 Lele Shen
Lele Shen Yao Zhou
Yao Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 March 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1503985
This article is part of the Research TopicAdvances in skin immunologyView all 6 articles
Hypertrophic Scar (HS) is a common fibrotic disease of the skin, usually caused by injury to the deep dermis due to trauma, burns, or surgical injury. The main feature of HS is the thickening and hardening of the skin, often accompanied by itching and pain, which seriously affects the patient’s quality of life. Macrophages are involved in all stages of HS genesis through phenotypic changes. M1-type macrophages primarily function in the early inflammatory phase by secreting pro-inflammatory factors, while M2-type macrophages actively contribute to tissue repair and fibrosis. Despite advances in understanding HS pathogenesis, the precise mechanisms linking macrophage phenotypic changes to fibrosis remain incompletely elucidated. This review addresses these gaps by discussing the pathological mechanisms of HS formation, the phenotypic changes of macrophages at different stages of HS formation, and the pathways through which macrophages influence HS progression. Furthermore, emerging technologies for HS treatment and novel therapeutic strategies targeting macrophages are highlighted, offering potential avenues for improved prevention and treatment of HS.
Hypertrophic scar (HS) is a fibrotic disease of the skin, usually caused by abnormal tissue repair after burns, trauma, or surgery (1). The clinical manifestations of HS are bright red skin surface, protruding from the surrounding normal skin tissues, and localized thickening. Itching, localized numbness, and sensory abnormalities produced by HS have a severe impact on the quality of life and mental health of patients (1, 2). HS formation is a complex and challenging clinical problem that affects about 100 million patients in developed countries alone. The incidence of post-burn HS has been reported to range from 32% to 72% (3, 4). Although many studies have been devoted to exploring the mechanisms of HS, its exact pathophysiologic processes are still not fully characterized.
The formation of HS is a complex and multistage process that usually includes hemostasis, inflammation, proliferation, and remodeling phases. Macrophages are an essential component of the innate immune system. They play a crucial role in tissue repair and scar formation. Macrophages exhibit different phenotypes and functions at different stages of HS formation. M1 macrophages, also known as classically activated macrophages, primarily mediate pro-inflammatory and antimicrobial responses, producing cytokines such as TNF-α, IL-6, and IL-1β, which amplify inflammation and recruit additional immune cells (5, 6). As the inflammatory response subsides, macrophages transition to the M2 phenotype, which promotes tissue repair, extracellular matrix remodeling, and fibrosis through the secretion of anti-inflammatory cytokines and growth factors (7, 8). However, macrophage-targeted therapies for HS face significant challenges, including the complex plasticity of macrophage phenotypes, their dynamic changes during the different stages of HS formation, and the need for specific delivery systems to target macrophages without affecting surrounding tissues. Precision therapy targeting macrophages has made significant progress in tumors and rheumatic diseases (9, 10). However, the exact mechanism of macrophage action in HS remains under-revealed.
In this review, we summarize the pathological process of HS formation and the phenotypic changes that occur in macrophages at various stages of HS formation. In addition, we explore macrophage-influenced pathways in HS. Finally, we summarize the therapeutic strategies for HS, including emerging technologies and macrophage-targeted treatment approaches, and discuss the specific challenges associated with these strategies. This review systematically integrates research findings spanning from molecular mechanisms to therapeutic strategies, based on comprehensive searches of databases such as PubMed and Web of Science, to identify critical gaps and highlight potential advancements in the treatment of HS. We hope that it will help develop drugs of potential treatment value for HS and provide theoretical support for developing more effective therapeutic strategies.
This review is based on a systematic literature search conducted in the PubMed and Web of Science databases using keywords such as “hypertrophic scar,” “macrophages,” “fibrosis,” and “therapeutic strategies,” combined with Boolean operators (AND/OR). The search was performed on August 24, 2024. The inclusion criteria were as follows: (1) studies focusing on the role of macrophages in the mechanisms of HS and therapeutic strategies for HS; (2) original research, including in vivo, in vitro, and clinical studies; and (3) studies published in English to ensure accessibility and practical usability. The exclusion criteria were as follows: (1) studies unrelated to HS; (2) conference abstracts, pathological reports, or review articles; and (3) articles with insufficient data quality or poor study design. The screening process consisted of two steps: title and abstract screening followed by full-text evaluation. These steps were implemented to ensure the relevance, reliability, and quality of the included studies.
Routine wound healing consists of four phases: hemostasis, inflammation, proliferation, and remodeling, each of which overlaps and differs in time and space (1).The formation of HS is the result of an abnormally active and dysregulated event during the wound healing phase, leading to an abnormal accumulation of extracellular matrix (ECM). Next, we will explore the characteristics of HS at different stages of formation.
Hemostasis is the first step in wound repair (11). Damaged endothelial cells release substances such as endothelin to constrict vascular smooth muscle and reduce bleeding (12, 13). Generally, forming a blood clot involves primary and secondary hemostatic processes. During initial hemostasis, platelet receptors interact with ECM proteins, such as fibronectin and collagen, to form platelet plugs that promote adhesion to the vessel wall (14). Secondary hemostasis depends on a coagulation factor cascade reaction that activates thrombin (15). Thrombin induces the conversion of fibrinogen to fibrin, which results in the formation of a stable fibrin network. This fibrin network and fibronectin, vitronectin, and thrombospondin form an insoluble clot (16).
During hemostasis, platelet activation and release of growth factors, the intensity and duration of the inflammatory response, and deposited fibrin are closely related to the formation of HS (17). The initial hemostatic event occurs when platelets produce pro-fibrotic growth factors such as PDGF, VEGF, TGF-β1, and CTGF. Moderate release of these factors promotes hemostasis (11). Overproduction of pro-fibrotic factors can lead to excessive cell proliferation and fibrous tissue production. Previous studies have found that the use of platelet-rich plasma (PRP) for treating various types of scars is increasing. PRP attenuates the fibrotic process by decreasing the levels of pro-fibrotic markers Transforming Growth Factor Beta 1 (TGF-β1), smooth muscle actin α (α-SMA), Collagen Type I (COL-I), and Matrix Metalloproteinase-9 (MMP-9) (18, 19). In addition, these pro-fibrotic molecules may induce excessive inflammation. This may lead to the overproduction of ECM, abnormal fibroblast differentiation, and inappropriate matrix remodeling, which may promote the formation of HS (1). Overall, the release of abnormal pro-fibrotic molecules during the hemostatic phase, and the sustained inflammatory response lead to excessive fibrin network formation in HS (Figure 1A), which ultimately promotes HS (20).
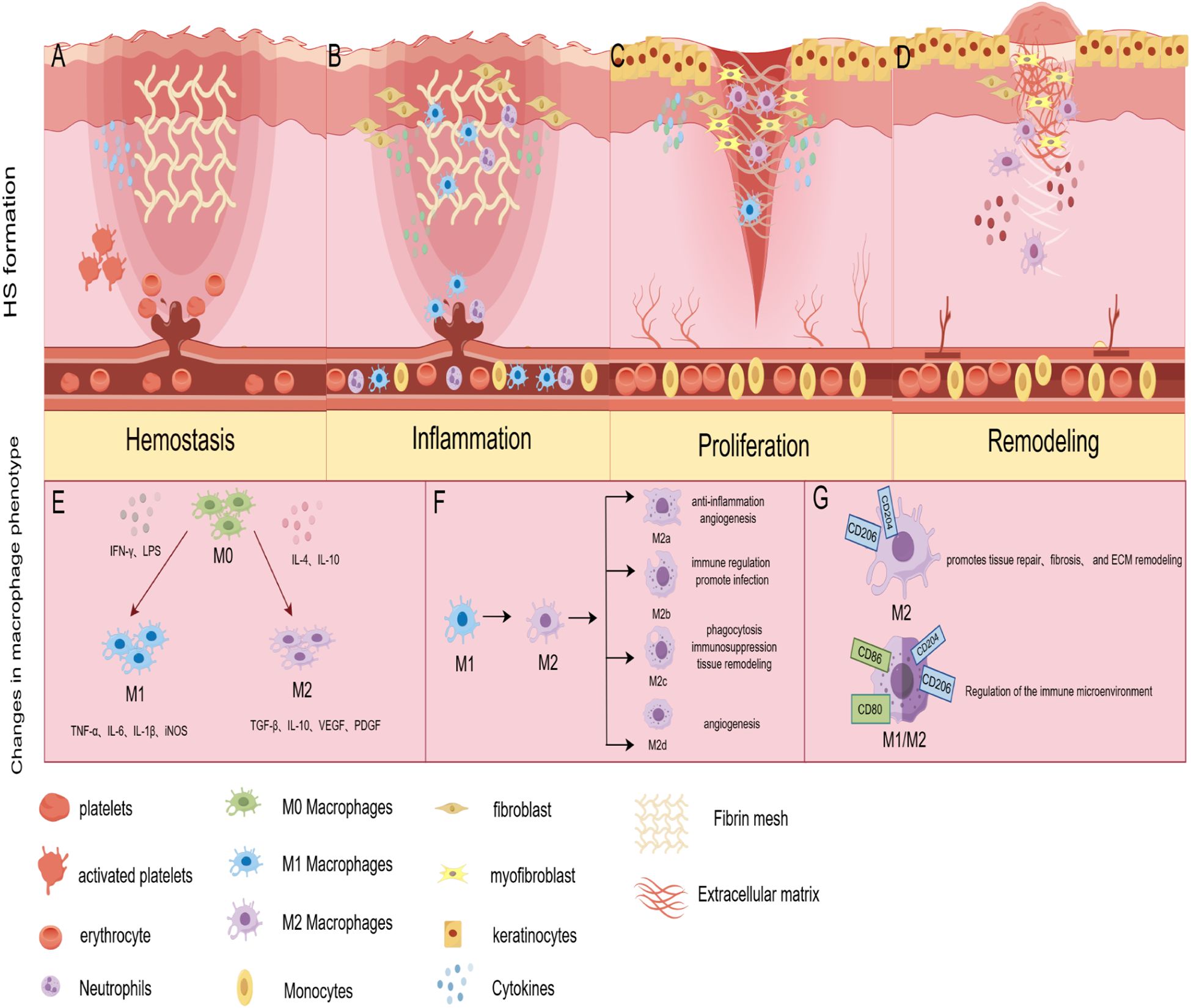
Figure 1. Pathological mechanisms of HS formation and phenotypic changes in macrophages during the HS formation phase. (A) Hemostasis: aberrant release of pro-fibrotic molecules leads to a sustained inflammatory response and excessive fibrin network formation. (B) Inflammation: high levels of inflammatory infiltrate leads to fibroblast activation. (C) Proliferation: overactive keratinocytes, dermal fibroblasts, and macrophages promote ECM deposition and excessive angiogenesis. (D) Remodeling: fibroblasts remodel the deposited ECM, myofibroblasts cause overall wound contraction; MMPs regulate fibroblast proliferation and are involved in ECM degradation. fibroblasts cause overall wound contraction; MMPs regulate fibroblast proliferation and participate in ECM degradation. (E) During HS formation, M0 macrophages respond to an initial stimulus to polarize to M1 and M2 phenotypes, with the M1 phenotype playing a significant role in the early stages. (F) Macrophages undergo an M1 to M2 phenotypic transition, or conversion of the M2 subtype, in the intermediate stages of HS formation; (G) Macrophages are dominated by the M2 phenotype in the later stages of injury and may develop a mixed M1/M2 phenotype. (By Figdraw).
HS is formed due to skin damage in the reticular dermis (21). The inflammatory response in the reticular dermis begins immediately at the time of injury and varies in duration depending on the degree of injury (12). The study suggests excessive inflammation is the pathologic basis for HS formation (22). Increased expression of interleukins, interferons, and growth factors released by immune cells, such as neutrophils and macrophages, can activate fibroblasts (Figure 1B), which are involved in the formation of HS (23, 24).
Previous studies have shown the most apparent effect of IL-6 in promoting human HS formation. IL-6 levels were significantly elevated in burned HS fibroblasts (25). IL-6 enhances VEGF expression in macrophages, keratinocytes, and fibroblasts, ultimately leading to scarring (26). In addition to IL-6, inflammatory factors such as IL-1β, IL-4, IL-17, and IL-13 were highly expressed in HS (27, 28). Interestingly, another point was made that pro-inflammatory cytokines contribute to wound healing. Inadequate pro-inflammatory responses slow the wound-healing process (29, 30). This delay in the inflammatory response during the acute phase of early healing and its role in HS formation deserves further investigation.
In contrast to IL-6, the expression of IL-10, IL-18, and IL-37 was lower in HS. IL-10 was found to regulate the TLR4/NF-κB pathway in dermal fibroblasts via the IL-10R/STAT3 axis. Through this mechanism, IL-10 attenuated the deleterious effects of LPS on wound healing, further reducing scar formation and skin fibrosis (31). This suggests that IL-10 may mediate the TLR4/NF-κB pathway to exert anti-scarring effects.IL-18 and IL-37 are members of the IL-1 family (32). The available evidence indicates that IL-18 and IL-37 have the potential as new treatments for pathologic scarring (33, 34). However, further research is required to fully ascertain their potential and elucidate these cytokines’ optimal targeting.
