 Yaoyao Wang
Yaoyao Wang Xiancong Yang
Xiancong Yang Yalin Liu1
Yalin Liu1 Youjie Li
Youjie Li- 1Department of Pediatrics of Yantai Affiliated Hospital, The Second Clinical Medical College of Binzhou Medical University, Yantai, Shandong, China
- 2Department of Biochemistry and Molecular Biology, Binzhou Medical University, Yantai, Shandong, China
- 3Laboratory Department, Qilu Hospital of ShanDong University Dezhou Hospital, Dezhou, Shandong, China
Acute myeloid leukemia (AML) is a highly aggressive hematological malignancy. Traditional chemotherapy methods not only bring serious side effects, but also lead to high recurrence rate and drug resistance in some patients. However, as an emerging therapeutic strategy, immunotherapy has shown great potential in the field of AML treatment in recent years. At present, common immunotherapy methods for AML include monoclonal antibodies, CAR-T cell therapy, and immune checkpoint inhibitors. With the deepening of research and technological progress, especially the application of nanotechnology in medicine, new immunotherapy is expected to become one of the important means for the treatment of acute myeloid leukemia in the future.
1 Introduction
Acute myeloid leukemia (AML) is a hematological malignancy that is prevalent worldwide. Its main feature is that myeloid progenitor cells or primitive granulocytes cannot differentiate normally, resulting in abnormal proliferation, accompanied by fever, anemia, bleeding and bone pain and other clinical (1, 2). With the in-depth study of the pathogenesis of AML, the prognosis of patients with acute myeloid leukemia has improved, but AML is still one of the cancer types with high recurrence rate and mortality (3–5).
The treatment of acute myeloid leukemia mainly uses chemotherapy, radiotherapy and hematopoietic stem cell transplantation, among which chemotherapy is regarded as the primary treatment. However, the chemotherapy methods have remained basically unchanged for decades, and the efficacy and prognosis are not optimistic. Most patients have failed to achieve complete remission or disease recurrence (6, 7). Immunotherapy has become an important research direction in the treatment of acute myeloid leukemia. The immune system plays an important role in cancer treatment, and immunotherapy has been widely used in B cell carcinoma and various solid cancers (8, 9). Common immunotherapy includes monoclonal antibodies, antibody-drug conjugates, radionuclide conjugates, bispecific antibodies, and chimeric antigen receptor T cells (CAR-T cells) (10, 11).
The research of new immunotherapy using nanotechnology in the field of cancer treatment has become a hot topic, and exploring the application of nanotechnology in cancer treatment has become a quite popular task (12). The rapid development of nanotechnology has promoted the research and application of nanomaterials and nanoparticles (NP). These tiny substances have shown broad application prospects in the fields of biology and medicine. As an ideal carrier for targeted drug delivery systems, nanoparticles can overcome the problem of tumor drug resistance caused by biochemical and physical barriers and cellular and non-cellular mechanisms in traditional chemotherapy (13).
The immune system can be divided into innate immunity (also known as non-specific immunity) and adaptive immunity (also known as specific immunity) (Figure 1). In adaptive immunity, it can be further subdivided into humoral immunity and cellular immunity. Innate immunity is the earliest immune response produced by human beings, which protects the human body through skin, mucosa, macrophages and natural killer cells. Adaptive immunity is an immune response produced after the first contact with pathogens, which can identify and eliminate pathogens that infect the human body (14, 15). The process of adaptive immunity includes four stages: perception, recognition, activation and execution. First, the body perceives the invading pathogens and recognizes and resists pathogens through immune cells and systems on the body surface and mucosa. The process of recognition is to produce specific antibodies and T cells to identify and attack foreign pathogens through the interaction of humoral immunity and cellular immunity. The process of activation is that immune cells release cytokines to regulate and activate other immune cells to form an immune response. The process is carried out by T cells and B cells synergistically to attack and eliminate pathogens by producing antibodies and cell-mediated effects (16, 17).
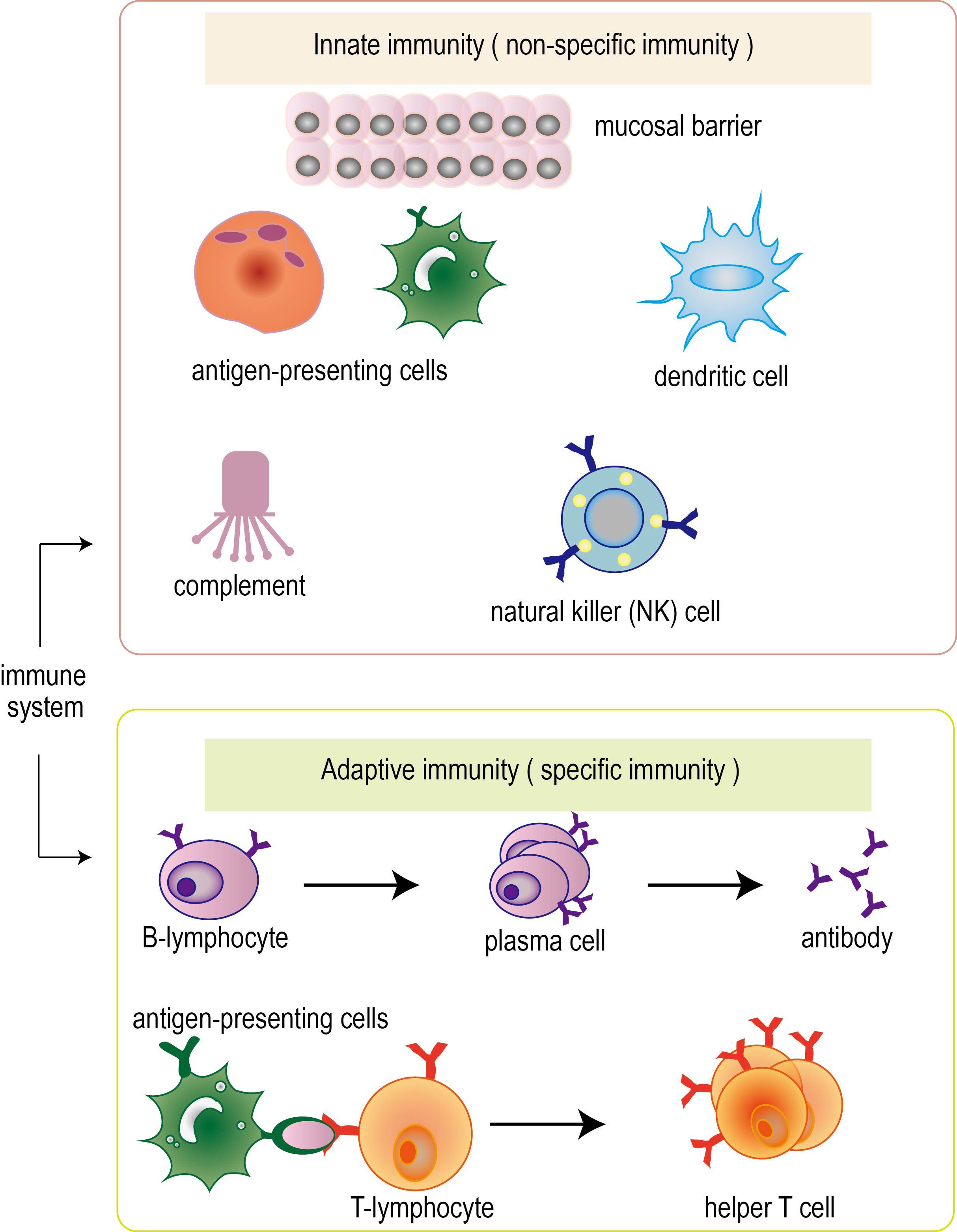
Figure 1. Classification of immune system. Immune system can be divided into innate immunity (also known as non-specific immunity) and adaptive immunity (also known as specific immunity). In adaptive immunity, it can be further subdivided into humoral immunity and cellular immunity. The immune system performs its function through the synergy of multiple immune cells.
There are a variety of immune cells and immune molecules in the human immune system that can resist the occurrence of cancer. For example, natural killer (NK) cells can directly recognize and kill cancer cells (18); T cells recognize and attack cancer cells by recognizing specific proteins on the surface of cancer cells; antibodies produced by B cells can specifically target and neutralize cancer cells. In addition, cytokines such as interferons and interleukins can promote the activation of immune cells and enhance their ability to target cancer cells. Chemokines can attract immune cells to the tumor site, and antibodies can bind to cancer cells and label them to be destroyed by immune cells. However, through development, cancer has been able to avoid being recognized by the immune system, so cancer immunotherapy can stimulate the patient ‘s immune system to recognize or destroy abnormally proliferating cells (19). In addition, with the emergence of nanotechnology, we are expected to inhibit tumor growth, reduce chemotherapy resistance and prevent metastasis, which provides new ideas and strategies for tumor immunotherapy.
2 Immune escape mechanism of acute myeloid leukemia
Acute myeloid leukemia cells evade the recognition and clearance of the immune system by constructing an immunosuppressive tumor microenvironment. The formation of this tumor microenvironment involves a variety of immune cells and immune factors (20, 21) (Figure 2).
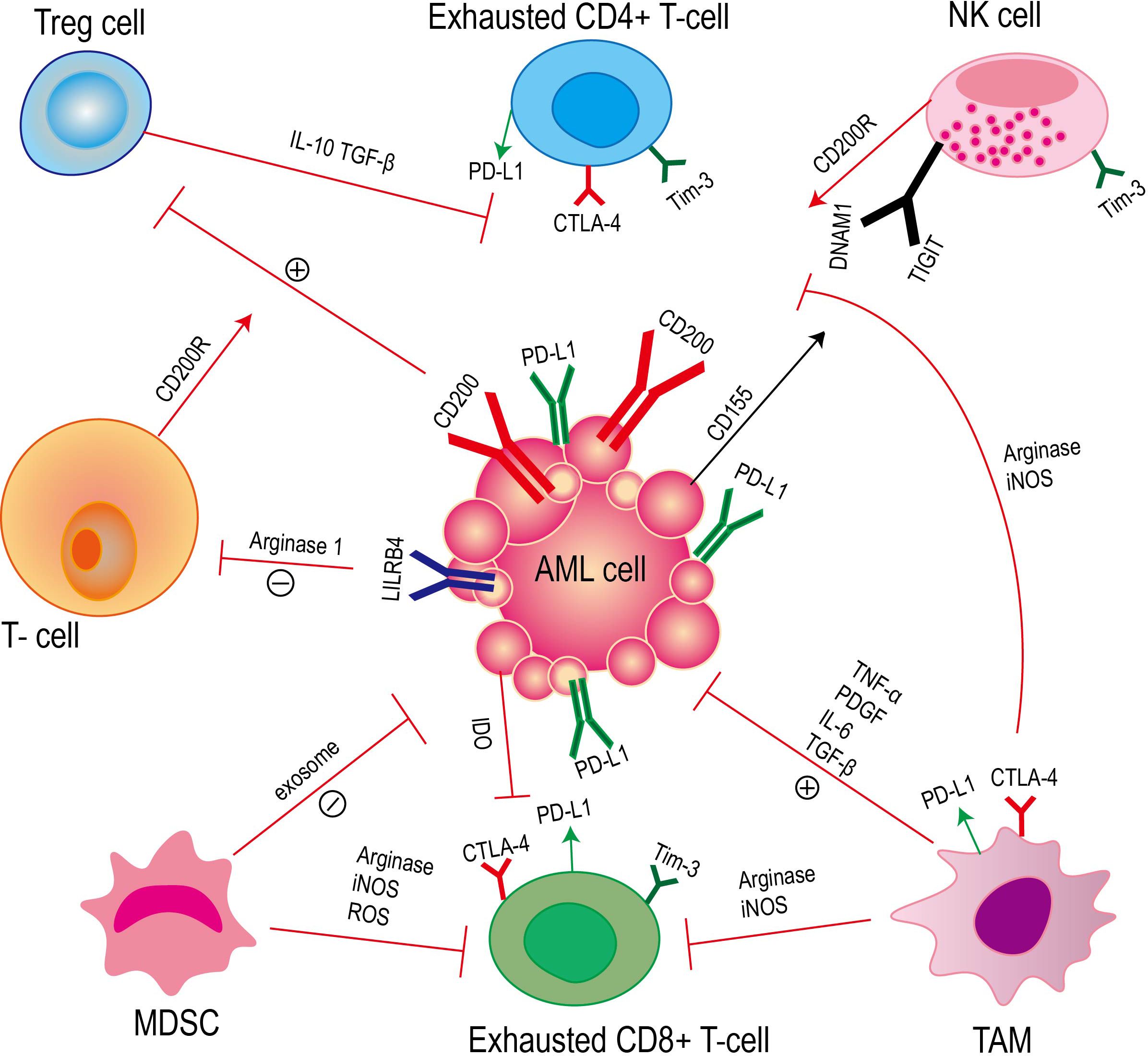
Figure 2. Acute myeloid leukemia achieves immune escape through the interaction between immune cells and immune factors. This process mainly involves T cells, natural killer cells (NK cells), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), as well as cytokines and related signaling pathways secreted by these cells.
