 Yi-Qian Liu
Yi-Qian Liu Zhan-Zhan Li2
Zhan-Zhan Li2 Yong-Li Han
Yong-Li Han
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 February 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1524058
Efferocytosis is the process by which various phagocytes clear apoptotic cells. In recent years, an increasing body of evidence has emphasized the importance of efferocytosis in maintaining internal homeostasis. Intestinal macrophages play a crucial role in modulating intestinal inflammation and promoting tissue repair. Inflammatory bowel disease (IBD) is a chronic, progressive, and relapsing condition, primarily marked by the presence of ulcers in the digestive tract. The exact mechanisms underlying IBD are not yet fully understood, and current treatment approaches mainly aim at repairing the damaged intestinal mucosa and reducing inflammatory responses to ease symptoms.This article provides new perspectives on IBD treatment and clinical management by examining the expression of macrophage efferocytosis-related molecules, the effects of efferocytosis on IBD development, the various roles of macrophage efferocytosis in IBD, and treatment strategies for IBD that focus on efferocytosis.
Efferocytosis refers to the process by which apoptotic cells are cleared by various phagocytes. Increasing evidence over recent years has emphasized its crucial role in maintaining internal homeostasis. Billions of cells undergo apoptosis daily in the human body, and any delay in their clearance can result in secondary necrosis, leading to the release of toxic intracellular contents. This can, in turn, trigger pathological inflammation and autoimmune responses (1). As a result, defects in efferocytosis are strongly associated with the development of several inflammatory and autoimmune disorders. Inflammatory bowel disease (IBD) is a chronic, progressive, and relapsing condition, characterized primarily by ulcers in the digestive tract. It includes ulcerative colitis (UC) and Crohn’s disease (CD), which together affect 6 to 8 million people worldwide, significantly impacting their quality of life and daily activities, with an increasing incidence in recent years (2). Although the pathogenesis of IBD is associated with various factors, such as environmental exposure, genetic predisposition, gut microbiota imbalances, and immune system defects, the exact mechanisms are still not fully understood. Current treatment strategies focus primarily on repairing damaged intestinal mucosa and reducing inflammation to alleviate symptoms (3). This article reviews the expression of macrophage efferocytosis-related molecules, the impact of efferocytosis on IBD, and the multifaceted roles of macrophage efferocytosis in the disease.
Macrophage efferocytosis differs from classical phagocytosis and requires the coordinated expression of multiple molecules to complete the process. It is generally divided into four stages: apoptotic cells release signals that attract macrophages, receptors on macrophages bind to these signals, macrophages then engulf the apoptotic cells, and the fusion of lysosomes and phagosomes leads to the digestion and degradation of the apoptotic cells and their contents (4).
The “find-me” signals emitted by apoptotic cells are primarily soluble molecules, which can be classified into three categories: nucleotides, membrane lipids, and chemokines. Nucleotides, such as adenosine triphosphate (ATP) and uridine triphosphate (UTP), are the most prominent “find-me” signals for apoptotic cells (5), and their release is regulated by the plasma membrane protein Pannexin-1 (PANX1) (6, 7). Membrane lipids, which serve as specific “find-me” signals for apoptotic cells, include lysophosphatidylcholine (LPC) and sphingosine-1-phosphate (S1P). The activation of LPC is controlled by caspase-3-activated phospholipase A2, which converts phosphatidylcholine to LPC (8). On the other hand, sphingosine kinase 1 (SphK1) is upregulated in apoptotic cells, driving the secretion of S1P (9). Chemokines, such as Fractalkine (CX3CL1), are membrane-bound proteins, and studies suggest that the release of CX3CL1 can enhance the chemotaxis of macrophages (10). These soluble signals are released by apoptotic cells into the surrounding environment, guiding macrophages to accumulate at the target site and stimulating their capacity to clear apoptotic cells. Phosphatidylserine (PS) is the most extensively studied “eat-me” signal, although other signals include calreticulin (Crt) (11), pentraxin 3 (PTX3) (12), and changes in surface protein glycosylation or surface charge (13). CD47 (11, 14) is the most well-known “don’t-eat-me” signal (15), and together, these signals help distinguish dying cells from adjacent healthy cells, enabling macrophages to effectively phagocytose apoptotic cells (16, 17).
In addition, research from the Ravichandran group has identified a novel family of solute carriers (SLC) that are specifically modified during efferocytosis, which plays a significant role in regulating the uptake of apoptotic cells. SLC2A1 supports the phagocytosis of apoptotic cells in vitro and in vivo, not only promoting the initial engulfment of the first apoptotic cell but also facilitating the continued uptake of additional apoptotic cells (18). Furthermore, the release of lactate via SLC16A1contributes to promoting an anti-inflammatory environment.
Phosphatidylserine (PS) is the most prominent “eat-me” signal found on apoptotic cells, capable of binding to macrophage surface receptors either directly or indirectly. In the direct method, PS binds directly to receptors such as T cell immunoglobulin mucin (TIM)–1, TIM-4 (19), brain-specific angiogenesis inhibitor 1 (BAI1) (20), stabilin-2 (21), and advanced glycation end product receptors (17). In the indirect method, PS binds to macrophage receptors via bridging molecules, including receptor tyrosine kinases Tyro-3, Axl, and Mertk (TAM), and integrins (22). These bridging molecules facilitate the connection between PS and macrophage receptors, including milk fat globule-EGF factor 8 protein (MFG-E8 or lactadherin) (23), growth arrest-specific factor 6 (Gas6) (24), and annexin 1 (ANXA1) (25), among others (26).
Furthermore, nucleotides such as ATP and UTP bind to the purinergic receptor P2Y2 on macrophage surfaces, while LPC and S1P bind to G protein-coupled receptors G2A and S1PR on macrophages, respectively (17). These interactions activate the macrophage efferocytosis receptors, which, through different signaling pathways, trigger cytoskeletal reorganization and enable the phagocytosis and clearance of apoptotic cells (26, 27).
When macrophage surface receptors bind to signals emitted by apoptotic cells, they activate the programmed clearance system within the macrophages. This process is initiated through the activation of key regulatory factors from the Rho family of small GTPases, such as Rac (28), via two primary mechanisms: the LDL receptor-related protein 1 (LRP1/CD91) (29) and the adaptor protein GULP (the mammalian equivalent of C. elegans ced-6) (30), or through the guanine nucleotide exchange factor (GEF) DOCK180 and the cell movement protein (ELMO) (20, 31). Activated Rac triggers actin remodeling through the WASP pathway, promoting actin polymerization to engulf or capture apoptotic cells, leading to membrane invagination and the internalization of the cells, which forms a phagosome. In addition to these pathways, a third potential signaling pathway may involve the tyrosine kinase Abl (ABL-1) inhibiting the Abl interactor AbI (ABI-1), which seems to counteract macrophage efferocytosis, though the precise mechanism remains unclear (32).
After the engulfment of apoptotic cells, the large GTPase Dynamin-Vps34 (a phosphatidylinositol (3)-kinase) pathway is activated, which in turn activates the small GTPase Rab5, promoting lysosome maturation (33). Following this, Mon1a (C. elegans SAND-1) and its partner Ccz1 recruit Rab5 and Rab7 to the phagosome, activating Rab7 and facilitating the fusion of phagosomes with lysosomes (34, 18). Concurrently, acid hydrolases and nucleases are activated to degrade and acidify the engulfed apoptotic cell remnants (35, 36). Table 1 presents the molecules involved in efferocytosis.
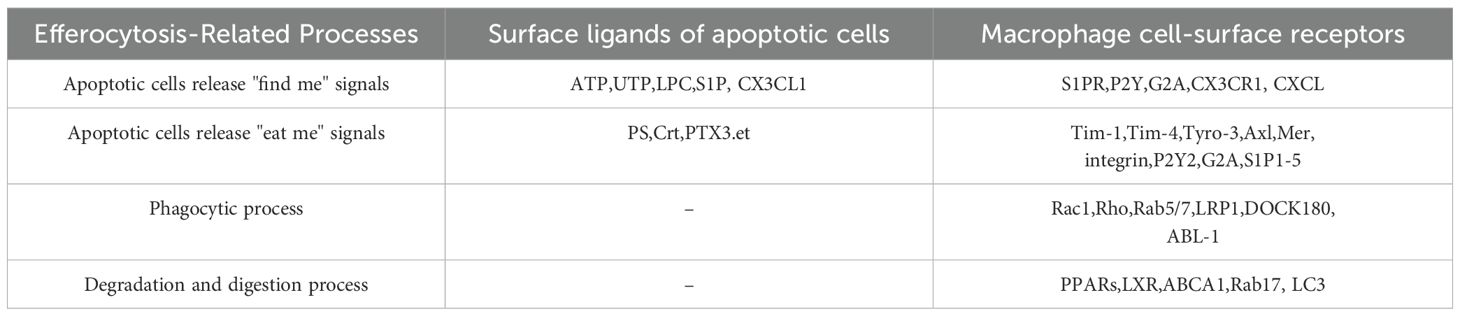
Table 1. presents the molecules involved in efferocytosis.
