
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 17 March 2025
Sec. Vaccines and Molecular Therapeutics
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1524477
This article is part of the Research TopicBroad Spectrum Viral VaccinesView all 5 articles
 Irene González-Domínguez1
Irene González-Domínguez1 Adam Abdeljawad1
Adam Abdeljawad1 Tsoi Ying Lai1
Tsoi Ying Lai1 Marta Boza1Stephen McCroskery1Nicholas Lemus1Stefan Slamanig1,2Gagandeep Singh1,3
Marta Boza1Stephen McCroskery1Nicholas Lemus1Stefan Slamanig1,2Gagandeep Singh1,3 Prajakta Warang1,3
Prajakta Warang1,3 Temima Yellin1
Temima Yellin1 Anass Abbad1
Anass Abbad1 Juan Manuel Carreño1Victoria Dolange1Jose Luis Martínez-Guevara1
Juan Manuel Carreño1Victoria Dolange1Jose Luis Martínez-Guevara1 Gagandeep Singh1Marina Barcena-Varela4
Gagandeep Singh1Marina Barcena-Varela4 Lauren A. Chang1,3
Lauren A. Chang1,3 Michael Schotsaert1,3,5,6
Michael Schotsaert1,3,5,6 Florian Krammer1,7,8,9
Florian Krammer1,7,8,9 Peter Palese1,10*
Peter Palese1,10* Weina Sun1*
Weina Sun1*Introduction: A new generation of mucosal vaccine against the ever-evolving SARS-CoV-2 is of great value to fight COVID-19. In previous studies, our groups developed a viral vector vaccine based on an avirulent Newcastle disease virus (NDV) expressing the prefusion-stabilized spike protein of SARS-CoV-2 (NDV-HXP-S).
Methods: Here we characterized the in vivo biodistribution and immunogenicity of a live mucosal NDV-HXP-S vaccine in animal models.
Results: NDV showed restricted replication in mice and hamsters. Despite limited replication, intranasal live NDV-HXP-S provided protection against SARS-CoV-2 challenge and direct-contact transmission in hamsters. Importantly, a trivalent live NDV-HXP-S vaccine (Wuhan, Beta, Delta) induced more cross-reactive antibody responses against the phylogenetically distant Omicron variant than the ancestral vaccine. Furthermore, intranasal trivalent live NDV-HXP-S boosted systemic and mucosal immunity in mice pre-immunized with mRNA vaccine.
Discussion: Overall, a mucosal multivalent live NDV-HXP-S vaccine shows great promise as a safe, next-generation vaccine conferring broad mucosal and systemic immunity against future SARS-CoV-2 variants.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of COVID-19 (Coronavirus Disease 2019). Since the beginning of the pandemic, the development of an effective vaccine has been a major effort. A successful vaccine must be safe and protective, but also cost-effective and accessible on a global scale. Furthermore, with the constant emergence of new SARS-CoV-2 variants of interest (VOIs), the vaccine needs either to be easily updated or to be able to confer broad protection. Although mRNA vaccines have been widely used in the US and the European Union [Our World in Data, as of September 6th 2023 (1)], a different scenario has occurred worldwide where cheaper vaccines dominated the market during the first vaccination campaigns (as of December 14th 2021) (2).
In previous work, our groups have developed a highly immunogenic vaccine based on Newcastle disease virus (NDV) as a viral vector expressing the prefusion-stabilized spike protein from SARS-CoV-2 (3). NDV-HXP-S (hexa proline-spike) can be produced at large-scale and at low-cost through the utilization of existing manufacturing facilities for influenza virus vaccines (4). NDV-HXP-S has proven to work as an intramuscular systemic vaccine and as a mucosal vaccine when administered intranasally (3). The safety and immunogenicity of the ancestral NDV-HXP-S vaccine has been tested in mice, hamsters, pigs and rats (3, 5, 6), and NDV-HXP-S has been used in clinical trials as a mucosal vaccine in the US (Mount Sinai, NCT05181709) or as a systemic vaccine (delivered intramuscularly) in Thailand (NCT04764422, HXP-GPOVac) (7), Vietnam (NCT04830800, COVIVAC) (8), Brazil (NCT04993209) and Mexico (NCT04871737, Patria) (9, 10). Although the safety profile of live NDV has been well studied as a human oncolytic vector and as a veterinary vaccine (11–13), less is known about the viral shedding of NDV after vaccination. To improve the breadth of protection, our previous study demonstrated that a multivalent formulation based on the inactivated NDV-HXP-S variant vaccines (Wuhan, Beta, Delta) given intramuscularly can confer a better protective immune response than the ancestral vaccine alone against a future Omicron variant (14).
In this work, we aim to study the tissue distribution, immunogenicity and in vivo protection against a homologous and heterologous SARS-CoV-2 variants of a live mucosal NDV-HXP-S vaccine in preclinical animal models. We used the ancestral NDV-HXP-S vaccine to study its biodistribution and its ability to induce mucosal immunity and to reduce viral transmission. Next, we characterized the multivalent formulation of the live NDV-HXP-S variant vaccine given via the mucosal route in terms of its breadth of protection against the mismatched Omicron BA.1 SARS-CoV-2 strain as an important addition to our previous study on the inactivated vaccine (15). We tested the immunity of this proof-of-principle mucosal multivalent vaccine in both naïve and pre-immunized models. Our results suggested that despite restricted replication of the vector in mice and hamsters, the live mucosal NDV-HXP-S conferred protection against viral replication and spread. Importantly, the mucosal multivalent vaccine retained the breadth of protection not only in naïve animals, but also in pre-immune animals.
NDV-HXP-S vectors expressing the pre-fusion stabilized spike protein of ancestral, Beta and Delta variants were rescued and produced in specific pathogen free embryonated (SPF) chicken eggs as described in a previous work (3, 15, 16). Purified NDV-HXP-S vaccines were re-suspended in Phosphate Buffered Saline (PBS) (pH 7.4) and the infectious titer in 50% egg infectious doses (EID50) and the total protein concentration was measured as previously described (14).
Mouse codon-optimized sequence of Firefly luciferase gene (GenBank ID: MN728548.1) was generated using the Integrated DNA Technologies (IDT) Codon Optimized Tool (IDT Technologies Inc, IA, USA). Luciferase sequence was synthesized using the gBlocks® Gene Fragments service (IDT) and inserted into pNDV_LS/L289A rescue plasmid (between P and M genes) by in-Fusion cloning (Clontech) and rNDV virus was rescued and quantified as previously described (17).
BSR-T7 monolayers were infected with rNDV-luc at 0.5 multiplicity of infection. Cells were harvested 48 hours post infection and an in vitro luciferase kit (Rapid Detection of Firefly Luciferase Activity, Promega, WI, USA) was used to measure the luciferase activity in infected cells following the manufacturer’s instructions.
All the animal experiments were performed in accordance with protocols approved by the Icahn School of Medicine at Mount Sinai (ISMMS) Institutional Animal Care and Use Committee (IACUC). All experiments with live SARS-CoV-2 were performed in the Centers for Disease Control and Prevention (CDC)/US Department of Agriculture (USDA)-approved biosafety level 3 (BSL-3) biocontainment facility of the Global Health and Emerging Pathogens Institute at the ISMMS, in accordance with institutional biosafety requirements.
For mice biodistribution studies, 8-to-10-week-old female BALB/c mice (RRID: IMSR_JAX:000651, The Jackson Laboratory) were used for these experiments. An infectious dose of 105 or 106 EID50 of NDV was diluted in PBS (pH 7.4, Gibco Cat#10010-023) in a total volume of 30 μl for IM or IN administration (3). In vivo bioluminescence imaging was performed using a Biophotonic IVIS©‐Spectrum system (Perkinelmer, MA, USA) located at the BioMedical Engineering and Imaging Institute at ISMMS. Mice anesthetized by isofluorane were imaged 5 minutes after intraperitoneal injection with fresh D-Luciferin (150 mg/kg; Thermo Fisher Scientific). Different set of mice from one group were analyzed each day to allow them for a complete recovery from anesthesia. Luciferase signal was quantified using Living Image software (Caliper LifeSciences) in three different regions of interest (ROI) of each mouse.
Eight- to ten-week-old female Golden Syrian hamsters (HsdHan®:AURA, Inotiv) were vaccinated with 107 EID50 of ancestral NDV-HXP-S vaccine or wildtype LaSota NDV (WT NDV) strains, as performed in previous work (3). Vaccines were diluted in PBS in a total volume of 20 µl or 50 µl per dose for its IN or IM administration (3), respectively. One- or seven-days post vaccination one subset of hamsters were euthanized, and several biological fluids including nasal washes, blood and urine and organs including lungs, brain, leg muscle at the site of the vaccination were collected. Nasal washes were collected in 0.4 mL of PBS with 100 µg/mL of P/S (pH 7.4, Gibco). Blood was retrieved using cardiac puncture and urine was extracted from the bladder. Nasal washes and urine were spun at 3,000 rpm for 20 min at 4°C and blood was spun at 5000 rpm for 30 min at 4°C and all samples were stored at -80 °C until further use. Right lung lobes were homogenized in 1 mL of PBS using a Lysing matrix A homogenization tubes (MP Biomedicals). Lung homogenate supernatant was collected after centrifugation (10,000 rpm x 10 min at room temperature, RT). Several organs including the left lung lobe, brain, leg muscle at the site of infection were collected and stored in 4% PFA (v/v) solution in PBS (pH 7.4, Gibco) overnight at RT and later embedded in paraffin as Formalin Fixed Paraffin Embedded tissue (FFPE) and stored at RT until use.
In the direct-contact transmission study, a subset of 8- to 10-week-old hamsters vaccinated at the same time as those in the biodistribution study were boosted five months later with a second dose of NDV-HXP-S IN or IM. The immunized animals were bled via gingival vein pre-boost and four-weeks after second boost. Thereafter, vaccinated hamsters (recipient) were placed in pairs with 8- to 10-week-old female Golden Syrian hamsters challenged with 105 PFU of WA1-USA/2020 SARS-CoV-2 strain (donors) in a direct contact transmission setting. A healthy control group in which the hamsters were mock-challenged was included as control. Weight changes were monitored for 5 days. Nasal washes were collected at day 1 and day 3 post challenge for both donors and recipients, as previously described. Animals from each group were euthanized day 5 post-challenge to harvest nasal turbinate and lungs. Nasal turbinate, upper right lung lobe and lower right lung lobes were homogenized as previously described (21). Left lung lobe was stored in 4% PFA (v/v) and latter embedded in FFPE as previously described.
RNA extraction was performed using Maxwell RSC RNA FFPE Kit protocol following the manufacturer’s instructions (Catalog number AS1440, Promega) in the Biorepository and Pathology Core (ISMMS). A total of 50 µm scrolls (3 µm thickness each) were cut from FFPE block for RNA extraction. The isolated RNA was quantified using a Qubit RNA HS Assay Kit and Qubit 3.0 Fluorometer (<ns/>Cat Q32855, Thermo Fisher Scientific).
A validated Reverse Transcriptase qPCR (RT-qPCR) method was used to quantify viral copies of the NP gene (Table 1) of WT NDV or NDV-HXP-S viruses in different tissues. Consensus sequences from the Golden Syrian hamster housekeeping genes β-actin (Gene ID: 101844587) and GAPDH (Gene ID: 121132788) were retrieved from NCBI and used as house-keeping controls. Primers were designed using SnapGene tool (Table 1) (18) and standards were generated in-house for each gene. Standards were 10-fold serially diluted in UltraPure™ DNase/RNase-Free Distilled Water (Thermo Fisher Scientific, MA, USA Cat<ns/> 10977015) resulting in 108 to 101 copies/sample. The efficiency, R2, slope and y-int used are present in Table 1. RT-qPCR was performed using the iTaq™ Universal SYBR Green One-Step Kit (BioRad, Hercules, CA) in the CFX Opus 96 Real-Time PCR Instrument 96-well (BioRad, Hercules, CA). A no-template control and a condition with 105 copies of the amplicon in triplicate were added as internal negative and positive control, respectively. RT-qPCR was performed following the manufacturer thermal cycling conditions, except for the annealing and extension time which was modified to 20 sec at 55°C. Melt curve analysis was also performed to confirm amplification specificity. The copies per sample were finally interpolated from the different standard curves, then, the results were normalized to copies/ng.