T cells play a crucial role in the immune response. The interaction between the expression of AML surface receptors (leukocyte immunoglobulin-like receptor B4, LILRB4) and T cells leads to the inhibition of T cell proliferation. However, LILRB4 directly inhibits T cell proliferation and cytotoxicity by mediating the release of arginase-1 (22). CD200 is a type I membrane glycoprotein of the immunoglobulin superfamily, which is up-regulated on the surface of AML cells (23). The interaction of CD200 with T cell CD200 receptor (CD200R) leads to an increase in regulatory T cells (Tregs) and a decrease in memory T cell function (24). Studies have confirmed that compared with healthy individuals, the number of CD4 + and CD8 + T cells in AML patients is significantly reduced, and these cells also show the characteristics of aging (25). At the same time, the high proportion of lymphocytes and T lymphocytes in bone marrow is related to the improvement of survival rate of AML patients (26).
NK cells are a kind of congenital lymphocytes (27). NK cells can effectively identify and eliminate AML cells in vivo, which can prevent the occurrence of diseases. NK cells regulate the immune response by secreting tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ). Interferon-γ can promote the maturation of dendritic cells, thereby promoting the formation of adaptive immunity (28). In AML blasts including leukemia stem cells (LSC), the up-regulation of the surface glycoprotein CD200 of immunosuppressive cells is associated with the functional inhibition of natural killer (NK) cells through their receptor CD200R (29, 30). In the DNAM1/TIGIT/CD96 signaling pathway, DNAM1 and TIGIT act as receptors for activating and inhibiting NK cells, respectively, and both target CD155, which is expressed in AML blasts. The relative expression levels of ligands and receptors in this signaling pathway are different, and this change in expression is related to the inhibition of NK cell function (31, 32).
Regulatory T cells (Tregs) play an immunosuppressive role in AML. In AML, Tregs are recognized as a factor that can be used by leukemia cells to evade immune monitoring (33). Studies have shown that in AML, the interaction between PD-1 and PD-L1 can promote the inhibition of Teffs by Tregs, thereby weakening the anti-tumor immune response (34). At the same time, Tregs can secrete immunosuppressive factors (such as TGF-β and interleukin-10) or direct cell contact to inhibit the effector T cell response and evade the recognition of the immune system (35–37). The number of Tregs with high inhibitory activity was increased in AML patients at diagnosis, suggesting that these cells may play an important role in the anti-tumor immune response (38). Recent studies have shown that Tregs are more abundant in the bone marrow of AML patients and are beneficial to the growth of leukemia cells. The large number of Tregs is not conducive to the treatment of AML patients (39). At the same time, a large number of studies have shown that the depletion of Treg cells in the tumor microenvironment can enhance the host ‘s anti-tumor immunity (40–42).
Myeloid-derived suppressor cells (MDSC) inhibit immune cell response through various mechanisms in AML. Myeloid-derived suppressor cells (MDSC) induce T cell tolerance in AML patients through a variety of mechanisms, such as PD-L1, arginase, indoleamine 2,3-dioxygenase (IDO), TGF-β and IL-10 (43–46). MDSC can directly inhibit anti-tumor T cell response, and MDSC can also indirectly inhibit antigen-specific T cell activation by inhibiting the function of antigen-presenting cells (APC). The presence of MDSC can effectively block Ag-specific CD8-mediated T cell function (47, 48). MDSC mainly exerts T cell inhibition by expressing ARG1, iNOS and ROS (49–51). Studies have shown that MDSC stops the cell cycle of T cells and blocks T cell proliferation, rather than directly killing T cells (52). MDSC also secretes various exosomes that promote tumor growth, which are transported to the tumor site and induce immunosuppression (53). In general, the mechanisms of MDSC in promoting tumor immunosuppression mainly include:1) reducing the amino acids required for T cell proliferation and activation; 2) To release immunosuppressive cytokines and promote the differentiation of regulatory B (Breg) cells and regulatory T cells (Treg); 3) Recruiting regulatory T cells; 4) binding to the inhibitory receptor PD1 to block the killing function of T cells/NK cells; 5) down-regulation of NK cell activating receptors; 6) Down-regulated STAT-3 and increased HIF1α to induce M2 macrophage differentiation; 7) Secretion of S100A8/9 promotes the polarization and chemotaxis of MDSC and M2 macrophages in the tumor microenvironment; 8) Inhibition of antigen presenting function of dendritic cells (DC) (54).
Tumor-associated macrophages (TAMs) play a key role in the immunosuppressive microenvironment of acute myeloid leukemia (AML). TAMs are derived from bone marrow mononuclear cells, including M1 macrophages with anti-tumor function and M2 macrophages with tumor-promoting properties (55). Studies have shown that there is a significant correlation between TAM and poor prognosis and recurrence of cancer patients (56). TAM can promote tumor formation and help tumor cells avoid being attacked by the body ‘s immune system by secreting growth factors and cytokines that support tumor cell proliferation, such as tumor necrosis factor (TNF)-α, platelet-derived growth factor (PDGF), TGF-β and IL-6 (57–63). TAMs can also regulate the killing function of T cells and NK cells (64).
For example, M1 macrophages regulate the immune microenvironment to activate NK cells, leading to apoptosis and tissue fibrosis (65). In malignant pleural mesothelioma, TAMs mainly have M2 phenotype, and there is a negative correlation between TAMs and T cells (66). In AML cells, ICOSL expression leads to the expansion of ICOS + Tregs, thereby promoting immune escape, and IL-10 secreted by ICOS + Tregs promotes the proliferation of AML cells (67). In addition, studies have shown that tumor-associated macrophages (TAMs) can directly inhibit the function of T cells by expressing immune checkpoint molecules such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD-1) (68–70). Most patients with acute myeloid leukemia (AML) lack arginine succinate synthase-1 (ASS1), which leads to a decrease in arginine synthesis. However, the depletion of extracellular arginine promotes macrophage polarization to M2 (71).
3 Common immunotherapy for acute myeloid leukemia
Immunotherapy shows potential ability to overcome relapse and drug resistance, which is particularly critical for patients with relapsed or refractory AML. These therapies play a key role in the treatment of AML and show broad prospects (Figure 3).
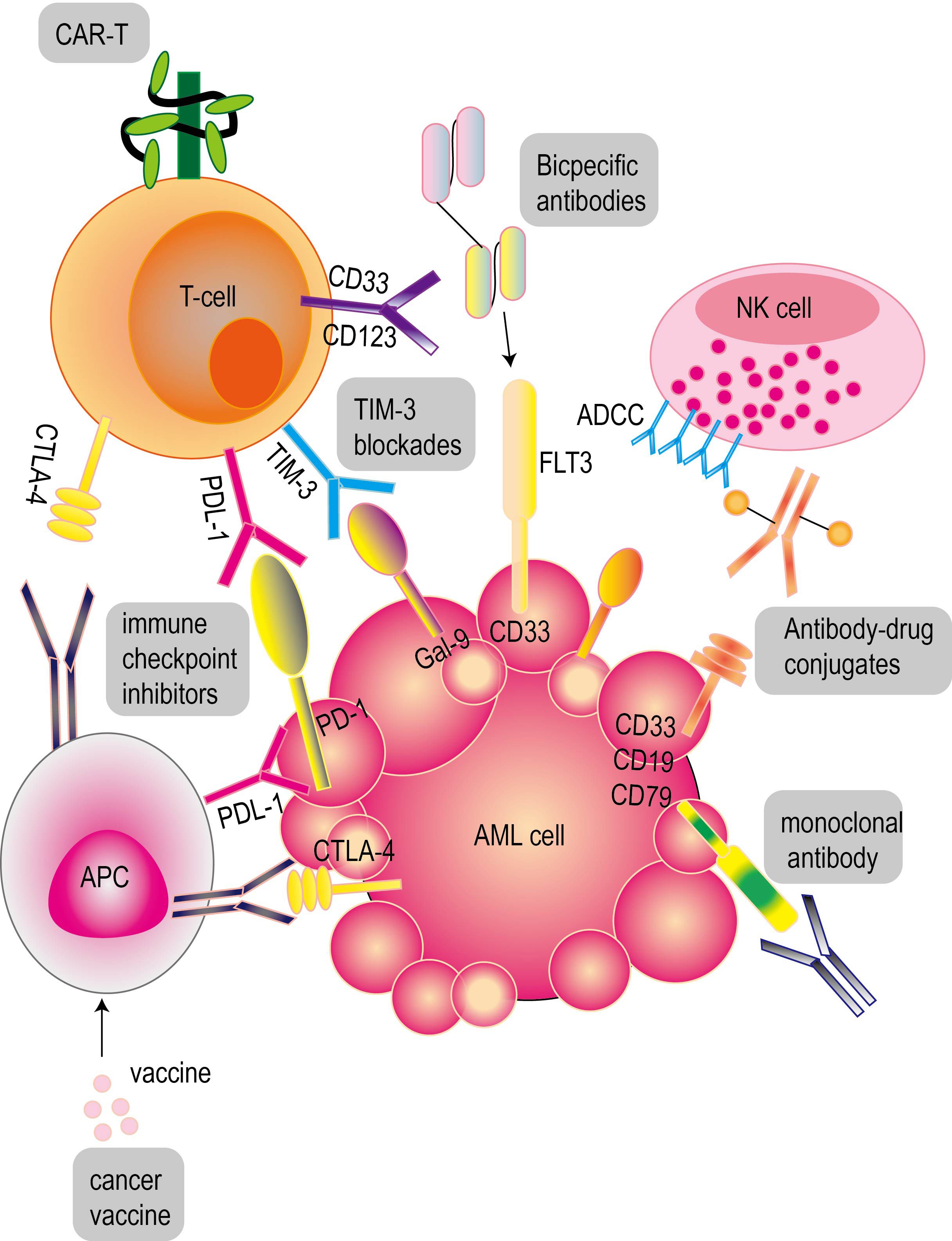
Figure 3. Common immunotherapy for acute myeloid leukemia. These therapies include antibody immunotherapy (monoclonal antibodies, bispecific antibodies, antibody-drug conjugates), CAR-T cell therapy and cancer vaccines for acute myeloid leukemia. PD-1 and CTLA-4 inhibitors are common immune checkpoint inhibitors (ICIs).
3.1 Antibody immunotherapy for acute myeloid leukemia
Antibody is a kind of protective protein secreted by plasma cells under the stimulation of antigen. The function of antibodies in acute myeloid leukemia depends on the following aspects: First, after the antibody binds to the surface antigen of tumor cells, it uses its Fc domain to recruit immune effector molecules, thereby triggering natural killer (NK) cell-mediated antibody-dependent cytotoxicity (ADCC), antibody-dependent phagocytosis or complement-dependent cytotoxicity; secondly, through receptor-mediated endocytosis of antibody-drug conjugates (ADCs) and radionuclide conjugates, it can effectively deliver toxic carriers to kill leukemia cells. The third is to use bispecific or multispecific antibodies to bind these cells to target leukemia cells, thereby enhancing the anti-leukemia effect of T cells or NK cells.
