 Zhidu Song1
Zhidu Song1 Jiaxin You
Jiaxin You- 1Department of Ophthalmology, The Second Hospital of Jilin University, Changchun, China
- 2Department of Anesthesiology, China-Japan Union Hospital of Jilin University, Changchun, China
Immunotherapy for cancer has made significant strides in the last several years. The prognosis for cancer patients has significantly improved as a result, particularly in hematological diseases. However, it was discovered that translating these achievements to solid tumors proved challenging. The peptide-loading complex (PLC), a temporary multisubunit membrane assembly in the endoplasmic reticulum (ER), is crucial for initiating a hierarchical immune response. Chaperones calreticulin and tapasin make up the PLC, unique to class I glycoproteins, thiooxido-reductase ERp57, and a transporter associated with antigen processing. The loading and editing of major histocompatibility complex class I (MHC-I) molecules with peptide translocation into the ER are synchronized by the PLC. One of the immune escape strategies revealed for tumors so far is changes in the expression of MHC molecules. This is because MHC antigens are crucial in presenting antigens to T-lymphocytes and controlling NK cell activity. Furthermore, decreased MHC-I expression has been linked to malignancies resistant to T-cell-based cancer immunotherapies (adoptive transfer of antitumor CD8 T-cells or checkpoint inhibition). The PLC is essential for T-cell priming, differentiation, and tumor growth control because it can bind to a wide range of MHC-I allomorphs. In this review, we have looked into PLC’s function and effects in all forms of cancer to improve cancer therapy techniques.
1 Introduction
Cancer affects people of all ages, socioeconomic levels, and ethnicities, making it a global health threat (1, 2). Cancer treatments include hormone therapy, radiation, chemotherapy, and surgical excision of the cancerous tissue (3). One popular method of treating cancer is chemotherapy, which involves the systemic administration of anticancer medications to patients to slow the unchecked growth of cancer cells (4). The effectiveness of immune checkpoint inhibitors in treating lung cancer (LC) (5) and melanoma (6) has sparked research into immunotherapy for a variety of tumor forms, including breast cancer (BC) (7). However, these trials have shown only a modest clinical benefit in many scenarios (8, 9). Therefore, to develop innovative treatment approaches that boost antitumor immunity in more resistant tumors, it is crucial to comprehend normal immunity and clarify resistance mechanisms that contribute to the evolution of cancer (3, 10, 11).
The capacity of immunotherapy to provide long-term effects in a substantial number of patients is crucial to its prospects. T-cell-based immunotherapeutic approaches, such as adoptive transfer of TCR-engineered T-cells and personalized cancer vaccines, can improve outcomes compared to immune checkpoint inhibition, which has demonstrated low rates of long-term tumor regression in individuals with recurring cancer (12). To implement these methods, T-cell antigen determination is necessary. Finding these antigens has been limited to researching a few tumor forms, mainly melanoma, in the most prevalent human leukocyte antigen (HLA) alleles (13). Although true immunogenic epitopes appear to be rare, recent advances in epitope discovery have the potential to open the door to more thorough protein and mutation searches, which could pave the way for tailored cancer immunotherapy for patients whose tumors retain all of their antigen and processing machinery (14). Unfortunately, there are many limitations with researchers’ current technology that make it hard to reliably identify an antigen that has been processed and presented in nature. According to recent research, epitope discovery has the potential to cause significant tumor shrinkage in individuals with advanced metastases (14). Future advancements in cancer treatment could be significantly aided by creating methods that facilitate precise and efficient antigen discovery, such as integrating peptide: HLA complex stability, which are essential components of TCR discovery for personalized cancer vaccine therapy and adoptive T-cell transfer (15–17).
Immunological investigations examine numerous immune responses, routes, control, recognition, and specificity, providing essential information for manipulating treatment outcomes (18). By using structural determination techniques, principally cryo-electron microscopy (cryoEM) or X-ray crystallography, on ligand-receptor complexes and different multiprotein assemblies, such as the peptide-loading complex (PLC), researchers may enhance their comprehension of functions and processes. A primary objective of structural immunology is to use this structural data to develop novel pharmaceuticals, therapies, immunogens, and vaccines (19).
Immunogenic peptides for cytotoxic CD8 + T-cell responses against cancerous or infected cells are provided by the major histocompatibility class I (MHC-I) molecules HLA (20). The MHC-I heavy chain consists of three extracellular domains called α chain which have three subunits α1, α2, α3, a transmembrane domain, and a cytoplasmic domain. The MHC-I antigen processing and presentation pathway selects peptides typically generated by proteasomal breakdown. In this pathway, two homologous peptide editors, TAPBPR and tapasin (Tsn), regulate and optimize the peptide repertoire before delivering it to the cell surface (21). The peptide transporter linked to antigen processing, ERp57, calreticulin (CRT), and MHC-I are all pieces of the puzzle in Tsn, a multiprotein PLC. Interactions within the PLC enable Tsn to load peptides onto MHC-I and keep MHC-I in a state that’s ready to receive peptides (22). A recent discovery is the Tsn-related molecule TAPBPR, which functions independently of the PLC. It is possible for TAPBPR to directly catalyze peptide exchange on MHC-I, in addition to recruiting UDP-glucose: glycoprotein glucosyltransferase 1 (UGT-1), which then reglucosylates peptide-receptive MHC-I and recycles it back to the PLC for Tsn-mediated peptide acquisition (23–26). In the PLC, TAPBPR and Tsn work together dynamically to contact MHC-I over a comprehensive interface with the MHC H chain and β2 microglobulin (β2m), influencing the global structure of MHC molecules and producing MHC molecules that are either peptide-free or peptide-receptive (27).
The origin of antigens meant for transfer onto MHC-I molecules consists of viral proteins or modified proteins, including oncogenes. Proteases produce peptides consisting of 8 to 10 amino acids within the cytoplasm, which are subsequently transported to the ER. The PLC plays a crucial role in the maintenance of MHC-I peptide assembly (28). The PLC consists of seven specific subunits that are meticulously organized within a singular macromolecular complex: the oxidoreductase ERp57, the chaperones Tsn and CRT, the nascent MHC-I molecule (a heterodimer made up of the α chain and β-microglobulin), the transporter associated with antigen processing (TAP) (comprising two subunits, TAP1 and TAP2, collectively known as TAP), and the oxidoreductase ERp57 (29). TAP plays a crucial role in the movement of antigens from the cytosol to the ER. PLC plays a vital role in stabilizing nascent MHC-I molecules, which remain unstable until the peptide antigen is precisely inserted into the antigen-binding cleft. When antigen peptides are correctly loaded, MHC-I molecules can exit the ER and arrive at the plasma membrane through the secretory pathway (30, 31).
The regulation of the immune system and the surveillance of cells that have undergone malignancy or infection depend on the display of antigenic peptides on MHC-I molecules. Peptides are moved from the cytosol to the ER, where peptide loading occurs through catalytic processes. The PLC verifies the accuracy of peptide-MHC-I (pMHC-I) complexes (32). The PLC oversees the complex process of generating pMHC-I complexes (33). As the complexes’ proofreader, the PLC is crucial for displaying kinetically stable pMHC-I complexes. Because MHC-I molecules linger in the secretory and recycling routes, pMHC-I stability is affected by suboptimal low-affinity peptide loading. Mutations, downregulation, or deletion of PLC components may change pMHC-I production, leading to a reduction in MHC-I molecules on the cell surface. Many different types of cancer cells and viruses employ this strategy to evade the immune system (29, 34). The PLC plays a critical role in the adaptive immune system by coordinating the transfer of peptides, loading them onto MHC-I, and optimizing their presentation. Two MHC-I-dedicated enzymes, Tsn, and the PLC-independent TAPBPR, choose stable pMHC-I complexes during peptide editing or proofreading and chaperone empty or poorly loaded MHC-I. This process is known as peptide loading and optimization. Recent structural and functional investigations of peptide editing have significantly advanced researchers’ understanding of this critical step in antigen processing and presentation (35).
In this review, we have summarized PLC and its elements. Subsequently, we have investigated the function of PLC and PLC components in different cancer types to improve cancer therapy techniques. We have now covered the shortcomings and prospects for this field of study in terms of clinical advancement and the creation of new cancer therapy approaches.
2 Peptide-loading complex
The ER is the site of peptide attachment to MHC-I. In addition to the MHC-I-β2m dimer, many other components are necessary for efficient peptide binding (33). These comprise Tsn, a membrane protein encoded by MHC, and the two antigen presentation transporter subunits (TAP1 and TAP2) critical for peptide entry into the ER from the cytosol. A massive multisubunit ER complex containing TAP and Tsn comprises MHC-I-β2m dimers before peptide binding. This complex also includes two soluble “housekeeping” proteins, CRT and ERp57, which are thiol oxidoreductases, in addition to these specialized components (36).
Most nucleated cells have MHC-I complexes on their surface that display peptides from both foreign and self-intracellular proteins. The formed heterotrimeric complexes comprise a polymorphism glycosylated HC, non-polymorphic β2m, and a peptide with an average length of nine amino acids (37). A multitude of chaperone molecules aid in assembling the class I complexes, which take place in the ER. The PLC, a multimolecular unit, is essential to this procedure. The PLC comprises the glycoprotein chaperone CRT, the class I-specific Tsn, the trioxide-reductase ERp57, and the peptide transporter. According to research, class I assembly entails an optimization phase in which the PLC modifies the complex’s peptide cargo (38, 39). Moreover, this selective peptide loading preferentially supports peptides that exhibit a longer off-rate from the established complex. Researchers have shown that Tsn is the central chaperone regulating PLC activity, while CRT and possibly ERp57 provide additional support (39).
Establishing a hierarchical immune response necessitates the presence of PLC, a transitory multisubunit membrane complex in the ER. The PLC coordinates the translocation of peptides into the ER and loads and modifies the MHC-I molecule. Final proofreading completed in the PLC triggers a T-cell response that explicitly targets cancerous or infected cells by methodically releasing stable peptide-MHC-I complexes to the cell surface. Coordinating seven distinct subunits within a solitary macromolecular assembly is necessary to sample various MHC-I allomorphs (40). These subunits include the MHC-I heterodimer, the oxidoreductase ERp57, the chaperones Tsn and CRT, and the TAP (41). Tsn, CRT, ERp57, and MHC-I circle TAP as two ER-resident editing modules with pseudo-symmetric orientations. A network of multivalent chaperones linking the editing modules creates a proofreading mechanism via two lateral binding platforms associated with MHC-I. The P domain of CRT reaches beyond the MHC-I peptide-binding pocket and extends toward ERp57, whereas the lectin-like domain interacts with the MHC-I glycan. This arrangement suggests that Tsn can clamp MHC-I, which might aid peptide editing. A large ER lumenal cavity is accessible via the membrane entry sites of Tsn and MHC-I, which restrict the translocation pathway of TAP. Because of two lateral windows, antigenic peptides are directed to MHC-I. PLC structures obtained at different stages of assembly provide a mechanistic understanding of MHC-I recruitment and release. Researchers clarified the molecular connection between an ER chaperone network and an ABC transporter in MHC-I construction and offered information on when the adaptive immune response begins (28).
The TAP feature has been well-studied (42, 43). TAP identifies the presence of aromatic, hydrophobic, or positively charged terminal amino acids such as phenylalanine, tyrosine, arginine, or leucine at the peptide’s C-terminus (44). The peptide binding is substantially influenced by the first three residues on the N-terminus. It is ideal to have an N-terminal arginine and aromatic and hydrophobic side chains (45). The C-terminal residue of proteasomes is not very particular, but it does conform to the constraints of MHC-I and TAP molecules (46). Peptides that TAP has translocated must undergo further processing in the ER before binding to MHC-I. The intraluminal processing of peptides is the responsibility of ER aminopeptidase 1 (ERAP1) and ERAP2, which are luminal components of the ER (47). To make peptides that can fit into the MHC-I binding groove, ERAP1 trims amino acids of the N-terminus. These peptides may be 8-11mers long. The structural changes in peptides of this size render ERAP1 incapable of cutting any more (15, 48–50).
The structure of the PLC editing module was revealed by Alexander Domnick et al. (51), who used cryogenic electron microscopy at a resolution of 3.7 Å to determine the PLC’s multi-chaperone-client interaction network. Interactions between multivalent chaperones, including the CRT-engulfed mono-glucosylated MHC-I glycan, and peptide-receptive MHC-I molecules stabilize them, as shown in conjunction with epitope-proofreading studies of the PLC in a near-native lipid environment. After suitable epitopes have been loaded, this glycan can only be processed by α-glucosidase II. Scientists have shown that glycan processing and pMHC-I assembly interact allosterically. This example of interprocess communication exemplifies the well-planned steps in ER quality control and determines when an adaptive immune response starts (51).
The PLC is essential for the effective immune identification of virally and malignantly altered cells because it plays a crucial role in Ag processing. Researchers outlined the relationship between Tsn and MHC-I molecules. Peptide editing was seen in real time by researchers after ultrafast photoconversion to pseudoempty MHC-I molecules. Tsn distinguishes between MHC-I bound to suboptimal cargo and MHC-I loaded with optimum payload. Grasping the kinetics of epitope proofreading requires a thorough understanding of this unique interaction. Researchers utilized all-atom molecular dynamics (MDs) simulations to elucidate the Tsn/MHC-I complex, providing insights into the underlying processes at the atomic level. When cargo is deficient, Tsn’s interaction with MHC-I leads to a reconfiguration of the energy landscape that favors MHC-I complexes with immunodominant epitopes, according to a catalytic working cycle outlined by researchers (52).
This large membrane-bound protein complex was recently crystallized using low-resolution cryo-electron microscopy (cryo-EM), and its atomistic model was based on these data. In an explicit lipid bilayer and water environment, with a total of 1.6 million atoms, the model states that researchers investigated the conformational dynamics of the PLC on a time scale of multimicroseconds using all-atom MDs simulations. An inert, catalytically active core surrounds a bendable protein belt created by two editing modules in the PLC’s layered structure. The PLC’s function involves Tns maintaining the MHC-I binding groove in a conformation similar to that of antigen-loaded MHC-I. The binding groove is approached via a Tns loop in peptide editing led by the MHC-I-linked glycan. Additionally, researchers revealed that CRT affects the dynamics of Tns confirmation via a conformational selection mechanism that helps attract MHC-I into the complex (53).
The process is contingent upon the PLC, which enables the movement of peptides from the cytosol to the ER and the loading and verification of pMHC-I complexes. The significance of specific PLC components on the pMHC-I complexes displayed remains inadequately comprehended. Researchers utilized stoichiometrically defined antibody–nanobody complexes to quantify distinct MHC-I allomorphs and generated soluble T-cell receptors (sTCRs) to characterize pMHC-I complexes. The researchers identified that different PLC components impacted the pMHC-I surface pool (29, 34, 54). The MHC-I surface composition was altered by knockouts of Tsn, ERp57, or CRT, three PLC editing module components. Researchers demonstrated that an increased ratio of HLA-B*40:01 molecules compensated for the decreased proportion of HLA-A*02:01 presentation. These mutants improved the display of HLA-A*02:01 complexes with inadequate loading and high-affinity peptides overexpressed in the cytosol. According to researchers, the PLC editing module’s components have two functions: they restrict the amount of abundant peptides and serve as peptide proofreaders. This dual purpose guarantees a wide range of antigenic peptides are presented (29) (Figure 1).
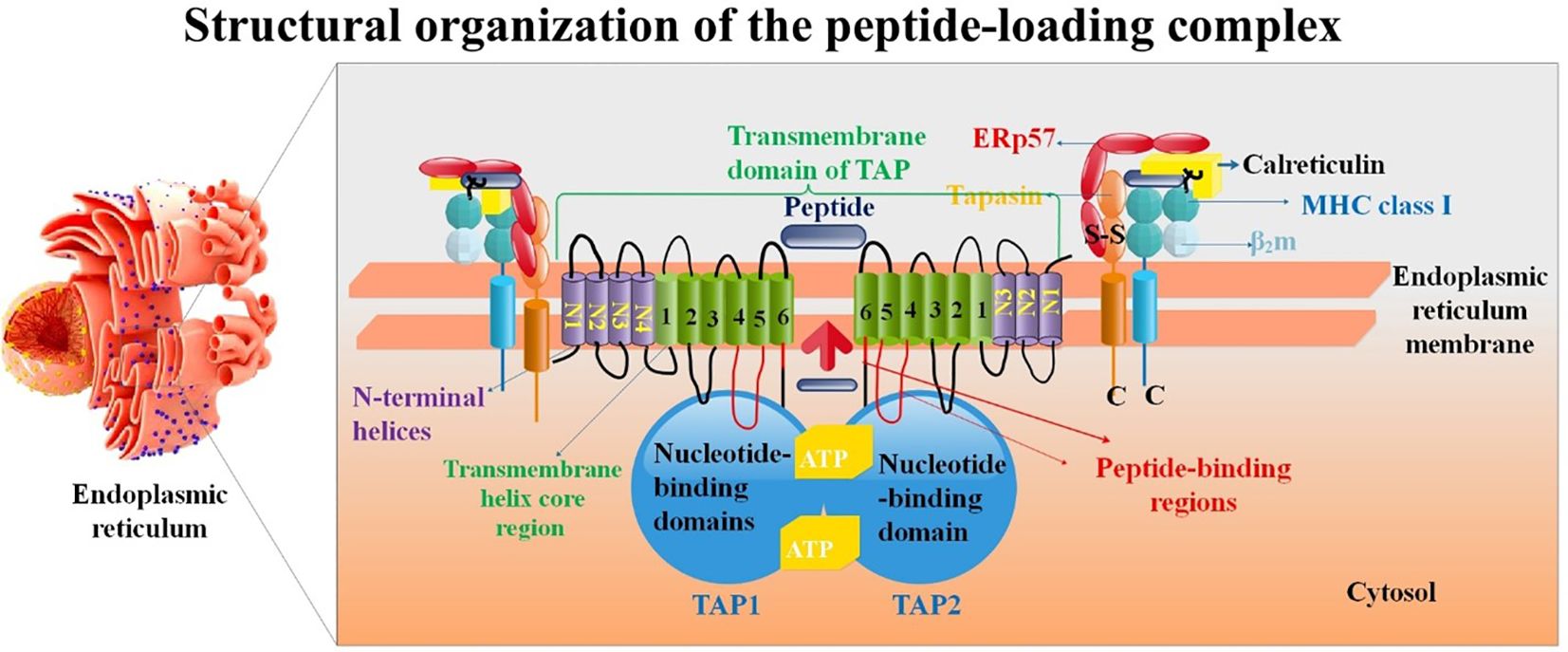
Figure 1. Structure of the PLC. Proteins such as the TAP1/TAP2 complex, ERp57, ER-resident aminopeptidase ERAP1/2, β2m, calreticulin, tapasin, and the MHC-I heavy chain are all part of the PLC. TAP has a translocation pore-aligning 6 + 6 TM core domain (H1-H6). Antigenic peptide transport is powered by ATP binding and hydrolysis, which is carried out by Walker A (A), Walker B (B), and C-loop (C) of the nucleotide-binding domain (NBD). It is believed that ATP binding/hydrolysis and peptide transport are connected via an interaction between the L-loop and the Q-loop (55).
2.1 Endoplasmic reticulum resident protein 57
The core of the MHC-I PLC is formed by the endoplasmic reticulum (ER)-resident proteins TAP, Tsn, and ERp57, which are essential for peptide loading by MHC-I-β2m dimers. The PLC is home to a stable heterodimer formed by disulfide connections between ERp57 and Tsn (56). Since ERp57 is involved in the formation of the heavy chain (HC) of MHC-I molecules, it has mainly been investigated for its function in the immune system (57). Enzymes such as phospholipase C, carnitine palmitoyl transferase, oxidase, and reductase rely on it, and it also helps ensure that freshly produced glycoproteins are of high quality in the ER (58).
Additionally, some ER-resident redox protein cofactors, such as protein disulfide isomerase (PDI) and its homologs ERp5, ERp57, and ERp72, are secreted (59, 60). In its role as a 1,25-dihydroxy vitamin D3 (1,25(OH)2D3) receptor, ERp57 regulates several processes, including the quality of newly synthesized glycoproteins, the assembly of MHC-I molecules, immune responses, the unfolded protein response (UPR), the NF-κB and STAT3 pathways, DNA repair, and cytoskeletal remodeling. It also controls immunogenic cell death (ICD) (61).
New evidence suggests that ERp57 is overexpressed in several human cancers (62–64). Additionally, abnormal ERp57 functioning and expression are linked to alterations in the chemosensitivity of tumors and their development and progression. To counteract the chemoresistance and growth of cancer, ERp57 may be a useful biomarker and therapeutic target (62). Quality control, steroid and phospholipid production, post-translational modification of newly generated proteins, Ca2+ homeostasis maintenance, and stress response modulation are just a few of the ER’s vital functions within the cell. The ER is populated with numerous proteins to assist in completing these innumerable duties. A thiol oxidoreductase exclusive to ER luminal glycoproteins, ERp57 is a PDI family member. Some of the names given to ERp57 include Grp58, ERp60, ERp61, PDI-Q2, Pdia3, and 1,25D3-MARRS (Membrane Associated, Rapid Response Steroid binding) (57).
The aggregation phenomenon is recognized in ERp57-deficient loading complexes, resulting from the expression of a Tsn mutant (C95A) within a Tsn-negative cell line. This mutant cannot establish a disulfide bond with ERp57. Researchers utilized cell lines that reliably expressed fluorescently tagged Tsn (wild type (WT) and C95A mutant) along with TAP1 to investigate the core loading complex’s assembly, stability, and aggregation. Researchers have indicated that the stability of core loading complexes depends on ERp57. Without ERp57, the core loading complexes will likely create stable aggregates within the ER (56).
Scientists discovered a temporary link between the loading complex and MHC-I molecules in ERp57-deficient cells. Hence, there were almost no MHC-I molecules in the loading complex in the stable state. The model antigen’s presentation and stability of the surface H-2Kb peptide were reduced. Though it does not affect the redox state of MHC-I molecules, researchers showed that ERp57 is a structural component crucial for the stable assembly of the PLC (65).
The 48 kDa glycoprotein known as Tsn was first shown to link the TAP heterodimer and peptide-responsive class I heterodimers. TAP and Tsn are the only accessory molecules and chaperones explicitly implicated in the class I assembly route (66). Recent investigations have demonstrated the necessity of covalent connections between Tsn and ERp57. Tsn forms a somewhat persistent disulfide bond with ERp57, which traps a significant quantity of cellular ERp57 in the PLC, in contrast to many other related chaperones. Recent research suggested that the functional unit in vivo responsible for peptide editing is the covalent ERp57–Tsn heterodimer (67).
2.1.1 ERp57 in cancer
Extracellular matrix (ECM) breakdown is necessary for tumor development and invasiveness, and the Src triggers it also drives the polypeptide N-acetylgalactosaminyltransferase (GALNT) activation (GALA) pathway, which causes protein O-glycosylation in the ER. Metalloproteases are needed for ECM breakdown; however, whether other enzymes are also required is not apparent. Studies reveal that in both liver and BC, GALA causes the ER-resident calnexin (Cnx) to become glycosylated (68–70). Because MMP14’s proteolytic activity is reliant on the addition of GalNAc, glycosylation of MMP14 is one mechanism by which GALA increases invasiveness in breast and liver cancer cells (68). ECM degradation sites known as invadosomes are the destinations of trafficked, glycosylated Cnx and its companion, ERp57. Disulfide bridges are shown to be prevalent in the connective and hepatic ECM (71). ECM breakdown requires the reduction of these extracellular disulfide bonds by cell surface Cnx–ERp57 complexes. Hepatocytes do not exhibit cell surface Cnx, although liver cancer cells do in vivo. Anti-Cnx antibodies block the formation of liver tumors and the lung metastasis of breast and liver cancer cells. Researchers revealed a Cnx-ERp57 side role at the cell surface that is essential for the formation of tumors and the disintegration of the extracellular matrix (71).