Overactive keratinocytes, dermal fibroblasts, vascular endothelial cells, and macrophages promote HS progression during proliferative (Figure 1C). The standard proliferative period of wound healing begins about three days after injury and may last 2 ~ 3 weeks. Indeed, from 12 hours after injury, keratinocytes are activated by changes in the microenvironment of hydrogen peroxide, pathogens, growth factors, and cytokines, which result in a high degree of activation, hyperproliferation, and aberrant differentiation in the HS (35). In this process, keratinocytes produce pro-fibrotic molecules such as TGF-βand PDGF (36). These molecules induce fibroblasts to respond to the pro-fibrotic environment by producing more extracellular matrix (ECM) proteins or differentiating into myofibroblasts, accelerating scar formation (11). Fibroblasts in the upper and deeper dermis have different functions (37). Where fibroblasts in the upper dermis contribute to re-epithelialization, fibroblasts in the deeper spectrum contribute to ECM deposition. In HS proliferative events, endothelial dysfunction and altered expression of angiogenic genes such as endothelin and angiopoietin can lead to excessive angiogenesis (38). Studies have shown that excessive angiogenesis can increase collagen deposition (39). Macrophages play an essential role in this process by participating in the remodeling of neovascularization, phagocytosis of excess blood vessels, and inhibition of the angiogenic response (40).
The main event during the HS remodeling phase is the dysregulation of the balance between ECM collagen synthesis and degradation (41). This is accomplished by regulating key MMPs (42). MMPs are expressed by macrophages, fibroblasts, and keratin-forming cells (43). Previous studies have found that MMP2 and MMP9, enzymes essential for remodeling the ECM, are significantly elevated in the pathological microenvironment of HS (44, 45). MMP1 and MMP7 are downregulated during HS formation (46). A study observed that fibroblast proliferation and migration can be inhibited by reducing MMP-9 expression, thereby reducing fibrotic scar formation (47). Altered expression of these MMPs results in reduced degradation of ECM components, including COL-1, COL-3, and fibronectin (48). Additionally, macrophage signaling pathways play a crucial role in regulating HS remodeling. The Notch signaling pathway controls the expression of Smad, α-SMA, and collagen in HS fibroblasts to some extent (49, 50). A study using RBP-J knockout mice demonstrated that inhibition of Notch signaling in macrophages suppresses the inflammatory response and reduces collagen deposition, leading to better wound healing and reducing fibrosis (51). The above studies suggest macrophage-derived MMPs and their intracellular signaling control tissue remodeling during skin wound repair (Figure 1D). However, the precise role of macrophages in scar formation remains insufficiently understood.
In response to initial stimuli such as pathogens, cytokines, or injury signals, macrophages typically polarize rapidly into either a classically activated (M1-type) or alternatively activated (M2-type) phenotype (52) (Figure 1E). This initial response typically occurs within a few hours to a few days, representing a rapid immune reaction to injury (53). M1-type macrophages are typically activated by IFNγ, either alone or in combination with LPS (54). Macrophages with the M1 phenotype highly express CD80, CD86, and CD16/32 (55), and are capable of producing pro-inflammatory cytokines such as IL-6, IL-12; chemokines such as CXCL9 and CXCL10; and NO (56). IL-4 and IL-13 induce the M2 macrophage phenotype, which is distinguished by its ability to produce vasoactive substances, exhibit anti-inflammatory properties, and promote tissue repair (57, 58). At the initial stage of HS formation, the phenotype of macrophages is predominantly of the M1 type. Under normal circumstances, the pro-inflammatory factors secreted by M1 macrophages function to clear pathogens and necrotic tissue in the initial wound environment. However, in HS, the overexpression of pro-inflammatory factors promotes the proliferation and differentiation of fibroblasts. Increasing evidence suggests that, during the early stages of wound healing, it is crucial to shift macrophages from the M1 pro-inflammatory phenotype to the M2 anti-inflammatory phenotype (59, 60).
Notably, the M1 and M2 classification model of macrophages is a simplified model for describing the different functional properties exhibited by macrophages during polarization. In addition, macrophages responding to the initial stimulus may exhibit a mixed phenotype between M1/M2, Mregs, and CXCL4-induced M4 type (53, 61, 62). However, these phenotypes have not been intensely studied in HS.
As the inflammatory response develops or environmental factors change, the macrophage phenotype shifts in the medium term, a process that usually occurs within a few days to a week (63). At this point, macrophages can shift from M1 to M2 type or between M2 subtypes (Figure 1F). M2-type macrophages express specific surface markers, such as CD206 and CD204, and secrete anti-inflammatory factors, thereby inhibiting inflammation and promoting tissue regeneration and repair. M2 macrophages can be further categorized into M2a, M2b, M2c, and M2d subtypes (64, 65) (Figure 1F). Although current data suggest that different phenotypes of M2 macrophage subpopulations play different roles, no reports have evaluated the role of M2 macrophage subtypes in HS, and further studies are warranted.
Interestingly, one study suggests that tumors are somehow characteristic of unhealed wounds. Significant similarities exist between many tumor markers and the wound-healing process’s biological markers (66). In the tumor microenvironment, macrophages typically exhibit specific phenotypes associated with tumor progression. Tumor-associated macrophages (TAMs), which are highly plastic, can adopt either an M1-like phenotype, contributing to anti-tumor immunity, or an M2-like phenotype, promoting tumor growth and immune escape (67, 68). This mixture of phenotypes and their dynamic changes reflect the complex response of macrophages in different environments. Recent advances in cancer immunotherapy have demonstrated that targeting TAMs can effectively modulate their function. For instance, PD-1+ TAMs exhibit impaired phagocytic capacity, which can be rescued by PD-1/PD-L1 blockade, leading to a reduction in tumor burden (69, 70). Given the functional plasticity of macrophages in both tumor progression and wound healing, it is plausible that a similar immunomodulatory approach could be relevant to HS treatment. Future research should explore whether macrophages undergo a TAM-like phenotypic shift during HS formation and whether targeting immune checkpoints such as PD-1/PD-L1 could regulate macrophage activity to control excessive fibrosis and pathological scar formation. Investigating these mechanisms may provide novel insights into macrophage-targeted therapies for HS.
Under prolonged inflammation lasting weeks to months, macrophages tend to adopt a phenotype suited to the specific tissue or pathological state, facilitating tissue stabilization and functional recovery. In this process, M2-type macrophages predominate (Figure 1G). By secreting anti-inflammatory cytokines, M2-type macrophages inhibit inflammatory responses and prevent tissue damage caused by excessive immune responses. In addition, M2-type macrophages secrete VEGF and PDGF during the recovery phase after injury, which promotes neovascularization and collagen production to support new tissue generation and repair. This M2-type phenotype tends to be persistent during the remodeling phase of HS formation. This results in excessive scar tissue formation via mechanisms such as prolonged anti-inflammatory response, fibrosis promotion, and ECM remodeling.
Besides, in long-standing chronic inflammatory or tumor environments, macrophages can also exhibit a mixture of M1 and M2 phenotypes (71) (Figure 1G). This mixed phenotype can sustain the fight against pathogens while modulating the local immune microenvironment to avoid excessive damage to normal tissues. Overall, the phenotypic changes of macrophages during the formative stages of HS further reflect their complex roles in maintaining immune homeostasis and regulating pathological states. Modulation of macrophage phenotypic switching is expected to be a new strategy for treating disease.
TNF-α is a pleiotropic cytokine secreted by various immune cells, including monocytes, T cells, dendritic cells, natural killer cells, and macrophages. M1-type macrophages are one of the significant sources of TNF-α (72). TNF-α regulates inflammatory responses and plays a crucial role in biological processes such as apoptosis, cell proliferation, and autophagy (73–75). A 16S rRNA sequencing showed that the expression of TNF-α was significantly higher in HS tissues than in normal tissues (76). TNF-α is involved in HS through the activation of relevant inflammatory pathways. The NF-kB pathway was shown to be activated in HS fibroblasts (77). Studies have shown that TNF-α induces ROS production in human dermal fibroblasts and upregulates the transcription factor NF-κB (78). In vitro experiments demonstrated that TNF-α induced up-regulation of MMP-1 and MMP-3 expression in human dermal fibroblasts. Resveratrol significantly inhibited this effect through the NF-κB pathway (79). Another study showed that blocking TNF-α interaction with TNFR effectively inhibits TNF-α-induced NF-κB activation (80). These studies suggest that TNF-α activates the NF-κB signaling pathway by TNFR, which alters the biological behavior of human dermal fibroblasts and may play a role in HS formation (Figure 2A).
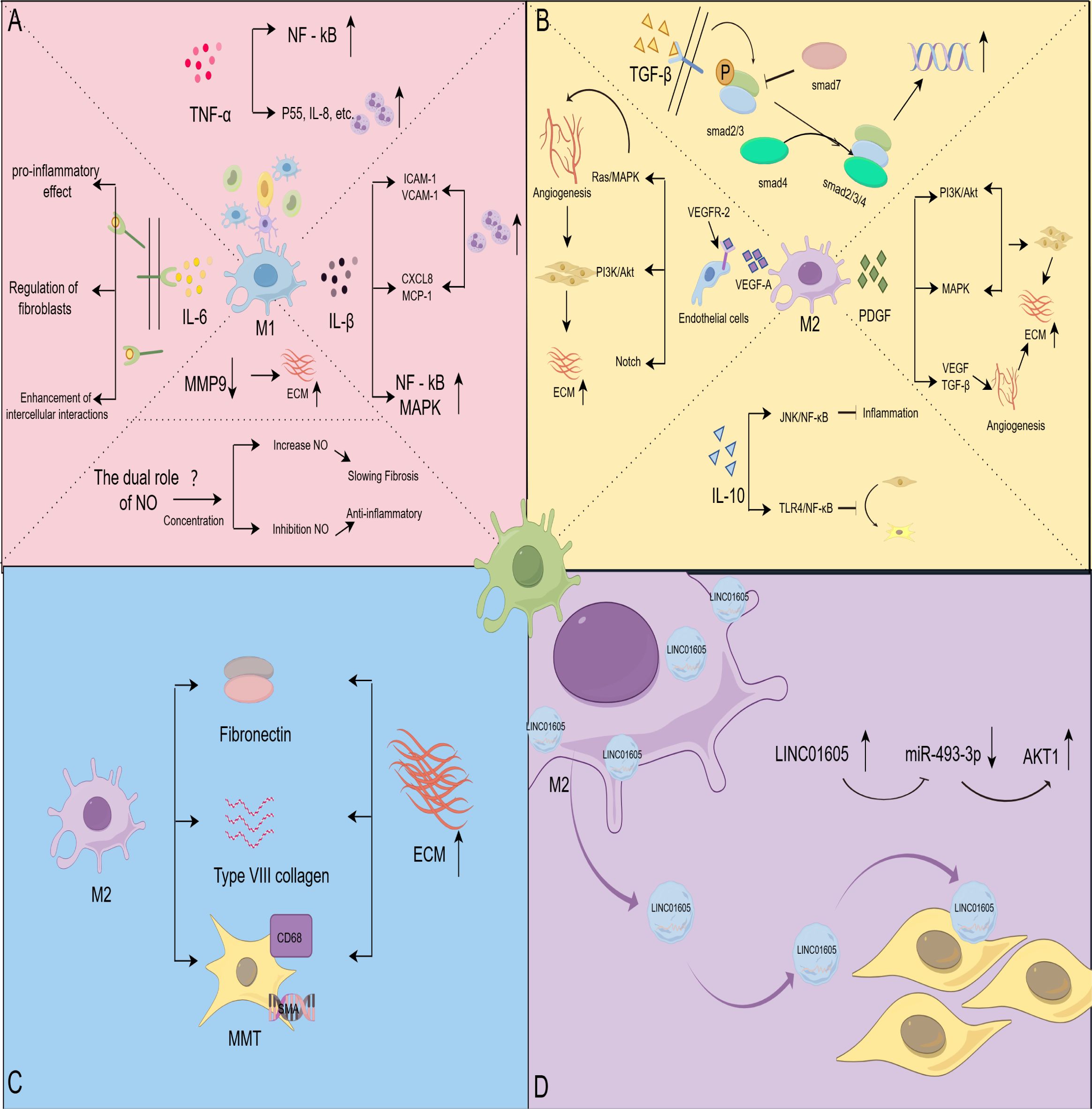
Figure 2. Macrophages affect HS through different pathways. (A) Effect of M1-type macrophage-associated signaling molecules on HS; TNF-α activates NF-κB and promotes inflammatory cell infiltration; IL-6 exhibits pro-inflammatory activity, regulates fibroblast behavior, and interacts with macrophages, fibroblasts, and endothelial cells; IL-1β recruits immune cells and activates the NF-κB and MAPK signaling pathways; NO regulates fibrotic disease in a concentration-dependent manner; MMP9 reduces ECM deposition. (B) Effect of M2-type macrophage-associated signaling molecules on HS; over-activation of the TGF-β pathway promotes fibroblast value-addition and differentiation, and Smad7 negatively regulates TGF-β signaling; IL-10 inhibits inflammatory responses and fibroblast differentiation through the JNK/NF-κB and TLR4/NF-κB pathways; VEGF activation Ras/MAPK, PI3K/Akt and Notch signaling pathways, which regulate the behavior of vascular endothelial cells and promote angiogenesis overgrowth; PDGFPI3K/Akt, MAPK and other signaling pathways enhance the secretory activity of fibroblasts, and synergistically act with VEGF and TGF-β to promote angiogenesis and promote the development of HS. (C) Macrophages were directly involved in matrix remodeling; macrophages secreted fibronectin and collagen type VIII and differentiated into myofibroblasts to directly intervene in ECM remodeling. (D) Macrophages mediate HS via exosome formation; exosomes derived from M2 macrophages were enriched in LINC01605, and high levels of LINC01605 caused a decrease in miR-493-3p and activated AKT to enhance the inflammatory response. (By Figdraw).