Since most leukemia cells are associated with the positive expression of specific antigens (such as CD123, CD33, CD96 and CLL-1) (72), targeted immunotherapy, including monoclonal antibodies (73) and bispecific antibodies, is currently in the clinical evaluation stage. These therapies aim to target multiple antigens expressed on AML blast cells, of which CD33 and CD123 are the most common targeted antigens (74). The bispecific T cell conjugative molecule (BiTE) is composed of a single variable fragment (Fv). Bispecific T cell conjugator (BiTE) targeting CD33 can activate and amplify T cells in autologous clinical samples of patients with AML, and mediate the lysis of primary AML cells and normal myeloid cells in vitro in a dose-dependent manner (75–77). The use of anti-CD19 BiTE triggered CD33-independent activation, resulting in CD33 expression on a small number of T cells. This phenomenon is related to the self-killing of T cells, but has little effect on the function of cytotoxic T cells associated with AML (78). The single-chain Fv trisomics (sctb) based on CD16 single-chain Fv fragment (scFv) targeting CD123 exhibits significant anti-leukemia activity, but its effect is not as good as the dual-targeting sctb targeting both CD33 and CD123 (79). 7370 anti-FLT3 bispecific IgG can activate T cells in patients with acute myeloid leukemia, thereby inducing cytotoxicity to autologous blasts. The results of in vivo experiments showed that the bispecific antibody could also effectively guide human T cells to target AML cell lines and produce killing effects in vivo. This effect depends on the expression of FLT3 antigen on the surface of AML cells, and is not affected by FLT3 mutation status. Both double-targeted single-chain Fv trisomy (sctb) and single-targeted sctb use isolated monocytes (MNC) as effector cells, which can effectively induce antibody-dependent cytotoxicity (ADCC) of two different acute myeloid leukemia (AML) -derived CD33 and CD123 double-positive cell lines at low concentrations (80). In addition, a bispecific single-chain Fv (bsscFv) was formed by binding another scFv that is specific for low-affinity Fcγ receptor III (CD16). This bsscFv can effectively mediate the cleavage of AML-derived leukemia cell lines in a specific concentration of ADCC reaction (81). Bispecific antibodies produced by chemical coupling of anti-CD3 and anti-CD13 Fab ‘ fragments enhance the cytotoxicity of peripheral blood mononuclear cells (PBMC) to CD13-positive AML cells stimulated by IL2 or IL7 (82). At the same time, the clinical potential of monoclonal antibodies (mAbs) against CD123 in the treatment of AML has been supported by relevant evidence (83). BI836858 is an antibody against CD33 with an Fc domain. Decitabine can increase the expression of NKG2D ligands on the surface of leukemia cells, thereby enhancing BI836858-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) (84). Evorpacept (ALX148) is mainly composed of a modified SIRPαD1 domain targeting CD47 and can bind to the inactivated human IgG1 fragment (Fc) (85). At present, the molecule is being studied in an ongoing phase I/II clinical trial (NCT04755244) to evaluate its efficacy in combination with the BCL2 inhibitor venetox and AZA, especially for patients with relapsed/refractory acute myeloid leukemia (R/R AML) who have not been treated or are not suitable for standard induction chemotherapy (86). At the same time, in vivo experimental results showed that under the action of activated human T cells, AMG 330 targeting CD33 could inhibit the growth of xenografts of humanized mouse subcutaneous AML cell lines, thereby significantly improving the survival rate (76, 77). In addition, treatment with anti-CD28 activation antibody not only enhanced the killing effect of AMG 330 on human AML cell lines, but also increased the cytotoxicity of primary AML samples from patients with refractory leukemia (87). Lintuzumab (SGN-33) is a monoclonal antibody (mAb) against CD33. Studies have shown that lintuzumab may have an anti-leukemia effect, which can induce remission after cytoreductive surgery with low-dose cytarabine in untreated AML patients (88). Studies on AML animal models have shown that MGD024 combined with cytarabine or venetox can almost completely eliminate tumor cells. Therefore, a drug dose escalation study was proposed to evaluate the safety of MGD024 in refractory/recurrent hematological malignancies (including AML and BPDCN).
A few years ago, researchers developed an antibody-drug conjugate (ADC) treatment strategy, which showed good therapeutic effect and ideal side effects (89). ADC has gradually become an emerging chemotherapeutic drug for the treatment of cancers including AML (90). IMGN632 is an antibody-drug conjugate (ADC) targeting CD123, which uses a new IGN payload (91). In addition, compared with X-ADC, IMGN632 showed cytotoxic effects on AML samples at doses that had no adverse effects on normal myeloid progenitor cells (92). Clinical studies have shown that IMGN632 and venetoclax (a BCL-2 inhibitor for patients with acute myeloid leukemia) combined with azacitidine showed a consistent anti-leukemia synergistic effect, and the synergistic anti-leukemia effect between IMGN632 and venetoclax was verified in leukemia cell lines and xenotransplantation (PDX) derived from patients with acute myeloid leukemia. Mylotarg is a kind of ADC, which is used to treat newly diagnosed or refractory CD33 positive AML. Compared with Mylotarg, ADC targeting AML-specific antigens is expected to provide a more effective treatment for AML, and has a wider range of indications (93). SGN-CD123A is an antibody-drug conjugate (ADC) based on humanized CD123 antibody, which can effectively induce the apoptosis of leukemia cells expressing CD123. The drug shows a significant effect on promoting the apoptosis of leukemia cells in myeloid leukemia cell lines, primary AML blasts and patient-derived xenograft models (94). Antibody therapy has made significant progress in the clinical research of acute myeloid leukemia, but it still needs to be continuously optimized and improved to provide more effective and safe treatment for patients.
3.2 Immune checkpoint inhibitors
Immune checkpoint inhibitors (ICIs) relieve the inhibition of tumor cells on the immune system by interfering with the signal transmission mechanism between tumor cells and immune cells, thereby enhancing the immune system ‘s ability to attack tumor cells. Specifically, immune checkpoint inhibitors can effectively kill tumor cells by blocking the inhibitory molecules on the surface of T cells and transforming T cells from an ‘ exhausted ‘ state to an ‘ activated ‘ state, which has been proved to be a promising therapeutic option (95). But ICIs are not very promising drugs in AML treatment, and can be consider only as an addition to the treatment, do not work in monotherapy. In the current study, PD-1 and CTLA-4 are the two most active checkpoint receptors in the current study. They play a key role in different stages of anti-tumor immune response. At the same time, inhibition of CTLA-4 and PD-1 is the most widely used two immune checkpoint blocking strategies in clinical practice.
PD-1 inhibitors commonly used in clinical research for the treatment of AML include drugs such as ipidilizumab, nivolumab, pembrolizumab, durvalumab and atezolizumab (96). The results showed that in patients with AML, the complete remission rate (CR) of anti-PD-1 antibody nivolumab combined with azacitidine was 18%, and the hematological improvement rate was 15% (97). Studies have shown that the combination of azacitidine, nivolumab and ipilimumab (an anti-CTLA-4 antibody) has achieved complete remission (CR) or complete remission with incomplete hematological recovery (CRi) in 3% of patients with AML (98). The application of nivolumab combined with conventional induction chemotherapy (such as idarubicin plus cytarabine) in newly diagnosed AML patients is feasible (99). The efficacy of Pembrolizumab combined with decitabine or azacitidine in patients with R/R AML was similar to that of azacitidine combined with nivolumab. Tiragolumab is an anti-TIGIT antibody that can improve the prognosis of lung cancer patients when combined with atezolizumab. Anti-TIGIT antibody reshapes the tumor microenvironment by enhancing the blocking effect of PD-L1 on bone marrow cells and Treg cells, thereby improving the prognosis of tumor patients. The study found that tumor patients with higher baseline levels of macrophages and regulatory T cells in tumors had a better prognosis when treated with atezolizumab combined with tiragolumab, while atezolizumab alone did not have this effect. This indicates that TIGIT checkpoint inhibitors can play a role by reshaping the immunosuppressive tumor microenvironment (100). And the study reported the safety of azacitidine combined with duralizumab in patients with MDS and AML.
CTLA-4 (CD152) transmits immunosuppressive signals to terminate the immune response by interacting with CD80 and CD86 ligands. In the study of the treatment of melanoma patients, we found that anti-CTLA-4 antibody ipilimumab can effectively increase the proportion of Teff/Tregs, enhance the activity of NK cells, and restore the function of T effector cells, thereby significantly prolonging the survival of patients (101). Anti-CTLA-4 can enhance the function of AML-specific T cells, which is manifested in increasing its frequency, cytotoxicity and IFN-γ secretion (102). When ipilimumab monotherapy was used in patients undergoing hematopoietic stem cell transplantation (HCT) for recurrent hematological malignancies, a complete remission (CR) rate of 23% and a partial remission (PR) rate of 9% were observed (103). A phase I clinical trial for recurrent or refractory AML and myelodysplastic syndrome (MDS) is underway to evaluate the efficacy of ipilimumab in combination with decitabine (DAC) in patients who have received or have not received allogeneic hematopoietic stem cell transplantation (HCT). At present, the trial has not yet begun to recruit subjects (clinical trial registration number: NCT2890329).
3.3 Chimeric antigen receptor T cell therapy
The concept of chimeric antigen receptor (CAR) T cell therapy was first proposed in 1993 (104). This therapy has achieved remarkable results in the treatment of lymphatic and hematological malignancies by targeting unique targets such as CD19, CD22 and BCMA (105). CAR-T cells are genetically engineered from autologous peripheral blood T cells and allogeneic CAR-T cells. These cells have specific extracellular antigen recognition domains, which are usually composed of single-chain variable fragments of monoclonal antibodies and are connected to the intracellular signal transduction domain (106). The advantages of chimeric antigen receptor (CAR) T cell therapy are reflected in the following aspects: (a) CAR-T cells are activated only when recognizing specific targets (107); (b) CAR-modified cells can be effectively eliminated after disease eradication by using non-persistent cell types or safe switching mechanisms. The idea of safety switch is to trigger it when a high grade of CRS occurs to save the patient’s life; if the patient is safe without a serious toxicity, keeping CAR T cells in the body provides the treatment for many more years after disease eradication; the CAR T cells not active sitting in the body, ready to attack the cancer cells again when needed. “The safety switch” works like “an insurance policy” for the patient, in case of the relapse of the disease, high T cells activation followed by a high grade of CRS. (c) Genome editing of hematopoietic stem cells (HSCs) to generate hematopoietic systems that are resistant to CD33 (destruction of normal bone marrow) (108).
At present, a variety of strategies have been developed to improve the safety and reliability of CAR-T cell therapy in AML treatment (Figure 4) (Table 1). These strategies include using inducible caspase-9 to control the death of CAR-T cells and adjusting the affinity of CAR-T cells to only target cells with high expression levels (109). A large number of studies have shown that anti-CD123 CAR-T cells exhibit significant anti-leukemia activity before clinical practice. However, some studies have also raised concerns about the toxicity of hematopoietic stem cells (HSCs) (110). A variety of anti-CD33 CAR-T cell constructs have shown significant preclinical therapeutic effects on primary acute myeloid leukemia (AML) cells in vitro and in humanized animal models, and their toxic effects on leukemia cells and non-leukemia myeloid cells have also been observed (111). At present, in a clinical trial report of anti-CD33 CAR-T cell therapy, the treatment of a patient with refractory acute myeloid leukemia (AML) was recorded in detail. After receiving autologous anti-CD33 CAR-T cell infusion, the patient not only developed cytokine release syndrome (CRS), but also observed a temporary decrease in the original cells in the bone marrow (BM) (112). Studies have shown that human peripheral blood T lymphocytes transduced with CD19 CAR can completely eliminate lymphoma and leukemia in immunodeficient mice (113). CAR-T cells targeting CD19 in combination with T cell inhibitors can destroy pathological B cells and regulate T cell response, thereby inducing remission of refractory antisynthase syndrome (114). In a phase I clinical trial (ClinicalTrials.gov identifier:NCT02842138) for patients with B-cell lymphoma,11 patients received CD19-BBz (86) CAR-T cell therapy. Each patient received a dose of 2×108 to 4×108 cells of CD19-BBz (86) CAR-T cells, of which 6 patients achieved complete remission (115). A study showed that 23 of 27 adult and pediatric ALL patients (including 11 patients with extramedullary diseases) achieved complete remission after the first CD19 CAR-T treatment (116). CAR-T cell therapy has been reported to show great potential in improving the treatment of relapsed or refractory B-cell malignancies (117–119). According to relevant reports, a bispecific split CAR (BissCAR) T cell targeting CD13 and TIM-3 has a significant effect in clearing patient-derived acute myeloid leukemia (AML). At the same time, in mouse and patient-derived xenograft models, this treatment method is less toxic to normal hematopoietic stem cells (HSC), myeloid cells, and healthy organ systems (120). CD123 CAR-T cells have shown significant anti-leukemia effects in vitro on leukemia cell lines and primary patient leukemia cells, as well as in vivo using leukemia mouse models. However, it is unclear whether CD123 CAR-T cells affect the normal hematopoietic function of bone marrow (121, 122). A study selected ADGRE2 and CLEC12A as the target antigens by analyzing samples from patients with resistant AML and doing a series of tests in mouse models beginning in 2018 (123). They showed that CAR T cells designed for these antigens were safe and effective against human AML cells implanted in mice. Based on these results, the clinical trial was approved to go forward.CRISPR-Cas9 technology has been proved to be feasible for gene ablation of CD33 antigen in human HSPCs, and this technology shows multi-lineage hematopoietic recovery ability in in vivo model system (124). Based on this result, we will initiate a clinical trial that will combine allogeneic HSCT using transgenic CD33-negative HSCs and CD33-targeted CAR-T cell therapy (125). In addition to CD33 and CD123, targets such as folate receptor β (126), FLT3 (127), NKG2D ligand (128) and CD70 (129) are also being tested for the development of CAR-T cells.
