
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 February 2025
Sec. Alloimmunity and Transplantation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1520619
 Paolo Durigutto1†
Paolo Durigutto1† Maria Cristina Grimaldi2†
Maria Cristina Grimaldi2† Sara Bozzer2
Sara Bozzer2 Elena Raschi1
Elena Raschi1 Pierluigi Meroni1
Pierluigi Meroni1 Francesco Tedesco1
Francesco Tedesco1 Paolo Macor2*
Paolo Macor2*Anti-phospholipid syndrome (APS) is a systemic autoimmune disease characterized by thrombotic vascular occlusion and maternal morbidity. Anti-coagulants remain pivotal drugs for the management of APS, but a significant proportion of patients do not benefit from long-term anti-coagulation and may require an alternative therapy to prevent antibody deposition and vascular thrombosis. We have developed a therapeutic approach based on the use of safe polymeric nanoparticles that selectively target beta2-glycoprotein I (β2GPI) deposited on endothelial cells (tNPs). Their efficacy was tested in a rat model of APS developed by infusing patients’ sera containing medium–high titer antibodies against domain I of β2GPI. The tNPs bearing a CH2-deleted anti-β2GPI recombinant antibody as a targeting agent recognize β2GPI deposited on endothelial cells but failed to induce blood clot formation. The tNPs infused into rats immediately before APS sera competed with patients’ antibodies, preventing their binding to deposited β2GPI and, as a consequence, resulted in thrombus formations and occlusion of mesenteric vessels. Similar results were obtained by injecting tNPs 24 hours before the administration of patients’ sera to induce blood clot formation. Our findings suggest that β2GPI-targeted polymeric nanoparticles represent a stable and safe approach to prevent thrombus formation and vessel occlusion in a rat model of APS and may be used to control thrombosis developing in APS patients as a result of acute triggering events.
Vascular thrombosis and pregnancy complications are the main clinical manifestations of anti-phospholipid syndrome (APS), an autoimmune disease that affects relatively young individuals with important clinical and socio-economic implications (1). Although blood clots can form in arteries and veins of different sizes in the vascular tree, they tend to develop more often in vessels of some districts, leading to stroke, deep vein thrombosis, pulmonary embolism, and, less frequently, myocardial infarction (2). Occasionally, the disease manifests as catastrophic APS (CAPS), a very aggressive variant of the syndrome, characterized by small-vessel occlusion and multiple organ failure that develops over a short period of time (3, 4).
Anti-phospholipid antibodies (aPL) detected in the sera of patients with APS are mainly directed against beta2-glycoprotein I (β2GPI), currently considered the most relevant target antigen of aPL (5, 6). The anti-β2GPI antibodies, now included among diagnostic tests and classification criteria of the disease, have been shown to play a key role in the development of vascular thrombosis and pregnancy loss. This conclusion is supported by the finding that IgG, purified from the sera of patients with medium–high titers of anti-β2GPI antibodies, were able to induce thrombus formation in a rat model of APS and that the prothrombotic effect was prevented by the depletion of anti-β2GPI antibodies via affinity chromatography (7). The management of patients with APS, aimed at preventing vascular thrombi, is based on the use of anti-coagulant drugs, such as heparin or warfarin, and a combination of low-dose aspirin and low-molecular-weight heparin (8, 9). However, despite the satisfactory results obtained with these preventive measures, a significant proportion of patients do not benefit from long-term anti-coagulant therapy and develop recurrent thrombosis (1, 10). Drugs that interfere with the immune mechanism responsible for thrombus formation in APS, starting from the binding of antibodies to their target antigen β2GPI and proceeding with complement activation, represent an alternative therapeutic strategy. Complement inhibition with eculizumab (Soliris) has been recommended for the treatment of patients with CAPS, following anecdotal reports of beneficial effects of this antibody in a few patients refractory to standard treatment (11, 12). However, there is no general consensus on the use of complement inhibitors as first-line therapy in these patients (13). Inhibition of antibody binding to β2GPI is another therapeutic option that can be obtained with a monoclonal antibody that competes with the patients’ antibodies, preventing their deposition on cell-bound β2GPI. We have previously shown that this approach is feasible using an anti-β2GPI single-chain fragment variable isolated from a human phase display library and subsequently engineered to contain an IgG1 hinge-CH2-CH3 domain (MBB2) (14). This antibody recognizes the DI domain of β2GPI with a higher affinity than patients’ antibodies, and the CH2-deleted variant of this antibody (MBB2ΔCH2) was shown to prevent their ability to induce thrombus formation in a rat model of APS (13).
We now present a therapeutic approach to prevent thrombotic vascular occlusion using nanoparticles coated with MBB2ΔCH2 to target β2GPI deposited on the endothelium, allowing the β2GPI-targeted nanostructures to act as a protective shield against the prothrombotic effect of patients’ antibodies in a rat model of APS.
Poly(lactic-co-glycolic) acid (PLGA)–polyvinyl alcohol (PVA) nanoparticles (NPs) were synthesized by the double-emulsion solvent evaporation method described by Vasir and Labhasetwar (15). Briefly, PVA (1 mg/mL in 2-Morpholinoethanesulfonic acid monohydrate (MES) buffer) (Sigma-Aldrich Co., St. Louis, MO, USA; m.w. 30,000–70,000, 87%–90% hydrolyzed) was covalently linked to MBB2ΔCH2 (300 μg) through the EDC/sulfo-NHS Crosslinking protocol (Thermo-Fisher Scientific, Waltham, MA, USA) and then added to 10 mL of 2.0% (w/v) PVA solution in cold Tris-EDTA pH 8 (TE buffer). Afterward, 30 mg of PLGA (Sigma-Aldrich Co.; m.w. 30,000–60,000, lactide:glycolide 50:50) were dissolved in 1 mL of chloroform. To start the synthesis, 300 μL of bovine serum albumin (BSA) or Fluorescein isothiocyanate (Fitc)-BSA (Sigma-Aldrich Co.; 20 mg/mL) were added to the PLGA solution and vortexed and sonicated for 30 s at 15 W of the maximum power using Bandelin Sonopuls HD 2070 (Bandelin electronic GmbH & Co. KG, Berlin, Germany) to form the primary emulsion. The emulsion was added to the PVA solution and sonicated for 1 minute as described above to form a secondary emulsion. The resulting emulsion was stirred (750 rpm) overnight to allow for chloroform evaporation. NPs were collected by centrifugation at 12,000g for 30 minutes at 4°C, and the pellet was washed in TE buffer. Finally, NPs were resuspended in 2 mL of phosphate-buffered saline (PBS) filtered 0.2 μm and stored at 4°C. Untargeted NPs were produced following the same protocol, skipping the antibody coupling step.
Nanoparticle tracking analysis (NTA) was performed using NanoSight LM10 (Malvern Panalytical, Malvern, UK) diluting NP samples 1:4,000 (v/v) in Milli-Q water immediately before the measurements.
Hydrodynamic diameter (Z-Ave), polydispersity index (PDI), and surface charge (zeta potential) analyses were performed with the dynamic light scattering technique using Zetasizer Nano ZSP (Malvern Panalytical, Malvern, UK). NPs were diluted 1:200 (v/v) in H2O Milli-Q filtered 0.2 μm, and measurements were performed at 37°C and a scattering angle of 173°.
The morphology of NPs was analyzed by scanning electron microscopy (SEM). NPs were diluted 1:50 v/v in H2O Milli-Q filtered 0.2 μm. Then, a drop of the sample was deposited on a glass coverslip and left to dry at room temperature. Glass coverslips were mounted on aluminum stubs coated with double-sided carbon tape. Samples were carbon coated using the Q150T ES plus sputter coater (Quorum, Sacramento, CA, USA). Samples were analyzed by scanning electron microscopy using a Gemini 300 SEM (Zeiss, Oberkochen, Germany), working in secondary electron mode, at an acceleration voltage of 2 kV and a working distance of 3 mm.