In addition, TNF-α interacts with neutrophils to promote HS formation (Figure 2A). Research suggests that TNF-α activates neutrophils via IL-8, p55, secretory vesicles, and specific granules, enhancing their bactericidal activity and releasing inflammatory mediators (81, 82). In the early stages of infection and inflammation, TNF-α regulates the activity of neutrophil surface receptors, inhibits apoptosis, and prolongs the lifespan of neutrophils through signaling pathways such as JNK and NF-κB (83, 84). Notably, TNFR1 and TNFR2 initiate pro-inflammatory signaling, but only TNFR1 triggers a pro-apoptotic response (85). Moreover, TNF exhibits dual properties that activate neutrophil apoptosis under specific conditions (86, 87).
Although the mechanism of TNF-α has been extensively studied, TNF-α inhibitors are widely used in treating psoriasis, rheumatoid arthritis, and other immune-related diseases (88, 89). However, their specific role in HS lacks support from large-scale research data. Understanding the interactions between TNF-α and other inflammatory factors and cell types could help clarify its overall role in HS formation and potentially optimize treatment strategies.
IL-6 is a multifunctional cytokine that affects the fibrotic process by promoting monocyte recruitment, M2-type macrophage polarization, and increased ECM deposition (90). After binding to its receptor, IL-6 exerts its biological effects primarily through three signaling modes: cis, trans, and cluster signaling (91). Studies indicate that IL-6 is one of the most significant cytokines promoting human HS formation, highlighting its critical role in fibrotic pathology (25). Trans signaling by IL-6 exhibits pro-inflammatory activity and is associated with various pathological changes (92). Previous studies have demonstrated that activation of trans IL-6 signaling accelerates fibrosis (93). Inhibition of IL-6 and its downstream pathways positively impacts the clinical management of fibrotic diseases (94, 95). Interestingly, a study of renal scarring found no significant differences in trans IL-6 signaling markers between patients with renal scarring and those without scarring (96). This suggests that there may be some degree of dysfunction in the trans-IL-6 signaling pathway during scarring that deserves further investigation.
IL-6 regulates fibroblast behavior through autocrine mechanisms. In vitro experiments demonstrated that IL-6 autocrine activity drove scar fibroblasts to exhibit significant anisotropy and altered ECM arrangement, resulting in a directional matrix structure (97). This change in matrix structure and orientation may contribute to the rigid and irregular morphology of scar tissue. This study suggests that IL-6 not only promotes HS formation via its pro-inflammatory effects but also influences ECM structure by modulating fibroblast behavior. This highlights the importance of IL-6 signaling in inhibiting fibrosis progression as a potential therapeutic target.
Additionally, elevated IL-6 levels promote HS formation through intercellular interactions. Specifically, IL-6 stimulates macrophage polarization towards the M2 type, upregulates TGF-β and promotes fibroblast differentiation towards myofibroblasts (98). Furthermore, IL-6 amplifies the inflammatory response in vascular endothelial cells, regulating angiogenesis and immune cell recruitment. These effects are mediated by classical and trans signals activating PI3K-Akt, ERK1/2, and gp130 signaling pathways (99, 100). In summary, IL-6 plays a key role in HS formation through multiple signaling pathways and intercellular interactions (Figure 2A). However, the complexity of the IL-6 signaling pathway has not been fully clarified. Exploring the causes of dysregulation of the IL-6 signaling pathway and its specific role in the fibrosis process will help develop more precise therapeutic strategies.
IL-1β is also highly expressed in HS (27). IL-1β belongs to the IL-1 family, which consists of 11 cytokines.IL-1β is one of these ligands with proinflammatory activity and is produced mainly by macrophages. In the early stages of scar formation, IL-1β attracts immune cells to migrate to the damaged site by activating the inflammatory response. It has been shown that IL-1β enhances vascular permeability and promotes the aggregation of inflammatory cells to the area of injury by upregulating the expression of ICAM-1 and VCAM-1, adhesion molecules of vascular endothelial cells (101, 102). IL-1β also stimulates endothelial cells to secrete CXCL8 and monocyte chemotactic protein-1, which further attracts immune cells, such as neutrophils and monocytes, to the site of infection or injury (103). In addition, IL-1β activates the NF-κB and MAPK signaling pathways, amplifying the inflammatory response and exacerbating tissue fibrosis (104, 105). Studies have shown that anti-IL-1β therapy is effective in inhibiting the course of chronic progressive fibrosis. This effect may be attributed to its inhibition of IL-1β-mediated pro-inflammatory responses (106, 107).
Overall, IL-1β exacerbates tissue fibrosis by enhancing the inflammatory response and attracting immune cell aggregation (Figure 2A). Although some preliminary studies have suggested a possible role for anti-IL-1β therapy in organ fibrotic diseases (107, 108), more clinical studies and trials are needed to determine the efficacy and applicability of anti-IL-1β therapy.
Endogenous nitric oxide (NO) is produced by three different types of enzymes: neuronal NOS (nNOS; NOS1), inducible NOS (iNOS; NOS2), and endothelial NOS (eNOS; NOS3) (109). Of these, iNOS plays a critical role in the fibrotic process. iNOS activity is upregulated by cytokines, such as IFN-γ and LPS, secreted by M1-type macrophages. This leads to the production of large amounts of NO, which is involved in regulating fibrosis and the inflammatory response. Recent studies have found that NO exhibits different effects in various fibrotic diseases through different concentrations and mechanisms (110) (Figure 2A). NO inhibition of myofibroblast activation and collagen I production in renal fibrosis, using nanocarrier-delivered NO, slows the progression of renal fibrosis (111). In a phase 2 clinical trial, pulsed inhaled NO demonstrated favorable safety and tolerability in treating interstitial lung disease and improved patients’ physical activity (112). These studies suggest the potential benefit of NO supplementation in fibrotic diseases.
In contrast, other studies have demonstrated that inhibiting NO production in LPS-stimulated RAW 264.7 macrophages exerts an anti-inflammatory effect (113). Huseyin Gungor’s study supports the notion that inhibiting NO production helps improve liver fibrosis (114). Another in vitro study showed that p53 knockdown mesenchymal stem cells (MSCs) promoted fibroblast proliferation by increasing NO production. This phenomenon was reversed by inhibiting NO production (115). It is thus clear that the role of NO in fibrotic diseases is dual, and its specific effect depends mainly on the concentration level of NO. Further research on the application of NO in fibrotic diseases is of critical significance for guiding HS treatment.
MMPs are a class of metal ion-dependent proteases capable of degrading a wide range of components in the ECM. The human genome contains 24 MMP genes, two of which encode the MMP23 protein, resulting in 23 distinct MMPs. Under normal conditions, MMP activity is low, but it increases significantly during tissue repair and inflammation. MMPs regulate tissue degradation and remodeling by cleaving ECM components such as collagen, fibronectin, laminin, and gelatin (116). Among MMPs, MMP-9 plays a crucial role in HS formation (Figure 2A). Previous studies have found that upregulated gene expression of MMP-2, MMP-9, and TIMP-1 is strongly associated with proliferative scarring (117). In experiments with a rabbit ear scar model, elevated MMP-2 and MMP-9 expression significantly reduced the Scar Elevation Index, Epidermal Thickness Index, and collagen deposition (118). Recent in vitro studies have demonstrated that capacitive resistive electro transfer therapy alters MMP-9 expression in human myofibroblast cultures, potentially benefiting fibrotic pathology treatment (119). These findings suggest that the activity of MMP-9 has a critical role in tissue repair. The mechanism of macrophage-MMP-ECM interactions warrants further investigation. Targeted interventions against this interaction may offer new strategies for treating HS.
TGF possesses the ability to induce a transformed phenotype in untransformed cells, and it was first discovered by De Larco and Todaro in 1978. Mammalian cells express three isoforms of TGF: TGF-β1, TGF-β2, and TGF-β3. These isoforms are widely involved in biological processes, such as inflammation, matrix generation, matrix remodeling, cell proliferation, and the regulation of apoptosis, all of which play an important regulatory role in HS formation (120, 121).
Recent studies have confirmed that TGF-β, especially the TGF-β1 isoform, significantly promotes HS formation. Specifically, TGF-β1 promotes fibroblast activation by activating the downstream Smad protein signaling pathway (Figure 2A). Simultaneously, it induces fibroblasts to secrete large amounts of collagen and fibronectin, which leads to the excessive accumulation of scar tissue (122). Studies have shown that inhibition of the activation of the TGF-β1-Smad2/3/4 signaling pathway promotes apoptosis of fibroblasts, thereby alleviating HS production (123). In addition, TGF-β1 promotes the differentiation of fibroblasts into myofibroblasts by regulating the expression of α-SMA, further enhancing contractility and fibrosis at the trauma site (124).
Several studies have shown that moderate TGF-β signaling during the early stages of wound repair helps maintain the tissue repair balance and promotes wound healing. In contrast, when TGF-β signaling is overactivated, it leads to fibrosis formation. Smad7 is an inhibitory factor in the TGF-β signaling pathway and plays a key role in the negative regulation of fibrosis. It inhibits the excessive transmission of TGF-β signaling by competitively inhibiting the phosphorylation and nuclear translocation of Smad2/3, thereby limiting the progression of fibrosis (125) (Figure 2B). These findings highlight the complex role of TGF-β signaling in wound repair, including its positive role in promoting wound healing but also the risk of fibrosis. Previous studies have shown that the critical role of TGF-β in scar formation has been extensively researched, and that TGF-β inhibitors have demonstrated potential efficacy in preclinical studies (126). However, since the TGF-β signaling pathway is critical for various normal physiological processes, such as wound healing and immune regulation, treatments directly targeting TGF-β may lead to significant side effects. Therefore, future studies may need to develop more selective TGF-β inhibitory strategies to minimize side effects while preserving therapeutic efficacy.
IL-10 is an anti-inflammatory cytokine, mainly secreted by M2-type macrophages, and it is involved in scar formation and fibrosis-related diseases. However, the anti-fibrotic molecular mechanism of IL-10 in skin scarring remains unclear. Early studies found that a lack of IL-10 in fetal skin triggered scar formation. This intrinsic lack of IL-10 may lead to a sustained amplification of inflammatory cytokines, persistent stimulation of fibroblasts, and abnormal collagen deposition (127). Other studies have demonstrated that IL-10 is highly expressed in fetal skin during mid-gestation and is absent in human skin after birth (128). These findings tentatively suggest that IL-10 is involved in wound healing and scar formation. Recently, several studies have shown that IL-10-modified BMSCs inhibited inflammatory progression through the JNK/NF-κB pathway and prevented the formation of HS in a rabbit ear model (129). This suggests that IL-10 may mediate the JNK/NF-κB pathway to exert an anti-scarring effect. The study by Xie Fang et al. demonstrated that IL-10-modified Adipose-Derived Mesenchymal Stem Cells prevented HS formation by modulating fibroblast biology and inflammation (130). Another in vitro study demonstrated that IL-10 regulates the TLR4/NF-κB pathway in dermal fibroblasts via the IL-10R/STAT3 axis, which in turn reduced ECM deposition and fibroblast-to-myofibroblast transformation, thereby attenuating LPS-induced HS formation (31).
In summary, IL-10 may take on an anti-scarring role through various mechanisms, such as reducing the inflammatory response and modulating the biological behavior of fibroblasts (Figure 2B). Although IL-10 has the potential to inhibit HS formation, it is poorly stable in vivo and requires an effective delivery system to ensure adequate concentration in target tissues. Existing delivery systems (e.g., nanocarriers, hydrogels, and other methods) are effective, but their effectiveness and safety must be further validated. Future research may focus on developing controlled-release IL-10 systems that can be combined with antifibrotic drugs, laser therapy, or other cytokines to develop a combination therapy strategy for preventing HS.
The process of HS formation is closely related to dysregulated angiogenesis. VEGF is the major pro-angiogenic factor, generating different mRNA variants through alternative splicing. These variants are translated to produce protein subtypes of different lengths and biological functions (131). Specifically, VEGF-A binds to VEGFR-2, forming a dimer and activating the receptor’s tyrosine kinase activity. This process triggers the autophosphorylation of tyrosine residues on the receptor. The phosphorylated tyrosine residues become binding sites for various downstream signaling molecules, activating multiple signaling pathways such as Ras/MAPK, PI3K/Akt, and Notch (132, 133) (Figure 2B). These pathways act synergistically to regulate the behavior of vascular endothelial cells and ultimately promote angiogenesis. However, overactivation of these signaling pathways can lead to excessive angiogenesis. The overproduced blood vessels provide sufficient nutritional support for the abnormal proliferation of fibroblasts and collagen deposition, thus exacerbating scarring. Numerous studies have shown that downregulation of VEGF signaling can alleviate HS formation (134, 135). Moreover, VEGF signaling also upregulates the expression of MMPs, promoting endothelial cell migration in the stroma and neovascularization (136, 137). Thus, VEGF and its associated signaling pathways play a multifaceted role in the formation of HS, making it an essential target for understanding and treating pathological scarring.