Salvia miltiorrhiza, or Dan Shen, is a famous traditional Chinese herb used to treat BC and many other diseases. That being said, little is known about its mechanics. Salvia miltiorrhiza extract (SME) contains the active component dihydrotanshinone I (DHT), which binds ERp57. DHT may have reduced ERp57 expression in both the RNA and protein forms and ERp57 function. A molecular docking simulation shows that DHT and the ERp57 catalytic site could form a hydrogen bond. Furthermore, in MDA-MB-231 cells, ERp57 overexpression reduced DHT-induced cytotoxicity. Following that, scientists looked into the signaling chain that ERp57 is downstream of. According to the molecular analysis, DHT therapy activated the UPR, ER stress, and cellular death. The researchers suggested that DHT inhibited ERp57, caused ER stress, and activated UPR, all leading to the death of BC cells (72).
One possible biomarker for the early identification of Hepatocellular carcinoma (HCC) is ERp57 expression, which varies considerably across HCC patients, at-risk patients, and healthy persons (73). The overexpression of ERp57 in HCC, colorectal cancer (CRC) (74), and BC (75) has been linked to carcinogenesis and cancer development. ERp57 expression is also connected with cancer cells’ ability to metastasize. High expression of ERp57 is associated with poor overall survival (OS) and high recurrence-free survival rates in adenocarcinoma patients, and it is overexpressed in 73% of cervical malignancies, particularly adenocarcinoma. Cancer metastasis and invasiveness are inhibited in HeLa cells when ERp57 is knocked down in vitro (62, 76, 77). There is a correlation between ERp57 levels and prostate cancer progression to the malignant stages (62, 78).
2.2 Tapasin in cancer
In the past two decades, the role of Tsn, commonly called TAP binding protein, has undergone thorough investigation and detailed characterization. Tsn plays multiple roles in peptide loading, such as stabilizing TAP, bridging peptide-receptive MHC-I to the TAP transporters, maintaining MHC-I in a peptide-receptive conformation, and facilitating peptide editing, which involves replacing low-affinity cargo with higher-affinity peptides. The implications of Tsn’s role in immune response, especially regarding antigen presentation and CD8+ T-cell differentiation, underscore its significance (79). The exact outcome of MHC-I molecules following peptide loading remains unclear. Peptide-loaded MHC-I molecules do not undergo immediate export from the ER following their dissociation from TAP, indicating that additional regulatory inspections and sorting processes occur after the PLC. UDP-glucose is a critical component in the post-PLC quality control process, specifically through the action of UDP-glucuronosyltransferases 1 (UGT 1), an enzyme located in the ER/cis-Golgi responsible for overseeing the proper folding of glycoproteins (80). By reglucosylating MHC-I molecules linked to less-than-ideal ligands, UGT1 can make CRT recognize these molecules again. This enables them to reconnect with the PLC and has been shown to affect the best possible peptide choice (81).
Loss of Tsn helps malignancies evade the immune system and diminish their immunogenicity (82). It was first shown in vivo in mice tumor models how crucial Tsn is for antigen presentation in cancer. An LC cell line that downregulated many MHC I pathway components was implanted into mice. Against this backdrop, Tsn transfection was enough to improve antigen presentation, boost the immune response specific to the antigen, slow tumor development, and improve survival (83). Researchers determined whether there is a relationship between Tsn expression and the presence of CD8+ cytotoxic T lymphocytes (CTLs) in CRC, as well as OS. Scientists have discovered that Tsn aids in antigen presentation, tumor immune detection, and CD8+ CTL destruction; hence, a decrease in Tsn expression is linked to tumor growth in CRC (84).
Shionoya et al. (85) found a favorable correlation between Tsn expression and patient survival in their study of 85 primary tumor lesions from NSCLC patients. CD8+ T-cell infiltration of tumor lesions was shown to be favorably linked with survival and to have a synergistic effect with Tsn expression. Researchers used the CRISPR/Cas9 technology to specifically target the Tsn gene to prove a causal relationship between Tsn deletion and CTL recognition in cancer models derived from humans. Their goal was to create human lung and colon cancer cells lacking in Tsn. To test the efficacy of each Tsn-proficient WT, researchers stimulated CTLs that recognize endogenous tumor-associated antigens (TAA), survivin, or cep55. However, despite expressing the antigen, neither CTL line paid any attention to the Tsn-deficient mutants. Also, animals with the Tsn-deficient version still developed tumors even after receiving the cep55-specific CTL line by adoptive transfer. The inability to process TAA antigens due to Tsn loss probably allowed the escape from CTL identification that was unique to TAAs. Expression of Tsn is, consequently, crucial for CTL monitoring of human malignancies (85).
The three oral squamous cell carcinoma (OSCC) cell lines that were studied showed evidence of Tsn promoter methylation. Additionally, the levels of Tsn mRNA and protein were shown to rise significantly after treatment with 5-aza-2′-deoxycytidine. Potentially useful as a predictive biomarker, the downregulation of Tsn is linked to worse clinical outcomes in individuals with OSCC. The downregulation of Tsn in OSCC may be influenced by promoter methylation (86).
Immune recognition and antigen presentation depend on the catalytic chaperone Tsn in the PLC loading MHC-I molecules with peptides. A significant limitation to mechanistic understanding has been the absence of comprehensive structural data for Tsn–MHC–I. Researchers revealed crystal structures of human Tsn complexed with both anti-Tsn antibodies and the MHC-I protein HLA-B*44:05. HLA-B*44:05’s Tsn-stabilized peptide-receptive state is characterized by peptide binding groove distortion and β2m interaction instability, resulting in peptide release. The migration of the membrane-proximal immunoglobulin-like domains of Tsn, HLA-B*44:05, and β2M synchronizes with the change to a peptide-receptive state. Combined, these crystal structures provide light on a unique mechanism of Tsn-mediated peptide exchange (87).
2.3 MHC-I peptide processing and presentation
The process of antigen presentation through MHC-I molecules represents a complex and essential protective mechanism employed by the adaptive immune system to combat pathogens and malignant cells (88). Part of the superfamily of ATP-binding cassette (ABC) proteins, the heterodimeric TAP is an essential component of the antigen presentation pathway. A nucleotide-binding domain is connected to six transmembrane helices in the core transporter region of the TAP subunits TAP1 and TAP2 (ABCB2 and ABCB3, respectively). Additionally, each core subunit is N-terminally attached to TMD0, an additional transmembrane domain with four helices (89). TAP is an integral part of the ER-resident PLC because it serves as a gatekeeper for the reservoir of antigenic peptides created by cytosolic proteasomal degradation. TAP carries peptides with overlapping lengths and sequence specificities to engage with MHC-I molecules and ensure MHC-I antigen presentation. The MHC-I peptide-binding pocket is aligned with the aminopeptidases ERAP1/2 because they can digest longer peptides in the ER lumen. Tsn carries out peptide proofreading, whereas peptide loading is aided by the chaperones CRT and ERp57, which guarantee the proper assembly of MHC-I molecules and the editing module (90–92). Researchers found a way to control antigen presentation by adding a photo-caged amino acid to TAP’s catalytic ATP-binding domain, as our knowledge of MHC-I trafficking pathways is still restricted. If researchers can start TAP-dependent antigen translocation by optical manipulation, they may learn more about TAP’s function in live cell PLC and MHC-I trafficking. This adaptable approach can be used to investigate other cellular pathways that are affected by P-loop ATP/GTPases (92).
Dendritic cells (DCs) present antigenic peptides on MHC molecules to T-cells, thereby orchestrating immunological responses. The fundamental component of the supramolecular structure referred to as the PLC, essential for antigen processing and presentation through MHC-I, is the peptide transporter located in the ER membrane, commonly known as TAP (93, 94). According to the research, the PLC attracts extra proteins such as B-cell receptor-associated protein 31, vesicle-associated membrane protein-associated protein A (VAPA), and extended synaptotagmin-1 (ESYT1) as DC develops and matures. Scientists have found the proteins that bind to specific sites and export cargo from the ER to nearby TAP, which is located 40 nm from the PLC. Based on these findings, the APM and the locations of membrane contact and ER departure are closely related. The functions of BAP31, VAPA, and ESYT1 were similar within the framework of DC MHC-I antigen processing. Nevertheless, the examination of single-gene deletions of the recognized PLC interaction partners revealed that the MHC-I surface expression was considerably decreased when TAP and Tsn were deleted using CRISPR/Cas9 technology. Researchers demonstrated the dynamic and adaptable nature of the PLC composition in DCs, an aspect not identified through cell line studies (32).
Furthermore, researchers investigate the role of the chaperone Tsn-binding protein-related (TAPBPR) in facilitating peptide loading. According to the researchers, TAPBPR attaches to the F pocket and allosterically changes the structures of the distant pocket B to produce a peptide-receptive conformation ideal for tolerating the arriving N-terminus. Researchers elucidated the molecular mechanisms involved in peptide loading onto MHC-I, both in the presence and absence of chaperones (95).
Also, scientists showed that the binding of TAPBPR at the MHC-I pocket-F may facilitate the N-terminal loading of the following peptide by considerably influencing the distant pocket-B via allosteric modulation. Researchers demonstrated that the partly loaded peptide has the potential to significantly decrease the stability of the TAPBPR-MHC complex by inducing the SL (TAPBPR scoop-loop) region to detach from the pocket-F and adopt a more solvent-exposed conformation. Additional structural investigations indicate that the peptide loading may indirectly use direct contacts and allosteric perturbations to affect the SL binding site. Moreover, it was discovered that the jack hairpin region, another structural feature of TAPBPR, assisted in facilitating peptide editing. Researchers identified the critical structural elements that control the kinetics of peptide loading and provide insight into the intricate molecular processes behind the process of peptide loading into TAPBPR-bound MHC-I (96).
Polymorphisms in the endoplasmic reticulum aminopeptidase (ERAP) genes, namely ERAP1 and ERAP2, have recently been shown to be much more prevalent in individuals with ankylosing spondylitis (AS), psoriasis, or Behçet’s disease (BD), according to genetic association studies. The fact that ERAP is in epistasis with the risk alleles of the MHC-I allele is significant under these settings. Nevertheless, it has come to light that not all AS or BD patients possess the MHC-I risk allele. This finding has sparked debates over the significance of this commonality and whether it indicates a common illness origin (97–99) Jonas J W Kuiper et al. mapped the ERAP1 and ERAP2 haplotypes in 84 cases and 890 controls from the Netherlands (100). A relationship was found at variation rs10044354, which led to a significant upregulation of ERAP2 expression, according to the researchers. Separately, researchers found and cloned an ERAP1 haplotype (tagged by rs2287987) that was related to over 50% of the cases; this ERAP1 haplotype serves as both a risk factor and a protective factor for additional MHC-I-opathies. Based on transcriptome data (n = 360), researchers found that the risk ERAP1 haplotype caused a dramatic change in ERAP1 isoform expression, leading to reduced protein expression and unique enzymatic activity. A separate Spanish cohort consisting of 46 cases and 2,103 controls confirmed and confirmed the direction of impact for both rs10044354 [meta-analysis: odds ratio (OR) [95% CI]=2.07[1.58-2.71], P = 1.24 × 10(−7)] and rs2287987 [OR[95% CI]: =2.01[1.51-2.67], P = 1.41 × 10(−6)]. The rs2287987-rs10044354 haplotype was more strongly related to Birdshot in both cohorts compared to the individual variants. At last, after analyzing three European populations (n = 3353), researchers found that the ERAP1 background influences the expression of the ERAP2 protein. Lastly, Birdshot is characterized by a functionally different mix of ERAP1 and ERAP2, which supports the idea that techniques may be developed to rectify ERAP function in order to cure Birdshot and other MHC-I-opathies (100).
2.3.1 MHC I in cancer
Numerous innate and adaptive immune cells, such as DCs, macrophages, neutrophils, natural killer cells (NK), γδ T-cells, CD4+ T-cells, and CD8+ T-cells (cytotoxic T-cells), may infiltrate solid tumors. Some of these immune cells can even eliminate particular tumors. Tumor cells may effectively evade the immune system’s regulation by activating several internal processes and modifying external variables in return. It is now thought that immune escape is one of many tactics cancers use to protect their survival and growth—also referred to as the cancer hallmarks. These molecular immune escape mechanisms involve the following: A) obstructing MHC-mediated antigen presentation to hinder T-cell surveillance; B) releasing broadly immunosuppressive cytokines like TGF-β and IL-10; and C) displaying inhibitory receptors on the cell surface such as PD-L1 and PD-L2, which limit T-cell activation (101).
Using a human melanoma cell line, researchers conducted a genome-wide CRISPR screen. Contrary regulators of MHC-I transcription were the deubiquitinating enzyme BRCA1-Associated Protein 1 (BAP1) and the polycomb repressive complex 1 (PRC1) component Polycomb Group RING Finger Protein 1 (PCGF1). PCGF1 facilitates ubiquitin deposition at H2AK119, which silences the MHC-I promoters. BAP1 reverses this change, restoring MHC-I expression. Despite PCGF1’s extensive-expression in malignancies, certain tumor lines, particularly MHC-I low cancers, demonstrated enhanced MHC-I expression with a decrease in PCGF1. Both PRC1 and PRC2 work together to suppress low transcription in cells with low MHC-I expression. Rest assured, MHC-I expression was amplified, and T-cell-mediated tumor cell killing was reinstated after PCGF1 elimination. Researchers light up an additional mechanism by which malignancies regulate MHC-I expression: epigenetic repression by the PRC1 component PCGF1 (102).
The HLA-B27 subtypes that are related to spondyloarthritis (SpA), namely B*27:02, B*27:05, and B*27:07, have a higher propensity to aggregate in cytoplasmic vesicles and form intracellular oligomers compared to the non-SpA-associated subtypes HLA-B*07:02 and HLA-B*27:06. Better to understand the relationship between SpA propensity and HLA-B-containing vesicles, researchers set out to describe their composition and nature. Subtypes of HLA-B27 prone to SpA have an accumulation of misfolded HLA-B HC, β2m, and ER chaperones from vesicles produced from the ER that are distinct from the PLC. Through a noncanonical route, this behavior may contribute to the pathogenicity of HLA-B27 (103).
Peptides containing proline at position 2 bind preferentially to MHC-I molecules of the HLA-B7 supertype. Two subpeptidomes are found in HLA-B*51:01 and B*51:08, with one subpeptidome enriched at position 1 with Ala2 and Asp and the other with Pro2 and hydrophobic residues at P1. To explore the existence of subpeptidomes across various allotypes, researchers provided a meta-analysis of the peptidomes offered by molecules of the B7 supertype. The presence or absence of a Pro or other residue at position 2 differentiated the subpeptidomes of many allotypes. Glu1 was present in Ala2-containing ligands, whereas Asp1 was chosen by the Ala2 subpeptidomes in HLA-B*54:01. Researchers suggested that subpeptidomes may be present at MHC HC sites 45 and 67 based on sequence alignment and crystal structure analysis. Antigen presentation in other MHC-I molecules may be better understood if the principles behind the existence of subpeptidomes can be deciphered—efficacy of HLA-B7 supertype subpeptidome selection (104).
Due to gene loss or epigenetic silencing, cancers may lose the production of non-essential molecules and are often genetically unstable. Molecules belonging to the MHC-I antigen presentation pathway, including MHC-I, are not essential for cell survival and proliferation. Consequently, tumors may inhibit or eliminate MHC-I antigen presentation, rendering them less stimulatory or invisible to CD8 T-cells without compromising tumor growth and metastasis (88). The researchers examined the therapeutic implications, mode of action, and frequency of MHC-I deletion in cancers. Because of their clinical significance, this overview mainly focuses on human malignancies, except those mentioned. Cancers may also avoid immune clearance by expressing HLA-E and HLA-G, two “non-classical” MHC-Ib molecules. Studies examining MHC-I expression in primary patient samples have mainly used immunohistochemistry (IHC). Traditional MHC-I molecules, including HLA-A, HLA-B, and HLA-C, and monomorphic determinants on their HC may be targeted by antibodies to perform IHC. This has led to a downregulation of MHC-I antigen expression in several cancers. There have also been reports of a single MHC-I molecule losing its expression. Many investigations have shown MHC-I negative malignancies; however, due to IHC’s sensitivity limitations, some patients could display some MHC-I molecules (88).
Along with HLA-F and HLA-H, HLA-G and HLA-E are members of the non-classical HLA-class Ib family. Numerous studies have examined these molecules’ function in regulating the immune response in both healthy and diseased settings. Different immune cell types, including T-cells, antigen-presenting cells, and immunoregulatory cell populations, including mesenchymal stem cells, may also express HLA-G. However, HLA-G is up-regulated in a variety of clinical circumstances, including inflammatory disorders, malignancies, viral infections, and transplantation. Transformed cells (tumor and virus-infected cells) that express HLA-G have an immune escape mechanism that prevents them from being recognized and lysed by cytotoxic immune effectors such as NK cells and cytotoxic T lymphocytes (105–107).
Furthermore, HLA-E binds peptides isolated from the leader sequence of HLA-class I molecules (HLA-A, -B, -C, and -G) and delivers them to NK cells via the inhibitory receptor CD94/NKG2A. This prevents NK cell lysis of cells expressing normal amounts of HLA-class I molecules. When HLA-class I expression is low, cells produce fewer HLA-I-derived peptides, which means they have less HLA-E and are more susceptible to NK cell lysis. When HLA-E binds to peptides produced by HLA-G, it may cause an interaction with the CD94/NKG2C activating receptor on NK cells. When HLA-G+ trophoblast cells invade the placenta, this property is used to trigger NK cell lysis, which in turn causes tissue remodeling (106). Furthermore, Researchers showed that mesenchymal stromal cells produced from gestational tissue, namely from cord blood, are not very immunogenic. This is associated with the fact that these cells co-express HLA-G and HLA-E (106, 108). DCs release exosomes (EXOs), which carry antigens and include MHC-I and MHC-II, along with other components. Immunogenicity of EXOs MHC-I has been shown by scientific studies when these molecules are “indirectly” loaded onto DC cells using peptide addition. A peptide binding study was not carried out to establish a relationship between the quantity of MHC-I/peptide complexes on the EXOs and the T-cell-stimulating function. Scientists tested the EXOs’ activation potencies in T-cell activation assays and analyzed peptide binding to MHC-I under different loading conditions (109–112).
Researchers showed that peptides may be directly and much more strongly loaded onto pure EXOs’ MHC-I than by indirect loading. Even without exogenous β2m, the moderately acidic circumstances in which the direct loading approach was carried out proved efficient. EXO potency was significantly increased by this increase in peptide binding, enabling researchers to investigate EXO biologic activity in vitro. When combined with APC, EXOs carrying the HLA-A2/MART1 tumor peptide stimulated a T-cell line targeting HLA-A2/MART1. Using HLA-A2neg APC, the T-cells reacted to EXOs, indicating that functional MHC-I/peptide complexes were transferred to APC rather than peptides alone. Under the same circumstances as MHC-I, MHC-II molecules—highly expressed on DC EXOs—were likewise functionally loaded. This property facilitates the transmission of a wide range of peptide antigens capable of activating CD4+ T helper cells and CD8+ cytotoxic T-cells, both essential for an effective antitumor response. The ideal loading conditions and capacity for transmitting both MHC-I and MHC-II antigens to antigen-presenting cells have resulted in the advancement of extracellular vesicles as a “acellular” immunotherapy approach, which is presently under investigation in clinical trials (113, 114) (Figure 2).
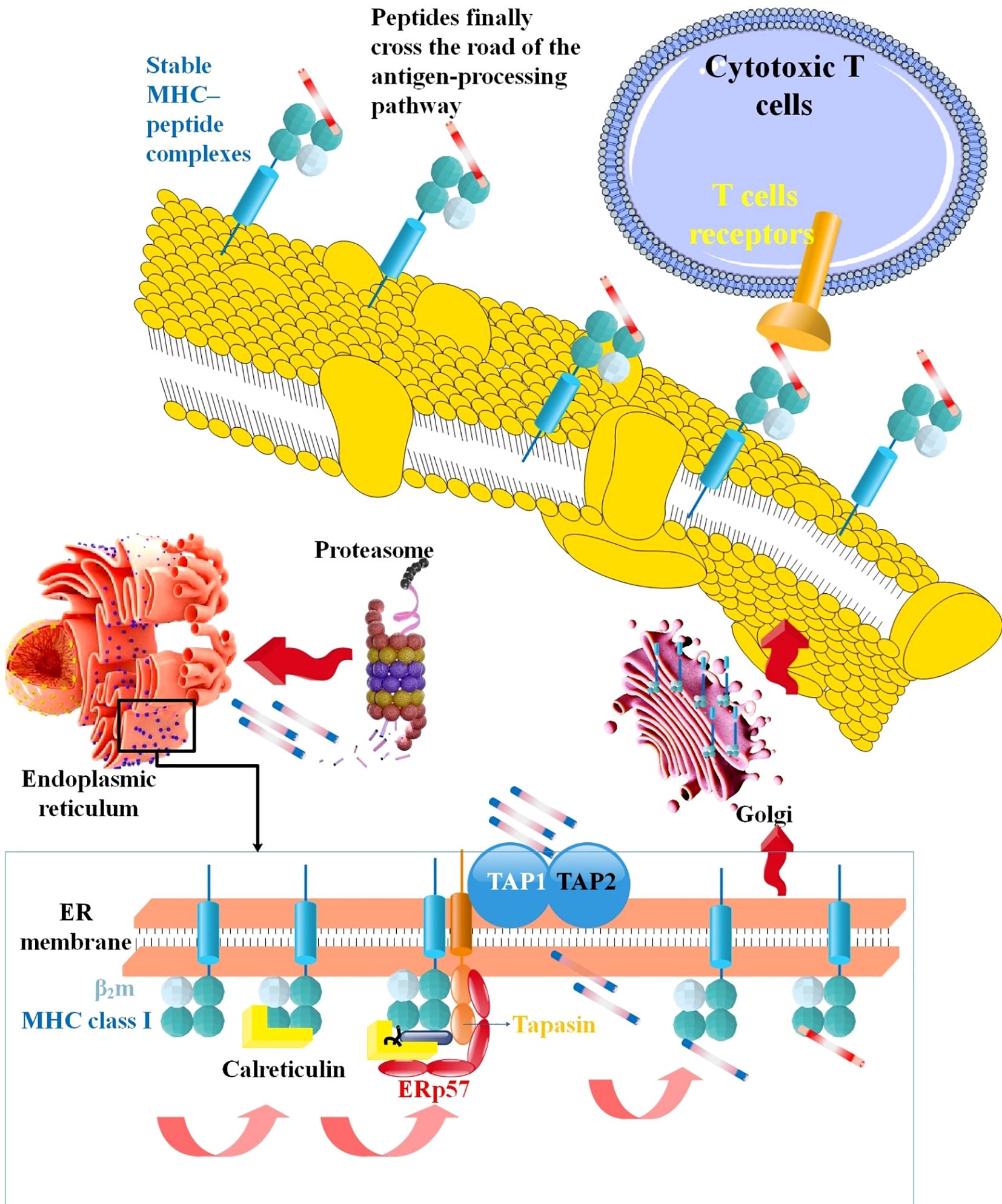
Figure 2. The mechanism via which MHC-I processes antigens. TAP carries them into the ER lumen to load antigenic peptides produced by the proteasome in the cytosol onto MHC-I, and TAP carries them into the ER lumen. CTL recognize stable MHC-peptide complexes when they exit ER via the Golgi network and reach the cell surface (115).