Functional activity of complement was determined by measuring the activity of the classical pathway (CH50) on sensitized mutton erythrocytes (EA, Microbiol s.r.l., Cagliari, CA, Italy) as previously described (16, 17). Briefly, NHS (100 μL) and NPs (8 μL) were incubated for 2 hours at 37°C under shaking (750 rpm). The samples were centrifuged for 5 minutes at 8,000g, and the supernatants were diluted in complement fixation diluent (CFD; NaCl 142 mM, Na-5-5-diethylbarbiturate 5 mM, MgCl2 0.5 mM, agar 0.05%, NaN3 0.01%, and Ethyleneglycol- bis(β-aminoethyl)-N,N,Nʹ,Nʹ-tetraacetic Acid (EGTA) 10 mM) at 1:50, 1:100, 1:200, and 1:400 (v/v) in a final volume of 200 μL and incubated with 50 μL of antibody-sensitized erythrocytes (EA) 1% for 30 minutes to activate the classical complement pathway. The total lysis is represented by EA 1% diluted in H2O, while the blank is represented by EA 1% diluted in CFD. The reaction was then blocked by the addition of EDTA 20 mM. The % lysis was measured at 415 nm with ELISA Reader TECAN Infinite M200 (Tecan Italia, Milan, Italy) after centrifugation at 12,000g for 1 minute. Complement activation was checked by preparing a standard curve using NHS as a positive control. The % lysis was calculated using the following formula:
The % lysis versus the serum dilution was plotted, and the dilution required for 50% hemolysis was calculated. The CH50 (U/mL) (hemolytic complement 50) provides the number of hemolytic units present in mL of NHS.
Direct erythrocyte cell lysis was performed by incubating EA 1% with NPs (8 μL) diluted in CFD for 2 hours at 37°C under shaking (750 rpm). The % lysis was calculated as described in the section “CH50 screening assay”.
NPs (4 μL/well) and normal human plasma (NHP; 80 μL/well) were seeded in a 96-well plate. To assess the clotting capacity of the nano-devices, CaCl2 20 mM was added to initiate the clotting reaction. The eventual coagulation causes an increase in the turbidity of the well, which has been read at 405 nm every 2 minutes for 70 minutes with ELISA Reader TECAN Infinite M200 (Tecan Italia S.r.l.).
Serum samples were obtained from five APS patients (Table 1) with medium–high titer antibodies to domain I of β2GPI after obtaining informed consent and were previously shown to induce clot formation in rats (18). Normal human serum was the pool of at least 30 different healthy donors. NHP was the pool of at least 30 different healthy donors using EDTA as an anti-coagulant. The ethical committee of Istituto Auxologico Italiano approved the study; the patients/participants provided written informed consent to participate in this study. Total IgG were purified from human sera using protein A affinity chromatography and then labeled with Fitc, as already described by our group (19).

Table 1. Characterization of APS sera.
MBB2ΔCH2 was produced as previously described (14). Briefly, stable transfected CHO cells were cultured in a serum-free medium, and the recombinant antibody was purified from cell supernatant by protein A affinity chromatography.
In vivo experimental models were established in groups of three male Wistar rats (290–320 g) kept under standard conditions in the Animal House of the University of Trieste, Italy. The in vivo procedures were performed in compliance with the guidelines of European (86/609/EEC) and Italian (Legislative Decree 116/92) laws and were approved by the Italian Ministry of University and Research (Prot. No. 910/2018PR). The study was conducted in accordance with the Declaration of Helsinki.
Rats were intraperitoneally injected with lipopolysaccharide (LPS) (2.5 mg/kg) and then anesthetized with Zoletil (Virbac, Carros, France; 25 mg/kg) and xylazine (Rompun; 7.5 mg/kg). Afterward, they received a slow infusion of Rhodamine 6G (Sigma) into the jugular vein to stain leukocytes and platelets. Targeted or untargeted BSA-Fitc (Sigma)-filled NPs (400 μL of a suspension of approximately 1.9 × 1012 NPs/mL) were then infused into the carotid artery. The deposition of NPs on mesenteric endothelium was monitored by intravital microscopy for 90 minutes. The amount of NPs bound to the endothelial surface was evaluated by measuring the fluorescence of BSA-Fitc encapsulated in NPs and tNPs. First, four different regions of interest (ROIs) were drawn in the background of the picture, and the mean fluorescence intensity of the background was calculated. This value was subtracted from the starting picture, and then this was converted into a binary picture in order to create a different ROI set on the fluorescent signal. Finally, the Fitc and the background signals were calculated and employed to measure the corrected total cell fluorescence (CTCF):
Rats were intraperitoneally injected with LPS (2.5 mg/kg) and then anesthetized with Zoletil (Virbac, 25 mg/kg) and xylazine (Rompun, 7.5 mg/kg). Afterward, they received a slow infusion of Rhodamine 6G (Sigma) into the jugular vein to stain leukocytes and platelets. Targeted or untargeted NPs (400 μL of a suspension of 1.9 × 1012 NPs/mL) or saline (400 μL) were then infused into the carotid artery. Ten minutes later, they were then infused into the carotid artery with 6.5 mg of Fitc (Sigma)-labeled IgG purified from five APS patients with medium–high titer antibodies to the Domain I (DI) domain of β2GPI. The deposition of IgG on mesenteric endothelium was monitored by intravital microscopy for 90 minutes. The rate of IgG bound to endothelium was evaluated by comparing the fluorescence intensity on the endothelial surface to the fluorescence intensity of the vascular lumen. First, four ROIs were drawn in the background of the picture, and the mean fluorescence intensity of the background was calculated. This value was subtracted from the starting picture, and then this was converted into a binary picture in order to create a different ROI set on the fluorescent signal. After that, four ROIs were drawn on the blood vessel lumen, and as performed before, this signal was subtracted from the original picture in order to isolate only the most fluorescent spots representing the deposited IgG. Finally, the Fitc and the background signals were calculated and employed to measure the spots-CTCF.
The APS model (7) was established in rats that were intraperitoneally injected with LPS (2.5 mg/kg) and then anesthetized with Zoletil (Virbac, 25 mg/kg) and xylazine (Rompun, 7.5 mg/kg). Afterward, they received a slow infusion of Rhodamine 6G (Sigma) into the jugular vein to stain leukocytes and platelets; tNPs, NPs (400 μL of a solution of 1.9 × 1012 NPs/mL), and saline were then infused into the carotid artery. Ten minutes later, 1 mL of pooled sera from five APS patients containing antibodies to β2GPI (18) was infused into the carotid artery to cause thrombus formation. The evolution of hemodynamics in the mesenteric vessels was monitored by intravital microscopy for 90 minutes. The number of thrombi observed in the vascular tree and the percentage of occluded vessels were evaluated by measuring the blood flow. A fourth group of rats received 400 μL of a solution of 1.9 × 1012 tNPs/mL intravenously 1 day before the above-described procedure, and the same parameters were evaluated.
Statistical significance was determined using the Prism (GraphPad) software. Statistical analysis of means was performed using the two-way ANOVA for the in vivo experiments regarding the ability of tNPs to bind activated endothelyum and to block thrombus formation or one-way ANOVA for the in vitro tests of NPs and TNPs safety and for the in vivo experiment about the ability of tNPs to block APS' IgG endothelial deposition. Variance was similar between groups, and p-values <0.05 were considered statistically significant.