The process of efferocytosis not only acts as a waste disposal mechanism to remove apoptotic cells but also leads to various biological outcomes. It can drive macrophage polarization toward a pro-resolution phenotype, release pro-resolving factors and anti-inflammatory mediators, promote self-tolerance, and help resolve inflammation. Upon completion of the degradation and phagocytosis of apoptotic cells, efferocytosis encourages macrophages to adopt a pro-resolution phenotype by decreasing the production of pro-inflammatory cytokines and increasing the levels of pro-resolving mediators. At the same time, it releases significant amounts of anti-inflammatory mediators, such as IL-10 (37) and TGF-β (38), as well as pro-resolving factors that help mitigate inflammation in IBD. These factors also stimulate the proliferation and differentiation of intestinal epithelial cells and fibroblasts, promoting healing of the intestinal mucosa in IBD (39). Besides these effects, efferocytosis also interacts with the immune system. For example, regulatory T cells have been shown to enhance efferocytosis in both in vitro and in vivo models (40). However, studies also indicate that TH17 cells play a critical role in the pathogenesis of IBD, with elevated numbers of Th17 cells present in the intestinal tissues of IBD patients (41). Infected apoptotic cells can induce Th17 differentiation, triggering intestinal inflammation. Pathogens often induce cell apoptosis, which may preferentially activate Th17-mediated immunity, contributing to IBD (42, 43). This suggests that while efferocytosis generally promotes anti-inflammatory and resolution responses, it may also lead to inflammation under specific conditions. Further research is required to better understand the connection between IBD and efferocytosis (Figure 1).
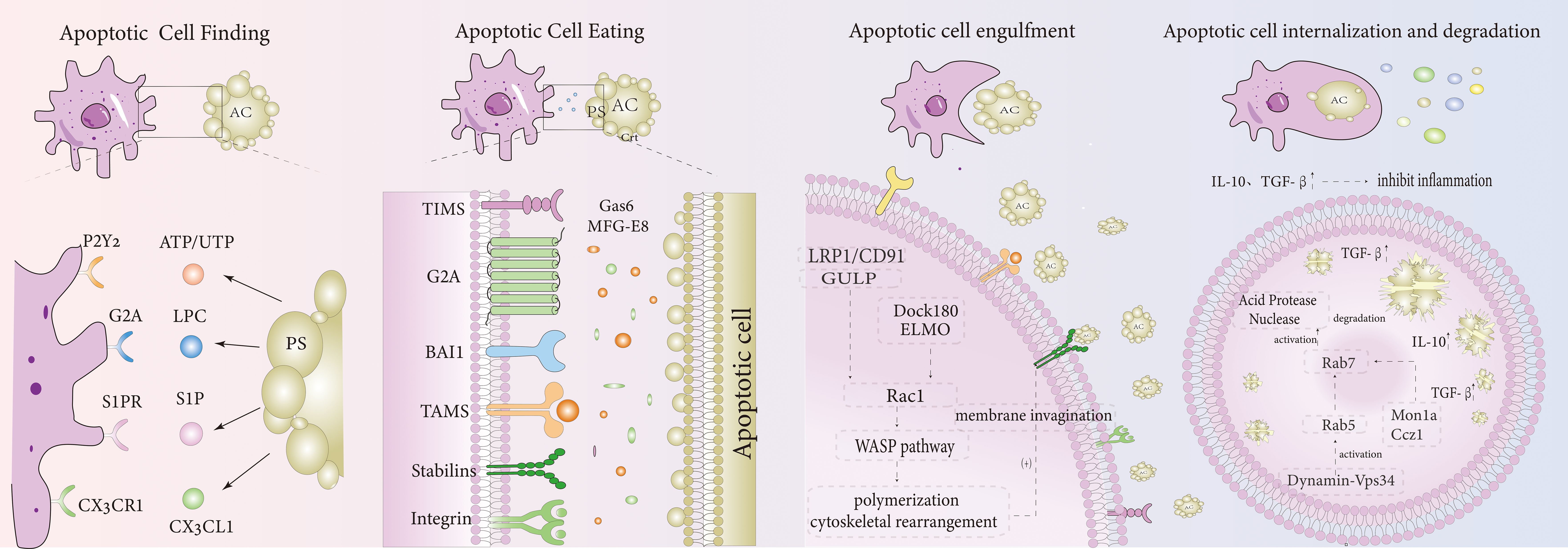
Figure 1. Efferocytosis mechanism diagram. Apoptotic Cell Finding: Apoptotic cells release recognition signals such as ATP/UTP, LPC, S1P, and CX3CL1, which bind to receptors on the macrophage surface, including P2Y2, G2A, S1PR, and CX3CR1, respectively. Apoptotic Cell Eating: Apoptotic cells express “eat me” signals like PS, which can bind directly to macrophage receptors such as TIMS, BAI1, and Stabilins. Additionally, PS can also bind to macrophage receptors indirectly through bridging molecules such as Gas6 and MFG-E8, which connect to TAMS and integrins. Apoptotic Cell Engulfment: After binding, apoptotic cells activate Rac1 through two pathways: LRP1/CD91/GULP and Dock180/ELMO. This activation triggers actin polymerization via the WASP pathway, leading to the formation of a phagocytic cup and the invagination of the plasma membrane. Apoptotic Cell Internalization and Degradation: After internalization, apoptotic cells are processed through the large GTPase Dynamin-Vps34 pathway, which activates Rab5 and promotes lysosomal maturation. Mon1a and Ccz1 subsequently recruit Rab5 and activate Rab7 on the phagosome. This Rab7 activation leads to the activation of acid hydrolases and nucleases, which degrade and acidify the engulfed apoptotic cell remnants. Anti- inflammatory mediators, such as TGF-β and IL-10, are released, dampening the inflammatory response.
IBD is an idiopathic condition primarily characterized by symptoms like rectal bleeding and weight loss, stemming from chronic and excessive inflammation of the gastrointestinal tract (44). IBD development involves a range of factors, both genetic and environmental. Genetic studies, including GWAS, have revealed more than 200 genetic loci associated with IBD (45), with the nucleotide-binding oligomerization domain 2 (NOD2) being one of the most prominent loci. Mutations in this locus can activate the NF-κB inflammatory pathway and initiate autophagy (46), and studies suggest that this factor can suppress macrophage function. Consequently, the accumulation of apoptotic cells that enhances efferocytosis might play a crucial role in terminating the inflammatory response (47). Moreover, the gut microbiota has been shown to contribute significantly to IBD pathogenesis, likely due to abnormal host-microbiota interactions affecting the immune system (48). Immune dysfunction in IBD is marked by epithelial damage and a failure to properly regulate immune responses (49), which include both innate and adaptive immune dysfunctions. This leads to abnormalities in intestinal epithelial cells (IECs), dendritic cells (DCs), macrophages, T cells, and other factors. In this process, several molecules such as TNF-α (50), IL-6 (49), IL-9 (51), IL-23 (52), and IL-17 (53) may be dysregulated, but efferocytosis can suppress these molecules while promoting the release of anti-inflammatory mediators (39). The following discussion will describe how efferocytosis can limit the progression of IBD from multiple angles (Figure 2).
