggered by the release of hemoglobin activates astrocytes, leading to an increased expression of matrix metalloproteinase-9 (MMP-9). This contributes to the disruption of the BBB, increasing vascular permeability, which in turn exacerbates neuroinflammation and secondary brain injury (34). Following ICH, inhibiting the CD147 receptor on astrocytes has been shown to reduce MMP-9 levels in the surrounding hematoma, leading to decreased tissue degradation and improved neurological recovery. Additionally, enhancing the expression of aquaporin-4 (AQP4) on astrocytes may mitigate cerebral edema by regulating water homeostasis in the brain, thereby protecting against secondary injury and further functional decline. Therefore, modulating the activity of astrocytes may serve as a promising therapeutic approach to reduce neuroinflammation and cerebral edema following ICH. Modulating astrocyte function may help alleviate the progression of secondary brain injury and improve patient outcomes. Astrocytes also express the NLRP3 inflammasome (34). Within hypoxic-ischemic encephalopathy (HIE), transient receptor potential vanilloid 1 (TRPV1) intensifies astrocyte activation and promotes the secretion of IL-1β by astrocytes via the JAK2-STAT3 signaling pathway and activation of NLRP3 inflammasomes (56). Additionally, studies have shown that in rats with thalamic hemorrhage-induced central post-stroke pain (CPSP), around the thalamic injury area, the expression of NLRP3 is significantly increased with microglia and astrocytes activation. Thalamic administration of MCC950, a small-molecule NLRP3 inflammasome inhibitor, significantly diminishes microglia and astrocytes activation and lowers the secretion of pro-inflammatory cytokines (57). Neuroinflammation is characterized by an increase in the secretion of inflammatory mediators and microglia and astrocytes activation. The study’s findings highlight the critical interplay between astrocyte activation, the NLRP3 inflammasome, and neuroinflammation. However, research on the interaction between astrocyte activation, NLRP3 inflammasome activity, and neuroinflammation following ICH remains limited, which will be one of our future research focuses. Pyroptosis is a form of caspase-1-dependent programmed cell death, leading to the release of IL-1β and IL-18, which trigger a strong neuroinflammatory response. In a mouse model of ischemic stroke induced by middle cerebral artery occlusion (MCAO), the expression of lipocalin-2 (LCN2) in astrocytes markedly increases after 24 hours, which significantly promotes neuroinflammation. MCC950 inhibition of NLRP3 inflammasome activation reduces astrocyte pyroptosis, which is exacerbated by rLCN2 injection in mice (58). Both ICH and cerebral infarction are classified as strokes and exhibit similar pathological mechanisms. Therefore, exploring the link between astrocyte activation, NLRP3 inflammasome, and neuroinflammation in ICH could provide valuable insights for future therapeutic development.
The NLRP3 inflammasome is regulated through multiple pathways, and this section highlights the latest insights into its regulation. In the following section, we will elaborate on the regulatory mechanisms involving ROS and thioredoxin-interacting protein (TXNIP), P2X7 receptor (P2X7R), tumor necrosis factor receptor-associated factor 6 (TRAF6), and nuclear factor erythroid-2 related factor 2 (Nrf2) (Figure 2, Table 1).
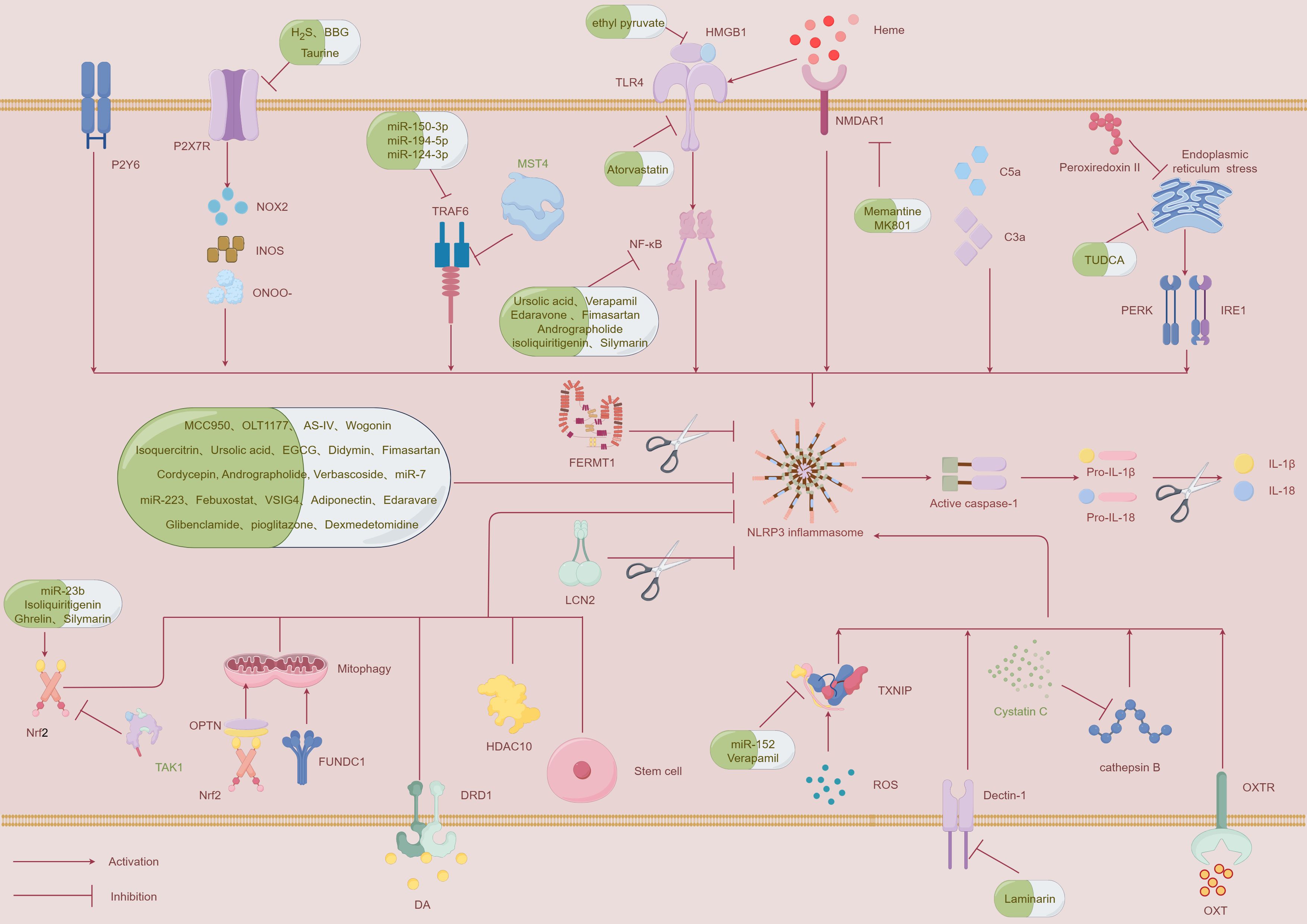
Figure 2. A schematic diagram illustrating the mechanisms that regulate the NLRP3 inflammasome and potential therapeutic strategies to inhibit its activation following ICH. This figure was created by Figdraw.

Table 1. Potential drugs for blocking NLRP3 inflammasome after ICH.
Upon erythrocyte destruction, substantial levels of hemoglobin are released into the extracellular space, where it undergoes oxidation. The oxidized hemoglobin then releases heme, a classical DAMP. When released from hemoproteins, heme acts as a potentially harmful molecule by catalyzing the production of reactive ROS (59, 60). Given that DAMPs and ROS are upstream signals for NLRP3 inflammasome activation, it suggests that heme may be involved in its activation. Hemin, derived from hemoglobin, becomes unstable at physiological temperatures after release. Ferrous heme (Fe2+) spontaneously oxidizes to ferric heme (Fe3+), dissociating from globin, and is classified as a DAMP. Earlier studies showed that in a mouse model of ICH with hemin injection, NLRP3 and IL-1β expression levels were significantly elevated. Moreover, the synergistic interaction with N-methyl-D-aspartate receptor 1 (NMDAR1) led to the activation of NLRP3, ultimately contributing to inflammatory damage (61, 62). Memantine, an antagonist of NMDAR, inhibits nitric oxide synthase ser1412 phosphorylation through an NMDAR-related antagonistic mechanism, thereby reducing peroxynitrite, MMP-9, and NLRP3 production, which in turn mitigates BBB disruption and neurological dysfunction (63). However, the precise mechanisms underlying NMDAR antagonism remain unclear, and this will be a focus of future research. Similarly, Weng et al. observed that administering the NMDAR antagonist MK801 reduces the expression of NLRP3 in heme-treated microglia (62). In a recent study, the injection of heme into the mouse striatum was shown to increase phosphorylation levels of extracellular signal-regulated kinase (ERK) and p38, indicating MAPK pathway activation associated with inflammation. Such activation triggers the secretion of pro-IL-1β, leading to lipid peroxidation and initiating an inflammatory response within the brain. Notably, the knockout of the NLRP3 gene in mice significantly mitigated behavioral impairments and reduced the release of inflammatory cytokines (64). These results imply that heme triggers neuroinflammation through a pathway reliant on NLRP3 activation. Lin et al. additionally found that in a mouse model of ICH, heme amplified microglial activation through TLR4, which in turn triggered NF-κB activation via the TLR4/MyD88/TRIF pathway, thus exacerbating inflammatory damage (65). The study did not thoroughly investigate whether the TLR4/MyD88/TRIF signaling pathway, following the activation of NF-κB, participates in the upregulation process of NLRP3. Given the upregulation of NLRP3 by NF-κB in the priming signals of inflammasome activation, we hypothesize that heme may promote the expression of NLRP3 through this pathway. Nevertheless, further research is required to validate this hypothesis. However, a previous study appears to contradict the conclusions of the aforementioned research. Tan et al. observed that while hemin-treated primary microglial cells could promote the activation of NLRP3 inflammasome and caspase-1, there was no corresponding increase in IL-1β secretion (61). Although the role of heme in activating the NLRP3 inflammasome has been relatively well-studied, the precise connection between NLRP3 activation and the subsequent secretion of inflammatory cytokines remains an unresolved issue in current research. Therefore, further investigation in future experiments is necessary to explore this relationship in greater detail. While collagenase and autologous blood injection are the standard methods for modeling ICH, the study suggests that heme injection may offer a novel model for exploring post-ICH neuroinflammation.
Dopamine is a monoamine neurotransmitter classified within the catecholamine family. It is essential for the regulation of motor function, behavior, endocrine responses, and cardiovascular activity, and acts as a critical link between the nervous and immune systems (66, 67). Yan et al. showed that dopamine regulates systemic inflammation through suppressing NLRP3 inflammasome activation in the hippocampus. Dopamine D1 receptor (DRD1) signaling mediates a negative regulation of hippocampal NLRP3 via the second messenger cyclic adenosine monophosphate (cAMP). This pathway promotes the ubiquitination of NLRP3 by the E3 ubiquitin ligase MARCH7, resulting in NLRP3 degradation and, consequently, controlling the inflammatory response both locally and systemically (67). In a mouse model of ICH, activation of DRD1 was found to suppress microglial activation and downregulate the expression of NLRP3, caspase-1, and IL-1β, while upregulating IFN-β. This modulation significantly reduced NLRP3-driven inflammatory responses, attenuated cerebral edema, and improved neurological outcomes (68). IFN-β is a well-known immunoregulatory cytokine, recognized for its potent anti-inflammatory effect. IFN-β inhibits NLRP3 inflammasome activation and promotes T cell polarization through STAT1 phosphorylation to exert these effects (69, 70).
