 Hua Bai
Hua Bai Dongming Zhou
Dongming Zhou Jinwen Liu1
Jinwen Liu1 Zhou Min
Zhou Min Wenyong Fan
Wenyong Fan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 28 January 2025
Sec. Cytokines and Soluble Mediators in Immunity
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1520470
This article is part of the Research TopicBiomarker-Driven Strategies for Personalized Management of Systemic Inflammatory Response SyndromeView all 4 articles
Schnitzler syndrome (SchS) is a rare acquired systemic autoinflammatory disorder, characterized by chronic urticarial rash and immunoglobulin M (IgM) monoclonal gammopathy. Anti-interleukin-1 (IL-1) therapies have been shown to be more effective in managing the clinical symptoms of SchS compared to anti-IL-6 therapies. In this case report, we present a male patient with urticarial rash, fever, and arthralgia. Laboratory tests identified the presence of IgMκ monoclonal protein, and the absence of IL-1β in serum. Whole exome sequencing (WES) did not reveal any pathological variants associated with monogenic autoinflammatory diseases or the MYD88 L265P mutation. He met the diagnostic criteria for SchS and was treated with bortezomib, leading to a significant improvement in clinical symptoms and a decline in IgMκ monoclonal protein levels. The patient tolerated the treatment well. This case suggests that bortezomib may be considered as a potential treatment option for SchS, in addition to anti-IL-1 therapies and bruton tyrosine kinase (BTK) inhibitors.
Schnitzler syndrome (SchS) is an adult-onset, rare, autoinflammatory disorder characterized by urticarial rash and monoclonal gammopathy (usually of the immunoglobulin M, IgM, rarely IgG). This disease usually manifests around the age of 50 and is clinically marked by urticarial rash, fever, arthralgia, lymphadenopathy, and elevated inflammatory markers (1, 2). The central pathogenesis of SchS is dysregulation of interleukin (IL)-1β and the inflammasome pathway. The long-term follow-up of IL-1 blockade in SchS demonstrates that canakinumab treatment effectively reduces clinical signs and symptoms, decreases inflammatory markers, and improves quality of life over a four-year period (3). However, as observed in a comprehensive study of 281 cases of SchS, the efficacy of anti-IL-1 or anti-IL-6 therapies may decrease over time in some patients, underscoring the importance of continuous monitoring and possible modifications to the therapeutic approach (2).
While the rarity and complexity of SchS can make its initial diagnosis challenging and sometimes delayed, 20% of SchS patients often have lymphoproliferative disorders, including multiple myeloma (MM) and lymphoplasmacytic lymphoma (LPL), and its specific variant, Waldenstrom's macroglobulinemia (WM), which is the most frequent form of LPL (4). Moreover, IL-1 or IL-6 targeting therapy cannot prevent the occurrence of lymphoproliferative diseases. Current evidence indicates that while these treatments can control symptoms and reduce inflammation, they may not alter the underlying monoclonal gammopathy or plasma cell dyscrasia, which are associated with the development of such disorders (4, 5). So we need to explore new therapeutic drugs. MYD88 is at the crossroad of toll-like receptor, IL-1R, and B-cell receptor pathways and may participate in uncontrolled inflammation. A key pathogenic factor is the MYD88 L265P gain-of-function mutation, which promotes cell survival by forming a protein complex of IL-1 receptor-associated kinase 1 (IRAK1) and IRAK4, contributing to up-regulation of IL-1 signaling and other pro-inflammatory pathways (6). BTK is involved in toll-like receptor signaling, which also involves the adaptor protein MYD88. The activating MYD88 L265P mutation causes MYD88 to spontaneously assemble protein complexes that trigger pro-survival signaling along multiple pathways (7, 8). Considering the obvious specificity of MYD88 L265P mutation for inflammasome activation, and the occurrence of this mutation in WM, IgM monoclonal gammopathy of undetermined significance (MGUS) and SchS patients. Treatment with bruton tyrosine kinase (BTK) inhibitor ibrutinib has proven effective in two cases involving the MYD88 L265P mutation (9–11). However, efficacy in MYD88 wild-type patients is uncertain (12). Preliminary exploratory therapy may highlight the pathogenic effect of underlying B-cell clone in the development of SchS and suggest a potential universal therapeutic value of clone-targeted therapy.
Moreover, even a minimal plasma-cell clone can lead to severe clinical manifestations due to the toxicity of the monoclonal immunoglobulin or other mechanisms (4). Indication for therapy in monoclonal gammopathy of clinical significance (MGCS) is driven by organ damage due to the small plasma cell clone (13). The anti-plasma cell therapy, such as bortezomib, could target both the inflammasome activation and the underlying plasma cell clone (14). To our knowledge, this case report presents the first documented use of bortezomib in SchS patients.