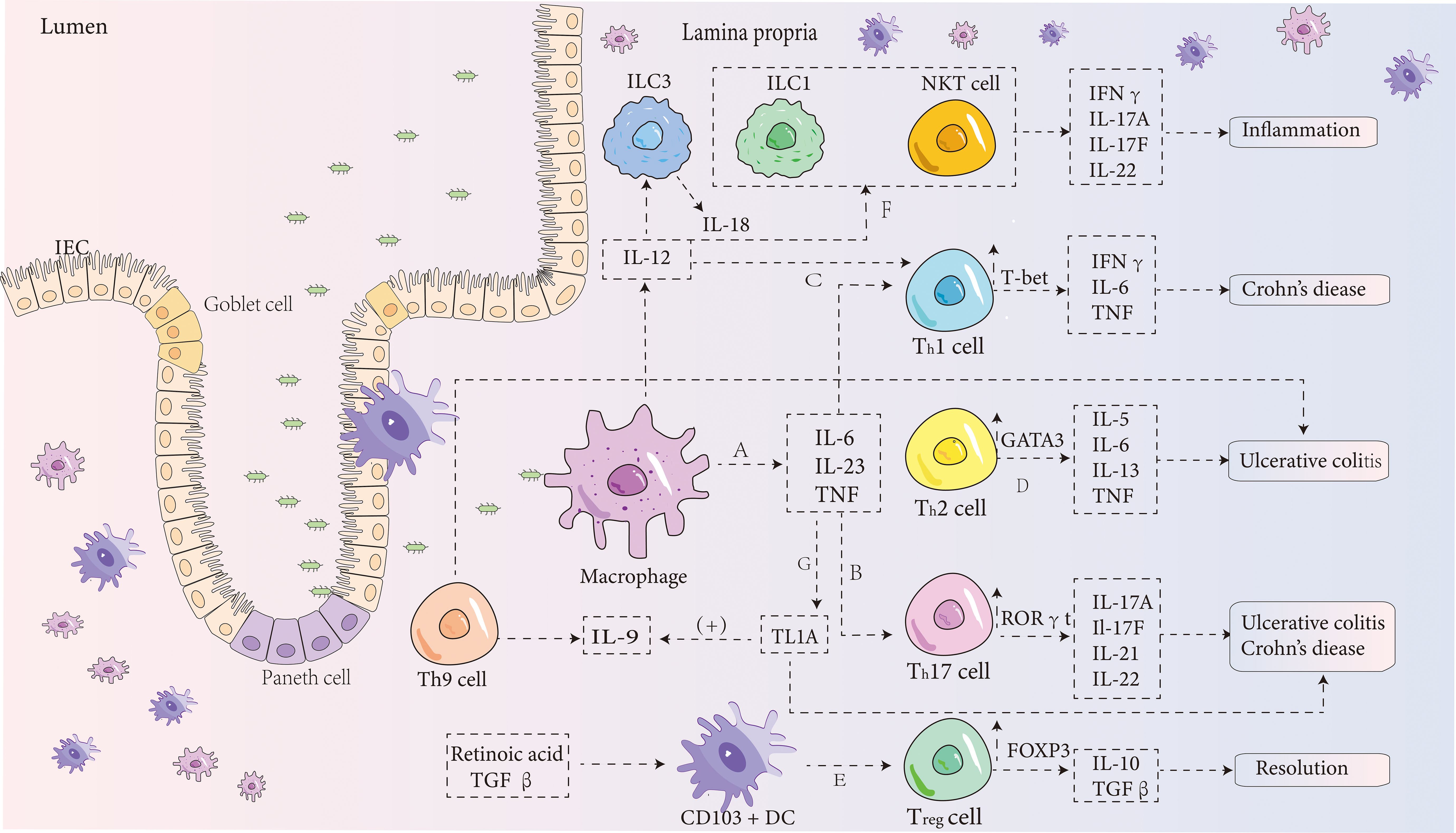
Figure 2. IBD pathogenesis diagram. (A) In the intestinal lamina propria, macrophages can stimulate the production of pro-inflammatory cytokines such as IL-6, IL-23, and TNF. IL-6 binds to its receptor in a soluble form, activating T cells and triggering the translocation of STAT-3. This process then promotes the transcription of anti-apoptotic genes such as Bcl-2 and Bcl-xl, which reduces T-cell apoptosis, thereby intensifying intestinal inflammation (54). (B) IL-6 also induces the differentiation of CD4+ T cells into Th17 cells (55), which release cytokines such as IL-17A, IL-17F (56), IL-21 (57), and IL-22. These cytokines, together with IL-23, contribute significantly to the development of IBD (56). During this process, the expression of the transcription factor RORγt in Th17 cells is enhanced. (C) Under the combined influence of IL-12 and IL-6, CD4+ T cells differentiate into Th1 cells (55), which mainly produce IFNγ, IL-6, and TNF, while increasing the expression of T-bet and STAT4. This leads to an exaggerated Th1 cell response, which is a major contributor to the development of Crohn’s disease. (D) Stimulation by IL-4 leads to the differentiation of CD4+ T cells into Th2 cells, which release cytokines such as IL-5, IL-6, IL-13, and TNF (58). Research has shown that NKT cells in ulcerative colitis release more IL-13, a Th2 cytokine, than T cells in Crohn’s disease (58). Thus, Crohn’s disease is typically associated with a Th1 response, while ulcerative colitis is thought to be driven by Th2 cells. However, this distinction is still debated. (E) Besides adaptive immune responses, innate immune mechanisms also play a role in IBD. CD103+ DCs, which depend on retinoic acid and TGF-β, promote the development of Foxp3+ Tregs, which release IL-10 and TGF-β (59). These cytokines have a suppressive effect on inflammation and protect against the development of IBD. (F) NKT cells, a subset of lymphocytes, can be activated indirectly by cytokines such as IL-12 and IL-18 secreted by innate lymphoid cell group 3 (60). These cells rapidly produce Th1, Th2, and Th17 cytokines, contributing to inflammation. (G) TNF is a key pro-inflammatory cytokine involved in IBD pathogenesis through several pathways. Recent studies indicate that TNF-like ligand 1A (TL1A) is a crucial mediator of intestinal inflammation (61). TL1A enhances the ability of IL-12, IL-4, or IL-23 to promote the differentiation of Th1, Th2, and Th17 cells (62, 63). It may also induce Th9 differentiation and increase IL-9 secretion by upregulating TGF-β and IL-4 expression, exacerbating DSS-induced colitis in mice (64).
Macrophages are essential for maintaining intestinal immune homeostasis and have recently been identified as a potential target for treating inflammatory bowel disease (IBD). Several studies have highlighted the strong connection between macrophage activation and cellular metabolism (65). In the context of IBD, various immunometabolic alterations influence macrophage phenotype and function by regulating metabolic reprogramming and transcription, which in turn affects disease progression. Research indicates that during efferocytosis, a specific family of solute carrier (SLC) genes undergoes modification. At different stages of efferocytosis, various SLCs and other molecules are activated, facilitating SLC2A1-mediated aerobic glycolysis while suppressing oxidative phosphorylation. Furthermore, SLC16A1 contributes to the release of lactate, which promotes anti- inflammatory effects (18, 66). Through this process, SLC2A1-mediated glucose uptake and glycolytic intermediates enhance actin polymerization during efferocytosis, facilitating the uptake and clearance of apoptotic cells (18). Recent findings suggest that activation of 6-phosphofructo-2-kinase/fructose- 2,6-bisphosphatase 2 (PFKFB2) boosts glycolysis, increasing lactate release and regulating the expression of efferocytosis receptors like MerTK and LRP1, which extend the efferocytosis process (67). Thus, whether through SLC or PFKFB2-mediated glycolysis, there is a clear link between glycolysis and efferocytosis, although additional mechanisms remain to be explored.
The resolution of inflammation in efferocytosis is often closely tied to cellular metabolic processes. Studies have pointed to a unique relationship between apoptotic cell breakdown, fatty acid oxidation (FAO), and mitochondrial respiration during efferocytosis. One study observed that after efferocytosis, long-chain fatty acids accumulated in macrophages, activating the respiratory chain and generating metabolic intermediates that promoted macrophage anti-inflammatory responses (68). Although the direct link between this pathway and IBD was not identified, this non-classical mitochondrial response plays a crucial role in tissue repair and healing during damage and may offer new therapeutic avenues for IBD in the future. Other research suggests that efferocytosis can activate the tryptophan (TRP) pathway, triggering several pro-resolving programs, such as the induction of pro-resolving proteins, which further enhance efferocytosis (69). This study, using atherosclerosis as a model, connects the resolution of inflammation with efferocytosis and provides a theoretical basis for treating similar chronic inflammatory diseases like IBD. Additionally, macrophage arginase 1 (Arg1) and ornithine decarboxylase (ODC) convert apoptotic cell-derived arginine and ornithine into agmatine, thereby promoting efferocytosis (70). This research, using atherosclerosis as a model, demonstrates that disruption of this metabolic pathway can impair efferocytosis, leading to further inflammation and tissue necrosis in chronic inflammatory conditions. Conversely, enhancing this pathway could break the pathological cycle, offering new therapeutic possibilities for chronic inflammatory diseases.
Defects in efferocytosis can lead to the development of various inflammatory and autoimmune disorders. Macrophage nuclear receptors, such as peroxisome proliferator-activated receptor (PPARγ) and liver X receptor (LXR), are involved in these processes. Research has shown that LXR expression is notably reduced in the intestinal tissues of IBD patients (71, 72). In vivo stimulation of apoptotic cells can activate LXR, leading to an increase in the expression of the apoptotic cell recognition receptor tyrosine kinase Mertk. This activation enhances efferocytosis, helping prevent and treat DSS-induced colitis and ulcerative colitis (71, 73). PPARγ and LXR have similar roles, and their mechanisms of action share notable similarities. Studies have suggested that activating PPARγ can effectively reduce the signs and symptoms of IBD and its progression (74). During macrophage efferocytosis, PPARγ functions as a transcriptional sensor of dying cells, responding to apoptotic cell signals and coordinating their clearance, which promotes self-tolerance and alleviates IBD (75). In this process, macrophage clearance of apoptotic cells reduces the production of reactive oxygen species, which are pro-inflammatory mediators activated by PPARγ (76). Additionally, PPARγ activation further enhances macrophage efferocytosis (77).
In the early phase of efferocytosis recognition, the externalization of intracellular phospholipids and extracellular molecules on apoptotic cells leads to the binding of proteins S and Gas6, activated by bridging molecules like TIM4, to phosphatidylserine (PS), and subsequently to the TAM family receptors. This binding activates downstream transcription factors such as LXRα, LXRβ, and PPARγ, which suppress inflammatory signaling pathways in macrophages (78, 79), thereby inhibiting the inflammatory response and preventing the continued development of IBD.