PLGA–PVA uncoated and targeted nanoparticles (indicated as NPs and tNPs, respectively) were produced using a standardized approach that guarantees a final concentration of approximately 1.9 × 1012 NPs/mL, as measured by NTA. Both preparations consisted of an aqueous inner core and a polymeric outer shell, covalently decorated with MBB2ΔCH2, targeting β2GPI, in tNP (20). The core was loaded with Fitc-BSA, to allow visualization of NPs bound to blood vessels by fluorescence microscopy, or BSA, to have comparable nanostructures in experiments aimed at evaluating their therapeutic effect. The physicochemical characterization of NPs is reported in Figure 1A. All preparations exhibited a similar size with an average diameter lower than 300 nm (279 ± 19.6 nm for NP and 283 ± 6.63 nm for tNP), a negative surface charge (−13 mV for NP and −10 mV for tNP), and a good index of heterogeneity as confirmed by the low PDI (0.15 for NP and 0.17 for tNP). The regular round shape of the NPs was verified by SEM (Figure 1B), showing that MBB2ΔCH2 coupling does not significantly alter NP diameter and morphology. All formulations exhibited very good stability when stored at 4°C for 6 months; no variation in size and zeta potential and no NP aggregation were detected when analyzing preparations by dynamic light scattering (DLS) (data not shown).
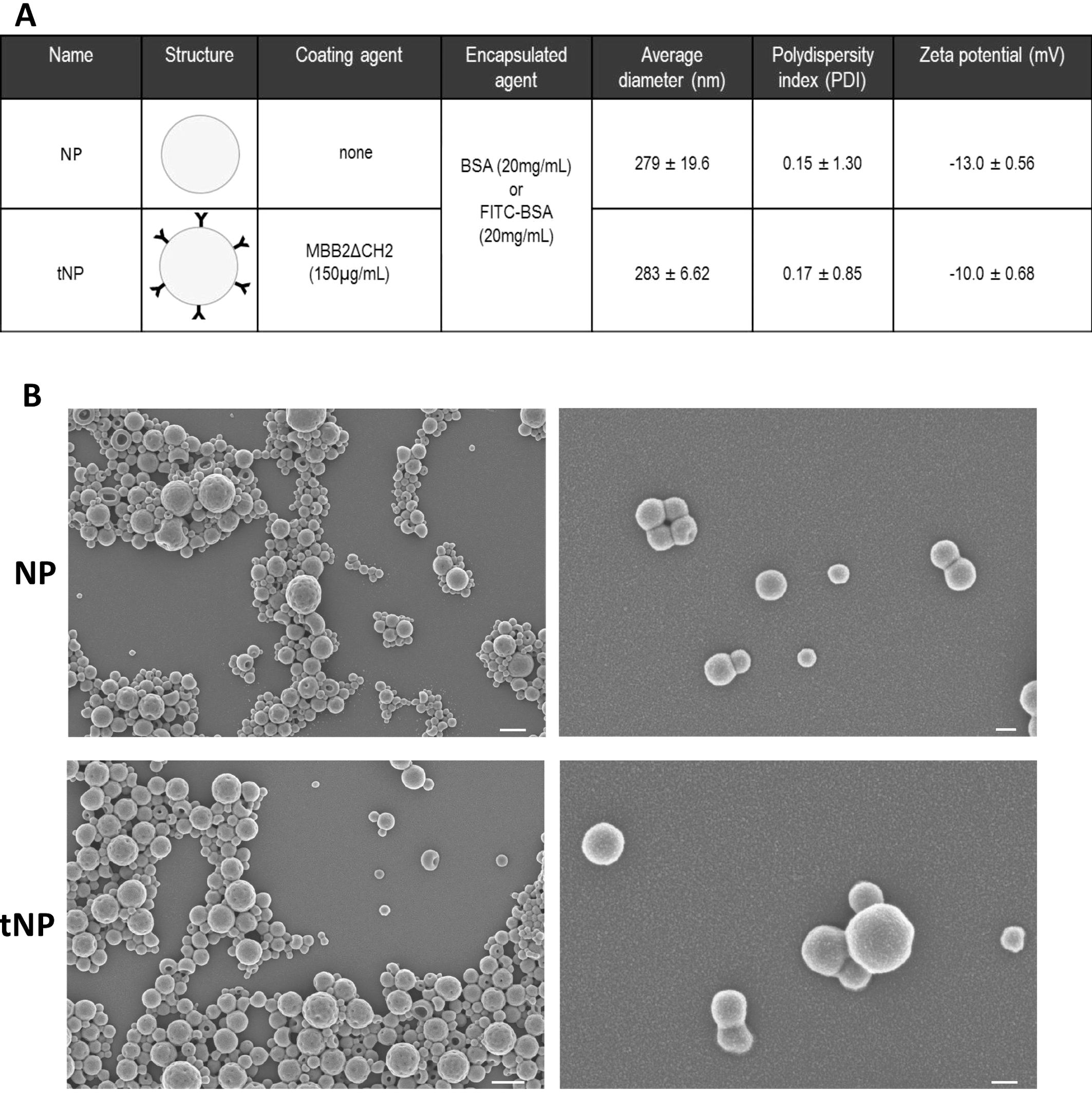
Figure 1. Physicochemical characteristics of untargeted and targeted nanoparticles. (A) Untargeted (NP) and targeted nanoparticles (tNP) analyzed by dynamic light scattering (DLS) share similar values of average diameter, polydispersity index (PDI), and particle charge (zeta potential). (B) SEM images (scale bars: 500 nm on the left and 100 nm on the right images) show similar morphology of NP and tNP. MBB2ΔCH2 denotes the recombinant CH2-deleted scFv-Fc miniantibody against the DI domain of β2-GPI on tNP.
PLGA–PVA NPs were tested in vitro to evaluate their potential safety in circulating blood (Figure 2). The possibility that NPs may cause mechanical disruption of red blood cells was investigated by incubating an erythrocyte suspension with NP or tNP for 2 hours at 37°C. No lysis was observed in the supernatant collected at the end of the experiment, thus excluding a direct damaging effect of NPs on red cells. The possible effect of NPs and tNPs on complement activation was then evaluated. Briefly, pooled normal human serum was incubated with each NP at a concentration similar to that used in in vivo experiments, and the residual complement activity was analyzed by a hemolytic assay (CH50). The results showed a modest, non-significant reduction of the complement activity, and no difference was found in the effect of the two NPs. A turbidity assay was finally performed to ascertain whether NPs interfere with blood clot formation. To this end, NPs were incubated with pooled NHP, and the sample turbidity was analyzed over time following the addition of a solution containing Ca2+ to trigger the coagulation process. The data revealed a similar trend either in the presence or in the absence of NPs, and no significant difference was found between NPs and control groups in the half coagulation time extrapolated from the curves.