However, the specific mechanism by which dopamine induces the upregulation of IFN-β was not addressed in the study. Investigating this regulatory pathway in future studies may provide valuable insights for more effectively managing inflammation associated with ICH.
The endoplasmic reticulum (ER), a vital organelle within the cell, fulfills several essential functions, including lipid biosynthesis, promoting protein folding and maturation, and regulating intracellular calcium homeostasis. After ICH, ER stress is induced, which is closely associated with NLRP3 inflammasome activation (71). In obese mice, ER stress triggers the activation of key proteins such as protein kinase RNA-like ER kinase (PERK) and inositol-requiring enzyme-1α (IRE1α), which subsequently activate the NLRP3 inflammasome. This activation results in an increase in caspase-1 expression, which in turn triggers pyroptotic cell death (72). In summary, NLRP3 inflammasome activation induced by ER stress requires both PERK and IRE1 (71). Studies have shown that tauroursodeoxycholic acid (TUDCA) can inhibit ER stress, reducing NLRP3 inflammasome activation and the expression of inflammatory markers in the ICH model (73). Recent studies have also indicated that Peroxiredoxin II reduces neuroinflammation after intracerebral hemorrhage by alleviating neuronal pyroptosis and suppressing NLRP3 inflammasome activation through counteracting ER stress via the PI3K/AKT pathway (74). While research on ER stress-induced inflammasome activation in ICH remains limited, current evidence suggests that targeting ER stress could offer a promising therapeutic approach to reduce post-hemorrhagic inflammation.
After ICH, high mobility group box 1 (HMGB1) is released from the nucleus into the cytoplasm, where it interacts with toll-like receptors (TLRs) on immune cells, initiating an inflammatory cascade. Experimental studies have demonstrated that knocking down HMGB1 or TLR4 significantly reduces associated neurological impairments (75). In a rat model of ICH, elevated expression of NLRP3 inflammasome and IL-1β were observed. Notably, inhibition of HMGB1 or TLR4 led to a substantial decrease in NLRP3 expression (76). Although this study did not address whether the HMGB1/TLR4 axis activates NF-κB expression, previous research has shown that intraperitoneal injection of ethyl pyruvate in diabetic ICH mice can reduce the level of NLRP3 inflammasome via the HMGB1/TLR4 pathway, while concurrently inhibiting NF-κB activation, thereby alleviating neuroinflammation (77). A series of studies have demonstrated that NLRP3, IL-1β are target genes of NF-κB (30, 78). Based on this evidence, we hypothesize that the HMGB1/TLR4 pathway promotes NLRP3 expression through NF-κB activation, exacerbating neuroinflammation following intracerebral hemorrhage. Although there is currently limited research on this pathway in the context of ICH, we hypothesize that targeting the HMGB1/TLR4 axis could serve as a potential therapeutic approach to mitigate post-ICH inflammation. Further studies are needed to validate this mechanism and its potential for improving post-ICH neurological outcomes. Similarly, another study indicated that the TLR4/MyD88 pathway can activate NF-κB, subsequently promoting NLRP3 expression. However, atorvastatin has been shown to interrupt this process, alleviating neuroinflammation following intracerebral hemorrhage (79). Taken together with the aforementioned findings, we hypothesize that directly or indirectly modulating TLR4 may serve as an effective approach for reducing neuroinflammation post-hemorrhage.
The P2Y6 receptor (P2Y6R), part of the P2Y purinergic receptor family, is extensively expressed in the nervous system. It is very important during a range of pathological processes, particularly in immune responses and inflammatory reactions (80, 81). Recent researches have indicated that following ICH, there is a significant increase in the levels of P2Y6R and NLRP3 inflammasome. P2Y6R exacerbates NLRP3-mediated cell apoptosis by activating the PI3K/AKT pathway. Furthermore, inhibition of P2Y6R decreases the expression of key inflammatory factors, highlighting its potential role in the regulation of neuroinflammation (82). Zhu et al. also discovered that a highly selective P2Y6R antagonist (compound 50) effectively suppressed NLRP3 inflammasome activation in intestinal tissues, thereby alleviating symptoms of ulcerative colitis (83). Given that both P2Y6R and P2X7R, as previously discussed, belong to the same purinergic receptor family, they may share similar mechanisms in regulating NLRP3 inflammasome activation. Alternatively, these two receptors could synergistically activate NLRP3 inflammasomes. Exploring this potential interaction may represent a promising future research direction. The development of a dual-target inhibitor capable of blocking both receptors could offer a novel therapeutic approach to mitigate neuroinflammation after ICH.
In rats with ICH, complement activation was first demonstrated, revealing its involvement in promoting brain edema and injury through multiple pathways (84). Complement is closely linked to inflammation in ICH, with the complement cascade representing the initial phase of sterile inflammation (85). Recent research has shown that during ICH-induced neuroinflammation, the levels of complement components C3a and C5a are elevated, and their supplementation can induce IL-1β expression. In contrast, inhibiting NLRP3 mitigates this effect, highlighting the crucial role of NLRP3 in complement-mediated neuroinflammation after ICH (86). However, studies in this area remain limited, and the precise mechanisms connecting complement activation with NLRP3 inflammasome activation have yet to be fully elucidated.
The activation of the NLRP3 inflammasome is closely associated with the occurrence of neuroinflammation. Therefore, targeting the NLRP3 inflammasome is of great significance for alleviating neuroinflammation following intracerebral hemorrhage. Accordingly, we have outlined several effective therapeutic strategies targeting NLRP3 inflammasome activation(Figure 2, Table 1).
Thioredoxin-interacting protein (TXNIP), as a critical component of the α-arrestin superfamily, plays an essential regulatory role in inflammation triggered by oxidative stress. TXNIP interacts with thioredoxin (TRX), and upon dissociation, it triggers NLRP3 inflammasome activation. Oxidative stress is a primary factor that disrupts the interaction between TXNIP and TRX, thereby activating the inflammasome (87, 88). Recent research has demonstrated that in thrombin-induced brain injury, thrombin enhances NLRP3 inflammasome expression via ROS/TXNIP signaling (87). Hu et al. additionally demonstrated that MicroRNA-152 can markedly inhibit TXNIP expression induced by ICH and disrupt the interaction between TXNIP and NLRP3. In microglial cells, overexpression of MicroRNA-152 reduced hemin-induced neuroinflammation and lowered ROS production, offering protective effects to co-cultured neurons. In vivo, overexpression of MicroRNA-152 in rat brains significantly mitigated neurological damage, cerebral edema, and promoted hematoma clearance following ICH (89). Verapamil, a commonly used phenylalkylamine L-type calcium (Ca2+) channel blocker (CCB), is pharmacologically recognized for its antihypertensive, anti-inflammatory, and BBB-modulating effects. Recent studies have demonstrated its benefits across various neurovascular conditions. In a collagenase-induced ICH mouse model, verapamil administration has demonstrated efficacy in reducing TXNIP and NLRP3 expression around the hematoma region, inhibiting NF-κB activation, facilitating hematoma clearance, and improving both neuroinflammation and blood-brain barrier dysfunction (88). Furthermore, in mouse models of hyperglycemic stroke, TXNIP promotes the activation of the NLRP3 inflammasome, thereby aggravating neuronal injury (90). Recent research indicates that in cerebral ischemia/reperfusion (I/R) injury, COG1410, an agonist of Low-density lipoprotein receptor-related protein-1 (LRP1), inhibits the TXNIP/NLRP3 signaling pathway and promotes the shift in microglial polarization from the M1 phenotype to the M2 phenotype, thereby contributing to anti-inflammatory effects (91). While the latter two studies do not focus specifically on ICH, an integrated analysis of these findings suggests that the ROS/TXNIP/NLRP3 pathway plays a significant pro-inflammatory role in cerebrovascular diseases. This suggests that further investigation into the inhibition of the ROS/TXNIP/NLRP3 pathway could be crucial for reducing neuroinflammation and improving outcomes following ICH.
The P2X7 receptor (P2X7R) is a member of the P2X purinergic receptor family, which includes subtypes P2X1 to P2X7. P2X7R is a non-selective cation channel activated by ATP and is highly expressed in microglial cells and astrocytes (92, 93). Downregulation of the P2X7R gene effectively inhibits the NLRP3 inflammasome activation and reduces the secretion of IL-1 and IL-18, which helps to remarkably decrease brain edema and improve neurological impairments. Treatment with the specific P2X7R inhibitor Blue Brilliant G (BBG) has been shown to significantly reduce the levels of NADPH oxidase 2 (NOX2), inducible nitric oxide synthase (iNOS), and peroxynitrite (ONOO-), all of which are associated with neurotoxicity in the CNS following ICH. The application of peroxynitrite (ONOO-) decomposing catalysts significantly inhibits the NLRP3 inflammasome activation and reduces the release of IL-1β and IL-18 (94). Emerging research indicates that H2S offers considerable neuroprotection across a range of CNS conditions, including Alzheimer’s disease, subarachnoid hemorrhage, and ischemic stroke. After ICH, endogenous H2S levels decrease; however, exogenous supplementation of H2S can alleviate the P2X7R/NLRP3 inflammatory cascade, reducing brain edema, neurological deficits, and microglial accumulation following ICH (93). Moreover, a study suggested that high doses of taurine can mitigate post-ICH dysfunction, brain edema, and hematoma volume by upregulating H2S in brain tissue and downregulating P2X7R expression (95). Recent findings also showed that puerarin suppresses ferroptosis by inhibiting the P2X7R/NLRP3 pathway and mitigates Staphylococcus aureus-induced endometritis (96). While research on the P2X7R/NLRP3 pathway in the context of ICH is still limited, significant progress has been made in reducing inflammation via this pathway in other disease models. Therefore, developing more inhibitors or drugs targeting this signaling pathway will be one of our future research directions.
Tumor necrosis factor receptor-associated factor 6 (TRAF6), a key adaptor protein in the TNF and TLR superfamilies, is well recognized for its strong connection to neural damage and NLRP3-driven inflammation. TRAF6 levels increase 24 hours post-ICH and bind to NLRP3, facilitating inflammasome activation (97, 98). Mammalian sterile 20-like kinase 4 (MST4), part of the GCK subfamily, functions as a key inhibitor of inflammation. MST4 alleviates the inflammatory response by directly phosphorylating TRAF6. Increased expression of the MST4 gene suppresses NLRP3 inflammasome activation following ICH, leading to a decrease in pro-inflammatory cytokine release, which in turn helps to reduce cerebral edema and improve neurological function (99). However, the study did not clarify the underlying mechanisms by which MST4 upregulation impacts TRAF6, leaving this question open for future investigation. Furthermore, the role of TRAF6 in regulating microglia-mediated secondary inflammatory responses post-ICH via NLRP3 inflammasome activation was not addressed. Consequently, exploring the interactions between TRAF6, NLRP3 inflammasome activation, and microglia-driven secondary neuroinflammation will be a primary focus in our future research endeavors.