The PDGF family consists of PDGF-aa, PDGF-bb, PDGF-ab, PDGF-cc, and PDGF-dd, which are composed of five members that form disulfide-linked homo- or heterodimers (138). The primary sources of PDGF are platelets and fibroblasts, and M2 macrophages can secrete small amounts of PDGF during the proliferation phase of wound healing. PDGF regulates cell proliferation, migration, and differentiation by initiating downstream signaling pathways through binding to its specific receptor, PDGFR (139).
PDGF’s ability to promote fibroblast activity plays a crucial role in HS formation. Studies have shown that PDGF expression in HS tissue is significantly higher than in normal skin, which correlates directly with the hyperactivation of fibroblasts (140). The overexpression of PDGF leads to abnormal proliferation and migration of fibroblasts, resulting in collagen production exceeding normal levels and causing thickened and hardened scar tissue. Another in vitro study found that adding PDGF-BB to fibroblasts cultured in vitro significantly increased the proliferation rate of the cells (141). This effect was more pronounced in HS fibroblasts (142), suggesting an essential role of PDGF in promoting cell proliferation in this process.
PDGF also enhances the secretory activity of fibroblasts by activating the PI3K/Akt, MAPK, and other signaling pathways (Figure 2B). This results in the overproduction of collagen, fibronectin, and other ECM components, ultimately leading to the persistent and abnormal proliferation of scar tissue (143, 144). In addition, PDGF promotes HS generation through synergistic effects with VEGF and TGF-β (145, 146) (Figure 2B). This suggests that PDGF affects scar formation by directly acting on fibroblasts and also indirectly contributes to the development of HS by regulating angiogenesis. Inhibiting the PDGF/PDGFR signaling pathway can effectively block the aberrant signal transduction of various growth factors, thereby preventing the onset and progression of diseases such as fibrosis (147). Given the critical role of PDGF in HS, targeting the PDGF/PDGFR pathway is a promising therapeutic strategy.
Macrophages are not only indirectly involved in HS formation by secreting signaling molecules but also directly influence HS formation by remodeling the ECM. Macrophages directly intervene in ECM remodeling by both secreting collagen and differentiating into myofibroblasts (Figure 2C). As early as 1999, Weitkamp et al. used fluorescence techniques to demonstrate that macrophages can synthesize type VIII collagen themselves (148). Another study found that macrophages secrete fibronectin and collagen type VIII to promote ECM formation and express almost all known collagen and collagen-related mRNAs (149). These findings suggest a direct role for macrophages in HS formation.
Notably, macrophages undergo macrophage-to-myofibroblast trans differentiation (MMT) in chronic inflammation and fibrotic pathological environments. A study collected human liver specimens at different stages of hepatic fibrosis and found MMT cells, which co-expressed macrophage (CD68) and myofibroblast (a-SMA) markers. Moreover, this result was validated in an animal model of liver fibrosis (150). In another study, researchers found that macrophages are involved in the formation of subretinal fibrosis through MMT changes (151). This suggests that MMT is involved in the progression of multiple fibrotic diseases. However, current research on direct collagen secretion by macrophages has primarily focused on organ fibrosis. Therefore, there is an urgent need to improve the understanding of macrophage-secreted ECM components in skin scarring.
Macrophages secrete different extracellular vesicles, including Exosomes, Microvesicles, and Apoptotic Bodies. Exosomes are important extracellular vesicles that contain various bioactive molecules such as microRNA, proteins, and lipids, which can regulate intercellular communication and influence the behavior of recipient cells.
Exosomes are secreted by prokaryotic and eukaryotic cells, with a diameter of about 30-150 nm, and are essential carriers of paracrine signaling (152). Macrophages initiate the process through membrane endocytosis, forming endosomes. Subsequently, the endosomes generate intraluminal vesicles in the cytoplasm, which transform into multivesicular bodies (MVBs). Finally, the MVBs fuse with the cell membrane, releasing exosomes (152, 153). A study co-culturing M2 macrophages with human dermal fibroblasts found that exosomes derived from M2 macrophages promoted the proliferation and migration of human dermal fibroblasts by delivering LINC01605 (154) (Figure 2D). Another study showed that M2 macrophage-derived exosomes were enriched in long-stranded noncoding RNA, specifically lncRNA-ASLNCS5088. This lncRNA can be efficiently transferred to fibroblasts, increasing α-SMA expression (155). Interestingly, recent studies have shown that macrophage filamentous pseudopods can produce filopodia tip vesicles. Such vesicles detach from the tips of the cell’s filamentous pseudopods and deliver many molecular signals to fibroblasts (156). These studies suggest that macrophage-derived exosomes play a role in HS formation by influencing fibroblast behavior.
Although studies of macrophage-mediated HS formation via exosomes have shown potential, their high cost and shortcomings in delivery efficiency and specificity have limited their application. In the future, new breakthroughs in therapeutic HS should be achieved by studying standardized production and optimizing delivery systems.
Compared with traditional conservative treatments, such as local drug injections and physical pressure therapy, photoelectric technology offers non-invasiveness, high precision, a rapid onset of action, and shorter treatment durations. Common photoelectric therapy modalities used in clinical practice include laser therapy (ablative and non-ablative), microplasma radiofrequency technology, and photodynamic therapy (PDT).Recently, PDT has emerged as a promising non-surgical strategy for treating HS in both cellular studies and animal models. PDT primarily relies on the cytotoxic effects of photosensitizers to achieve its therapeutic action. When photosensitizers accumulate around proliferating fibroblasts, laser irradiation triggers the production of reactive oxygen species (ROS).ROS exert cytotoxic effects on fibroblasts, inducing apoptosis and ultimately leading to the necrosis of scar tissue (157, 158). However, the detailed mechanism underlying PDT’s scar-inhibitory effects remains unclear. In clinical applications, PDT is often combined with microneedling techniques, which enhance drug penetration while amplifying PDT’s anti-scarring effects (159, 160). There is no doubt that PDT is an effective strategy for the prevention and treatment of HS. Notably, the selection of photosensitizers, the presence of side effects such as pain during treatment and how to effectively combine photodynamic therapy with other therapies are current challenges.
Controlled-release materials significantly enhance the continuity and efficacy of therapy due to their unique drug slow-release properties. Hydrogel is a network structure composed of hydrophilic polymer chains linked by various chemical bonds and forces, featuring diverse cross-linking modes. In recent years, it has been used as a bioscaffold to promote wound healing, demonstrating promising therapeutic effects in the treatment of HS. Zivari-Ghader T et al. showed that hydrogel wound dressings made of chitosan/alginate scaffolds loaded with HPCE effectively prevented HS formation. This hydrogel exhibited antimicrobial, antioxidant, and anti-inflammatory properties, effectively inhibiting excessive collagen deposition and reducing inflammation (161). Similarly, numerous studies support these findings (162–164). Fu et al. demonstrated that hydrogels possess tension-shielding capabilities, which reduce wound tension via shape fixation and ultimately minimize HS formation (165). Zhang et al. utilized a bioglass/alginate composite hydrogel, which significantly inhibited scar formation in a rabbit ear scar model. The main mechanism involves stimulating the expression of the integrin subunit Alpha 2 in dermal fibroblasts, which accelerates wound healing and modulates fibroblast behavior (166). This indicates that different functional hydrogels can inhibit HS formation through multiple mechanisms (Table 1).
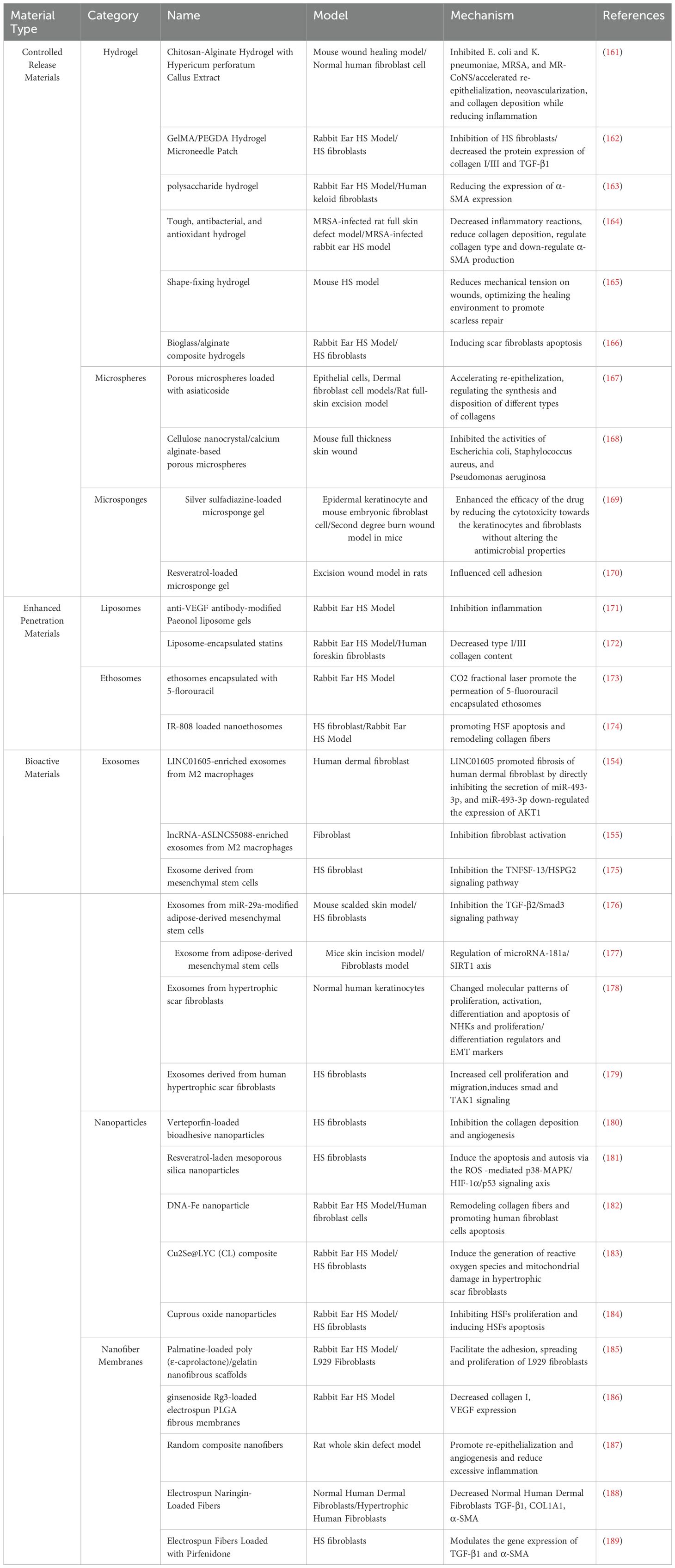
Table 1. Novel drug delivery systems for HS.
Microspheres are small spherical multiparticulate drug delivery systems with diameters ranging from 1 to 1,000 μm, capable of enhancing the bioavailability, stability, and efficacy of traditional drugs while ensuring good safety. Zhang et al. constructed asiaticoside microspheres to achieve efficient drug loading and sustained release, providing regenerative healing and anti-scarring effects (167). Another study prepared hemostatic porous microspheres, which demonstrated high fluid absorption capacity and excellent coagulation properties, accelerating wound healing and highlighting their potential in scar treatment (168).
Microsponges are porous structures composed of polymerized particles, typically ranging from 5 to 300 μm in diameter. The porous structure of microsponges enables controlled drug release. In a study investigating a microsponge gel of silver sulfadiazine for burn wound treatment, loading silver sulfadiazine into a microsponge incorporated in a gel matrix enhanced drug potency, enabled sustained drug release, reduced dosing frequency, improved adherence in burn patients, and minimized cytotoxicity (169). Furthermore, incorporating microsponges into a hydrogel allows for sustained drug delivery to the wound site, while the gel matrix maintains a moist environment and enhances cell adhesion, promoting wound healing and offering a novel approach for HS treatment (170).
The stratum corneum is the primary barrier to drug delivery through the skin to the scar tissue. Both liposomes and ethosomes possess bilayer membrane structures that resemble biological membranes. This structure enables them to mimic the properties of biological membranes, facilitating their fusion with cell membranes, penetration of the stratum corneum, and enhancement of drug permeation. SHI et al. developed an anti-VEGF antibody-modified liposome gel containing salvinorin. It exhibited excellent skin permeability, delayed drug release, and promoted high drug accumulation in the dermis. In vivo studies demonstrated that it reduced VEGF, TGF-β1, and TNF-α levels, inhibited cell proliferation, and exhibited therapeutic effects on HS in a rabbit ear model (171). Xie et al. developed new statin-loaded liposomes with enhanced skin penetration, which were successfully delivered topically and significantly reduced HS formation in a rabbit ear model (172). Zhang et al. prepared 5-FU-encapsulated ethosomes for HS treatment in combination with CO2 fractional laser therapy. The nanoscale ethosomes penetrated scar tissue through narrow and tightly connected cellular gaps. Fractional laser reduces the required drug dose, facilitating drug penetration into deeper skin layers, achieving higher local concentrations, and effectively inhibiting HS formation (173). Similarly, Yu et al. utilized the transdermal delivery capability of ethosomes to prepare an IR-808-loaded nanoethosome system as a novel photosensitizer for HS treatment with transdermal photodynamic therapy (174).