2.3.2 HLA associated with cancer
CTLs are exposed to antigenic peptides via HLA-I. An essential phase in the creation of CTL responses is this one. The degree of HLA-I expression directly influences NK-cell responses, although peptide-HLA-I (pHLA-I) complex quantity and quality are also crucial for CTL responses. Antigen processing machinery (APM) proteins have a role in choosing which peptides are given and in the maturation of HLA-I. These proteins thus have a significant role in determining how the immune system reacts to cells in both health and illness. Scholars specifically focus on the multifunctional protein Tsn, which is peculiar to HLA-I. In light of the varying Tsn-dependencies across HLA-I allomorphs, researchers also examine the unique characteristics of each allomorph in terms of development, structure, and association with malignant illnesses, namely brain tumors (116).
Autologous treatments are hindered by polymorphic and unstable MHC-I and MHC-like molecules with inadequate peptides, metabolites, and glycolipids. These features complicate antigen-specific TCR and disease-relevant antigen identification (117). To bind to the MHC-I, researchers linked conserved epitopes across the HC/β2m interface using an engineered disulfide linkage, therefore enabling positive allosteric interaction between the peptide and light chain (β2m) subunits. These let researchers create “open MHC-I,” conformationally stable peptide-receptive molecules (117).
Furthermore, loaded with low- to moderate-affinity peptides, open MHC-I molecules are well-folded and more thermally stable than WT. Researchers investigated how the disulfide link influences MHC-I kinetics and conformation by use of solution Nuclear Magnetic Resonance (NMR). Observed were long-term effects on the α2-1 helix and α3 domain as well as local modifications in the β2m-interacting sections of the peptide-binding groove. The interchain disulfide bond stabilizes MHC-I molecules in an open conformation, therefore enabling peptide interchange across five HLA-A supertypes, six HLA-B supertypes, and oligomorphic HLA-Ib molecules. Stable and ready-to-load MHC-I systems may be built by researchers using structure-guided design and conditional β-peptide ligands. This enables highly polymorphic HLA-I allotypes and oligomorphic nonclassical molecules to screen libraries of antigenic epitopes and polyclonal TCR repertoires (117).
Researchers investigated the possibility that peptides directly associated with β2m may form a complex with the human HLA-I HC and be identified as components of the cell surface and soluble reagents by human CTL. The N terminus of human β2m was physically associated with a peptide epitope limited by HLA-A2. When this fusion protein was multimerized, it formed complexes called “fusamers” that linked exclusively to specific CTL clones. The HLA-A2 HC also refolded quickly in a laboratory setting. These fused peptide/MHC complexes were equally effective as traditional tetrameric peptide/MHC complexes in detecting CTL specific to antigens. After delivering the fusion protein via a retroviral vector, appropriate CTL clones identified and eliminated the target cells. β2m-negative, TAP-negative and unmutated B cell lines showed adequate sensitization to CTL lysis, suggesting these constructs might help trigger CTL even when the MHC-I pathway is disrupted. Retroviral vectors carrying specific peptides covalently bound to β2m might be helpful for in vivo CTL priming against therapeutically relevant epitopes (118).
When CTL detects pMHC-I complexes on target cells, they lyse them. The lack of Tsn, a critical component of the PLC, impacts the surface repertoire of MHC-I peptides. Analysis of 85 primary tumor lesions from non-small cell lung cancer (NSCLC) patients revealed a statistically significant association between Tsn expression and OS. Together with Tsn expression, CD8+ T-cell infiltration of tumor lesions was seen as positively linked with survival. Researchers used the CRISPR/Cas9 technology to target the Tsn gene, resulting in Tsn-deficient human lung and colon cancer cells. This was done to demonstrate that in human cancer models, there is a clear association between the loss of Tsn and CTL recognition. The researchers primed the CTLs to respond to endogenous tumor-associated antigens (TAA), survivin, or cep55, and each Tsn-proficient WT. On the other hand, even though the Tsn-deficient mutants expressed antigen, both CTL lines disregarded them. Furthermore, in mice with the Tsn-deficient version, adoptive transfer of the cep55-specific CTL line could not stop tumor development. Tsn loss most likely restricted TAA antigen processing and allowed escape from CTL recognition specific to TAAs. To protect human malignancies from CTL surveillance, Tsn expression is crucial (85).
Tumor cell death after CTL recognition of TAAs on tumor cells is thought to be directly correlated with TAA protein production and reliant on the amount of peptide present in the HLA-I molecule’s binding site (117, 119). On human tumor cell lines, researchers looked for evidence of a connection between the expression levels of the Her-2/neu protein and the presentation and recognition of CTL by the HLA-A*0201/Her2/neu peptide. Scientists developed a TCR mimic (TCRm) monoclonal antibody (mAb) called 1B8 to locate the Her2(369)-A2 complex, which is represented by the HLA-A2.1/Her2/neu peptide(369-377) (117). A possible explanation for the observed quantitative differences in the Her2(369)-A2 complex levels on the five human tumor cell lines studied is the variable degree of TCRm mAb labeling. No significant correlation was seen between the levels of Her2/neu Ag, HLA-A2 molecule, and Her2(369)-A2 complex expression in tumor cell lines that were pretreated with IFN-γ and TNF-α for Her2/neu protein and HLA-A2 molecule expression. Additionally, there was a clear correlation between the enhanced tumor cell mortality and the elevation in Her2(369)-A2 epitope density in cytokine-treated cell lines compared to untreated cells. Her2(369)-A2 complex level for untreated cells showed a trend with tumor cell lysis, although the relationship was not statistically significant. These results imply that rather than the overall amount of TAA expression, the vulnerability of tumor cells to CTL-mediated lysis may be anticipated based on the degree of particular pMHC-I expression. These investigations also demonstrated the potential of the TCRm mAb for validating endogenous HLA-peptide epitopes on tumor cells (120).
Mutations in the p53 gene typically result in an excess of the WT p53 protein, representing the most common genetic alterations observed in human cancers. The potential for T lymphocytes to react to tumor cells that overexpress WT or mutant p53-derived peptides supports the application of these epitopes in cancer immunotherapies. The researchers employed two separate flow cytometry-based assay techniques to assess the binding capabilities of WT and mutant p53 peptides to HLA-A2.1: the T2 MHC-I peptide stabilization assay and the peptide-induced MHC-I reconstitution test. The reason is that antigen-specific T-cell recognition requires peptide binding to MHC-I molecules. Twenty WT sequences were selected because they all met the criteria for the HLA-A2.1 peptide-binding motif. From the WT peptides attached to HLA-A2.1 that were previously chosen, seven WT p53 and two out of thirteen mutant p53 peptides were generated for the stabilization and reconstitution assays. The observed lower affinity for HLA-A2.1 could account for detecting only six additional WT and six mutant p53 peptides in the reconstitution experiment. Hypothesized to serve as immunogens for the in vitro and in vivo generation of cytolytic T-cells specific to HLA-A2.1, these p53 peptides bind HLA-A2.1 (121) (Figure 3, Table 1).
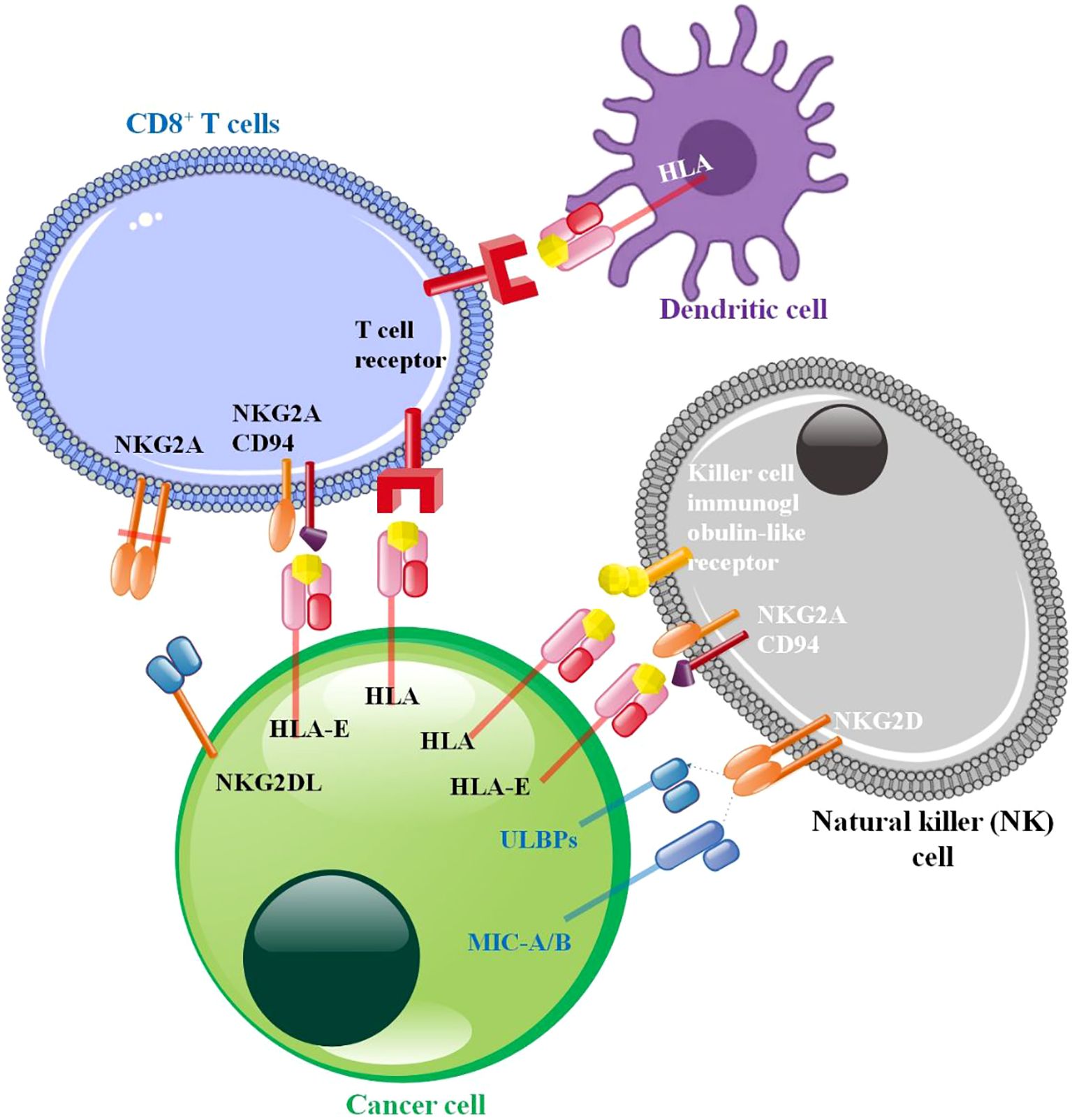
Figure 3. Cancer cell-immune cell interaction in tumor microenvironment. The NK cell illustrates well. NK cell surface immunoglobulin-like receptor (KIR), a conventional MHC-I inhibitory receptor. NK cell activation is inhibited by KIR and MHC-I molecules in normal cells. Their findings suggest that HLA-E: CD94-NKG2A blockage may cure cancer. HLA-G suppresses NK cell function by binding to its receptors ILT2 and KIR2DL4. The best-studied activating receptor is NKG2D, which recognizes MIC and various UL16-binding proteins (ULBPs). Continuous activating receptor activation on NK cells promotes desensitization. NK cell desensitization occurs when tumor cell ligands MHC class I chain-related protein A (MICA) and MICB directly interact with NKG2D. The anti-cancer response may be reduced by blocking TCR-MHC-I interaction. T and NK cells have an inhibitory receptor formed when NKG2A and CD94 dimerize. T-cells produce an inhibitory signal when they come into contact with peptide-loading HLA-E, which phosphorylates ITIM. Enhancement of NKG2A and HLA-E by cancer cells (122–126).
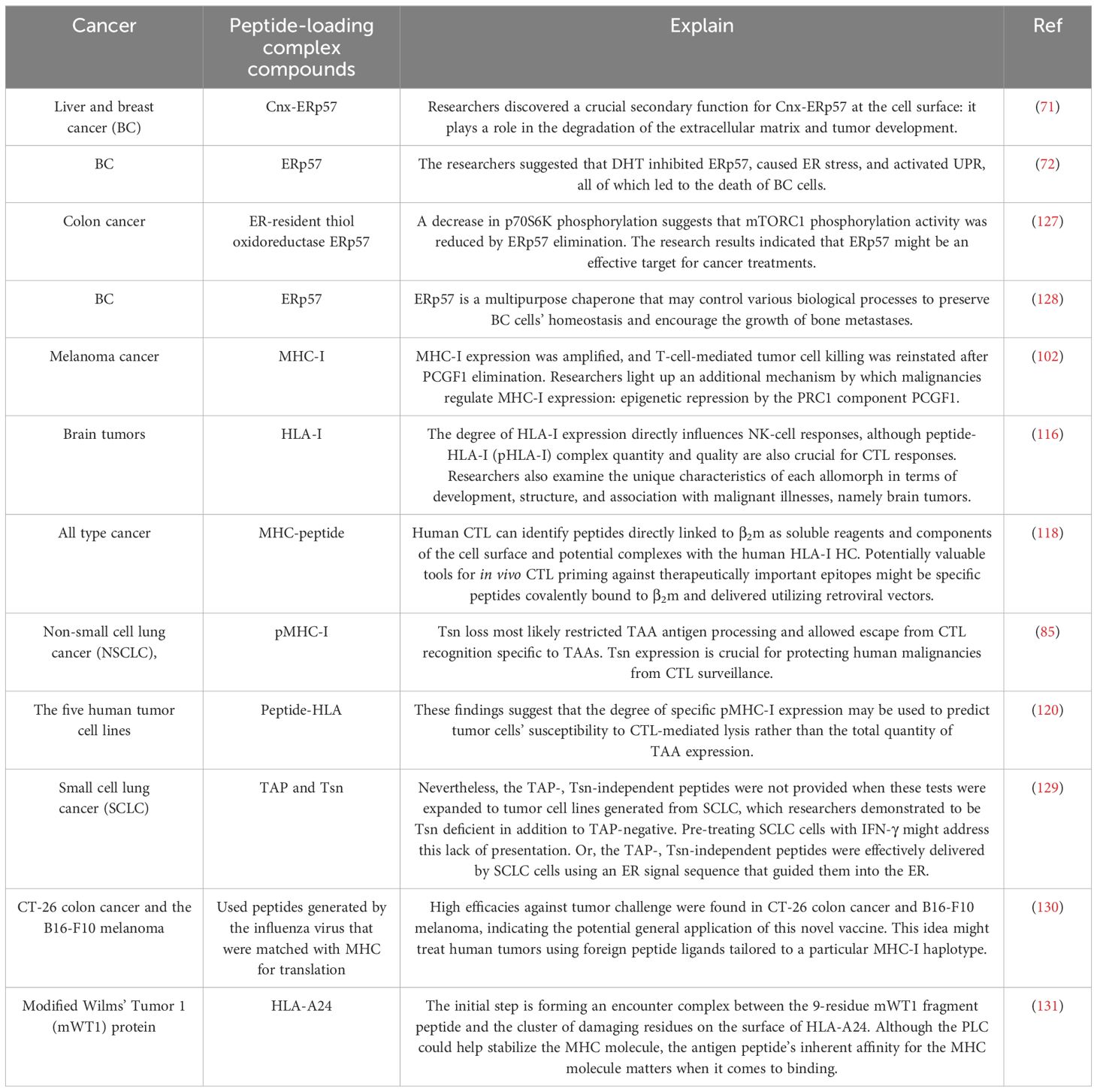
Table 1. The function of complicated peptide loading compounds in various cancer types.
Both the conserved and invariant c-type lectin CD94/NKG2A inhibitory receptor (henceforth referred to as NKG2A, which stands for NK cell protein Group 2-A) that binds non-classical HLA-E and the polymorphic killer-cell immunoglobulin-like receptors (KIR) that bind classical HLA-I molecules HLA-A, HLA-B, and HLA-C are significant inhibitory receptors that can instruct NK cells. HLA-E requires the input of peptides from classical HLA-A, HLA-B, or HLA-C for correct folding and cell surface trafficking (132, 133). A meta-analysis conducted by Norman Shreeve et al. found that effects such as suboptimal maternal vascular responses during pregnancy, altered placental gene expression, decreased fetal weight, increased rates of smaller fetuses with asymmetric growth, and abnormal brain development were caused by NKG2A genetic ablation in dams mated with wild-type males. These symptoms are seen in pregnant women with pre-eclampsia. A genome-wide association study of 7,219 pre-eclampsia patients found that when the maternal HLA-B allele did not boost NKG2A education, there was a 7% higher relative risk. Researchers’ findings showed that the mother’s HLA-B→HLA-E→NKG2A pathway contributes to a healthy pregnancy and may affect the children’s health, showing the physiological significance of NK cell education (134).
3 Peptide loading complex in liquid tumors
The binding of peptides to MHC-I in the ER is enhanced by the presence of PLC, which includes CRT. CRT binds to MHC-I through a conserved glycan, facilitating its movement to the PLC for peptide binding. Somatic frameshift mutations in CRT lead to a specific subgroup of myeloproliferative neoplasms characterized by ongoing blood malignancies, resulting in proliferation (CRT-FS) (135). All CRT-FS proteins exhibit a net negatively charged C-terminal sequence instead of a positively charged one, and they lack the typical ER-retention signal. Najla Arshad and colleagues examined the influence of CRT-FS on antigen presentation via MHC-I in human cells. Research indicates that CRT-FS cannot facilitate the PLC peptide-loading function of CRT. Although CRT-FS expression did not enhance high-affinity peptide binding, the observed reduction in surface MHC-I levels in CRT-deficient cells was associated with a decrease in high-affinity peptide binding. Despite the secretion of CRT-FS and its lack of binding to the PLC, it could still bind to MHC-I glycan independently. This observation explained the suboptimal recruitment of MHC-I. The introduction of the ER-retention sequence to CRT-FS successfully restored its interaction with the PLC; however, it did not enhance surface expression or MHC-I recruitment. This suggests that the mutations in CRT-FS significantly impair the functionality of the PLC. Since tumor cells may avoid immune monitoring when MHC-I is down-regulated, these results could be crucial in developing immunotherapies that effectively manage myeloproliferative neoplasms (136).
New medications have been discovered, and various therapy techniques have been developed in recent years as our understanding of the pathophysiology of acute myeloid leukemia (AML) has advanced (137). The development of AML immunotherapies and their clinical translation has been sluggish. Intrinsic AML characteristics hinder the process of moving from immunobiology to immunotherapy. Genetic/epigenetic heterogeneity and subclonal contribute to biologic variance in AML, a family of distinct cancers rather than a single illness (138, 139). Immunosuppression in the AML microenvironment is linked to global alterations in the profiles of immune cells in the bone marrow. Additionally, the microenvironment shields leukemic stem cells—which may cause relapse—from being destroyed by therapy or the immune system (140).
The absence of specific immune targets is the fundamental reason why novel immunotherapies have failed to improve outcomes for patients with AML. Scientists isolated 58 tumor-specific antigens (TSAs) by using a novel proteogenomic strategy to the immunopeptidome associated with MHC-I in 19 primary AML samples. Most of these TSAs (86%, to be exact) originated from purportedly non-coding genomic areas and did not include any mutations. Intron retention, epigenetic alterations, and TSA biogenesis were all aided by two AML-specific abnormalities. Both blasts and leukemic stem cells exhibited AML TSA-coding transcripts, which were substantially shared across individuals. Improved survival, immunoediting, accumulation of activated cytotoxic T-cells, and spontaneous expansion of cognate TCR clonotypes were all associated with the expected amount of TSAs in AML patients. Immunotherapy against AML could be a promising strategy if it targets these TSAs (141). The discovery of MHC-I-associated peptides originating from any reading frame of any genomic region (not only exons) has been made possible using a proteogenomic strategy that was recently developed. Eighteen primary human AML samples were analyzed, and 82 TSAs were discovered. Even though traditional EXO-based methods would have missed these TSAs, they were all produced from commonly expressed transcripts with mutations. Consistent with earlier findings that increased intron retention is an essential characteristic of AML, researchers discovered that introns were the primary source of TSAs. Approximately 98.04 percent of the global population has one of the 24 TSA-presenting HLA allotypes. While all of them were either expressed at deficient levels or not expressed at all in normal tissues (from the “Genotype-Tissue Expression” (GTEx) project), the majority of them were represented by at least 50% of AML samples in the “The Cancer Genome Atlas” (TCGA) cohort. The precise epigenetic modifications that cause cancer cells to produce TSA remain unknown to researchers. According to the researchers, there is no concern about collateral harm to healthy normal tissues, and immunization against aberrantly expressed TSAs might be utilized to cure most patients (142).
Particular proteins involved in the processing pathway of HLA-I and HLA-II complexes are responsible for antigen presentation. The invariant chain (II) peptide Class II-associated Invariant Chain Peptide (CLIP) is necessary for HLA-II-mediated antigen presentation since it stabilizes HLA-II molecules before antigen loading by transiently and non-specifically attaching to various HLA-II receptor grooves. Scientists proved that CLIP binds to leukemic cells’ surface HLA-I molecules differently. Scientists discovered that CLIP peptide display in HLA-II-negative AML cells (143). A direct involvement of CLIP in the HLA-I antigen presentation pathway was shown when HLA-I cell surface display was diminished in AML cells by silencing Ii. Five Ii-derived peptides, including two from the CLIP region, were detected in HLA-I-specific peptide eluates from B-LCLs. According to peptide binding experiments, the eluted CLIP peptide RMATPLLMQALPM is firmly bound to four different HLA-I supertypes (-A2, -B7, -A3, -B40) in vitro. In addition, these four supertypes are bound by shorter versions of this CLIP peptide, even if in silico methods only foretold binding to HLA-A2 or -B7. No CTL responses were induced when these peptides were administered to HLA-A2 transgenic mice. Researchers demonstrated, taken as a whole, that CLIP binds to a wide variety of HLA-I molecules with surprising promiscuity. The discovery of CLIP’s involvement in the HLA-I antigen presentation pathway raises the possibility of a new processing route or immune escape mechanism, or it may indicate an abnormal process in leukemic cells (144).
The antigen delivery processing pathway of HLA-I and HLA-II complexes involves specific proteins. For HLA-II to deliver antigens, the invariant chain (II) peptide CLIP must stabilize HLA-II molecules by transiently and non-specifically adhering to different grooves on HLA-II. Scientists demonstrated an alternate mechanism for CLIP binding to HLA-I molecules on the surface of leukemic cells. Scientists found that AML cells that did not express HLA-II displayed the CLIP peptide on their plasma membrane. It was postulated that CLIP is directly involved in the HLA-I antigen presentation pathway due to the reduced HLA-I cell surface display in AML cells after Ii silencing. The HLA-I-specific peptide eluates from B-LCLs included five peptides derived from Ii, including two within the CLIP region. Experiments involving the binding of eluted CLIP peptide RMATPLLMQALPM to four distinct HLA-I supertypes (-A2, -B7, -A3, -B40) revealed an astonishing result. Shorter CLIP peptides demonstrated binding to all four supertypes, unlike in silico methods that solely anticipated binding to HLA-A2 or -B7. The administration of these peptides to HLA-A2 transgenic mice did not activate CTL responses. The results indicate that CLIP can bind to a diverse range of HLA-I molecules, which is noteworthy. The involvement of CLIP in the HLA-I antigen presentation pathway suggests a potential malfunction in leukemic cells. Still, it may also provide light on new processing routes or immune escape strategies (143, 145).