A 60-year-old Chinese male was admitted to Nanjing Drum Tower Hospital in 2023 due to recurrent fever, urticarial rash, and arthralgia for 3 months. Laboratory evaluation revealed leukocytosis (10.7*109/L) with neutrophilia (81.6%), elevated C-reactive protein (CRP) (43.6 mg/L), and monoclonal IgM (6.61 g/L). Serum protein electrophoresis demonstrated the presence of IgMκ monoclonal gammopathy (Figure 1A). Bone marrow aspiration showed no morphologic evidence of LPL, but flow cytometry identified an abnormal population of plasma cells expressing CD138, CD38, CD81, CD27, CD45, and cytoplasmic κ light chain. No abnormality was detected on lymphocyte immunophenotyping, excluding a diagnosis of LPL/WM (Figure 1B). Cytogenetic analysis revealed a normal male karyotype. Imaging showed osteosclerotic bone lesions (Figure 1C). Skin biopsy showed perivascular infiltration of neutrophils, without vasculitis change, consistent with neutrophilic dermatosis (Figure 1D). The combination of recurrent urticaria rash, IgMκ gammopathy, elevated CRP level, neutrophilic dermatosis without vasculitis, osteosclerotic bone lesions, and fever met two major and four minor criteria of the Strasbourg criteria for the diagnosis of SchS (15).
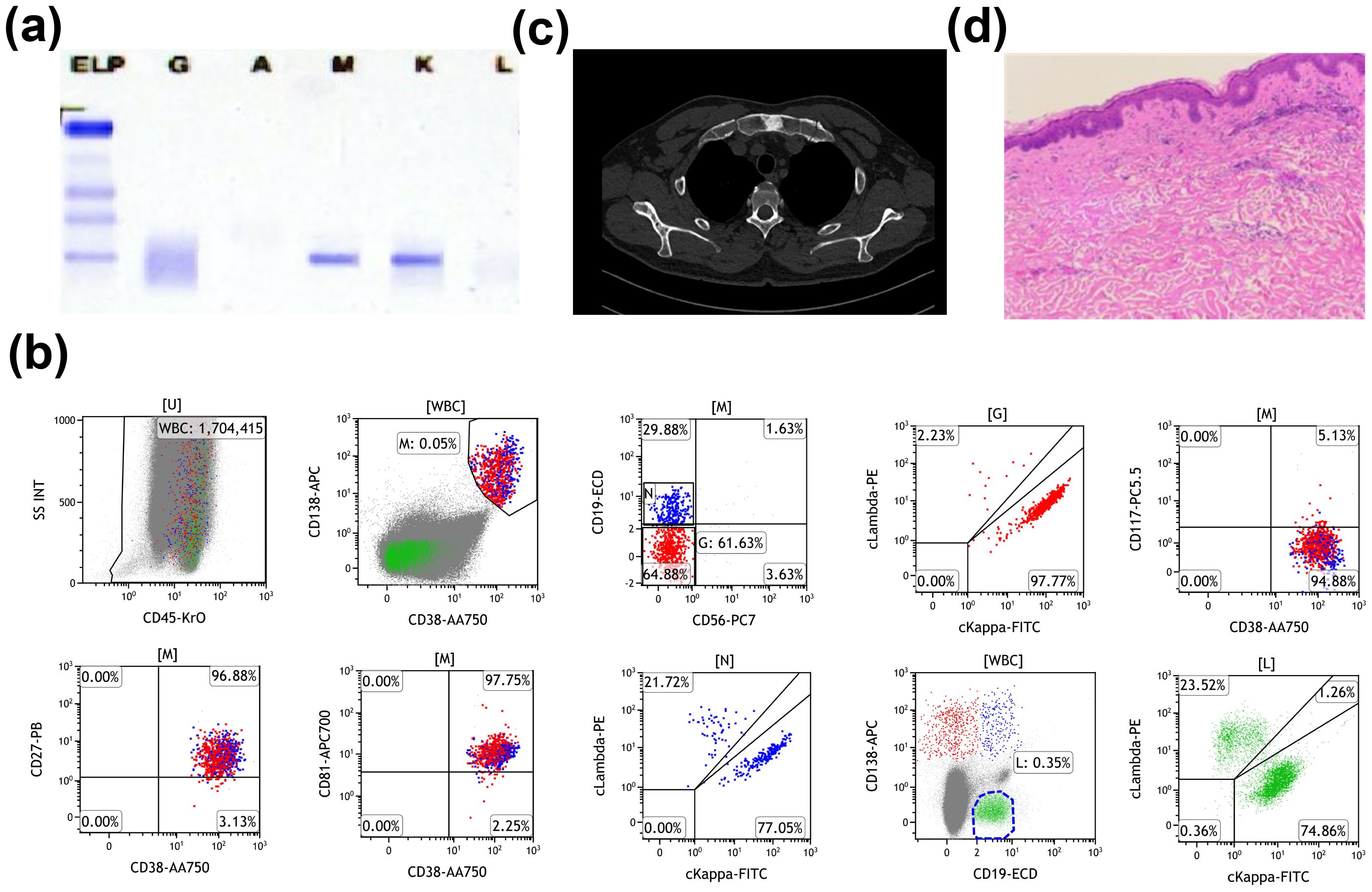
Figure 1. Summary of laboratory findings. (A) Serum protein electrophoresis before treatment. (B) Flow cytometry analysis of bone marrow showing 0.03% monoclonal B cells, expressing CD38, CD138, CD81, CD27, CD45, and cytoplasmic kappa light chain, with no expression of CD19, CD56, CD117, or cytoplasmic lambda light chain. (C) Computed tomography scans demonstrating osteosclerotic bone lesions. (D) Cutaneous biopsy revealing perivascular infiltration of neutrophils, consistent with neutrophilic dermatosis, without evidence of vasculitis.