In recent years, with the advancement of regenerative medicine, using stem cells and exosomes to promote neuroprotection and recovery after ICH has shown great potential. Sun et al. reported that exosomal miR-150-3p derived from stem cells can bind directly to TRAF6, exhibiting an inverse correlation with its expression levels. Mechanistically, exosomal miR-150-3p alleviates injury and neurological deficits after ICH by modulating the TRAF6/NF-κB axis (100). MST4, an anti-inflammatory regulator, directly phosphorylates the adaptor protein TRAF6, thereby inhibiting NLRP3 inflammasome activation. This process reduces neuroinflammation and helps alleviate brain injury after ICH. In contrast, Hesperadin, a selective inhibitor of MST4, increases NLRP3 expression, exacerbating neurological deficits and brain edema (99).Additionally, two microRNAs also regulate TRAF6 expression. miR-124-3p effectively suppresses the microglia-mediated secondary inflammatory response following ICH by modulating TRAF6 expression and inhibiting NLRP3 inflammasome activation (97). Likewise, the overexpression of miR-194-5p diminishes the interaction between TRAF6 and NLRP3, thereby suppressing NLRP3 inflammasome activation and alleviating neuroinflammation following ICH (98).
Nuclear factor erythroid-2 related factor 2 (Nrf2) is a nuclear transcription factor that responds to oxidative stress by activating the expression of antioxidant and detoxifying enzymes, including NAD(P)H: quinone oxidoreductase-1 (NQO1) and catalase (CAT). This activation is essential for preserving intracellular homeostasis, as it regulates the redox balance within cells (101, 102). In animal models of ICH, Nrf2 is upregulated, which activates protective genes regulated by antioxidant response elements (AREs). This increase in expression suppresses reactive ROS production and inflammation, resulting in a neuroprotective effect (103). Previous researches have confirmed that Nrf2 primarily inhibits the NLRP3 inflammasome through reducing ROS levels and/or suppressing NF-κB activity, which helps alleviate early brain injury. In Nrf2 knockout animals, oxidative damage and leukocyte infiltration following ICH are significantly higher than in control groups (103, 104).
Transforming Growth Factor-beta-activated kinase 1 (TAK1), as a member of the MAP3K family within the mitogen-activated protein kinase (MAPK) cascade, plays a critical role in various disease models, especially in the generation of ROS and the amplification of oxidative damage. In a rat model of ICH, inhibiting TAK1 alleviated BBB disruption, neuroinflammation, and oxidative damage following ICH. Mechanistically, inhibition of TAK1 facilitates the nuclear translocation of NRF2, which in turn reduces ROS production and oxidative stress. This mechanism effectively suppresses NLRP3-mediated neuroinflammation, contributing to the mitigation of neuroinflammatory damage (105). Phosphatase and tensin homolog (PTEN) plays a crucial role in the regulation of oxidative stress and inflammation. PTEN can directly interact with NLRP3 and dephosphorylate it, thereby promoting the interaction between NLRP3 and ASC, which leads to inflammasome activation. In a rat with ICH, the intravenous injection of exosomal miR-23b derived from bone marrow mesenchymal stem cells (BMSCs) significantly reduced brain edema and led to improved behavioral outcomes. BMSC-exosomal miR-23b alleviates NLRP3-mediated neuroinflammation by promoting NRF2 nuclear translocation and inhibiting the interaction between PTEN and NLRP3 (106). Another study revealed that Nrf2 interacts with the mitophagy receptor optineurin (OPTN), regulating mitophagy to remove dysfunctional mitochondria following ICH. This interaction prevents the NLRP3 inflammasome activation, resulting in a reduction in neuroinflammation and mitigating secondary brain injury in rat models of ICH (103). Ghrelin has the ability to cross the BBB and provides neuroprotective effects in various CNS diseases, including subarachnoid hemorrhage and traumatic cerebral damage. In a mouse with ICH, intraperitoneal injection of Ghrelin reduced neuroinflammation, brain edema, and promoted hematoma clearance. Mechanistically, ghrelin activates the Nrf2/ARE pathway, enhancing antioxidant enzymes expression like NQO1. This results in reduced ROS levels and suppression of NLRP3 inflammasome activation (104). Zeng et al. also demonstrated that intraperitoneal injection of Isoliquiritigenin significantly inhibits the NF-κB and NLRP3 inflammasome pathways while activating the Nrf2-mediated antioxidant system, thereby alleviating cerebral edema, neuronal deformation, and neurological deficits following ICH (102). Silymarin has been shown to downregulate NF-κB expression, thereby inhibiting caspase-1 and IL-1β expression triggered by inflammasome activation. Additionally, it promotes the activation of the Nrf2/HO-1 signaling pathway. These lead to increased overall cellular protection and reduced inflammatory activation after ICH (101).
However, there is some controversy regarding whether Nrf2 can suppress NLRP3 expression. In several disease models, Nrf2 has been associated with NLRP3 inflammasome activation, promoting IL-1-mediated vascular inflammation and potentially aggravating conditions such as atherosclerosis (107). Recent researches reported that hydrogen gas can attenuate NLRP3 inflammasome activation through inhibiting Nrf2, which in turn reduces inflammation and helps alleviate sepsis-associated encephalopathy (108). It is possible that Nrf2 modulates NLRP3 via different mechanisms, either activating or inhibiting it, or that Nrf2 specifically inhibits NLRP3 expression in ICH models. Further studies are necessary to clarify this issue.
MCC950, a potent selective NLRP3 inflammasome inhibitor, effectively suppresses its activation even at nanomolar concentrations. In a mouse model of ICH, MCC950 significantly decreased IL-1β secretion, alleviated cerebral edema, and enhanced neurological recovery (109). A recent study further showed that in a mouse with ischemic stroke, MCC950 effectively lowered the levels of caspase-1 and IL-1β, leading to reduced brain edema and a smaller infarct area (110). A new NLRP3 inflammasome inhibitor, OLT1177, has recently emerged. In mice with intracerebral hemorrhage (ICH), treatment with OLT1177 significantly inhibited Caspase-1 activation and IL-1β release, notably reducing brain edema and improving neurological deficits following ICH (15). These results, along with earlier studies on MCC950, indicate that both OLT1177 and MCC950 may serve as promising therapeutic options for mitigating post-stroke neuroinflammation by inhibiting NLRP3 inflammasome activation.
In recent years, traditional Chinese herbs and plant extracts have shown promising potential in mitigating neuroinflammation following ICH. Astragaloside IV(AS-IV), a monomer extracted from Radix Astragali, has demonstrated neuroprotective effects by modulating protein expression and reducing apoptosis, thereby alleviating neuroinflammation. In preclinical studies using a mouse model of ICH, AS-IV was found to inhibit NLRP3 inflammasome expression by upregulating Kruppel-like factor 2 (KLF2), which resulted in a reduction in brain edema, attenuation of neuroinflammation, and improvements in neurological function (17). Methylation of the NLRP3 gene is essential for regulating apoptosis in the context of ICH. NLRP3 mRNA methylation plays a crucial role in regulating cell apoptosis in brain hemorrhage. Wogonin, a monomer derived from traditional Chinese medicine, reduces pyroptosis following ICH by inhibiting the methyltransferase METTL14, which mediates NLRP3 m6A methylation. This action helps restore neurological function and decreases pro-inflammatory factor release (111). METTL14’s role in NLRP3 m6A methylation has demonstrated significant potential in regulating inflammasome activation. In an acute lung injury model, METTL14 promotes the activation of the NLRP3 inflammasome through NLRP3 mRNA m6A methylation, while METTL14 knockout alleviates injury (112). Targeting the methylation of NLRP3 may represent an important therapeutic approach to mitigate neuroinflammation following ICH. Isoquercitrin, found in functional foods such as mulberries, exerts its anti-inflammatory effects by scavenging reactive oxygen species, enhancing antioxidant enzyme activity, and inhibiting inflammatory factors. It can reduce the expression of the ion channel Piezo1, thereby modulating calcium influx and suppressing NLRP3 expression, ultimately alleviating neuronal damage following intracerebral hemorrhage in rats (113). Ursolic acid, a type of pentacyclic triterpene found in several plant species, is recognized for its potent anti-inflammatory properties. In a collagenase-induced rat ICH model, ursolic acid inhibits the transformation of microglia into the M1 phenotype. This was accompanied by a significant decrease in the levels of NF-κB, TNF-α, IL-6, IL-1β, and caspase-1. This reduction contributed to the alleviation of neuroinflammation following ICH. Mechanistically, ursolic acid exerts its neuroprotective effects primarily by inhibiting the NF-κB/NLRP3/GSDMD pathway (9). Bao et al. found that Epigallocatechin-3-gallate (EGCG) can reduce neuroinflammation after ICH by increasing the expression of heme oxygenase-1 (HO-1) and decreasing the levels of caspase-1, NLRP3, IL-1β, and IL-18. Additionally, EGCG enhances microglial polarization towards the M2 phenotype, which is known for its anti-inflammatory effects. This dual action of reducing inflammatory markers and promoting beneficial microglial phenotypes indicates that EGCG can effectively improve neuroinflammation after ICH (46). Didymin, a citrus-derived flavonoid, exhibits potent antioxidant, anti-inflammatory, and neuroprotective properties. Raf kinase inhibitor protein (RKIP) has been shown to bind to ASC, thereby preventing the assembly of the NLRP3 inflammasome. Didymin alleviates neuroinflammation following ICH by upregulating RKIP and downregulating the expression of NLRP3, caspase-1, IL-1β, TNF-α, and MPO (10, 114). Moreover, herbal compounds such as Cordycepin, Andrographolide, and Verbascoside have been shown to attenuate inflammation following ICH by inhibiting the NLRP3 inflammasome. Andrographolide can also inhibit the NF-κB signaling pathway (115–117). These studies suggest that traditional Chinese medicines and plant-derived extracts exhibit remarkable anti-inflammatory effects in the context of ICH. Future research into identifying additional herbal compounds or plant extracts capable of mitigating post-hemorrhagic inflammation holds significant therapeutic potential.
MicroRNAs (miRNAs) are one of the most extensively studied classes of non-coding RNAs. They regulate gene expression by blocking mRNA translation and mediating mRNA degradation, playing significant roles in cell proliferation, apoptosis, differentiation, and metabolism (118–120). A recent study found that MicroRNA-7 is capable of inhibiting NLRP3 expression in microglia and macrophages. This action reduces both the protein and mRNA expression of pro-inflammatory cytokines, thereby offering protection to neurological function following ICH (14). Similarly, Yang et al. discovered that MicroRNA-223 binds to specific sites on NLRP3, leading to a reduction in its expression. This, in turn, decreases the expression of caspase-1 and IL-1β, ultimately alleviating cerebral edema and improving neurological function after ICH (121).