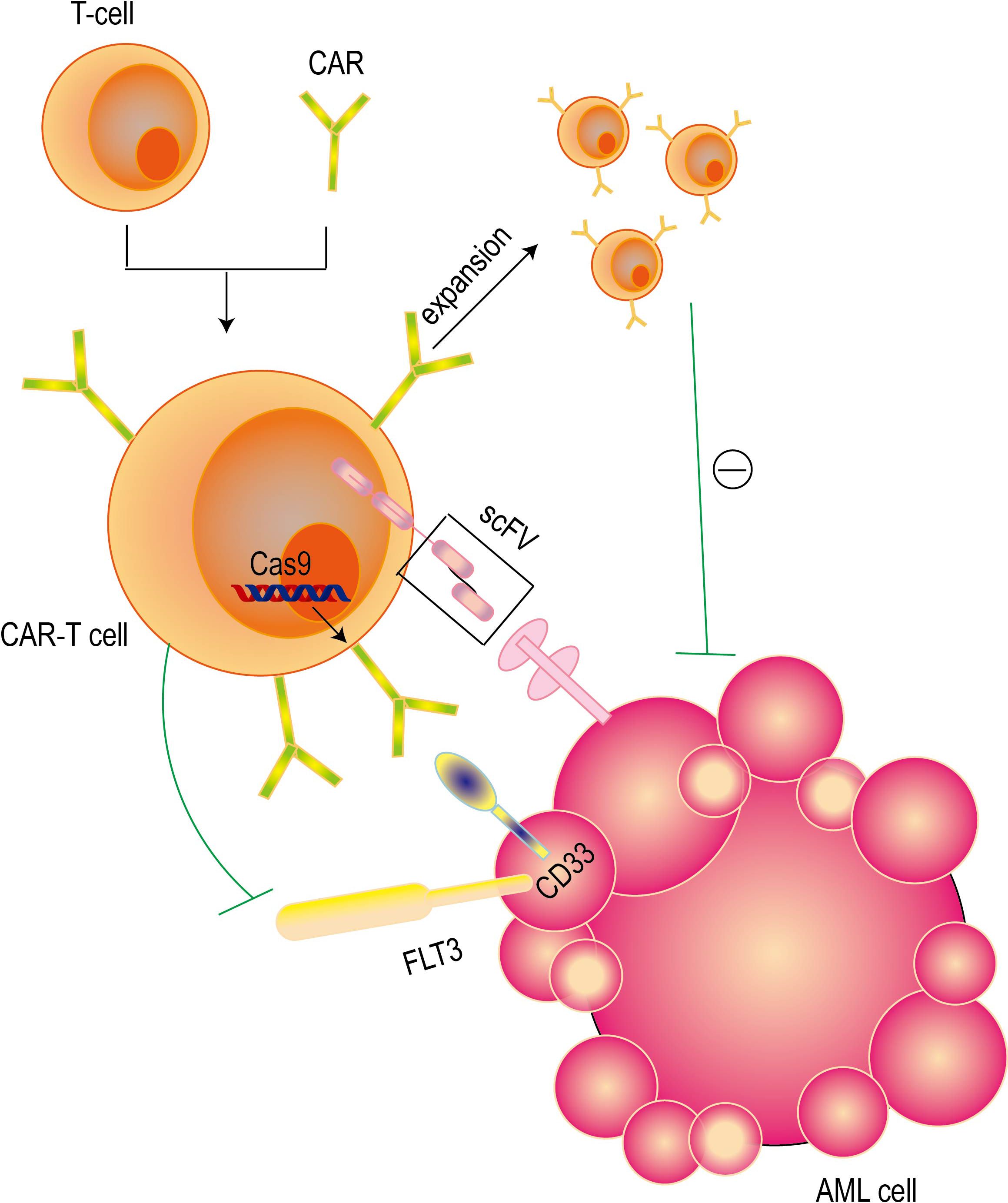
Figure 4. Mechanisms of CAR-T cell therapy. CAR-T cell therapy achieves precise targeted elimination of AML cells by combining the killing ability of T cells and the high specificity of antibodies. Single-chain variable fragments (ScFv) give this therapy the ability to accurately identify targets, while the intracellular signaling domain is responsible for activating the cytotoxic effects of T cells. At the same time, CRISPR-Cas9 technology can be used to perform gene ablation on CD33 antigen to kill AML cells.
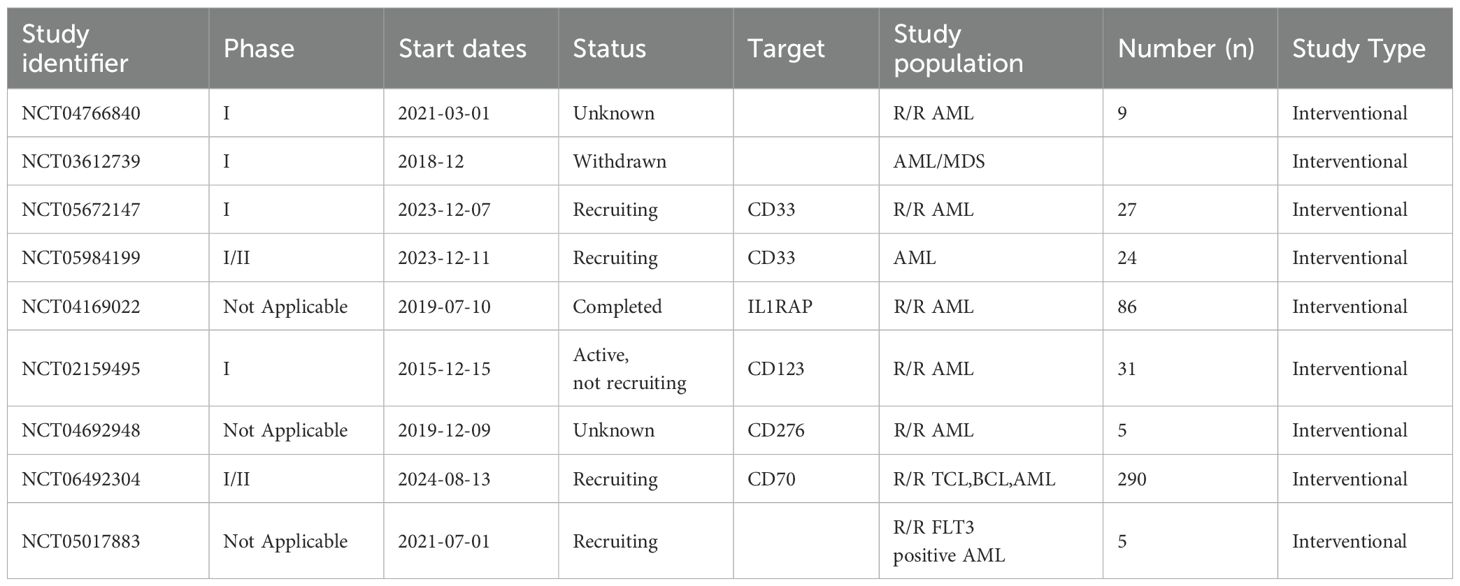
Table 1. Clinical trial of CAR-T in the treatment of acute myeloid leukemia.
The latest research provides strong evidence for the effectiveness of the combination therapy of anti-FLT3 CAR-T cells and crenolanib. It has been found that pretreatment with crenolanib can significantly increase the expression level of FLT3 on the cell surface, thereby enhancing the ability of T cells to recognize AML cell lines. This effect has been observed in both in vitro experiments and animal models (130). The efficacy and safety of CAR-T therapy in AML are not convincing enough, which is mainly due to the lack of ideal therapeutic targets, a series of complex factors related to AML microenvironment, and CAR-T cell exhaustion (131). At present, the most promising candidate targets for CAR-T treatment of AML include CD33, CD123 and CLL1.A large number of literatures have reviewed these targets and other possible targets in detail (132). The resistance of AML to CAR-T therapy involves a variety of mechanisms, including antigen escape, inhibition of tumor immune microenvironment, CAR-T cell dysfunction, and tumor heterogeneity. In response to these challenges, CAR-T cells that simultaneously target multiple antigens can be developed to reduce the risk of antigen escape; the persistence and efficacy of CAR-T cells are enhanced by genetic modification or combined use of immune checkpoint inhibitors. Although CAR-T therapy has a significant effect, it may also cause side effects such as cytokine release syndrome (CRS) and immune effector cell-related neurotoxicity syndrome (ICANS).
3.4 Vaccine
The cancer vaccines currently studied mainly include Peptide vaccines and dendritic cell (DC) vaccines (133) (Table 2). Peptide vaccines mainly target leukemia-associated antigens including nephroblastoma 1 (WT1), protease 3 (PR3), hyaluronic acid-mediated motor receptor (RHAMM), and mucin 1 (MUC1) (134). Mutations in the WT1 gene may cause abnormalities in cell growth and differentiation, which play an important role in the occurrence of leukemia (135). At the same time, it has been reported that the overexpression of WT1-specific T cells is significantly associated with a variety of hematological diseases (136). In AML patients, humoral immune response and cytotoxic response to WT1 protein have been observed (137). The above findings provide a theoretical basis for the application of WT1 protein in cancer vaccine therapy. The multivalent WT1 peptide vaccine galinpepimut-S (GPS) is considered to induce a specific immune response and is associated with the 5-year survival rate of AML patients participating in this clinical trial (138). CV-501 is a different HLA class II restricted peptide vaccine based on WT1, which has been studied in patients with acute myeloid leukemia (AML). However, the test results of this vaccine did not show any significant immune response (139). High-dose RHAMM-R3 peptide vaccine can effectively stimulate the body ‘s immune response and has a good effect in the treatment of hematological malignancies (140). Studies have explored the effects of combined use of PADRE, adjuvants, and WT1 or PR3 vaccines in AML patients. The results showed that the combination failed to produce clinical or immune responses (141).
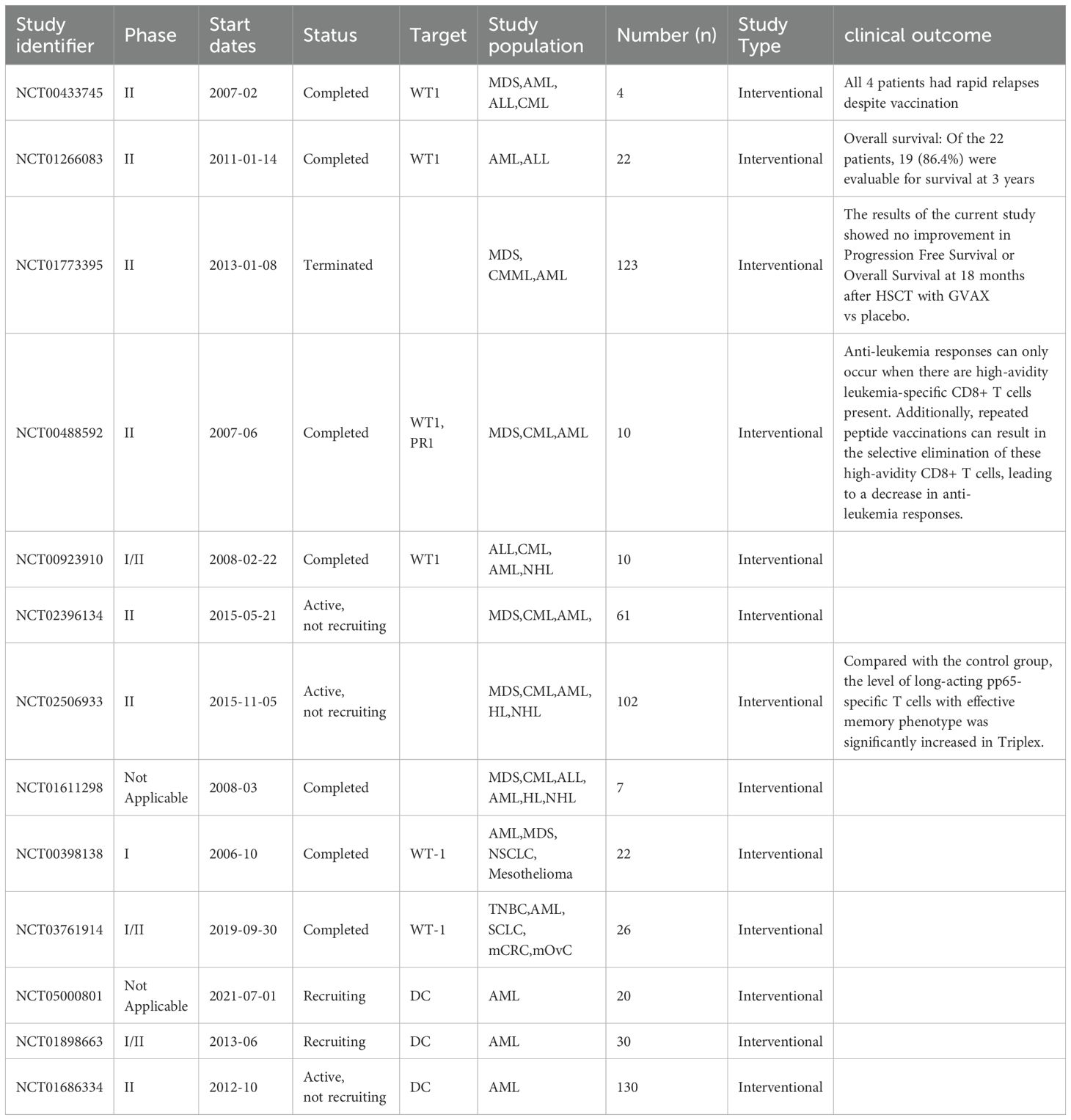
Table 2. Application of cancer vaccine in AML clinical trials.