Clinically, SchS closely mimics cryopyrin-associated periodic syndromes (CAPS), which are due to gain-of-function mutations in the NLRP3 gene, critical for NLRP3-inflammasome activity and IL-1β production (16). The NLRP3 inflammasome plays vital effects in the innate immune system in response to a variety of stimuli. Upon the formation of the NLRP3 inflammasome, caspase-1 is released and activates the pro-inflammatory cytokines IL-1β and IL-18 (17). However, previous studies do not support a role for somatic NLRP3 mosaicism in SchS pathogenesis (5). To identify the two diseases, whole exome sequencing (WES) was performed to search for somatic NLRP3 mutations and 32 genes linked to inherited autoinflammatory diseases, with no pathological variants identified. Analysis of the 32 genes showed no mutations in TNFRSF1A, NOD2, or NLRC4 (genes associated with dominant autoinflammatory diseases with rash) and did not identify common susceptibility factors for SchS (Figure 2A) (5). Gene Ontology (GO) enrichment analysis of aberrant genes (PRDM16, KLF4, TP73, BCL2, HIF1A, and CHD4) indicated associations with DNA-binding transcription factor and NF-κB binding activity (Figure 2B). In addition, to distinguish SchS from lymphoproliferative disorders, which often feature monoclonal gammopathy (2), MYD88 mutations were sought, predicting the development of WM (18), and the results were negative. Now VEXAS syndrome, a newly discovered syndrome driven by somatic mutations in UBA1, has been identified as a link between autoinflammatory and malignant hematologic diseases (19). In order to distinguish between SchS and VEXAS, we further tested for UBA1 mutation, and the result indicated negative (Figure 2A).
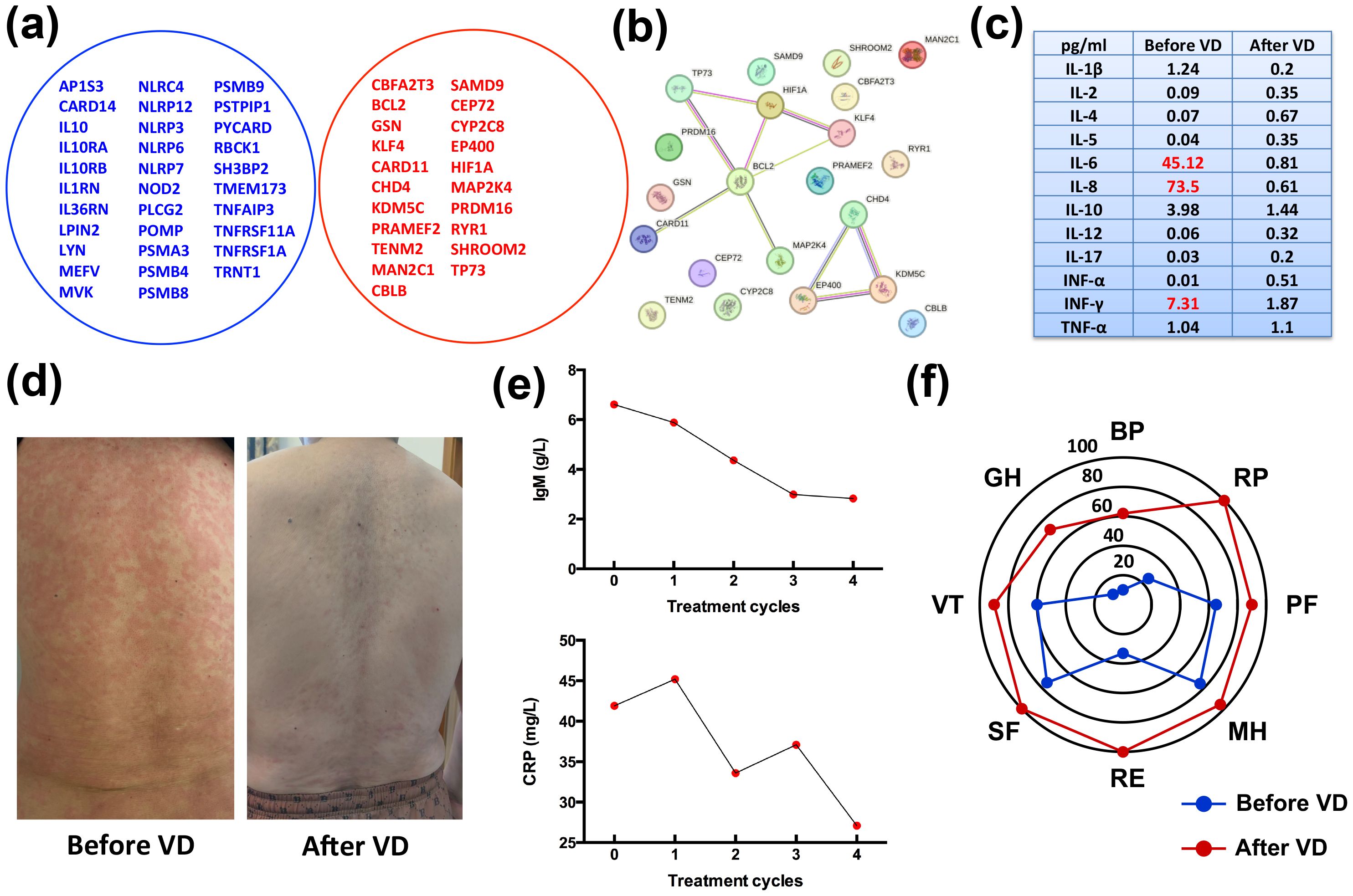
Figure 2. Clinical manifestations and laboratory findings after four cycles of VD treatment. (A) Venn diagram illustrating no overlap between 32 autoinflammatory genes and the patient’s gene mutations. (B) Protein interaction network constructed using the STRING online tool, with GO enrichment analysis identifying associations of aberrant genes (PRDM16, KLF4, TP73, BCL2, HIF1A, and CHD4) with DNA-binding transcription factor and NF-κB binding activity. (C) Inflammatory cytokine levels at different therapeutic time points. Initial levels of IL-6, IL-8 and INF-γ were elevated at diagnosis, but returned to normal after four cycles of VD treatment. (D) Clinical presentation of the patient before and after four treatment cycles. (E) Serum CRP and IgM levels showing significant reductions after treatment. (F) Quality of life (QoL) survey results before starting VD and after four treatment cycles. A statistically significant improvement in all domains was observed (a change of 10 points or more is considered clinically significant). BP, bodily pain; RP, role physical; PF, physical function; MH, mental health; RE, role emotional; SF, social function; VT, vitality; GH, general health.