Febuxostat, a xanthine oxidoreductase inhibitor, has been previously used in the treatment of chronic gout. In ICH mice pretreated with Febuxostat, inhibition of the NLRP3/ASC/caspase-1 pathway led to a reduction in neuronal deformation and cell death, thereby alleviating neurological deficits (122). V-set and immunoglobulin domain-containing 4 (VSIG4), which belongs to the B7 superfamily, is involved in transmitting inflammatory signals and has been implicated in various inflammatory conditions. Injection of VSIG4 into ICH mice can suppress NLRP3 expression through the JAK2-STAT3-A20 pathway, thereby reducing BBB disruption and cerebral edema (123). Adiponectin (APN) and Edaravone exert effects comparable to those of MCC950 through decreasing the levels of NLRP3, IL-1β, and IL-18, thus reducing brain injury and enhancing recovery following ICH. Edaravone can also inhibit the NF-κB signaling pathway (124, 125).Recently, two drugs commonly used for treating diabetes have also been applied to mitigate NLRP3-mediated neurological deficits following ICH. Glibenclamide has been shown to preserve the integrity of the BBB by inhibiting NLRP3 inflammasome activation in microvascular endothelial cells, thereby preventing ICH-associated damage (126). Meanwhile, pioglitazone exerts neuroprotective effects post-ICH by suppressing NLRP3 expression and enhancing anaerobic glycolysis, leading to increased lactate production and reduced cerebral edema (127).
Dexmedetomidine, a highly selective α2-adrenergic receptor agonist frequently employed for anesthesia, sedation, and analgesia, has also been found to downregulate NLRP3 expression following ICH. This promotes hematoma clearance, reduces cerebral edema, and alleviates neuroinflammation (128). Chronic hypertension is a leading risk factor for ICH, and fimasartan is commonly prescribed for managing this condition (129). Research has demonstrated that low doses of fimasartan significantly reduce NLRP3 inflammasome and NF-κB pathways activation, alleviating neuroinflammation and brain injury post-ICH without affecting blood pressure (130). Given that fimasartan’s antihypertensive effect is dose-dependent, future studies could investigate the potential impact of increasing fimasartan dosage on the prognosis of hypertensive ICH patients.
Autophagy serves as a highly conserved cellular mechanism, playing an essential role in the removal of damaged, aging, or surplus cytoplasmic components, directing them to lysosomes for degradation and recycling. Mitophagy, a specialized subtype of autophagy, is tasked with the selective removal of impaired or malfunctioning mitochondria via lysosomal degradation. Emerging studies indicate that mitophagy helps to inhibit NLRP3 inflammasome activation, thereby regulating inflammatory responses (131, 132). FUN14 domain containing 1 (FUNDC1) is a key mitophagy receptor. Zheng and colleagues demonstrated that FUNDC1 mitigates neuroinflammation following ICH by promoting mitophagy, which in turn suppresses the expression of NLRP3 inflammasome (133). Studies have also found that Nrf2 interacts with the mitochondrial autophagy receptor optineurin (OPTN), regulating mitophagy to clear dysfunctional mitochondria following intracerebral hemorrhage (ICH). This process inhibits NLRP3 inflammasome activation, thereby reducing neuroinflammation (103).However, conflicting evidence exists, with earlier studies showing that TLR4 can activate microglia and exacerbate inflammatory damage in ICH through autophagy pathways (134). This suggests ongoing debate about the precise role of autophagy in regulating inflammation post-ICH. Despite this, numerous studies have indicated that both autophagy and mitophagy can attenuate inflammation after ICH (135). So further studies are needed to elucidate the detailed mechanisms underlying these effects.
Stem cell therapy offers significant promise for promoting neuroprotection and enhancing neurorecovery (136). BMSC possess multi-differentiation and self-renewal abilities, offering significant potential in treating neurological sequelae following ICH (137, 138). Extracellular vesicles derived from BMSC have demonstrated the potential to enhance neurological function, decrease brain edema, and mitigate neuroinflammation in diabetic ICH, primarily through the delivery of miR-183-5p. Mechanistically, miR-183-5p exerts its effects by targeting PDCD4 and suppressing the NLRP3 pathway (139). Furthermore, hypoxic preconditioning of mesenchymal stem cells has been shown to suppress microglial activation, decrease IL-1β and TNF-α levels, and downregulate NLRP3 and Caspase-1 expression, thereby reducing pyroptosis and neuroinflammation following ICH (140). At present, stem cell-based therapies for ICH are gaining significant attention, with various types of stem cells being explored, including mesenchymal stem cells, embryonic stem cells, hematopoietic stem cells, and neural stem cells (136). However, most studies on inhibiting neuroinflammation after ICH have concentrated on mesenchymal stem cells. In the future, we may explore the potential of different stem cell types in alleviating neuroinflammation following ICH.
We found that knocking out certain genes can also inhibit NLRP3 expression and thereby reduce neuroinflammation following ICH. Zhao et al. found that LCN2 knockout in an ICH rat model significantly diminished the secretion of inflammatory cytokines including IL-1β, IL-18, and TNF-α. Additionally, it reduced NLRP3 inflammasome expression, facilitated hematoma clearance, and alleviated brain edema and inflammatory cell infiltration (141).Additionally, the FERM domain-containing kindlin 1 (FERMT1) is an integrin-associated protein. Knocking out FERMT1 reduced brain edema after ICH, promoted hematoma clearance, and improved behavioral outcomes. FERMT1 knockout was also found to suppress NLRP3 inflammasome activation, leading to a reduction in the release of pro-inflammatory cytokines such as IL-1β and IL-18 (142). The commonly used gene editing technologies, such as CRISPR-Cas9, have developed rapidly, and successful clinical trials have already been conducted. For example, Lu et al. used CRISPR-Cas9 technology to knock out Programmed Cell Death Protein 1 (PD-1) on human T cells to treat patients with non-small cell lung cancer, demonstrating strong safety and feasibility (143). However, the clinical translation of gene editing technology still faces significant obstacles. The application of gene editing in humans raises ethical controversies, particularly in cases involving the editing of human germline cells, which could lead to severe ethical debates. Additionally, gene editing technologies carry the risk of off-target effects, where the editing tool may mistakenly cut non-target genes during DNA cleavage, potentially leading to the deletion of essential genes and causing severe consequences. Therefore, the effectiveness of specific gene knockout treatments for post-ICH neuroinflammation remains to be determined.
Studies have shown that cathepsin B (CTSB) is crucial in regulating NLRP3 expression. The activation of the CTSB/NLRP3 signaling pathway exacerbates neurological damage, especially in cases of hippocampal injury (144). In a study conducted on rats with ICH, Zhou et al. reported that Cystatin C effectively inhibits CTSB-mediated activation of the NLRP3 inflammasome within microglia, consequently mitigating secondary brain damage after ICH (16). Oxytocin (OXT), secreted by neurons in the hypothalamus, plays a multifunctional role by promoting parturition and exhibiting anti-inflammatory and antioxidant properties through the stimulation of oxytocin receptors (OXTR). Yang et al. found that following ICH, endogenous OXT levels were decreased, while the expression of OXTR was significantly upregulated. Intranasal administration of OXT has been shown to suppress the secretion of NLRP3, caspase-1, IL-1, and IL-18. Mechanistically, OXT exerts its neuroprotective effects and attenuates neuroinflammation after ICH through activation of the OXTR/p-PKA/DRP1 signaling pathway (145). Dendritic cell-associated C-type lectin-1 (Dectin-1) is a pattern recognition receptor known to initiate inflammatory responses. Research has shown that Dectin-1 expression significantly increases in microglia following ICH. Laminarin-mediated inhibition of Dectin-1 has been shown to downregulate the expression of NLRP3, IL-1, and IL-18. This results in a reduction of brain edema and a decrease in neurological damage after ICH, suggesting that targeting Dectin-1 could be a promising approach for alleviating neuroinflammation and improving outcomes following ICH (146). Protein tyrosine phosphatase non-receptor type 22 (PTPN22) has been linked to the development of several inflammatory disorders. It plays a role in enhancing NLRP3 inflammasome activation and promoting the release of IL-1β, contributing to the inflammatory response (147). Wang et al. demonstrated that histone deacetylase 10 (HDAC10) can mitigate neuroinflammation after ICH by blocking the interaction between PTPN22 and NLRP3. This inhibition prevents inflammasome activation and reduces the inflammatory damage that follows (148).
Acute spontaneous ICH represents the most prevalent subtype among ICH types. Cases of acute ICH typically require prompt and aggressive management, commonly involving surgical intervention or pharmacological therapy (149, 150).
For patients with cerebellar hemorrhage and rapid deterioration due to brainstem compression, surgical hematoma evacuation is broadly recognized as the standard of care. In cases of supratentorial hemorrhage, early craniotomy (within 24 hours of onset) to remove the hematoma is considered a critical life-saving intervention (151, 152). Following ICH, blood degradation products can induce inflammatory meningitis and hydrocephalus. Ventriculostomy facilitates the rapid evacuation of hemorrhage from the ventricles, significantly enhancing survival rates in patients with deteriorating neurological status (153, 154). In China, minimally invasive hematoma decompression surgery, guided by non-contrast CT with catheter or probe placement, has become the standard approach. In developed countries, this technique has demonstrated better prognostic outcomes compared to craniotomy and conservative treatment in brain hemorrhage management (152, 155, 156).
Hypertension is a significant risk factor for ICH. Consequently, effective blood pressure control is essential during both the preoperative and postoperative periods. Consistent regulation of blood pressure can help prevent ICH. Furthermore, findings from an international clinical trial indicate that early reduction of systolic blood pressure to below 140 mmHg in patients with ICH can modestly improve functional outcomes (157, 158). Additionally, strong evidence suggests that maintaining blood pressure within the range of 130–140 mmHg within six hours of ICH onset can enhance clinical prognosis (159). Hemostatic therapy is widely applied in the management of ICH, with the primary goal of mitigating the early expansion of hematomas. For patients who present within 4 hours of ICH onset, recombinant factor VIIa (rFVIIa) serves as a key therapeutic option (160, 161). Additionally, a large-scale international trial investigating the treatment of ultra-acute primary ICH demonstrated that intravenous administration of tranexamic acid within 8 hours of ICH onset significantly reduced the rates of mortality and severe adverse events within 7 days compared to the placebo group, while also modestly limiting hematoma expansion (159). Furthermore, the reversal of anticoagulant and antiplatelet therapies constitutes a critical component of ICH management. Patients with a history of anticoagulant or antiplatelet use generally exhibit poorer prognoses, underscoring the importance of timely intervention. For those with prior antiplatelet therapy, platelet transfusion has been shown to improve clinical outcomes. In cases involving warfarin-related anticoagulation, reversal strategies include the administration of vitamin K, prothrombin complex concentrates, fresh frozen plasma, or rFVIIa (150, 162).