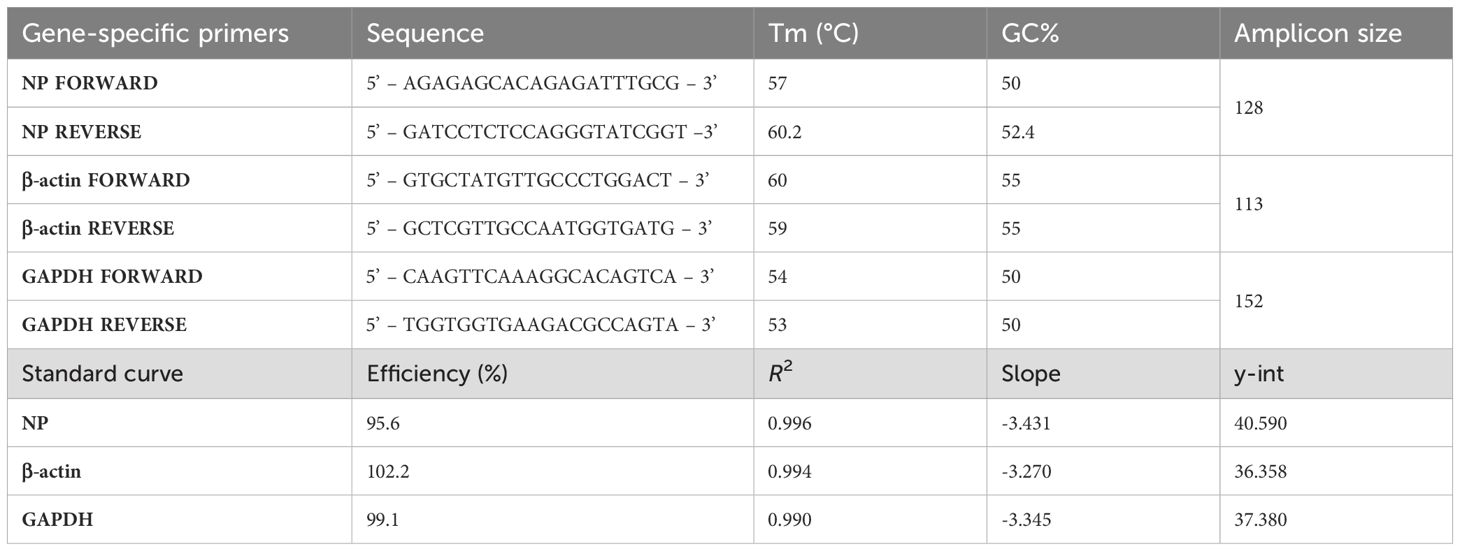
Table 1. Primers and standard curve of the Golden Syrian hamster tissue RT-qPCR analysis.
Vaccinations were performed in female BALB/c mice (RRID: IMSR_JAX:000651, The Jackson Laboratory) using a two-dose regimen as indicated in each experiment. For IN vaccination, 8-to-10-week-old mice were anesthetized with a ketamine/xylazine cocktail before administration of 10 µl of total volume split between each nostril. For intramuscular (IM) vaccination, vaccines were prepared in 100 µl total volume and 50 µl were administered into the thigh muscle of each leg. The live concentrated NDV-HXP-S and its trivalent version were used at a total infectious dose of 106 EID50, being a dose of 3.3x105 EID50 for each variant vaccine when a trivalent formulation was used diluted in PBS (19).
Mucosal samples were collected after mouse euthanasia. Mice nasal washes and bronchioalveolar lavage fluid (BALF) were collected in 1 mL total volume of PBS with 100 units (U)/mL of penicillin-streptomycin (P/S; Gibco Cat<ns/>15140122). Oral washes were collected in a total volume of 0.4 mL of PBS with 100 U/mL P/S (pH 7.4, Gibco). Vaginal lavages and intestinal lavages were collected in a total volume of 0.4 mL or 5 mL, respectively of PBS with 1% (v/v) anti-protease cocktail (Sigma Cat<ns/> P8340). Nasal washes, BALF, oral washes, intestinal lavages, and vaginal lavages samples were spun at 3000 rpm for 20 min at 4°C then the supernatant was collected and stored at -20°C until further use. Fecal homogenates were generated by diluting the feces to a concentration of 0.1 g/mL of PBS with 1% (v/v) anti-protease cocktail. Feces were then homogenized and centrifuged for 10 min at maximum speed (~14,000 rpm) at 4°C. The supernatant was then collected and stored at -20°C.
129 mice (RRID: IMSR_JAX:002448, The Jackson 212 Laboratory) were used in the heterologous vaccine platform study (mRNA vaccination followed by NDV booster). 129 mice were vaccinated with Pfizer mRNA vaccine at 0.25 µg IM twice 3 weeks apart as performed in previous work (19, 20). Ten-months after the second dose, animals were boosted with NDV-based vaccines via IN or IM route as previously described (14, 19). Twenty-one days later animals were euthanized, and blood, spleen and mucosal samples were collected. The live concentrated NDV-HXP-S and its trivalent version were used at 106 EID50 diluted in PBS. The inactivated monovalent and trivalent NDV-HXP-S vaccines were used at 1 µg of total protein diluted in PBS as previously described (14).
Naïve 8- to 10-week-old female Golden Syrian hamsters were vaccinated with a two-dose vaccination regimen at 106 EID50/animal of ancestral NDV-HXP-S, trivalent NDV-HXP-S vaccines, or wild type LaSota NDV (WT NDV) strain in a 12-week interval (21). Vaccines were diluted in PBS (Gibco) in a total volume of 20 µl or 100 µl per dose for its IN or IM administration, respectively. Hamsters were bleed 12-weeks post boost and challenged with 5x104 PFU of Omicron BA.1 SARS-CoV-2 strain. Weight changes were monitored for 5 days. Throat swabs were collected and viruses were lysed in 0.3 mL Qiagen lysis buffer at days 1, 3 and 5 post challenge and stored at -80 °C until further processing. Animals from each group were euthanized day 5 post-challenge to harvest nasal turbinate and lungs. Nasal turbinate, upper right lung lobe and lower right lung lobes were homogenized as previously described (21).
FFPE left lung tissues obtained from hamsters were cut into 5 μm sections and stained by the Biorepository and Pathology Core (ISMMS). The presence of NDV viral antigens was detected with a rabbit polyclonal serum (1:2000 dilution, brown) generated by Covance (labcorp, Princeton, NJ, USA) through immunization rabbit with adjuvanted inactivated NDV viruses and a human mAb 1A9 (Cat<ns/> MA5-35946, Thermofisher) anti SARS-CoV-2 spike antibody (1:2000 dilution, pink). SARS-CoV-2 nucleoprotein (N) was stained using a mouse anti-N monoclonal antibody 1C7C7 (1:50 dilution, brown) kindly provided by Dr. James Duty at ISMMS (Millipore Sigma, cat. no. ZMS1075) (3, 21). Briefly, IHC staining was performed using VENTANA DISCOVERY ULTRA automated slide staining instrument (Roche, Basilea, Switzerland). Single staining (automatic or semi-automatic) was performed using the selected primary antibody and the and Discovery OMNIMap anti-host-HRP (Roche) as secondary antibody. The signal was obtained using Discovery ChromoMap DAB RUO (760–2513) (depicted in brown signal). For double staining, the same procedure was performed sequentially. Briefly, after the first staining (depicted in brown), slides went throughout a step of inhibition, heat denaturation and neutralization and then the second primary antibody was applied followed a secondary and a developing kit (depicted in pink). Tissues were counterstained with Hematoxylin to visualize the nuclei. Whole tissue sections on the slide will be converted into high-resolution digital data using a NanoZoomer S210 Digital slide scanner (Hamamatsu). The HALO image analysis platform was used for quantitative tissue analysis (Indica Labs, Inc.), using random forest algorithm classifier. Multiplex IHC module and color deconvolution were used to separate chromogenic stains together with nuclei segmentation to set up the system for quantitative analysis.
RNA was extracted from throat swab samples using the QIAamp Viral RNA Mini Kit (QIAGEN, Germantown, MD) according to the manufacturer’s instructions. Genomic copies of SARS-CoV-2 Nucleocapsid (N) gene were measured as previously described (22). Briefly, Forward Primer 5’-GACCCCAAAATCAGCGAAAT-3’ and Reverse Primer 5’-TCTGGTTACTGCCAGTTGAATCTG-3’ were used, yielding a 72 bp amplicon. Standard curves were generated using the 2019-nCoV_N_Positive Control plasmid (IDT, Cat No.10006625), which encodes the N gene of SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank: NC_045512.2) (Table 2). RT-qPCR was performed using the iTaq™ Universal SYBR Green One-Step Kit and the CFX Opus 96 Real-Time PCR System following the manufacturer recommended thermal cycling conditions (39 cycles of 30 sec at 55°C of anneal and extension). Copies per sample were afterwards calculated (obtained copies/5 µl of RNA extracted per reaction x 60 µl of total RNA extracted per sample).

Table 2. Standard curve of the SARS-CoV-2 N RT-qPCR analysis.
Spike-specific IgG and IgA antibody titers in mice and hamster samples were measured by ELISA as described previously (14, 16, 23). Proteins were coated onto Immulon® 4 HBX 96-well microtiter plates (Thermo Fisher Scientific, Cat<ns/>3855) at 2 μg/mL in 1x coating buffer (SeraCare Life Sciences Inc. USA Cat<ns/>5150-0014 (50–84–00), MA, USA) at 50 μL/well overnight at 4°C. All plates were washed 3 times with 225 μL PBS containing 0.1% (vol/vol) Tween-20 (PBST) and 220 μL blocking solution (3% goat serum, 0.5% non-fat dried milk powder, 96.5% PBST) was added to each well and incubated for 1 hour at RT. Samples were serially diluted in blocking solution followed by a 2-hour incubation at RT at a starting dilution of 1:30 for serum samples and 1:2 dilution for mucosal samples. ELISA plates were afterwards washed 3 times with PBST and 50 μL of anti- IgG (1:3,000) or anti -IgA (1:2,000)-horseradish peroxidase (HRP) conjugated antibody (Cytiva, GE Healthcare) was diluted in blocking solution. After 1 hour, plates were washed 3 times with PBST and developed using SigmaFast OPD (Sigma-Aldrich Cat<ns/> P9187, MI, USA) for 10 minutes. Reactions were stopped by adding 50 μL 3M hydrochloric acid and absorbance at 492 nm was determined on a Synergy 4 plate reader (BioTek, Agilent Technologies inc., CA, USA) or similar. For each ELISA plate, the blank average absorbance plus 3 standard deviations were used as a cutoff to determine endpoint titers and the area under the curve (AUC) using GraphPad Prism.
Microneutralization assays using the authentic SARS-CoV-2 viruses were performed as described previously in Vero E6-TMPRSS2 (24). Vero-TMPRSS2 cells were seeded in 96-well high binding cell culture plates (Costar Cat<ns/> 07620009, Corning) at a density of 20,000 cells/well in complete Dulbecco’s modified Eagle medium (cDMEM Cat<ns/>10-013-CV, Corning) one day prior to the infection. Heat-inactivated serum samples (56°C for 1 hour) were serially diluted (3-fold) in minimum essential media (MEM Cat<ns/>11430-030, Gibco) supplemented with 2 mM L-glutamine (Gibco Cat<ns/>25030081), 0.1% sodium bicarbonate (w/v, HyClone Cat<ns/>SH30033.01), 10 mM 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Cat<ns/> 15630080 Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin (P/S; Gibco) and 0.2% BSA (MP Biomedicals Cat<ns/>810063, CA, USA) starting at 1:10. Remdesivir (Medkoo Bioscience inc. Cat<ns/> 329511, NC, USA) was included to monitor assay variation. Serially diluted sera were incubated with 10,000 TCID50 per mL of Wuhan-like ancestral USA-WA1/2020 and PV44488/2021 (B.1.1.529, Omicron) for one hour at RT, followed by the transfer of 120 μl of the virus-sera mix to Vero E6-TMPRSS2 plates. Infection proceeded for one hour at 37°C and inoculum was removed. One hundred μl/well of the corresponding antibody dilutions plus 100 μl/well of infection media supplemented with 2% Fetal Bovine Serum (FBS, Cat<ns/>10423-028, Gibco) were added to the cells. Plates were incubated for 48 h at 37°C followed by fixation overnight at 4°C in 200 μl/well of a 10% formaldehyde solution. For staining of the nucleoprotein, formaldehyde solution was removed, and cells were washed with PBS (pH 7.4 Gibco) and permeabilized by adding 150 μl/well of PBS, 0.1% Triton X-100 (Fisher Bioreagents Cat<ns/> BP151-100, MA, USA) for 15 min at RT. Permeabilization solution was removed, plates were washed with 200 μl/well of PBS (Gibco) twice and blocked with PBS, 3% BSA for 1 hour at RT. During this time the primary antibody was biotinylated according to manufacturer protocol (Thermo Scientific EZ-Link NHS-PEG4-Biotin). Blocking solution was removed and 100 μl/well of biotinylated mAb 1C7C7 at a concentration of 1μg/ml in PBS, 1% BSA was added for 1 hour at RT. Cells were washed with 200 μl/well of PBS twice and 100 μl/well of HRP-conjugated streptavidin (Thermo Fisher Scientific) diluted in PBS, 1% BSA were added at a 1:2,000 dilution for 1 hour at RT. Cells were washed twice with PBS, and 100 μl/well of SigmaFast OPD (Sigma-Aldrich) were added for 10 min at RT, followed by addition of 50 μl/well of a 3 M HCl solution (Thermo Fisher Scientific). Optical density (OD) was measured (490 nm) using a microplate reader (Synergy H1; Biotek). Analysis was performed using GraphPad Prism 7 software. After subtraction of background and calculation of the percentage of neutralization with respect to the “virus only” control, a nonlinear regression curve fit analysis was performed to calculate the 50% inhibitory dilution (ID50), with top and bottom constraints set to 100% and 0% respectively. All samples were analyzed in a blinded manner.