Cytokine profiling was performed to assess immune responses and guide treatment choices. At disease onset, the levels of IL-6, IL-8, and INF-γ were elevated, with 45.12 pg/ml, 73.5 pg/ml, and 7.31 pg/ml, respectively, as indicated in Figure 2C. Due to the IL-1β was not detected and limited access to IL-1 antagonist agents (anakinra or canakinumab) in China, IL-1 blockade therapies were not pursued (20–22). The patient also declined IL-6 blockade therapy with tocilizumab for financial reasons. Recently, SchS has been incorporated into the spectrum of MGCS with a characterized small plasma cell clone (13). Therefore, this patient underwent treatment with the bortezomib-dexamethasone (VD) regimen, consisting of bortezomib (1.3 mg/m2) on days 1, 4, 8, and 11, and dexamethasone (20 mg) on days 1, 2, 4, 5, 8, 9, 11, and 12, aiming for anti-plasma cell therapy. Following four cycles of treatment, the patient’s urticarial rash resolved completely (Figure 2D), and serum levels of lgM and CRP showed marked declines (Figure 2E), and there were moderate declines in the levels of serum CRP (27.1 mg/L) and IgM (2.83 g/L). These declines of IL-6 and CRP correspond to the patient’s improving condition (Figure 2C). At the four-month follow-up, the patient’s responses were accompanied by improved quality of life (QoL) (Figure 2F). The patient was closely monitored for side effects during VD regimen, and no infectious complications and peripheral neuropathy occurred. Follow-up consisted of monthly visits for the first four months, followed by bi-monthly assessments, which included comprehensive clinical evaluations and laboratory tests for CRP and IgM levels. These assessments were crucial in monitoring the patient's response to treatment and overall disease activity. The stable values of CRP and IgM during the follow-up period indicate a sustained response to the bortezomib therapy, suggesting effective disease control and a positive impact on the patient's clinical status. As of the last follow-up, the patient continues on bortezomib therapy, with a dosing adjustment to 1.3 mg/m2 monthly, reflecting a good response and tolerability to the treatment.
The pathophysiology of SchS remains enigmatic, with evidence indicating it involves an aberrant autoinflammatory response characterized by inflammatory cytokines such as IL-1β, IL-6, and TNF-α (23). It is hypothesized that IL-1β may trigger IL-18, facilitating the differentiation of B-lymphocytes into plasma cells and leading to monoclonal gammopathy (24). IL-18, another cytokine of the IL-1 family that is cleaved by caspase-1, which cleaves pro-IL-1β and triggers the formation of gasdermin D pores. Gasdermin D pores allow for the secretion of active IL-1β and IL-18 initiating the organism-wide inflammatory response (25, 26). The different conventional therapeutic approaches (interferon, corticosteroids, anti-rheumatic medications, and immunosuppressants) get highly variable responses, often partial and temporary (27, 28). SchS is recognized as a late-onset acquired autoinflammatory syndrome with a significant inflammatory component. Central to its pathogenesis are the proinflammatory cytokines IL-1 and IL-6, with IL-1 being particularly crucial in the syndrome's development (29, 30). Given the established role of IL-1 in the pathogenesis of the auto-inflammatory component, IL-1 blockade therapies (anakinra and canakinumab) are recommended as the first-line treatments for SchS. These treatments effectively manage symptoms and reduce inflammation in most SchS patients, but they are not disease-modifying, and discontinuation typically results in relapse (4). Moreover, IL-1 blockade dose not alter the levels of monoclonal proteins, and IgM MGUS may progress during treatment (31). The connection between IgM monoclonal gammopathy and inflammasome activation in SchS is not yet established, necessitating the exploration of treatments that can address both issues simultaneously.