The exceptional biocompatibility and functionality of bioactive materials facilitate the effective repair of scar tissue. Exosomes, as critical mediators of intercellular communication, can deliver biologically active substances and have demonstrated significant therapeutic potential in the treatment of HS. Recent research on adipose stem cell exosomes (ADSC-exos) has yielded increasing evidence that ADSC-exos not only promote wound repair but also possess therapeutic potential for HS. By carrying specific microRNAs, ADSC-exos regulate target gene expression, suppress fibrosis-related signaling pathways such as TGF-β/Smad, reduce fibroblast proliferation, migration, and collagen deposition, and promote HS tissue repair. This demonstrates the great potential of ADSC-exos in the treatment of HS (175–177). Moreover, fibroblast exosomes have shown a beneficial role in HS treatment. They affect HS formation by regulating fibrotic signaling pathways, promoting cell proliferation migration and epithelial-mesenchymal transition (178, 179). Notably, lncRNAs enriched in M2-type macrophage-derived exosomes were found to influence HS formation through a mechanism potentially linked to fibroblast activation (154, 155).
Bioactive nanomaterials exhibit tremendous potential for treating proliferative scarring. Studies have utilized nanoparticles as encapsulants, effectively inhibiting scar tissue formation. For example, Rerteporfin, Resveratrol, and Doxorubicin hydrochloride were encapsulated into nanoparticles to enhance drug stability and targeting, reduce side effects, and inhibit scar fibroblast proliferation effectively (180–182). Additionally, nanoparticles have been used to deliver photosensitizers in combination with near-infrared light therapy to induce mitochondrial damage and cell death (183). Furthermore, nanoparticles have been found to regulate the proliferation and apoptosis of HS fibroblasts, providing a scientific basis for developing novel therapeutic strategies for HS (184). Beyond nanoparticles, nanofiber membranes play a critical role in HS treatment. One study prepared Palmatine-loaded electrospun poly(ϵ-caprolactone)/gelatin nanofibrous scaffolds. These scaffolds exhibited strong antimicrobial and antioxidant activities, significantly inhibited scar formation, and accelerated wound healing (185). Another study developed ginsenoside Rg3-loaded electrospun PLGA fibrous membranes using electrostatic spinning and pressure-driven infiltration techniques. These membranes promoted tissue repair during the early stage of wound healing and inhibited scar formation during the later stage (186). In addition, nanofibrous membranes can influence scar formation by regulating macrophage function and promoting macrophage polarization. In a study, dendritic mesoporous bioglass nanoparticles loaded with VR23 were blended with poly (ester-curcumin-urethane) urea to prepare random composite nanofibers with bi-directional modulation. The dressing effectively promoted scarless healing of chronic wounds (187).
In addition to serving as drug carriers, the structural and mechanical properties of nanofiber membranes play a crucial role in scar treatment. One study developed electrospun fibers loaded with naringin. The fibers featured an innovative rounded texture, which effectively minimized HS formation during early wound healing (188). Another biofiber loaded with Pirfenidone exhibited outstanding elongation and toughness, enabling it to effectively treat HS during the established wound healing phase (189).
Clinical treatment of HS has made significant progress fueled by advancements in optoelectronics and novel drug delivery systems, but these emerging technologies still face multiple challenges in practical application. With its non-invasive and minimally invasive characteristics, photofacial technology provides an innovative and promising approach to treating scarring. However, the technical complexity, high equipment costs, and stringent requirements for operator expertise have restricted its widespread adoption. Moreover, the efficacy of photoelectric treatment is often unpredictable due to patient-specific variability, and its long-term effects require further validation. Therefore, reducing treatment costs, improving operational simplicity, and ensuring treatment efficacy have become pressing challenges for the application of photoelectric technology in HS therapy.
On the other hand, novel drug delivery systems offer a more targeted and efficient method of administering drugs for HS therapy. However, the complex preparation processes of these novel delivery systems impose higher requirements on the biocompatibility, targeting, and stability of the materials. Poor biocompatibility may trigger immune responses, while inadequate targeting can result in non-specific drug distribution in normal tissues, raising the risk of side effects. Additionally, the stability of novel delivery systems in vivo is a critical determinant of their therapeutic efficacy. Therefore, optimizing the preparation process and enhancing delivery efficiency while ensuring material safety and efficacy have become major challenges for novel drug delivery systems in HS therapy.
Current treatment of HS focuses on controlling the inflammatory response with steroids and NSAIDs, as well as managing scar tissue through physical interventions such as surgery (190, 191). Although these methods are effective to a certain extent, they still have the limitations of significant side effects and high recurrence rates. Therefore, the development of new treatment modalities is urgently necessary. The role of macrophages in scar formation is gaining attention. Macrophages play a dual role in tissue repair, promoting both the inflammatory response and facilitating tissue remodeling. Therefore, therapeutic strategies targeting macrophages have emerged as a potential approach to mitigate proliferative scar formation. This review will discuss the strategies and prospects of targeting macrophages to treat proliferative scarring from four aspects (Figure 3, Table 2).
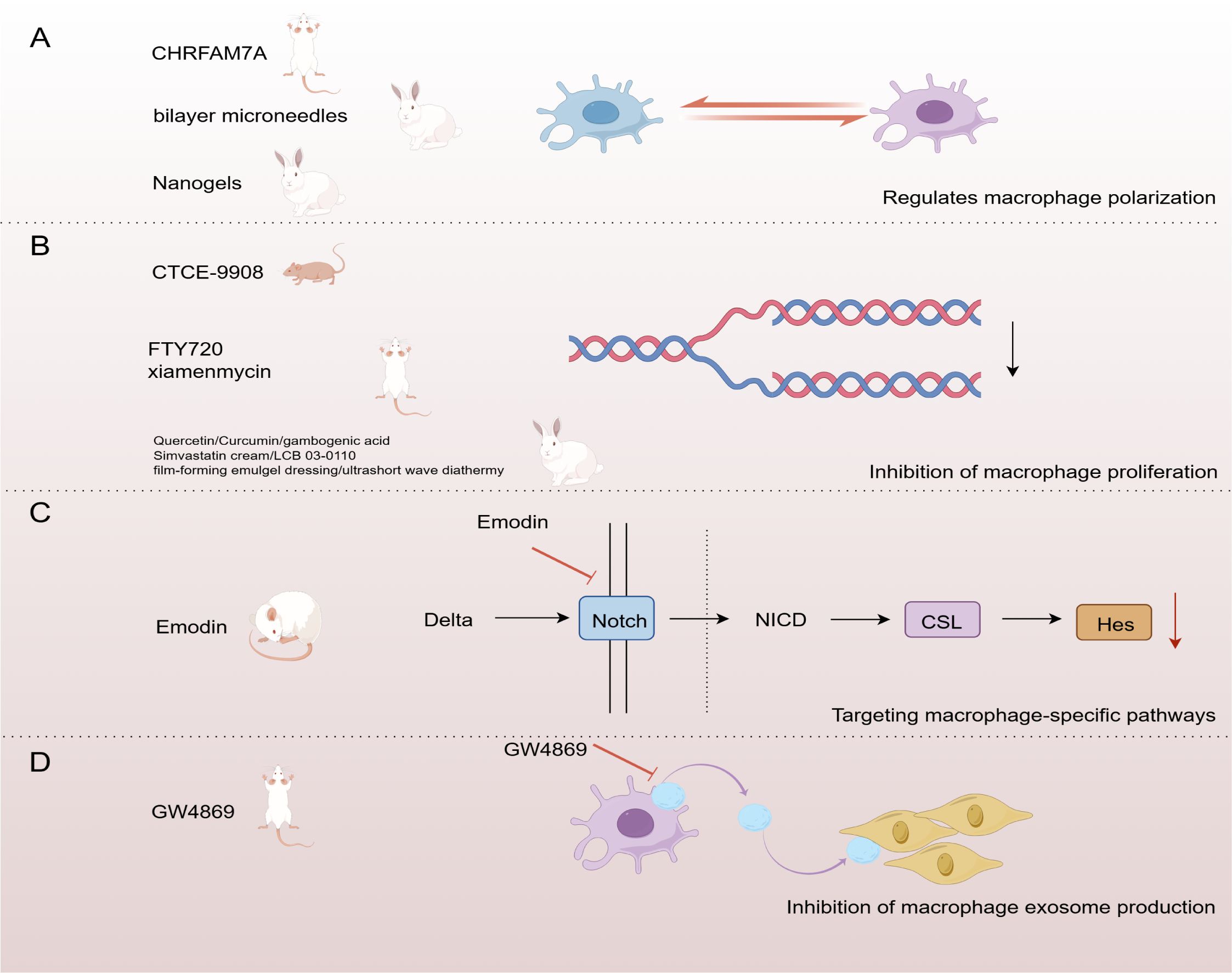
Figure 3. Strategies for targeting macrophages in the treatment of HS. (A) Regulation of macrophage polarization (B) Inhibition of macrophage proliferation (C)Targeting macrophage-specific pathways (D) Inhibition of macrophage exosome production. (By Figdraw).
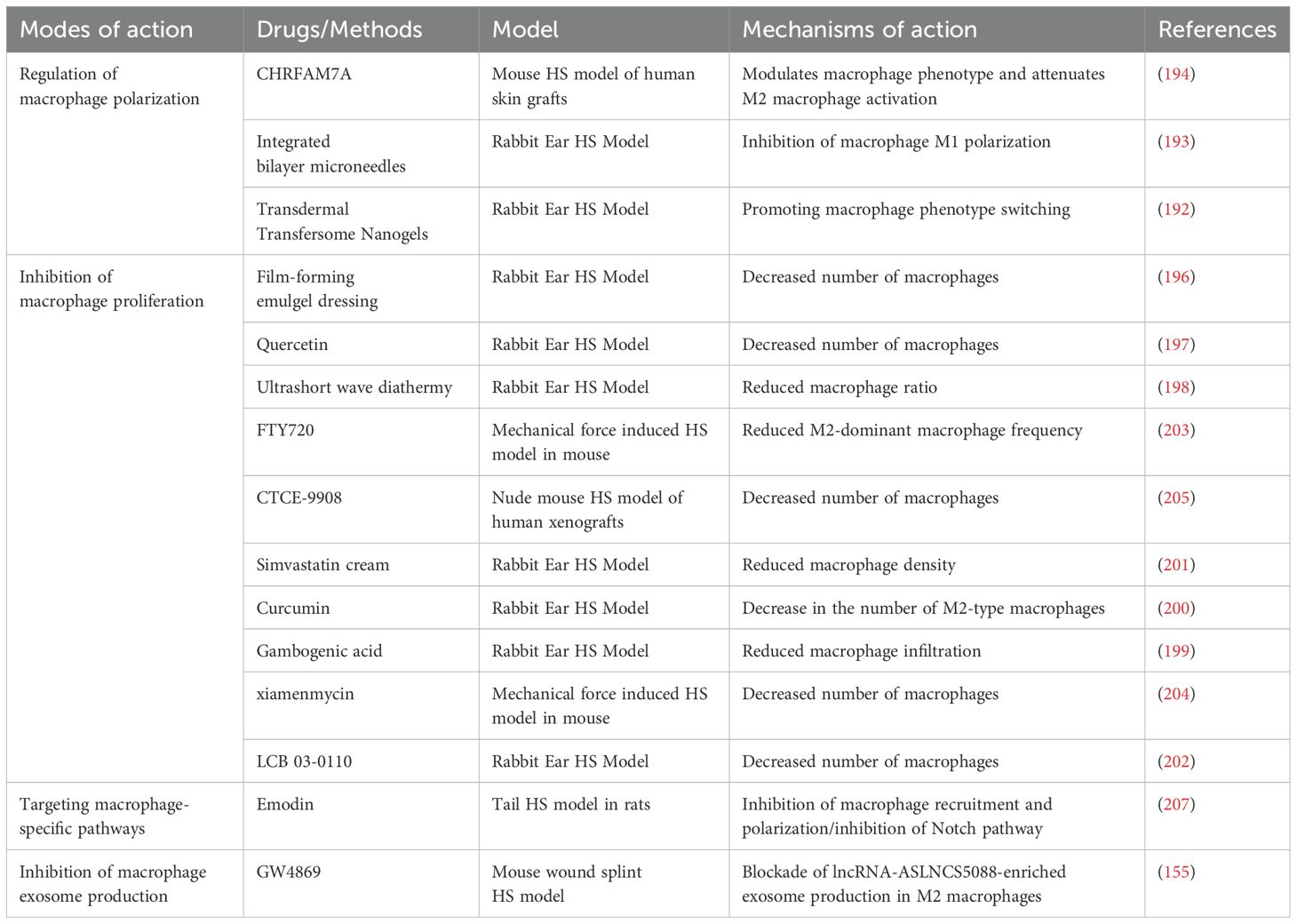
Table 2. Strategies for targeting macrophages in the treatment of HS.