HLA-II antigen presentation is impacted by the regulation of normal and malignant cell peptide repertoires by the peptide editor HLA-DM and its antagonist HLA-DO. Balancing HLA-DM and HLA-DO means alloreactive T-cells may present leukemia-associated antigens and peptides differently, impacting graft versus host disease and anti-leukemia immunity. Utilizing a substantial quantity of bulk and single-cell RNA sequencing data from many archives, researchers examined the distribution and abundance of HLA-DM and HLA-DO in various human cell types and organs. Researchers attempting to analyze the dual function of HLA-II peptide editing in allogeneic hematopoietic cell transplantation (alloHCT) and their possible influence on its clinical success will benefit from the expression atlas produced (146).
The HLA-I and HLA-II complexes carry antigens through their processing routes using different proteins. By transiently and non-specifically attaching to different HLA-II peptide grooves, the peptide CLIP from the invariant chain (II) stabilizes HLA-II molecules before antigen loading, enabling antigen presentation. CLIP binds differently to HLA-I molecules on leukemic cells, according to researchers. In AML cells lacking HLA-II, the CLIP peptide was found on plasma membranes. The decrease in HLA-I cell surface expression in AML cells after Ii silencing shows that CLIP directly affects the HLA-I antigen presentation pathway. Five peptides from Ii and two from the CLIP area were found in B-LCL HLA-I peptide eluates. Four HLA-I supertypes (-A2, -B7, -A3, -B40) bound well to the eluted CLIP peptide RMATPLLMQALPM in vitro. Even while in silico approaches suggested exclusive binding to HLA-A2 or -B7, shorter CLIP peptides bound to all four supertypes. HLA-A2 transgenic mice were immunized with these peptides but did not produce CTLs. The findings showed CLIP’s versatility in interacting with HLA-I molecules. That CLIP is implicated in the HLA-I antigen presentation pathway implies a disturbance in leukemic cell processes. It may also reveal novel processing channels or immune escape methods (147).
A small number of malignant Hodgkin Reed-Sternberg (HRS) cells are present in primary classical Hodgkin lymphomas (cHL), which are characterized by a diverse infiltrate of immune and inflammatory cells (148). Several methods, primarily resulting from certain genetic defects, allow HRS cells to evade antitumor immunity. These include altered antigen presentation and enhanced PD-1 signaling. Researchers identified β2M mutations in both primary and HRS cell lines that hinder the production of the β2M/MHC-I dual protein complex on the cell surface (149). The expression of PD-1 ligands in HRS cells is influenced by the copy number variations of 9p24.1/CD274(PD-L1)/PDCD1LG2(PD-L2). A more advanced clinical stage and poorer progression-free survival (PFS) are associated with PD-L1/PD-L2 amplification after first-line (induction) therapy. Few details are known about the relationships between PD-L1/PD-L2 amplification, clinical outcome in cHL, and the enhanced expression of β2M, MHC-I, and MHC-II by HRS cells (150). The variables were examined in diagnostic biopsy specimens from 108 patients with cHL who were treated uniformly and followed up for an extended period. Researchers demonstrated that out of these patients, 79% (85/108) had reduced or nonexistent expression of β2m/MHC-I, and 67% (72/108) had reduced or nonexistent expression of MHC-II. Subjects without PD-L1/PD-L2 amplification or advanced stage showed a lower PFS if their β2m/MHC-I was diminished or nonexistent. There was no correlation between the result and reduced or nonexistent MHC-II. These findings provide further evidence that the biological response to conventional chemo and radiation treatment relies on MHC-I-mediated antigen presentation by HRS cells. Speculation about other ways of PD-1 blockage working in cHL is also prompted by the lack of β2m/MHC-I expression on HRS cells (151).
4 Peptide loading complex in breast cancer
As the cause of almost 41,000 deaths and 30% of all new cancer diagnoses in the US, BC continues to pose a danger to women’s health and well-being (152). The mortality rate from BC has decreased by 38% due to improvements in early identification and treatment, yet almost all patients who get metastatic illness will still die from it. These alarming numbers highlight the urgent need for new strategies in BC treatment to lessen the likelihood of recurrence and mortality (153). Evidence has been mounting regarding the immune system’s essential involvement in predicting BC patients’ responses to conventional treatment and their chances of long-term survival in recent years (154).
The sample for meta-analysis research included 3590 patients from eight different studies. Research on conventional HLA-Ia molecules (HLA-ABC) was the only one taken into consideration. Improved disease-free survival (DFS) was correlated with elevated HLA-I protein expression (HR 0.58, 95% CI 0.35-0.95, P = 0.03), especially in patients with triple-negative breast cancer (TNBC) (HR 0.31, 95% CI 0.18-0.52, P < 0.001). However, researchers revealed that there was no correlation with OS. Improvements in DFS, positivity for human epidermal growth factor receptor 2 (HER2), TNBC, high Ki-67 indices, and nuclear grades were all found to be associated with increased expression of HLA-I proteins, according to this meta-analysis. One possible predictive marker for BC is the immunological target HLA-I (155).
NLRC5 plays a pivotal role in cancer immunosurveillance by transactivating MHC-I, which is typically HLA-ABC in humans. However, the expressional and functional abnormalities of NLRC5 are a significant mechanism of immune evasion in many malignancies. In cancer immunotherapy and the fight against cancer immune evasion, NLRC5 promotion in conjunction with MHC-I augmentation is essential (156, 157). Scientists showed that the groups given 50 U/ml and 100 U/ml of IFN-γ had relative levels of NLRC5 mRNA, β2m mRNA, and HLA-ABC α HC mRNA that were dose-dependently higher than the control group. Protein levels of NLRC5 and β2m were noticeably more excellent in the groups treated with 50 U/ml and 100 U/ml IFN-γ, as well as HLA-ABC (positive rates) at different doses of IFN-γ, compared to the control group. Additionally, there was a dose-dependent tendency for both NLRC5 and HLA-ABC. This might be useful in cancer immunotherapy since it prevents cancer cells from escaping immunosurveillance, as NLRC5 is boosted in SKBR3 BC cells by IFN-γ along with overexpression in MHC-I (HLA-ABC) expression (158).
The protooncogene ERBB2 codes for a 185-kDa transmembrane glycoprotein that belongs to the epidermal growth factor receptor (EGFR) family of tyrosine kinases; it is more often known as HER2 or Neu (159). Since 20-30% of BCs have amplified and overexpressed HER2, it is a potential target for immunotherapeutic treatments using T-cell-based methods. Additionally, PD98059, an inhibitor of mitogen-associated protein kinases, elevated MHC-I expression on BC cell lines in a dosage-dependent way. BC cells may express more MHC-I if prescribed a medicine that inhibits the MAPK signaling pathway. A RAS/MAPK pathway-involved mechanism causes MHC-I downregulation in response to HER2-overexpression. Researchers provided further evidence that inhibitors of the RAS/MAPK pathway may enhance the production of MHC-I in BC cells (160, 161).
5 Peptide loading complex in lung cancer
LC is one of the most common cancers in the globe. Based on its shape and histology, LC may be classified into two main categories (1, 162). A little over 15% of newly diagnosed LC cases are small cell lung cancer (SCLC), but the vast majority (about 85%) are NSCLC. SCLC may be either extensive-stage (ES-SCLC) or limited-stage (LS-SCLC). Patients with LS-SCLC, which accounts for about 30% of SCLC cases, often undergo radiation therapy and chemotherapy based on platinum. The standard first treatment for ES-SCLC is chemotherapy alone, which consists of four to six cycles of carboplatin or cisplatin with etoposide. Approximately 70% of all SCLCs are ES-SCLC (163). The majority of NSCLCs were adenocarcinomas, squamous cell carcinomas, and large cell carcinomas (3, 164, 165). LC is caused by several factors, including smoking, radon gas, air pollution, asbestos, and heredity (166). LC is diagnosed using tumor markers such as cytokeratin 19 (Cyfra 21-1), neuron-specific enolase (NSE), and carcinoembryonic antigen (CEA). However, tumor markers are not appropriate for early diagnosis (167).
NSCLC with ALK translocations is currently treated with targeted therapy because of the molecular understanding of LC, which includes mutations in the EGFR TK domain and crizotinib (168). Given the known link between increased tumor- and stromal-infiltrating CTL numbers and better disease-specific survival in LC patients, immunotherapy that stimulates a CTL response against the cancer, such as peptide vaccines, may be a promising treatment approach. Researchers looked at HLA-I and II-presented peptides and gene expression patterns in surgically excised LC tissues to find (new) tumor antigens. Researchers also looked at the possibility that healthy donors can develop a CTL response to HLA ligands derived from these antigens. Researchers identified several potential candidates for tailored CTL recognition among the 170 HLA ligands they have examined. These targets might generate peptides from cyclin D1 or protein kinase, DNA-activated polypeptides, catalytic polypeptides, and lysed tumor cells loaded with peptides into CD8+ T-cells. This is the first ex vivo molecular characterization of HLA-I and II ligands taken from human LC tissues using the efficient identification of both known and new tumor antigens that may induce a CTL response (169).
A crucial gene in antigen-presenting cells, human leukocyte antigen-DP alpha 1 (HLA-DPA1), is involved in immunological modulation. Researchers set out to thoroughly examine the functions of HLA-DPA1 and how it relates to lung adenocarcinoma (LUAD). The effects of HLA-DPA1 expression on LUAD development and immunity were investigated by Ke Shi et al. (170) by a meta-analysis and bioinformatics. To further confirm HLA-DPA1’s roles in LUAD, researchers used the CCK-8, wound healing, and Transwell assays. Overexpression of HLA-DPA1 reduces the proliferation and development of cancer cells, while decreased expression is linked to a poor prognosis and immune infiltration in LUAD. Thus, HLA-DPA1 may be helpful as a treatment target for LUAD and a biomarker for prognosis (170).
It is increasingly evident that most malignancies exhibit deficiencies in the MHC-I antigen processing pathway and low levels of TAP and Tsn. Therefore, solutions to address these deficiencies are needed in immunotherapy efforts that target such tumors using CD8+ CTL. Researchers have discovered an antigen processing route that allows hydrophobic peptides produced cytosolically to be delivered without TAP. Researchers have demonstrated that numerous hydrophobic TAP-independent peptides can also be delivered in a Tsn-independent manner, as shown by the Tsn-negative cell line 721.220. However, the TAP-, Tsn-independent peptides were not seen when these tests were expanded to tumor cell lines generated from SCLC, which researchers demonstrated to be Tsn deficient in addition to TAP-negative. IFN-γ pretreatment of SCLC cells might address this lack of presentation. Alternatively, SCLC cells effectively presented the TAP-, Tsn-independent peptides by guiding them into the ER via an ER signal sequence. These findings led researchers to conclude that the TAP-independent mechanism for presenting hydrophobic peptides produces a low peptide concentration in the ER. This antigenic peptide concentration is insufficient for tumor cells lacking Tsn to bind to MHC-I molecules. Consequently, when creating immunotherapeutic strategies to target SCLC and other malignancies with anomalies in the MHC-I antigen processing pathway, it will be imperative to consider strategies that treat Tsn-defects (171).
Most tumors progressively show signs of abnormalities in the MHC-I antigen processing pathway, including low levels of TAP and Tsn. Therefore, methods to address these flaws are needed in immunotherapy efforts that use CD8+ CTL to target such tumors. Hydrophobic peptides produced from cytosol might be delivered without TAP thanks to a previously discovered antigen processing mechanism. Researchers showed that some of these hydrophobic TAP-independent peptides may also be given in a Tsn-independent fashion using the Tsn-negative cell line 721.220. Nevertheless, the TAP-, Tsn-independent peptides were not provided when these tests were expanded to tumor cell lines generated from SCLC, which researchers demonstrated to be Tsn deficient in addition to TAP-negative. Pre-treating SCLC cells with IFN-γ might address this lack of presentation. Or, the TAP-, Tsn-independent peptides were effectively delivered by SCLC cells using an ER signal sequence that guided them into the ER. Researchers deduced from these findings that the ER produces a low quantity of peptide in the TAP-independent route for the presentation of hydrophobic peptides and that this concentration of antigenic peptide is inadequate for tumor cells that also lack Tsn to load onto MHC-I molecules. Therefore, techniques that treat Tsn-abnormalities will be crucial to take into account for immunotherapeutic efforts to target SCLC and other tumors with defects in the MHC-I antigen processing pathway (129).
The highly aggressive neuroendocrine tumor SCLC exhibits moderate benefits with immune checkpoint blocking (ICB) and early-developed treatment resistance. A significant factor contributing to resistance against T-cell-based immunotherapies is the suppression of MHC-I expression. Using human SCLC cell lines and immunocompetent animal models, researchers assessed Lysine-Specific Demethylase 1 (LSD1)’s impact on MHC-I expression, functional antigen presentation, and immune activation in SCLC in vitro and in vivo. By explicitly blocking LSD1, researchers were able to promote the transcription of genes related to the antigen presentation pathway and reestablish MHC-I cell surface expression in SCLC. Blocking LSD1 makes tumor cells more intrinsically immunogenic, increases interferon signaling, and makes SCLC cells more susceptible to cytolysis by T lymphocytes that are limited to MHC-I. In refractory SCLC models, combining ICB with an LSD1 inhibitor enhances the anti-tumor immune response. The results collectively indicate that LSD1 serves as a significant regulator of MHC-I antigen presentation, supporting the hypothesis that the combination of LSD1 inhibitors and immune checkpoint blockade could enhance therapeutic responses in patients with SCLC. A possible explanation for SCLC’s poor response to ICB might be the epigenetic suppression of MHC-I. Recently, researchers showed that LSD1 regulates MHC-I antigen presentation in SCLC, a largely unknown activity. In SCLC, blocking LSD1 allows for MHC-I-restricted T-cell cytolysis, immunological activation, and an improved anti-tumor immune response to ICB (172).
6 Peptide loading complex in other cancer
Regarding cancer diagnoses worldwide, CRC is the third most common diagnosis, with stomach cancer taking the fifth position. As a result of advancements in early detection techniques and longer lifespans, synchronous and metachronous cancers—whose estimated incidences vary from 2.5% to 3.4%—have become more common. Interestingly, CRC is the most common synchronous and metachronous event in individuals with gastric cancer, with lung and liver cancers coming in second and third, respectively (173).
W Schmidt et al. have devised a method to produce a highly effective immunogenic vaccine: This method, which scientists call “trans-loading,” entails combining MHC-I-positive cancer cells with foreign, nonself peptide ligands. Cancer CT-26 (H2-Kd), melanoma M-3 (H2-Db haplotype), and H2-Kd were given as irradiation vaccines, together with influenza virus peptides that were MHC-matched. Mice treated with this method successfully eradicated a tumor containing live M-3 melanoma cells. The CT-26 colon cancer and the B16-F10 melanoma showed high efficacies against tumor challenge, suggesting that this new vaccination might have broad applicability. This idea might be used to treat human tumors using foreign peptide ligands tailored to a particular MHC-I haplotype (130, 174).
Giving a patient with gastric cancer a composition including activated T-cells—a population of lymphocytes that can identify and specifically target patient cells that produce a peptide aberrantly—is one way to treat the patient. Activated T-cells that can identify patient cells that aberrantly express a peptide are included in a pharmaceutical composition. The composition is intended to treat gastric cancer patients by having the T-cells bind to the peptide in a complex with an MHC-I molecule and a pharmaceutically acceptable carrier. One way to treat a patient with stomach cancer is to give them a composition that has a peptide in a salt that their doctor approves. This will trigger a T-cell response to the gastric cancer (175).
Using a novel monoclonal anti-pan HLA-I HC antibody (EMR 8-5), which interacted with paraffin-fixed sections, researchers examined the predictive significance of HLA-I molecules in patients undergoing radical cystectomy for muscle-invasive bladder cancer. Researchers demonstrated that subjects from 65 patients who had radical cystectomy, pelvic lymph node dissection, and clinically confirmed muscle-invasive bladder cancer were immunohistochemically stained for HLA-I molecules using the mAb EMR 8-5. Neoadjuvant treatment was not administered to these patients. Researchers examined the clinicopathological and predictive importance of HLA-I expression. According to immunohistochemical examination, 22 invasive bladder tumors had HLA-I down-regulation. This downregulation and clinicopathological parameters, including grade, nodal status, or pathologic stage, were not correlated, and compared to patients whose tumors were downregulated, those whose tumors were HLA-I positive had a much better recurrence-free survival rate. After a cystectomy, individuals with bladder cancer who had HLA-I expression had a substantial impact on their chance of not having a recurrence, according to multivariate analysis. Researchers showed that HLA-I was down-regulated in tumor cells in around a third of the patients. People who develop muscle-invasive bladder cancer after a cystectomy may be able to use HLA-I expression as a prediction (176).
A meta-analysis was conducted by Hadis Najafimehr et al. to (177) ascertain the function of classical HLA-I in patient survival prediction. Furthermore, researchers assessed the potential association between HLA-I and several clinicopathological variables. Researchers identified any associations between HLA-I and OS or RFS in gastrointestinal cancer patients by reviewing published research that examined the influence of HLA-I expression on these malignancies. Effect sizes with a 95% confidence interval were hazard ratios (HR) and odds ratios (OR). The analysis included 10 trials with a total of 1,307 patients. Overexpression of HLA-I was favorably associated with OS (HR:0.72; 95% CI:0.53-0.96) and showed little connection with RFS (HR:0.70; 95% CI:0.46-0.08), according to the combined findings. Overexpression of HLA-I is negatively linked to lower tumor differentiation [OR: 0.53; 95% CI (0.43-0.81)] and advanced cancer stages [OR: 0.29; 95% CI (0.13-0.64)]. There was a positive correlation between HLA-I overexpression and OS prognosis, although there was likely little effect on RFS (177) (Table 1).
The prognostic significance of human leukocyte antigen-G (HLA-G) is still up for debate despite reports of abnormal expression in CRC. Accordingly, researchers used the literature and the datasets from The Cancer Genome Atlas (TCGA) to conduct a meta-analysis that will evaluate the predictive usefulness of HLA-G in patients with CRC. Researchers meta-analysis found a strong correlation between LA-G expression and poor OS in CRC. Nevertheless, when comparing researchers’ findings to the TCGA data in CRC, researchers discovered that the prognostic significance did not align. That is why researchers still need further studies to prove that HLA-G has a predictive function in CRC (178).
In order to determine if there is a correlation between HLA-G expression and solid tumor outcomes, Jorge Bartolome et al. conducted a meta-analysis and systematic review. The inclusion criteria were satisfied by 25 research. Data were collected from 4871 patients who reported OS and 961 patients who reported DFS. There was a greater risk of gastric, pancreatic, and CRCs being related to HLA-G expression, which was associated with poorer OS (HR 2.09, 95% CI = 1.67 to 2.63; P <.001). The most used antibody (4H84) and other detection techniques showed no statistically significant changes, according to the researchers. An association between HLA-G expression and DFS was observed. However, it did not reach statistical significance (179).
7 Compare the PLC-based approaches with CAR-T cell therapy in cancer
Recently, chimeric antigen receptor (CAR) T-cell therapy—which the U.S. Food and Drug Administration has authorized—has shown promise in treating mantle cell lymphoma (MCL), DLBCL, and r/r B-ALL. Research on the B-cell maturation antigen (BCMA) in multiple myeloma has shown promising results, with reversible toxic consequences such as pancytopenia and cytokine release syndrome (CRS). Nevertheless, there has been uneven toxicity and effectiveness (180). CAR-T cells must initially travel to tumor locations in order to attach to their target proteins on the tumor surface. This is a basic need for the effective use of T-cell immunotherapy. In contrast to hematological malignancies, the immunosuppressive microenvironment often significantly restricts T-cell trafficking to and infiltration into tumor sites in solid tumors. Additionally, some chemokines released by solid tumors, such as CXCL1, CXCL12, and CXCL5, inhibit T-cells from migrating to and penetrating the tumor lesions, in contrast to hematological malignancies that are simple for CAR-T cells to target and reach. CAR-T cells’ intended immuno-cytotoxicity to kill tumor cells is severely hampered by their inability to traffic and infiltrate into tumor locations due to the absence of matching chemokine receptors present in T-cells. T-cells must thus be altered to express a chemokine receptor that corresponds to the relevant tumor-derived chemokine in order to get beyond this barrier (181).
An overactive immune system and systemic inflammatory response are the results of the vast quantities of cytokines released when CAR-T cells destroy tumor cells. Fever, chills, and headache are signs of mild CRS; hypotension, respiratory distress, and organ insufficiency are indications of severe CRS, which may be fatal. Further complications include organ function degradation due to CAR-T cell persistent cell growth and anemia, thrombocytopenia, and leukopenia caused by CRS, which may increase the risk of infection and spontaneous bleeding. Reduced numbers of lymphocytes and other immune cells may result from cytokine release. The total number of immune cells, including T-cells, B cells, and NK cells, decreases due to lymphodepletion, resulting in cytopenia (182–184).
One way that CAR T-cells avoid some of the problems with the TCR-based method is by targeting cells in a way that is not reliant on MHC and DCs. A CAR differs significantly from a TCR in its ability to attach to a target and activate T-cells. Here are a few examples: the amount of MHC molecules, the existence of co-receptors, the TCR avidity for a particular pMHC, and the moderate TCR affinity for the associated MHC peptide complex, as opposed to the high antibody affinity in a CAR. The TCR’s variability is restricted since it can only identify peptide antigens within the setting of a certain MHC. One advantage of CAR T-cells based on antibodies is their ability to target antigens with varying shapes and compositions, including peptides, carbohydrates, and inorganic substances. Furthermore, TCRs have an innate cross-reactivity with endogenous antigens. As of right now, there are considerably more possible CAR targets than their MHC-presented counterparts that TCR-modified T-cells can identify. Researchers examined the benefits and drawbacks of CAR T-cells’ ability to recognize targets without MHC and highlighted the most critical advancements in cancer treatment early-stage clinical studies (185).
Problems with poor antigen density, tonic signaling, and antigen loss are obstacles to CAR-T cell treatment. Traditional CAR T-cell treatment cannot reach most tumor-specific antigens because they are either secreted or located inside cells. Researchers utilized alpha-fetoprotein (AFP), a specific marker for liver cancer, to test the hypothesis that peptide-MHC complexes could be targets for CAR T-cell therapy against solid tumors. This is because all intracellular and secreted proteins are processed into peptides and presented by MHC-I on the surface of tumor cells (184, 186, 187).
Researchers created ET1402L1-CAR (AFP-CAR), an entirely human chimeric antigen receptor, which has exceptional selectivity and specificity for the AFP158–166 peptides complexed with HLA-A*02:01. T-cells expressing AFP-CAR spared cells from various tissue types that were negative for either expressed protein. Still, they specifically degranulated, produced cytokines, and lysed liver cancer cells that were HLA-A*02:01+/AFP+. In SCID-Beige mice (n = 8 per group), intratumoral injection of AFP-CAR T-cells markedly reduced both Hep G2 and AFP158-expressing SK-HEP-1 tumors. Furthermore, Hep G2 tumor-bearing NSG mice that receive intravenous AFP-CAR T-cell injection have a significant and quick suppression of tumor development (n = 6). Lastly, AFP-CAR T-cells demonstrated strong antitumor efficacy in a well-established intraperitoneal liver cancer xenograft paradigm (n = 6). Researchers showed that a potent antitumor response may be produced by CAR T-cell immunotherapy that targets intracellular/secreted solid tumor antigens. Researchers’ method provides a potential new path for liver cancer immunotherapy and broadens the range of antigens accessible for redirected T-cell treatment against solid tumors (187).