Neutrophils have a dual role in inflammation. During acute inflammation, they are rapidly recruited to the affected site to eliminate pathogens and mediate inflammatory responses through various pathways. In chronic inflammation, the role of neutrophils has also gained attention. Studies have shown that neutrophil extracellular traps (NETs) are present in the inflamed colon, where they can worsen tissue damage and contribute to thrombotic tendencies during active IBD (80). Additionally, NETs may disrupt the intestinal mucosal barrier through several mechanisms (81). NETs represent a distinctive form of cell death, occurring when dying neutrophils release their nuclear DNA, which combines with cytoplasmic proteins to form a unique structure (82). Efferocytosis can regulate the release of NETs by DNase I-dependent digestion, reducing their accumulation (83), which is significant in inhibiting the progression of IBD. Furthermore, research indicates that in autoimmune vasculitis (AAV), NETs accumulate, and blocking CD47 can prevent disease progression by restoring efferocytosis (84). Although this study did not directly examine this pathway in IBD, the similarities in the pathogenesis of AAV and IBD may offer valuable insights for IBD research. In addition to releasing NETs, apoptotic neutrophils also secrete reparative proteins, such as Annexin A1, which interacts with FPR2. This interaction can inhibit the recruitment of inflammatory cells by reducing integrin activation triggered by chemokines, while simultaneously enhancing macrophage efferocytosis (85, 86). Neutrophils also release antimicrobial peptides like α-defensins, which suppress macrophage mRNA translation, thereby increasing macrophage phagocytic activity and reducing the production of pro-inflammatory cytokines (87, 88), which helps to limit the development of IBD.
The complex pathogenesis of IBD results from the intricate interactions between the gut microbiota, environmental factors, and the immune system. Immunological mechanisms underlying IBD involve dysregulation of both innate and adaptive immunity. TH17 cells are critical in IBD development, as demonstrated by elevated levels of IL-17A and IL-17F in the inflamed intestinal mucosa of IBD patients (56). In addition to producing IL-17, TH17 cells also promote the secretion of other effector cytokines, including IL-9 and IL-22. These cytokines, along with TH9/IL-9, perpetuate a pro- inflammatory loop that contributes to IBD progression (89). Efferocytosis, the process of clearing apoptotic cells, plays a key role in the production of anti-inflammatory mediators such as TGF-β, PGE2, and IL-10. IL-25 has been shown to regulate TH1/TH17 immune responses in an IL-10- dependent manner (90), thereby slowing IBD progression. This highlights IL-25 as a promising candidate for therapeutic strategies in IBD treatment. Furthermore, Treg cells have been observed to enhance IL-13 secretion during the resolution of inflammation, which stimulates macrophages to release IL-10. The released IL-10 activates macrophage Vav1 through autocrine or paracrine signaling, resulting in Rac1 activation and promoting macrophage efferocytosis. This series of events aids in controlling inflammation and provides a potential new therapeutic approach for treating chronic inflammatory diseases (40).
Autophagy plays a role in enhancing macrophage phagocytosis of apoptotic cells, and efferocytosis shares several regulatory factors with classical autophagy, particularly in terms of fusion and degradation with lysosomes. There is a strong association between deficiencies in autophagy genes and the enhancement of efferocytosis in various diseases (91). Research has shown that mice deficient in the autophagy-related gene NRBF2 experience impaired fusion of phagosomes containing apoptotic cells and lysosomes in macrophages through the MON1-CCZ1-Rab7 module. These mice are more susceptible to DSS-induced colitis, exhibiting severe intestinal inflammation and an accumulation of apoptotic cells (92). Thus, the lack of autophagy genes may lead to defective macrophage efferocytosis, contributing to the worsening of symptoms in several inflammatory diseases, including IBD.
Glucocorticoids (GCs) are frequently used to treat inflammatory conditions such as IBD. GCs activate the glucocorticoid receptor (GR), which suppresses pro-inflammatory genes while activating anti-inflammatory genes (93). The glucocorticoid-induced leucine zipper (GILZ), a well-know target of GR, mediates anti-inflammatory and pro-resolution effects, promoting macrophage reprogramming and enhancing efferocytosis. This reprogramming leads to the polarization of macrophages toward the M2 phenotype, initiating pro-resolution programs (94), which reduce inflammatory responses. IL-23 is also a significant pro-inflammatory cytokine in the pathogenesis of ulcerative colitis (UC) and Crohn’s disease (CD) (95). The uptake of apoptotic neutrophils by macrophages can reduce IL-23 transcription, thereby inhibiting inflammatory responses (96). Moreover, apoptotic cells heavily depend on TGF-β to coordinate anti-inflammatory responses in macrophages and to suppress the production of inflammatory mediators (97).
In the DSS-induced colitis model, BAI1-deficient mice display more severe colitis, characterized by an increased accumulation of unresolved apoptotic cells and higher levels of inflammatory cytokines. Targeted overexpression of BAI1 in colonic epithelial cells, which enhances efferocytosis, can significantly reduce colonic inflammation (98). Paneth cells, specialized epithelial cells in the intestine, function as phagocytes, facilitating the uptake of surrounding apoptotic cells to help maintain intestinal homeostasis (99). These cells play a crucial role in the progression of IBD. The autophagy-related gene ATG16L1 is a genetic risk factor that plays a key role in CD. Paneth cells with a deficiency in Atg16L1 exhibit significant abnormalities in the efferocytosis pathway and are closely linked to adipokines, leptin and adiponectin, which directly influence responses to intestinal damage, thus affecting the intestinal epithelium and contributing to the progression of CD (100, 101). Consequently, promoting efferocytosis in Paneth cells and intestinal epithelial cells may effectively reduce intestinal inflammation in patients with mucosal inflammation, offering potential therapeutic implications for IBD and other inflammatory bowel diseases (Figure 3).
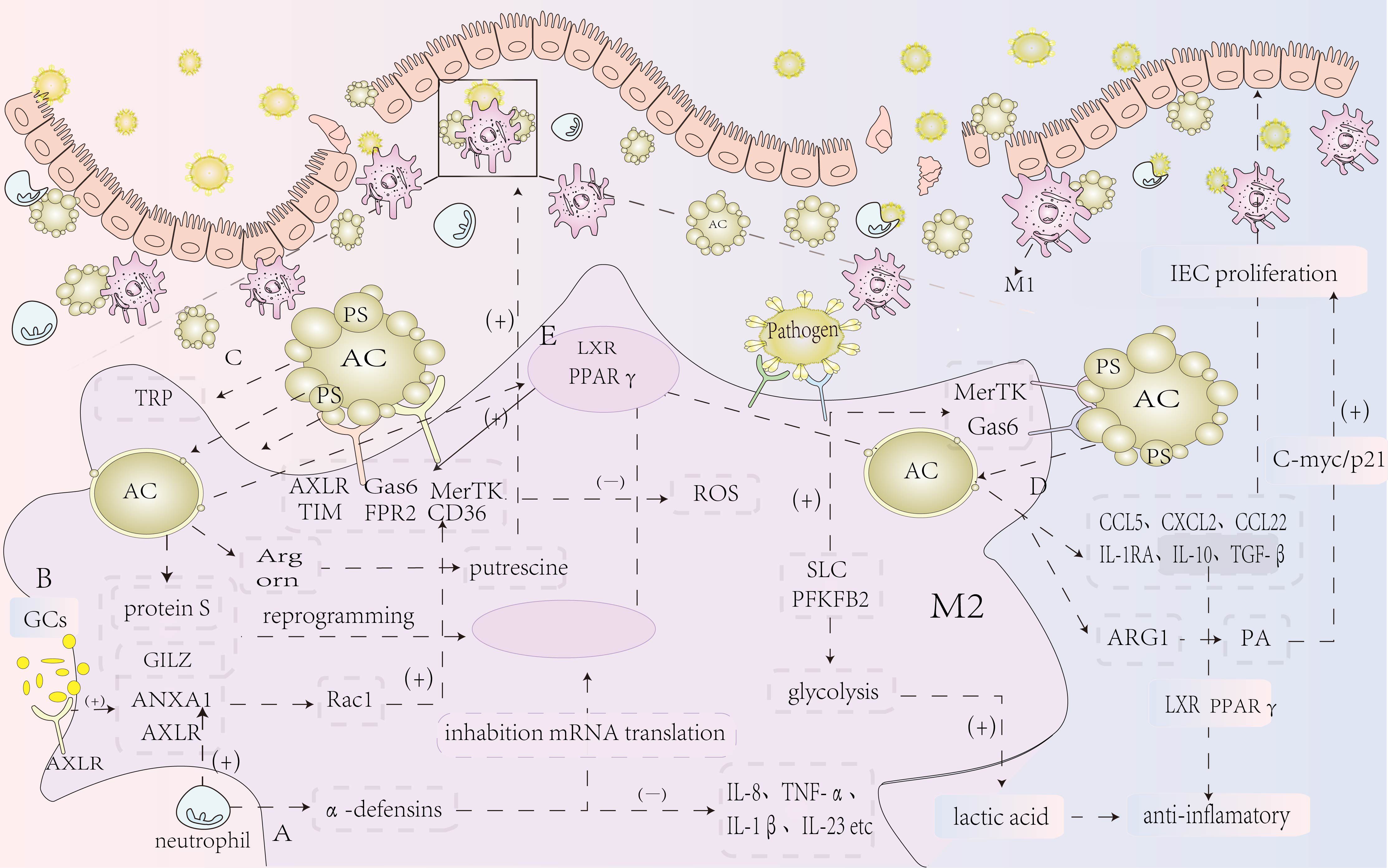
Figure 3. Summary of the mechanism of efferocytosis in IBD. (A) Neutrophils release antimicrobial peptides, such as α-defensins, which inhibit macrophage mRNA translation, thereby reducing the production of pro-inflammatory cytokines and lessening the inflammatory response. (B) Glucocorticoid treatment increases the expression of AnxA1 and its receptor ALXR, which regulate the inflammatory response, boost Rac1 activity in macrophages, and promote efferocytosis through CD36, reducing intestinal inflammation and maintaining homeostasis. Additionally, glucocorticoids elevate the expression of GILZ, a leucine zipper protein, which, together with protein S secreted by neutrophils, mediates anti-inflammatory and pro-resolution effects, enhancing macrophage reprogramming and efferocytosis. (C) Efferocytosis triggers the activation of the SLC gene family, promoting lactate release, which exerts anti-inflammatory effects. It also activates the tryptophan (TRP) pathway to promote key pro-resolution programs, including the induction of pro-resolving proteins, thereby enhancing efferocytosis. Moreover, macrophage arginase 1 (Arg1) and ornithine decarboxylase (ODC) convert apoptotic cell-derived arginine and ornithine into putrescine, further enhancing efferocytosis and reducing the inflammatory response. (D) Following efferocytosis, various chemokines (CCL5, CXCL2, CCL22) and anti-inflammatory mediators (IL-10 and TGF-β) are released, helping to suppress the inflammatory response and repair damaged intestinal mucosal epithelium. (E) Nuclear genes in macrophages regulate the transcription of surface receptors, enhancing efferocytosis and suppressing inflammation. Deficiencies in macrophage nuclear receptor genes may promote the development of IBD, while efferocytosis can activate nuclear receptors to prevent IBD development.