Dysregulation of the macrophage M1/M2 phenotypic transition is one of the major causes of HS formation. Thus, regulating the polarization state of macrophages is expected to inhibit fibroplasia and reduce scar formation. A study used nanogels as carriers to deliver tretinoin and 5-fluorouracil subcutaneously to a rabbit ear HS model. The results showed that this method effectively modulated macrophage phenotypic switching and had an antifibrotic effect, providing a promising therapeutic strategy for HS (192). Other studies have shown that a single dose of a two-layer microneedle system enhances the therapeutic effect against HS. The mechanism may be related to the anti-inflammatory drug dexamethasone, released from the outer layer, which inhibits the polarization of macrophages into a pro-inflammatory phenotype (193). Research by Tianya Li et al. also demonstrated that inhibiting the excessive polarization of M2 macrophages can effectively reduce scar formation (194) (Figure 3A). These findings suggest that therapeutic strategies targeting the modulation of macrophage polarization status have significant potential for preventing and treating HS. This provides new ideas for developing more effective anti-scarring therapies in the future.
Macrophage proliferation and accumulation have been noted as critical factors in the formation of HS (195). Therefore, inhibition of macrophage proliferation is considered a promising therapeutic strategy. Multiple studies have explored the effects of different drugs on macrophage proliferation and accumulation using a rabbit ear HS model. SFN/ASA-containing gel dressing and quercetin have been shown to inhibit scar formation by reducing macrophage numbers in a rabbit ear HS model (196, 197). In addition, ultrashort-wave hyperthermia reduced the macrophage ratio, while gambogenic acid decreased macrophage infiltration. Both demonstrated significant anti-scarring effects in this model (198, 199). Finally, simvastatin cream, curcumin, and LCB 03-0110 in a rabbit ear HS model also showed that reducing macrophage accumulation significantly inhibited fibrosis (200–202).
In a mechanical force-induced mouse model of HS, FTY720 significantly inhibited scar formation by reducing M2 -dominant macrophages (203). Xiamenmycin showed sound anti-scarring effects in this model by reducing macrophage retention (204) (Figure 3B).
Another study used a nude mouse model of HS generated by human xenografts and found that CTCE-9908 effectively controlled scar formation by reducing macrophage accumulation (205). These studies suggest that HS formation can be effectively controlled by targeting macrophage proliferation and accumulation, providing a new direction for future anti-scarring therapies.
Targeting macrophage-specific pathways is one of the critical strategies for treating HS. The Notch signaling pathway plays a role in dermal fibrosis by regulating fibroblast proliferation and activation, influencing inflammatory responses, and controlling ECM remodeling (206). It was shown that emodin significantly attenuated HS formation in the rat tail by inhibiting macrophage recruitment and polarization, an effect associated with inhibition of the Notch signaling pathway. This study revealed that down-regulation of Notch1, Notch4, and Hes1 could inhibit macrophage polarization and attenuate HS formation (207) (Figure 3C). This suggests that targeting the Notch pathway may be an effective intervention strategy for HS formation.
Therapeutic strategies to inhibit exosome production have shown potential in various diseases, including cancer, neurodegenerative diseases, and cardiovascular diseases (208, 209). GW4869 is a selective neutral sphingomyelinase inhibitor widely used to study the role of exosomes in disease by blocking their production (210). Studies have shown that GW4869 successfully inhibited the activation of fibroblasts by M2 macrophages by blocking the production of exosomes enriched with the long-chain lncRNA ASLNCS5088 from M2 macrophages. This mechanism was validated in an in vitro macrophage-fibroblast co-culture model and a mouse wound splint HS model, demonstrating its potential therapeutic value in inhibiting scar formation (155) (Figure 3D). These findings suggest that inhibition of exosome production may be a new direction for treating HS.
In summary, therapeutic strategies targeting macrophages demonstrate significant potential in managing HS. Modulating macrophage polarization status, inhibiting their proliferation, targeting specific signaling pathways, and inhibiting macrophage exosome production effectively reduce scar formation. Future studies should optimize strategies for targeting macrophages through further research and technological improvements to achieve more effective scar management in clinical practice.
HS formation is a complex pathological process involving the interaction of multiple cell types and signaling pathways. Macrophages play a key role at different stages as an important part of the innate immune system. This paper reviews the phenotypic changes of macrophages during HS formation, the effects of key signaling molecules on HS, and the potential of macrophages as therapeutic targets, revealing the importance of macrophages in forming and regulating scarring. However, there are still some questions about the role of macrophages in HS.
First, most studies have shown that M1-type macrophages promote the maintenance and expansion of the inflammatory response, while M2-type macrophages drive the formation and remodeling of scar tissue. However, the diverse roles of macrophages in HS are influenced by various factors, including the severity of the condition and variations in disease models. Currently, there is a lack of a standardized model for HS. Therefore, standardizing the methods for HS modeling is necessary to study the role of macrophages in HS better. Our review emphasizes the complex role of macrophages in HS formation, highlighting their dynamic phenotypic changes and their interactions with the scar microenvironment. These findings provide valuable guidance for the development of physiologically relevant and standardized HS models. By simulating the phenotypic changes of macrophages at different stages and their effects on the scar microenvironment, the physiological relevance of HS models and the translational value in preclinical research can be significantly enhanced. This not only facilitates the study of HS pathophysiological mechanisms but also provides new directions for the evaluation and optimization of emerging therapeutic strategies.
Second, this paper reviews the direct effects of macrophages on HS formation through various signaling molecules. However, macrophage-associated signaling molecules such as TNF-α, IL-6, TGF-β, and IL-10 may play dual roles in different pathological settings. Therefore, further studies on the mechanisms by which macrophage-associated signaling molecules function are necessary.
Third, macrophage-targeted therapies are considered a promising strategy for managing HS due to their critical role in scar formation and remodeling. By precisely regulating macrophage polarization and function, these therapies can reduce scar formation and improve wound healing outcomes. The integration of macrophage-targeted therapies into existing HS management practices has the potential to enhance treatment efficacy. For instance, combining macrophage-targeted therapies with laser therapy or novel delivery systems can provide more precise, localized, and sustained therapeutic effects while minimizing the systemic side effects commonly associated with traditional treatments. However, successful clinical application still requires addressing individual variability in patient responses, challenges related to cost-effectiveness and affordability, and rigorous clinical trials to validate their efficacy and safety. Overall, targeting macrophages is expected to be a new and effective therapeutic strategy for preventing HS.
LS: Writing – review & editing. YZ: Writing – original draft. JG: Writing – original draft. HF: Writing – review & editing. LL: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation for Youth (82205128); Open Fund for the Cultivation Base of National Key Laboratory of Chinese Medicine Powder and Innovative Drugs Co-constructed by the Ministry and Province(23PTKF1004); Innovative Projects for Graduate Students of Hunan University of Traditional Chinese Medicine(2024CX009). General Program of Health Research Project of Hunan Provincial Health Commission (B202304136861).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mony MP, Harmon KA, Hess R, Dorafshar AH, Shafikhani SH. An updated review of hypertrophic scarring. Cells. (2023) 12:678. doi: 10.3390/cells12050678
2. Ogawa R. The most current algorithms for the treatment and prevention of hypertrophic scars and keloids: A 2020 update of the algorithms published 10 years ago. Plast Reconstr Surg. (2022) 149:79e–94e. doi: 10.1097/PRS.0000000000008667
3. Marshall CD, Hu MS, Leavitt T, Barnes LA, Lorenz HP, Longaker MT. Cutaneous scarring: basic science, current treatments, and future directions. Adv Wound Care. (2018) 7:29–45. doi: 10.1089/wound.2016.0696
4. Goverman J, He W, Martello G, Whalen A, Bittner E, Schulz J, et al. The presence of scarring and associated morbidity in the burn model system national database. Ann Plast Surg. (2019) 82:S162–8. doi: 10.1097/SAP.0000000000001826
5. Chen S, Saeed A, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
6. Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps - myth or reality. Clin Sci (Lond). (2023) 137:1067–93. doi: 10.1042/CS20220531
7. Long H, Lichtnekert J, Andrassy J, Schraml BU, Romagnani P, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes account for organ injury, regeneration or atrophy. Front Immunol. (2023) 14:1194988. doi: 10.3389/fimmu.2023.1194988
8. Li X, Liu Y, Tang Y, Xia Z. Transformation of macrophages into myofibroblasts in fibrosis-related diseases: emerging biological concepts and potential mechanism. Front Immunol. (2024) 15:1474688. doi: 10.3389/fimmu.2024.1474688
9. Gu Q, Qi A, Wang N, Zhou Z, Zhou X. Macrophage dynamics in prostate cancer: Molecular to therapeutic insights. BioMed Pharmacother. (2024) 177:117002. doi: 10.1016/j.biopha.2024.117002
10. Guthridge JM, Wagner CA, James JA. The promise of precision medicine in rheumatology. Nat Med. (2022) 28:1363–71. doi: 10.1038/s41591-022-01880-6
11. Wilkinson HN, Hardman MJ. Wound healing: cellular mechanisms and pathological outcomes. Open Biol. (2020) 10:200223. doi: 10.1098/rsob.200223
12. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: A cellular perspective. Physiol Rev. (2019) 99:665–706. doi: 10.1152/physrev.00067.2017
13. Wang Z, Qi F, Luo H, Xu G, Wang D. Inflammatory microenvironment of skin wounds. Front Immunol. (2022) 13:789274. doi: 10.3389/fimmu.2022.789274
14. Chaudhary PK, Kim S, Kim S. An insight into recent advances on platelet function in health and disease. Int J Mol Sci. (2022) 23:6022. doi: 10.3390/ijms23116022
15. Lim HY, O’Malley C, Donnan G, Nandurkar H, Ho P. A review of global coagulation assays - Is there a role in thrombosis risk prediction. Thromb Res. (2019) 179:45–55. doi: 10.1016/j.thromres.2019.04.033
16. Scridon A. Platelets and their role in hemostasis and thrombosis-from physiology to pathophysiology and therapeutic implications. Int J Mol Sci. (2022) 23:12772. doi: 10.3390/ijms232112772
18. Pixley JN, Cook MK, Singh R, Larrondo J, McMichael AJ. A comprehensive review of platelet-rich plasma for the treatment of dermatologic disorders. J Dermatol Treat. (2023) 34:2142035. doi: 10.1080/09546634.2022.2142035
19. Zhang Y, Wang Z, Zong C, Gu X, Fan S, Xu L, et al. Platelet-rich plasma attenuates the severity of joint capsule fibrosis following post-traumatic joint contracture in rats. Front Bioeng Biotechnol. (2022) 10:1078527. doi: 10.3389/fbioe.2022.1078527
20. Bharadia SK, Burnett L, Gabriel V. Hypertrophic scar. Phys Med Rehabil Clin N Am. (2023) 34:783–98. doi: 10.1016/j.pmr.2023.05.002
21. Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. (2017) 18:606. doi: 10.3390/ijms18030606
22. Dong X, Mao S, Wen H. Upregulation of proinflammatory genes in skin lesions may be the cause of keloid formation (Review). BioMed Rep. (2013) 1:833–6. doi: 10.3892/br.2013.169
23. Kidzeru EB, Lebeko M, Sharma JR, Nkengazong L, Adeola HA, Ndlovu H, et al. Immune cells and associated molecular markers in dermal fibrosis with focus on raised cutaneous scars. Exp Dermatol. (2023) 32:570–87. doi: 10.1111/exd.14734
24. Chitturi P, Leask A. The role of positional information in determining dermal fibroblast diversity. Matrix Biol. (2024) 128:31–8. doi: 10.1016/j.matbio.2024.02.009
25. Xue H, McCauley RL, Zhang W, Martini DK. Altered interleukin-6 expression in fibroblasts from hypertrophic burn scars. J Burn Care Rehabil. (2000) 21:142–6. doi: 10.1097/00004630-200021020-00010
26. Zhu M, Yang M, Yang Q, Liu W, Geng H, Pan L, et al. Chronic hypoxia-induced microvessel proliferation and basal membrane degradation in the bone marrow of rats regulated through the IL-6/JAK2/STAT3/MMP-9 pathway. BioMed Res Int. (2020) 2020:9204708. doi: 10.1155/2020/9204708
27. Zhang D, Li B, Zhao M. Therapeutic strategies by regulating interleukin family to suppress inflammation in hypertrophic scar and keloid. Front Pharmacol. (2021) 12:667763. doi: 10.3389/fphar.2021.667763
28. Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. (2020) 312:81–92. doi: 10.1007/s00403-019-01972-3
29. Liu C, Lu Y, Du P, Yang F, Guo P, Tang X, et al. Mesenchymal stem cells pretreated with proinflammatory cytokines accelerate skin wound healing by promoting macrophages migration and M2 polarization. Regener Ther. (2022) 21:192–200. doi: 10.1016/j.reth.2022.06.009
30. Chen W, Zhao S, Ita M, Li Y, Ji J, Jiang Y, et al. An Early Neutrophil Recruitment into the Infectious Site Is Critical for Bacterial Lipoprotein Tolerance-Afforded Protection against Microbial Sepsis. J Immunol. (2020) 204:408–17. doi: 10.4049/jimmunol.1801602
31. Shi J, Shi S, Xie W, Zhao M, Li Y, Zhang J, et al. IL-10 alleviates lipopolysaccharide-induced skin scarring via IL-10R/STAT3 axis regulating TLR4/NF-κB pathway in dermal fibroblasts. J Cell Mol Med. (2021) 25:1554–67. doi: 10.1111/jcmm.16250
32. Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in health and disease. Int J Mol Sci. (2019) 20:649. doi: 10.3390/ijms20030649
33. Le X, Wu WW. The therapeutic effect of Interleukin-18 on hypertrophic scar through inducing Fas ligand expression. Burns. (2021) 47:430–8. doi: 10.1016/j.burns.2020.07.008
34. Khattab FM, Samir MA. Correlation between serum IL 37 levels with keloid severity. J Cosmet Dermatol. (2020) 19:2428–31. doi: 10.1111/jocd.13290
35. Shaw TJ, Martin P. Wound repair: a showcase for cell plasticity and migration. Curr Opin Cell Biol. (2016) 42:29–37. doi: 10.1016/j.ceb.2016.04.001
36. Lian N, Li T. Growth factor pathways in hypertrophic scars: Molecular pathogenesis and therapeutic implications. BioMed Pharmacother. (2016) 84:42–50. doi: 10.1016/j.biopha.2016.09.010
37. Varkey M, Ding J, Tredget EE. Differential collagen-glycosaminoglycan matrix remodeling by superficial and deep dermal fibroblasts: potential therapeutic targets for hypertrophic scar. Biomaterials. (2011) 32:7581–91. doi: 10.1016/j.biomaterials.2011.06.070
38. Ogawa R, Akaishi S. Endothelial dysfunction may play a key role in keloid and hypertrophic scar pathogenesis - Keloids and hypertrophic scars may be vascular disorders. Med Hypotheses. (2016) 96:51–60. doi: 10.1016/j.mehy.2016.09.024
39. Shi Z, Yao C, Shui Y, Li S, Yan H. Research progress on the mechanism of angiogenesis in wound repair and regeneration. Front Physiol. (2023) 14:1284981. doi: 10.3389/fphys.2023.1284981
40. Sun K, Li YY, Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. (2021) 6:79. doi: 10.1038/s41392-020-00455-6
41. Kanchanawong P, Calderwood DA. Organization, dynamics and mechanoregulation of integrin-mediated cell-ECM adhesions. Nat Rev Mol Cell Biol. (2023) 24:142–61. doi: 10.1038/s41580-022-00531-5
42. Kümper M, Steinkamp J, Zigrino P. Metalloproteinases in dermal homeostasis. Am J Physiol Cell Physiol. (2022) 323:C1290–303. doi: 10.1152/ajpcell.00450.2021
43. Koller FL, Dozier EA, Nam KT, Swee M, Birkland TP, Parks WC, et al. Lack of MMP10 exacerbates experimental colitis and promotes development of inflammation-associated colonic dysplasia. Lab Invest. (2012) 92:1749–59. doi: 10.1038/labinvest.2012.141
44. Ulrich D, Ulrich F, Unglaub F, Piatkowski A, Pallua N. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with different types of scars and keloids. J Plast Reconstr Aesthet Surg. (2010) 63:1015–21. doi: 10.1016/j.bjps.2009.04.021
45. Xu H, Zhu Z, Hu J, Sun J, Wo Y, Wang X, et al. Downregulated cytotoxic CD8(+) T-cell identifies with the NKG2A-soluble HLA-E axis as a predictive biomarker and potential therapeutic target in keloids. Cell Mol Immunol. (2022) 19:527–39. doi: 10.1038/s41423-021-00834-1
46. Travis TE, Ghassemi P, Prindeze NJ, Moffatt LT, Carney BC, Alkhalil A, et al. Matrix metalloproteinases are differentially regulated and responsive to compression therapy in a red duroc model of hypertrophic scar. Eplasty. (2018) 18:e1.