Although mHAg-specific TCRs have been created in genetically engineered T-cells, their functionality may be jeopardized if endogenous and exogenous TCR chains generate mismatched chimeric TCRs. A different strategy is the creation of chimeric antigen receptor (CAR) T-cells with TCR-like specificity, whose intracellular and transmembrane domains send their activation signals instead of competing with endogenous TCRs for CD3 complexes. However, it has been shown that high-affinity CAR-T cells have poor sensitivity and specificity when it comes to recognizing low-density antigens (188, 189). The groove of an HLA molecule is where a minor histocompatibility antigen (mHAg) may be found, which is made up of different genes from the donor and the recipient. The graft-vs.-leukemia effect, which occurs after donor lymphocyte infusion, has demonstrated the therapeutic potential of T-cells specific to mHAgs against recurring hematologic malignancies. Still, this effect only applies to recipient-target hematopoietic cells, such as leukemia cells (190). According to current research, the most crucial goal in adoptive cell therapy is the rapid development of TCR-like CAR-T cells that use an all-in-one chimeric receptor with adjustable intracellular signaling domains. Chimeric antigen receptor-T cells outperform modified TCR gene-introduced T-cells in situations where costimulatory signals from target cells are either not present or are downregulated. This is due to their built-in signaling domain. This alternative is safer and more promising than CAR-T therapies; nevertheless, due to a number of advancements in TCR gene-introduced T-cells (190, 191).
The development of efficient screening systems, including a range of panel peptides, HLA-typed cell lines, and animal models to assess the efficacy and toxicity of TCR-like antibodies, along with meticulous preclinical experiment planning, is essential for the acquisition of TCR-like antibodies that could have therapeutic uses. In contrast to gene-modified T-cells and other passive immunotherapies, current studies are evaluating active immunotherapies such as DC-based peptide or DNA vaccines with or without adjuvants. Since gene-modified cells are subject to stringent controls, these methods are believed to be more practical and cheaper. Nevertheless, due to the lack of publicly available phase I/II clinical data, including a phase I dosage assessment trial for a HA-1 mHAg vaccination, it is premature to compare the two main methods. To determine the best approaches and patient groups for mHAg-targeted immunotherapy, more intervention-specific studies are required (189, 191–193) (Table 2).
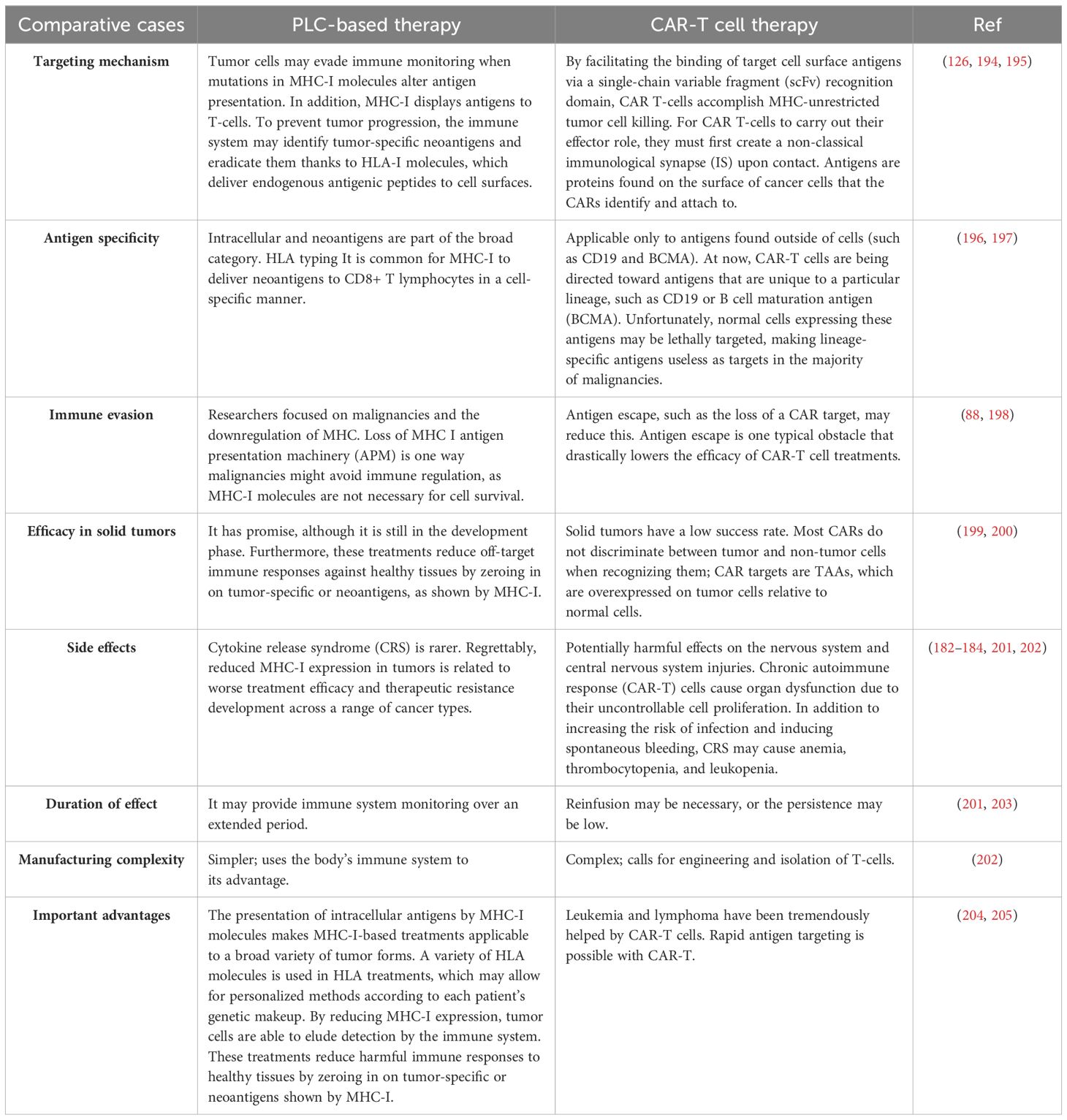
Table 2. Compare the PLC-based approaches with other emerging cancer therapies, including CAR-T cell therapy.
8 Clinical studies related to PLC-based approaches in the treatment of cancers
The ability of DC loaded with wild-type vs modified gp100 peptides with more remarkable binding affinities to elicit an immunological and clinical response in patients with advanced melanoma was examined in this research by Lesterhuis WJ et al. In patients with metastatic HLA-A2.1(+) melanoma, mature DC loaded with keyhole limpet hemocyanin (KLH), tyrosinase peptide, and either wild-type (15 patients) or modified (12 patients) gp100 peptides were administered intravenously (on average 25 × 10(6) DC) and intradermally (on average 11 × 10(6) DC). Every patient who received the vaccination had a strong humoral or proliferative T-cell response against KLH. Tetramer and functional analysis were used to track Gp100-specific T-cell responses in post-treatment delayed-type hypersensitivity (DTH) skin biopsies. Two out of fifteen patients who received the wild-type gp100-loaded DC vaccine had antigen-specific T-cells, compared to one out of twelve who received the modified peptide-loaded DC vaccine. With two patients in each group exhibiting long-term (>8 years) complete responses, these three patients also showed the most significant clinical response. Researchers find that a small percentage of patients with metastatic melanoma may have long-term clinical responses after receiving peptide-loaded DC immunization and that using modified gp100 peptides for DC loading instead of wild-type peptides does not provide a meaningfully stronger immune response (206).
In patients with metastatic melanoma, the DCs vaccination identified hitherto unidentified HLA-I-restricted neoantigens and increased naturally existing neoantigen-specific immunity. Mass spectrometry verified the presence of neoantigens by HLA-A*02:01 in human melanoma. In terms of TCR-β use and clonal composition, vaccination facilitated a varied repertoire of neoantigen-specific TCRs. Researchers’ findings showed that antitumor immunity’s clonal diversity and antigenic breadth are enhanced by vaccination targeting tumor-encoded amino acid changes (ClinicalTrials.gov NCT00683670) (207).
In order to link the findings with transcriptional and genomic studies as well as the clinical response to anti-CTLA-4, anti-PD-1, or combination therapy, researchers looked at the expression of MHC-I and II proteins on tumor cells from melanoma patients who had not received treatment before. Primary resistance to anti-CTLA-4, but not anti-PD-1, treatment was predicted by the majority (>50% of cells) or total loss of melanoma MHC-I membrane expression, which was seen in 78 out of 181 cases (43%). It was linked to transcriptional suppression of HLA-A, HLA-B, HLA-C, and β2M. 55 out of 181 patients (30%) had melanoma MHC-II membrane expression on >1% cells, which was linked to IFN-γ and IFN-γ-mediated gene signatures and indicated a response to anti-PD-1 treatment but not anti-CTLA-4 therapy. Researchers inferred that strong melanoma MHC-I expression is necessary for the primary reaction to anti-CTLA-4. Contrarily, in cases where MHC-I is compromised, the primary response to anti-PD-1 is associated with pre-existing immune activation via IFN-γ, which includes tumor-specific MHC-II expression and components of innate immunity. The fact that antitumor immunity has various requirements for melanoma-specific antigen presentation might be one reason why combination checkpoint blockage is more effective (208).
One of the most challenging side effects of unrelated donor HCT is graft-versus-host disease (GVHD) (209). The highly polymorphic MHC class I chain-related protein A (MICA), encodes a stress-induced glycoprotein that is mainly generated on epithelia. The cytotoxic lymphocyte-expressed invariant activating receptor NKG2D interacts with MICA, which is located in the MHC next to HLA-B. Thus, MICA has all the essential qualities of a valid antigen for transplantation. Using high-resolution sequence-based MICA genotyping, researchers retrospectively studied the clinical effect of MICA mismatches in a multicenter cohort of 922 unrelated donors of HLA-A, HLA-B, HLA-C, HLA-DRB1, and HLA-DQB1 alleles in 10/10 HCT couples. There were 113 mismatched pairs (12.3%) out of 922 in MICA. An increased risk of nonrelapse mortality and acute GVHD grade III–IV were significantly associated with MICA mismatches. A potential graft-versus-leukemia impact was suggested by the fact that the decreased risk of relapse matched the elevated risk for GVHD. In conclusion, choosing a donor who is MICA-matched wherever feasible has a major impact on significant clinical outcomes of HCT when a considerable decrease in GVHD is crucial. Because of the close linkage disequilibrium between MICA and HLA-B, finding a donor who matches MICA is easily achievable in clinical settings (210).
Immune checkpoint blockers (ICBs) cause either fast progression (primary resistance) or long-lasting benefits (secondary resistance) in patients with advanced NSCLC. Following ICB failure, the cancer vaccine OSE2101 may boost antitumor-specific immune responses. Assessing ATALANTE-1’s safety and effectiveness in these patients was its primary goal. The purpose of the two-step open-label ATALANTE-1 research was to compare the safety and efficacy of OSE2101 to standard-of-care (SoC) chemotherapy (CT). Individuals with advanced NSCLC who tested positive for the HLA-A2 and had no discernible changes, failing either sequential or concurrent CT and ICB, were randomized 2:1 to either OSE2101 or SoC (docetaxel or pemetrexed). OS was the primary outcome. An interim OS futility analysis was scheduled in line with Fleming’s design. The decision to immediately stop the accumulation caused by the 2019 coronavirus infection (COVID-19) was decided at the interim review in April 2020. The final analysis included all patients as well as a subgroup with ICB secondary resistance, which was defined as failure after 12 weeks of second-line ICB monotherapy without improvement. When compared to CT, OSE2101 increased survival. It decreased safety for patients with advanced NSCLC who tested positive for HLA-A2 and who had developed resistance to previous treatments. Additional investigation into this group of people is required (211).
Melanoma-associated Antigen A4 (MAGE-A4) is only expressed in immune-privileged tissues; it belongs to the MAGE protein family of cancer/testis antigens (212, 213). Patients with relapsed/refractory metastatic solid tumors expressing MAGE-A4, such as head and neck cancer, ovarian cancer, and synovial sarcoma (SS), participated in a multicenter, dose-escalation, phase 1 study by David S. Hong et al. (NCT03132922) (214). According to exploratory research, afami-cel infiltrates tumors and activates adaptive immune responses via an interferon-γ-driven mode of action. Furthermore, afami-cel has a favorable benefit-risk profile, exhibiting prompt and long-lasting effects, particularly in patients with metastatic SS. MAGE-A4-specific T-cells had a satisfactory safety profile in a phase 1 dose-escalation experiment, including patients with nine distinct solid tumor types. Patients with synovial sarcoma had a promising overall response rate, according to the researchers. The findings merit further testing in larger research, even if the limited trial size restricts the inferences that can be made (214).
Patients who test positive for HLA-A2 or -A24 are the primary focus of peptide-based cancer vaccines currently under development by researchers. A phase I clinical trial of peptide vaccines targeting six distinct HLA-A types in cancer patients was thus undertaken to circumvent this restriction. Patients have to test positive for the HLA-A2, -A24, or -A3 (A3, A11, A31, or A33) supertype and had previously failed traditional cancer treatments to be considered. Patients with HLA-A2(+), HLA-A24(+), and HLA-A3(+) alleles were given a different set of eight candidate peptides for a total of twenty-four peptides prior to vaccination. By taking into account the patients’ HLA types and pre-existing levels of IgGs to the candidate peptides, the vaccine peptides were personalized from the candidate pool. This research included seventeen patients. No patients had serious adverse effects as a result of the peptide vaccines, and they were well-tolerated overall. Eleven out of thirteen instances showed an increase in CTL responses. In contrast, ten out of thirteen cases showed an increase in IgG responses specific to the vaccine peptides. Due to its immunological reactions to the vaccinated peptides and their tolerability, this novel vaccine is suggested for phase II clinical trials (215).
Tumors expressing New York esophageal squamous cell carcinoma-1 (NY-ESO-1) are responsive to TCR-T cell treatment; however, a safe and effective TCR-T cell therapeutic regimen is still needed (212). Results from a phase 1 IND clinical trial, including TCR affinity-enhanced specific T-cell therapy (TAEST160001) for the treatment of NY-ESO-1, were published by Pan et al. After 3 days of dose-reduced lymphodepletion with cyclophosphamide (15 mg/kg/day) and fludarabine (20 mg/m2/day), patients who are enrolled in the study get an infusion of TAEST16001 cells (216). After the adoptive transfer, modest doses of IL-2 injection are used to maintain the TCR-T cells. Researchers demonstrated that there were no significant adverse effects associated with the medication in the 12 individuals who were given the regimen. A total of 41.7% of people have responded. The median response length is 13.1 months, while progression-free survival is 7.2 months on average. Pan et al. proved that TAEST16001 cells were safe, effective, and survived in patients with advanced soft tissue sarcoma who had the HLA-A∗02:01 gene mutation. According to ClinicalTrials.gov (NCT04318964), researchers demonstrated that TAEST16001 cell treatment for advanced soft tissue sarcoma expressing the NY-ESO-1 antigen is well-tolerated and has potential anti-tumor actions (216).
An essential component of fetal-maternal immunological tolerance is the non-classical MHC-I protein known as human leukocyte antigen-G (HLA-G) (217). As a first-of-its-kind immunoglobulin (Ig)G1 bispecific antibody, JNJ-78306358 was created by researchers to treat advanced solid cancers that express HLA-G. By binding to both the α3 domain of HLA-G isoforms on tumor cells and the CD3 receptor complex on T-cells at the same time, JNJ-78306358 facilitates the creation of immunological synapses, which allows cytotoxic T-cells to destroy tumor cells, and it also reduces the immunosuppressive tumor microenvironment (218, 219). This dose-escalation research assessed the pharmacokinetics, pharmacodynamics, and safety of JNJ-78306358 in patients with advanced solid tumors, as well as its preliminary anticancer efficacy. The study included adult patients whose solid tumors had spread or were not amenable to surgery and who also had a high frequency of HLA-G expression. Researchers showed that inducing cytokines and T-cell activation were pharmacodynamic effects of JNJ-78306358. The ability to raise the dosage of JNJ-78306358 to levels where it may be effective was hindered by CRS-related toxicities, such as pneumonitis and elevated transaminases (NCT04991740) (220).
9 Future directions
Peptides are increasingly being developed for use as therapeutics to treat many diseases, including cancer. The benefits of therapeutic peptides include reduced toxicity and target selectivity. A peptide’s ability to inhibit cancer may stem from its direct binding to its target or via its conjugation with a radionuclide or chemotherapeutic treatment to direct the agent’s actions specifically toward cancer cells. Peptides may penetrate the cell membrane directly or be directed to proteins on the cell surface, where peptide-protein interactions can start the internalization of the complex. Peptides can cause cell death by various methods, such as immunological modulation, disruption of cell signaling pathways, cell cycle control, DNA repair pathways, apoptosis, tumor angiogenesis inhibition, membrane breakdown, consequent necrosis, or cell death pathways. While there are numerous benefits to employing peptides as treatments, peptides have the drawback of being readily broken down by proteases after they are provided and sometimes having trouble getting absorbed into the bloodstream, depending on how they are supplied (221). The advances in modification chemistry and analytical technology over the last several decades have significantly advanced the creation of peptide medicines. The newly developed peptide pharmaceuticals have been enhanced through various biochemical techniques in therapeutic, diagnostic, and drug-delivery tactics. Scholars discovered that it supports overcoming the intrinsic constraints of peptides and advancing their applications further. The development of peptide-drug conjugates (PDCs) has laid the groundwork for the new era of tailored peptide pharmaceuticals by using target-oriented peptide moieties as a vehicle for lethal payloads by conjugation with cleavable chemical agents (222, 223).
The development of therapeutic CD8+ T-cell-based cancer vaccines and adoptive T-cell therapies is hindered by the lack of tumor-specific antigens (TSAs) that bind to MHC-I but do not evade T-cell self-tolerance. Most cancers have mutations, and the present approaches primarily focus on producing tolerance-breaking TSAs with a greater affinity for either MHC-I molecules or TCRs by predicting “neoantigens” in silico. These neoantigens are the product of alterations in annotated genes. The approach’s poor success ignited the search for TSAs derived from non-tolerized retroelements, genetic fusion events, or mRNA splice variants. Furthermore, T-cells collaborate with other immune cells to perform immunosurveillance. Lysing MHC-I-deficient cells, NK prevents MHC-I-mediated escape. Although they may not have as much direct effect, other immune cells such as B cells, DCs, macrophages, and CD4+ T-cells may have a significant pro-tumor and antitumor effect. Modulating several populations of innate and adaptive immune cells will be necessary to optimize immunotherapy. Comprehending and regulating therapeutic T-cell responses and immunoevasion need knowledge of immunity down to unique tumor-specific peptides. In cancer, changes to antigen presentation may influence some peptides but not MHC-I levels or the specific allomorph of MHC-I that presents the antigen. This means that the most extreme cases of immunoediting may be all that is now understood about immunoevasion. At the same time, many more nuanced alterations in the synthesis of tumor-specific peptides go undetected. The outcomes of these endeavors will have far-reaching implications for cancer immunotherapy. Still, they will also inform important questions about protein synthesis, degradation, trafficking, and all aspects of MHC-I immunosurveillance, including infections, autoimmune diseases, and tissue transplantation (224).
Important players in the tumor microenvironment include MHC-I and its associated molecules. MHC-I’s traditional antigen presentation roles preserve CD8+ T-cells’ fundamental anticancer immunity, which is vital in fighting against tumor cells. However, recent research has typically shown that MHC-I is cancer’s primary immunological escape mechanism. In the context of cancer, many elements cogently alter MHC-I expression and function. Pathways connected to MHC-I might be a promising target for cancer immunotherapy collaboration. Nonetheless, it is necessary to identify and clarify the key molecules that alter the MHC-I working orientations and their precise processes (126). Limitations mean that not all peptide/MHC complexes associated with a disease state will be found using indirect discovery techniques, and some unrelated peptide/MHC complexes may even be found. For example, an immune-centric approach to indirect discovery identified a peptide/HLA-A*02:01 complex unique to melanoma, which contains the human telomerase peptide hTERT540–548 (ILAKFLHWL). This result has not been replicated using other techniques. Vaccination or in vitro activation of CTLs resulted in CTL clones that specifically recognized hTERT540–548/HLA-A*02:01; nevertheless, results concerning the clones’ ability to lyse hTERT-expressing tumor cell lines are conflicting. Additionally, the hTERT540–548 peptide was not detected by mass spectrometry analysis of peptides that were directly isolated from the HLA-A*02:01 molecules of several hTERT-positive tumor cell lines, and proteasome processing experiments conducted in vitro did not result in the production of the hTERT540–548 peptide (unpublished data). The hTERT540–548/HLA-A2 ligand generated controversy among researchers, who emphasized the need to verify epitopes discovered indirectly (225).
One significant element assisting in tumor immune evasion is the downregulation of MHC-I. However, immune evasion of cancers is a complex process, as shown by the demonstrated resistance of solid tumors to CAR-therapy and by IFN-γ-mediated induction of PD-L. The authors thus postulate that in order to prevent tumor immune evasion, combined treatment that targets several tumor-immunomodulatory pathways at the same time would be required. Numerous immunomodulatory medication combinations, such as CPI in conjunction with STAT3 inhibitors or STING agonists, have encouraging outcomes in (pre-)clinical research (201, 226–228).
Tumors may avoid immune monitoring when they lose the ability to display MHC-I antigens, which is a typical occurrence in cancer. Even while structural genetic changes like β2m deletion cause MHC-I loss that cannot be undone, downregulation of MHC-I after transcription can be undone. Hence, for immunotherapies to work better, new ways to increase tumor-specific MHC-I (tsMHC-I) expression are needed (205). The IFN signaling pathway is one possible method by which tsMHC-I is upregulated. Numerous antigen presentation genes, including MHC-I and TAP, have been shown to express more when IFN-γ is present (205, 229). A transcriptional regulator of MHC-I and associated genes is NLRC5, a member of the NLR protein family (nucleotide-binding domain, leucine-rich repeat). Additionally, NLRC5 nuclear distribution and an intact nuclear localization signal are prerequisites for MHC-I gene activation via NLRC5-mediated mechanisms. Immediate effects on MHC-I expression and MHC-I-mediated antigen presentation are anticipated to result from adjustments to the cellular location of NLRC5. Therefore, NLRC5 could be an excellent target for transplant medicine-related modification of MHC-I antigen presentation (230). In SK-BR-3 BC cells, IFN-γ promotes NLRC5 with overexpression of HLA-ABC, which may help prevent cancer evasion from immunosurveillance and aid in cancer immunotherapy (158). The potentially fatal toxicities experienced by patients make the use of IFN in cancer treatment limited despite the fact that they may improve immunotherapy response. Through antibody-directed, dose-dependent cytokine release at the tumor-specific location, one potential way to use IFNs for therapeutic overexpression of tsMHC-I without the accompanying toxicity is to use this strategy. However, large-scale clinical studies have not yet explored this method (205).
The expression of MHC-I, β2M, and other APM components in cancer has been shown to be influenced by a number of oncogenic pathways, including the c-MYC, n-MYC, HER2, MAPK, and EGFR. It is hypothesized that stimulation of the MAPK pathway reduces IRF1 activity and STAT1 expression, which in turn affects MHC-I expression. It has been shown that the MEK inhibitors trametinib and cobimetinib boost STAT1 phosphorylation and IRF1 expression in human keratinocytes (201). Scientists examined the effects of EGFR tyrosine kinase inhibitors (TKIs) on MHC-I expression in NSCLC cell lines. Treatment with certain EGFR-TKIs increased MHC-I expression on both the mRNA and cell surface protein levels in cells that tested positive for EGFR mutations, including cells with the T790M secondary mutation. Since the phosphatidylinositol 3-kinase (PI3K) inhibitor buparlisib failed to increase MHC-I expression in response to EGFR activation, it seems that the down-regulation of MHC-I expression is mediated via the MEK-ERK pathway. The ERK kinase MEK inhibitor trametinib, however, was successful. Upon immunohistochemical analysis of EGFR-mutated NSCLC samples collected both before and after EGFR-TKI therapy, it was shown that MHC-I, the quantity of infiltrating CD8+ T-cells, phosphorylated EGFR, and ERK were all down-regulated, whilst PD-1 ligand one expression was up-regulated. Immunotherapy is ineffective against NSCLC tumors in part because the MEK-ERK pathway is blocked by EGFR mutational activation, which prevents MHC-I synthesis. Further research is required to fully understand the immune response to EGFR-mutated NSCLC and the signaling cascade including EGFR, MEK, and ERK (231).