The plaque assay was performed in the BSL-3 facility of the Icahn School of Medicine at Mount Sinai. The day before the assay, 3 x 105 cells of Vero-E6 cells or Vero-E6 TMPRSS2 T2A ACE2 cells were seeded in 12-well plates (Thermo Fisher Scientific) as previously described (3, 19). Lung and nasal turbinate homogenates were 10-fold serially diluted in infection medium (DMEM + 2% FBS + 1% P/S + 10 mM HEPES). After removing the media from the cells, 200 μL of each tissue homogenate dilution were inoculated onto each well. The dilutions used range from 10-1 to 10-6. The plates were incubated at 37°C for 1h with rocking every 15 min. Subsequently, the inoculum was removed and 1 mL of agar overlay consisting of 0.7% agar in 2x MEM + 2% FBS was placed onto each well. Once the agar was solidified, the plates were incubated at 37°C with 5% CO2. Two days later, the plates were fixed with 4% formaldehyde in PBS overnight before being taken out of the BSL-3 facility for subsequent staining in BSL-2 facility. The plaques were immuno-stained with an anti-SARS-CoV-2 NP primary mouse monoclonal antibody 1C7C7 kindly provided by Dr. James Duty at ISMMS (3, 21). Amersham ECL sheep anti-mouse IgG Horseradish Peroxidase (HRP)-linked whole secondary antibody (Cytiva Cat<ns/> NA931, RRID: AB_772210) was used at 1:2,000 and the plaques were visualized using TrueBlue Peroxidase Substrate (SeraCare Life Sciences Inc., <ns/>5510-0030).
Splenocyte isolation and intracellular cytokine staining was performed adapted from (25). Briefly, single suspension splenocytes were resuspended in R10 media and seeded in U-bottom 96-well plates (CELLSTAR, Greiner Bio-One North America Inc., Monroe, NC, USA) at an average of 2 ×106 cells/well containing anti-mouse CD28 (1:500, BD Biosciences Cat<ns/> 557393), brefeldin A (1:1000, GolgiPlug™, BD Biosciences Cat<ns/> 555029), and monensin (1:1,000, GolgiStop™, BD Biosciences Cat<ns/> 554724). Splenocytes were stimulated with PepMix™ SARS-CoV-2 or PepMixTM SARS-Cov-2 (Spike B.1.1.529/BA.1/Omicron, Cat<ns/>PM-WCPV-2, Cat<ns/>PM-SARS2-SMUT08-1, JPT Peptides), at a final individual peptide concentration of 5 µg/mL at 37 °C with 5% CO2 for 8-10 hours. Negative control cells were stimulated with an equivalent volume of DMSO. Positive control cells were stimulated with a cocktail containing phorbol 12-myristate 13-acetate (PMA, 0.5 mg/mL, Sigma-Aldrich) and ionomycin (1 mg/mL, Sigma-Aldrich). The unstimulated control cells were only treated with the R10 media. After stimulation, cells were washed with PBS containing 2% FBS and centrifuged at 350 x g for 5 min and then stained with Zombie Red™ diluted in PBS (1:500, BioLegend Cat<ns/> 423109) for 15 min at RT in the dark. Cells were washed in PBS containing 2% FBS (350 x g for 5 min) and incubated with surface staining cocktail containing Fc Block CD16/CD32 1:50 (BD Biosciences Cat<ns/> 553141, RRID: AB_394656) and the anti-mouse antibodies BV 711 CD3 (1:300, clone 17A2, BD, Cat<ns/> 100241, RRID: AB_256394), Pacific Blue CD4 (1:400, clone GK1.5, BioLegend, Cat<ns/> 100428, RRID: AB_493647), PerCP/Cy5.5 CD8 (1:200 clone 53-6.7, BioLegend, Cat<ns/>100734, RRID: AB_2075238) for 30 min at 4 °C in FACS buffer. Cells were washed in FACS buffer and then incubated in fixation/permeabilization buffer (CytoFix, BD Biosciences) for 5 min at 4°C. After fixation, cells were washed in 1 x permeabilization buffer (CytoPerm, BD Biosciences), then incubated with the intracellular staining cocktail containing anti-mouse antibodies Alexa Fluor 647 IFN-γ (1:400, clone XMG1.2, BioLegend, Cat<ns/> 505814, RRID: AB_49331), Alexa Fluor 488 TNF-α (1:300, clone MP6-XT22, BioLegend, Cat<ns/> 506313, RRID: AB_493328), PE/Cy7 IL-2 (1:300, clone JES6-5H4, BioLegend, Cat<ns/> 503832, RRID: AB_2561750), in 1x permeabilization buffer for 1 h at 4 °C. Samples were then washed in 1x permeabilization buffer and resuspended in PBS buffer for acquisition. Samples were acquired on an Aurora spectral cytometer (Cytek, Fremont, CA, USA) using SpectroFlo® software (Cytek). Analysis was performed with FCS Express 7 (DeNovo Software) and GraphPad Prism 9.5.1 (GraphPad Software 10.4.0).
One volume of blood was collected into four volumes of 0.5 M EDTA in centrifuge tubes to prevent coagulation, then pooled by group as previously described (19). Whole blood was stained with the direct addition of 1 μL of PE-conjugated H-2K(b) VNFNFNGL Tetramer (1:100, NIH Tetramer Core Facility). Tubes were flicked to mix reagents then incubated for 40 min at RT in the dark. After incubation, the following antibodies or fluorescent dyes were added directly to tubes containing tetramer-stained whole blood: 1 μL of Fc Block (BD, clone 2.4G2, RRID: AB_394656), 1 μL of CD8α PerCP (clone 53-6.7, BD, RRID: AB_394573), 1 μL of CD3ϵ Alexa Fluor 700 (clone 17A2, BioLegend, RRID: AB_493696), 0.5 μL of MHC II eFluor 450 (clone M5/115.15.2, Invitrogen, RRID: AB_1272204), and 0.5 μL of Fixable Viability Dye eFluor 450 (Invitrogen). Tubes were flicked to mix and incubated at RT for 20 min in the dark. After incubation, 1 mL of staining buffer was added to each tube to quench staining, then tubes were centrifuged at 400 x g for 5 min at 4°C. Supernatants were removed using a pipette, then to fix and lyse RBCs, cell pellets were resuspended with 200 μL of 1x eBioscience Foxp3/Transcription Factor Fixation/Permeabilization Buffer (Invitrogen), prepared according to manufacturer instructions. Cells were incubated for 10 min at RT in the dark. After incubation, cells were washed twice by adding 1 mL of staining buffer to each tube, centrifuging at 400 x g for 5 min at 4°C, then removing supernatants. After the final wash, cell pellets were resuspended with 200 μL of staining buffer. To prepare compensation controls, 1μL of each antibody was added to 1 drop of UltraComp eBeads Plus (Invitrogen) and incubated for 15 minutes before 200μL of staining buffer was added to the tube. Stained blood samples were measured on a Beckman Coulter Gallios flow cytometer equipped with Kaluza data acquisition software. Analysis was performed using FlowJo 10.8.1 (Treestar) and compensated using the built-in AutoSpill algorithm.
Paired t-test, one-way ANOVA, two-way-ANOVA or non-parametric Kruskal Wallis test were used to compare the different experiments. Statistical analyses were performed using Prism software (GraphPad 10.4.0).
A bioluminescence-based live imaging assay was first used to investigate the NDV tissue distribution in the mouse model. A recombinant NDV vector expressing an intracellular Firefly luciferase was used as a reporter virus for in vivo imaging (rNDV-luc, Supplementary Figure S1A) (17). The activity of the luciferase expressed by the rNDV-luc was confirmed in BSR-T7 cells upon infection (Supplementary Figure S1B) and in mice that were intranasally infected with rNDV-luc (FigureS1C-D). For live-imaging biodistribution study, rNDV-luc was administered intranasally (IN) or intramuscularly (IM) at 106 EID50 or 105 EID50 into BALB/c mice. Bioluminescence was measured every day until the signal was no longer detected. Luciferase activity peaked at 24 hours primarily at the site of administration and quickly declined to baseline after three days (Supplementary Figure S1E-I). We observed a very high luminescent signal in the lungs of mice that received rNDV-luc IN at 24 hours post inoculation (Supplementary Figure S1E, G). A transient increase of signal, that was much lower than that in the lungs, was observed in the abdomen of mice that received rNDV-luc IN, likely due to the swallowing of the material (Supplementary Figure S1H). In addition, we observed a slight increase of luminescence signal in the legs of mice that received the high-dose vaccine IM - also at 24 h post inoculation (Supplementary Figure S1E, I), although the difference was not significant. Overall, NDV was observed to be restricted in replication in mice (26–28).
Next, we investigated biodistribution of the live NDV-HXP-S vaccines. The prototype ancestral NDV-HXP-S was chosen as a representative since similar replication kinetics have been reported for all NDV-HXP-S variant vaccines (14, 21).Golden Syrian hamsters were selected due to their natural expression of the ACE2 receptor and their natural susceptibility to SARS-CoV-2 infection (29, 30). Hence, NDV-HXP-S virus tropism compared to the wildtype vector could be evaluated. The ancestral NDV-HXP-S vaccine was administered IN or IM at a dose of 107 EID50. The presence of NDV and the expression of the spike protein was evaluated after 1 day and 7 days post vaccination. A group vaccinated with the wild type NDV LaSota (WT NDV) and an unvaccinated group were included as controls (Figure 1a). The presence of infectious NDV was evaluated in lung homogenates, nasal washes, blood serum and urine samples by injecting 100 µl of the biological fluid into specific-pathogen free (SPF) embryonated chicken eggs. If infectious NDV is present, it is expected to present hemagglutination (HA) activity after incubation, which can be measured by HA assay. Then, the titer could be quantified by EID50 titrations as previously described (3) (Figures1b-e). Among all the biological samples collected, infectious NDV was only detected in two out of four lung homogenate samples of the WT NDV IN group and in one out of four lung homogenate samples from the NDV-HXP-S IN group, one day post-administration (Figure 1b). Importantly, these titers were much lower than the dose administered, suggesting a restricted replication of both WT NDV and NDV-HXP-S vaccine in the hamster model. Of note, NDV-HXP-S appeared to be attenuated as compared to the WT NDV based on the viral titers in the lung homogenates.