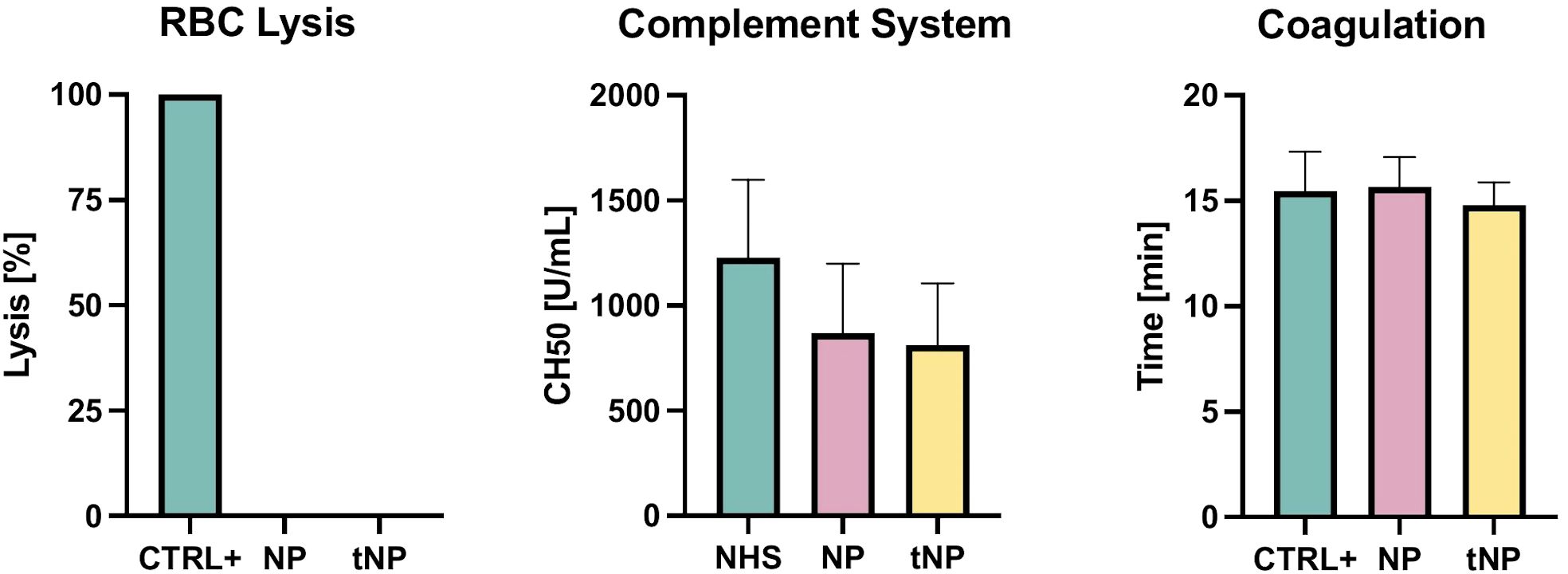
Figure 2. Evaluation of NP safety. RBC Lysis NPs and tNPs did not cause mechanical disruption of red blood cells after 2 hours at 37°C incubation. Complement System NPs were incubated with pooled NHS for 2 hours at 37°C; residual activity of the classical pathway was measured through a hemolytic assay (CH50), showing no significant direct activation of the complement system. Coagulation A turbidity assay, consisting of giving Ca2+ to pooled NHP and analyzing turbidity over time, was performed to clarify whether NPs interfere with clotting formation; no significant difference in the half-coagulation time of both NPs and control was documented. The results are presented as mean ± SD. Ns = p-value ≥0.05; one-way ANOVA test.
Preliminary experiments were conducted to verify the in vivo ability of anti-β2GPI antibodies (recombinant or present in APS patients) to bind to activated endothelium. Serum samples were obtained from five APS patients selected for the presence of medium–high titer IgG to cardiolipin, β2GPI, and, in particular, domain I of β2GPI (Table 1); the sera were pooled before use.
The rats received an intraperitoneal injection of LPS in order to induce binding of β2GPI to endothelial cells, as previously described (21). Fitc-labeled MBB2ΔCH2 or Fitc-labeled APS IgG were administered into the carotid artery, and their localization on mesenteric vessels was monitored over time. Intravital fluorescence microscopy analysis showed a patchy distribution of the fluorescent signal on the mesenteric endothelium of rats receiving either fluorescent MBB2ΔCH2 or aPL IgG (Figure 3). As expected, aPL IgG caused vascular occlusions (Figure 3), while MBB2ΔCH2, which does not activate the complement system, was able to target β2GPI on endothelial cells but failed to trigger the coagulation process and the vessel occlusion.
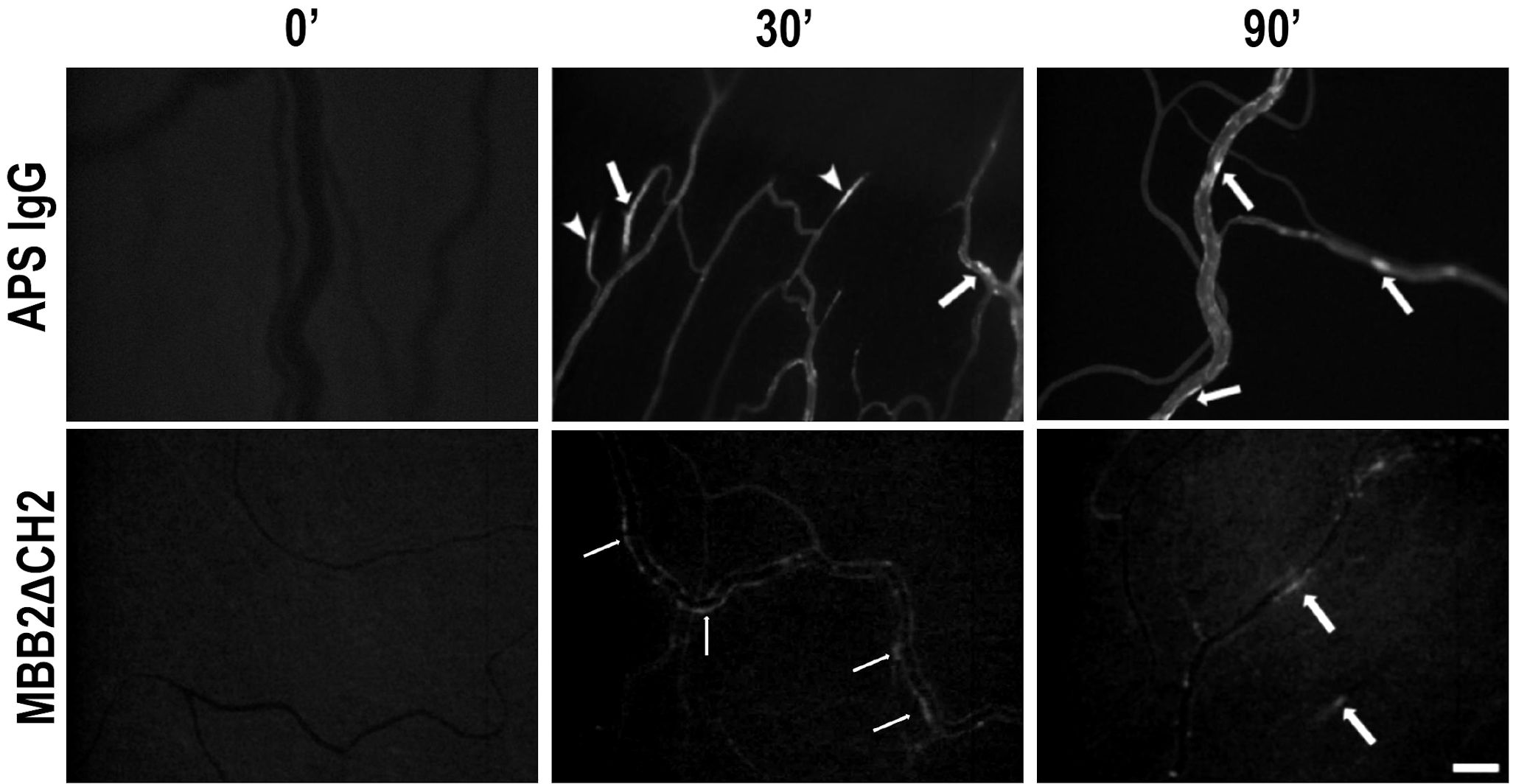
Figure 3. Deposition of anti-phospholipid syndrome (APS) IgG or MBB2ΔCH2 on mesenteric vessels of rats treated with lipopolysaccharide (LPS). The animals were treated with LPS followed by the injection of Fitc-labeled IgG from APS patients or Fitc-labeled MBB2ΔCH2. Antibody deposition in mesenteric vessels was constantly monitored by intravital microscopy for 90 minutes (representative images were obtained at 0, 30, and 90 minutes after antibody infusion). Arrows indicate antibodies bound to the endothelium, while arrowheads indicate formed thrombi. Original magnification ×100. Scale bar 50 μm.
A similar distribution pattern of BSA-Fitc loaded tNPs on the mesenteric endothelial cell was observed in rats treated with LPS, which stimulated cell deposition of β2GPI, recognized by the recombinant anti-β2GPI antibody on NP surface. On the contrary, no relevant fluorescent signal was seen on the mesenteric vessels of rats receiving fluorescent untargeted NPs (Figure 4A). The deposition of both types of nanoparticles on vascular endothelium was analyzed by measuring the fluorescence signal of BSA-Fitc encapsulated in the NPs. The results presented in Figure 4B show a progressive increase in the deposition of tNPs on the endothelium of blood vessels that started within a few minutes from NP injection, reaching the highest level in 30 minutes and slightly decreased up to 90 minutes. A marginal localization of untargeted NPs was observed on vascular endothelium, and the extent of their deposition was significantly lower than that of tNPs.