Drugs targeting neuroinflammation after brain hemorrhage are currently under development or undergoing clinical evaluation (55). Statins, for instance, have demonstrated the ability to inhibit microglial activation in the CNS, reduce ROS and MMP production, and suppress inflammatory progression (163). In a clinical trial on rosuvastatin, statin therapy was associated with a reduced mortality rate in brain hemorrhage patients (164). Furthermore, a Danish registry study indicated that statin therapy could lower the risk of brain hemorrhage (165). We mentioned earlier that, in preclinical animal models, atorvastatin was shown to suppress NLRP3 expression and neuroinflammation via the TLR4/MYD88 signaling pathway (79). Minocycline, a tetracycline-class drug, has demonstrated anti-inflammatory effects by inhibiting MMP production and promoting the secretion of neurotrophic factors (166). In a randomized, blinded clinical trial, high-dose administration of minocycline within 12 hours of ICH symptom onset significantly reduced MMP-9 levels compared to the placebo group, which helped to mitigate inflammation (167). Similarly, preclinical studies in SAH rat models have shown that minocycline suppresses NLRP3 inflammasome expression and reduces IL-1β release, thereby alleviating inflammation (168). Although direct evidence on minocycline’s role in suppressing NLRP3 inflammasome expression in ICH is limited, the shared pathophysiological mechanisms between ICH and SAH suggest its potential for clinical translation based on findings from both clinical and preclinical studies. As previously discussed, pioglitazone exerts neuroprotective effects and alleviates cerebral edema following ICH in mice by inhibiting NLRP3 expression and enhancing anaerobic glycolysis to increase lactate production (127). The anti-inflammatory drug pioglitazone is currently undergoing a Phase I clinical trial for ICH, although the related results appear to remain unpublished (55). In preclinical studies using animal models, glibenclamide has been shown to protect the integrity of the blood-brain barrier by inhibiting the activation of NLRP3 inflammasome in microvascular endothelial cells, thereby preventing ICH-induced damage (126). Clinical trials investigating the use of glibenclamide in ICH are currently recruiting participants (55). Hypertension is a major risk factor for ICH, and the use of antihypertensive drugs has proven therapeutic value both before and after surgery (157, 158). As discussed earlier, fimasartan, an antihypertensive agent, significantly suppresses the activation of the NLRP3 inflammasome and the NF-κB pathway in rat models of ICH at low doses, thereby reducing neuroinflammation and brain injury (130). Although clinical trials for fimasartan in ICH have not yet begun, its preclinical results suggest considerable potential for clinical application in the comprehensive treatment of ICH.
In this review, we have highlighted various drugs capable of directly or indirectly inhibiting NLRP3 inflammasome activation. Among them, several are currently in clinical trials and show promising prospects for clinical translation based on trial outcomes.
ICH remains a significant challenge due to its high incidence and mortality rates. Neuroinflammation following ICH exacerbates inflammatory injury, contributing to brain edema and increasing intracranial pressure, which may further lead to ischemic damage. Thus, targeting neuroinflammation post-ICH is crucial for improving patient outcomes. NLRP3 inflammasome activation is pivotal in this process, typically leading to microglial activation and the subsequent secretion of pro-inflammatory cytokines. NLRP3 is expressed in both microglia and astrocytes, cells that play a crucial role in mediating neuroinflammatory responses. Pro-inflammatory M1 microglia and A1 astrocytes drive inflammation, whereas anti-inflammatory M2 microglia and A2 astrocytes are involved in tissue repair. Numerous studies indicate that inhibiting NLRP3 in microglia significantly reduces inflammation, cerebral edema, and neurological deficits after ICH. Additionally, models of HIE and CPSP show a clear association between NLRP3 upregulation and astrocyte activation. While research focusing on astrocytic NLRP3 inhibition in ICH is limited, it is plausible that jointly targeting NLRP3 in both microglia and astrocytes could represent a novel approach to mitigating post-ICH neuroinflammation.
With ongoing research in this area, multiple pathways have been uncovered that directly or indirectly downregulate NLRP3 expression. Notably, the ROS/TXNIP/NLRP3, P2X7R/NLRP3, TRAF6/NLRP3, and Nrf2/NLRP3 pathways have been extensively characterized. Nevertheless, considerable opportunities remain to explore alternative mechanisms for inhibiting NLRP3. For example, the increasing focus on stem cell therapies suggests that we might investigate the potential of different stem cell types, in combination with exosome therapy, to mitigate post-ICH inflammation. Although the knockout of specific genes in animal models can indeed alleviate neuroinflammation following ICH, its clinical translational potential remains uncertain. Furthermore, it is critical to investigate how both established and novel pathways that inhibit NLRP3 could be leveraged to modulate the phenotypes of microglia and astrocytes, a topic that has received limited attention in current research.
In summary, several drugs previously discussed that inhibit NLRP3 inflammasome activation are now in clinical trials, with promising preliminary findings highlighting their potential for clinical application. Modulating NLRP3 to reduce neuroinflammation after ICH represents a promising therapeutic avenue and may pave the way for innovative strategies to manage post-hemorrhagic inflammation in the future.
KH: Funding acquisition, Investigation, Methodology, Writing – original draft. XY: Methodology, Writing – original draft. LX: Methodology, Writing – review & editing. YX: Investigation, Writing – review & editing. EQ: Investigation, Writing – review & editing. LW: Investigation, Writing – review & editing. XH: Investigation, Writing – review & editing. CJ: Funding acquisition, Investigation, Methodology, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the National Natural Science Foundation of China (Grant Nos. 82271314 and 82471319).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ren H, Pan Y, Wang D, Hao H, Han R, Qi C, et al. CD22 blockade modulates microglia activity to suppress neuroinflammation following intracerebral hemorrhage. Pharmacol Res. (2023) 196:106912. doi: 10.1016/j.phrs.2023.106912
2. Yan XL, Xu FY, Ji JJ, Song P, Pei YQ, He MJ, et al. Activation of UCP2 by anethole trithione suppresses neuroinflammation after intracerebral hemorrhage. Acta Pharmacol Sin. (2022) 43:811–28. doi: 10.1038/s41401-021-00698-1
3. Dai S, Wei J, Zhang H, Luo P, Yang Y, Jiang X, et al. Intermittent fasting reduces neuroinflammation in intracerebral hemorrhage through the Sirt3/Nrf2/HO-1 pathway. J Neuroinflamm. (2022) 19:122. doi: 10.1186/s12974-022-02474-2
4. Bautista W, Adelson PD, Bicher N, Themistocleous M, Tsivgoulis G, Chang JJ. Secondary mechanisms of injury and viable pathophysiological targets in intracerebral hemorrhage. Ther Adv Neurol Disord. (2021) 14:17562864211049208. doi: 10.1177/17562864211049208
5. Duan Z, Zhou W, He S, Wang W, Huang H, Yi L, et al. Intranasal delivery of curcumin nanoparticles improves neuroinflammation and neurological deficits in mice with intracerebral hemorrhage. Small Methods. (2024) 8:e2400304. doi: 10.1002/smtd.202400304
6. Wang Y, Tian M, Tan J, Pei X, Lu C, Xin Y, et al. Irisin ameliorates neuroinflammation and neuronal apoptosis through integrin αVβ5/AMPK signaling pathway after intracerebral hemorrhage in mice. J Neuroinflamm. (2022) 19:82. doi: 10.1186/s12974-022-02438-6
7. Tschoe C, Bushnell CD, Duncan PW, Alexander-Miller MA, Wolfe SQ. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J Stroke. (2020) 22:29–46. doi: 10.5853/jos.2019.02236
8. Xia S, Zheng Y, Yan F, Chen G. MicroRNAs modulate neuroinflammation after intracerebral hemorrhage: Prospects for new therapy. Front Immunol. (2022) 13:945860. doi: 10.3389/fimmu.2022.945860
9. Lei P, Li Z, Hua Q, Song P, Gao L, Zhou L, et al. Ursolic Acid Alleviates Neuroinflammation after Intracerebral Hemorrhage by Mediating Microglial Pyroptosis via the NF-κB/NLRP3/GSDMD Pathway. Int J Mol Sci. (2023) 24:14771. doi: 10.3390/ijms241914771
10. Gu L, Sun M, Li R, Zhang X, Tao Y, Yuan Y, et al. Didymin suppresses microglia pyroptosis and neuroinflammation through the Asc/Caspase-1/GSDMD pathway following experimental intracerebral hemorrhage. Front Immunol. (2022) 13:810582. doi: 10.3389/fimmu.2022.810582
11. Wu CH, Shyue SK, Hung TH, Wen S, Lin CC, Chang CF, et al. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase reduces brain damage and attenuates neuroinflammation after intracerebral hemorrhage. J Neuroinflamm. (2017) 14:230. doi: 10.1186/s12974-017-1005-4
12. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
13. Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. (2012) 11:720–31. doi: 10.1016/S1474-4422(12)70104-7
14. Luo B, Li L, Song XD, Chen HX, Yun DB, Wang L, et al. MicroRNA-7 attenuates secondary brain injury following experimental intracerebral hemorrhage via inhibition of NLRP3. J Stroke Cerebrovasc Dis. (2024) 33:107670. doi: 10.1016/j.jstrokecerebrovasdis.2024.107670
15. Fang M, Xia F, Wang J, Wang C, Teng B, You S, et al. The NLRP3 inhibitor, OLT1177 attenuates brain injury in experimental intracerebral hemorrhage. Int Immunopharmacol. (2024) 131:111869. doi: 10.1016/j.intimp.2024.111869
16. Zhou Y, Dong W, Wang L, Ren S, Wei W, Wu G. Cystatin C attenuates perihematomal secondary brain injury by inhibiting the cathepsin B/NLRP3 signaling pathway in a rat model of intracerebral hemorrhage. Mol Neurobiol. (2024) 61:9646–62. doi: 10.1007/s12035-024-04195-4