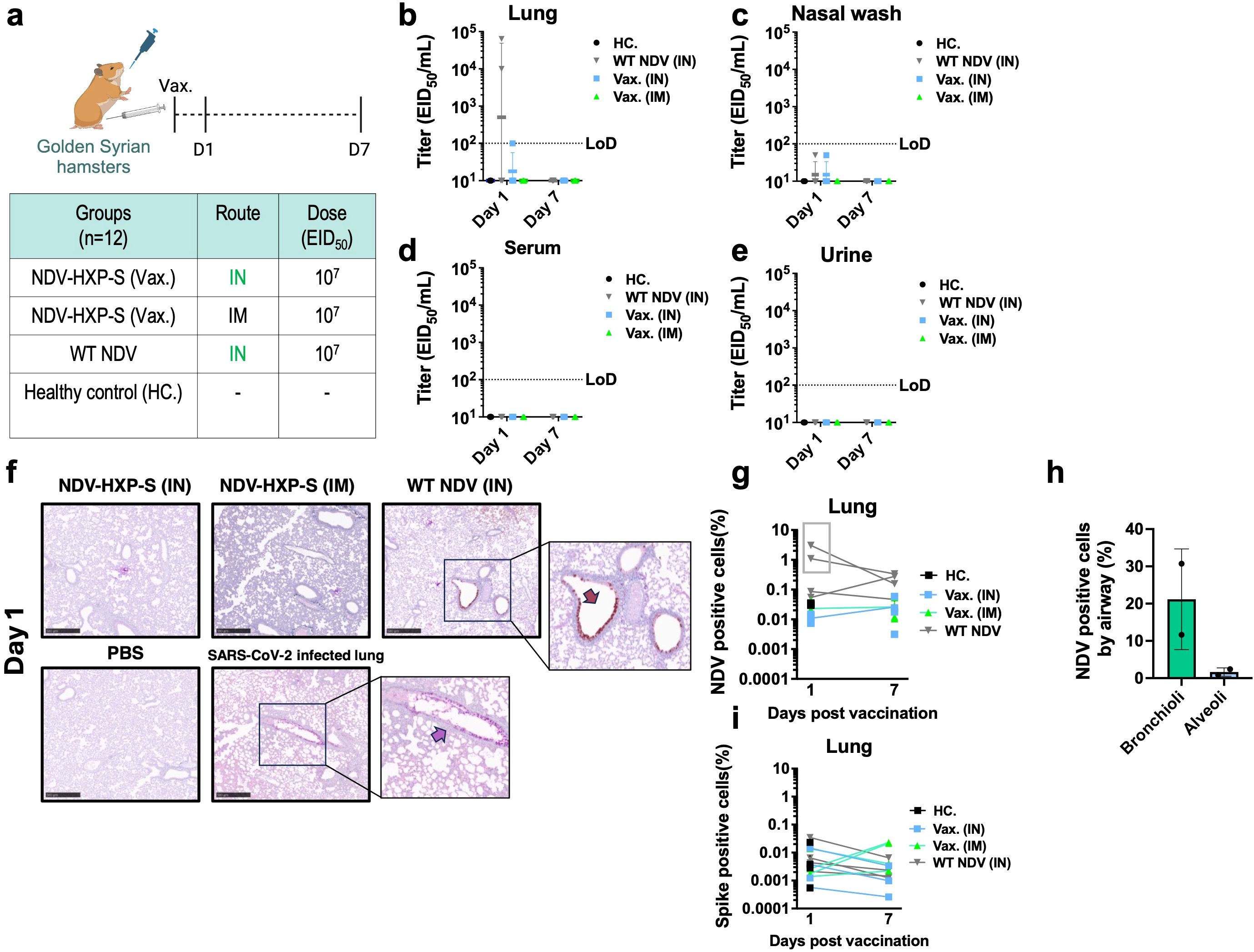
Figure 1. Biodistribution of live NDV-HXP-S vaccine in Golden Syrian hamsters. (a) Experimental design and vaccination groups. Golden Syrian hamsters (n=12) were immunized with a total dose of 107 EID50 of ancestral NDV-HXP-S via IN or IM route. Two more groups vaccinated with NDV LaSota (WT) via IN and PBS were used as controls. A total of 4 hamsters were analyzed per time point (b–e) At day 1 and day 7 after the vaccination, (b) lung homogenates, (c) nasal wash, (d) blood serum and (e) urine were collected and presence of infectious NDV was checked by injecting 100 µL of each biological fluid into each of the specific pathogen-free (SPF) embryonated chicken eggs. Viral titers were subsequently measured by EID50. (limit of detection equals to 100 EID50/mL; a titer of 10 EID50/mL was assigned to HA negative samples, and a titer of 50 EID50/mL was assigned to HA positive samples with no EID50/mL titer). (f-i) Left lobe was fixed in 4% paraformaldehyde in PBS (4% PFA, v/v) overnight and analyzed by immunohistochemistry (IHC) at day 1 and day 7 post vaccination. Presence of NDV viral protein or spike expression was measured with a rabbit polyclonal anti-NDV serum 1:2000 (shown in brown) or a human anti-SARS-CoV-2 spike 1A9 antibody 1:2000 (shown in pink). Examples of positively stained regions are indicated with arrows. Golden Syrian hamster lungs infected with SARS-CoV-2 (USA-WA1/2020), were used as spike staining positive control. The percentage of (g) NDV and (i) spike positive cells in each slide and (h) the percentage of NDV positive cells by airway in the two lung samples from the 1-day post administration of WT NDV group were measured with HALO software.
To examine the tropism of NDV-HXP-S relative to WT NDV in the respiratory tract, double staining of spike and NDV antigens by immunohistochemistry (IHC) was performed on formaldehyde-fixed parafilm embedded (FFPE) lungs on day 1 and day 7 after vaccination (Figure 1f). The presence of NDV infected cells was only detected in two samples in the WT NDV group on day 1 post-administration (highlighted within the grey rectangle, Figures 1f, g). In the WT NDV positive samples, bronchioles presented a higher degree of infection than the alveoli (Figure 1h). No presence of spike expression was detected in any of the samples (Figure 1i). In addition, viral genomic copies of NDV were quantified by reverse transcription-quantitative PCR in different tissues (RT-qPCR, Supplementary Figure S2). In agreement with replicative NDV shedding, high levels of viral RNA were only detected in the two lung homogenate samples from the WT NDV group that had measurable titers of infectious NDV (Supplementary Figure S2A). No viral RNA was detected in the brain (Supplementary Figure S2D). Very low levels of RNA were detected in leg muscles of animals that received the NDV-HXP-S IM (Supplementary Figure S2G).
A direct-contact transmission study was set up to test whether IN administration of NDV-HXP-S would enhance mucosal immunity and reduce viral shedding and transmission (31). To do so, a subset of Golden Syrian hamsters that were previously vaccinated with 107 EID50 of the live NDV-HXP-S (IM or IN, Figure 1a) were boosted again with the same regimen (Figure 2a). A robust humoral immune response was observed in serum samples even only after 1 dose (Figure 2b) in agreement with previous data (3, 21). Four weeks after the second boost, naïve Golden Syrian hamsters (Donors) were challenged with ancestral-like virus (USA-WA1/2020) at a dose of 105 plaque forming units (PFU) and co-housed with previously vaccinated hamsters (Recipients) (Figure 2a). Infected naïve donors showed weight loss after challenge (Figure 2c). In recipient hamsters, weight loss was only observed in WT NDV (grey line) or vaccinated IM groups (green line). But no weight loss was observed in hamsters vaccinated IN. (Figure 2c). SARS-CoV-2 shedding was measured in nasal washes on day 1 and day 3 post challenge (Figure 2d). Viral titers were detected in all donors (D) as well as WT NDV recipients (R), but not in the vaccinated ones. Viral titers at day 5 post-challenge measured in the nasal turbinate showed the greatest reduction in IN NDV-HXP-S vaccinated hamsters compared to the control group (WT NDV) (Figure 2e). Lower respiratory tract was equally protected by both IN and IM vaccinations, showing no detectable infectious SARS-CoV-2 in the lungs (Figure 2f) or SARS-CoV-2 antigens as measured by IHC (Figure 2g). This study demonstrated that the same live NDV-HXP-S vaccine that was administered via the mucosal route is better at preventing viral transmission than it administered systemically.
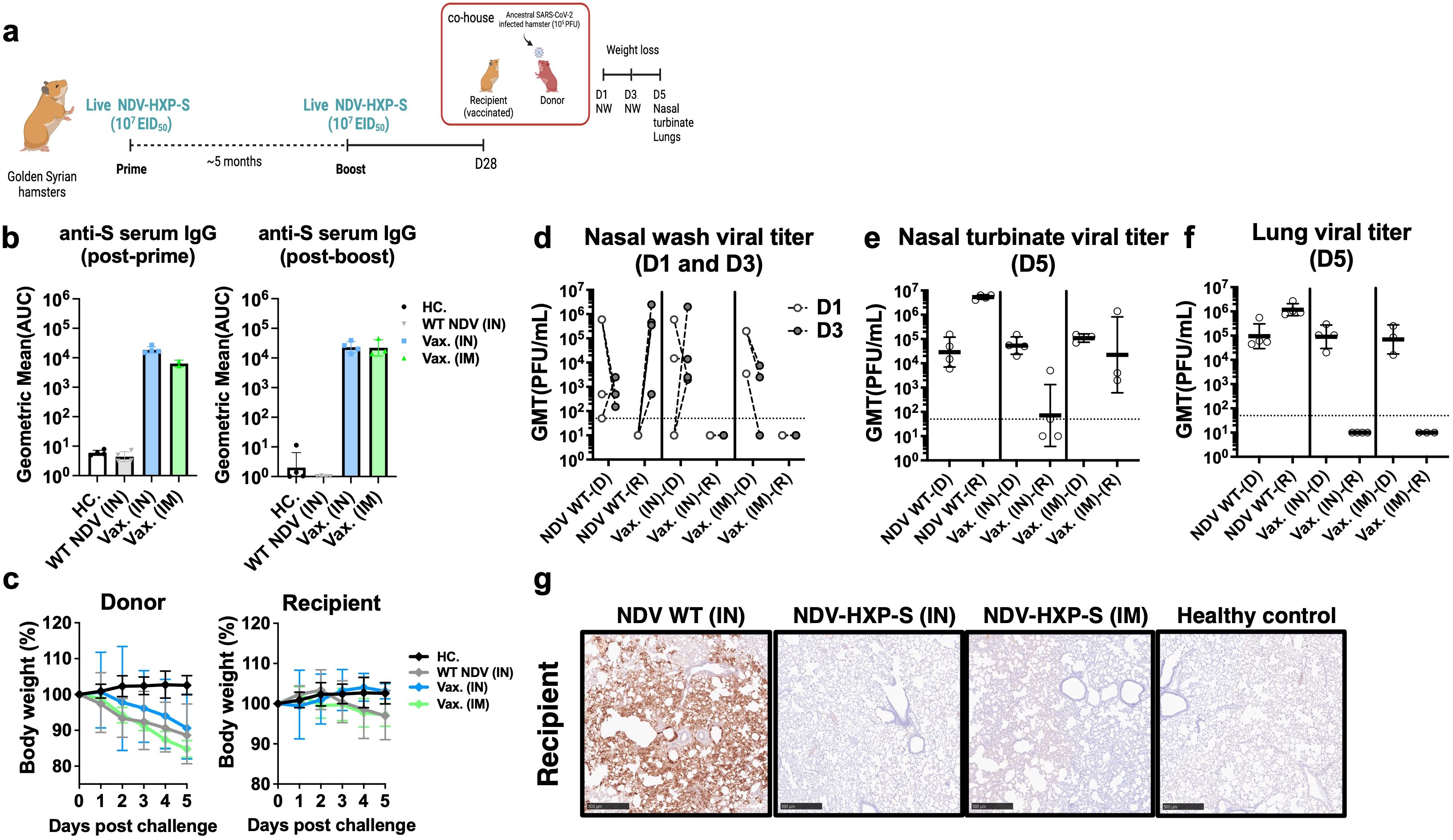
Figure 2. Live intranasal NDV-HXP-S vaccination reduces SARS-CoV-2 viral shedding in the upper respiratory tract in direct-contact transmission study. (a) Experimental design. Eight- to ten-week-old female Golden Syrian hamsters (n= 3 or 4) were either vaccinated with 107 EID50 of live NDV-HXP-S intranasally (IN) or intramuscularly (IM), or WT NDV IN (negative control). Two immunizations were performed with 5-month interval. Twenty-eight days after second boost, naïve hamsters were challenged with 105 PFU of ancestral (USA-WA1/2020) strain (Donors) and co-housed in pairs with vaccinated hamsters from different vaccinated groups (Recipient) and transmission was monitored in a direct-contact model. (b) Spike-specific IgG serum antibody titers were measured 5 months post-prime and 28 days post-boost, respectively. The geometric mean titer (GMT) of the area under the curve (AUC) ± the standard deviation (SD) is depicted. (c) Weight loss was monitored for 5 days. (d) Nasal washes were collected 1 and 3 days post challenge. At 5 days post challenge, (e) nasal turbinate and (f) right lung lobes were harvested and homogenized in 1 mL PBS and SARS-CoV-2 titer was measured by plaque assay. (D: Donor, R: recipient) (g) Left lobes were fixed in 4% PFA (v/v) and analyzed by IHC with a mouse anti-N monoclonal (1C7C7) (1:50, brown). Viral titers were measured by plaque assay on Vero E6 cells and were plotted as GMT of PFU/mL (limit of detection equals to 50 PFU/mL; a titer of 25 PFU/mL was assigned to negative samples).