IBD is a prevalent inflammatory condition affecting individuals globally, with various treatment options available. Traditionally, IBD has been managed through drug therapies aimed at controlling symptoms, such as aminosalicylates (102), corticosteroids (CS) (103), immunomodulators, and biologics (104, 105), with surgical resection and other general measures utilized as needed. In recent years, new treatment strategies have emerged to improve clinical outcomes, including apheresis therapy (106), modulation of gut microbiota (107), and cellular therapies (108). Besides Western medical treatments, traditional Chinese medicine (TCM) offers distinct advantages in treating IBD. For example, baicalin has been shown to increase interferon factor 4 protein expression and reverse LPS-induced macrophage activation, thereby modulating macrophage polarization and enhancing inflammatory responses in DSS-induced colitis in mice (109). Indirubin, a key component of Qingdai, has been found to alleviate DSS-induced colitis in mice when administered orally (110). However, research on how TCM regulates the immune microenvironment remains insufficient, with a primary focus on anti-inflammatory effects and a lack of novel approaches. As treatment strategies continue to advance, the focus of IBD management has shifted from simply addressing clinical symptoms and preventing complications to promoting healing of the colonic mucosa, evidenced by the resolution of endoscopic ulcers (111, 112). Additionally, new and emerging treatment approaches continue to offer promising avenues for IBD therapy.
Research suggests that a single infusion of apoptotic cells can significantly improve the clinical symptoms of DSS-induced colitis (47). Apoptotic cells can inhibit pro-inflammatory signaling pathways and serve as a primary mechanism for resolving inflammation, mainly through their suppression of the transcription factor NF-κB and inflammatory bodies. NF-κB plays a central role in regulating gene expression during inflammation, particularly in the production and secretion of IL-1β (113). In DSS-induced colitis models, treatments such as anti-IL-1β therapy and the inhibition of inflammatory bodies (114, 115) have shown that NF-κB can reduce the activity of macrophages and dendritic cells in the lamina propria, along with inflammatory bodies. Therefore, the accumulation of apoptotic cells at the inflammation site is likely a key mechanism in halting the inflammatory response. The development of IBD is influenced by both genetic and environmental factors, with DSS exposure contributing to disease onset. Additionally, studies have demonstrated that extracellular vesicles from apoptotic cells can promote TGF-β production in macrophages and suppress inflammatory responses in experimental colitis models (116). Consequently, a single infusion of apoptotic cells to trigger the resolution phase of inflammation via efferocytosis presents a promising treatment approach for IBD, marking a new area of exploration. However, there remains a lack of extensive research and practical experience in this field. Overall, it holds considerable potential for future IBD therapies.
Efferocytosis is a continuous process in the body, where the phagocytic capacity is both expansive and constrained. Studies have demonstrated that enhancing efferocytosis in intestinal epithelial cells and Paneth cells can improve the clearance of apoptotic cells and mitigate inflammatory responses (98, 99). In severe IBD cases, intestinal tissues often show an elevated number of apoptotic colonic epithelial cells (117, 118). This suggests that an increase in apoptotic cells or a malfunction in their clearance may contribute to the intensification of inflammation. Therefore, boosting efferocytosis in the early phases of IBD, accelerating the removal of apoptotic cells, and promoting efferocytosis by intestinal epithelial cells could be crucial in reducing inflammation later in the disease. Recent advancements have introduced a “chimeric efferocytosis receptor” that enhances macrophage efferocytosis, allowing them to engulf more apoptotic cells and initiate anti-inflammatory signaling, alleviating colitis symptoms. However, the findings are still preliminary, and the effectiveness is restricted to BAI1 and TIM-4 receptors, which have specific limitations. It remains uncertain whether this approach will function effectively in human inflammatory conditions, but it provides a new avenue for IBD treatment (119). In conclusion, from a pharmacological perspective, developing agents to enhance apoptotic cell clearance, including strategies that boost efferocytosis by intestinal epithelial cells and Paneth cells, represents a promising therapeutic approach for IBD that deserves further investigation.
As mentioned earlier, the pro-resolving factors secreted following efferocytosis can stimulate the proliferation and differentiation of various intestinal cells, aiding in the repair of the intestinal mucosa, while also releasing anti-inflammatory mediators to help resolve inflammation. These macrophage-secreted pro-resolving factors are collectively known as SuperMApo, a novel biological complex that includes all cytokines released by macrophages during efferocytosis, such as TGF-β, IL-10, IL-1RA, and others. These factors primarily coordinate the actions of fibroblasts and intestinal epithelial cells (IECs) to support mucosal healing and pro-resolution responses (120–122). This has been confirmed through three experimental models: the initial T-cell transfer model (123), the DSS-induced colitis model, and the biopsy forceps injury model of the colonic mucosa (124). From a resolution pharmacology perspective, introducing these pro-resolving factors in the early stages of IBD can enhance the clearance of apoptotic cells by IECs, thereby reducing inflammation in the later stages of the disease. Additionally, this process helps trigger mechanisms involved in mucosal healing (39) and promotes myofibroblast contraction to facilitate wound closure (125), making it a promising potential treatment approach for IBD (39).
Macrophages play a multifaceted role in the immune system and inflammatory responses, making them an attractive target for the treatment of various inflammatory diseases. Inspired by macrophages’ ability to selectively engulf apoptotic cells while leaving healthy cells unharmed, Zheng Ying and colleagues created a biomimetic drug delivery system (Effero-RLP) by hybridizing apoptotic red blood cell membranes with liposomes. This system targets inflammation via macrophage-mediated efferocytosis. The researchers encapsulated rosiglitazone (ROSI), an agonist of the anti-inflammatory peroxisome proliferator-activated receptor γ (PPAR-γ), within the delivery system. Through efferocytosis, ROSI is directed to the site of inflammation. In a DSS-induced colitis model, Effero-RLP significantly enhanced macrophage efferocytosis, induced polarization of macrophages to an anti-inflammatory phenotype, and accumulated in inflamed colonic tissues to treat IBD (126). Additionally, β-cyclodextrin, a biomimetic nanodrug sensitive to reactive oxygen species and coated with macrophage membranes, has shown high efficiency in treating UC (127). These biomimetic drug therapies offer several benefits, including enhanced clinical efficacy for IBD and precise drug delivery targeting. However, challenges such as material preparation and binding efficiency remain, and the approach still faces several hurdles before reaching clinical trials.
COL is a biased agonist that partially occupies the ligand-binding pocket of formyl peptide receptor 2 (FPR2), a receptor involved in regulating inflammation. COL enhances efferocytosis by directly binding to FPR2 and has shown significant therapeutic effects in DSS-induced colitis (38). Additionally, macrophage efferocytosis receptors Axl and MerTK, which are IL-4-dependent, play a role in repairing damaged intestinal tissue, suggesting that targeting Axl/MerTK on macrophages could offer a new therapeutic approach for IBD treatment (128). These findings suggest that enhancing macrophage efferocytosis pharmacologically can alleviate colitis symptoms, and IBD is a disease that could benefit from such treatment. Therefore, this research provides valuable insights for developing new therapies for various IBD conditions.