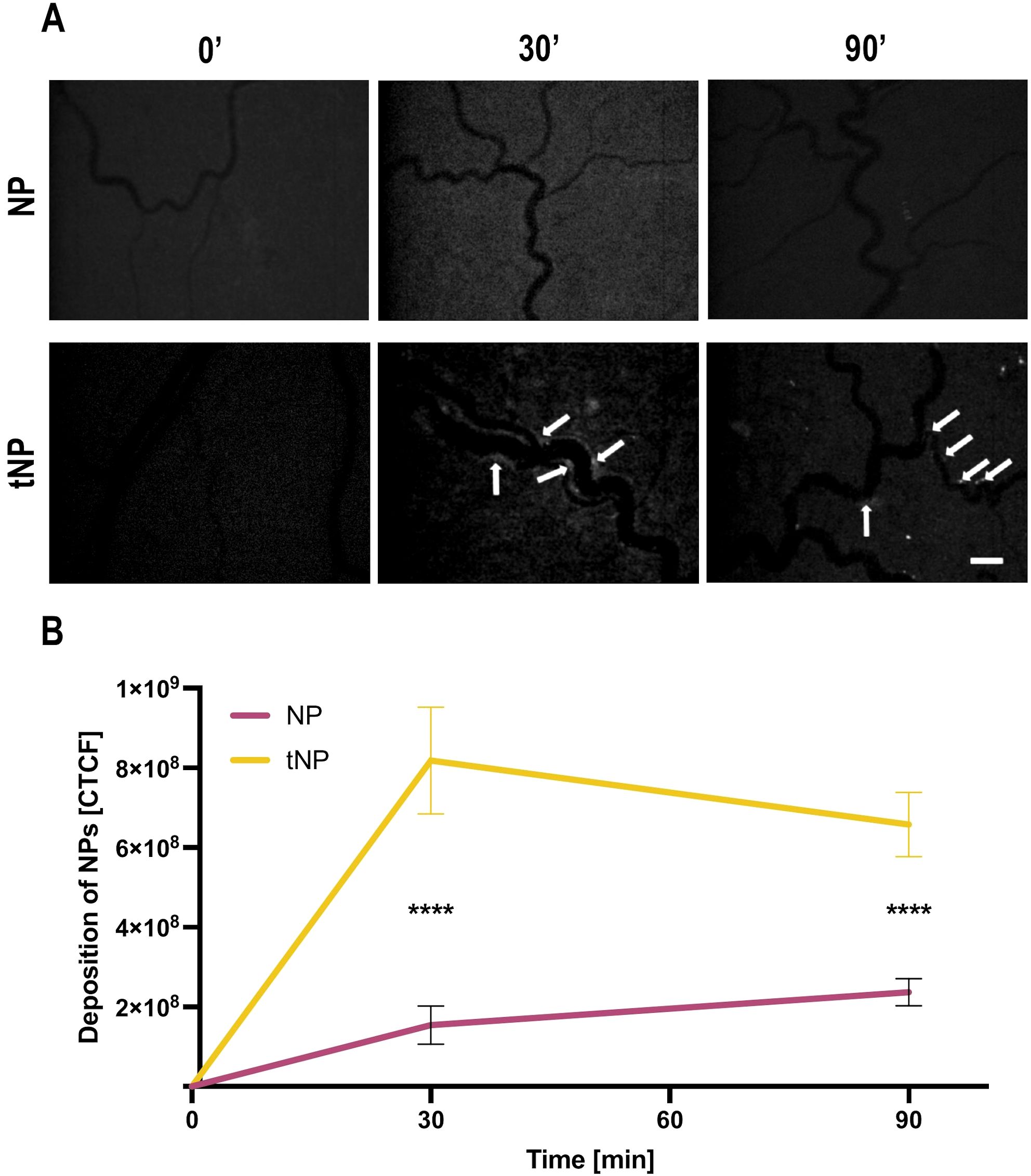
Figure 4. Anti-b2 GPI-targeted NPs are able to bind activated rat mesenteric vascular endothelium. The animals were treated with lipopolysaccharide (LPS) followed by the injection of fluorescent untargeted (NPs) or fluorescent targeted nanoparticles (tNPs). NP and tNP deposition in mesenteric vessels were constantly monitored by intravital microscopy for 90 minutes. (A) Representative images obtained at 0, 30, and 90 minutes after nanoparticle infusion. tNPs bound to the endothelium are indicated with arrows. Note the absence of untargeted NBs in mesenteric vasculature. Original magnification ×100. Scale bar 50 μm. (B) Quantification of NPs bound to endothelial surface evaluated by measuring the fluorescence of BSA-Fitc encapsulated in NPs and tNPs. A significant deposition of tNPs but not of NPs was measured. The results are presented as mean ± SD. ****p-value ≤0.0001; two-way ANOVA test.
It is important to underline that neither NPs nor tNPs were able to activate the coagulation cascade in blood, and intravascular thrombi were not documented during the analysis.
To examine the effect of tNPs in preventing the binding of aPL to blood vessels, LPS-treated rats received targeted or untargeted NPs, or saline as a control, followed by a pool of Fitc-labeled IgG purified from five APS patients, containing antibodies to β2GPI. The deposition of IgG on mesenteric blood vessels was monitored over time by intravital microscopy (Figure 5A). The analysis showed again an irregular distribution of Fitc-labeled IgG on endothelial cells, and the sites along the vessels, where the labeled IgG were deposited, had decreased in number in rats treated with tNPs compared to those seen in rats receiving saline or untargeted NPs (Figure 5A). Aware of the fact that the number of deposits alone was not exhaustive in describing the phenomenon observed, a quantitative analysis of fluorescence intensity was performed as indicated above. As shown in Figure 5B, the deposits of aPL IgG on endothelial cells were markedly reduced and nearly undetectable 90 minutes after the administration of tNP compared to those seen in animals receiving saline. Interestingly, some inhibition of IgG binding to endothelial cells was observed in rats treated with untargeted NPs, most likely due to non-specific absorption of circulating IgG by the NPs rather than the result of competitive inhibition of IgG binding to endothelial cells.