47. Xu Y, He X, Wang Y, Jian J, Peng X, Zhou L, et al. 5-Fluorouracil reduces the fibrotic scar via inhibiting matrix metalloproteinase 9 and stabilizing microtubules after spinal cord injury. CNS Neurosci Ther. (2022) 28:2011–23. doi: 10.1111/cns.13930
48. Menchaca AD, Style CC, Lazar DA, Mushin O, Olutoye OO. Serum amyloid P attenuates hypertrophic scarring in large animal models. J Surg Res. (2023) 290:285–92. doi: 10.1016/j.jss.2023.05.013
49. Wang P, Shu B, Xu Y, Zhu J, Liu J, Zhou Z, et al. Basic fibroblast growth factor reduces scar by inhibiting the differentiation of epidermal stem cells to myofibroblasts via the Notch1/Jagged1 pathway. Stem Cell Res Ther. (2017) 8:114. doi: 10.1186/s13287-017-0549-7
50. Li B, Gao C, Diao JS, Wang DL, Chu FF, Li Y, et al. Aberrant Notch signaling contributes to hypertrophic scar formation by modulating the phenotype of keratinocytes. Exp Dermatol. (2016) 25:137–42. doi: 10.1111/exd.2016.25.issue-2
51. He T, Bai X, Jing J, Liu Y, Wang H, Zhang W, et al. Notch signal deficiency alleviates hypertrophic scar formation after wound healing through the inhibition of inflammation. Arch Biochem Biophys. (2020) 682:108286. doi: 10.1016/j.abb.2020.108286
52. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.v233.9
53. Unuvar Purcu D, Korkmaz A, Gunalp S, Helvaci DG, Erdal Y, Dogan Y, et al. Effect of stimulation time on the expression of human macrophage polarization markers. PloS One. (2022) 17:e0265196. doi: 10.1371/journal.pone.0265196
54. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
55. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
56. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
57. Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction. Annu Rev Pharmacol Toxicol. (2011) 51:267–88. doi: 10.1146/annurev.pharmtox.010909.105812
58. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
59. Wang T, Xue Y, Zhang W, Zheng Z, Peng X, Zhou Y. Collagen sponge scaffolds loaded with Trichostatin A pretreated BMSCs-derived exosomes regulate macrophage polarization to promote skin wound healing. Int J Biol Macromol. (2024) 269:131948. doi: 10.1016/j.ijbiomac.2024.131948
60. Liang X, Ding L, Ma J, Li J, Cao L, Liu H, et al. Enhanced mechanical strength and sustained drug release in carrier-free silver-coordinated anthraquinone natural antibacterial anti-inflammatory hydrogel for infectious wound healing. Adv Healthc Mater. (2024) 13:e2400841. doi: 10.1002/adhm.202400841
61. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
62. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. (2010) 184:4810–8. doi: 10.4049/jimmunol.0901368
63. Zhang Z, Tang J, Cui X, Qin B, Zhang J, Zhang L, et al. New insights and novel therapeutic potentials for macrophages in myocardial infarction. Inflammation. (2021) 44:1696–712. doi: 10.1007/s10753-021-01467-2
64. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
65. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
66. Swanton C, Bernard E, Abbosh C, André F, Auwerx J, Balmain A, et al. Embracing cancer complexity: Hallmarks of systemic disease. Cell. (2024) 187:1589–616. doi: 10.1016/j.cell.2024.02.009
67. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
68. Ley K. M1 means kill; M2 means heal. J Immunol. (2017) 199:2191–3. doi: 10.4049/jimmunol.1701135
69. Zhang H, Liu L, Liu J, Dang P, Hu S, Yuan W, et al. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. (2023) 22:58. doi: 10.1186/s12943-023-01725-x
70. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. (2021) 6:127. doi: 10.1038/s41392-021-00506-6
71. Liu H, Lv Z, Zhang G, Yan Z, Bai S, Dong D, et al. Molecular understanding and clinical aspects of tumor-associated macrophages in the immunotherapy of renal cell carcinoma. J Exp Clin Cancer Res. (2024) 43:242. doi: 10.1186/s13046-024-03164-y
72. Xia T, Fu S, Yang R, Yang K, Lei W, Yang Y, et al. Advances in the study of macrophage polarization in inflammatory immune skin diseases. J Inflammation (Lond). (2023) 20:33. doi: 10.1186/s12950-023-00360-z
73. Yao Q, He L, Bao C, Yan X, Ao J. The role of TNF-α in osteoporosis, bone repair and inflammatory bone diseases: A review. Tissue Cell. (2024) 89:102422. doi: 10.1016/j.tice.2024.102422
74. Yang Y, Wang J, Lin X, Zhang Z, Zhang M, Tang C, et al. TNF-α-licensed exosome-integrated titaniumaccelerated T2D osseointegration by promoting autophagy-regulated M2 macrophage polarization. Biochem Biophys Res Commun. (2024) 727:150316. doi: 10.1016/j.bbrc.2024.150316
75. Feng Z, Jie L, Guimin L, Xi W. Mixed lineage leukemia 1 promoted neuron apoptosis in ischemic penumbra via regulating ASK-1/TNF-α Complex. Front Neuroanat. (2020) 14:36. doi: 10.3389/fnana.2020.00036
76. Yu J, Mao Z, Zhou Z, Yuan B, Wang X. Microbiome dysbiosis occurred in hypertrophic scars is dominated by S. aureus colonization. Front Immunol. (2023) 14:1227024. doi: 10.3389/fimmu.2023.1227024
77. Shin JY, Beckett JD, Bagirzadeh R, Creamer TJ, Shah AA, McMahan Z, et al. Epigenetic activation and memory at a TGFB2 enhancer in systemic sclerosis. Sci Transl Med. (2019) 11:eaaw0790. doi: 10.1126/scitranslmed.aaw0790
78. Kim KS, Choi YJ, Jang DS, Lee S. 2-O-β-d-glucopyranosyl-4,6-dihydroxybenzaldehyde isolated from morus alba (Mulberry) fruits suppresses damage by regulating oxidative and inflammatory responses in TNF-α-induced human dermal fibroblasts. Int J Mol Sci. (2022) 23:14802. doi: 10.3390/ijms232314802
79. Lu YE, Chen YJ. Resveratrol inhibits matrix metalloproteinase-1 and -3 expression by suppressing of p300/NFκB acetylation in TNF-α-treated human dermal fibroblasts. Chem Biol Interact. (2021) 337:109395. doi: 10.1016/j.cbi.2021.109395
80. Xu H, Gan C, Xiang Z, Xiang T, Li J, Huang X, et al. Targeting the TNF-α-TNFR interaction with EGCG to block NF-κB signaling in human synovial fibroblasts. BioMed Pharmacother. (2023) 161:114575. doi: 10.1016/j.biopha.2023.114575
81. Liang Y, Lou X, Xu Y, Zheng Z. Protection of neutrophils by bone marrow mesenchymal stromal cells is enhanced by tumor-associated inflammatory cytokines. Front Immunol. (2024) 15:1361596. doi: 10.3389/fimmu.2024.1361596
82. Zhang L, Yao LN, Liu W, Chen AQ, He SM, Wei ML, et al. N-acetylcholine receptors regulate cytokines expression and neutrophils recruitment via MAPK/ERK signaling in zebrafish. Dev Comp Immunol. (2022) 128:104328. doi: 10.1016/j.dci.2021.104328
83. Noseykina EM, Schepetkin IA, Atochin DN. Molecular mechanisms for regulation of neutrophil apoptosis under normal and pathological conditions. J Evol Biochem Physiol. (2021) 57:429–50. doi: 10.1134/S0022093021030017
84. Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
85. Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood. (2011) 117:5953–62. doi: 10.1182/blood-2010-11-322206
86. Dyugovskaya L, Polyakov A, Ginsberg D, Lavie P, Lavie L. Molecular pathways of spontaneous and TNF-α-mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol. (2011) 45:154–62. doi: 10.1165/rcmb.2010-0025OC
87. Luo HR, Loison F. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol. (2008) 83:288–95. doi: 10.1002/ajh.21078
88. de Vries AC, Thio HB, de Kort WJ, Opmeer BC, van der Stok HM, de Jong EM, et al. A prospective randomized controlled trial comparing infliximab and etanercept in patients with moderate-to-severe chronic plaque-type psoriasis: the Psoriasis Infliximab vs. Etanercept Comparison Evaluation (PIECE) study. Br J Dermatol. (2017) 176:624–33. doi: 10.1111/bjd.14867
89. Constantin A, Caporali R, Edwards CJ, Fonseca JE, Iannone F, Keystone E, et al. Efficacy of subcutaneous vs intravenous infliximab in rheumatoid arthritis: a post-hoc analysis of a randomized phase III trial. Rheumatol (Oxford). (2023) 62:2838–44. doi: 10.1093/rheumatology/keac689
90. Juhl P, Bondesen S, Hawkins CL, Karsdal MA, Bay-Jensen AC, Davies MJ, et al. Dermal fibroblasts have different extracellular matrix profiles induced by TGF-β, PDGF and IL-6 in a model for skin fibrosis. Sci Rep. (2020) 10:17300. doi: 10.1038/s41598-020-74179-6
91. Wang MJ, Zhang HL, Chen F, Guo XJ, Liu QG, Hou J. The double-edged effects of IL-6 in liver regeneration, aging, inflammation, and diseases. Exp Hematol Oncol. (2024) 13:62. doi: 10.1186/s40164-024-00527-1
92. Odell ID, Agrawal K, Sefik E, Odell AV, Caves E, Kirkiles-Smith NC, et al. IL-6 trans-signaling in a humanized mouse model of scleroderma. Proc Natl Acad Sci U S A. (2023) 120:e2306965120. doi: 10.1073/pnas.2306965120
93. Gunes A, Schmitt C, Bilodeau L, Huet C, Belblidia A, Baldwin C, et al. IL-6 trans-signaling is increased in diabetes, impacted by glucolipotoxicity, and associated with liver stiffness and fibrosis in fatty liver disease. Diabetes. (2023) 72:1820–34. doi: 10.2337/db23-0171
94. Seneschall C, Law S, Roufosse C, Woodham S, Kousios A. Tocilizumab (anti-IL-6) treatment for AA renal amyloidosis in a patient with advanced chronic kidney disease, a case report. J Nephrol. (2024) 37:1147–52. doi: 10.1007/s40620-023-01845-z
95. Kaur S, Bansal Y, Kumar R, Bansal G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorg Med Chem. (2020) 28:115327. doi: 10.1016/j.bmc.2020.115327
96. Gupta S, Junquera GY, Nicassio L, Becknell B, Ching CB. Trans IL-6 signaling does not appear to play a role in renal scarring after urinary tract infection. J Pediatr Urol. (2020) 16:586–91. doi: 10.1016/j.jpurol.2020.05.010
97. Kenny FN, Marcotti S, De Freitas DB, Drudi EM, Leech V, Bell RE, et al. Autocrine IL-6 drives cell and extracellular matrix anisotropy in scar fibroblasts. Matrix Biol. (2023) 123:1–16. doi: 10.1016/j.matbio.2023.08.004
98. Sanmarco LM, Ponce NE, Visconti LM, Eberhardt N, Theumer MG, Minguez ÁR, et al. IL-6 promotes M2 macrophage polarization by modulating purinergic signaling and regulates the lethal release of nitric oxide during Trypanosoma cruzi infection. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:857–69. doi: 10.1016/j.bbadis.2017.01.006