Dendritic cells may be derived from different progenitor cells, but in the study of vaccine therapy, the most widely discussed sources are acute myeloid leukemia dendritic cells (AML-DC) (142) and monocyte-derived dendritic cells (mo-DC) (143). Most DC-based vaccine therapies are carried out in the state of minimal residual disease (MRD) to prevent the recurrence of leukemia. Leukemia-derived dendritic cell-based vaccines have shown good tolerance and a low probability of adverse reactions (144). Mo-DC can effectively load complete apoptotic leukemia cells, leukemia cell lysates or RNA/mRNA derived from leukemia cells, thereby further enhancing its therapeutic effect (145, 146). WT1 mRNA-electroporated DCs can improve the overall survival rate of AML patients with high risk of recurrence and further promote vaccine-induced WT1-specific CD8 T cell response (147). The efficacy of allogeneic dendritic cell vaccine DCP-001 in AML patients was studied. The results showed that DCP-001 had good safety and feasibility, and could induce cellular and humoral immune responses (148).
Telomerase is considered to be an important target in the study of cancer vaccine therapy. This is because the expression of telomerase is more in AML patients, and it plays a key role in maintaining the microenvironment of leukemia stem cells, especially in the context of high-risk cytogenetics. Studies have shown that telomerase (human telomerase reverse transcriptase, hTERT) and lysosomal-associated membrane protein (LAMP) can significantly enhance immunoreactivity by encoding mRNA, and this signal specifically targets lysosomes (149).
Exosome-based vaccines are a new approach to cancer treatment. The first study on tumor suppression using exosome-based vaccines was reported in 1998 (150). Some studies have evaluated the therapeutic effect of a novel interferon-modified exosome vaccine on prostate cancer (151). The inoculation of exosome vaccine not only reduces the expression level of vascular endothelial growth factor receptor 2, but also effectively reduces the ability of tumor metastasis (152). A study has shown that trastuzumab emtansine (T-DM1) can specifically bind to exosomes derived from HER2-positive cells, while some studies have explored the potential of exosomes as HER2-positive tumor vaccines by targeting and activating CD4 + and CD8 + T cells, and promoting long-term immune response through CTL memory cells (153, 154). Studies have found that exosomes derived from human adipose-derived mesenchymal stem cells (haMSC) can effectively induce apoptosis of ovarian cancer cells by blocking the cell cycle, up-regulating the molecular levels of BAX, CASP9 and CASP3, and down-regulating the anti-apoptotic protein BCL2 (155). A number of clinical studies have confirmed that vaccine therapy has shown significant efficacy in improving patient survival and reducing the risk of recurrence (156, 157). Leukemia-derived exosomes can induce the polarization of regulatory T cells and macrophages, and these exosomes promote the formation of tumor microenvironment in bone marrow, and activate HF-1α, AKT, VEGF, c-Myc, IL-8 and cyclin D1 signaling pathways by transporting tyrosine kinase receptor MET. AML-derived exosomes can transform the bone marrow microenvironment into conditions conducive to the development of leukemia by regulating multiple molecules (158). In addition, circulating exosomes produced by AML can also carry immunosuppressive substances, thereby inhibiting the body ‘s anti-tumor immune response (159). Exosomes can efficiently deliver drugs and antigens and accurately target malignant tumor cells in the blood system. At present, there are few reports on the application of MSC-derived exosome vaccines in acute myeloid leukemia.
4 Application of nanoimmunotherapy
Nano immunotherapy is an innovative targeted therapy in the field of cancer treatment. Nanotechnology refers to the use of biomolecules in the range of 5 to 500 nm in size for medical treatment and diagnosis. By improving the solubility and stability of drugs, prolonging the half-life of drugs in the blood, reducing toxic and side effects, and achieving precise targeting of drugs at specific sites, nanotechnology breaks the limitations of traditional therapies and gives full play to their anti-cancer effects (160–162). At present, nanomedicines for a variety of indications are being studied in clinical trials (163, 164). Nanoparticles (NP) used in nanopharmaceutical formulations currently cover a wide variety of types, including liposomes, polymers, micelles, nanocrystals, metal/metal oxides, and other inorganic materials and proteins (165). These NP can alter the biochemical, electronic, magnetic or optical properties of pharmaceutical preparations, allowing them to play an important role in therapeutic applications (166).
Compared with conventional immunotherapy alone, nanomedicines can enhance the immune response. By precisely targeting specific immune cells, the nanoparticle system activates and enhances their ability to recognize and attack cancer cells, thereby enhancing the immune system ‘s response (167). Targeting drug delivery is achieved by loading immune checkpoint inhibitors or immune stimulators onto NP, thereby enhancing the activation of immune cells and anti-tumor effects in TME. By loading tumor-specific antigens or immune stimulators onto NP to stimulate specific immune responses against tumors, thereby inhibiting tumor growth and metastasis, an effective cancer vaccine is prepared (168). Tumors usually lack a functional lymphatic system, which leads to long-term retention of macromolecules in tumors. This enhanced permeability and retention (EPR) effect is the main theoretical basis for current nano-drug design (169). With the help of the EPR effect, nanodrugs with a diameter greater than 8 nm can penetrate the blood vessel wall and enter the tumor cells, thereby achieving efficient drug delivery (170). Nano-diamonds can not only specifically target tumor cells, but also directly transport doxorubicin to mitochondria in cells, effectively cutting off the energy supply of cells, thereby inhibiting the growth and reproduction of tumor cells without affecting the function of normal cells (171).
The effect of cancer treatment can be significantly enhanced by using NP-based treatments. Nanogels enhance the anti-tumor effect by promoting the secretion of dendritic cells, cytotoxic T lymphocytes and immune stimulating cytokines, thereby improving the therapeutic effect on melanoma (172). In combination with anti-PD-L1 antibody therapy, the self-assembled core-shell nanosystem loaded with oxaliplatin and dihydroartemisinin can effectively induce T cell activation and reduce inhibitory cell infiltration in a mouse colorectal tumor model, thereby achieving a lasting enhancement of anti-tumor immunity (173). Studies have found that when α-PD-L1 and nanoparticles were injected into mice with lung cancer, the expression of IL-12 was significantly increased, while the levels of IL-10, arginase I and CCL22 were decreased, and the number of TREG cells was reduced, thus effectively inhibiting tumor growth (174). It has been reported that the use of PD-L1 siRNA-loaded folic acid (FA) modified polyethyleneimine nanoparticles can enhance the uptake of nanoparticles by ovarian cancer cells (175). As a carrier of mRNA vaccine, lipid nanoparticles (LNP) can not only efficiently deliver mRNA vaccine in liver tumor mouse model, but also stimulate specific immune response against tumor antigens (176, 177). Studies have shown that half of patients with unresectable pancreatic ductal adenocarcinoma (PDAC) showed a significant ability to induce antigen-specific T cells by using a novel personalized antigen vaccine developed by uridine-modified mRNA-lipid nanoparticle technology (178). The combination of CAR-T cell therapy and nanoparticles can improve the anti-tumor effect and enhance the targeting ability in cancer treatment, which provides a new research direction for the treatment of hepatocellular carcinoma (179). It has been reported that a NLS peptide-functionalized gold nanoparticle loaded with AS1411 and anti-221 can accurately bind AS1411 and anti-221 in vitro and in vivo, and target the key molecules in the NCL/miR-221/NFκB/DNMT1 signaling pathway, thereby effectively inhibiting the growth of AML cells (180). CPX-351 is a classic nanoliposome. Compared with traditional therapy, its toxicity to normal cells is significantly reduced, and it has a lower IC50 value. In addition, CPX-351 can promote the accumulation of daunorubicin and cytarabine in patients with acute myeloid leukemia (AML), thereby effectively improving the therapeutic effect of anti-leukemia (181, 182). The drug was approved by the US Food and Drug Administration (FDA) in 2017 for the treatment of newly diagnosed treatment-related AML (t-AML) or AML patients with myelodysplastic changes (183). The researchers developed a nanoparticle based on poly (lactic-co-glycolic acid) (PLGA), which was loaded with idarubicin (IDA) and achieved sustained release of IDA by methoxypolyethylene glycol-b-PLGA (mPEG-PLGA) technology. This design not only maintains the stable release of IDA, but also increases its anti-leukemia activity by 2 to 4 times compared to free IDA (184). In addition, studies have pointed out that by combining PLGA nanoparticles with anti-d44 antibody to form PLGA-antid44-PTL complexes and encapsulating parthenolide, an effective nuclear factor kappa B (NF-κB) inhibitor, the cellular uptake efficiency of drugs on acute myeloid leukemia cells can be significantly improved, thereby more effectively inhibiting the proliferation of these cells (185). Researchers have successfully developed an innovative ferritin dendrimer nanoparticle for precise delivery of miRNA to NB4 cells overexpressing the CD71 receptor. This technology can not only significantly induce leukemia cells to show phenotypic and morphological changes similar to early differentiation, but also effectively inhibit the cytotoxicity caused by free PAMAM dendrimers, and ensure the stability of nucleic acid during transmission to avoid its degradation (186). It is worth noting that miR-150, as a key tumor suppressor, plays an important role by negatively regulating FLT3. Experiments have shown that when PAMAM dendrimers are combined with FLT3 ligands and loaded with miR-150, selective clearance of FLT3-overexpressing acute myeloid leukemia (AML) cells can be achieved, and extremely low side effects are shown in vivo (187).
5 Conclusion
In this review, we summarize the progress of conventional immunotherapy and nanoimmunotherapy for AML, and highlight several representative emerging strategies (Table 3). AML is regarded as a disease with poor prognosis. In recent years, significant progress has been made in the molecular mechanism of tumor immunology and the clinical application of immunomodulators, which brings new hope for the treatment of AML. Because AML cells have immune escape characteristics, it is difficult for the immune system to effectively identify and attack these cancer cells. With the deepening understanding of tumor immunology, it provides an important theoretical basis for the development of more effective AML treatment. In order to improve the efficacy of immunotherapy for AML, we need to identify drug resistance mechanisms as early as possible and formulate corresponding strategies to overcome these mechanisms, thereby reducing the recurrence rate. Although there are few reports on the application of nano immunotherapy in AML, this field shows great potential and is expected to become a research hotspot in the near future. The current research mainly focuses on the combination therapy strategy, which shows a good development prospect in the future.