17. Wu H, Chen S, You G, Lei B, Chen L, Wu J, et al. The mechanism of astragaloside IV in NOD-like receptor family pyrin domain containing 3 inflammasome-mediated pyroptosis after intracerebral hemorrhage. Curr Neurovasc Res. (2024) 21:74–85. doi: 10.2174/0115672026295640240212095049
18. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
19. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
20. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
21. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
22. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. (2018) 113:5. doi: 10.1007/s00395-017-0663-9
23. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. (2018) 17:158. doi: 10.1186/s12943-018-0900-3
24. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. (2015) 6:262. doi: 10.3389/fphar.2015.00262
25. Luo Y, Reis C, Chen S. NLRP3 inflammasome in the pathophysiology of hemorrhagic stroke: A review. Curr Neuropharmacol. (2019) 17:582–9. doi: 10.2174/1570159X17666181227170053
26. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
27. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
28. Zhang L, Tang Y, Huang P, Luo S, She Z, Peng H, et al. Role of NLRP3 inflammasome in central nervous system diseases. Cell Biosci. (2024) 14:75. doi: 10.1186/s13578-024-01256-y
29. Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. (2021) 18:1141–60. doi: 10.1038/s41423-021-00670-3
30. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
31. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflammation Res. (2015) 8:15–27. doi: 10.2147/JIR.S51250
32. Rabeony H, Pohin M, Vasseur P, Petit-Paris I, Jegou JF, Favot L, et al. IMQ-induced skin inflammation in mice is dependent on IL-1R1 and MyD88 signaling but independent of the NLRP3 inflammasome. Eur J Immunol. (2015) 45:2847–57. doi: 10.1002/eji.2015.45.issue-10
33. Kim EH, Park MJ, Park S, Lee ES. Increased expression of the NLRP3 inflammasome components in patients with Behcet’s disease. J Inflammation (Lond). (2015) 12:41. doi: 10.1186/s12950-015-0086-z
34. Xiao L, Wang M, Shi Y, Xu Y, Gao Y, Zhang W, et al. Secondary white matter injury mediated by neuroinflammation after intracerebral hemorrhage and promising therapeutic strategies of targeting the NLRP3 inflammasome. Curr Neuropharmacol. (2023) 21:669–86. doi: 10.2174/1570159X20666220830115018
35. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. (2014) 111:E4110–8. doi: 10.1073/pnas.1405023111
36. Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. (2014) 75:209–19. doi: 10.1002/ana.24070
37. Zhou X, Zhao R, Lv M, Xu X, Liu W, Li X, et al. ACSL4 promotes microglia-mediated neuroinflammation by regulating lipid metabolism and VGLL4 expression. Brain Behav Immun. (2023) 109:331–43. doi: 10.1016/j.bbi.2023.02.012
38. Woodburn SC, Bollinger JL, Wohleb ES. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J Neuroinflamm. (2021) 18:258. doi: 10.1186/s12974-021-02309-6
39. Olude MA, Mouihate A, Mustapha OA, Farina C, Quintana FJ, Olopade JO. Astrocytes and microglia in stress-induced neuroinflammation: the African perspective. Front Immunol. (2022) 13:795089. doi: 10.3389/fimmu.2022.795089
40. Yang R, Yang B, Liu W, Tan C, Chen H, Wang X. Emerging role of non-coding RNAs in neuroinflammation mediated by microglia and astrocytes. J Neuroinflamm. (2023) 20:173. doi: 10.1186/s12974-023-02856-0
41. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. (2017) 18:2135. doi: 10.3390/ijms18102135
42. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.v173.4
43. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
44. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. (2015) 35:3384–96. doi: 10.1523/JNEUROSCI.2620-14.2015
45. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron. (2022) 110:3458–83. doi: 10.1016/j.neuron.2022.10.020
46. Bao B, Yin XP, Wen XQ, Suo YJ, Chen ZY, Li DL, et al. The protective effects of EGCG was associated with HO-1 active and microglia pyroptosis inhibition in experimental intracerebral hemorrhage. Neurochem Int. (2023) 170:105603. doi: 10.1016/j.neuint.2023.105603
47. Xia DY, Yuan JL, Jiang XC, Qi M, Lai NS, Wu LY, et al. SIRT1 promotes M2 microglia polarization via reducing ROS-mediated NLRP3 inflammasome signaling after subarachnoid hemorrhage. Front Immunol. (2021) 12:770744. doi: 10.3389/fimmu.2021.770744
48. Nakano-Kobayashi A, Canela A, Yoshihara T, Hagiwara M. Astrocyte-targeting therapy rescues cognitive impairment caused by neuroinflammation via the Nrf2 pathway. Proc Natl Acad Sci U S A. (2023) 120:e2303809120. doi: 10.1073/pnas.2303809120
49. Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflamm. (2022) 19:206. doi: 10.1186/s12974-022-02565-0
50. Lawrence JM, Schardien K, Wigdahl B, Nonnemacher MR. Roles of neuropathology-associated reactive astrocytes: a systematic review. Acta Neuropathol Commun. (2023) 11:42. doi: 10.1186/s40478-023-01526-9
51. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. (2020) 9:42. doi: 10.1186/s40035-020-00221-2
52. Fan YY, Huo J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem Int. (2021) 148:105080. doi: 10.1016/j.neuint.2021.105080
53. Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
54. Fei X, Dou YN, Wang L, Wu X, Huan Y, Wu S, et al. Homer1 promotes the conversion of A1 astrocytes to A2 astrocytes and improves the recovery of transgenic mice after intracerebral hemorrhage. J Neuroinflamm. (2022) 19:67. doi: 10.1186/s12974-022-02428-8
55. Xue M, Yong VW. Neuroinflammation in intracerebral haemorrhage: immunotherapies with potential for translation. Lancet Neurol. (2020) 19:1023–32. doi: 10.1016/S1474-4422(20)30364-1
56. Yang XL, Wang X, Shao L, Jiang GT, Min JW, Mei XY, et al. TRPV1 mediates astrocyte activation and interleukin-1beta release induced by hypoxic ischemia (HI). J Neuroinflamm. (2019) 16:114. doi: 10.1186/s12974-019-1487-3
57. Shi ZM, Jing JJ, Xue ZJ, Chen WJ, Tang YB, Chen DJ, et al. Stellate ganglion block ameliorated central post-stroke pain with comorbid anxiety and depression through inhibiting HIF-1alpha/NLRP3 signaling following thalamic hemorrhagic stroke. J Neuroinflamm. (2023) 20:82. doi: 10.1186/s12974-023-02765-2
58. Li J, Xu P, Hong Y, Xie Y, Peng M, Sun R, et al. Lipocalin-2-mediated astrocyte pyroptosis promotes neuroinflammatory injury via NLRP3 inflammasome activation in cerebral ischemia/reperfusion injury. J Neuroinflamm. (2023) 20:148. doi: 10.1186/s12974-023-02819-5
59. Salgar S, Bolivar BE, Flanagan JM, Anum SJ, Bouchier-Hayes L. The NLRP3 inflammasome fires up heme-induced inflammation in hemolytic conditions. Transl Res. (2023) 252:34–44. doi: 10.1016/j.trsl.2022.08.011
60. Bozza MT, Jeney V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front Immunol. (2020) 11:1323. doi: 10.3389/fimmu.2020.01323
61. Tan Y, Tan SW, Fan BY, Li L, Zhou YG. Hemin induces the activation of NLRP3 inflammasome in N9 microglial cells. Iran J Immunol. (2018) 15:122–32. doi: 10.22034/iji.2018.39376
62. Weng X, Tan Y, Chu X, Wu XF, Liu R, Tian Y, et al. N-methyl-D-aspartic acid receptor 1 (NMDAR1) aggravates secondary inflammatory damage induced by hemin-NLRP3 pathway after intracerebral hemorrhage. Chin J Traumatol. (2015) 18:254–8. doi: 10.1016/j.cjtee.2015.11.010
63. Chen X, Xiang X, Xie T, Chen Z, Mou Y, Gao Z, et al. Memantine protects blood-brain barrier integrity and attenuates neurological deficits through inhibiting nitric oxide synthase ser1412 phosphorylation in intracerebral hemorrhage rats: involvement of peroxynitrite-related matrix metalloproteinase-9/NLRP3 inflammasome activation. Neuroreport. (2021) 32:228–37. doi: 10.1097/WNR.0000000000001577
64. Vasconcellos LRC, Martimiano L, Dantas DP, Fonseca FM, Mata-Santos H, Travassos L, et al. Intracerebral injection of heme induces lipid peroxidation, neuroinflammation, and sensorimotor deficits. Stroke. (2021) 52:1788–97. doi: 10.1161/STROKEAHA.120.031911
65. Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflamm. (2012) 9:46. doi: 10.1186/1742-2094-9-46
66. Possemato E, La Barbera L, Nobili A, Krashia P, D’Amelio M. The role of dopamine in NLRP3 inflammasome inhibition: Implications for neurodegenerative diseases. Ageing Res Rev. (2023) 87:101907. doi: 10.1016/j.arr.2023.101907
67. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. (2015) 160:62–73. doi: 10.1016/j.cell.2014.11.047
68. Wang T, Nowrangi D, Yu L, Lu T, Tang J, Han B, et al. Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflamm. (2018) 15:2. doi: 10.1186/s12974-017-1039-7
69. Kasper LH, Reder AT. Immunomodulatory activity of interferon-beta. Ann Clin Transl Neurol. (2014) 1:622–31. doi: 10.1002/acn3.84
70. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
71. Mohammed Thangameeran SI, Tsai ST, Hung HY, Hu WF, Pang CY, Chen SY, et al. A role for endoplasmic reticulum stress in intracerebral hemorrhage. Cells. (2020) 9:750. doi: 10.3390/cells9030750
72. Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. (2015) 6:e1879. doi: 10.1038/cddis.2015.248
73. Chen G, Gao C, Yan Y, Wang T, Luo C, Zhang M, et al. Inhibiting ER stress weakens neuronal pyroptosis in a mouse acute hemorrhagic stroke model. Mol Neurobiol. (2020) 57:5324–35. doi: 10.1007/s12035-020-02097-9
74. Jin MH, Liu XD, Sun HN, Han YH, Kwon T. Peroxiredoxin II exerts neuroprotective effects by inhibiting endoplasmic reticulum stress and oxidative stress-induced neuronal pyroptosis. Mol Biol Rep. (2024) 51:607. doi: 10.1007/s11033-024-09568-5
75. Lei C, Li Y, Zhu X, Li H, Chang X. HMGB1/TLR4 induces autophagy and promotes neuroinflammation after intracerebral hemorrhage. Brain Res. (2022) 1792:148003. doi: 10.1016/j.brainres.2022.148003
76. Lei C, Chen K, Gu Y, Li Y, Wang L, Zhu X, et al. HMGB1/TLR4 axis promotes pyroptosis after ICH by activating the NLRP3 inflammasome. J Neuroimmunol. (2024) 393:578401. doi: 10.1016/j.jneuroim.2024.578401
77. Wang Y, Li K, Liu Z, Sun Y, Wang J, Liu Q, et al. Ethyl pyruvate alleviating inflammatory response after diabetic cerebral hemorrhage. Curr Neurovasc Res. (2022) 19:196–202. doi: 10.2174/1567202619666220602153937
78. Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. (1993) 13:6231–40. doi: 10.1128/MCB.13.10.6231
79. Chen D, Sui L, Chen C, Liu S, Sun X, Guan J. Atorvastatin suppresses NLRP3 inflammasome activation in intracerebral hemorrhage via TLR4- and MyD88-dependent pathways. Aging (Albany NY). (2022) 14:462–76. doi: 10.18632/aging.203824
80. Anwar S, Pons V, Rivest S. Microglia purinoceptor P2Y6: an emerging therapeutic target in CNS diseases. Cells. (2020) 9:1595. doi: 10.3390/cells9071595
81. Liu GD, Ding JQ, Xiao Q, Chen SD. P2Y6 receptor and immunoinflammation. Neurosci Bull. (2009) 25:161–4. doi: 10.1007/s12264-009-0120-3
82. Li Y, Tu H, Zhang S, Ding Z, Wu G, Piao J, et al. P2Y6 receptor activation aggravates NLRP3-dependent microglial pyroptosis via downregulation of the PI3K/AKT pathway in a mouse model of intracerebral hemorrhage. Mol Neurobiol. (2024) 61:4259–77. doi: 10.1007/s12035-023-03834-6
83. Kira S, Yoshiyama M, Tsuchiya S, Shigetomi E, Miyamoto T, Nakagomi H, et al. P2Y(6)-deficiency increases micturition frequency and attenuates sustained contractility of the urinary bladder in mice. Sci Rep. (2017) 7:771. doi: 10.1038/s41598-017-00824-2
84. Holste K, Xia F, Garton HJL, Wan S, Hua Y, Keep RF, et al. The role of complement in brain injury following intracerebral hemorrhage: A review. Exp Neurol. (2021) 340:113654. doi: 10.1016/j.expneurol.2021.113654
85. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
86. Yao ST, Cao F, Chen JL, Chen W, Fan RM, Li G, et al. NLRP3 is required for complement-mediated caspase-1 and IL-1beta activation in ICH. J Mol Neurosci. (2017) 61:385–95. doi: 10.1007/s12031-016-0874-9
87. Ye X, Zuo D, Yu L, Zhang L, Tang J, Cui C, et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem Biophys Res Commun. (2017) 485:499–505. doi: 10.1016/j.bbrc.2017.02.019
88. Ismael S, Patrick D, Salman M, Parveen A, Stanfill AG, Ishrat T. Verapamil inhibits TXNIP-NLRP3 inflammasome activation and preserves functional recovery after intracerebral hemorrhage in mice. Neurochem Int. (2022) 161:105423. doi: 10.1016/j.neuint.2022.105423
89. Hu L, Zhang H, Wang B, Ao Q, He Z. MicroRNA-152 attenuates neuroinflammation in intracerebral hemorrhage by inhibiting thioredoxin interacting protein (TXNIP)-mediated NLRP3 inflammasome activation. Int Immunopharmacol. (2020) 80:106141. doi: 10.1016/j.intimp.2019.106141
90. Ismael S, Nasoohi S, Yoo A, Ahmed HA, Ishrat T. Tissue plasminogen activator promotes TXNIP-NLRP3 inflammasome activation after hyperglycemic stroke in mice. Mol Neurobiol. (2020) 57:2495–508. doi: 10.1007/s12035-020-01893-7
91. Yang CJ, Li X, Feng XQ, Chen Y, Feng JG, Jia J, et al. Activation of LRP1 ameliorates cerebral ischemia/reperfusion injury and cognitive decline by suppressing neuroinflammation and oxidative stress through TXNIP/NLRP3 signaling pathway in mice. Oxid Med Cell Longev. (2022) 2022:8729398. doi: 10.1155/2022/8729398
92. Zhao H, Chen Y, Feng H. P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol. (2018) 16:1282–95. doi: 10.2174/1570159X16666180516094500
93. Zhao H, Pan P, Yang Y, Ge H, Chen W, Qu J, et al. Endogenous hydrogen sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation by suppressing the P2X7 receptor after intracerebral haemorrhage in rats. J Neuroinflamm. (2017) 14:163. doi: 10.1186/s12974-017-0940-4
94. Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflamm. (2015) 12:190. doi: 10.1186/s12974-015-0409-2
95. Zhao H, Qu J, Li Q, Cui M, Wang J, Zhang K, et al. Taurine supplementation reduces neuroinflammation and protects against white matter injury after intracerebral hemorrhage in rats. Amino Acids. (2018) 50:439–51. doi: 10.1007/s00726-017-2529-8
96. Yuan C, Liu L, Zhao Y, Wang K. Puerarin inhibits Staphylococcus aureus-induced endometritis through attenuating inflammation and ferroptosis via regulating the P2X7/NLRP3 signalling pathway. J Cell Mol Med. (2024) 28:e18550. doi: 10.1111/jcmm.v28.14
97. Fang Y, Hong X. miR-124-3p inhibits microglial secondary inflammation after basal ganglia hemorrhage by targeting TRAF6 and repressing the activation of NLRP3 inflammasome. Front Neurol. (2021) 12:653321. doi: 10.3389/fneur.2021.653321
98. Wan SY, Li GS, Tu C, Chen WL, Wang XW, Wang YN, et al. MicroNAR-194-5p hinders the activation of NLRP3 inflammasomes and alleviates neuroinflammation during intracerebral hemorrhage by blocking the interaction between TRAF6 and NLRP3. Brain Res. (2021) 1752:147228. doi: 10.1016/j.brainres.2020.147228
99. Wu X, Zhang Y, Zhang Y, Xia L, Yang Y, Wang P, et al. MST4 attenuates NLRP3 inflammasome-mediated neuroinflammation and affects the prognosis after intracerebral hemorrhage in mice. Brain Res Bull. (2021) 177:31–8. doi: 10.1016/j.brainresbull.2021.09.006
100. Sun J, Xu G. Mesenchymal stem cell-derived exosomal miR-150-3p affects intracerebral hemorrhage by regulating TRAF6/NF-κB axis, gut microbiota and metabolism. Stem Cell Rev Rep. (2023) 19:1907–21. doi: 10.1007/s12015-023-10541-1
101. Yuan R, Fan H, Cheng S, Gao W, Xu X, Lv S, et al. Silymarin prevents NLRP3 inflammasome activation and protects against intracerebral hemorrhage. BioMed Pharmacother. (2017) 93:308–15. doi: 10.1016/j.biopha.2017.06.018
102. Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang S, et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J Neuroinflamm. (2017) 14:119. doi: 10.1186/s12974-017-0895-5
103. Cheng Y, Liu M, Tang H, Chen B, Yang G, Zhao W, et al. iTRAQ-based quantitative proteomics indicated Nrf2/OPTN-mediated mitophagy inhibits NLRP3 inflammasome activation after intracerebral hemorrhage. Oxid Med Cell Longev. (2021) 2021:6630281. doi: 10.1155/2021/6630281
104. Cheng Y, Chen B, Xie W, Chen Z, Yang G, Cai Y, et al. Ghrelin attenuates secondary brain injury following intracerebral hemorrhage by inhibiting NLRP3 inflammasome activation and promoting Nrf2/ARE signaling pathway in mice. Int Immunopharmacol. (2020) 79:106180. doi: 10.1016/j.intimp.2019.106180
105. Zhao J, Chen C, Ge L, Jiang Z, Hu Z, Yin L. TAK1 inhibition mitigates intracerebral hemorrhage-induced brain injury through reduction of oxidative stress and neuronal pyroptosis via the NRF2 signaling pathway. Front Immunol. (2024) 15:1386780. doi: 10.3389/fimmu.2024.1386780
106. Hu LT, Wang BY, Fan YH, He ZY, Zheng WX. Exosomal miR-23b from bone marrow mesenchymal stem cells alleviates oxidative stress and pyroptosis after intracerebral hemorrhage. Neural Regener Res. (2023) 18:560–7. doi: 10.4103/1673-5374.346551
107. Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. (2011) 41:2040–51. doi: 10.1002/eji.201041316
108. Xie K, Zhang Y, Wang Y, Meng X, Wang Y, Yu Y, et al. Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Inflammation Res. (2020) 69:697–710. doi: 10.1007/s00011-020-01347-9
109. Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, et al. Selective NLRP3 (Pyrin domain-containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke. (2018) 49:184–92. doi: 10.1161/STROKEAHA.117.018904
110. Ismael S, Zhao L, Nasoohi S, Ishrat T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci Rep. (2018) 8:5971. doi: 10.1038/s41598-018-24350-x
111. Li L, Gong J, Zhang W. Treatment of intracerebral hemorrhage with traditional Chinese medicine monomer wogonin by modifying NLRP3 with METTL14 to inhibit neuronal cell pyroptosis. Appl Biochem Biotechnol. (2024) 196:6174–88. doi: 10.1007/s12010-023-04849-4
112. Cao F, Chen G, Xu Y, Wang X, Tang X, Zhang W, et al. METTL14 contributes to acute lung injury by stabilizing NLRP3 expression in an IGF2BP2-dependent manner. Cell Death Dis. (2024) 15:43. doi: 10.1038/s41419-023-06407-6
113. Guo T, Chen G, Yang L, Deng J, Pan Y. Piezo1 inhibitor isoquercitrin rescues neural impairment mediated by NLRP3 after intracerebral hemorrhage. Exp Neurol. (2024) 379:114852. doi: 10.1016/j.expneurol.2024.114852
114. Gu L, Sun M, Li R, Tao Y, Luo X, Xu J, et al. Activation of RKIP binding ASC attenuates neuronal pyroptosis and brain injury via caspase-1/GSDMD signaling pathway after intracerebral hemorrhage in mice. Transl Stroke Res. (2022) 13:1037–54. doi: 10.1007/s12975-022-01009-4
115. Cheng Y, Wei Y, Yang W, Song Y, Shang H, Cai Y, et al. Cordycepin confers neuroprotection in mice models of intracerebral hemorrhage via suppressing NLRP3 inflammasome activation. Metab Brain Dis. (2017) 32:1133–45. doi: 10.1007/s11011-017-0003-7
116. Li X, Wang T, Zhang D, Li H, Shen H, Ding X, et al. Andrographolide ameliorates intracerebral hemorrhage induced secondary brain injury by inhibiting neuroinflammation induction. Neuropharmacology. (2018) 141:305–15. doi: 10.1016/j.neuropharm.2018.09.015
117. Zhou H, Zhang C, Huang C. Verbascoside attenuates acute inflammatory injury caused by an intracerebral hemorrhage through the suppression of NLRP3. Neurochem Res. (2021) 46:770–7. doi: 10.1007/s11064-020-03206-9
118. Ul Hussain M. Micro-RNAs (miRNAs): genomic organisation, biogenesis and mode of action. Cell Tissue Res. (2012) 349:405–13. doi: 10.1007/s00441-012-1438-0
119. Großhans H, Chatterjee S. MicroRNases and the regulated degradation of mature animal miRNAs. Adv Exp Med Biol. (2011) 700:140–55. doi: 10.1007/978-1-4419-7823-3_12
120. Cabarcas SM, Thomas S, Zhang X, Cherry JM, Sebastian T, Yerramilli S, et al. The role of upregulated miRNAs and the identification of novel mRNA targets in prostatospheres. Genomics. (2012) 99:108–17. doi: 10.1016/j.ygeno.2011.11.007
121. Yang Z, Zhong L, Xian R, Yuan B. MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol Immunol. (2015) 65:267–76. doi: 10.1016/j.molimm.2014.12.018
122. Bai Y, Shi H, Zhang Y, Zhang C, Wu B, Wu X, et al. Febuxostat attenuates secondary brain injury caused by cerebral hemorrhage through inhibiting inflammatory pathways. Iran J Basic Med Sci. (2024) 27:740–6. doi: 10.22038/IJBMS.2024.74655.16212
123. Ji N, Wu L, Shi H, Li Q, Yu A, Yang Z. VSIG4 Attenuates NLRP3 and Ameliorates Neuroinflammation via JAK2-STAT3-A20 Pathway after Intracerebral Hemorrhage in Mice. Neurotox Res. (2022) 40:78–88. doi: 10.1007/s12640-021-00456-5