In previous studies, we described the development of novel NDV-HXP-S variant-based vaccines (14, 19) and the possibility to combine them in a multivalent formulation to extend protection to mismatched strains not present in the formulation (14). Here, we further investigated the use of this prototype multivalent NDV-HXP-S formulation as a mucosal vaccine. This vaccine combines ancestral (Wuhan), Beta and Delta NDV-HXP-S in equal amounts (14). BALB/c mice were vaccinated in a 2-dose regimen with live trivalent NDV-HXP-S vaccine and spike-specific IgA titers were examined at different mucosal surfaces (Supplementary Figure S3) (14, 32, 33). The live trivalent vaccine induced mucosal spike-IgA at local mucosae (nasal wash, BAL, mouthwash) (Supplementary Figure S3C-E), but also at distal mucosae (vaginal lavage, intestinal lavage and feces samples) (Supplementary Figure S3F-H), demonstrating that the live NDV vector is a strong stimulator of mucosal immunity (34).
We then investigated the kinetics of humoral immune responses induced by IN vaccination in mice. BALB/c mice were vaccinated with live monovalent or trivalent NDV-HXP-S at 106 EID50 in a 1-dose or 2-dose regimen (Figures 3a, b). Spike-specific IgG antibody titers against ancestral Wuhan and Omicron BA.1 spike proteins (as a surrogate of cross-reactivity) were measured every other week for 30 weeks (Figures 3c–f). A dose-dependent and formulation-dependent kinetics was observed. Spike-specific IgG serum antibody titers presented an exponential increase in the first 6 weeks that later plateaued (Figures 3c, d). In the case of the 2-dose regimen, antibody titers were boosted by the second dose given at week-10 (Figures 3e, f). At the early time point of week-6, 1-dose of trivalent vaccine already presented higher antibody titers than 1-dose of monovalent vaccine (Figures 3g, k). The high spike and RBD antibody titers against both Wuhan (Figures 3h, i) and Omicron BA.1 (Figures 3l, 3m) showed by 2-dose trivalent NDV-HXP-S correlated with high neutralizing capacity against both viruses (Figures 3j, n), confirming the great potential of this new mucosal formulation.
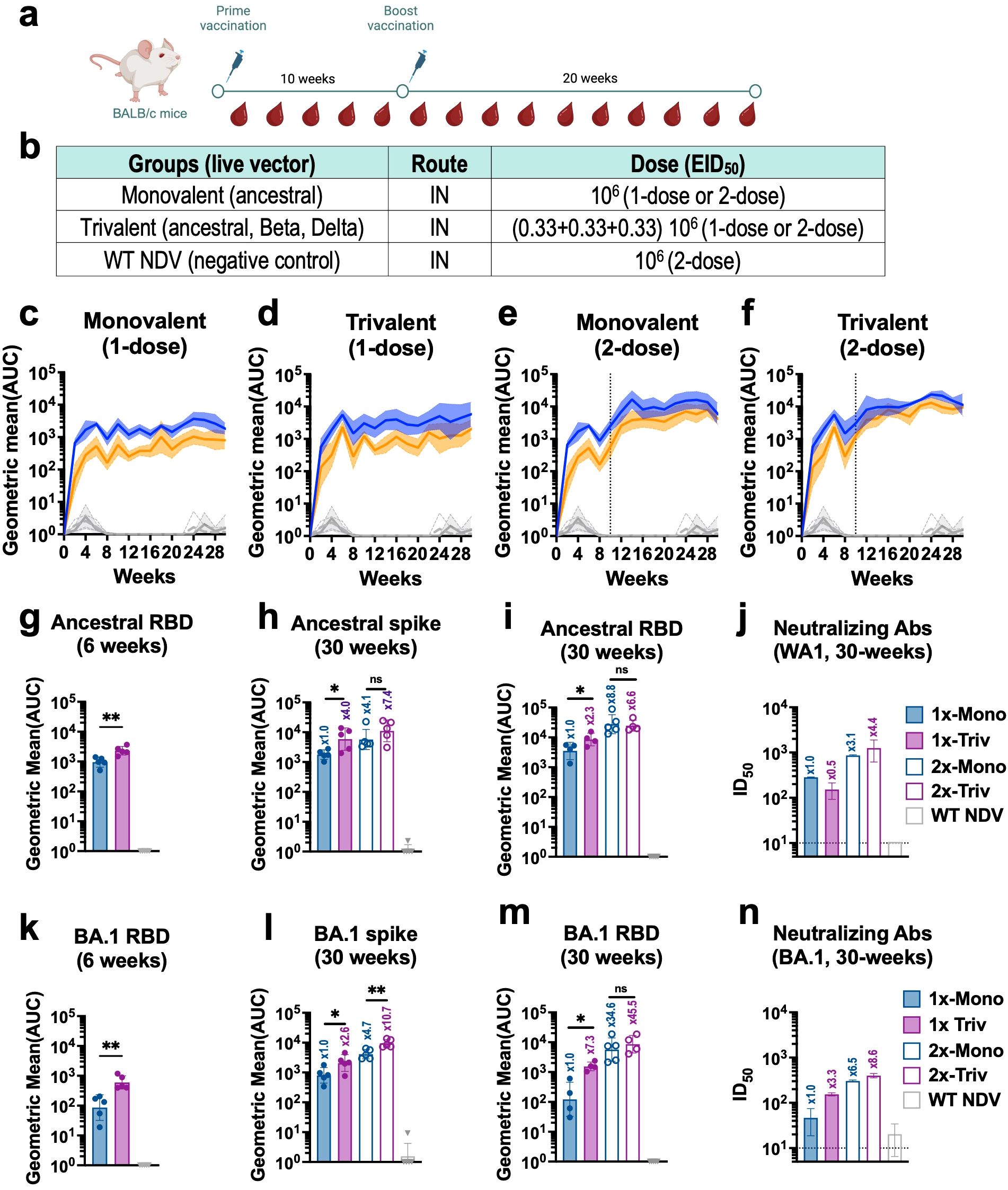
Figure 3. Live intranasal trivalent NDV-HXP-S vaccination induces superior cross-reactive humoral immune responses against phylogenetically distant SARS-CoV-2 variants in mice. (a) Experimental design and (b) vaccination groups. Eight- to ten-week-old female BALB/c mice were vaccinated with 106 EID50 of live NDV-HXP-S monovalent or trivalent vaccines administered intranasally (IN) in a 1- or 2-dose regimen with a ten-week interval between doses. Mice vaccinated with WT NDV at a dose of 106 EID50 were used as negative control. (c-f) Spike-specific IgG serum antibody titer kinetic (n=5) against ancestral (Wuhan, blue) or Omicron BA.1(B.1.1.529, orange) spike was measured every other week by ELISA for thirty -weeks. Individual kinetics for (c) monovalent 1-dose, or (e) 2-doses, (d) trivalent 1-dose or (F) 2-doses, are shown respectively. Spike-specific serum IgG of NDV WT vaccinated group against ancestral (light grey dotted line) or Omicron BA.1 (dark grey line) are also depicted in these graphs for comparison. Ancestral serum IgG antibody titers against (g) RBD at 6-weeks, (h) Spike at 30-weeks, (i) RBD at 30-weeks and (j) microneutralization assays against ancestral virus (USA-WA1/2020) at 30-weeks. Omicron BA.1 serum IgG antibody titers against (k) RBD at 6-weeks, (l) Spike at 30-weeks, (m) RBD at 30-weeks and (n) microneutralization assays against Omicron BA.1. One-tailed unpaired t-test are depicted (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Microneutralization assays against ancestral (USA-WA1/2020) (j) and Omicron BA.1 (n) were performed in technical duplicated from pooled sera. GMT ± SD is depicted.
To test the in vivo protection of the IN trivalent NDV-HXP-S vaccine, Golden Syrian hamsters were vaccinated at a final dose of 106 EID50 to observe greater differences between formulations in an in vivo challenge model (Figure 4a), as previously shown (21). Four different vaccination regimens were compared as shown in Figure 4b: live monovalent NDV-HXP-S given IN or IM and live trivalent NDV-HXP-S given IN or IM. A group vaccinated IN with WT NDV was included as negative control. Hamsters were vaccinated in a 12-week interval to allow for the immune response to plateau as shown in Figure 3. Twelve weeks after the second boost, hamster serum samples were collected. Ancestral spike-specific (Figure 4c), Omicron BA.1 spike-specific (Figure 4d) and RBD-specific (Figure 4e) antibody titers, and neutralizing antibody titers against ancestral (Figure 4f) and Omicron BA.1 (Figure 4g) were measured. While both IN live monovalent and trivalent NDV-HXP-S vaccines elicited the highest levels of binding-antibody titers (Figure 4c-e), trivalent formulations provided more cross-reactive neutralizing antibody titers against Omicron BA.1 than the monovalent ones (Figure 4g).