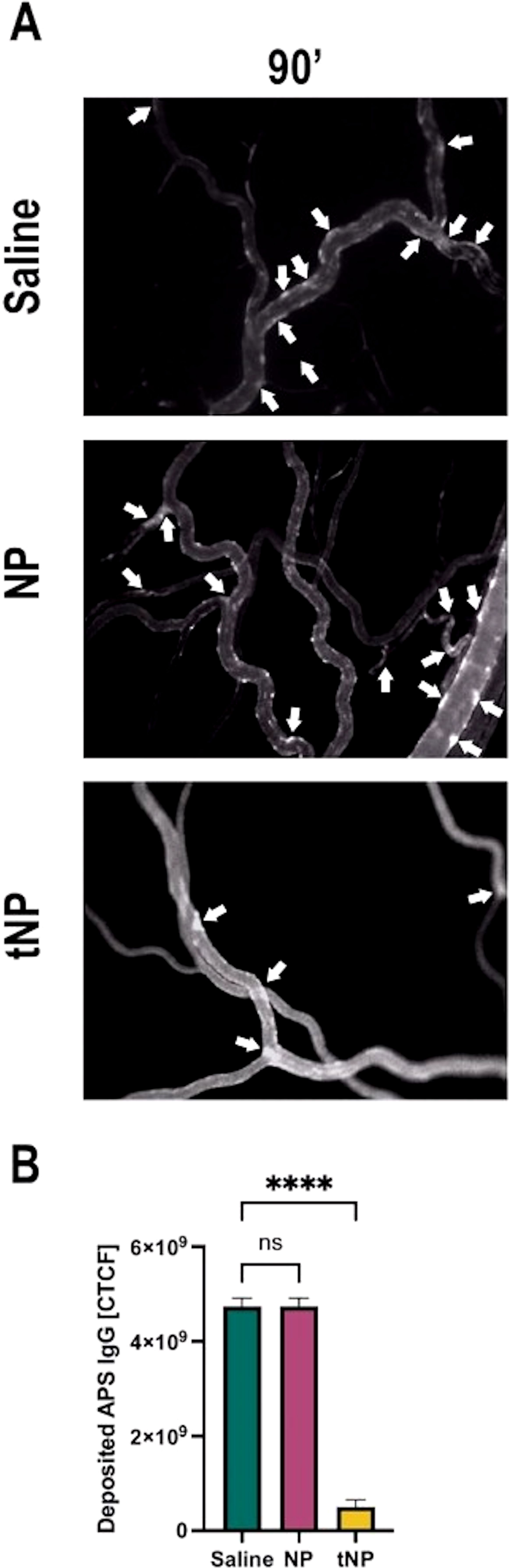
Figure 5. Anti-β2-GPI-targeted NPs prevent anti-phospholipid syndrome (APS) IgG deposits in APS rat model. The animals, after treatment with lipopolysaccharide, received the injection of saline solution, untargeted (NP) or targeted nanoparticles (tNP), followed by the injection of Fitc-labeled IgG purified from APS patients. IgG deposits (indicated with arrows) were constantly observed by intravital microscopy. (A) Representative images were collected 90 minutes after the injection of labeled IgG. (B) The amount of IgG bound to the endothelial cell surface was evaluated by measuring the fluorescence of Fitc-labeled IgG. The results in panel B are presented as mean ± SD. ****p-value ≤0.0001; one-way ANOVA test. NS, Not significant.
The ability of NPs to prevent the thrombotic activity of deposited IgG anti-β2GPI was then examined. To this end, rats primed with LPS, to promote deposition of β2GPI on endothelial cells, received either targeted or untargeted NPs, or saline as a control prior to administration of pooled sera from five APS patients containing antibodies to β2GPI to trigger coagulation. Analysis of the mesenteric vasculature, monitored over time by intravital microscopy, revealed a marked decrease in the number of thrombi in tNP-treated rats compared to animals receiving saline (Figure 6A). A reduced number of blood clots were also seen following administration of untargeted NPs, consistent with the results of passive absorption of circulating aPL IgG shown in Figure 5B, although the decrease was less marked than that observed in tNP-treated rats (Figure 6A). Interestingly, the analysis of the blood clot dimension, which may be responsible for vascular occlusion, revealed that only tNPs have a significant effect in reducing the number of occluded vessels while untargeted NPs were almost ineffective (Figure 6B).
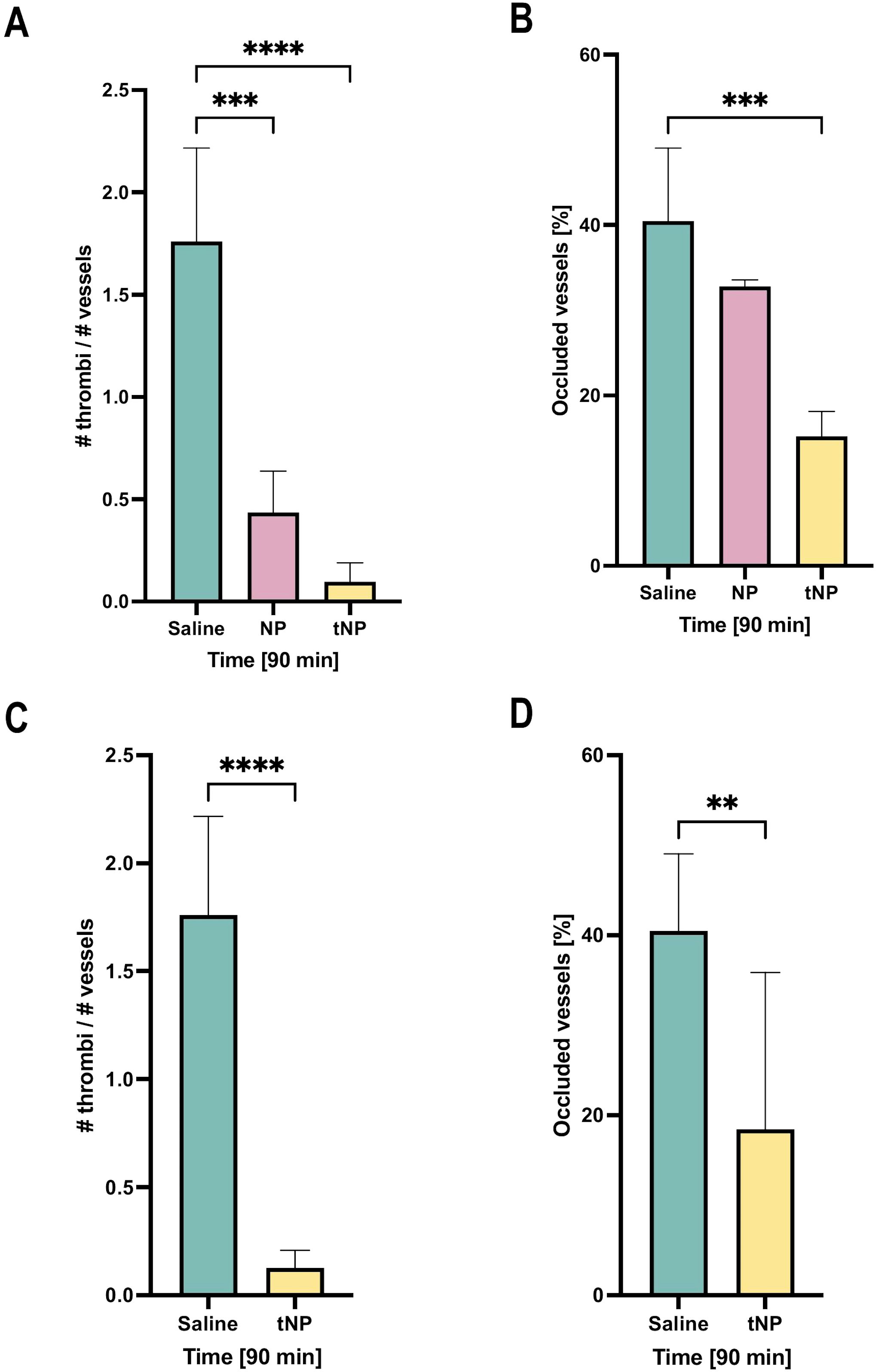
Figure 6. Effect of targeted NBs on thrombus formation in the rat model of anti-phospholipid syndrome (APS). A first group of rats, after treatment with lipopolysaccharide, received the injection of saline solution, and untargeted (NP) and targeted nanoparticles (tNP) followed by the injection of APS sera; thrombus formation and vascular occlusions were observed by intravital microscopy for 90 minutes, showing that NPs markedly reduced the formation (A) and the dimension (B) of intravascular thrombi compared to saline treatment. A second group of rats received the injection of saline solution and tNPs 24 hours before lipopolysaccharide (LPS) and APS serum treatments; animals were observed by intravital microscopy showing that tNPs still reduced the formation (C) and the dimension (D) of intravascular thrombi compared to saline treatment, more significantly after 90 minutes after sera injection. The results are presented as mean ± SD. ****p-value ≤0.0001; ***p-value ≤0.001; **p-value ≤0.01; two-way ANOVA test.