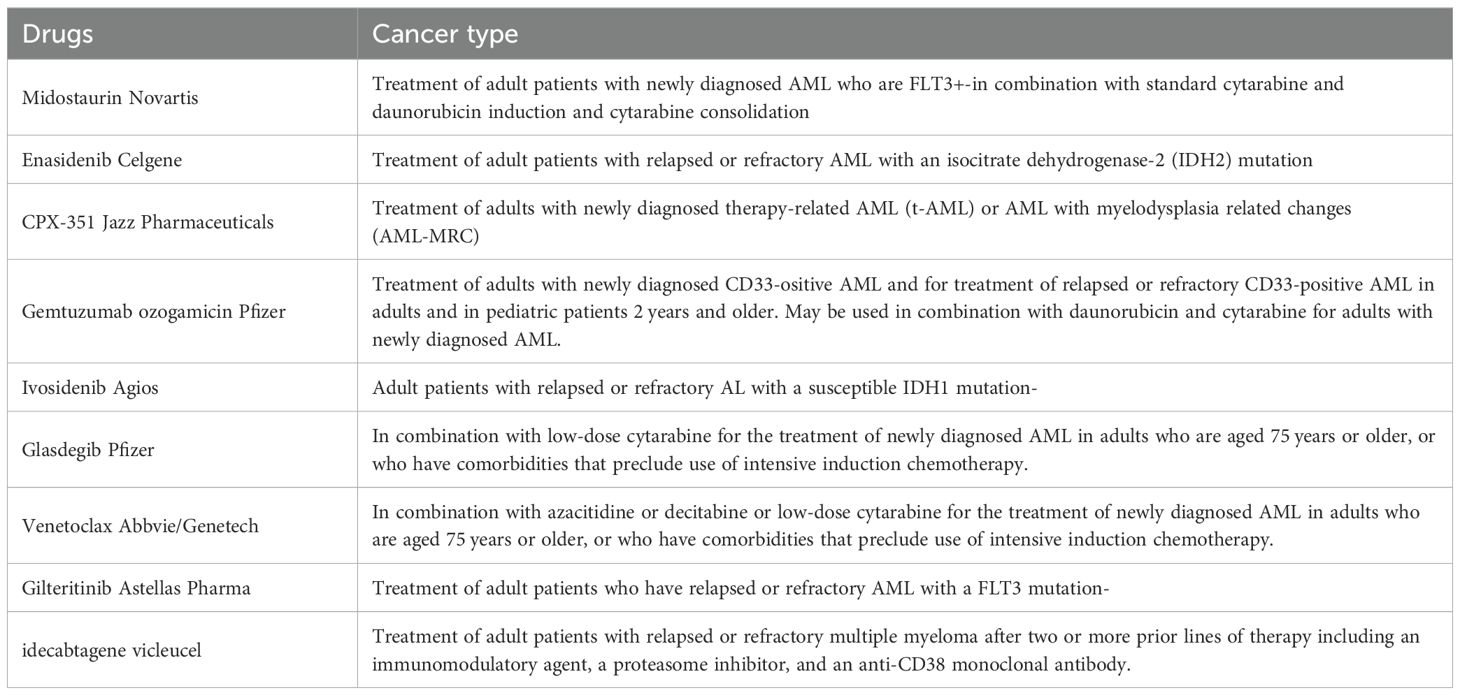
Table 3. FDA approved drugs for the treatment of hematological malignancies.
Author contributions
YW: Investigation, Methodology, Software, Visualization, Writing – original draft. XY: Investigation, Software, Writing – review & editing. YLL: Conceptualization, Data curation, Investigation, Software, Writing – review & editing. YJL: Data curation, Supervision, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The present study was supported by The Shandong Science and Technology Committee (grant nos. ZR2023MH223).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ. (2021) 375:n2026. doi: 10.1136/bmj.n2026
2. Shimony S, Stahl M, Stone RM. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. (2023) 98:502–26. doi: 10.1002/ajh.26822
3. Tosic N, Marjanovic I, Lazic J. Pediatric acute myeloid leukemia: Insight into genetic landscape and novel targeted approaches. Biochem Pharmacol. (2023) 215:115705. doi: 10.1016/j.bcp.2023.115705
4. Obszanski P, Kozlowska A, Wancowiat J, Twardowska J, Lejman M, Zawitkowska J. Molecular-targeted therapy of pediatric acute myeloid leukemia. Molecules. (2022) 27(12):3911. doi: 10.3390/molecules27123911
5. Singh V, Uddin MH, Zonder JA, Azmi AS, Balasubramanian SK. Circular RNAs in acute myeloid leukemia. Mol Cancer. (2021) 20:149. doi: 10.1186/s12943-021-01446-z
6. Vago L, Gojo I. Immune escape and immunotherapy of acute myeloid leukemia. J Clin Invest. (2020) 130:1552–64. doi: 10.1172/JCI129204
7. Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet 2013 Feb. (9865) 9:381. doi: 10.1016/S0140-6736(12)61727-9
8. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. (2020) 27:S87–97. doi: 10.3747/co.27.5223
9. Akkin S, Varan G, Bilensoy E. A review on cancer immunotherapy and applications of nanotechnology to chemoimmunotherapy of different cancers. Molecules. (2021) 26(11):3382. doi: 10.3390/molecules26113382
10. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
11. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
12. Merlin JPJ, Li X. Role of nanotechnology and their perspectives in the treatment of kidney diseases. Front Genet. (2021) 12:817974. doi: 10.3389/fgene.2021.817974
13. Saleh RO, Yuseran H, Mansouri S, Kareem AH, Shakir MN, Alasheqi MQ, et al. Two effective factors in cancer: Investigating the effect of ncRNAs in cancer and also the effect of nanotherapy in its treatment. Pathol Res Pract. (2024) 256:155218. doi: 10.1016/j.prp.2024.155218
14. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe. (2019) 25:13–26. doi: 10.1016/j.chom.2018.12.006
15. Ciraci C, Janczy JR, Sutterwala FS, Cassel SL. Control of innate and adaptive immunity by the inflammasome. Microbes Infect. (2012) 14:1263–70. doi: 10.1016/j.micinf.2012.07.007
16. Mantovani A, Garlanda C. Humoral innate immunity and acute-phase proteins. N Engl J Med. (2023) 388:439–52. doi: 10.1056/NEJMra2206346
17. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
18. Mujal AM, Delconte RB, Sun JC. Natural killer cells: from innate to adaptive features. Annu Rev Immunol. (2021) 39:417–47. doi: 10.1146/annurev-immunol-101819-074948
19. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
20. Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. (2018) 3(21):e120974. doi: 10.1172/jci.insight.120974
21. Knaus HA, Kanakry CG, Luznik L, Gojo I. Immunomodulatory drugs: immune checkpoint agents in acute leukemia. Curr Drug Targets. (2017) 18:315–31. doi: 10.2174/1389450116666150518095346
22. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. (2018) 562:605–9. doi: 10.1038/s41586-018-0615-z
23. Hao F, Sholy C, Wang C, Cao M, Kang X. The role of T cell immunotherapy in acute myeloid leukemia. Cells. (2021) 10(12):3376. doi: 10.3390/cells10123376
24. Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, et al. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia. (2012) 26:2146–8. doi: 10.1038/leu.2012.75
25. Tang L, Wu J, Li CG, Jiang HW, Xu M, Du M, et al. Characterization of immune dysfunction and identification of prognostic immune-related risk factors in acute myeloid leukemia. Clin Cancer Res. (2020) 26:1763–72. doi: 10.1158/1078-0432.CCR-19-3003
26. Ismail MM, Abdulateef NAB. Bone marrow T-cell percentage: A novel prognostic indicator in acute myeloid leukemia. Int J Hematol. (2017) 105:453–64. doi: 10.1007/s12185-016-2153-5
27. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
28. Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. (2002) 195:327–33. doi: 10.1084/jem.20010938
29. Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. (2011) 25:792–9. doi: 10.1038/leu.2011.1
30. Ho JM, Dobson SM, Voisin V, McLeod J, Kennedy JA, Mitchell A, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. (2020) 4:5402–13. doi: 10.1182/bloodadvances.2020001802
31. Kearney CJ, Ramsbottom KM, Voskoboinik I, Darcy PK, Oliaro J. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. Oncoimmunology. (2016) 5:e1196308. doi: 10.1080/2162402X.2016.1196308
32. Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Sanchez-Correa B, Guerrero B, et al. Characterization of the DNAM-1, TIGIT and TACTILE axis on circulating NK, NKT-like and T cell subsets in patients with acute myeloid leukemia. Cancers (Basel). (2020) 12(8):2171. doi: 10.3390/cancers12082171
33. Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. (2011) 118:5084–95. doi: 10.1182/blood-2011-07-365817
34. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. (2010) 116:2484–93. doi: 10.1182/blood-2010-03-275446
35. Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. (2003) 170:2516–22. doi: 10.4049/jimmunol.170.5.2516
36. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
37. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. (2003) 170:1240–8. doi: 10.4049/jimmunol.170.3.1240
38. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. (2009) 15:3325–32. doi: 10.1158/1078-0432.CCR-08-3010
39. Delia M, Carluccio P, Mestice A, Brunetti C, Albano F, Specchia G. Impact of bone marrow aspirate tregs on the response rate of younger newly diagnosed acute myeloid leukemia patients. J Immunol Res. (2018) 2018:9325261. doi: 10.1155/2018/9325261
40. Linehan DC, Goedegebuure PS. CD25+ CD4+ regulatory T-cells in cancer. Immunol Res. (2005) 32:155–68. doi: 10.1385/IR:32:1-3:155
41. Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ, et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol. (2006) 13:1252–8. doi: 10.1245/s10434-006-9015-y
42. Curti A, Trabanelli S, Salvestrini V, Baccarani M, Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood. (2009) 113:2394–401. doi: 10.1182/blood-2008-07-144485
43. Sun H, Li Y, Zhang ZF, Ju Y, Li L, Zhang BC, et al. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int J Hematol. (2015) 102:579–86. doi: 10.1007/s12185-015-1865-2
44. Wang L, Jia B, Claxton DF, Ehmann WC, Rybka WB, Mineishi S, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. (2018) 7:e1469594. doi: 10.1080/2162402X.2018.1469594
45. Lu T, Gabrilovich DI. Molecular pathways: tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin Cancer Res. (2012) 18:4877–82. doi: 10.1158/1078-0432.CCR-11-2939
46. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791–801. doi: 10.1182/blood-2016-07-730614
47. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. (2001) 166:5398–406. doi: 10.4049/jimmunol.166.9.5398
48. Solito S, Bronte V, Mandruzzato S. Antigen specificity of immune suppression by myeloid-derived suppressor cells. J Leukoc Biol. (2011) 90:31–6. doi: 10.1189/jlb.0111021
49. Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. (2022) 21:184. doi: 10.1186/s12943-022-01657-y
50. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
51. Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, et al. Targeting MDSC for immune-checkpoint blockade in cancer immunotherapy: current progress and new prospects. Clin Med Insights Oncol. (2021) 15:11795549211035540. doi: 10.1177/11795549211035540
52. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. (2002) 168:689–95. doi: 10.4049/jimmunol.168.2.689
53. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. (2009) 124:2621–33. doi: 10.1002/ijc.24249
54. Joshi S, Sharabi A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther. (2022) 235:108114. doi: 10.1016/j.pharmthera.2022.108114
55. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
56. Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C. Tumor-associated macrophages in cancers. Clin Transl Oncol. (2016) 18:251–8. doi: 10.1007/s12094-015-1373-0
57. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. (2021) 6:75. doi: 10.1038/s41392-021-00484-9
58. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
59. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. (2019) 18:177. doi: 10.1186/s12943-019-1102-3
60. Wang L, Wang FS. Clinical immunology and immunotherapy for hepatocellular carcinoma: current progress and challenges. Hepatol Int. (2019) 13:521–33. doi: 10.1007/s12072-019-09967-y
61. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. (2020) 182:886–900.e17. doi: 10.1016/j.cell.2020.07.013
62. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. (2016) 16:447–62. doi: 10.1038/nrc.2016.54
63. Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. (2010) 101:1913–9. doi: 10.1111/j.1349-7006.2010.01614.x
64. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
65. Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. (2017) 67:770–9. doi: 10.1016/j.jhep.2017.05.022
66. Lievense LA, Cornelissen R, Bezemer K, Kaijen-Lambers ME, Hegmans JP, Aerts JG. Pleural effusion of patients with Malignant mesothelioma induces macrophage-mediated T cell suppression. J Thorac Oncol. (2016) 11:1755–64. doi: 10.1016/j.jtho.2016.06.021
67. Han Y, Dong Y, Yang Q, Xu W, Jiang S, Yu Z, et al. Acute myeloid leukemia cells express ICOS ligand to promote the expansion of regulatory T cells. Front Immunol. (2018) 9:2227. doi: 10.3389/fimmu.2018.02227
68. Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. (2013) 36:382–9. doi: 10.1097/CJI.0b013e31829fb7a2
69. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
70. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. (2009) 206:1327–37. doi: 10.1084/jem.20082173
71. Ou Y, Yang Y, Li X, Zhang X, Zhao L, Yang C, et al. Arginine metabolism key enzymes affect the prognosis of myelodysplastic syndrome by interfering with macrophage polarization. Cancer Med. (2023) 12:16444–54. doi: 10.1002/cam4.6287
72. Sun S, Zou H, Li L, Liu Q, Ding N, Zeng L, et al. CD123/CD33 dual-antibody modified liposomes effectively target acute myeloid leukemia cells and reduce antigen-negative escape. Int J Pharm. (2019) :568:118518. doi: 10.1016/j.ijpharm.2019.118518
73. Mahalleh M, Shabani M, Rayzan E, Rezaei N. Reinforcing the primary immunotherapy modulators against acute leukemia; monoclonal antibodies in AML. Immunotherapy. (2019) 11:1583–600. doi: 10.2217/imt-2019-0043
74. Gauthier L, Virone-Oddos A, Beninga J, Rossi B, Nicolazzi C, Amara C, et al. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nat Biotechnol. (2023) 41:1296–306. doi: 10.1038/s41587-022-01626-2