The prognosis of SchS is influenced by the development of lymphoproliferative complications associated with dysglobulinemia component, such as IgM MGUS, lymphoma, and WM (32, 33). IgM monoclonal gammopathy is a defining feature of SchS according to the Strasbourg criteria. If the cell clone is lymphoplasmacytic or corresponds to chronic lymphocytic leukemia or B cell lymphoma, treatment should be adjusted accordingly, often involving an anti-CD20 monoclonal antibody (34). However, in cases where the clone does not match these conditions, such as this patient discussed, an anti-CD20 monoclonal antibody is not indicated (35). Genetic testing has revealed the presence of the MYD88 L265P mutation in peripheral blood samples of 30% of SchS patients (36). The link between MYD88 and BTK lies in their involvement in the signaling pathways that lead to B cell activation and the production of immunoglobulins. In certain B cell malignancies and lymphoproliferative disorders, the MYD88 L265P mutation leads to constitutive activation of the NF-κB pathway, resulting in uncontrolled B cell proliferation and survival (37, 38). This mutation is also associated with the production of monoclonal gammopathy, which can be a feature of SchS. BTK inhibitors, such as ibrutinib, target the BCR signaling pathway, which is downstream of MYD88 in the B cell activation process (39, 40). Of note is that several SchS patients have been successfully treated with the BTK inhibitor ibrutinib, which may target both IgM monoclonal gammopathy and the NLRP3 inflammasome (11, 41, 42). Compared to the successful ibrutinib-treated cases, our patient had no MYD88 L265P mutation (7). It also highlights the importance of exploring clone-directed therapy. Above observations support the inclusion of SchS within the spectrum of MGCS, where organ damage is related to monoclonal protein or other paraneoplastic mechanisms (13).
The cytokine profiles of this patient align with our treatment options. The direct activation of IL-6 production by IL-1β, as well as the direct production of IL-6 by the NF-κB signaling, accounts for the involvement of these two cytokine pathways in the increase of CRP (43–45). Indeed, elevated IL-6 and CRP account for constitutional symptoms of SchS. Furthermore, the GO analysis has shown the functional enrichments (DNA-binding transcription factor and NF-κB binding activity) in this patient’s network (46). As we all know, NF-κB regulates the transcription of numerous genes involved in immune response, cell apoptosis, differentiation, proliferation, adhesion, and angiogenesis (47, 48). The expression of IL-1, IL-6, and CRP is mediated by activation of NF-κB signaling pathways (43, 49). Multiple kinds of NF-κB pathway mutations have been identified in various inflammatory diseases, lymphomas, and MM. Even a small plasma cell clone can produce severe clinical manifestations due to toxicity of the monoclonal immunoglobulin. To enhance early diagnosis and management of these small plasma cell clone-related disorders, the concept of MGCS has been proposed (13). Treatment in MGCS is driven by organ damage caused by the secreting plasma clone. The current treatment strategy primarily relies on repeated courses of bortezomib combined with dexamethasone, which produces rapid and deep hematological responses in various MGCSs (14) [POEMS syndrome (50), Light chain deposition disease (51), TEMPI (52), Cryoglobulinemia (53), Scleromyxedema (54), and Clarkson disease (55)]. Bortezomib, a proteasome inhibitor (PI) targeting at the ubiquitin-proteasome pathway, is the first clinically approved PI for the treatment of MM (56). The ubiquitin-proteasome system (UPS) is recognized as the major pathway for degradation of intracellular proteins, many of which play an important role in the regulation of pro-inflammatory cytokines. UPS deregulation is involved in several pathological processes such as malignant hematologic diseases, viral infections, and autoimmunities (57, 58). Bortezomib was demonstrated to be a rapid and potent inhibitor of NF-κB-inducible cytokines, including TNF-α, IL-1β, IL-6, and IL-10 (59). Hence, it was supposed that bortezomib can inhibit NF-κB activation process and indirectly produces a potential anti-inflammatory response for treating SchS. It is known that glucocorticosteroid treatment could result in diminution of symptoms and normalization of the cytokine responses in SchS patients (2). It should be noted that this patient was started on prednisone 30 mg daily by an immunologist. However, his clinical symptoms did not improve, and the rash got worse. Above all, the direct or indirect NF-κB inhibitory effects of VD regimen reflect the effectiveness of this combined treatment on both entities, regardless of the potential trigger role of one on the other. Bortezomib represents a novel and unique therapeutic option for SchS patients, targeting both the underlying IgM monoclonal gammopathy and inflammasome activation. While it is recognized that even minimal plasma cell clones can lead to severe clinical manifestations due to the potential toxicity of monoclonal immunoglobulins or other mechanisms, the precise role of these clones and IgM paraprotein secretion in the pathogenesis of SchS remains a subject of investigation (60). Some researchers posit that the presence of such clones may be an epiphenomenon, not directly contributing to the disease's core pathophysiology (61, 62). However, others suggest that they may play a more active role, possibly influencing the inflammatory process (63–65). The variability in clinical presentations and responses to treatment among SchS patients supports the need for further exploration into the significance of these clones and the potential mechanisms by which they might contribute to the disease. Future studies are warranted to clarify the relationship between minimal plasma cell clones, IgM paraprotein secretion, and the development of SchS.