To investigate whether the tNPs, administered several hours prior to patients’ aPL IgG, circulate in sufficient numbers to prevent coagulation, the rats were challenged with tNPs or saline 24 hours before LPS priming followed by administration of aPL IgG. The results presented in Figures 6C, D show that tNPs were still able to significantly reduce both the number of blood clots and the percentage of occluded vessels.
Polymeric nanoparticles coated with a monoclonal antibody to β2GPI selectively targeting endothelial cell-bound β2GPI were effective in preventing thrombus formation and vessel occlusion in a rat model of APS. These results extend our previous observation of rapid elimination of vessel-occluding clots obtained with nanobubbles coated with recombinant tissue plasminogen activator and a monoclonal antibody to β2GPI but delivered only after thrombus formation (20).
The nanoplatform proposed in this study is based on PLGA/PVA, biodegradable and biocompatible synthetic polymers, already approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) as delivery systems for therapeutic agents (22) and has been subjected to clinical trial (23); today, drugs that use this platform as nano/microparticle structure are commercially available for cancer therapy, to reduce inflammation, to treat pain or neurological diseases, or for vaccination (24). Nanoparticles showed a regular round shape with a diameter of approximately 280 nm surrounded by a polymeric outer shell, as previously documented (17, 25, 26). The aqueous inner core can be loaded with fluorescent dyes (16), for their visualization, or drugs (17, 27–29), for specific delivery to selected targets. The safety of NPs was confirmed by their failure to lyse red blood cells or to interfere with coagulation or complement systems and, in vivo, following their injection in rat circulation. In this study, we focused on the results obtained by the injection of 0.4 mL of NP suspension containing approximately 0.76 × 1012 NPs. The dose was derived from a previous study in mice (30, 31) and was adjusted taking into consideration the different body weights of the animals.
The in vivo biodistribution of Fitc-labeled aPL IgG in rats was observed by intravital microscopy that revealed their deposition on the mesenteric endothelium with a patchy distribution. This fluorescent pattern is consistent with our previous findings (18, 19) and is most likely the result of the uneven deposition of circulating β2GPI on endothelial cells. Studies have shown that the binding of β2GPI to endothelial cells occurs independently of the presence of antibodies and requires priming with LPS, followed by the interaction of cell-bound β2GPI with specific auto-antibodies preformed in the serum, providing another hit for activation of the complement activation and the coagulation cascade (7, 21). A similar priming effect can be induced by pregnancy, characterized by β2GPI deposition on trophoblast and decidual endothelial cells (21, 32). Antibody clustering in areas of β2GPI localization favors complement activation that plays a critical role in triggering activation of the coagulation cascade, as suggested by the failure of aPL IgG to induce thrombus formation in C6-deficient rats and animals treated with a neutralizing antibody to C5 (7).
The distribution pattern of the monoclonal antibody to β2GPI (MBB2ΔCH2) on endothelial cells was similar to that of aPL IgG, further supporting the conclusion that β2GPI deposited on endothelium is a potential target of circulating aPL antibodies in APS patients. MBB2ΔCH2 was selected to coat NP as a targeting agent for cell-bound β2GPI for several reasons. First, this monoclonal antibody recognizes a neoepitope on domain I of bound β2GPI, regarded as the main target of pathogenic aPL antibodies responsible for thrombosis and fetal loss in animal models of APS (7, 14, 18, 33). MBB2ΔCH2 binds to β2GPI equally well as MBB2 but, unlike the parent molecule, is unable to activate the complement system due to the deletion of the C1q fixing CH2 domain of the antibody and therefore fails to trigger the coagulation cascade (14). Finally, the recombinant antibody has a higher affinity than patients’ antibodies for the target antigen and can compete with them for their interaction with cell-bound β2GPI (14).
NPs coated with MBB2ΔCH2, targeting β2GPI, bound to endothelial cells within 30 minutes from injection and were effective in preventing in vivo binding of IgG purified from APS patients to endothelial cells and their prothrombotic activity. These results were obtained using an amount of NP-bound MBB2ΔCH2 20 times lower than that of soluble antibody required to obtain a similar effect. Moreover, the tNPs were persistently present on the endothelium through the experimental procedure and contributed to protection from thrombosis induced by patients’ IgG by competing with them, as a result of the higher affinity of MBB2ΔCH2 and also by steric hindrance, as they cover a larger surface area than the soluble antibodies.
The medical need behind the project is to develop a novel targeted therapy to prevent thrombus formation in APS. The use of tNPs for long-term treatment of APS patients, in the absence of events triggering coagulation, is probably not preferable to the standard anti-coagulant therapy, although the latter treatment is not free from recurrent thrombosis reported in 30%–40% of high-risk patients with triple APL positivity (10). The tNPs may be better indicated as first-line therapy of APS patients in acute situations, such as inflammatory processes, acute infections, and vascular surgery, that promote coagulation by activating the endothelial cells (34). We have previously shown that re-thrombosis was successfully prevented by treating an APS patient undergoing femoral popliteal bypass for thrombotic vascular occlusion with eculizumab to inhibit complement activation (6). MBB2ΔCH2-coated NPs have the advantage of acting upstream of complement inactivation, competing with aPL for binding to endothelial cells and remaining cell-bound throughout the experimental procedure. This approach could also be effective in other surgical situations involving APS patients, like the preparation of arteriovenous fistulas in dialysis patients, cardiac valvular diseases, or diagnostic biopsies.
A preventive approach is typically studied by injecting the drug just before the stimulus, which is not always feasible in a clinical setting, considering also the drawbacks of long-term anti-coagulation (35).
For these reasons, we also tested the preventive effect of injecting tNPs 24 hours before the challenge with antibodies from APS patients; this period represents also the initial elimination of polymeric NPs, mainly dependent on liver macrophages (36), that can induce a fast clearance from the circulation; a single injection of tNPs was still able to prevent thrombus formation and vascular occlusion for all durations of the study.
In conclusion, anti-β2GPI-targeted polymeric nanoparticles represent a stable and safe approach to preventing thrombus formation and vessel occlusion in a rat model of APS caused by patients’ antibodies.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by the ethical committee of Istituto Auxologico Italiano. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
PD: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing. SB: Writing – original draft, Writing – review & editing. ER: Writing – original draft, Writing – review & editing. PLM: Writing – original draft, Writing – review & editing. FT: Writing – original draft, Writing – review & editing. PM: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funds from the University of Trieste (to PM). The paper was also supported by the Italian Ministry of Health, IRCCS Istituto Auxologico Italiano, Ricerca Corrente (to PLM).