75. Walter RB. Biting back: BiTE antibodies as a promising therapy for acute myeloid leukemia. Expert Rev Hematol. (2014) 7:317–9. doi: 10.1586/17474086.2014.896190
76. Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, et al. and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. (2013) 27:1107–15. doi: 10.1038/leu.2012.341
77. Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther. (2014) 13:1549–57. doi: 10.1158/1535-7163.MCT-13-0956
78. Hernandez-Caselles T, Martinez-Esparza M, Perez-Oliva AB, Quintanilla-Cecconi AM, Garcia-Alonso A, Alvarez-Lopez DM, et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. J Leukoc Biol. (2006) 79:46–58. doi: 10.1189/jlb.0205096
79. Stein C, Kellner C, Kugler M, Reiff N, Mentz K, Schwenkert M, et al. Novel conjugates of single-chain Fv antibody fragments specific for stem cell antigen CD123 mediate potent death of acute myeloid leukaemia cells. Br J Haematol. (2010) 148:879–89. doi: 10.1111/j.1365-2141.2009.08033.x
80. Kugler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. (2010) 150:574–86. doi: 10.1111/j.1365-2141.2010.08300.x
81. Singer H, Kellner C, Lanig H, Aigner M, Stockmeyer B, Oduncu F, et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J Immunother. (2010) 33:599–608. doi: 10.1097/CJI.0b013e3181dda225
82. Piedfer M, Dauzonne D, Tang R, N’Guyen J, Billard C, Bauvois B. Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. FASEB J. (2011) 25:2831–42. doi: 10.1096/fj.11-181396
83. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. (2009) 5:31–42. doi: 10.1016/j.stem.2009.04.018
84. Vasu S, He S, Cheney C, Gopalakrishnan B, Mani R, Lozanski G, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood. (2016) 127:2879–89. doi: 10.1182/blood-2015-11-680546
85. Kauder SE, Kuo TC, Harrabi O, Chen A, Sangalang E, Doyle L, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PloS One. (2018) 13:e0201832. doi: 10.1371/journal.pone.0201832
86. Gallazzi M, Ucciero MAM, Faraci DG, Mahmoud AM, Al Essa W, Gaidano G, et al. New frontiers in monoclonal antibodies for the targeted therapy of acute myeloid leukemia and myelodysplastic syndromes. Int J Mol Sci. (2022) 23(14):7542. doi: 10.3390/ijms23147542
87. Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J. (2015) 5:e340. doi: 10.1038/bcj.2015.68
88. Jurcic JG. Targeted alpha-particle therapy for hematologic Malignancies. J Med Imaging Radiat Sci. (2019) 50:S53–7. doi: 10.1016/j.jmir.2019.05.008
89. Miller ML, Fishkin NE, Li W, Whiteman KR, Kovtun Y, Reid EE, et al. A new class of antibody-drug conjugates with potent DNA alkylating activity. Mol Cancer Ther. (2016) 15:1870–8. doi: 10.1158/1535-7163.MCT-16-0184
90. Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. (2019) 394:793–804. doi: 10.1016/S0140-6736(19)31774-X
91. Angelova E, Audette C, Kovtun Y, Daver N, Wang SA, Pierce S, et al. CD123 expression patterns and selective targeting with a CD123-targeted antibody-drug conjugate (IMGN632) in acute lymphoblastic leukemia. Haematologica. (2019) 104:749–55. doi: 10.3324/haematol.2018.205252
92. Kovtun Y, Jones GE, Adams S, Harvey L, Audette CA, Wilhelm A, et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. (2018) 2:848–58. doi: 10.1182/bloodadvances.2018017517
93. Anami Y, Deng M, Gui X, Yamaguchi A, Yamazaki CM, Zhang N, et al. LILRB4-targeting antibody-drug conjugates for the treatment of acute myeloid leukemia. Mol Cancer Ther. (2020) 19:2330–9. doi: 10.1158/1535-7163.MCT-20-0407
94. Li F, Sutherland MK, Yu C, Walter RB, Westendorf L, Valliere-Douglass J, et al. Characterization of SGN-CD123A, A potent CD123-directed antibody-drug conjugate for acute myeloid leukemia. Mol Cancer Ther. (2018) 17:554–64. doi: 10.1158/1535-7163.MCT-17-0742
95. Liao D, Wang M, Liao Y, Li J, Niu T. A review of efficacy and safety of checkpoint inhibitor for the treatment of acute myeloid leukemia. Front Pharmacol. (2019) 10:609. doi: 10.3389/fphar.2019.00609
96. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) :62:29–39. doi: 10.1016/j.intimp.2018.06.001
97. Saxena K, Herbrich SM, Pemmaraju N, Kadia TM, DiNardo CD, Borthakur G, et al. A phase 1b/2 study of azacitidine with PD-L1 antibody avelumab in relapsed/refractory acute myeloid leukemia. Cancer. (2021) 127:3761–71. doi: 10.1002/cncr.33690
98. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discovery. (2019) 9:370–83. doi: 10.1158/2159-8290.CD-18-0774
99. Ravandi F, Assi R, Daver N, Benton CB, Kadia T, Thompson PA, et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a single-arm, phase 2 study. Lancet Haematol. (2019) 6:e480–8. doi: 10.1016/S2352-3026(19)30114-0
100. Guan X, Hu R, Choi Y, Srivats S, Nabet BY, Silva J, et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and T(reg) cells. Nature. (2024) 627:646–55. doi: 10.1038/s41586-024-07121-9
101. Kim N, Kim HS. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front Immunol. (2018) 9:2041. doi: 10.3389/fimmu.2018.02041
102. Zhong RK, Loken M, Lane TA, Ball ED. CTLA-4 blockade by a human MAb enhances the capacity of AML-derived DC to induce T-cell responses against AML cells in an autologous culture system. Cytotherapy. (2006) 8:3–12. doi: 10.1080/14653240500499507
103. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Leukemia, lymphoma society blood cancer research P. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. (2016) 375:143–53. doi: 10.1056/NEJMoa1601202
104. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
105. Liu H. Emerging agents and regimens for AML. J Hematol Oncol. (2021) 14:49. doi: 10.1186/s13045-021-01062-w
106. Tabata R, Chi S, Yuda J, Minami Y. Emerging immunotherapy for acute myeloid leukemia. Int J Mol Sci. (2021) 22(4):1944. doi: 10.3390/ijms22041944
107. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
108. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013
109. Perales MA, Kebriaei P, Kean LS, Sadelain M. Reprint of: building a safer and faster CAR: seatbelts, airbags, and CRISPR. Biol Blood Marrow Transplant. (2018) 24:S15–9. doi: 10.1016/j.bbmt.2017.12.789
110. Li S, Tao Z, Xu Y, Liu J, An N, Wang Y, et al. CD33-specific chimeric antigen receptor T cells with different co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum Gene Ther. (2018) 29:626–39. doi: 10.1089/hum.2017.241
111. Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol. (2012) 2012:683065. doi: 10.1155/2012/683065
112. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
113. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. (2003) 9:279–86. doi: 10.1038/nm827
114. Pecher AC, Hensen L, Klein R, Schairer R, Lutz K, Atar D, et al. CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome. JAMA. (2023) 329:2154–62. doi: 10.1001/jama.2023.8753
115. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
116. Liu S, Deng B, Yin Z, Lin Y, An L, Liu D, et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. (2021) 96:671–9. doi: 10.1002/ajh.26160
117. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
118. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
119. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
120. He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. (2020) 135:713–23. doi: 10.1182/blood.2019002779
121. Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. (2013) 122:3138–48. doi: 10.1182/blood-2012-12-474056
122. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. (2014) 123:2343–54. doi: 10.1182/blood-2013-09-529537
123. Haubner S, Mansilla-Soto J, Nataraj S, Kogel F, Chang Q, de StanChina E, et al. Cooperative CAR targeting to selectively eliminate AML and minimize escape. Cancer Cell. (2023) 41:1871–1891.e6. doi: 10.1016/j.ccell.2023.09.010
124. Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid Malignancies. Proc Natl Acad Sci U S A. (2019) 116:11978–87. doi: 10.1073/pnas.1819992116
125. Liu Y, Wang S, Schubert ML, Lauk A, Yao H, Blank MF, et al. CD33-directed immunotherapy with third-generation chimeric antigen receptor T cells and gemtuzumab ozogamicin in intact and CD33-edited acute myeloid leukemia and hematopoietic stem and progenitor cells. Int J Cancer. (2022) 150:1141–55. doi: 10.1002/ijc.33865
126. Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. (2015) 125:3466–76. doi: 10.1182/blood-2014-11-612721
127. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. (2018) 32:1168–79. doi: 10.1038/s41375-018-0009-0
128. Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. (2006) 66:5927–33. doi: 10.1158/0008-5472.CAN-06-0130
129. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. (2021) 138:318–30. doi: 10.1182/blood.2020008221
130. Kamezaki K, Luchsinger LL, Snoeck HW. Differential requirement for wild-type Flt3 in leukemia initiation among mouse models of human leukemia. Exp Hematol. (2014) 42:192–203.e1. doi: 10.1016/j.exphem.2013.11.008
131. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
132. Beyar-Katz O, Gill S. Novel approaches to acute myeloid leukemia immunotherapy. Clin Cancer Res. (2018) 24:5502–15. doi: 10.1158/1078-0432.CCR-17-3016
133. Wu Y, Li Y, Gao Y, Zhang P, Jing Q, Zhang Y, et al. Immunotherapies of acute myeloid leukemia: Rationale, clinical evidence and perspective. BioMed Pharmacother. (2024) 171:116132. doi: 10.1016/j.biopha.2024.116132
134. Barbullushi K, Rampi N, Serpenti F, Sciume M, Fabris S, De Roberto P, et al. Vaccination therapy for acute myeloid leukemia: where do we stand? Cancers (Basel). (2022) 14(12):2994. doi: 10.3390/cancers14122994
135. Sugiyama H. Wilms’ tumor gene WT1: its oncogenic function and clinical application. Int J Hematol. (2001) 73:177–87. doi: 10.1007/BF02981935
136. Oka Y, Tsuboi A, Elisseeva OA, Nakajima H, Fujiki F, Kawakami M, et al. WT1 peptide cancer vaccine for patients with hematopoietic Malignancies and solid cancers. ScientificWorldJournal. (2007) 7:649–65. doi: 10.1100/tsw.2007.119
137. Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, et al. CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood. (2002) 100:2132–7. doi: 10.1182/blood-2002-01-0163
138. Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. (2018) 2:224–34. doi: 10.1182/bloodadvances.2017014175
139. Naoe T, Saito A, Hosono N, Kasahara S, Muto H, Hatano K, et al. Immunoreactivity to WT1 peptide vaccine is associated with prognosis in elderly patients with acute myeloid leukemia: follow-up study of randomized phase II trial of OCV-501, an HLA class II-binding WT1 polypeptide. Cancer Immunol Immunother. (2023) 72:2865–71. doi: 10.1007/s00262-023-03432-4
140. Greiner J, Schmitt A, Giannopoulos K, Rojewski MT, Gotz M, Funk I, et al. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica. (2010) 95:1191–7. doi: 10.3324/haematol.2009.014704
141. Kuball J, de Boer K, Wagner E, Wattad M, Antunes E, Weeratna RD, et al. Pitfalls of vaccinations with WT1-, Proteinase3- and MUC1-derived peptides in combination with MontanideISA51 and CpG7909. Cancer Immunol Immunother. (2011) 60:161–71. doi: 10.1007/s00262-010-0929-7
142. Houtenbos I, Westers TM, Ossenkoppele GJ, van de Loosdrecht AA. Leukemia-derived dendritic cells: towards clinical vaccination protocols in acute myeloid leukemia. Haematologica. (2006) 91:348–55.