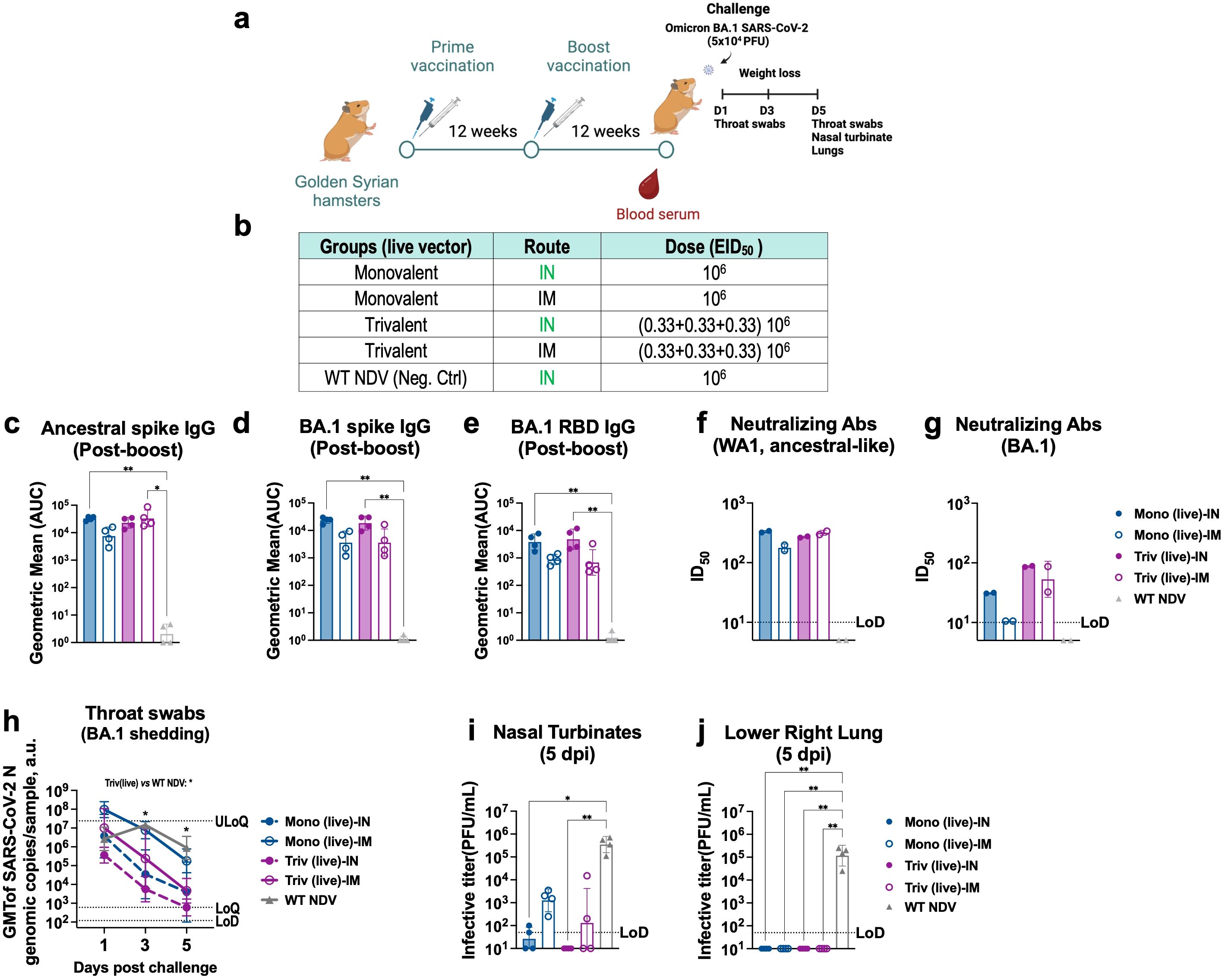
Figure 4. Intranasal and intramuscular vaccination of live trivalent NDV-HXP-S confer better protections against BA.1 than the monovalent vaccines in hamsters. (a) Experimental design and (b) groups. Eight- to ten-week-old female Golden Syrian hamsters (n= 4) were either vaccinated with live monovalent (Mono) or trivalent (Triv) NDV-HXP-S or WT NDV (negative control), intranasally (IN, green) or intramuscularly (IM, blue). Two immunizations were performed with 12-week interval. Twelve weeks after the second boost, hamsters were challenged with 5x104 PFU of BA.1 (B.1.1.529) strain and infection was monitored for five days. (c) Ancestral Spike-specific (d) Omicron BA.1 Spike-specific or (e) RBD-specific serum IgG antibody titers, neutralizing antibodies against (f) ancestral-like and (g) Omicron BA.1strains were measured 12 weeks post-boost vaccinations, respectively. The geometric mean titer (GMT) of the area under the curve (AUC) ± the standard deviation (SD) is depicted. (h) Throat swabs were collected 1, 3 and 5 days post-challenge and SARS-CoV-2 N genomic copies per sample were measured by RT-qPCR (limit of detection [LoD] equals to 120 copies; limits of quantification [LoQ] equals 600 copies and upper limit of quantification [ULoQ] equals to 2.4 x107 copies). (i) Nasal turbinate and (j) lower right lung lobes were harvested and homogenized in 1 mL PBS. Viral titers were measured by plaque assay on Vero E6-TMPRSS2-ACE2 cells and were plotted as GMT of PFU/mL (limit of detection [LoD] equals to 50 PFU/mL; a titer of 10 PFU/mL was assigned to negative samples). Non-parametric Kluskal-Wallis test is depicted (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Hamsters were next challenged with Omicron BA.1 strain and throat swab samples were taken 1, 3 and 5-days post challenge to monitor viral oral shedding (Figure 4h). Among the different regimens tested, IN trivalent NDV-HXP-S vaccine showed the fastest elimination of Omicron BA.1 viral copies in the throat (Figure 4h, dark pink dotted line, p value<0.05 versus. NDV WT group at 3 and 5 dpi), followed by IN monovalent vaccine (Figure 4g, dark blue dotted line), again demonstrating the advantages of the mucosal vaccine at preventing viral shedding. The live trivalent vaccine given IM (Figure 4h, dark pink solid line) also reduced oral viral copies more efficiently than the monovalent vaccine given IM (Figure 4h, dark blue solid line). The lowest level of throat viral copies in animals IN vaccinated with the live trivalent vaccine correlated with the absence of infectious virus in nasal turbinate measured by plaque assay (Figure 4i). Infection was detected in the other vaccinated groups being the highest in animals given IM monovalent NDV-HXP-S vaccine. No infectious viruses were detected in the lungs of vaccinated animals (Figure 4j). Collectively, these data show that the presence of mucosal immunity provided by IN vaccination and the elicitation of cross-reactive antibodies by the trivalent formulation additively contribute to reduce Omicron BA.1 in the upper respiratory tract. Overall, an improved in vivo protection against Omicron BA.1 infection was observed with a live trivalent NDV-HXP-S vaccine formulation, administered intranasally or intramuscularly, in that intranasal vaccination is more effective than intramuscular vaccination.
By the third quarter of 2022, ninety six percent of the population older than 16 years had SARS-CoV-2 antibodies from previous infection or vaccination (35). Despite of this high pre-existing immunity, the emergence of new VOCs, like XBB.1.5, JN.1 or KP.2 which evade previous immunity, still poses a public health problem in the post-pandemic era. We evaluated whether a trivalent NDV-HXP-S vaccine would be able to boost the same cross-reactive immune response in the context of mass pre-existing immunity. To do so, 8- to 10-week-old 129 mice were vaccinated with two doses of Comirnaty (Pfizer BioNTech) mRNA vaccine at a dose of 0.25 µg per mouse to reflect real world scenario in human populations. The 129-mouse model has shown great promise to characterize SARS-CoV-2 immunogenicity (20, 36). Ten months later, we vaccinated them with either inactivated NDV-HXP-S given IM (Supplementary Figure S4A) or live/inactivated NDV-HXP-S given IN (Figure 5a). Naïve age-matched mice were vaccinated in a 2-dose regimen of monovalent or trivalent NDV-HXP-S vaccine as positive vaccination controls. A no booster control group and naïve mice negative control group were included. For pre-immune mice, three-weeks after the booster, animals were euthanized, and spike-specific antibody levels were measured. A boost in ancestral and Omicron BA.1 spike-specific and RBD-specific antibody titers, was obtained with the IM inactivated vaccine boosters (Supplementary Figure S4C). This increase correlated with enhanced neutralizing antibody titers against both viruses, proving the capacity of NDV-HXP-S vaccine to work as a third booster (Supplementary Figure S4E). However, in any case, the trivalent booster showed advantage (even less) over the ancestral vaccine at inducing cross-reactive antibodies against BA.1. This suggested that a strong immune imprinting by the ancestral mRNA vaccine might restrain the de novo Omicron-specific responses induced by the trivalent vaccines in the model tested.
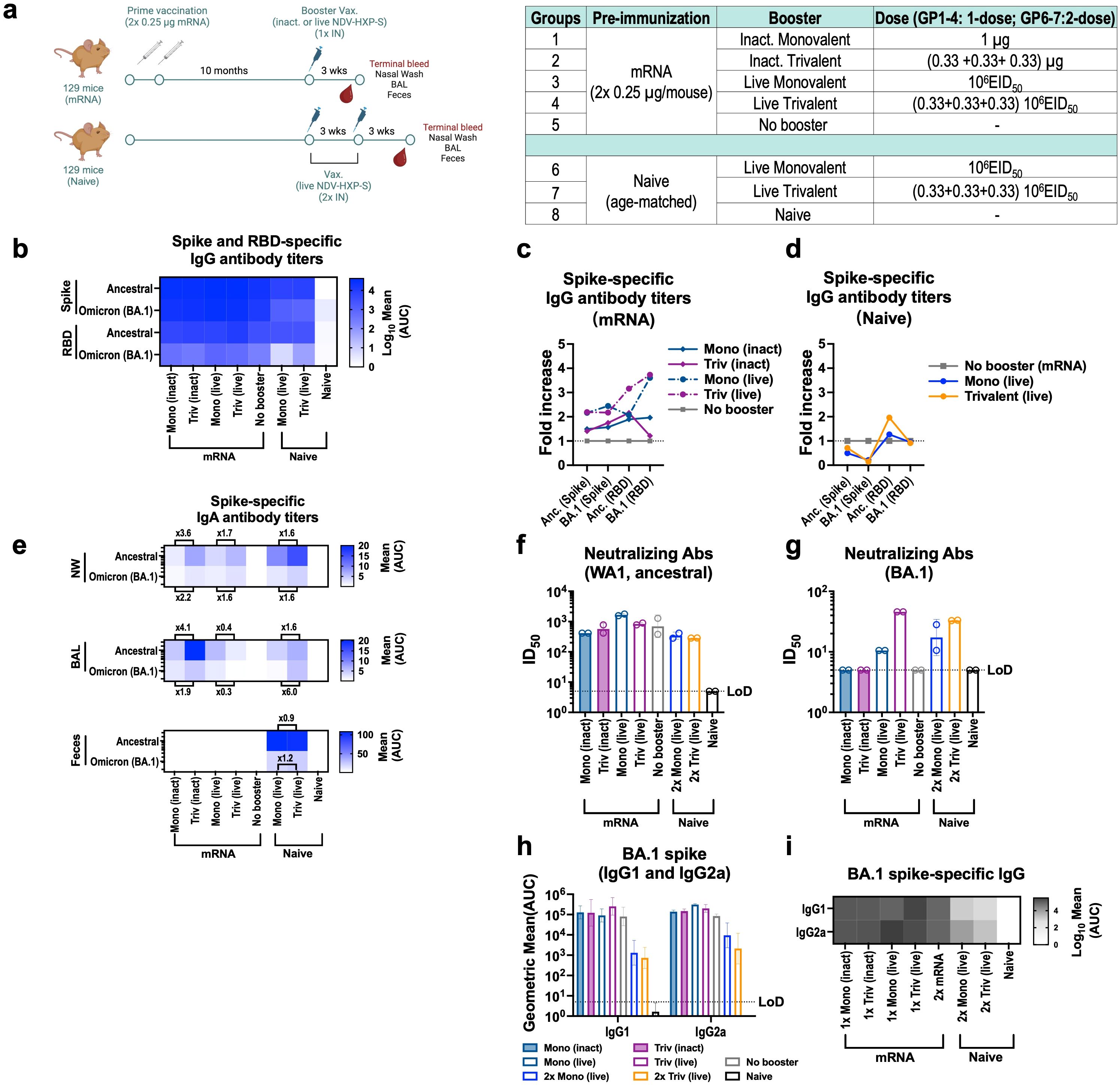
Figure 5. Intranasal live NDV-HXP-S vaccines boost humoral and mucosal immunity after two doses of 0.25 µg mRNA in mice. (a) Experimental design and vaccination groups. Eight- to ten-week-old female 129 mice were vaccinated with two doses 0.25 µg of Comirnaty mRNA vaccine (Pfizer) and 10 months later, mice were intranasally (IN) boosted with either 1 µg of inactivated or 106 EID50 of live monovalent or trivalent NDV-HXP-S vaccine. Naïve age-matched mice were vaccinated in a two-dose regimen with 106 EID50 total dose of monovalent or trivalent NDV-HXP-S vaccine as positive vaccination controls and a no booster group and complete naïve mice were used as negative controls. Blood serum, nasal washes, BAL, feces and spleen were harvested 3-weeks after the boost. (b) Heatmap of mean spike and RBD-specific IgG serum antibody titers and (c-d) fold-increase in serum antibody titers of boosted groups over no booster control against ancestral (Wuhan) or Omicron BA.1 proteins (n=4) measured by ELISAs 21 days after last boost. Inactivated groups are shown in continuous lines whereas live are shown in discontinuous lines in Fig (c) The GMT of fold increase of serum antibody titers over two doses 0.25 µg (grey squares) of Comirnaty mRNA vaccine (Pfizer) are depicted. (e) Heatmap of mean spike-specific IgA antibody titers against ancestral (Wuhan) or Omicron BA.1 spike (n=4) measured 21 days after last boost by ELISAs in nasal washes (NW), bronchioalveolar lavage (BAL) and feces samples. (f-g) Neutralizing activity of post-boost pooled sera was tested in microneutralization (MNT) assays against USA-WA1/2020 strain, and Omicron BA.1variant in technical duplicates. GMT serum dilutions inhibiting 50% of the infection (ID50) is plotted (limit of detection equals to 10 and a value of 5 and was assigned to negative samples). GMT ID50 ± SD are depicted. (h-i) Omicron BA.1 serum IgG1 and IgG2a antibody titers (n=3-4).
Next, IN booster vaccines were compared in the same model (Figure 5). Four formulations were tested: inactivated monovalent, inactivated trivalent NDV-HXP-S vaccine and their live-vectored version, all administered IN. We included the inactivated versions to examine the contribution of the live nature of the vector to the vaccine humoral, mucosal and cellular immunity (Figure 5a). Three weeks after the booster, spike-specific and RBD-specific serum antibodies against ancestral Wuhan and Omicron BA.1 were measured, shown as a heatmap (Figure 5b) and fold-increase of titers over no booster group was calculated (Supplementary Figure S5C-D). IN live boosters presented a higher increase in serum IgG antibody titers against the ancestral and BA.1 spike/RBD (Figure 5c, dark blue and dark pink dotted lines) than IN inactivated vaccines (Figure 5c, dark blue and dark pink solid lines). The induction of mucosal immunity after the booster was next measured in the upper and lower respiratory tract and in the gut by collecting nasal washes (NW), BAL fluid (BALF) and feces, respectively (Figure 5e). mRNA immunized mice without the booster showed no spike-specific IgA antibodies whereas mice receiving any of the four IN boosters developed spike specific IgA titers in both nasal washes and BALF. Interestingly, inactivated vaccines showed higher IgA values than the live versions. This result needs to be explored in future studies to understand its biological relevance. Of note, in contrast to the IM inactivated booster in Supplementary Figure S4F, live trivalent NDV-HXP-S vaccine induced more Omicron BA.1 neutralizing antibody titers than the live monovalent vaccine (Figure 5g), while both formulations induced marginal increase of neutralizing antibodies against the ancestral virus (Figure 5f). This suggested a mucosal booster after a systemic primary vaccination might be able to generate de novo antibody responses. Finally, IgG antibody subclasses were examined (Figures 5h-i), where a boost in these two subclasses was observed for all boosters, with the highest one reported in the live trivalent group.