We thank the staff of the Animal Facility of the University of Trieste, directed by Dr. Paola Zarattini, for their valuable work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SB declared a parent affiliation with the authors PD, ER, and PLM to the handling editor at the time of review.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Knight JS, Erkan D. Rethinking antiphospholipid syndrome to guide future management and research. Nat Rev Rheumatol. (2024) 20:377–88. doi: 10.1038/s41584-024-01110-y
2. Gaspar P, Sciascia S, Tektonidou MG. Epidemiology of antiphospholipid syndrome: macro- and microvascular manifestations. Rheumatol (Oxford). (2024) 63:SI24–36. doi: 10.1093/rheumatology/kead571
3. Cervera R, Rodriguez-Pinto I, Espinosa G. The diagnosis and clinical management of the catastrophic antiphospholipid syndrome: A comprehensive review. J Autoimmun. (2018) 92:1–11. doi: 10.1016/j.jaut.2018.05.007
4. Jacobs L, Wauters N, Lablad Y, Morelle J, Taghavi M. Diagnosis and management of catastrophic antiphospholipid syndrome and the potential impact of the 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Antibodies (Basel). (2024) 13:365–74. doi: 10.3390/antib13010021
5. Meroni PL, Borghi MO, Grossi C, Chighizola CB, Durigutto P, Tedesco F. Obstetric and vascular antiphospholipid syndrome: same antibodies but different diseases? Nat Rev Rheumatol. (2018) 14:433–40. doi: 10.1038/s41584-018-0032-6
6. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
7. Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. (2005) 106:2340–6. doi: 10.1182/blood-2005-03-1319
8. Chighizola CB, Meroni PL. Thrombosis and anti-phospholipid syndrome: a 5-year update on treatment. Curr Rheumatol Rep. (2018) 20:44. doi: 10.1007/s11926-018-0741-5
9. Kostopoulou M, Mukhtyar CB, Bertsias G, Boumpas DT, Fanouriakis A. Management of systemic lupus erythematosus: a systematic literature review informing the 2023 update of the EULAR recommendations. Ann Rheum Dis. (2024) 1–13. doi: 10.1136/ard-2023-225319
10. Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. (2015) 74:1011–8. doi: 10.1136/annrheumdis-2013-204838
11. Chaturvedi S, Braunstein EM, Brodsky RA. Antiphospholipid syndrome: Complement activation, complement gene mutations, and therapeutic implications. J Thromb Haemost. (2021) 19:607–16. doi: 10.1111/jth.15082
12. Tedesco F, Borghi MO, Gerosa M, Chighizola CB, Macor P, Lonati PA, et al. Pathogenic role of complement in antiphospholipid syndrome and therapeutic implications. Front Immunol. (2018) 9:1388. doi: 10.3389/fimmu.2018.01388
13. Venturelli V, Maranini B, Tohidi-Esfahani I, Isenberg DA, Cohen H, Efthymiou M. Can complement activation be the missing link in antiphospholipid syndrome? Rheumatol (Oxford). (2024) 63:3243–54. doi: 10.1093/rheumatology/keae178
14. Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to beta2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. (2014) 123:3478–87. doi: 10.1182/blood-2013-11-537704
15. Vasir JK, Labhasetwar V. Preparation of biodegradable nanoparticles and their use in transfection. CSH Protoc. (2008) 2008:pdb.prot4888. doi: 10.1101/pdb.prot4888
16. Bozzer S, Dal Bo M, Grimaldi MC, Toffoli G, Macor P. Nanocarriers as a delivery platform for anticancer treatment: biological limits and perspectives in B-cell Malignancies. Pharmaceutics. (2022) 14:1–24. doi: 10.3390/pharmaceutics14091965
17. Capolla S, Colombo F, De Maso L, Mauro P, Bertoncin P, Kähne T, et al. Surface antibody changes protein corona both in human and mouse serum but not final opsonization and elimination of targeted polymeric nanoparticles. J Nanobiotechnology. (2023) 21:376. doi: 10.1186/s12951-023-02134-4
18. Durigutto P, Grossi C, Borghi MO, Macor P, Pregnolato F, Raschi E, et al. New insight into antiphospholipid syndrome: antibodies to beta2glycoprotein I-domain 5 fail to induce thrombi in rats. Haematologica. (2019) 104:819–26. doi: 10.3324/haematol.2018.198119
19. Fischetti F, Durigutto P, Macor P, Marzari R, Carretta R, Tedesco F. Selective therapeutic control of C5a and the terminal complement complex by anti-C5 single-chain Fv in an experimental model of antigen-induced arthritis in rats. Arthritis Rheumatol. (2007) 56:1187–97. doi: 10.1002/art.22492
20. Macor P, Durigutto P, Argenziano M, Smith-Jackson K, Capolla S, Di Leonardo V, et al. Plasminogen activator-coated nanobubbles targeting cellbound beta2-glycoprotein I as a novel thrombus-specific thrombolytic strategy. Haematologica. (2023) 108:1861–72. doi: 10.3324/haematol.2022.281505
21. Agostinis C, Biffi S, Garrovo C, Durigutto P, Lorenzon A, Bek A, et al. In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood. (2011) 118:4231–8. doi: 10.1182/blood-2011-01-333617
22. Danhier F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. (2016) 244:108–21. doi: 10.1016/j.jconrel.2016.11.015
23. Rezvantalab S, Keshavarz Moraveji M. Microfluidic assisted synthesis of PLGA drug delivery systems. RSC Adv. (2019) 9:2055–72. doi: 10.1039/C8RA08972H
24. Yang J, Zeng H, Luo Y, Chen Y, Wang M, Wu C, et al. Recent applications of PLGA in drug delivery systems. Polymers (Basel). (2024) 16:1–25. doi: 10.3390/polym16182606
25. Bozzer S, De Maso L, Grimaldi MC, Capolla S, Dal Bo M, Toffoli G, et al. Zebrafish: A useful animal model for the characterization of drug-loaded polymeric NPs. Biomedicines. (2022) 10:1–13. doi: 10.3390/biomedicines10092252
26. Bozzer S, Grimaldi MC, De Maso L, Manfredi M, Toffoli G, Dal Bo M, et al. Stealth-engineered albumin-coated nanoparticles for targeted therapy: effective drug delivery and tumor suppression in xenograft-zebrafish model. Int J Nanomed. (2024) 19:13267–86. doi: 10.2147/IJN.S476241
27. Jiang Z, Xu Y, Du G, Sun X. Emerging advances in delivery systems for mRNA cancer vaccines. J Control Release. (2024) 370:287–301. doi: 10.1016/j.jconrel.2024.04.039
28. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. (2017) 17:20–37. doi: 10.1038/nrc.2016.108
29. Yao Y, Zhou Y, Liu L, Xu Y, Chen Q, Wang Y, et al. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front Mol Biosci. (2020) 7:193. doi: 10.3389/fmolb.2020.00193
30. Colombo F, Durigutto P, De Maso L, Biffi S, Belmonte B, Tripodo C, et al. Targeting CD34(+) cells of the inflamed synovial endothelium by guided nanoparticles for the treatment of rheumatoid arthritis. J Autoimmun. (2019) 103:102288. doi: 10.1016/j.jaut.2019.05.016
31. Mezzaroba N, Zorzet S, Secco E, Biffi S, Tripodo C, Calvaruso M, et al. New potential therapeutic approach for the treatment of B-Cell Malignancies using chlorambucil/hydroxychloroquine-loaded anti-CD20 nanoparticles. PloS One. (2013) 8:e74216. doi: 10.1371/journal.pone.0074216
32. La Rosa L, Meroni PL, Tincani A, Balestrieri G, Faden D, Lojacono A, et al. Beta 2 glycoprotein I and placental anticoagulant protein I in placentae from patients with antiphospholipid syndrome. J Rheumatol. (1994) 21:1684–93.
33. Radin M, Cecchi I, Roccatello D, Meroni PL, Sciascia S. Prevalence and thrombotic risk assessment of anti-beta2 glycoprotein I domain I antibodies: A systematic review. Semin Thromb Hemost. (2018) 44:466–74. doi: 10.1055/s-0037-1603936
34. Raschi E, Borghi MO, Tedesco F, Meroni PL. Antiphospholipid syndrome pathogenesis in 2023: an update of new mechanisms or just a reconsideration of the old ones? Rheumatol (Oxford). (2024) 63:SI4–SI13. doi: 10.1093/rheumatology/kead603
35. McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, et al. The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev. (2020) 39:100610. doi: 10.1016/j.blre.2019.100610
Keywords: nanoparticles, thrombosis, anti-phospholipid syndrome, β2GPI, rat model
Citation: Durigutto P, Grimaldi MC, Bozzer S, Raschi E, Meroni P, Tedesco F and Macor P (2025) β2GPI-targeted polymeric nanoparticles form a protective layer to prevent vascular thrombosis in an anti-phospholipid syndrome model. Front. Immunol. 16:1520619. doi: 10.3389/fimmu.2025.1520619
Received: 31 October 2024; Accepted: 06 January 2025;
Published: 27 February 2025.
Edited by:
Aurore Saudemont, Xap Therapeutics, United KingdomReviewed by:
Stefania Biffi, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyCopyright © 2025 Durigutto, Grimaldi, Bozzer, Raschi, Meroni, Tedesco and Macor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Macor, cG1hY29yQHVuaXRzLml0
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.