According to Yi Bao et al. (232), the expression of ubiquitin-like modifier activating enzyme 1 (UBA1) had the most negative correlation with effector CD8+ T-cell-related characteristics. In ICB cohorts, poor survival and treatment resistance were highly predicted by high UBA1 expression. According to functional tests, UBA1 stimulated tumor development by mediating immune escape. Additionally, Uba1 overexpression or deletion significantly reduced or boosted functional intratumoral CD8+ T-cells, respectively, according to immune profile analysis. Crucially, in some syngeneic animals, TAK-243, a specific UBA1 inhibitor, greatly enhanced ICB’s synergy. Mechanistically, the UBA1-STUB1 axis’s reduction or inactivation increased important immune modulators such as CXCL9, CXCL10, and MHC-I, improved IFN-signaling and stabilized a crucial interferon pathway component (JAK1) (232).
Research into other methods of increasing tsMHC-I expression in an effort to reduce immunotherapy resistance is ongoing. Radiotherapy and chemotherapy, the gold standards of cancer treatment today, are recognized to stimulate immunogenic cell death without inducing anti-tumor immune responses. Researchers have shown that several chemotherapeutic drugs increase tumor-specific CD8+ T-cell infiltration and tsMHC-I expression in vitro studies involving different tumor cell types. Even though it does not cause immunogenic cell death in vitro, the popular chemotherapeutic drug docetaxel may enhance the expression of antigen presentation pathway components like TAP and Tsn (205). When tumor cells were treated with docetaxel, neither ATP nor high-mobility group box 1 (HMGB1) production nor cell death was seen. Nevertheless, after chemotherapy, CRT exposure was seen in every cell line analyzed. Following docetaxel therapy, killing by CEA, MUC-1, or PSA-specific CD8+ CTLs was markedly increased. Functional knockdown of CRT, PERK, or CRT-blocking peptide revealed that this killing was primarily caused by CRT membrane translocation and was linked to increases in antigen-processing machinery components. Continuous treatment with docetaxel resulted in the selection of a docetaxel-resistant cell line (MDR-1+, CD133+). These cells were not immune to the chemomodulatory effects that increased CTL killing, even if they were resistant to the direct cytostatic impacts of docetaxel. Here, scientists provide an operational description of “immunogenic modulation,” which is the process by which tumor cells exposed to nonlethal or sublethal chemotherapeutic dosages change their phenotypic, making the tumor more vulnerable to CTL destruction. Researchers provided a manner by which chemotherapy and immunotherapy may be utilized in conjunction, and they are different from and complementary to immunogenic cell death (233).
Allogeneic CAR-T cells may address autologous T-cells’ low quantity and poor quality. When HLA divergence occurs, the host immune system rejects allogeneic T-cells. Generating negative HLA T-cells through the CRISPR/Cas9 could address this issue. The activation of NK cells is a primary factor contributing to the mortality of CAR-T cells, potentially initiated by negative MHC-I interactions. Consequently, additional analysis is necessary to identify methods to inhibit their ability to eliminate CAR-T cells (234).
The adaptive immune system must be strongly activated for peptide-based cancer vaccines to exert their effector role. Although they are very safe and specific, they have not yet been demonstrated to be a successful cancer therapy in clinical settings. Tumor heterogeneity, self-tolerance, and immune suppression are a few causes of this. The general architecture of peptide-based cancer vaccines has been given significant attention. These vaccines have progressed from basic peptide derivatives of a cancer antigen to sophisticated medications that use conjugates, overlapping regions, and delivery mechanisms to target and activate various APC components and enhance antigen cross-presentation. Peptide-based cancer vaccinations are often used in conjunction with current cancer therapies and are getting more and more tailored to a person’s repertoire of tumor antigens (235–239). Ultimately, this approach helps counteract the drawbacks of a more universal vaccination. It offers a thorough course of therapy that considers the variety of cancer cells and their capacity to evade immune interrogation. Furthermore, as of May 2021, there were about 80 phase I or II clinical studies using a peptide-based vaccination method for cancer; 20 were ongoing at the time of writing, and another 20 had been completed since the beginning of 2019. summarizes the ongoing Phase I and II peptide-based cancer vaccination studies recruiting or actively taking participants. Peptide-based cancer vaccines are high on the list; they protect against blood, brain, lung, and BC, to mention a few. Researchers were demonstrating the variety of targets being tested for peptide-based cancer vaccines. The absence of phase II studies, however, is visible and highlights the present problems with effectiveness that peptide-based cancer vaccines encounter. Nonetheless, there is a positive trend in the pool of ongoing studies towards a more individualized approach to patient neoepitope selection, with a stronger emphasis on using peptide-based cancer vaccines in conjunction with other cancer therapy approaches. Bezu L. et al. have meticulously compiled and examined studies for peptide-based cancer vaccines until 2018 to provide a more thorough study (240–242).
The above information suggests that peptide vaccinations may stimulate robust CD8+ Tcell responses, which can sometimes help patients clinically. Vaccines based on peptides provide many benefits as a cancer immunotherapy approach. The flexible architecture of these vaccines enables the inclusion of multiple peptide epitopes within a single dose. This facilitates the incorporation of numerous MHC-I epitopes that provoke a T-cell response. It is crucial to include peptides that bind to single alleles (like HLA-A2 or HLA-A24) and those that can bind to multiple alleles (like HLA-A2 and HLA-A24) in the same formulation since people have different MHC alleles. This suggests that people with a wide range of MHC allele variations may benefit from a vaccination made from peptides that have been naturally processed. Furthermore, by eliciting a more diffuse and oligoclonal immune response, a vaccination with many epitopes may be able to prevent tumors from developing resistance to antigen downregulation. Although many epitopes from the same antigen may evade HLA restriction (e.g., MAGE-n, survivin, and CEA), the epitopes used in this kind of vaccination must come from different parent proteins. This method will prevent tumor cells from reducing the expression of a specific protein, which will increase the number of clonal T-cells and prevent them from responding to the vaccination. Peptide-based vaccines have the potential to include both B cell and MHC-II-restricted epitopes, which may activate CD4+ T-cells and T helper and antibody-mediated responses, respectively. A more dependable and effective way of eliminating cancers may be possible with a combined complete adaptive immune response. Antigen processing differs substantially among cells even though there are similar reasons why a protein-based vaccine might be attractive (243) (Table 3).
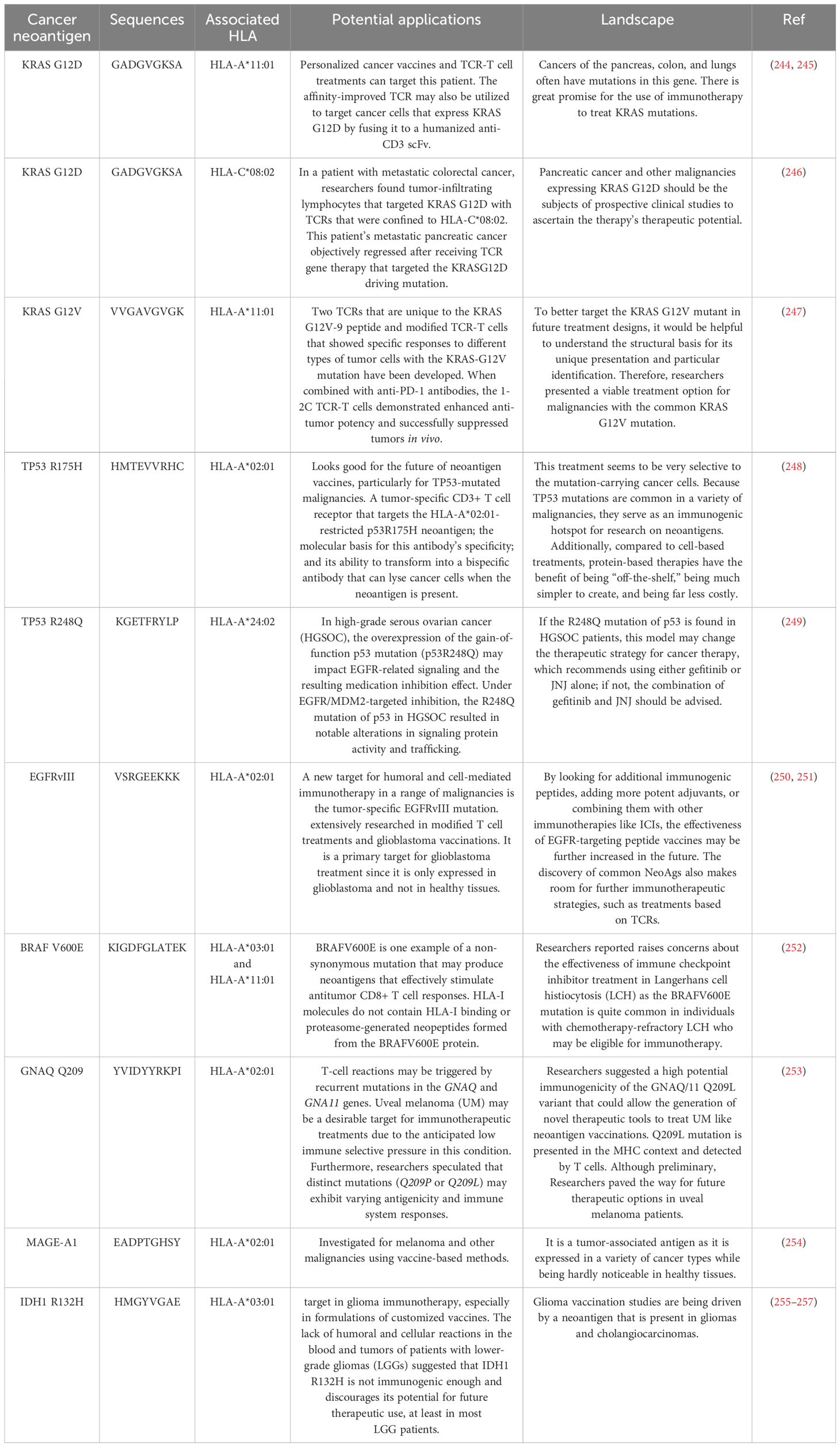
Table 3. A brief overview of some prominent cancer neoantigens together with the HLA alleles, sequences, and possible uses for which they are linked.
10 Conclusion
The production of pMHC-I complexes, which are necessary for identifying cancer cells, is coordinated by the PLC and presented on the cell surface. Cancer cells employ various strategies to evade detection by the immune system, including suppressing MHC-I molecule synthesis and other proteins that play a role in the processing and presentation of antigens. They also employ various strategies to inhibit the expression of TAP, Tsn, CRT, ERp57, MHC HC, or β2m, resulting in the diminished or absent expression of MHC-I on the cell surface. The accuracy and side effects of cancer treatment approaches are reduced, and medicines are advanced due to understanding the impact of PLC components on different forms of cancer. Furthermore, vaccinations based on peptides provide several benefits as a cancer immunotherapy strategy. These vaccines have a versatile architecture that allows them to include many peptide epitopes in a single dosage. This makes adding many MHC-I epitopes that trigger a T-cell response possible. The absence of a comprehensive and coordinated mobilization of the MHC system is a fundamental drawback of existing protein-based standalone vaccination methods, which often results in an inability to produce potent anti-tumor therapeutic effects. The vaccine’s clinical restriction may be addressed by concentrating research on the role of the PLC component in various cancer types. As a result, more thorough research on all cancer types—including blood, lung, prostate, and BC—should be conducted.
Author contributions
ZS: Conceptualization, Formal analysis, Software, Writing – original draft. YT: Data curation, Methodology, Resources, Visualization, Writing – original draft. JY: Data curation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yasamineh S, Nikben N, Hamed Ahmed M, Abdul Kareem R, Kadhim Al-Aridhy A, Hosseini Hooshiar M. Increasing the sensitivity and accuracy of detecting exosomes as biomarkers for cancer monitoring using optical nanobiosensors. Cancer Cell Int. (2024) 24(1):189. doi: 10.1186/s12935-024-03379-1
2. Faghihkhorasani A, Dalvand A, Derafsh E, Tavakoli F, Khairi Younis N, Yasamineh S, et al. The role of oncolytic virotherapy and viral oncogenes in the cancer stem cells: a review of virus in cancer stem cells. Cancer Cell Int. (2023) 23:250. doi: 10.1186/s12935-023-03099-y
3. Sadeghi MS, Lotfi M, Soltani N, Farmani E, Fernandez JHO, Akhlaghitehrani S, et al. Recent advances on high-efficiency of microRNAs in different types of lung cancer: a comprehensive review. Cancer Cell Int. (2023) 23(1):284. doi: 10.1186/s12935-023-03133-z
4. Jabir NR, Tabrez S, Ashraf GM, Shakil S, Damanhouri GA, Kamal MA. Nanotechnology-based approaches in anticancer research. Int J Nanomedicine. (2012) 7:4391–408. doi: 10.2147/IJN.S33838
5. Onoi K, Chihara Y, Uchino J, Shimamoto T, Morimoto Y, Iwasaku M, et al. Immune checkpoint inhibitors for lung cancer treatment: a review. J Clin Med. (2020) 9:1362. doi: 10.3390/jcm9051362
6. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet. (2021) 398:1002–14. doi: 10.1016/S0140-6736(21)01206-X
7. Lopci E. Immunotherapy monitoring with immune checkpoint inhibitors based on [18F] FDG PET/CT in metastatic melanomas and lung cancer. J Clin Med. (2021) 10:5160. doi: 10.3390/jcm10215160
8. Brüstle K, Heidecker B. Checkpoint inhibitor induced cardiotoxicity: Managing the drawbacks of our newest agents against cancer. Oncotarget. (2017) 8:106165. doi: 10.18632/oncotarget.22579
9. Tan S, Li D, Zhu X. Cancer immunotherapy: Pros, cons and beyond. Biomedicine Pharmacotherapy. (2020) 124:109821. doi: 10.1016/j.biopha.2020.109821
11. Saeedi F, Salehi M, Kamali MJ, Mir MA, Kazemi S, Shirafkan F, et al. Evaluation of the cytotoxic activities of the essential oil from Pistacia atlantica Desf. oleoresin on human gastric cancer cell lines. Med Oncol. (2024) 41:1–9. doi: 10.1007/s12032-024-02339-z
12. Klener P, Otahal P, Lateckova L. Immunotherapy approaches in cancer treatment. Curr Pharm Biotechnol. (2015) 16:771–81. doi: 10.2174/1389201016666150619114554
13. Chowell D, Krishna C, Pierini F, Makarov V, Rizvi NA, Kuo F, et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med. (2019) 25:1715–20. doi: 10.1038/s41591-019-0639-4
14. Schaap-Johansen A-L, Vujoviæ M, Borch A, Hadrup SR, Marcatili P, et al. T cell epitope prediction and its application to immunotherapy. Front Immunol. (2021) 12:712488. doi: 10.3389/fimmu.2021.712488
15. Lee MY, Jeon JW, Sievers C, Allen CT. Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer. (2020) 8(2):e001111. doi: 10.1136/jitc-2020-001111
16. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. New Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
17. Salehi M, Kamali MJ, Rajabzadeh A, Minoo S, Mosharafi H, Saeedi F, et al. tRNA-derived fragments: Key determinants of cancer metastasis with emerging therapeutic and diagnostic potentials. Arch Biochem Biophysics. (2024) 753:109930. doi: 10.1016/j.abb.2024.109930
18. Varadé J, Magadán S, González-Fernández Á. Human immunology and immunotherapy: main achievements and challenges. Cell Mol Immunol. (2021) 18:805–28. doi: 10.1038/s41423-020-00530-6
19. Jiang J, Natarajan K, Margulies DH. Chaperone-mediated MHC-I peptide exchange in antigen presentation. IUCrJ. (2024) 11:287–98. doi: 10.1107/S2052252524002768
20. Harig S, Witzens M, Krackhardt AM, Trojan A, Barrett P, Broderick R, et al. Induction of cytotoxic T-cell responses against immunoglobulin V region–derived peptides modified at human leukocyte antigen–A2 binding residues. Blood J Am Soc Hematol. (2001) 98:2999–3005. doi: 10.1182/blood.V98.10.2999
21. Truong HV, Sgourakis NG. Dynamics of MHC-I molecules in the antigen processing and presentation pathway. Curr Opin Immunol. (2021) 70:122–8. doi: 10.1016/j.coi.2021.04.012
22. Lan H, Becker M, Freund C. The mode of action of tapasin on major histocompatibility class I (MHC-I) molecules. J Biol Chem. (2023) 299(4):102987. doi: 10.1016/j.jbc.2023.102987
23. Morozov GI, Zhao H, Mage MG, Boyd LF, Jiang J, Dolan MA, et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc Natl Acad Sci. (2016) 113:E1006–15. doi: 10.1073/pnas.1519894113
24. Sagert L, Winter C, Ruppert I, Zehetmaier M, Thomas C, Tampé R. The ER folding sensor UGGT1 acts on TAPBPR-chaperoned peptide-free MHC I. Elife. (2023) 12:e85432. doi: 10.7554/eLife.85432
25. Ilca FT, Neerincx A, Wills MR, de la Roche M, Boyle LH. Utilizing TAPBPR to promote exogenous peptide loading onto cell surface MHC I molecules. Proc Natl Acad Sci. (2018) 115:E9353–61. doi: 10.1073/pnas.1809465115
26. Altenburg AF, Morley JL, Bauer J, Walz JS, Boyle LH. Reanalysis of Immunopeptidomics Datasets Provides Mechanistic Insight into TAPBPR-Mediated Peptide Editing on HLA-A,-B and-C Molecules. Wellcome Open Res. (2024) 9:113. doi: 10.12688/wellcomeopenres.20738.1
27. Margulies DH, Jiang J, Ahmad J, Boyd LF, Natarajan K. Chaperone function in antigen presentation by MHC class I molecules—tapasin in the PLC and TAPBPR beyond. Front Immunol. (2023) 14:1179846. doi: 10.3389/fimmu.2023.1179846
28. Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, et al. Structure of the human MHC-I peptide-loading complex. Nature. (2017) 551:525–8. doi: 10.1038/nature24627
29. Brunnberg J, Barends M, Frühschulz S, Winter C, Battin C, de Wet B, et al. Dual role of the peptide-loading complex as proofreader and limiter of MHC-I presentation. Proc Natl Acad Sci. (2024) 121:e2321600121. doi: 10.1073/pnas.2321600121
30. Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. (2003) 110:163–9. doi: 10.1046/j.1365-2567.2003.01738.x
31. Calligaris M, Spanò DP, Bonelli S, Müller SA, Carcione C, D’apolito D, et al. iRhom2 regulates ectodomain shedding and surface expression of the major histocompatibility complex (MHC) class I. Cell Mol Life Sci. (2024) 81:163. doi: 10.1007/s00018-024-05201-7
32. Barends M, Koller N, Schölz C, Durán V, Bošnjak B, Becker J, et al. Dynamic interactome of the MHC I peptide loading complex in human dendritic cells. Proc Natl Acad Sci. (2023) 120:e2219790120. doi: 10.1073/pnas.2219790120
33. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
34. Trowitzsch S, Tampé R. Multifunctional chaperone and quality control complexes in adaptive immunity. Annu Rev biophysics. (2020) 49:135–61. doi: 10.1146/annurev-biophys-121219-081643
35. Thomas C, Tampé R. MHC I assembly and peptide editing—chaperones, clients, and molecular plasticity in immunity. Curr Opin Immunol. (2021) 70:48–56. doi: 10.1016/j.coi.2021.02.004
36. Cresswell P, Bangia N, Dick T, Diedrich G. The nature of the MHC class I peptide loading complex. Immunol Rev. (1999) 172:21–8. doi: 10.1111/j.1600-065X.1999.tb01353.x
37. Šimurina M, de Haan N, Vuèkoviæ F, Kennedy NA, Štambuk J, Falck D, et al. Glycosylation of immunoglobulin G associates with clinical features of inflammatory bowel diseases. Gastroenterology. (2018) 154:1320–1333.e10. doi: 10.1053/j.gastro.2018.01.002
38. Halenius A, Gerke C, Hengel H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver? Cell Mol Immunol. (2015) 12:139–53. doi: 10.1038/cmi.2014.105
39. Elliott T, Williams A. The optimization of peptide cargo bound to MHC class I molecules by the peptide-loading complex. Immunol Rev. (2005) 207:89–99. doi: 10.1111/j.0105-2896.2005.00311.x
40. Saini SK, Tamhane T, Anjanappa R, Saikia A, Ramskov S, Donia M, et al. Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci Immunol. (2019) 4:eaau9039. doi: 10.1126/sciimmunol.aau9039
41. Thomas C, Tampé R. MHC I chaperone complexes shaping immunity. Curr Opin Immunol. (2019) 58:9–15. doi: 10.1016/j.coi.2019.01.001
42. Lankat-Buttgereit B, Tampe R. The transporter associated with antigen processing: function and implications in human diseases. Physiol Rev. (2002) 82:187–204. doi: 10.1152/physrev.00025.2001
43. Eggensperger S, Tampé R. The transporter associated with antigen processing: a key player in adaptive immunity. Biol Chem. (2015) 396:1059–72. doi: 10.1515/hsz-2014-0320
44. Rudolph MG, Stevens J, Speir JA, Trowsdale J, Butcher GW, Joly E, et al. Crystal structures of two rat MHC class Ia (RT1-a) molecules that are associated differentially with peptide transporter alleles TAP-A and TAP-B. J Mol Biol. (2002) 324:975–90. doi: 10.1016/S0022-2836(02)01095-1
45. Ritz U, Seliger B. The transporter associated with antigen processing (TAP): structural integrity, expression, function, and its clinical relevance. Mol Med. (2001) 7:149–58. doi: 10.1007/BF03401948
46. Vigneron N, Van den Eynde BJ. Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules. (2014) 4:994–1025. doi: 10.3390/biom4040994
47. Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci U.S.A. (2005) 102(47):17107–12. doi: 10.1073/pnas.0500721102
48. Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. (2002) 419:480–3. doi: 10.1038/nature01074
49. Nguyen TT, Chang S-C, Evnouchidou I, York IA, Zikos C, Rock KL, et al. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol. (2011) 18:604–13. doi: 10.1038/nsmb.2021
50. Kochan G, Krojer T, Harvey D, Fischer R, Chen L, Vollmar M, et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc Natl Acad Sci. (2011) 108:7745–50. doi: 10.1073/pnas.1101262108
51. Domnick A, Winter C, Sušac L, Hennecke L, Hensen M, Zitzmann N, et al. Molecular basis of MHC I quality control in the peptide loading complex. Nat Commun. (2022) 13:4701. doi: 10.1038/s41467-022-32384-z
52. Fleischmann G, Fisette O, Thomas C, Wieneke R, Tumulka F, Schneeweiss C, et al. Mechanistic basis for epitope proofreading in the peptide-loading complex. J Immunol. (2015) 195:4503–13. doi: 10.4049/jimmunol.1501515
53. Fisette O, Schröder GF, Schäfer LV. Atomistic structure and dynamics of the human MHC-I peptide-loading complex. Proc Natl Acad Sci. (2020) 117:20597–606. doi: 10.1073/pnas.2004445117
54. Olson E, Raghavan M. Major histocompatibility complex class I assembly within endolysosomal pathways. Curr Opin Immunol. (2023) 84:102356. doi: 10.1016/j.coi.2023.102356
55. Schölz C, Tampé R. The intracellular antigen transport machinery TAP in adaptive immunity and virus escape mechanisms. J Bioenergetics Biomembranes. (2005) 37:509–15. doi: 10.1007/s10863-005-9500-1
56. Stepensky D, Bangia N, Cresswell P. Aggregate formation by ERp57-deficient MHC class I peptide-loading complexes. Traffic. (2007) 8:1530–42. doi: 10.1111/j.1600-0854.2007.00639.x
57. Coe H, Michalak M. ERp57, a multifunctional endoplasmic reticulum resident oxidoreductase. Int J Biochem Cell Biol. (2010) 42:796–9. doi: 10.1016/j.biocel.2010.01.009
58. Yi MC, Melkonian AV, Ousey JA, Khosla C. Endoplasmic reticulum–resident protein 57 (ERp57) oxidatively inactivates human transglutaminase 2. J Biol Chem. (2018) 293(8):2640–9. doi: 10.1074/jbc.RA117.001382
59. Schulman S, Bendapudi P, Sharda A, Chen V, Bellido-Martin L, Jasuja R, et al. Extracellular thiol isomerases and their role in thrombus formation. Antioxidants Redox Signaling. (2016) 24:1–15. doi: 10.1089/ars.2015.6530
60. Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. (2002) 193:154–63. doi: 10.1002/jcp.v193:2
61. Garbi N, Hämmerling G, Tanaka S. Interaction of ERp57 and tapasin in the generation of MHC class I–peptide complexes. Curr Opin Immunol. (2007) 19:99–105. doi: 10.1016/j.coi.2006.11.013