In recent years, an increasing number of studies have highlighted the connection between efferocytosis and IBD. The role of efferocytosis in IBD primarily involves two aspects: promoting pro-resolving responses and supporting the repair of the intestinal mucosa. This article outlines the efferocytosis process and its associated molecules, and reviews the key mechanisms by which efferocytosis affects IBD. These mechanisms include the secretion of pro-resolving factors that encourage the proliferation and differentiation of fibroblasts and intestinal epithelial cells (IECs) to repair the damaged mucosa (39), as well as the release of anti-inflammatory mediators such as TGF-β (38) and IL-10 (37), which help suppress intestinal inflammation. Furthermore, macrophages and other cells enhance efferocytosis by modifying metabolism and activating receptors, indicating the potential and feasibility of boosting efferocytosis to treat diseases like IBD. During the literature review, it was also found that intestinal epithelial cells and Paneth cells are involved in efferocytosis (98, 99), affecting the progression of IBD. Additionally, the immunological mechanisms underlying IBD are related to efferocytosis (41, 42). However, the current research still lacks direct studies linking IBD mechanisms with efferocytosis, with most studies focusing on inflammatory and pro-resolving responses. There is a need for more research exploring the relationship between IBD-associated immune mechanisms and efferocytosis.
A considerable body of evidence demonstrates a strong association between defects in macrophage efferocytosis and the progression of IBD (129), indicating the potential of macrophages as a novel therapeutic target for IBD. The present review examines various potential therapies targeting macrophage efferocytosis for IBD treatment, though most of these approaches lack practical application and corresponding advancements in drug research and development. Furthermore, questions regarding the long-term efficacy and real-world feasibility of these treatment methods remain unresolved, necessitating further in-depth and comprehensive investigations.
Research also emphasizes the link between IBD and the onset of colon cancer and other malignancies, with factors such as genetic predisposition, impaired intestinal barrier function, and immunological dysregulation underlying persistent intestinal inflammation in IBD (130). Both deficiencies and enhancements in efferocytosis have been implicated in promoting cancer progression (26). The defining features of UC and CD include chronic, unresolved gastrointestinal inflammation, which over time may lead to complications such as fibrosis, organ damage, and subsequent organ failure, including CD-associated strictures, abscesses, and fistula formation (131). Consequently, achieving a balance in the transition between M1 and M2 macrophages and optimizing the regulation of efferocytosis remains a critical area for further investigation, with significant implications for improving the clinical management of IBD patients.
Y-QL: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. Z-ZL: Funding acquisition, Methodology, Supervision, Validation, Visualization, Writing – review & editing. Y-LH: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. Q-BW: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Software, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was granted and supported by the Scientific research project of traditional Chinese medicine in Henan Province (No. 2024ZY3040), the 75th General Program of China Postdoctoral Science Foundation (No. 2024M750840), The Scientific research project of traditional Chinese medicine in Henan Province (No. 2021JDZX2005), Henan Medical Science and Technology Research Program in 2024 (No.SBGJ202403039), Zhengzhou Medical and health science and Technology Innovation Guidance Program (No.2024YLZDJH072), and key scientific research projects of Henan Universities in 2025 (No.25A360025).
We appreciate all the reviewers who participated in the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. (2019) 20:254–67. doi: 10.1038/s41577-019-0240-6
3. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. (2019) 2019:7247238. doi: 10.1155/2019/7247238
4. Ge Y, Huang M, Yao YM. Efferocytosis and its role in inflammatory disorders. Front Cell Dev Biol. (2022) 25:839248. doi: 10.3389/fcell.2022.839248
5. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
6. Imamura H, Sakamoto S, Yoshida T, Matsui Y, Penuela S, Laird DW, et al. Single-cell dynamics of pannexin-1-facilitated programmed ATP loss during apoptosis. Elife. (2020) 9:e61960. doi: 10.7554/eLife.61960
7. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. (2010) 467:863–7. doi: 10.1038/nature09413
8. Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. (2003) 113:717–30. doi: 10.1016/s0092-8674(03)00422-7
9. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. (2008) 22:2629–38. doi: 10.1096/fj.08-107169
10. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. (2008) 112:5026–36. doi: 10.1182/blood-2008-06-162404
11. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell- surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. (2005) 123:321–34. doi: 10.1016/j.cell.2005.08.032
12. Jaillon S, Jeannin P, Hamon Y, Frémaux I, Doni A, Bottazzi B, et al. Endogenous PTX3 translocates at the membrane of late apoptotic human neutrophils and is involved in their engulfment by macrophages. Cell Death Differ. (2009) 16:465–74. doi: 10.1038/cdd.2008.173
13. Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, et al. Decrease of sialic acid residues as an eat-me signal on the surface of apoptotic lymphocytes. J Cell Sci. (2010) 123:3347–56. doi: 10.1242/jcs.066696
14. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. (2000) 288:20514. doi: 10.1126/science.288.5473.2051
15. Liao CY, Li G, Kang FP, Lin CF, Xie CK, Wu YD, et al. Necroptosis enhances ‘don’t eat me’ signal and induces macrophage extracellular traps to promote pancreatic cancer liver metastasis. Nat Commun. (2024) 15:6043. doi: 10.1038/s41467-024-50450-6
16. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
17. Morioka S, Maueröder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
18. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. (2018) 563:714–8. doi: 10.1038/s41586-018-0735-5
19. Tajbakhsh A, Yousefi F, Abedi SM, Rezaee M, Savardashtaki A, Teng Y, et al. The cross-talk between soluble “Find me” and “Keep out” signals as an initial step in regulating efferocytosis. J Cell Physiol. (2022) 237:3113–26. doi: 10.1002/jcp.30770
20. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. (2007) 450:430–4. doi: 10.1038/nature06329
21. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. (2008) 15:192–201. doi: 10.1038/sj.cdd.4402242
22. Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. (2010) 1209:23–9. doi: 10.1111/j.1749-6632.2010.05744.x
23. Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. (2004) 11:943–5. doi: 10.1038/sj.cdd.4401421
24. Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, et al. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. (1997) 272:29411–4. doi: 10.1074/jbc.272.47.29411
25. Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. (2003) 4:587–98. doi: 10.1016/s1534-5807(03)00090-x
26. Gheibi Hayat SM, Bianconi V, Pirro M, Sahebkar A. Efferocytosis: molecular mechanisms and pathophysiological perspectives. Immunol Cell Biol. (2019) 97:124–33. doi: 10.1111/imcb.12206
27. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front Immunol. (2020) 11:553. doi: 10.3389/fimmu.2020.00553
28. Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. (2001) 11:471–7. doi: 10.1016/s0962-8924(01)02153-5
29. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. (2001) 194:781–95. doi: 10.1084/jem.194.6.781
30. Liu QA, Hengartner MO. Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans Cell. (1998) 93:961–72. doi: 10.1016/s0092-8674(00)81202-7
31. Su HP, Nakada-Tsukui K, Tosello-Trampont AC, Li Y, Bu G, Henson PM, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/lowdensity lipoprotein receptor-related protein (LRP). J Biol Chem. (2002) 277:11772–9. doi: 10.1074/jbc.M109336200
32. Hurwitz ME, Vanderzalm PJ, Bloom L, Goldman J, Garriga G, Horvitz HR. Abl kinase inhibits the engulfment of apoptotic [corrected] cells in Caenorhabditis elegans. PloS Biol. (2009) 7:e99. doi: 10.1371/annotation/2259f958-a68e-4e57-92b5-2ef003070cf1
33. Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. (2008) 10:556–66. doi: 10.1038/ncb1718
34. Nordmann M, Cabrera M, Perz A, Bröcker C, Ostrowicz C, Engelbrecht-Vandré S, et al. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol. (2010) 20:1654–9. doi: 10.1016/j.cub.2010.08.002