In conclusion, while treatments for SchS have been reported with anti-IL-1 therapies and BTK inhibitors, there is no report on anti-plasma cell therapies. The successful remission of clinical symptoms, alongside a decrease in IgM monoclonal protein and improvement in quality of life, suggests that anti-plasma cell therapies such as bortezomib should be considered in the treatment of SchS.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by the Ethics Committee of the Nanjing Drum Tower Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
HB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DZ: Data curation, Formal analysis, Investigation, Writing – original draft. JL: Investigation, Methodology, Writing – original draft. JH: Conceptualization, Data curation, Investigation, Software, Writing – original draft. ZM: Data curation, Investigation, Methodology, Writing – original draft. WF: Formal analysis, Methodology, Software, Validation, Writing – original draft. BC: Data curation, Formal analysis, Investigation, Project administration, Validation, Writing – original draft, Writing – review & editing. YX: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This project was supported by grants from the National Natural Science Foundation of China (82000215), the Nanjing Medical Science and Technique Development Foundation (YKK21097), the Clinical Trials from Nanjing Drum Tower Hospital (2024-LCYJ-PY-32).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kacar M, Pathak S, Savic S. Hereditary systemic autoinflammatory diseases and Schnitzler's syndrome. Rheumatol (Oxford England). (2019) 58:vi31–43. doi: 10.1093/rheumatology/kez448
2. de Koning HD. Schnitzler's syndrome: lessons from 281 cases. Clin Trans Allergy. (2014) 4:41. doi: 10.1186/2045-7022-4-41
3. Krause K, Bonnekoh H, Ellrich A, et al. Long-term efficacy of canakinumab in the treatment of Schnitzler syndrome. J Allergy Clin Immunol. (2020) 145:1681–1686.e1685. doi: 10.1016/j.jaci.2019.12.909
4. de Koning HD, Schalkwijk J, van der Ven-Jongekrijg J, Stoffels M, van der Meer JW, Simon A. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler's syndrome. Ann rheumatic diseases. (2013) 72:1634–8. doi: 10.1136/annrheumdis-2012-202192
5. Rowczenio DM, Pathak S, Arostegui JI, et al. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood. (2018) 131:974–81. doi: 10.1182/blood-2017-10-810366
6. Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
7. Buske C, Jurczak W, Salem JE, Dimopoulos MA. Managing Waldenström's macroglobulinemia with BTK inhibitors. Leukemia. (2023) 37:35–46. doi: 10.1038/s41375-022-01732-9
8. Treon SP, Xu L, Guerrera ML, et al. Genomic landscape of waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. (2020) 38:1198–208. doi: 10.1200/JCO.19.02314
9. Goodman AM, Cohen PR, Li A, Hinds B, Kurzrock R. Schnitzler syndrome associated with MYD88 L265P mutation. JAAD Case Rep. (2019) 5:312–6. doi: 10.1016/j.jdcr.2019.02.002
10. Varettoni M, Matous JV. BTK inhibitors in the frontline management of waldenström macroglobulinemia. Hematology/oncology Clinics North America. (2023) 37:707–17. doi: 10.1016/j.hoc.2023.04.005
11. Huang Y, Wang Y, Yu F, et al. Case report: therapeutic use of ibrutinib in a patient with schnitzler syndrome. Front Immunol. (2022) 13:894464. doi: 10.3389/fimmu.2022.894464
12. Tam CS, Opat S, D'Sa S, et al. Biomarker analysis of the ASPEN study comparing zanubrutinib with ibrutinib for patients with Waldenström macroglobulinemia. Blood advances. (2024) 8:1639–50. doi: 10.1182/bloodadvances.2023010906
13. Fermand JP, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. (2018) 132:1478–85. doi: 10.1182/blood-2018-04-839480
14. Dispenzieri A. Monoclonal gammopathies of clinical significance. Hematol Am Soc Hematol Educ Program. (2020) 2020:380–8. doi: 10.1182/hematology.2020000122
15. Gellrich FF, Günther C. Schnitzler syndrome. Der Hautarzt; Z fur Dermatologie Venerologie und verwandte Gebiete. (2018) 69:761–72. doi: 10.1007/s00105-018-4250-2
16. de Koning HD, van Gijn ME, Stoffels M, et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol. (2015) 135:561–4. doi: 10.1016/j.jaci.2014.07.050
17. Blevins HM, Xu Y, Biby S, Zhang S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front Aging Neurosci. (2022) 14:879021. doi: 10.3389/fnagi.2022.879021
18. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. New Engl J Med. (2012) 367:826–33. doi: 10.1056/NEJMoa1200710
19. Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. New Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
20. de Koning HD, Schalkwijk J, van der Meer JW, Simon A. Successful canakinumab treatment identifies IL-1β as a pivotal mediator in Schnitzler syndrome. J Allergy Clin Immunol. (2011) 128:1352–4. doi: 10.1016/j.jaci.2011.05.023