124. Wang S, Yao Q, Wan Y, Wang J, Huang C, Li D, et al. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing NLRP3 inflammasome expression. Int J Neurosci. (2020) 130:301–8. doi: 10.1080/00207454.2019.1679810
125. Miao H, Jiang Y, Geng J, Zhang B, Zhu G, Tang J. Edaravone Administration Confers Neuroprotection after Experimental Intracerebral Hemorrhage in Rats via NLRP3 Suppression. J Stroke Cerebrovasc Dis. (2020) 29:104468. doi: 10.1016/j.jstrokecerebrovasdis.2019.104468
126. Xu F, Shen G, Su Z, He Z, Yuan L. Glibenclamide ameliorates the disrupted blood-brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome. Brain Behav. (2019) 9:e01254. doi: 10.1002/brb3.2019.9.issue-4
127. Kim H, Lee JE, Yoo HJ, Sung JH, Yang SH. Effect of pioglitazone on perihematomal edema in intracerebral hemorrhage mouse model by regulating NLRP3 expression and energy metabolism. J Korean Neurosurg Soc. (2020) 63:689–97. doi: 10.3340/jkns.2020.0056
128. Song HL, Zhang SB. Therapeutic effect of dexmedetomidine on intracerebral hemorrhage via regulating NLRP3. Eur Rev Med Pharmacol Sci. (2019) 23:2612–9. doi: 10.26355/eurrev_201903_17411
129. Saavedra JM. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond). (2012) 123:567–90. doi: 10.1042/CS20120078
130. Yang X, Sun J, Kim TJ, Kim YJ, Ko SB, Kim CK, et al. Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp Neurol. (2018) 310:22–32. doi: 10.1016/j.expneurol.2018.08.013
131. Bravo-San-Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. (2017) 120:1812–24. doi: 10.1161/CIRCRESAHA.117.311082
132. Moyzis AG, Sadoshima J, Gustafsson ÅB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol. (2015) 308:H183–92. doi: 10.1152/ajpheart.00708.2014
133. Zheng S, Jian D, Gan H, Wang L, Zhao J, Zhai X. FUNDC1 inhibits NLRP3-mediated inflammation after intracerebral hemorrhage by promoting mitophagy in mice. Neurosci Lett. (2021) 756:135967. doi: 10.1016/j.neulet.2021.135967
134. Yang Z, Liu B, Zhong L, Shen H, Lin C, Lin L, et al. Toll-like receptor-4-mediated autophagy contributes to microglial activation and inflammatory injury in mouse models of intracerebral haemorrhage. Neuropathol Appl Neurobiol. (2015) 41:e95–106. doi: 10.1111/nan.2015.41.issue-4
135. Fu K, Xu W, Lenahan C, Mo Y, Wen J, Deng T, et al. Autophagy regulates inflammation in intracerebral hemorrhage: Enemy or friend? Front Cell Neurosci. (2022) 16:1036313. doi: 10.3389/fncel.2022.1036313
136. Gao L, Xu W, Li T, Chen J, Shao A, Yan F, et al. Stem cell therapy: A promising therapeutic method for intracerebral hemorrhage. Cell Transpl. (2018) 27:1809–24. doi: 10.1177/0963689718773363
137. Ma Y, Qi M, An Y, Zhang L, Yang R, Doro DH, et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell. (2018) 17:e12709. doi: 10.1111/acel.2018.17.issue-1
138. Vaquero J, Otero L, Bonilla C, Aguayo C, Rico MA, Rodriguez A, et al. Cell therapy with bone marrow stromal cells after intracerebral hemorrhage: impact of platelet-rich plasma scaffolds. Cytotherapy. (2013) 15:33–43. doi: 10.1016/j.jcyt.2012.10.005
139. Ding H, Jia Y, Lv H, Chang W, Liu F, Wang D. Extracellular vesicles derived from bone marrow mesenchymal stem cells alleviate neuroinflammation after diabetic intracerebral hemorrhage via the miR-183-5p/PDCD4/NLRP3 pathway. J Endocrinol Invest. (2021) 44:2685–98. doi: 10.1007/s40618-021-01583-8
140. Liu J, He J, Huang Y, Ge L, Xiao H, Zeng L, et al. Hypoxia-preconditioned mesenchymal stem cells attenuate microglial pyroptosis after intracerebral hemorrhage. Ann Transl Med. (2021) 9:1362. doi: 10.21037/atm-21-2590
141. Zhao Y, Xiao Q, Sun T, Yu H, Luo M. Knockdown of LCN2 attenuates brain injury after intracerebral hemorrhage via suppressing pyroptosis. Neuropsychiatr Dis Treat. (2024) 20:83–99. doi: 10.2147/NDT.S440065
142. Wei J, Yin J, Cui Y, Wang K, Hong M, Cui J. FERM domain containing kindlin 1 knockdown attenuates inflammation induced by intracerebral hemorrhage in rats via NLR family pyrin domain containing 3/nuclear factor kappa B pathway. Exp Anim. (2023) 72:324–35. doi: 10.1538/expanim.22-0145
143. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. (2020) 26:732–40. doi: 10.1038/s41591-020-0840-5
144. Zhang H, Wang J, Ruan C, Gao Z, Zhu Q, Li S. Co-exposure of chronic stress and alumina nanoparticles aggravates hippocampal microglia pyroptosis by activating cathepsin B/NLRP3 signaling pathway. J Hazard Mater. (2022) 436:129093. doi: 10.1016/j.jhazmat.2022.129093
145. Yang M, Deng S, Jiang J, Tian M, Xiao L, Gong Y. Oxytocin improves intracerebral hemorrhage outcomes by suppressing neuronal pyroptosis and mitochondrial fission. Stroke. (2023) 54:1888–900. doi: 10.1161/STROKEAHA.123.043391
146. Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, et al. Inhibition of dectin-1 alleviates neuroinflammatory injury by attenuating NLRP3 inflammasome-mediated pyroptosis after intracerebral hemorrhage in mice: preliminary study results. J Inflammation Res. (2022) 15:5917–33. doi: 10.2147/JIR.S384020
147. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. (2023) 133:4388. doi: 10.1172/JCI169304
148. Wang L, Zheng S, Zhang L, Xiao H, Gan H, Chen H, et al. Histone deacetylation 10 alleviates inflammation after intracerebral hemorrhage via the PTPN22/NLRP3 pathway in rats. Neuroscience. (2020) 432:247–59. doi: 10.1016/j.neuroscience.2020.02.027
149. Schrag M, Kirshner H. Management of intracerebral hemorrhage: JACC focus seminar. J Am Coll Cardiol. (2020) 75:1819–31. doi: 10.1016/j.jacc.2019.10.066
150. Cordonnier C, Demchuk A, Ziai W, Anderson CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. (2018) 392:1257–68. doi: 10.1016/S0140-6736(18)31878-6
151. Luparello V, Canavero S. Treatment of hypertensive cerebellar hemorrhage–surgical or conservative management? Neurosurgery. (1995) 37:552–3. doi: 10.1227/00006123-199509000-00037
152. Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. (2005) 365:387–97. doi: 10.1016/S0140-6736(05)70233-6
153. Phan TG, Koh M, Vierkant RA, Wijdicks EF. Hydrocephalus is a determinant of early mortality in putaminal hemorrhage. Stroke. (2000) 31:2157–62. doi: 10.1161/01.STR.31.9.2157
154. Hemphill JC 3rd, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2015) 46:2032–60. doi: 10.1161/STR.0000000000000069
155. Li Y, Zhang H, Wang X, She L, Yan Z, Zhang N, et al. Neuroendoscopic surgery versus external ventricular drainage alone or with intraventricular fibrinolysis for intraventricular hemorrhage secondary to spontaneous supratentorial hemorrhage: a systematic review and meta-analysis. PloS One. (2013) 8:e80599. doi: 10.1371/journal.pone.0080599
156. Zhou X, Chen J, Li Q, Ren G, Yao G, Liu M, et al. Minimally invasive surgery for spontaneous supratentorial intracerebral hemorrhage: a meta-analysis of randomized controlled trials. Stroke. (2012) 43:2923–30. doi: 10.1161/STROKEAHA.112.667535
157. Anderson CS, Heeley E, Huang Y, Wang J, Stapf C, Delcourt C, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med. (2013) 368:2355–65. doi: 10.1056/NEJMoa1214609
158. Magid-Bernstein J, Girard R, Polster S, Srinath A, Romanos S, Awad IA, et al. Cerebral hemorrhage: pathophysiology, treatment, and future directions. Circ Res. (2022) 130:1204–29. doi: 10.1161/CIRCRESAHA.121.319949
159. Sprigg N, Flaherty K, Appleton JP, Al-Shahi-Salman R, Bereczki D, Beridze M, et al. Tranexamic acid for hyperacute primary IntraCerebral Haemorrhage (TICH-2): an international randomised, placebo-controlled, phase 3 superiority trial. Lancet. (2018) 391:2107–15. doi: 10.1016/S0140-6736(18)31033-X
160. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. (2005) 352:777–85. doi: 10.1056/NEJMoa042991
161. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. (2008) 358:2127–37. doi: 10.1056/NEJMoa0707534
162. Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. (2009) 373:1632–44. doi: 10.1016/S0140-6736(09)60371-8
163. Arefieva TI, Filatova AY, Potekhina AV, Shchinova AM. Immunotropic effects and proposed mechanism of action for 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (Statins). Biochem (Mosc). (2018) 83:874–89. doi: 10.1134/S0006297918080023
164. Tapia-Perez H, Sanchez-Aguilar M, Torres-Corzo JG, Rodriguez-Leyva I, Gonzalez-Aguirre D, Gordillo-Moscoso A, et al. Use of statins for the treatment of spontaneous intracerebral hemorrhage: results of a pilot study. Cent Eur Neurosurg. (2009) 70:15–20. doi: 10.1055/s-0028-1082064
165. Ribe AR, Vestergaard CH, Vestergaard M, Fenger-Grøn M, Pedersen HS, Lietzen LW, et al. Statins and risk of intracerebral haemorrhage in a stroke-free population: A nationwide Danish propensity score matched cohort study. EClinicalMedicine. (2019) 8:78–84. doi: 10.1016/j.eclinm.2019.02.007
166. Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. (2004) 3:744–51. doi: 10.1016/S1474-4422(04)00937-8
167. Chang JJ, Kim-Tenser M, Emanuel BA, Jones GM, Chapple K, Alikhani A, et al. Minocycline and matrix metalloproteinase inhibition in acute intracerebral hemorrhage: a pilot study. Eur J Neurol. (2017) 24:1384–91. doi: 10.1111/ene.2017.24.issue-11
Keywords: intracerebral hemorrhage, neuroinflammation, NLRP3 inflammasome, microglia, astrocyte, inflammation regulation, clinical translation
Citation: He K-l, Yu X, Xia L, Xie Y-d, Qi E-b, Wan L, Hua X-m and Jing C-h (2025) A new perspective on the regulation of neuroinflammation in intracerebral hemorrhage: mechanisms of NLRP3 inflammasome activation and therapeutic strategies. Front. Immunol. 16:1526786. doi: 10.3389/fimmu.2025.1526786
Received: 12 November 2024; Accepted: 10 February 2025;
Published: 27 February 2025.
Edited by:
Marcelo Ezquer, University for Development, ChileCopyright © 2025 He, Yu, Xia, Xie, Qi, Wan, Hua and Jing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao-hui Jing, amluZ2NoYW9odWlAeGluaHVhbWVkLmNvbS5jbg==
†Present address: Chao-hui Jing, MOE-Shanghai Key Laboratory of Children's Environmental Health, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.