143. Zhou LJ, Tedder TF. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci U S A. (1996) 93:2588–92. doi: 10.1073/pnas.93.6.2588
144. Yu J, Sun H, Cao W, Song Y, Jiang Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp Hematol Oncol. (2022) 11:3. doi: 10.1186/s40164-022-00257-2
145. Chevallier P, Saiagh S, Dehame V, Guillaume T, Peterlin P, Bercegeay S, et al. A phase I/II feasibility vaccine study by autologous leukemic apoptotic corpse-pulsed dendritic cells for elderly AML patients. Hum Vaccin Immunother. (2021) 17:3511–4. doi: 10.1080/21645515.2021.1943991
146. Stroopinsky D, Liegel J, Bhasin M, Cheloni G, Thomas B, Bhasin S, et al. Leukemia vaccine overcomes limitations of checkpoint blockade by evoking clonal T cell responses in a murine acute myeloid leukemia model. Haematologica. (2021) 106:1330–42. doi: 10.3324/haematol.2020.259457
147. Anguille S, Van de Velde AL, Smits EL, Van Tendeloo VF, Juliusson G, Cools N, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. (2017) 130:1713–21. doi: 10.1182/blood-2017-04-780155
148. van de Loosdrecht AA, van Wetering S, Santegoets S, Singh SK, Eeltink CM, den Hartog Y, et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol Immunother. (2018) 67:1505–18. doi: 10.1007/s00262-018-2198-9
149. Khoury HJ, Collins RH Jr., Blum W, Stiff PS, Elias L, Lebkowski JS, et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer. (2017) 123:3061–72. doi: 10.1002/cncr.30696
150. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. (1998) 4:594–600. doi: 10.1038/nm0598-594
151. Llorente A, Skotland T, Sylvanne T, Kauhanen D, Rog T, Orlowski A, et al. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim Biophys Acta. (2013) 1831:1302–9. doi: 10.1016/j.bbalip.2013.04.011
152. Patel G, Agnihotri TG, Gitte M, Shinde T, Gomte SS, Goswami R, et al. Exosomes: a potential diagnostic and treatment modality in the quest for counteracting cancer. Cell Oncol (Dordr). (2023) 46:1159–79. doi: 10.1007/s13402-023-00810-z
153. Wang L, Xie Y, Ahmed KA, Ahmed S, Sami A, Chibbar R, et al. Exosomal pMHC-I complex targets T cell-based vaccine to directly stimulate CTL responses leading to antitumor immunity in transgenic FVBneuN and HLA-A2/HER2 mice and eradicating trastuzumab-resistant tumor in athymic nude mice. Breast Cancer Res Treat. (2013) 140:273–84. doi: 10.1007/s10549-013-2626-7
154. Xie Y, Wu J, Xu A, Ahmeqd S, Sami A, Chibbar R, et al. Heterologous human/rat HER2-specific exosome-targeted T cell vaccine stimulates potent humoral and CTL responses leading to enhanced circumvention of HER2 tolerance in double transgenic HLA-A2/HER2 mice. Vaccine. (2018) 36:1414–22. doi: 10.1016/j.vaccine.2018.01.078
155. Reza A, Choi YJ, Yasuda H, Kim JH. Human adipose mesenchymal stem cell-derived exosomal-miRNAs are critical factors for inducing anti-proliferation signalling to A2780 and SKOV-3 ovarian cancer cells. Sci Rep. (2016) 6:38498. doi: 10.1038/srep38498
156. Massumoto C, Sousa-Canavez JM, Leite KR, Camara-Lopes LH. Stabilization of acute myeloid leukemia with a dendritic cell vaccine. Hematol Oncol Stem Cell Ther. (2008) 1:239–40. doi: 10.1016/s1658-3876(08)50011-2
157. Li L, Giannopoulos K, Reinhardt P, Tabarkiewicz J, Schmitt A, Greiner J, et al. Immunotherapy for patients with acute myeloid leukemia using autologous dendritic cells generated from leukemic blasts. Int J Oncol. (2006) 28:855–61. doi: 10.3892/ijo.28.4.855
158. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia. (2018) 32:575–87. doi: 10.1038/leu.2017.259
159. Hong CS, Sharma P, Yerneni SS, Simms P, Jackson EK, Whiteside TL, et al. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci Rep. (2017) 7:14684. doi: 10.1038/s41598-017-14661-w
160. Bhatia SN, Chen X, Dobrovolskaia MA, Lammers T. Cancer nanomedicine. Nat Rev Cancer. (2022) 22:550–6. doi: 10.1038/s41568-022-00496-9
161. Kramer S, Langhanki J, Krumb M, Opatz T, Bros M, Zentel R. HPMA-based nanocarriers for effective immune system stimulation. Macromol Biosci. (2019) 19:e1800481. doi: 10.1002/mabi.201800481
162. Mu W, Chu Q, Liu Y, Zhang N. A review on nano-based drug delivery system for cancer chemoimmunotherapy. Nanomicro Lett. (2020) 12:142. doi: 10.1007/s40820-020-00482-6
163. Havel HA. Where are the nanodrugs? An industry perspective on development of drug products containing nanomaterials. AAPS J. (2016) 18:1351–3. doi: 10.1208/s12248-016-9970-6
164. Havel H, Finch G, Strode P, Wolfgang M, Zale S, Bobe I, et al. Nanomedicines: from bench to bedside and beyond. AAPS J. (2016) 18:1373–8. doi: 10.1208/s12248-016-9961-7
166. Sainz V, Conniot J, Matos AI, Peres C, Zupancic E, Moura L, et al. Regulatory aspects on nanomedicines. Biochem Biophys Res Commun. (2015) 468:504–10. doi: 10.1016/j.bbrc.2015.08.023
167. Ventola CL. Cancer immunotherapy, part 1: current strategies and agents. P T. (2017) 42:375–83.
168. Li W, Wei H, Li H, Gao J, Feng SS, Guo Y. Cancer nanoimmunotherapy using advanced pharmaceutical nanotechnology. Nanomedicine (Lond). (2014) 9:2587–605. doi: 10.2217/nnm.14.127
169. Wu X, Xin Y, Zhang H, Quan L, Ao Q. Biopolymer-based nanomedicine for cancer therapy: opportunities and challenges. Int J Nanomedicine. (2024) 19:7415–71. doi: 10.2147/IJN.S460047
170. Khan H, Mirzaei HR, Amiri A, Kupeli Akkol E, Ashhad Halimi SM, Mirzaei H. Glyco-nanoparticles: New drug delivery systems in cancer therapy. Semin Cancer Biol. (2021) 69:24–42. doi: 10.1016/j.semcancer.2019.12.004
171. Chan MS, Liu LS, Leung HM, Lo PK. Cancer-cell-specific mitochondria-targeted drug delivery by dual-ligand-functionalized nanodiamonds circumvent drug resistance. ACS Appl Mater Interfaces. (2017) 9:11780–9. doi: 10.1021/acsami.6b15954
172. Song Q, Yin Y, Shang L, Wu T, Zhang D, Kong M, et al. Tumor microenvironment responsive nanogel for the combinatorial antitumor effect of chemotherapy and immunotherapy. Nano Lett. (2017) 17:6366–75. doi: 10.1021/acs.nanolett.7b03186
173. Duan X, Chan C, Han W, Guo N, Weichselbaum RR, Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat Commun. (2019) 10:1899. doi: 10.1038/s41467-019-09221-x
174. Zhang Y, Wu L, Li Z, Zhang W, Luo F, Chu Y, et al. Glycocalyx-mimicking nanoparticles improve anti-PD-L1 cancer immunotherapy through reversion of tumor-associated macrophages. Biomacromolecules. (2018) 19:2098–108. doi: 10.1021/acs.biomac.8b00305
175. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJ, et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthc Mater. (2015) 4:1180–9. doi: 10.1002/adhm.201500089
176. Huayamares SG, Lokugamage MP, Rab R, Da Silva Sanchez AJ, Kim H, Radmand A, et al. High-throughput screens identify a lipid nanoparticle that preferentially delivers mRNA to human tumors in vivo. J Control Release. (2023) 357:394–403. doi: 10.1016/j.jconrel.2023.04.005
177. Kheirolomoom A, Kare AJ, Ingham ES, Paulmurugan R, Robinson ER, Baikoghli M, et al. In situ T-cell transfection by anti-CD3-conjugated lipid nanoparticles leads to T-cell activation, migration, and phenotypic shift. Biomaterials. (2022) 281:121339. doi: 10.1016/j.biomaterials.2021.121339
178. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. (2023) 618:144–50. doi: 10.1038/s41586-023-06063-y
179. Ma W, Zhu D, Li J, Chen X, Xie W, Jiang X, et al. Coating biomimetic nanoparticles with chimeric antigen receptor T cell-membrane provides high specificity for hepatocellular carcinoma photothermal therapy treatment. Theranostics. (2020) 10:1281–95. doi: 10.7150/thno.40291
180. Deng R, Shen N, Yang Y, Yu H, Xu S, Yang YW, et al. Targeting epigenetic pathway with gold nanoparticles for acute myeloid leukemia therapy. Biomaterials. (2018) 167:80–90. doi: 10.1016/j.biomaterials.2018.03.013
181. Lim WS, Tardi PG, Dos Santos N, Xie X, Fan M, Liboiron BD, et al. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine:daunorubicin formulation, in bone marrow xenografts. Leuk Res. (2010) 34:1214–23. doi: 10.1016/j.leukres.2010.01.015
182. Feldman EJ, Kolitz JE, Trang JM, Liboiron BD, Swenson CE, Chiarella MT, et al. Pharmacokinetics of CPX-351; a nano-scale liposomal fixed molar ratio formulation of cytarabine:daunorubicin, in patients with advanced leukemia. Leuk Res. (2012) 36:1283–9. doi: 10.1016/j.leukres.2012.07.006
183. Lemoli RM, Montesinos P, Jain A. Real-world experience with CPX-351 in high-risk acute myeloid leukemia. Crit Rev Oncol Hematol. (2023) 185:103984. doi: 10.1016/j.critrevonc.2023.103984
184. Liang B, Li N, Zhang S, Qi A, Feng J, Jing W, et al. Idarubicin-loaded methoxy poly(ethylene glycol)-b-poly(l-lactide-co-glycolide) nanoparticles for enhancing cellular uptake and promoting antileukemia activity. Int J Nanomedicine. (2019) 14:543–56. doi: 10.2147/IJN.S190027
185. Darwish NHE, Sudha T, Godugu K, Bharali DJ, Elbaz O, El-Ghaffar HAA, et al. Novel targeted nano-parthenolide molecule against NF-kB in acute myeloid leukemia. Molecules. (2019) 24(11):2103. doi: 10.3390/molecules24112103
186. Palombarini F, Masciarelli S, Incocciati A, Liccardo F, Di Fabio E, Iazzetti A, et al. Self-assembling ferritin-dendrimer nanoparticles for targeted delivery of nucleic acids to myeloid leukemia cells. J Nanobiotechnology. (2021) 19:172. doi: 10.1186/s12951-021-00921-5
Keywords: acute myeloid leukemia, antibody therapy, CAR-T cell therapy, immune checkpoint inhibitors, nanoimmunotherapy
Citation: Wang Y, Yang X, Liu Y and Li Y (2025) A review of common immunotherapy and nano immunotherapy for acute myeloid leukemia. Front. Immunol. 16:1505247. doi: 10.3389/fimmu.2025.1505247
Received: 02 October 2024; Accepted: 24 February 2025;
Published: 10 March 2025.
Edited by:
Haluk Barbaros Oral, Bursa Uludağ University, TürkiyeReviewed by:
Adolfo Martinez, General Hospital of Mexico, MexicoIda Franiak-Pietryga, Poseida Therapeutic, United States
Copyright © 2025 Wang, Yang, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youjie Li, eW91amllMTk3OUAxNjMuY29t
†These authors have contributed equally to this work