62. Song D, et al. Insights into the role of ERp57 in cancer. J Cancer. (2021) 12:2456–64. doi: 10.7150/jca.48707
63. Li S, Zhao X, Chang S, Li Y, Guo M, Guan Y. ERp57−small interfering RNA silencing can enhance the sensitivity of drug−resistant human ovarian cancer cells to paclitaxel. Int J Oncol. (2019) 54(1):249–60. doi: 10.3892/ijo.2018.4628
64. Rahbar F, Fagheh AF, Khosravi A, Shariatzadeh S, Akhavan-Sigari R. A review of the role of ERp57 in cancerous and non-cancerous cell physiology and its potential as a therapeutic target. Mol Cell Biomechanics. (2021) 18:187. doi: 10.32604/mcb.2021.017750
65. Garbi N, Tanaka S, Momburg F, Hämmerling GJ. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. (2006) 7:93–102. doi: 10.1038/ni1288
66. Hulpke S, Tomioka M, Kremmer E, Ueda K, Abele R, Tampé R. Direct evidence that the N-terminal extensions of the TAP complex act as autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex. Cell Mol Life Sci. (2012) 69:3317–27. doi: 10.1007/s00018-012-1005-6
67. Purcell AW, Elliott T. Molecular maChinations of the MHC-I peptide loading complex. Curr Opin Immunol. (2008) 20:75–81. doi: 10.1016/j.coi.2007.12.005
68. Purcell AW, Elliott T. Molecular machinations of the MHC-I peptide loading complex. Cancer Cell. (2017) 32:639–53.e6. doi: 10.1016/j.ccell.2017.10.001
69. Gill DJ, Tham KM, Chia J, Wang SC, Steentoft C, Clausen H, et al. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci. (2013) 110:E3152–61. doi: 10.1073/pnas.1305269110
70. Chia J, Tham KM, Gill DJ, Bard-Chapeau EA, Bard FA. ERK8 is a negative regulator of O-GalNAc glycosylation and cell migration. elife. (2014) 3:e01828. doi: 10.7554/eLife.01828
71. Ros M, Nguyen AT, Chia J, Le Tran S, Le Guezennec X, McDowall R, et al. ER-resident oxidoreductases are glycosylated and trafficked to the cell surface to promote matrix degradation by tumour cells. Nat Cell Biol. (2020) 22:1371–81. doi: 10.1038/s41556-020-00590-w
72. Shi W, Han H, Zou J, Zhang Y, Li H, Zhou H, et al. Identification of dihydrotanshinone I as an ERp57 inhibitor with anti-breast cancer properties via the UPR pathway. Biochem Pharmacol. (2021) 190:114637. doi: 10.1016/j.bcp.2021.114637
73. Chignard N, Shang S, Wang H, Marrero J, Bréchot C, Hanash S, et al. Cleavage of endoplasmic reticulum proteins in hepatocellular carcinoma: Detection of generated fragments in patient sera. Gastroenterology. (2006) 130:2010–22. doi: 10.1053/j.gastro.2006.02.058
74. Yang Z, Liu J, Shi Q, Chao Y, Di Y, Sun J, et al. Expression of protein disulfide isomerase A3 precursor in colorectal cancer. OncoTargets Ther. (2018) p:4159–66. doi: 10.2147/OTT.S154452
75. Ramos F, Serino L, Carvalho C, Lima R, Urban C, Cavalli I, et al. PDIA3 and PDIA6 gene expression as an aggressiveness marker in primary ductal breast cancer. Genet Mol Res. (2015) 14:6960–7. doi: 10.4238/2015.June.26.4
76. Chay D, Cho H, Lim BJ, Kang ES, Oh YJ, Choi SM, et al. ER-60 (PDIA3) is highly expressed in a newly established serous ovarian cancer cell line, YDOV-139. Int J Oncol. (2010) 37(2):399–412. doi: 10.3892/ijo_00000688
77. Liao CJ, Wu TI, Huang YH, Chang TC, Wang CS, Tsai MM, et al. Glucose-regulated protein 58 modulates cell invasiveness and serves as a prognostic marker for cervical cancer. Cancer Sci. (2011) 102:2255–63. doi: 10.1111/j.1349-7006.2011.02102.x
78. Pressinotti NC, Klocker H, Schäfer G, Luu V-D, Ruschhaupt M, Kuner R, et al. Differential expression of apoptotic genes PDIA3 and MAP3K5 distinguishes between low-and high-risk prostate cancer. Mol Cancer. (2009) 8:1–12. doi: 10.1186/1476-4598-8-130
79. Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. (2007) 26:1681–90. doi: 10.1038/sj.emboj.7601624
80. Kutsuno Y, Hirashima R, Sakamoto M, Ushikubo H, Michimae H, Itoh T, et al. Expression of UDP-glucuronosyltransferase 1 (UGT1) and glucuronidation activity toward endogenous substances in humanized UGT1 mouse brain. Drug Metab Dispos. (2015) 43:1071–6. doi: 10.1124/dmd.115.063719
81. Neerincx A, Boyle LH. Properties of the tapasin homologue TAPBPR. Curr Opin Immunol. (2017) 46:97–102. doi: 10.1016/j.coi.2017.04.008
82. Lou Y, Vitalis TZ, Basha G, Cai B, Chen SS, Choi KB, et al. Restoration of the expression of transporters associated with antigen processing in lung carcinoma increases tumor-specific immune responses and survival. Cancer Res. (2005) 65:7926–33. doi: 10.1158/0008-5472.CAN-04-3977
83. Lou Y, Basha G, Seipp RP, Cai B, Chen SS, Moise AR, et al. Combining the antigen processing components TAP and Tapasin elicits enhanced tumor-free survival. Clin Cancer Res. (2008) 14:1494–501. doi: 10.1158/1078-0432.CCR-07-1066
84. Sokol L, Koelzer VH, Rau TT, Karamitopoulou E, Zlobec I, Lugli A. Loss of tapasin correlates with diminished CD8+ T-cell immunity and prognosis in colorectal cancer. J Trans Med. (2015) 13:1–6. doi: 10.1186/s12967-015-0647-1
85. Shionoya Y, Kanaseki T, Miyamoto S, Tokita S, Hongo A, Kikuchi Y, et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology. (2017) 6:e1274476. doi: 10.1080/2162402X.2016.1274476
86. Jiang Q, Pan H-Y, Ye D-X, Zhang P, Zhong L-P, Zhang Z-Y. Downregulation of tapasin expression in primary human oral squamous cell carcinoma: association with clinical outcome. Tumor Biol. (2010) 31:451–9. doi: 10.1007/s13277-010-0054-4
87. Jiang J, Taylor DK, Kim EJ, Boyd LF, Ahmad J, Mage MG, et al. Structural mechanism of tapasin-mediated MHC-I peptide loading in antigen presentation. Nat Commun. (2022) 13:5470. doi: 10.1038/s41467-022-33153-8
88. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
89. Corradi V, Singh G, Tieleman DP. The human transporter associated with antigen processing: molecular models to describe peptide binding competent states. J Biol Chem. (2012) 287:28099–111. doi: 10.1074/jbc.M112.381251
90. Koch J, Guntrum R, Heintke S, Kyritsis C, Tampé R. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J Biol Chem. (2004) 279:10142–7. doi: 10.1074/jbc.M312816200
91. Uebel S, Kraas W, Kienle S, Wiesmüller K-H, Jung G, Tampé R. Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc Natl Acad Sci. (1997) 94:8976–81. doi: 10.1073/pnas.94.17.8976
92. Brunnberg J, Herbring V, Günther Castillo E, Krüger H, Wieneke R, Tampé R. Light control of the peptide-loading complex synchronizes antigen translocation and MHC I trafficking. Commun Biol. (2021) 4:430. doi: 10.1038/s42003-021-01890-z
93. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. (2007) 7:543–55. doi: 10.1038/nri2103
94. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
95. Lin M, Xu H, Shi Y, Da L-T. Antigenic peptide loading into major histocompatibility complex class I is driven by the substrate N-terminus. CCS Chem. (2022) 4:910–25. doi: 10.31635/ccschem.021.202000657
96. Xu H, Song K, Da L-T. Dynamics of peptide loading into major histocompatibility complex class I molecules chaperoned by TAPBPR. Phys Chem Chem Phys. (2022) 24:12397–409. doi: 10.1039/D2CP00423B
97. Giza M, Koftori D, Chen L, Bowness P. Is Behçet’s disease a ‘class 1-opathy’? The role of HLA-B* 51 in the pathogenesis of Behçet’s disease. Clin Exp Immunol. (2018) 191:11–8. doi: 10.1111/cei.13049
98. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet’s disease and epistasis between HLA-B* 51 and ERAP1. Nat Genet. (2013) 45:202–7. doi: 10.1038/ng.2520
99. Cortes A, Pulit SL, Leo PJ, Pointon JJ, Robinson PC, Weisman MH, et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun. (2015) 6:7146. doi: 10.1038/ncomms8146
100. Kuiper JJW, Setten JV, Devall M, Cretu-Stancu M, Hiddingh S, Ophoff RA, et al. Functionally distinct ERAP1 and ERAP2 are a hallmark of HLA-A29-(Birdshot) Uveitis. Hum Mol Genet. (2018) 27:4333–43. doi: 10.1093/hmg/ddy319
101. Jongsma M, Neefjes J, Spaapen R. Playing hide and seek: Tumor cells in control of MHC class I antigen presentation. Mol Immunol. (2021) 136:36–44. doi: 10.1016/j.molimm.2021.05.009
102. Wijdeven RH, Luk SJ, Schoufour TA, van der Zanden SY, Cabezuelo M, Heemskerk MH, et al. Balanced epigenetic regulation of MHC class I expression in tumor cells by the histone ubiquitin modifiers BAP1 and PCGF1. J Immunol. (2024) 212:446–54. doi: 10.4049/jimmunol.2300263
103. Jah N, Jobart‐Malfait A, Ermoza K, Noteuil A, Chiocchia G, Breban M, et al. HLA–B27 subtypes predisposing to ankylosing spondylitis accumulate in an endoplasmic reticulum–derived compartment apart from the peptide-loading complex. Arthritis Rheumatol. (2020) 72:1534–46. doi: 10.1002/art.41281
104. Tirado-Herranz A, Guasp P, Pastor-Moreno A, Area-Navarro M, Alvarez I. Analysis of the different subpeptidomes presented by the HLA class I molecules of the B7 supertype. Cell Immunol. (2023) 387:104707. doi: 10.1016/j.cellimm.2023.104707
105. Fainardi E, Castellazzi M, Stignani M, Morandi F, Sana G, Gonzalez R, et al. Emerging topics and new perspectives on HLA-G. Cell Mol Life Sci. (2011) 68:433–51. doi: 10.1007/s00018-010-0584-3
106. Morandi F, Pistoia V. Interactions between HLA-G and HLA-E in physiological and pathological conditions. Front Immunol. (2014) 5:394. doi: 10.3389/fimmu.2014.00394
107. Shahed Shoarishoar S, KaboodMehri R, Fakor F, Sorouri ZR, Mansour-Ghanaei M, Darkhaneh RF, et al. Assessment of decreased ovarian reserve and systemic inflammatory markers. Cell Mol Biol. (2024) 70(11):144–9. doi: 10.14715/cmb/2024.70.11.21
108. Stubbendorff M, Deuse T, Hua X, Phan TT, Bieback K, Atkinson K, et al. Immunological properties of extraembryonic human mesenchymal stromal cells derived from gestational tissue. Stem Cells Dev. (2013) 22:2619–29. doi: 10.1089/scd.2013.0043
109. Gao D, Jiang L. Exosomes in cancer therapy: a novel experimental strategy. Am J Cancer Res. (2018) 8:2165–75.
110. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell derived exosomes. Nat Med. (1998) 4:594–600. doi: 10.1038/nm0598-594
111. Vincent-Schneider H, Stumptner‐Cuvelette P, Lankar D, Pain S, Raposo G, Benaroch P, et al. Exosomes bearing HLA-DR1 molecules need dendritic cells to efficiently stimulate specific T cells. Int Immunol. (2002) 14:713–22. doi: 10.1093/intimm/dxf048
112. Wakim LM, Bevan MJ. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature. (2011) 471:629–32. doi: 10.1038/nature09863
113. Hsu D-H, Paz P, Villaflor G, Rivas A, Mehta-Damani A, Angevin E, et al. Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J Immunotherapy. (2003) 26:440–50. doi: 10.1097/00002371-200309000-00007
114. Salehi M, Kamali MJ, Arab D, Safaeian N, Ashuori Z, Maddahi M, et al. Exosomal microRNAs in regulation of tumor cells resistance to apoptosis. Biochem Biophysics Rep. (2024) 37:101644. doi: 10.1016/j.bbrep.2024.101644
115. Herget M, Tampé R. Intracellular peptide transporters in human–compartmentalization of the “peptidome. Pflügers Archiv-European J Physiol. (2007) 453:591–600. doi: 10.1007/s00424-006-0083-4
116. Darabi A, Thuring C, Paulsson KM. HLA-I antigen presentation and tapasin influence immune responses against Malignant brain tumors–considerations for successful immunotherapy. Anti-Cancer Agents Medicinal Chem (Formerly Curr Medicinal Chemistry-Anti-Cancer Agents). (2014) 14:1094–100. doi: 10.2174/1871520614666140825110001
117. Sun Y, Young MC, Woodward CH, Danon JN, Truong HV, Gupta S, et al. Universal open MHC-I molecules for rapid peptide loading and enhanced complex stability across HLA allotypes. Proc Natl Acad Sci U.S.A. (2023) 120:e2304055120. doi: 10.1073/pnas.2304055120
118. Tafuro S, Meier UC, Dunbar PR, Jones EY, Layton GT, Hunter MG, et al. Reconstitution of antigen presentation in HLA class I-negative cancer cells with peptide-β2m fusion molecules. Eur J Immunol. (2001) 31:440–9. doi: 10.1002/1521-4141(200102)31:2<440::AID-IMMU440>3.0.CO;2-0
119. Zhao Q. Identification and characterization of tumor-specific antigens for ovarian cancer immunotherapy. (2023).
120. Weidanz JA, Nguyen T, Woodburn T, Neethling FA, Chiriva-Internati M, Hildebrand WH, et al. Levels of specific peptide-HLA class I complex predicts tumor cell susceptibility to CTL killing. J Immunol. (2006) 177:5088–97. doi: 10.4049/jimmunol.177.8.5088
121. Stuber G, Leder GH, Storkus WJ, Lotze MT, Modrow S, Székely L, et al. Identification of wild-type and mutant p53 peptides binding to HLA-A2 assessed by a peptide loading-deficient cell line assay and a novel major histocompatibility complex class I peptide binding assay. Eur J Immunol. (1994) 24:765–8. doi: 10.1002/eji.1830240341
122. Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol. (2023) 23:90–105. doi: 10.1038/s41577-022-00732-1
123. Eugène J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Modern Pathol. (2020) 33(3):468–82. doi: 10.1038/s41379-019-0322-9
124. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
125. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
126. Wu X, Li T, Jiang R, Yang X, Guo H, Yang R. Targeting MHC-I molecules for cancer: function, mechanism, and therapeutic prospects. Mol Cancer. (2023) 22:194. doi: 10.1186/s12943-023-01899-4
127. Hussmann M, Janke K, Kranz P, Neumann F, Mersch E, Baumann M, et al. Depletion of the thiol oxidoreductase ERp57 in tumor cells inhibits proliferation and increases sensitivity to ionizing radiation and chemotherapeutics. Oncotarget. (2015) 6:39247. doi: 10.18632/oncotarget.v6i36
128. Santana-Codina N, Carretero R, Sanz-Pamplona R, Cabrera T, Guney E, Oliva B, et al. A transcriptome-proteome integrated network identifies endoplasmic reticulum thiol oxidoreductase (ERp57) as a hub that mediates bone metastasis. Mol Cell Proteomics. (2013) 12:2111–25. doi: 10.1074/mcp.M112.022772
129. Aladin F, Lautscham G, Humphries E, Coulson J, Blake N. Targeting tumour cells with defects in the MHC Class I antigen processing pathway with CD8+ T cells specific for hydrophobic TAP-and Tapasin-independent peptides: the requirement for directed access into the ER. Cancer Immunology Immunotherapy. (2007) 56:1143–52. doi: 10.1007/s00262-006-0263-2
130. Schmidt W, Steinlein P, Buschle M, Schweighoffer T, Herbst E, Mechtler K, et al. Transloading of tumor cells with foreign major histocompatibility complex class I peptide ligand: a novel general strategy for the generation of potent cancer vaccines. Proc Natl Acad Sci. (1996) 93:9759–63. doi: 10.1073/pnas.93.18.9759
131. Bekker GJ, Numoto N, Kawasaki M, Hayashi T, Yabuno S, Kozono Y, et al. Elucidation of binding mechanism, affinity, and complex structure between mWT1 tumor-associated antigen peptide and HLA-A* 24: 02. Protein Sci. (2023) 32:e4775. doi: 10.1002/pro.v32.10
132. Le Luduec J-B, Boudreau JE, Freiberg JC, Hsu KC. Novel approach to cell surface discrimination between KIR2DL1 subtypes and KIR2DS1 identifies hierarchies in NK repertoire, education, and tolerance. Front Immunol. (2019) 10:734. doi: 10.3389/fimmu.2019.00734
133. Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood J Am Soc Hematol. (2010) 116:3853–64. doi: 10.1182/blood-2010-04-281675
134. Shreeve N, Depierreux D, Hawkes D, Traherne JA, Sovio U, Huhn O, et al. The CD94/NKG2A inhibitory receptor educates uterine NK cells to optimize pregnancy outcomes in humans and mice. Immunity. (2021) 54:1231–1244.e4. doi: 10.1016/j.immuni.2021.03.021
135. Wearsch PA, Peaper DR, Cresswell P. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc Natl Acad Sci U.S.A. (2011) 108(12):4950–5. doi: 10.1073/pnas.1102524108
136. Arshad N, Cresswell P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J Biol Chem. (2018) 293:9555–69. doi: 10.1074/jbc.RA118.002836
137. Isidori A, Cerchione C, Daver N, DiNardo C, Garcia-Manero G, Konopleva M, et al. Immunotherapy in acute myeloid leukemia: where we stand. Front Oncol. (2021) 11:656218. doi: 10.3389/fonc.2021.656218
138. Li S, Mason CE, Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev. (2016) 36:100–6. doi: 10.1016/j.gde.2016.03.011
139. Bodini M, Ronchini C, Giacò L, Russo A, Melloni GE, Luzi L, et al. The hidden genomic landscape of acute myeloid leukemia: subclonal structure revealed by undetected mutations. Blood J Am Soc Hematol. (2015) 125:600–5. doi: 10.1182/blood-2014-05-576157
140. Khaldoyanidi S, et al. Immune biology of acute myeloid leukemia: implications for immunotherapy. J Clin Oncol. (2021) 39:419–32. doi: 10.1200/JCO.20.00475
141. Ehx G, Larouche J-D, Durette C, Laverdure J-P, Hesnard L, Vincent K, et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity. (2021) 54:737–752.e10. doi: 10.1016/j.immuni.2021.03.001
142. van Luijn MM, Chamuleau ME, Ressing ME, Wiertz EJ, Ostrand-Rosenberg S, Souwer Y, et al. Alternative Ii-independent antigen-processing pathway in leukemic blasts involves TAP-dependent peptide loading of HLA class II complexes. Cancer Immunology Immunotherapy. (2010) 59:1825–38. doi: 10.1007/s00262-010-0908-z
143. van Luijn MM, van de Loosdrecht AA, Lampen MH, van Veelen PA, Zevenbergen A, Kester MG, et al. Promiscuous binding of invariant chain-derived CLIP peptide to distinct HLA-I molecules revealed in leukemic cells. PloS One. (2012) 7:e34649. doi: 10.1371/journal.pone.0034649
144. Ehx G, et al. The tumor-specific antigen landscape of acute myeloid leukemia. Mol Immunol. (2022) 150:8. doi: 10.1016/j.molimm.2022.05.035
145. Shoarishoar SS, Milani F, Adineh S, Sorouri ZR, Attari SM. Comparison of pregnancy outcomes in amniocentesis recipients with normal and abnormal maternal serum analytes. Cell Mol Biol. (2024) 70(11):109–14.
146. Budeus B, Álvaro-Benito M, Crivello P. HLA-DM and HLA-DO interplay for the peptide editing of HLA class II in healthy tissues and leukemia. Best Pract Res Clin Haematology. (2024) 37(2):101561. doi: 10.1016/j.beha.2024.101561
147. Nikopaschou M, et al. ERAP1 activity modulates the immunopeptidome but also affects the proteome, metabolism and stress responses in cancer cells. bioRxiv. (2024). doi: 10.1101/2024.09.20.613815
148. Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. (2009) 9(1):15–27. doi: 10.1038/nrc2542
149. Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood J Am Soc Hematol. (2015) 125:1061–72. doi: 10.1182/blood-2014-11-610436
150. Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. (2016) 34:2690–7. doi: 10.1200/JCO.2016.66.4482
151. Roemer MG, Advani RH, Redd RA, Pinkus GS, Natkunam Y, Ligon AH, et al. Classical Hodgkin lymphoma with reduced β2M/MHC class I expression is associated with inferior outcome independent of 9p24. 1 status. Cancer Immunol Res. (2016) 4:910–6. doi: 10.1158/2326-6066.CIR-16-0201
152. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
153. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. (2016) 13:228–41. doi: 10.1038/nrclinonc.2015.215
154. Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. (2018) 24:511–20. doi: 10.1158/1078-0432.CCR-16-3001
155. Qiao W, Jia Z, Guo W, Liu Q, Guo X, Deng M. Prognostic and clinical significance of human leukocyte antigen class I expression in breast cancer: A meta-analysis. Mol Diagn Ther. (2023) 27:573–82. doi: 10.1007/s40291-023-00664-z
156. Meissner TB, Li A, Biswas A, Lee K-H, Liu Y-J, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci. (2010) 107:13794–9. doi: 10.1073/pnas.1008684107
157. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. (2012) 22:836–47. doi: 10.1038/cr.2012.56
158. Zhao M-Z, Sun Y, Jiang X-F, Liu L, Liu L, Sun L-X. Promotion on NLRC5 upregulating MHC-I expression by IFN-γ in MHC-I–deficient breast cancer cells. Immunologic Res. (2019) 67:497–504. doi: 10.1007/s12026-019-09111-w
159. Coussens L, Yang-Feng TL, Liao Y-C, Chen E, Gray A, McGrath J, et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. (1985) 230:1132–9. doi: 10.1126/science.2999974
160. Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology. (2012) 1:1104–10. doi: 10.4161/onci.21056
161. Salehi M, Kamali MJ, Ashuori Z, Ghadimi F, Shafiee M, Babaei S, et al. Functional significance of tRNA-derived fragments in sustained proliferation of tumor cells. Gene Rep. (2024) p:101901. doi: 10.1016/j.genrep.2024.101901
162. Waqar SN, Morgensztern D. Treatment advances in small cell lung cancer (SCLC). Pharmacol Ther. (2017). doi: 10.1016/j.pharmthera.2017.06.002
163. Santarpia M, Daffinà MG, Karachaliou N, González-Cao M, Lazzari C, Altavilla G, et al. Targeted drugs in small-cell lung cancer. Transl Lung Cancer Res. (2016) 5(1):51–70. doi: 10.3978/j.issn.2218-6751.2016.01.12
164. Sampsonas F, Ryan D, McPhillips D, Breen DP. Molecular testing and personalized treatment of lung cancer. Curr Mol Pharmacol. (2014) 7:22–32. doi: 10.2174/187446720701150105171219
165. Yasamineh S, Gholizadeh O, Ghafouri Kalajahi H, Yasamineh P, Firouzi-Amandi A, Dadashpour M. Future prospects of natural polymer-based drug delivery systems in combating lung diseases. In: Natural Polymeric Materials Based Drug Delivery Systems in Lung Diseases. Singapore: Springer (2023). p. 465–82.