Cellular immunity was measured by MHC-I spike-specific VNFNFNGL-tetramer staining of peripheral blood and intracellular cytokine staining (ICS) of splenocytes 3 weeks after the booster (Supplementary Figure S5). For ICS, ancestral and Omicron BA.1 spike-peptide pool stimulated polyfunctional T cells expressing interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α) and interleukne-2 (IL-2) were compared. Mice vaccinated with two doses of mRNA developed antigen-specific CD4+ and CD8+ T cells (Supplementary Figure S5A-D), as well as circulating SARS-CoV-2 tetramer-specific CD8+ T cells (Supplementary Figure S5E). A recall of polyfunctional CD4+ T cells was observed for all the booster formulations to various degrees among which the live monovalent vaccine induced the most robust response (Supplementary Figure S5A, C). The live monovalent vaccine also increased polyfunctional CD8+ T cells more predominantly than the other groups (Supplementary Figure S5B, D).
NDV has proven to be a safe viral vector vaccine in several preclinical and clinical studies (5–8, 11, 37). Here, biodistribution of the avirulent live NDV LaSota vector has shown to be limited to the site of administration in mice and hamsters. As an avian paramyxovirus, NDV predominantly enters cells by binding to cell receptors with terminal 2,3α sialic acids (38, 39). Presence of 2,3α sialic acid has been described in several cell types in the respiratory airways of BALB/c mice, Golden Syrian hamsters, and humans, which support the NDV entry in those tissues (38, 40–42). Despite this restricted replication, the live NDV-HXP-S proved to be highly immunogenic even after only one dose in mice and hamsters. Furthermore, it was able to reduce SARS-CoV-2 viral shedding in the upper respiratory tract when given IN. This proof of concept shows great promise toward the use of NDV-HXP-S to reduce SARS-CoV-2 transmission in high-risk populations like the elderly and people with comorbidities for whom a faster decay of antibodies after prime vaccination has been reported (43–45).
Aiming to improve the breadth of protection, we optimized a trivalent formulation combining the ancestral, Beta and Delta spikes, which previously had shown to induce better neutralizing antibodies than the ancestral monovalent vaccine against mismatched VOIs, using IM vaccination (14). Here, we investigated the immune responses induced by the live multivalent vaccine with the same variant components. We demonstrated that the live IN trivalent NDV-HXP-S vaccine was superior at generating cross-neutralizing antibodies against Omicron BA.1 over the ancestral monovalent vaccine in the two animal models tested. These cross-reactive antibodies seem to target the RBD region (i.e. higher BA.1 RBD-binding antibodies with trivalent vaccines) (46).
Due to rapid evolution of the SARS-CoV-2 variants and their immune evasion, several health agencies have recommended booster vaccinations to be changed to match the circulating variant (47–49). In this work, we explored the use of a broadly cross-reactive multivalent vaccine as an alternative to strain-specific boosters. In mRNA vaccinated mice via the intramuscular route, we saw no difference between the ancestral NDV-HXP-S vaccine versus the trivalent formulation when the inactivated vaccine booster was given IM, likely due to immune imprinting (50, 51). On the other hand, the IN administration of the live vaccine appeared to show a different stimulation of the humoral immune responses that might induce de novo variant-specific antibodies (25). Previously, we observed that an Omicron NDV-HXP-S IN booster could induce a higher level of antibodies against the Omicron variant, than the ancestral NDV-HXP-S, in ancestral mRNA vaccine pre-immune mice (25). These two studies combined suggest that a mucosal booster might better overcome the immune imprinting barrier induced by a previous systemic vaccine than a systemic booster. But future studies are imperative to validate such a hypothesis.
Overall, these results provided proof of concept for the development of mucosal multivalent NDV-HXP-S as next-generation COVID-19 boosters. Nonetheless, this study has several limitations. Preclinical studies in mice and hamsters may present a different immunogenic response than those expected in humans. The mechanisms of the difference between 1-dose and 2-dose intranasal live trivalent vaccination remain to be investigated. In addition, future studies investigating multivalent NDV-HXP-S formulations including more recent variants will be necessary to further extend the cross-protection against new variants like JN.1 or KP.2.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Icahn School of Medicine at Mount Sinai (ISMMS) Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
IG-D: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. AAbd: Formal Analysis, Investigation, Methodology, Writing – review & editing. TL: Investigation, Methodology, Writing – review & editing. MB: Investigation, Methodology, Writing – review & editing. SM: Investigation, Methodology, Writing – review & editing. NL: Investigation, Methodology, Writing – review & editing. SS: Methodology, Resources, Writing – review & editing. GS(8th author): Methodology, Resources, Writing – review & editing. PW: Methodology, Resources, Writing – review & editing. TY: Methodology, Resources, Writing – review & editing. AAba: Methodology, Resources, Writing – review & editing. JC: Methodology, Resources, Writing – review & editing. VD: Investigation, Writing – review & editing. JM-G: Investigation, Writing – review & editing. GS(15th author): Writing – review & editing, Resources. MB-V: Methodology, Writing – review & editing. LC: Writing – review & editing, Methodology. MS: Funding acquisition, Resources, Writing – review & editing. FK: Funding acquisition, Resources, Writing – review & editing. PP: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. WS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was partially funded by the NIAID Centers of Excellence for Influenza Research and Response (CEIRR) subcontract 75N93021C00014 (to P.P.) and by the NIAID Collaborative Vaccine Innovation Centers contract 75N93019C00051 (to F.K., W.S. and P.P.), by NIAID grant R01 AI145870 (to P.P.) and by institutional funding from the Icahn School of Medicine at Mount Sinai (to P.P., W.S). SARS-CoV-2 work in the Schotsaert lab is funded by NIH/NIAID R01AI160706, NIH/NIAID R21AI176069 and NIH/NIDDK R01DK130425 (to M.S.).
Animal workflows were created in BioRender. Sun, W. (2022) BioRender.com/b87k447. We kindly appreciate the work of Randy Albrecht, Carlos Franco and the CCMS team overseeing the CBSL-3 and ABSL-3 facilities (ISMMS, New York). FFPE block preparation, FFPE RNA extraction and immunohistochemistry analyses were performed in the Biorepository and Pathology Core, where the help of Dr. Monica Garcia-Barrios is the optimization of the IHC analyses is greatly appreciated (ISMMS, New York). The feedback of Dr. Ester Munera Maravilla and Dr. Rebeca Blanch to set up the RT-qPCR method is greatly appreciated. The Conventional Biocontainment Facility (CBF) and IMI Suite 13 ABSL-3 vivarium are NIHBSL3/BSL3 facilities that are part of the BSL-3 Biocontainment CoRE. This core is supported by subsidies from the ISMMS Dean’s Office and by investigator support through a cost recovery mechanism. Research reported in this publication was supported by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Number G20AI174733 (R.A. Albrecht). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The Icahn School of Medicine at Mount Sinai has filed patent applications entitled “RECOMBINANT NEWCASTLE DISEASE VIRUS EXPRESSING SARS-COV-2 SPIKE PROTEIN AND USES THEREOF” which names IG-D, PP, FK and WS as inventors. Mount Sinai is seeking to commercialize this vaccine; therefore, the institution and its faculty inventors could benefit financially. Mount Sinai has spun out a company, CastleVax to commercialize the NDV-based SARS-CoV.2 vaccine. PP, FK and WS serve on the scientific advisory board of CastleVax and are listed as co-founders of the company. FK has consulted for Merck, Seqirus, Curevac and Pfizer, and is currently consulting for Pfizer, Third Rock Ventures, GSK and Avimex. The FK laboratory is also collaborating with Pfizer on animal models of SARS-CoV-2. The M.S. laboratory has received unrelated research funding in sponsored research agreements from 7Hills Pharma, ArgenX N.V., Moderna and Phio Pharmaceuticals, which has no competing interest with this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1524477/full#supplementary-material
1. Mathieu E, Ritchie H, Rodés-Guirao L, Appel C, Giattino C, Hasell J, et al. Coronavirus pandemic (COVID-19) (2020). Available online at: https://ourworldindata.org/covid-vaccinationscitation (Accessed July 11, 2023).