35. Odaka C, Mizuochi T. Role of macrophage lysosomal enzymes in the degradation of nucleosomes of apoptotic cells. J Immunol. (1999) 163:5346–52. doi: 10.4049/jimmunol.163.10.5346
36. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. (2006) 443:998–1002. doi: 10.1038/nature05245
37. Schreiber S, Heinig T, Thiele HG, Raedler A. Immunoregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology. (1995) 108:1434–44. doi: 10.1016/0016-5085(95)90692-4
38. Del Zotto B, Mumolo G, Pronio AM, Montesani C, Tersigni R, Boirivant M. TGF-beta1 production in inflammatory bowel disease: differing production patterns in Crohn’s disease and ulcerative colitis. Clin Exp Immunol. (2003) 134:120–6. doi: 10.1046/j.1365-2249.2003.02250.x
39. Martin-Rodriguez O, Gauthier T, Bonnefoy F, Couturier M, Daoui A, Chagué C, et al. Pro- resolving factors released by macrophages after efferocytosis promote mucosal wound healing in inflammatory bowel disease. Front Immunol. (2021) 12:754475. doi: 10.3389/fimmu.2021.754475
40. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr, Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–677.e6. doi: 10.1016/j.immuni.2018.07.015
41. Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med. (2009) 15:199–207. doi: 10.1016/j.molmed.2009.03.002
42. Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. (2009) 458:78–82. doi: 10.1038/nature07781
43. Dejani NN, Orlando AB, Niño VE, Penteado LA, Verdan FF, Bazzano JMR, et al. Intestinal host defense outcome is dictated by PGE production during efferocytosis of infected cells. Proc Natl Acad Sci U S A. (2018) 115:E8469–78. doi: 10.1073/pnas.1722016115
44. Lee SH, Kwon JE, Cho ML. Immunological pathogenesis of inflammatory bowel disease. Intest Res. (2018) 16:26–42. doi: 10.5217/ir.2018.16.1.26
45. Peters LA, Perrigoue J, Mortha A, Iuga A, Song WM, Neiman EM, et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet. (2017) 49:1437–49. doi: 10.1038/ng.3947
46. Strober W, Kitani A, Fuss I, Asano N, Watanabe T. The molecular basis of NOD2 susceptibility mutations in Crohn’s disease. Mucosal Immunol. (2008) 1 Suppl 1:S5–9. doi: 10.1038/mi.2008.42
47. Grau A, Tabib A, Grau I, Reiner I, Mevorach D. Apoptotic cells induce NF-κB and inflammasome negative signaling. PloS One. (2015) 10:e0122440. doi: 10.1371/journal.pone.0122440
48. Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. (2010) 8:564–77. doi: 10.1038/nrmicro2403
49. Gross V, Andus T, Caesar I, Roth M, Schölmerich J. Evidence for continuous stimulation of interleukin-6 production in Crohn’s disease. Gastroenterology. (1992) 102:514–9. doi: 10.1016/0016-5085(92)90098-j
50. Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. (1999) 116:22–8. doi: 10.1016/s0016-5085(99)70224-6
51. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. (2014) 15(7):676–86. doi: 10.1038/ni.2920
52. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. (2010) 33:279–88. doi: 10.1016/j.immuni.2010.08.010
53. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. (2003) 52:65–70. doi: 10.1136/gut.52.1.65
54. Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. (2000) 6:583–8. doi: 10.1038/75068
55. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. (2010) 238:247–62. doi: 10.1111/j.1600-065X.2010.00951.x
56. Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflammation Bowel Dis. (2012) 18:180–6. doi: 10.1002/ibd.21677
57. Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology. (2008) 134:1038–48. doi: 10.1053/j.gastro.2008.01.041
58. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. (2005) 129:550–64. doi: 10.1016/j.gastro.2005.05.002
59. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. (2007) 204:1775–85. doi: 10.1084/jem.20070602
60. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol. (2007) 5:405–17. doi: 10.1038/nrmicro1657
61. Meng F, Jiang X, Wang X, Zheng Q, Wang XN, Mei C, et al. Tumor necrosis factor-like cytokine 1A plays a role in inflammatory bowel disease pathogenesis. Proc Natl Acad Sci U SA. (2023) 120:e2120771120. doi: 10.1073/pnas.2120771120
62. Kamada N, Hisamatsu T, Honda H, Kobayashi T, Chinen H, Takayama T, et al. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn’s disease. Inflammation Bowel Dis. (2010) 16:568–75. doi: 10.1002/ibd.21124
63. Sidhu-Varma M, Shih DQ, Targan SR. Differential levels of tl1a affect the expansion and function of regulatory T cells in modulating murine colitis. Inflammation Bowel Dis. (2016) 22:548–59. doi: 10.1097/MIB.0000000000000653
64. Wang D, Li H, Duan YY, Han F, Luo YX, Wu MY, et al. TL1A modulates the severity of colitis by promoting Th9 differentiation and IL-9 secretion. Life Sci. (2019) 231:116536. doi: 10.1016/j.lfs.2019.06.011
65. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
66. Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, et al. Myeloid slc2a1- deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J Immunol. (2019) 202:1265–86. doi: 10.4049/jimmunol.1800002
67. Schilperoort M, Ngai D, Katerelos M, Power DA, Tabas I. PFKFB2-mediated glycolysis promotes lactate-driven continual efferocytosis by macrophages. Nat Metab. (2023) 5:431–44. doi: 10.1038/s42255-023-00736-8
68. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004
69. Sukka SR, Ampomah PB, Darville LNF, Ngai D, Wang X, Kuriakose G, et al. Efferocytosis drives a tryptophan metabolism pathway in macrophages to promote tissue resolution. Nat Metab. (2024) 6:1736–55. doi: 10.1038/s42255-024-01115-7
70. Yurdagul A Jr, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–533.e10. doi: 10.1016/j.cmet.2020.01.001
71. Jakobsson T, Vedin LL, Hassan T, Venteclef N, Greco D, D’Amato M, et al. The oxysterol receptor LXRβ protects against DSS- and TNBS-induced colitis in mice. Mucosal Immunol. (2014) 7:1416–28. doi: 10.1038/mi.2014.31
72. Miranda-Bautista J, Rodríguez-Feo JA, Puerto M, López-Cauce B, Lara JM, González-Novo R, et al. Liver X receptor exerts anti-inflammatory effects in colonic epithelial cells via ABCA1 and its expression is decreased in human and experimental inflammatory bowel disease. Inflammation Bowel Dis. (2021) 27:1661–73. doi: 10.1093/ibd/izab034
73. Heimerl S, Moehle C, Zahn A, Boettcher A, Stremmel W, Langmann T, et al. Alterations inintestinal fatty acid metabolism in inflammatory bowel disease. Biochim Biophys Acta. (2006) 1762:341–50. doi: 10.1016/j.bbadis.2005.12.006
74. Pompili S, Vetuschi A, Latella G, Smakaj A, Sferra R, Cappariello A. PPAR-gamma orchestrates EMT, AGE, and cellular senescence pathways in colonic epithelium and restrains the progression of IBDs. Int J Mol Sci. (2023) 24:8952. doi: 10.3390/ijms24108952
75. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. (2009) 15:1266–72. doi: 10.1038/nm.2048
76. Johann AM, von Knethen A, Lindemann D, Brüne B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ. (2006) 13:1533–40. doi: 10.1038/sj.cdd.4401832
77. Majai G, Sarang Z, Csomós K, Zahuczky G, Fésüs L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. (2007) 37:1343–54. doi: 10.1002/eji.200636398
78. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
79. Yanagihashi Y, Segawa K, Maeda R, Nabeshima YI, Nagata S. Mouse macrophages show different requirements for phosphatidylserine receptor Tim4 in efferocytosis. Proc Natl Acad Sci U SA. (2017) 114:8800–5. doi: 10.1073/pnas.1705365114
80. Li T, Wang C, Liu Y, Li B, Zhang W, Wang L, et al. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohns Colitis. (2020) 14:240–53. doi: 10.1093/ecco-jcc/jjz132
81. Sun S, Duan Z, Wang X, Chu C, Yang C, Chen F, et al. Neutrophil extracellular traps impair intestinal barrier functions in sepsis by regulating TLR9-mediated endoplasmic reticulum stress pathway. Cell Death Dis. (2021) 12:606. doi: 10.1038/s41419-021-03896-1
82. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PloS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
83. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun. (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
84. Shiratori-Aso S, Nakazawa D, Kudo T, Kanda M, Ueda Y, Watanabe-Kusunoki K, et al. CD47 blockade ameliorates autoimmune vasculitis via efferocytosis of neutrophil extracellular traps. JCI Insight. (2023) 8:e167486. doi: 10.1172/jci.insight.167486
85. Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. (2007) 178:4595–605. doi: 10.4049/jimmunol.178.7.4595
86. MaChado MG, Tavares LP, Souza GVS, Queiroz-Junior CM, Ascenção FR, Lopes ME, et al. The Annexin A1/FPR2 pathway controls the inflammatory response and bacterial dissemination in experimental pneumococcal pneumonia. FASEB J. (2020) 34:2749–64. doi: 10.1096/fj.201902172R
87. Brook M, Tomlinson GH, Miles K, Smith RW, Rossi AG, Hiemstra PS, et al. Neutrophil-derived alpha defensins control inflammation by inhibiting macrophage mRNA translation. Proc Natl Acad Sci U.S.A. (2016) 113:4350–5. doi: 10.1073/pnas.1601831113