166. Rivera MP. Lung cancer in women: differences in epidemiology, biology, histology, and treatment outcomes. Semin Respir Crit Care Med. (2013) 34:792–801. doi: 10.1055/s-0033-1358550
167. Simsek C, Sonmez O, Keyf AI, Yurdakul AS, Ozturk C, Gulbahar O, et al. Importance of serum SELDI-TOF-MS analysis in the diagnosis of early lung cancer. Asian Pac J Cancer Prev. (2013) 14:2037–42. doi: 10.7314/APJCP.2013.14.3.2037
168. Gelsomino F, Facchinetti F, Haspinger ER, Garassino MC, Trusolino L, De Braud F, et al. Targeting the MET gene for the treatment of non-small-cell lung cancer. Crit Rev Oncol Hematol. (2014) 89:284–99. doi: 10.1016/j.critrevonc.2013.11.006
169. Neumann A, Hörzer H, Hillen N, Klingel K, Schmid-Horch B, Bühring H-J, et al. Identification of HLA ligands and T-cell epitopes for immunotherapy of lung cancer. Cancer Immunology Immunotherapy. (2013) 62:1485–97. doi: 10.1007/s00262-013-1454-2
170. Shi K, Li QY, Zhang YQ, Huang H, Ding DX, Luo WM, et al. HLA-DPA1 overexpression inhibits cancer progression, reduces resistance to cisplatin, and correlates with increased immune infiltration in lung adenocarcinoma. Aging (Albany NY). (2023) 15:11067–91. doi: 10.18632/aging.205082
171. Costantini A, Ranjan K, Ferrone S, Schalper K. Role of HLA class-I antigen peptide loading complex components in immune evasion and treatment sensitivity in human lung cancer. USA: American Society of Clinical Oncology (2023).
172. Nguyen EM, Taniguchi H, Chan JM, Zhan YA, Chen X, Qiu J, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility complex class I antigen presentation and overcomes programmed death-ligand 1 blockade resistance in SCLC. J Thorac Oncol. (2022) 17:1014–31. doi: 10.1016/j.jtho.2022.05.014
173. Campuzano N, Fernandez Trokhimtchouk T, Flores LF, Otanez ES, Guallasamín E. Synchronous gastric and colon cancer. Cureus. (2023) 15:e48437. doi: 10.7759/cureus.48437
174. Farhani I, Yamchi A, Madanchi H, Khazaei V, Behrouzikhah M, Abbasi H, et al. Designing a multi-epitope vaccine against the SARS-CoV-2 variant based on an Immunoinformatics Approach. Curr Computer-Aided Drug Design. (2024) 20:274–90. doi: 10.2174/1573409919666230612125440
175. Fritsche J, Weinschenk T, Walter S, Lewandrowski P, Singh H. Immunotherapy against several tumors including gastrointestinal and gastric cancer. USA: Google Patents (2018).
176. Homma I, Kitamura H, Torigoe T, Tanaka T, Sato E, Hirohashi Y, et al. Human leukocyte antigen class I down-regulation in muscle-invasive bladder cancer: its association with clinical characteristics and survival after cystectomy. Cancer Sci. (2009) 100:2331–4. doi: 10.1111/j.1349-7006.2009.01329.x
177. Najafimehr H, Hajizadeh N, Nazemalhosseini-Mojarad E, Pourhoseingholi MA, Abdollahpour-Alitappeh M, Ashtari S, et al. The role of Human leukocyte antigen class I on patient survival in Gastrointestinal cancers: a systematic review and meta- analysis. Sci Rep. (2020) 10:728. doi: 10.1038/s41598-020-57582-x
178. Zhang Y, He S, Yu L, Shi C, Zhang Y, Tang S. Prognostic significance of HLA-G in patients with colorectal cancer: a meta-analysis and bioinformatics analysis. BMC Cancer. (2023) 23:1024. doi: 10.1186/s12885-023-11522-w
179. Bartolome J, Molto C, Benitez-Fuentes JD, Fernandez-Hinojal G, Manzano A, Perez-Segura P, et al. Prognostic value of human leukocyte antigen G expression in solid tumors: a systematic review and meta-analysis. Front Immunol. (2023) 14:1165813. doi: 10.3389/fimmu.2023.1165813
180. Hu Y, Li J, Ni F, Yang Z, Gui X, Bao Z, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic Malignancies. Nat Commun. (2022) 13:5313. doi: 10.1038/s41467-022-32960-3
181. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. (2019) 15:2548. doi: 10.7150/ijbs.34213
182. Bupha-Intr O, Haeusler G, Chee L, Thursky K, Slavin M, Teh B. CAR-T cell therapy and infection: a review. Expert Rev Anti-Infective Ther. (2021) 19:749–58. doi: 10.1080/14787210.2021.1855143
183. Jain T, Olson TS, Locke FL. How I treat cytopenias after CAR T-cell therapy. Blood J Am Soc Hematol. (2023) 141:2460–9. doi: 10.1182/blood.2022017415
184. Sun D, Shi X, Li S, Wang X, Yang X, Wan M. CAR−T cell therapy: A breakthrough in traditional cancer treatment strategies. Mol Med Rep. (2024) 29(3):47. doi: 10.3892/mmr.2024.13171
185. Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. (2013) 4:371. doi: 10.3389/fimmu.2013.00371
186. Corti C, Venetis K, Sajjadi E, Zattoni L, Curigliano G, Fusco N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: preclinical and clinical progress. Expert Opin Investigational Drugs. (2022) 31:593–605. doi: 10.1080/13543784.2022.2054326
187. Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, et al. Targeting alpha-fetoprotein (AFP)–MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. (2017) 23:478–88. doi: 10.1158/1078-0432.CCR-16-1203
188. Marijt WE, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, Van der Hoorn MA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1-or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci. (2003) 100:2742–7. doi: 10.1073/pnas.0530192100
189. Spierings E. Minor histocompatibility antigens: past, present, and future. Tissue Antigens. (2014) 84:374–60. doi: 10.1111/tan.2014.84.issue-4
190. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood J Am Soc Hematol. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
191. Akatsuka Y. TCR-like CAR-T cells targeting MHC-bound minor histocompatibility antigens. Front Immunol. (2020) 11:257. doi: 10.3389/fimmu.2020.00257
192. Eldershaw SA, Pearce HAbbotts BE, Sirovica M, Ferguson P. Vaccination with a DNA and MVA ‘prime-boost’regimen induces strong CD8+ T-cell responses against the immunodominant minor histocompatibility antigen, HA-1: results of a first-in-man phase I clinical trial. Blood. (2017) 130:1908. doi: 10.1182/blood.V130.Suppl_1.1908.1908
193. Oostvogels R, Kneppers E, Minnema M, Doorn R, Franssen L, Aarts T, et al. Efficacy of host-dendritic cell vaccinations with or without minor histocompatibility antigen loading, combined with donor lymphocyte infusion in multiple myeloma patients. Bone Marrow Transplant. (2017) 52:228–37. doi: 10.1038/bmt.2016.250
194. Liu DH, Mou FF, An M, Xia P. Human leukocyte antigen and tumor immunotherapy (Review). Int J Oncol. (2023) 62(6):1–4. doi: 10.3892/ijo.2023.5594
195. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20(6):1283. doi: 10.3390/ijms20061283
196. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduction Targeted Ther. (2023) 8:9. doi: 10.1038/s41392-022-01270-x
197. Hosen N. Identification of cancer-specific cell surface targets for CAR-T cell therapy. Inflammation Regeneration. (2024) 44:17. doi: 10.1186/s41232-024-00329-2
198. Qian H, Tian L, Liu W, Liu L, Li M, Zhao Z, et al. Adenovirus type 5-expressing Gn induces better protective immunity than Gc against SFTSV infection in mice. NPJ Vaccines. (2024) 9:194. doi: 10.1038/s41541-024-00993-y
199. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. (2017) 8:90521–31. doi: 10.18632/oncotarget.19361
200. Shklovskaya E, Rizos H. MHC class I deficiency in solid tumors and therapeutic strategies to overcome it. Int J Mol Sci. (2021) 22(13):6741. doi: 10.3390/ijms22136741
201. Cornel AM, Mimpen IL, Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers. (2020) 12:1760. doi: 10.3390/cancers12071760
202. Guillaume J, Perzolli A, Boes M. Strategies to overcome low MHC-I expression in paediatric and adult tumours. Immunother Adv. (2024) 4:ltad028. doi: 10.1093/immadv/ltad028
203. Daei Sorkhabi A, Mohamed Khosroshahi L, Sarkesh A, Mardi A, Aghebati-Maleki A, Aghebati-Maleki L, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol. (2023) 14:1113882. doi: 10.3389/fimmu.2023.1113882
204. Zhang X, et al. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
205. Taylor BC, Balko JM. Mechanisms of MHC-I downregulation and role in immunotherapy response. Front Immunol. (2022) 13:844866. doi: 10.3389/fimmu.2022.844866
206. Lesterhuis WJ, Schreibelt G, Scharenborg NM, Brouwer HM, Gerritsen MJ, Croockewit S, et al. Wild-type and modified gp100 peptide-pulsed dendritic cell vaccination of advanced melanoma patients can lead to long-term clinical responses independent of the peptide used. Cancer Immunol Immunother. (2011) 60:249–60. doi: 10.1007/s00262-010-0942-x
207. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. (2015) 348:803–8. doi: 10.1126/science.aaa3828
208. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. (2018) 10(450):eaar3342. doi: 10.1126/scitranslmed.aar3342
209. McGuirk M, Shahzad M, Irfan S, Amin MK, Chaudhary SG, Anwar I, et al. Correlates of graft versus host disease following allogeneic hematopoietic stem cell transplantation. Blood. (2024) 144:7354. doi: 10.1182/blood-2024-193749
210. Carapito R, Jung N, Kwemou M, Untrau M, Michel S, Pichot A, et al. Matching for the nonconventional MHC-I MICA gene significantly reduces the incidence of acute and chronic GVHD. Blood. (2016) 128:1979–86. doi: 10.1182/blood-2016-05-719070
211. Besse B, Felip E, Garcia Campelo R, Cobo M, Mascaux C, Madroszyk A, et al. Randomized open-label controlled study of cancer vaccine OSE2101 versus chemotherapy in HLA-A2-positive patients with advanced non-small-cell lung cancer with resistance to immunotherapy: ATALANTE-1. Ann Oncol. (2023) 34:920–33. doi: 10.1016/j.annonc.2023.07.006
212. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. (2009) 100:2014–21. doi: 10.1111/j.1349-7006.2009.01303.x
213. Najafi M, Jahanbakhshi A, Finocchi Ghersi S, Giaccherini L, Botti A, Cavallieri F, et al. Clinical effects of immuno-oncology therapy on glioblastoma patients: A systematic review. Brain Sci. (2023) 13:159. doi: 10.3390/brainsci13020159
214. Hong DS, Van Tine BA, Biswas S, McAlpine C, Johnson ML, Olszanski AJ, et al. Autologous T cell therapy for MAGE-A4(+) solid cancers in HLA-A*02(+) patients: a phase 1 trial. Nat Med. (2023) 29:104–14. doi: 10.1038/s41591-022-02128-z
215. Yamada A, Noguchi M, Komatsu N, Suekane S, Yutani S, Moriya F, et al. Phase I clinical study of a personalized peptide vaccination available for six different human leukocyte antigen (HLA-A2, -A3, -A11, -A24, -A31 and -A33)-positive patients with advanced cancer. Exp Ther Med. (2011) 2:109–17. doi: 10.3892/etm.2010.177
216. Pan Q, Weng D, Liu J, Han Z, Ou Y, Xu B, et al. Phase 1 clinical trial to assess safety and efficacy of NY-ESO-1-specific TCR T cells in HLA-A∗02:01 patients with advanced soft tissue sarcoma. Cell Rep Med. (2023) 4:101133. doi: 10.1016/j.xcrm.2023.101133
217. Zhuang B, Shang J, Yao Y. HLA-G: An important mediator of maternal-fetal immune-tolerance. Front Immunol. (2021) 12:744324. doi: 10.3389/fimmu.2021.744324
218. Obermajer N, Zwolak A, Van De Ven K, Versmissen S, Brajic A, Petley T, et al. Abstract ND07: JNJ-78306358: A first-in-class bispecific T cell redirecting HLA-G antibody. Cancer Res. (2022) 82:ND07–7. doi: 10.1158/1538-7445.AM2022-ND07
219. Morandi F, Airoldi I. HLA-G and other immune checkpoint molecules as targets for novel combined immunotherapies. Int J Mol Sci. (2022) 23:2925. doi: 10.3390/ijms23062925
220. Geva R, Vieito M, Ramon J, Perets R, Pedregal M, Corral E, et al. Safety and clinical activity of JNJ-78306358, a human leukocyte antigen-G (HLA-G) x CD3 bispecific antibody, for the treatment of advanced stage solid tumors. Cancer Immunol Immunother. (2024) 73:205. doi: 10.1007/s00262-02403790-7
221. Li CM, Haratipour P, Lingeman RG, Perry JJP, Gu L, Hickey RJ, et al. Novel peptide therapeutic approaches for cancer treatment. Cells. (2021) 10:2908. doi: 10.3390/cells10112908
222. Rizvi SFA, Zhang H, Fang Q. Engineering peptide drug therapeutics through chemical conjugation and implication in clinics. Medicinal Res Rev. (2024) 44(6):2420–71. doi: 10.1002/med.22046
223. Milanifard M, Mehrabi S, Ahadi R, Nabiuni M, Souteh SA, Joghataei MT. Vitamin D receptor gene polymorphisms in patients with relapsing multiple sclerosis. Eur J Trans Myology. (2024). doi: 10.4081/ejtm.2024.12993
224. Dersh D, Hollý J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol. (2021) 21:116–28. doi: 10.1038/s41577-020-0390-6
225. Weidanz JA, Hawkins O, Verma B, Hildebrand WH. TCR-like biomolecules target peptide/MHC Class I complexes on the surface of infected and cancerous cells. Int Rev Immunol. (2011) 30:328–40. doi: 10.3109/08830185.2011.604880
226. Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, et al. STAT3 inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating type 1 interferon production by cancer cells. Cancer Res. (2015) 75:3812–22. doi: 10.1158/0008-5472.CAN-15-1122
227. Meric-Bernstam F, Sweis RF, Kasper S, Hamid O, Bhatia S, Dummer R, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. USA: American Society of Clinical Oncology (2019).
228. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. (2017) 5:676–84. doi: 10.1158/2326-6066.CIR-17-0049
229. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-γ induces tumor MHC expression in metastatic Malignant melanoma. Clin Cancer Res. (2003) 9(1):84–92.
230. Meissner TB, Li A, Liu Y-J, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem Biophys Res Commun. (2012) 418:786–91. doi: 10.1016/j.bbrc.2012.01.104
231. Watanabe S, Hayashi H, Haratani K, Shimizu S, Tanizaki J, Sakai K, et al. Mutational activation of the epidermal growth factor receptor down-regulates major histocompatibility complex class i expression via the extracellular signal-regulated kinase in non–small cell lung cancer. Cancer Sci. (2019) 110:52–60. doi: 10.1111/cas.2019.110.issue-1
232. Bao Y, Cruz G, Zhang Y, Qiao Y, Mannan R, Hu J, et al. The UBA1-STUB1 axis mediates cancer immune escape and resistance to checkpoint blockade. Cancer Discov. (2025) 15(2):363–81. doi: 10.1158/2159-8290.CD-24-0435
233. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang KY, Ferrone S, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. (2013) 133:624–36. doi: 10.1002/ijc.v133.3
234. Kim DW, Cho JY. Recent Advances in Allogeneic CAR-T Cells. Biomolecules. (2020) 10(2):263. doi: 10.3390/biom10020263
235. Jurewicz M, Nanaware P, Stern L. MHC class II peptide loading regulates selection and function of regulatory T cells. Mol Immunol. (2022) 150:3. doi: 10.1016/j.molimm.2022.05.021
236. Jähnisch H, Füssel S, Kiessling A, Wehner R, Zastrow S, Bachmann M, et al. Dendritic cell-based immunotherapy for prostate cancer. J Immunol Res. (2010) 2010:517493. doi: 10.1155/2010/517493
237. Emens LA. Breast cancer immunobiology driving immunotherapy: vaccines and immune checkpoint blockade. Expert Rev Anticancer Ther. (2012) 12:1597–611. doi: 10.1586/era.12.147
238. Burciu OM, Sas I, Popoiu T-A, Merce A-G, Moleriu L, Cobec IM. Correlations of imaging and therapy in breast cancer based on molecular patterns: an important issue in the diagnosis of breast cancer. Int J Mol Sci. (2024) 25:8506. doi: 10.3390/ijms25158506
239. Rozanov DV, Rozanov ND, Chiotti KE, Reddy A, Wilmarth PA, David LL, et al. MHC class I loaded ligands from breast cancer cell lines: A potential HLA-I-typed antigen collection. J Proteomics. (2018) 176:13–23. doi: 10.1016/j.jprot.2018.01.004
240. Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R, et al. Trial watch: peptide-based vaccines in anticancer therapy. Oncoimmunology. (2018) 7:e1511506. doi: 10.1080/2162402X.2018.1511506
241. Stephens AJ, Burgess-Brown NA, Jiang S. Beyond just peptide antigens: the complex world of peptide-based cancer vaccines. Front Immunol. (2021) 12:696791. doi: 10.3389/fimmu.2021.696791
242. Milanifard M, Mehrabi S, Ahadi R, Nabiuni M, Souteh SA, Joghataei MT. Evaluation relationship between VDR gene and clinical and inflammatory factors in patients with RRMS. Eur J Trans Myol. (2024) 34(4):12939. doi: 10.4081/ejtm.2024.12939
243. Comber JD, Philip R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther Adv Vaccines. (2014) 2:77–89. doi: 10.1177/2051013614525375
244. Poole A, Karuppiah V, Hartt A, Haidar JN, Moureau S, Dobrzycki T, et al. Therapeutic high affinity T cell receptor targeting a KRAS(G12D) cancer neoantigen. Nat Commun. (2022) 13:5333. doi: 10.1038/s41467-022-32811-1
245. Bannoura SF, Khan HY, Azmi AS. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front Oncol. (2022) 12:1013902. doi: 10.3389/fonc.2022.1013902
246. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y-P, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. New Engl J Med. (2022) 386:2112–9. doi: 10.1056/NEJMoa2119662
247. Lu D, Chen Y, Jiang M, Wang J, Li Y, Ma K, et al. KRAS G12V neoantigen specific T cell receptor for adoptive T cell therapy against tumors. Nat Commun. (2023) 14:6389. doi: 10.1038/s41467-023-42010-1
248. Hsiue EH-C, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, et al. Targeting a neoantigen derived from a common TP53 mutation. Science. (2021) 371:eabc8697. doi: 10.1126/science.abc8697
249. Lai ZY, Tsai KY, Chang SJ, Chuang YJ. Gain-of-function mutant TP53 R248Q overexpressed in epithelial ovarian carcinoma alters AKT-dependent regulation of intercellular trafficking in responses to EGFR/MDM2 inhibitor. Int J Mol Sci. (2021) 22(16):8784. doi: 10.3390/ijms22168784
250. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. (2008) 20(5):267–75. doi: 10.1016/j.smim.2008.04.001
251. Li F, Wu H, Du X, Sun Y, Rausseo BN, Talukder A, et al. Epidermal growth factor receptor-targeted neoantigen peptide vaccination for the treatment of non-small cell lung cancer and glioblastoma. Vaccines. (2023) 11:1460. doi: 10.3390/vaccines11091460
252. Kemps PG, Zondag TC, Steenwijk EC, Andriessen Q, Borst J, Vloemans S, et al. Apparent lack of BRAFV600E derived HLA class I presented neoantigens hampers neoplastic cell targeting by CD8+ T cells in langerhans cell histiocytosis. Front Immunol. (2020) 10:3045. doi: 10.3389/fimmu.2019.03045
253. García-Mulero S, Fornelino R, Punta M, Lise S, Varela M, Del Carpio LP, et al. Driver mutations in GNAQ and GNA11 genes as potential targets for precision immunotherapy in uveal melanoma patients. Oncoimmunology. (2023) 12:2261278. doi: 10.1080/2162402X.2023.2261278
254. Graff-Dubois S, Faure O, Gross DA, Alves P, Scardino A, Chouaib S, et al. Generation of CTL recognizing an HLA-A*0201-restricted epitope shared by MAGE-A1, -A2, -A3, -A4, -A6, -A10, and -A12 tumor antigens: implication in a broad-spectrum tumor immunotherapy. J Immunol. (2002) 169:575–80. doi: 10.4049/jimmunol.169.1.575
255. Andrulis M, Capper D, Meyer J, Penzel R, Hartmann C, Zentgraf H, et al. IDH1 R132H mutation is a rare event in myeloproliferative neoplasms as determined by a mutation. specific antibody. Haematologica. (2010) 95:1797–8. doi: 10.3324/haematol.2010.024430
256. Hosseinalizadeh H, Rahmati M, Ebrahimi A, O'Connor RS. Current status and challenges of vaccination therapy for glioblastoma. Mol Cancer Ther. (2023) 22:435–46. doi: 10.1158/1535-7163.MCT-22-0503
Keywords: peptide-loading complex, cancer, immunotherapy, major histocompatibility complex (MHC), cancer vaccine, treatment
Citation: Song Z, Tao Y and You J (2025) The potential applications of peptide-loading complex in cancer treatment. Front. Immunol. 16:1526137. doi: 10.3389/fimmu.2025.1526137
Received: 11 November 2024; Accepted: 05 February 2025;
Published: 03 March 2025.
Edited by:
Sonja Ludwig, University of Heidelberg, GermanyReviewed by:
Kiran Kundu, University of Texas MD Anderson Cancer Center, United StatesZhijian Huang, Fujian Medical University, China
Marco Cavaco, Universidade de Lisboa, Portugal
Copyright © 2025 Song, Tao and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaxin You, eW91anhAamx1LmVkdS5jbg==
†ORCID: Jiaxin You, orcid.org/0009-0000-4075-6599