2. Krammer F. The role of vaccines in the COVID-19 pandemic: what have we learned? Semin. (2023) 45(4-6):451–68. doi: 10.1007/s00281-023-00996-2
3. Sun W, Liu Y, Amanat F, Gonzalez-Dominguez I, McCroskery S, Slamanig S, et al. A Newcastle disease virus expressing a stabilized spike protein of SARS-CoV-2 induces protective immune responses. Nat Commun. (2021) 12:6197. doi: 10.1038/s41467-021-26499-y
4. Sparrow E, Wood JG, Chadwick C, Newall AT, Torvaldsen S, Moen A, et al. Global production capacity of seasonal and pandemic influenza vaccines in 2019. Vaccine. (2021) 39:512–20. doi: 10.1016/j.vaccine.2020.12.018
5. Lara-Puente JH, Carreno JM, Sun W, Suarez-Martinez A, Ramirez-Martinez L, Quezada-Monroy F, et al. Safety and immunogenicity of a newcastle disease virus vector-based SARS-coV-2 vaccine candidate, AVX/COVID-12-HEXAPRO (Patria), in pigs. mBio. (2021) 12:e0190821. doi: 10.1128/mBio.01908-21
6. Tcheou J, Raskin A, Singh G, Kawabata H, Bielak D, Sun W, et al. Safety and immunogenicity analysis of a newcastle disease virus (NDV-HXP-S) expressing the spike protein of SARS-coV-2 in sprague dawley rats. Front Immunol. (2021) 12:791764. doi: 10.3389/fimmu.2021.791764
7. Pitisuttithum P, Luvira V, Lawpoolsri S, Muangnoicharoen S, Kamolratanakul S, Sivakorn C, et al. Safety and immunogenicity of an inactivated recombinant Newcastle disease virus vaccine expressing SARS-CoV-2 spike: Interim results of a randomised, placebo-controlled, phase 1 trial. EClinicalMedicine. (2022) 45:101323. doi: 10.1016/j.eclinm.2022.101323
8. Duc Dang A, Dinh Vu T, Hai Vu H, Thanh Ta V, Thi Van Pham A, Thi Ngoc Dang M, et al. Safety and immunogenicity of an egg-based inactivated Newcastle disease virus vaccine expressing SARS-CoV-2 spike: Interim results of a randomized, placebo-controlled, phase 1/2 trial in Vietnam. Vaccine. (2022) 40:3621–32. doi: 10.1016/j.vaccine.2022.04.078
9. Ponce-de-Leon S, Torres M, Soto-Ramirez LE, Calva JJ, Santillan-Doherty P, Carranza-Salazar DE, et al. Interim safety and immunogenicity results from an NDV-based COVID-19 vaccine phase I trial in Mexico. NPJ Vaccines. (2023) 8:67. doi: 10.1038/s41541-023-00662-6
10. López-Macias C, Torres M, Armenta-Copca B, Wacher NH, Castro-Castrezana L, Colli-Dominguez AA, et al. Phase II study on the safety and immunogenicity of single-dose intramuscular or intranasal administration of the AVX/COVID-12 "Patria" recombinant Newcastle disease virus vaccine as a heterologous booster against COVID-19 in Mexico. Vaccine. (2025) 43(Pt 2):126511. doi: 10.1016/j.vaccine.2024.126511
11. Fulber JPC, Kamen AA. Development and scalable production of newcastle disease virus-vectored vaccines for human and veterinary use. Viruses. (2022) 14(5):975. doi: 10.3390/v14050975
12. Burman B, Pesci G, Zamarin D. Newcastle disease virus at the forefront of cancer immunotherapy. Cancers (Basel). (2020) 12(12):3552. doi: 10.3390/cancers12123552
13. Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. (2006) 13:221–8. doi: 10.1016/j.ymthe.2005.08.016
14. Gonzalez-Dominguez I, Martinez JL, Slamanig S, Lemus N, Liu Y, Lai TY, et al. Trivalent NDV-HXP-S vaccine protects against phylogenetically distant SARS-coV-2 variants of concern in mice. Microbiol Spectr. (2022) 10(3):e0153822. doi: 10.1128/spectrum.01538-22:e0153822
15. Gonzalez-Dominguez I, Martinez JL, Slamanig S, Lemus N, Liu Y, Lai TY, et al. Trivalent NDV-HXP-S vaccine protects against phylogenetically distant SARS-coV-2 variants of concern in mice. Microbiol Spectr. (2022) 10:e0153822. doi: 10.1128/spectrum.01538-22
16. Sun W, Leist SR, McCroskery S, Liu Y, Slamanig S, Oliva J, et al. Newcastle disease virus (NDV) expressing the spike protein of SARS-CoV-2 as a live virus vaccine candidate. EBioMedicine. (2020) 62:103132. doi: 10.1016/j.ebiom.2020.103132
17. Ayllon J, Garcia-Sastre A, Martinez-Sobrido L. Rescue of recombinant Newcastle disease virus from cDNA. J Vis Exp. (2013) (80):50830. doi: 10.3791/50830
18. Jin-xin M, Jian-yao W, Gh-r RN, Py SS-h, Ya-chao W, Sheng-nan H, et al. lidation of stable housekeeping genes for quantitative real time PCR in golden Syrian hamster. Indian J Anim Res. (2021) 55:1127–31. doi: 10.18805/IJAR.B-1237
19. Slamanig S, Gonzalez-Dominguez I, Chang LA, Lemus N, Lai TY, Martinez JL, et al. Intranasal SARS-CoV-2 Omicron variant vaccines elicit humoral and cellular mucosal immunity in female mice. EBioMedicine. (2024) 105:105185. doi: 10.1016/j.ebiom.2024.105185
20. Singh G, Garcia-Bernalt-Diego J, Warang P, Park SC, Chang LA, Noureddine M, et al. Outcome of SARS-CoV-2 reinfection depends on genetic background in female mice. Nat Commun. (2024) 15:10178. doi: 10.1038/s41467-024-54334-7
21. Slamanig S, Lemus N, Lai TY, Singh G, Mishra M, Abdeljawad A, et al. A single immunization with intranasal Newcastle disease virus (NDV)-based XBB.1.5 variant vaccine reduces disease and transmission in animals against matched-variant challenge. Vaccine. (2024) 45:126586. doi: 10.1016/j.vaccine.2024.126586
22. Dagotto G, Mercado NB, Martinez DR, Hou YJ, Nkolola JP, Carnahan RH, et al. Comparison of subgenomic and total RNA in SARS-coV-2 challenged rhesus macaques. J Virol. (2021) 95(8):e02370-20. doi: 10.1128/JVI.02370-20
23. Sun W, McCroskery S, Liu WC, Leist SR, Liu Y, Albrecht RA, et al. A newcastle disease virus (NDV) expressing a membrane-anchored spike as a cost-effective inactivated SARS-coV-2 vaccine. Vaccines (Basel). (2020) 8(4):771. doi: 10.1101/2020.07.30.229120
24. Carreño JM, Alshammary H, Tcheou J, Singh G, Raskin A, Kawabata H, et al. Activity of convalescent and vaccine serum against SARS-CoV-2 Omicron. Nature. (2021) 602(7898):682–8. doi: 10.1038/d41586-021-03846-z
25. Slamanig S, Gonzalez-Dominguez I, Chang L, Lemus N, Lai TY, Martinez JL, et al. Intranasal SARS-coV-2 omicron variant vaccines elicit humoral and cellular mucosal immunity in mice. EBioMedicine. (2024) 105:105185. doi: 10.1016/j.ebiom.2024.105185
26. López CB, García-Sastre A, Williams BR, Moran TM. Type I interferon induction pathway, but not released interferon, participates in the maturation of dendritic cells induced by negative-strand RNA viruses. J Infect Dis. (2003) 187:1126–36. doi: 10.1086/368381
27. Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park MS, Yuen T, et al. Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol. (2006) 80:6295–304. doi: 10.1128/JVI.02381-05
28. Park MS, Garcia-Sastre A, Cros JF, Basler CF, Palese P. Newcastle disease virus V protein is a determinant of host range restriction. J Virol. (2003) 77:9522–32. doi: 10.1128/JVI.77.17.9522-9532.2003
29. Suresh V, Parida D, Minz AP, Sethi M, Sahoo BS, Senapati S. Tissue distribution of ACE2 protein in Syrian golden hamster (Mesocricetus auratus) and its possible implications in SARS-coV-2 related studies. Front Pharmacol. (2020) 11:579330. doi: 10.3389/fphar.2020.579330
30. Tomris I, Bouwman KM, Adolfs Y, Noack D, van der Woude R, Kerster G, et al. Distinct spatial arrangements of ACE2 and TMPRSS2 expression in Syrian hamster lung lobes dictates SARS-CoV-2 infection patterns. PloS Pathog. (2022) 18:e1010340. doi: 10.1371/journal.ppat.1010340
31. Krammer F. SARS-CoV-2 vaccines in development. Nature. (2020) 586:516–27. doi: 10.1038/s41586-020-2798-3
32. Lavelle EC, Ward RW. Mucosal vaccines - fortifying the frontiers. Nat Rev Immunol. (2022) 22:236–50. doi: 10.1038/s41577-021-00583-2
33. van Splunter M, van Hoffen E, Floris-Vollenbroek EG, Timmerman H, de Bos EL, Meijer B, et al. Oral cholera vaccination promotes homing of IgA(+) memory B cells to the large intestine and the respiratory tract. Mucosal Immunol. (2018) 11:1254–64. doi: 10.1038/s41385-018-0006-7
34. Khattar SK, Manoharan V, Bhattarai B, LaBranche CC, Montefiori DC, Samal SK. Mucosal immunization with newcastle disease virus vector coexpressing HIV-1 env and gag proteins elicits potent serum, mucosal, and cellular immune responses that protect against vaccinia virus env and gag challenges. mBio. (2015) 6:e01005. doi: 10.1128/mBio.01005-15
35. Jones JM, Manrique IM, Stone MS, Grebe E, Saa P, Germanio CD, et al. Estimates of SARS-CoV-2 Seroprevalence and Incidence of Primary SARS-CoV-2 Infections Among Blood Donors, by COVID-19 Vaccination Status - United States, April 2021-September 2022. MMWR Morb Mortal Wkly Rep. (2023) 72(22):601–5. doi: 10.15585/mmwr.mm7222a3
36. Rathnasinghe R, Jangra S, Ye C, Cupic A, Singh G, Martinez-Romero C, et al. Characterization of SARS-CoV-2 Spike mutations important for infection of mice and escape from human immune sera. Nat Commun. (2022) 13:3921. doi: 10.1038/s41467-022-30763-0
37. Samuel-Ponce-de-León MT, Soto-Ramírez LE, Calva JJ, Santillán-Doherty P, Carranza-Salazar DE, Carreño JM, et al. Safety and immunogenicity of a live recombinant Newcastle disease virus-based COVID-19 vaccine (Patria) administered via the intramuscular or intranasal route: Interim results of a non-randomized open label phase I trial in Mexico. medRxiv. (2022) 8(1):67. doi: 10.1101/2022.02.08.22270676
38. Kumlin U, Olofsson S, Dimock K, Arnberg N. Sialic acid tissue distribution and influenza virus tropism. Influenza Other Respir Viruses. (2008) 2:147–54. doi: 10.1111/j.1750-2659.2008.00051.x
39. Villar E, Barroso IM. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: a minireview. Glycoconj J. (2006) 23:5–17. doi: 10.1007/s10719-006-5433-0
40. Zhao C, Pu J. Influence of host sialic acid receptors structure on the host specificity of influenza viruses. Viruses. (2022) 14(10):2141. doi: 10.3390/v14102141
41. Iwatsuki-Horimoto K, Nakajima N, Ichiko Y, Sakai-Tagawa Y, Noda T, Hasegawa H, et al. Syrian hamster as an animal model for the study of human influenza virus infection. J Virol. (2018) 92(4):e01693-17. doi: 10.1128/JVI.01693-17
42. Spruit CM, Nemanichvili N, Okamatsu M, Takematsu H, Boons GJ, de Vries RP. N-glycolylneuraminic acid in animal models for human influenza A virus. Viruses. (2021) 13(5):815. doi: 10.3390/v13050815
43. Shrotri M, Navaratnam AMD, Nguyen V, Byrne T, Geismar C, Fragaszy E, et al. Spike-antibody waning after second dose of BNT162b2 or ChAdOx1. Lancet. (2021) 398:385–7. doi: 10.1016/S0140-6736(21)01642-1
44. Levin EG, Lustig Y, Cohen C, Fluss R, Indenbaum V, Amit S, et al. Waning immune humoral response to BNT162b2 covid-19 vaccine over 6 months. N Engl J Med. (2021) 385:e84. doi: 10.1056/NEJMoa2114583
45. Canaday DH, Oyebanji OA, Keresztesy D, Payne M, Wilk D, Carias L, et al. Significant reduction in vaccine-induced antibody levels and neutralization activity among healthcare workers and nursing home residents 6 months following coronavirus disease 2019 BNT162b2 mRNA vaccination. Clin Infect Dis. (2022) 75:e884–7. doi: 10.1093/cid/ciab963
46. Carreno JM, Alshammary H, Singh G, Raskin A, Amanat F, Amoako A, et al. Evidence for retained spike-binding and neutralizing activity against emerging SARS-CoV-2 variants in serum of COVID-19 mRNA vaccine recipients. EBioMedicine. (2021) 73:103626. doi: 10.1016/j.ebiom.2021.103626
47. WHO. 18-05-2023-statement-on-the-antigen-composition-of-covid-19-vaccines (2023). Available online at: https://www.who.int/news/item/18-05-2023-statement-on-the-antigen-composition-of-covid-19-vaccines (Accessed February 1, 2024).
48. EMA. EMA and ECDC statement on updating COVID-19 vaccines to target new SARS-CoV-2 virus variants (2023). Available online at: https://www.ema.europa.eu/en/news/ema-ecdc-statement-updating-covid-19-vaccines-target-new-sars-cov-2-virus-variants (Accessed February 1, 2024).
49. (VRBPAC) FsVaRBPAC. Updated COVID-19 vaccines for use in the United States beginning in fall 2023 - VRBPAC 2023 (2023). Available online at: https://www.fda.gov/vaccines-blood-biologics/updated-covid-19-vaccines-use-united-states-beginning-fall-2023 (Accessed February 1, 2024).
50. Gagne M, Moliva JI, Foulds KE, Andrew SF, Flynn BJ, Werner AP, et al. mRNA-1273 or mRNA-Omicron boost in vaccinated macaques elicits similar B cell expansion, neutralizing responses, and protection from Omicron. Cell. (2022) 185:1556–1571.e18. doi: 10.1016/j.cell.2022.03.038
Keywords: transmission-proof, sterilizing immunity, viral shedding, COVID-19, Coronavirus, vector vaccine
Citation: González-Domínguez I, Abdeljawad A, Lai TY, Boza M, McCroskery S, Lemus N, Slamanig S, Singh G, Warang P, Yellin T, Abbad A, Carreño JM, Dolange V, Martínez-Guevara JL, Singh G, Barcena-Varela M, Chang LA, Schotsaert M, Krammer F, Palese P and Sun W (2025) Mucosal multivalent NDV-based vaccine provides cross-reactive immune responses against SARS-CoV-2 variants in animal models. Front. Immunol. 16:1524477. doi: 10.3389/fimmu.2025.1524477
Received: 07 November 2024; Accepted: 26 February 2025;
Published: 17 March 2025.
Edited by:
Diego Cantoni, MRC-University of Glasgow Centre For Virus Research (MRC), United KingdomReviewed by:
Gunnveig Grødeland, University of Oslo, NorwayCopyright © 2025 González-Domínguez, Abdeljawad, Lai, Boza, McCroskery, Lemus, Slamanig, Singh, Warang, Yellin, Abbad, Carreño, Dolange, Martínez-Guevara, Singh, Barcena-Varela, Chang, Schotsaert, Krammer, Palese and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weina Sun, d2VpbmEuc3VuQG1zc20uZWR1; Peter Palese, cGV0ZXIucGFsZXNlQG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.