21. Néel A, Henry B, Barbarot S, et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler's syndrome: a French multicenter study. Autoimmun Rev. (2014) 13:1035–41. doi: 10.1016/j.autrev.2014.08.031
22. Krause K, Weller K, Stefaniak R, et al. Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy. (2012) 67:943–50. doi: 10.1111/j.1398-9995.2012.02843.x
23. Masson Regnault M, Frouin E, Jéru I, et al. Cytokine signature in schnitzler syndrome: proinflammatory cytokine production associated to th suppression. Front Immunol. (2020) 11:588322. doi: 10.3389/fimmu.2020.588322
24. Li M, Chen YW, Shen A, et al. Case report: Successful treatment with tofacitinib and colchicine in a patient with Schnitzler syndrome. Int J rheumatic diseases. (2023) 26:160–3. doi: 10.1111/1756-185X.14457
25. Gupta S, Cassel SL, Sutterwala FS, Dagvadorj J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol Rev. (2024). doi: 10.1111/imr.13410
26. Migliorini P, Del Corso I, Tommasi C, Boraschi D. Free circulating interleukin-18 is increased in Schnitzler syndrome: a new autoinflammatory disease? Eur Cytokine network. (2009) 20:108–11. doi: 10.1684/ecn.2009.0164
27. de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis rheumatism. (2007) 37:137–48. doi: 10.1016/j.semarthrit.2007.04.001
28. Simon A, Asli B, Braun-Falco M, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy. (2013) 68:562–8. doi: 10.1111/all.2013.68.issue-5
29. Gusdorf L, Lipsker D. Schnitzler syndrome: a review. Curr Rheumatol Rep. (2017) 19:46. doi: 10.1007/s11926-017-0673-5
30. Möller B, Villiger PM. Inhibition of IL-1, IL-6, and TNF-alpha in immune-mediated inflammatory diseases. Springer Semin immunopathology. (2006) 27:391–408. doi: 10.1007/s00281-006-0012-9
31. Takimoto-Ito R, Kambe N, Kogame T, et al. Refractory serum immunoglobulin M elevation during anti-interleukin (IL)-1- or IL-6-targeted treatment in four patients with Schnitzler syndrome. J Dermatol. (2021) 48:1789–92. doi: 10.1111/1346-8138.16124
32. Verret JL, Leclech C, Rousselet MC, Hurez D, Schnitzler L. Schnitzler syndrome and Waldenström disease. Fatal outcome of the original case. Annales dermatologie venereologie. (1993) 120:459–60.
33. Aouba A, Pressiat C, Pricopi M, et al. Complete remission of Schnitzler syndrome and Waldenström macroglobulinemia under rituximab-cyclophosphamide-dexamethasone. Dermatol (Basel Switzerland). (2015) 230:18–22. doi: 10.1159/000368349
34. Castillo JJ, Treon SP. Toward personalized treatment in Waldenström macroglobulinemia. Hematol Am Soc Hematol Educ Program. (2017) 2017:365–70. doi: 10.1182/asheducation-2017.1.365
35. Roccatello D, Sciascia S, Baldovino S, et al. Improved (4 plus 2) rituximab protocol for severe cases of mixed cryoglobulinemia: A 6-year observational study. Am J nephrology. (2016) 43:251–60. doi: 10.1159/000445841
36. Pathak S, Rowczenio DM, Owen RG, et al. Exploratory study of MYD88 L265P, rare NLRP3 variants, and clonal hematopoiesis prevalence in patients with schnitzler syndrome. Arthritis Rheumatol (Hoboken NJ). (2019) 71:2121–5. doi: 10.1002/art.v71.12
37. Cea M, Cagnetta A, Acharya C, et al. Dual NAMPT and BTK targeting leads to synergistic killing of waldenström macroglobulinemia cells regardless of MYD88 and CXCR4 somatic mutation status. Clin Cancer Res. (2016) 22:6099–109. doi: 10.1158/1078-0432.CCR-16-0630
38. Gertz MA. Waldenström macroglobulinemia: 2023 update on diagnosis, risk stratification, and management. Am J hematology. (2023) 98:348–58. doi: 10.1002/ajh.26796
39. Ntanasis-Stathopoulos I, Gavriatopoulou M, Fotiou D, Dimopoulos MA. Current and novel BTK inhibitors in Waldenström's macroglobulinemia. Ther Adv hematology. (2021) 12:2040620721989586. doi: 10.1177/2040620721989586
40. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. (2018) 18:148–67. doi: 10.1038/nrc.2017.121
41. Jani P, Vissing MB, Ahmed S, et al. Ibrutinib for the management of schnitzler syndrome: A novel therapy for a rare condition. J Oncol practice. (2018) 14:387–8. doi: 10.1200/JOP.18.00050
42. Claves F, Siest R, Lefebvre C, Valmary-Degano S, Carras S. Dramatic efficacy of ibrutinib in a schnitzler syndrome case with indolent lymphoma. J Clin Immunol. (2021) 41:1380–3. doi: 10.1007/s10875-021-01038-y
43. Sun W, Wu Y, Gao M, et al. C-reactive protein promotes inflammation through TLR4/NF-κB/TGF-β pathway in HL-1 cells. Biosci Rep. (2019) 39(8):BSR20190888. doi: 10.1042/BSR20190888
44. Hatzimichael EC, Christou L, Bai M, Kolios G, Kefala L, Bourantas KL. Serum levels of IL-6 and its soluble receptor (sIL-6R) in Waldenström's macroglobulinemia. Eur J haematology. (2001) 66:1–6. doi: 10.1034/j.1600-0609.2001.00152.x