88. Miles K, Clarke DJ, Lu W, Sibinska Z, Beaumont PE, Davidson DJ, et al. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol. (2009) 183:2122–32. doi: 10.4049/jimmunol.0804187
89. Nalleweg N, Chiriac MT, Podstawa E, Lehmann C, Rau TT, Atreya R, et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut. (2015) 64:743–55. doi: 10.1136/gutjnl-2013-305947
90. Su J, Chen T, Ji XY, Liu C, Yadav PK, Wu R, et al. IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflammation Bowel Dis. (2013) 19:720–8. doi: 10.1097/MIB.0b013e3182802a76
91. Wang EJ, Wu MY, Ren ZY, Zheng Y, Ye RD, Tan CSH, et al. Targeting macrophage autophagy for inflammation resolution and tissue repair in inflammatory bowel disease. Burns Trauma. (2023) 11:tkad004. doi: 10.1093/burnst/tkad004
92. Wu MY, Liu L, Wang EJ, Xiao HT, Cai CZ, Wang J, et al. PI3KC3 complex subunit NRBF2 isrequired for apoptotic cell clearance to restrict intestinal inflammation. Autophagy. (2021) 17:1096–111. doi: 10.1080/15548627.2020.1741332
93. Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. (2012) 109:11776–81. doi: 10.1073/pnas.1206059109
94. Vago JP, Galvão I, Negreiros-Lima GL, Teixeira LCR, Lima KM, Sugimoto MA, et al. Glucocorticoid-induced leucine zipper modulates macrophage polarization and apoptotic cell clearance. Pharmacol Res. (2020) 158:104842. doi: 10.1016/j.phrs.2020.104842
95. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. (2006) 314:1461–3. doi: 10.1126/science.1135245
96. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. (2005) 22:285–94. doi: 10.1016/j.immuni.2005.01.011
97. Freire-de-Lima CG, Xiao YQ, Gardai SJ, Bratton DL, Schiemann WP, Henson PM. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J Biol Chem. (2006) 281:38376–84. doi: 10.1074/jbc.M605146200
98. Lee CS, Penberthy KK, Wheeler KM, Juncadella IJ, Vandenabeele P, Lysiak JJ, et al. Boosting apoptotic cell clearance by colonic epithelial cells attenuates inflammation in vivo. Immunity. (2016) 44:807–20. doi: 10.1016/j.immuni.2016.02.005
99. Shankman LS, Fleury ST, Evans WB, Penberthy KK, Arandjelovic S, Blumberg RS, et al. Efferocytosis by Paneth cells within the intestine. Curr Biol. (2021) 31:2469–2476.e5. doi: 10.1016/j.cub.2021.03.055
100. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. (2008) 456:259–63. doi: 10.1038/nature07416
101. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. (2013) 503:272–6. doi: 10.1038/nature12599
102. Murray A, Nguyen TM, Parker CE, Feagan BG, MacDonald JK. Oral 5-aminosalicylic acid for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. (2020) 8:CD000543. doi: 10.1002/14651858.CD000543.pub5
103. Truelove SC, Witts LJ. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br Med J. (1955) 2:1041–8. doi: 10.1136/bmj.2.4947.1041
104. Chande N, MacDonald JK, McDonald JW. Methotrexate for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. (2007) 4):CD006618. doi: 10.1002/14651858.CD006618.pub2
105. Naganuma M, Fujii T, Watanabe M. The use of traditional and newer calcineurin inhibitors in inflammatory bowel disease. J Gastroenterol. (2011) 46:129–37. doi: 10.1007/s00535-010-0352-z
106. Naganuma M, Yokoyama Y, Motoya S, Watanabe K, Sawada K, Hirai F, et al. Efficacy of apheresis as maintenance therapy for patients with ulcerative colitis in an open-label prospective multicenter randomised controlled trial. J Gastroenterol. (2020) 55:390–400. doi: 10.1007/s00535-019-01651-0
107. Ocansey DKW, Wang L, Wang J, Yan Y, Qian H, Zhang X, et al. Mesenchymal stem cell-gut microbiota interaction in the repair of inflammatory bowel disease: an enhanced therapeutic effect. Clin Transl Med. (2019) 8:31. doi: 10.1186/s40169-019-0251-8
109. Zhu W, Jin Z, Yu J, Liang J, Yang Q, Li F, et al. Baicalin ameliorates experimental inflammatory bowel disease through polarization of macrophages to an M2 phenotype. Int Immunopharmacol. (2016) 35:119–26. doi: 10.1016/j.intimp.2016.03.030
110. Tokuyasu N, Shomori K, Amano K, Honjo S, Sakamoto T, Watanabe J, et al. Indirubin, a constituent of the chinese herbal medicine qing-dai, attenuates dextran sulfate sodium-induced murine colitis. Yonago Acta Med. (2018) 61:128–36. doi: 10.33160/yam.2018.06.005
111. Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol. (2014) 7:6–19. doi: 10.1038/mi.2013.73
112. Nakase H, Hirano T, Wagatsuma K, Ichimiya T, Yamakawa T, Yokoyama Y, et al. Artificial intelligence-assisted endoscopy changes the definition of mucosal healing in ulcerative colitis. Dig Endosc. (2021) 33:903–11. doi: 10.1111/den.13825
113. Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. (1993) 13:6231–40. doi: 10.1128/mcb.13.10.6231-6240.1993
114. Bauer C, Duewell P, Lehr HA, Endres S, Schnurr M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis. (2012) 30 Suppl 1:82–90. doi: 10.1159/000341681
115. Cai H, Liu Z, Sun P, Zhou Y, Yan Y, Luo Y, et al. Discovery of a dual-acting inhibitor of interleukin-1β and STATs for the treatment of inflammatory bowel disease. RSC Med Chem. (2023) 15:193–206. doi: 10.1039/d3md00451a
116. Chen H, Kasagi S, Chia C, Zhang D, Tu E, Wu R, et al. Extracellular vesicles from apoptotic cells promote TGFβ Production in macrophages and suppress experimental colitis. Sci Rep. (2019) 9:5875. doi: 10.1038/s41598-019-42063-7
117. Di Sabatino A, Ciccocioppo R, Luinetti O, Ricevuti L, Morera R, Cifone MG, et al. Increased enterocyte apoptosis in inflamed areas of Crohn’s disease. Dis Colon Rectum. (2003) 46:1498–507. doi: 10.1007/s10350-004-6802-z
118. Hagiwara C, Tanaka M, Kudo H. Increase in colorectal epithelial apoptotic cells in patients with ulcerative colitis ultimately requiring surgery. J Gastroenterol Hepatol. (2002) 17:758–64. doi: 10.1046/j.1440-1746.2002.02791.x
119. Morioka S, Kajioka D, Yamaoka Y, Ellison RM, Tufan T, Werkman IL, et al. Chimeric efferocytic receptors improve apoptotic cell clearance and alleviate inflammation. Cell. (2022) 185:4887–4903.e17. doi: 10.1016/j.cell.2022.11.029
120. Neufert C, Becker C, Türeci Ö, Waldner MJ, Backert I, Floh K, et al. Tumor fibroblast-derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J Clin Invest. (2013) 123:1428–43. doi: 10.1172/JCI63748
121. Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. (2008) 14:348–53. doi: 10.3748/wjg.14.348
122. Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. (2009) 324:1666–9. doi: 10.1126/science.1172687
123. Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. (1994) 1:553–62. doi: 10.1016/1074-7613(94)90045-0
124. Neurath MF, Wittkopf N, Wlodarski A, Waldner M, Neufert C, Wirtz S, et al. Assessment of tumor development and wound healing using endoscopic techniques in mice. Gastroenterology. (2010) 139:1837–1843.e1. doi: 10.1053/j.gastro.2010.10.007
125. Montesano R, Orci L. Transforming growth factor beta stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proc Natl Acad Sci U S A. (1988) 85:4894–7. doi: 10.1073/pnas.85.13.4894
126. Han R, Ren Z, Wang Q, Zha H, Wang E, Wu M, et al. Synthetic biomimetic liposomes harness efferocytosis machinery for highly efficient macrophages-targeted drug delivery to alleviate inflammation. Adv Sci (Weinh). (2024) 11:e2308325. doi: 10.1002/advs.202308325
127. Sun T, Kwong CHT, Gao C, Wei J, Yue L, Zhang J, et al. Amelioration of ulcerative colitis via inflammatory regulation by macrophage-biomimetic nanomedicine. Theranostics. (2020) 10:10106–19. doi: 10.7150/thno.48448
128. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. (2017) 356:1072–6. doi: 10.1126/science.aai8132
129. Ho GT, Cartwright JA, Thompson EJ, Bain CC, Rossi AG. Resolution of inflammation and gut repair in IBD: translational steps towards complete mucosal healing. Inflammation Bowel Dis. (2020) 26:1131–43. doi: 10.1093/ibd/izaa045
130. Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. (2022) 162:715–730.e3. doi: 10.1053/j.gastro.2021.10.035
131. Zhang H, Shi Y, Lin C, He C, Wang S, Li Q, et al. Overcoming cancer risk in inflammatory bowel disease: new insights into preventive strategies and pathogenesis mechanisms including interactions of immune cells, cancer signaling pathways, and gut microbiota. Front Immunol. (2024) 14:1338918. doi: 10.3389/fimmu.2023.1338918
Keywords: efferocytosis, inflammatory bowel disease, macrophages, intestinal mucosal repair, inflammatory response
Citation: Liu Y-Q, Li Z-Z, Han Y-L and Wang Q-B (2025) The role of efferocytosis in inflammatory bowel disease. Front. Immunol. 16:1524058. doi: 10.3389/fimmu.2025.1524058
Received: 07 November 2024; Accepted: 16 January 2025;
Published: 18 February 2025.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Thierry Mp Gauthier, National Institutes of Health (NIH), United StatesCopyright © 2025 Liu, Li, Han and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Li Han, aHlsemhlbmppdUAxNjMuY29t; Qing-Bo Wang, d3FiMjJAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.