45. DuVillard L, Guiguet M, Casasnovas RO, et al. Diagnostic value of serum IL-6 level in monoclonal gammopathies. Br J haematology. (1995) 89:243–9. doi: 10.1111/j.1365-2141.1995.tb03296.x
46. Bai W, Wang H, Bai H. Identification of candidate genes and therapeutic agents for light chain amyloidosis based on bioinformatics approach. Pharmacogenomics personalized Med. (2019) 12:387–96. doi: 10.2147/PGPM.S228574
47. Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. (2004) 382:393–409. doi: 10.1042/BJ20040544
48. Sun XF, Zhang H. NFKB and NFKBI polymorphisms in relation to susceptibility of tumour and other diseases. Histol histopathology. (2007) 22:1387–98. doi: 10.14670/HH-22.1387
49. Yang M, Wang X, Wang L, Wang X, Li J, Yang Z. IL-1α Up-regulates IL-6 expression in bovine granulosa cells via MAPKs and NF-κB signaling pathways. Cell Physiol Biochem. (2017) 41:265–73. doi: 10.1159/000456091
50. Fang F, Lan XX, Hu RH, et al. Efficacy of bortezomib, cyclophosphamide, and dexamethasone for newly diagnosed POEMS syndrome patients. Ther Adv neurological Disord. (2024) 17:17562864231219151. doi: 10.1177/17562864231219151
51. Wada Y, Iyoda M, Saito T, et al. Light-chain deposition disease successfully treated with bortezomib in an elderly patient: A case report and review of the literature. Internal Med (Tokyo Japan). (2015) 54:2893–8. doi: 10.2169/internalmedicine.54.4994
52. Schroyens W, O'Connell C, Sykes DB. Complete and partial responses of the TEMPI syndrome to bortezomib. New Engl J Med. (2012) 367:778–80. doi: 10.1056/NEJMc1205806
53. Tocut M, Rozman Z, Biro A, Winder A, Tanay A, Zandman-Goddard G. The complexity of an overlap type resistant cryoglobulinemia: a case report and review of the literature. Clin Rheumatol. (2019) 38:1257–62. doi: 10.1007/s10067-018-04423-y
54. Win H, Gowin K. Treatment of scleromyxedema with lenalidomide, bortezomib and dexamethasone: A case report and review of the literature. Clin Case Rep. (2020) 8:3043–9. doi: 10.1002/ccr3.v8.12
55. Ramirez-Sandoval JC, Varela-Jimenez R, Morales-Buenrostro LE. Capillary leak syndrome as a complication of antibody-mediated rejection treatment: a case report. CEN Case Rep. (2018) 7:110–3. doi: 10.1007/s13730-018-0306-5
56. Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. (2008) 14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218
57. Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. (2009) 78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607
58. de Bettignies G, Coux O. Proteasome inhibitors: Dozens of molecules and still counting. Biochimie. (2010) 92:1530–45. doi: 10.1016/j.biochi.2010.06.023
59. van der Heijden JW, Oerlemans R, Lems WF, Scheper RJ, Dijkmans BA, Jansen G. The proteasome inhibitor bortezomib inhibits the release of NFkappaB-inducible cytokines and induces apoptosis of activated T cells from rheumatoid arthritis patients. Clin Exp Rheumatol. (2009) 27:92–8.
60. Pathak S, Rowczenio D, Lara-Reyna S, et al. Evidence of B cell clonality and investigation into properties of the igM in patients with schnitzler syndrome. Front Immunol. (2020) 11:569006. doi: 10.3389/fimmu.2020.569006
61. Ntanasis-Stathopoulos I, Kastritis E, Tzartos J, Terpos E, Dimopoulos MA, Gavriatopoulou M. Retinopathy in a patient with igM MGUS: causal association or an epiphenomenon? In Vivo (Athens Greece). (2024) 38:954–7. doi: 10.21873/invivo.13526
62. Al-Zuhairy A, Schrøder HD, Plesner T, Abildgaard N, Sindrup SH. Immunostaining of skin biopsy adds no diagnostic value in MGUS-associated peripheral neuropathy. J neurological Sci. (2015) 349:60–4. doi: 10.1016/j.jns.2014.12.026
63. Marinkovic A, Zypchen LN, Chan J, Chen LY, Parkin S. Monoclonal gammopathy of clinical significance: what the rheumatologist needs to know. Lancet Rheumatol. (2022) 4:e362–73. doi: 10.1016/S2665-9913(21)00348-9
64. Mondello P, Paludo J, Novak JP, et al. Molecular clusters and tumor-immune drivers of igM monoclonal gammopathies. Clin Cancer Res. (2023) 29:957–70. doi: 10.1158/1078-0432.CCR-22-2215
Keywords: Schnitzler syndrome, IgM monoclonal gammopathy, plasma cell, urticarial rash, inflammatory response, bortezomib
Citation: Bai H, Zhou D, Liu J, He J, Min Z, Fan W, Chen B and Xu Y (2025) Case report: Therapeutic use of bortezomib in a patient with Schnitzler syndrome. Front. Immunol. 16:1520470. doi: 10.3389/fimmu.2025.1520470
Received: 31 October 2024; Accepted: 06 January 2025;
Published: 28 January 2025.
Edited by:
Iolanda Jordan, Sant Joan de Déu Hospital, SpainReviewed by:
Bernhard Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2025 Bai, Zhou, Liu, He, Min, Fan, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Bai, YmFpaHVhOTJAMTI2LmNvbQ==; Bing Chen, Y2hlbmJpbmcyMDA0QDEyNi5jb20=; Yong Xu, ZmFuY3l4eXpAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.