99. Montgomery A, Tam F, Gursche C, Cheneval C, Besler K, Enns W, et al. Overlapping and distinct biological effects of IL-6 classic and trans-signaling in vascular endothelial cells. Am J Physiol Cell Physiol. (2021) 320:C554–65. doi: 10.1152/ajpcell.00323.2020
100. Lindkvist M, Zegeye MM, Grenegård M, Ljungberg LU. Pleiotropic, unique and shared responses elicited by IL-6 family cytokines in human vascular endothelial cells. Int J Mol Sci. (2022) 23:1448. doi: 10.3390/ijms23031448
101. Filippini A, Tamagnone L, D’Alessio A. Endothelial cell metabolism in vascular functions. Cancers (Basel). (2022) 14:1929. doi: 10.3390/cancers14081929
102. Fahey E, Doyle SL. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol. (2019) 10:1426. doi: 10.3389/fimmu.2019.01426
103. O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R, et al. Pro-inflammatory TNFα and IL-1β differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. (2015) 12:131. doi: 10.1186/s12974-015-0346-0
104. Jiang K, Zhang Y, He F, Zhang M, Li T, Tu Z, et al. A negative feedback loop involving NF-κB/TIR8 regulates IL-1β-induced epithelial- myofibroblast transdifferentiation in human tubular cells. J Cell Commun Signal. (2021) 15:393–403. doi: 10.1007/s12079-021-00620-8
105. Urban H, Little CB. The role of fat and inflammation in the pathogenesis and management of osteoarthritis. Rheumatol (Oxford). (2018) 57:iv10–21. doi: 10.1093/rheumatology/kex399
106. Lv T, Fan X, He C, Zhu S, Xiong X, Yan W, et al. SLC7A11-ROS/αKG-AMPK axis regulates liver inflammation through mitophagy and impairs liver fibrosis and NASH progression. Redox Biol. (2024) 72:103159. doi: 10.1016/j.redox.2024.103159
107. Li H, Li Q, Hao Z, Zhang L, Zheng X, Zhu L, et al. A recombinant IL-1β vaccine attenuates bleomycin-induced pulmonary fibrosis in mice. Vaccine. (2024) 42:3774–88. doi: 10.1016/j.vaccine.2024.04.091
108. Zhao R, Zhao H, Guo Q, Mu Y, Zhang J, Su Y, et al. Edaravone protects against liver fibrosis progression via decreasing the IL-1β secretion of macrophages. Chem Biol Interact. (2022) 368:110251. doi: 10.1016/j.cbi.2022.110251
110. Li A, Wu S, Li Q, Wang Q, Chen Y. Elucidating the molecular pathways and therapeutic interventions of gaseous mediators in the context of fibrosis. Antioxid (Basel). (2024) 13:515. doi: 10.3390/antiox13050515
111. Lee TY, Lu HH, Cheng HT, Huang HC, Tsai YJ, Chang IH, et al. Delivery of nitric oxide with a pH-responsive nanocarrier for the treatment of renal fibrosis. J Control Release. (2023) 354:417–28. doi: 10.1016/j.jconrel.2022.12.059
112. King CS, Flaherty KR, Glassberg MK, Lancaster L, Raghu G, Swigris JJ, et al. A phase-2 exploratory randomized controlled trial of INOpulse in patients with fibrotic interstitial lung disease requiring oxygen. Ann Am Thorac Soc. (2022) 19:594–602. doi: 10.1513/AnnalsATS.202107-864OC
113. Ma W, Ren FC, Wang XR, Li N. Anti-Inflammatory Effect of Xanthones from Hypericum beanii on Macrophage RAW 264.7 Cells through Reduced NO Production and TNF-α, IL-1β, IL-6, and COX-2 Expression. Molecules. (2024) 29:3705. doi: 10.3390/molecules29153705
114. Gungor H, Ekici M, Onder Karayigit M, Turgut NH, Kara H, Arslanbas E. Zingerone ameliorates oxidative stress and inflammation in bleomycin-induced pulmonary fibrosis: modulation of the expression of TGF-β1 and iNOS. Naunyn Schmiedebergs Arch Pharmacol. (2020) 393:1659–70. doi: 10.1007/s00210-020-01881-7
115. Liu YL, Liu WH, Sun J, Hou TJ, Liu YM, Liu HR, et al. Mesenchymal stem cell-mediated suppression of hypertrophic scarring is p53 dependent in a rabbit ear model. Stem Cell Res Ther. (2014) 5:136. doi: 10.1186/scrt526
116. Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. (2015) 44-46:113–21. doi: 10.1016/j.matbio.2015.03.002
117. Xie XF, He LX, Hao XF, Chen B, Jia CY, Sun ZG, et al. Expression of matrix metalloproteinase-2, -9 and their inhibitor-1 in hypertrophic scars. Zhonghua Shao Shang Za Zhi. (2007) 23:444–6.
118. Zarei H, Tamri P, Asl SS, Soleimani M, Moradkhani S. Hydroalcoholic extract of scrophularia striata attenuates hypertrophic scar, suppresses collagen synthesis, and stimulates MMP2 and 9 gene expression in rabbit ear model. J Pharmacopuncture. (2022) 25:258–67. doi: 10.3831/KPI.2022.25.3.258
119. Hernández-Bule ML, Toledano-Macías E, Pérez-González LA, Martínez-Pascual MA, Fernández-Guarino M. Anti-fibrotic effects of RF electric currents. Int J Mol Sci. (2023) 24:10986. doi: 10.3390/ijms241310986
120. Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: A historical perspective. Cold Spring Harb Perspect Biol. (2016) 8:a021865. doi: 10.1101/cshperspect.a021865
121. Zhang T, Wang XF, Wang ZC, Lou D, Fang QQ, Hu YY, et al. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. BioMed Pharmacother. (2020) 129:110287. doi: 10.1016/j.biopha.2020.110287
122. Ilieş RF, Aioanei CS, Cătană A, Halmagyi SR, Lukacs I, Tokes RE, et al. Involvement of COL5A2 and TGF-β1 in pathological scarring. Exp Ther Med. (2021) 22:1067. doi: 10.3892/etm.2021.10501
123. Qu Z, Chen Y, Du K, Qiao J, Chen L, Chen J, et al. ALA-PDT promotes the death and contractile capacity of hypertrophic scar fibroblasts through inhibiting the TGF-β1/Smad2/3/4 signaling pathway. Photodiagnosis Photodyn Ther. (2024) 45:103915. doi: 10.1016/j.pdpdt.2023.103915
124. Hinz B. The role of myofibroblasts in wound healing. Curr Res Transl Med. (2016) 64:171–7. doi: 10.1016/j.retram.2016.09.003
125. Liu C, Ni L, Li X, Rao H, Feng W, Zhu Y, et al. SETD2 deficiency promotes renal fibrosis through the TGF-β/Smad signaling pathway in the absence of VHL. Clin Transl Med. (2023) 13:e1468. doi: 10.1002/ctm2.1468
126. Kohavi L, Sprecher E, Zur E, Artzi O. The effect of tranilast 8% Liposomal gel versus placebo on post-cesarean surgical scars: A prospective double-blind split-scar study. Dermatol Surg. (2017) 43:1157–63. doi: 10.1097/DSS.0000000000001140
127. Liechty KW, Kim HB, Adzick NS, Crombleholme TM. Fetal wound repair results in scar formation in interleukin-10-deficient mice in a syngeneic murine model of scarless fetal wound repair. J Pediatr Surg. (2000) 35:866–72. doi: 10.1053/jpsu.2000.6868
128. Harsono AD, Prasetyono T, Dilogo IH. The role of interleukin 10 in keloid therapy: A literature review. Ann Plast Surg. (2022) 88:617–21. doi: 10.1097/SAP.0000000000003044
129. Xie F, Teng L, Xu J, Lu J, Zhang C, Yang L, et al. Interleukin-10 modified bone marrow mesenchymal stem cells prevent hypertrophic scar formation by inhibiting inflammation. Pharmazie. (2020) 75:571–5. doi: 10.1591/ph.2020.0572
130. Xie F, Teng L, Lu J, Xu J, Zhang C, Yang L, et al. Interleukin-10-modified adipose-derived mesenchymal stem cells prevent hypertrophic scar formation via regulating the biological characteristics of fibroblasts and inflammation. Mediators Inflamm. (2022) 2022:6368311. doi: 10.1155/2022/6368311
131. Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. (2018) 59:455–67.
132. Namjoo M, Ghafouri H, Assareh E, Aref AR, Mostafavi E, Hamrahi Mohsen A, et al. A VEGFB-based peptidomimetic inhibits VEGFR2-mediated PI3K/akt/mTOR and PLCγ/ERK signaling and elicits apoptotic, antiangiogenic, and antitumor activities. Pharm (Basel). (2023) 16:906. doi: 10.3390/ph16060906
133. Wang X, Bove AM, Simone G, Ma B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front Cell Dev Biol. (2020) 8:599281. doi: 10.3389/fcell.2020.599281
134. Hua Y, Wang K, Huo Y, Zhuang Y, Wang Y, Fang W, et al. Four-dimensional hydrogel dressing adaptable to the urethral microenvironment for scarless urethral reconstruction. Nat Commun. (2023) 14:7632. doi: 10.1038/s41467-023-43421-w
135. Li S, Fan H, Liu L, Ling J, Wu Y. Inhibition of Notch signaling pathway reduces angiogenesis in hypertrophic scar. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2021) 46:1195–202. doi: 10.11817/j.issn.1672-7347.2021.210234
136. Christoffersson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
137. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
139. Cheng MF, Abdullah FS, Buechler MB. Essential growth factor receptors for fibroblast homeostasis and activation: Fibroblast Growth Factor Receptor (FGFR), Platelet Derived Growth Factor Receptor (PDGFR), and Transforming Growth Factor β Receptor (TGFβR). F1000Res. (2024) 13:120. doi: 10.12688/f1000research
140. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.v214:2
141. Li Y, Ren HT. Endostatin inhibits fibrosis by modulating the PDGFR/ERK signal pathway: an in vitro study. J Zhejiang Univ Sci B. (2017) 18:994–1001. doi: 10.1631/jzus.B1700052
142. Wang XQ, Song F, Liu YK. Hypertrophic scar regression is linked to the occurrence of endothelial dysfunction. PloS One. (2017) 12:e0176681. doi: 10.1371/journal.pone.0176681
143. Wang J, You J, Gong D, Xu Y, Yang B, Jiang C. PDGF-BB induces conversion, proliferation, migration, and collagen synthesis of oral mucosal fibroblasts through PDGFR-β/PI3K/AKT signaling pathway. Cancer biomark. (2021) 30:407–15. doi: 10.3233/CBM-201681
144. Yamada K, Hamashima T, Ishii Y, Yamamoto S, Okuno N, Yoshida N, et al. Different PDGF receptor dimers drive distinct migration modes of the mouse skin fibroblast. Cell Physiol Biochem. (2018) 51:1461–79. doi: 10.1159/000495594
145. Tiede S, Ernst N, Bayat A, Paus R, Tronnier V, Zechel C. Basic fibroblast growth factor: a potential new therapeutic tool for the treatment of hypertrophic and keloid scars. Ann Anat. (2009) 191:33–44. doi: 10.1016/j.aanat.2008.10.001
146. Onimaru M, Yonemitsu Y, Fujii T, Tanii M, Nakano T, Nakagawa K, et al. VEGF-C regulates lymphangiogenesis and capillary stability by regulation of PDGF-B. Am J Physiol Heart Circ Physiol. (2009) 297:H1685–96. doi: 10.1152/ajpheart.00015.2009
147. Ai JY, Liu CF, Zhang W, Rao GW. Current status of drugs targeting PDGF/PDGFR. Drug Discovery Today. (2024) 29:103989. doi: 10.1016/j.drudis.2024.103989
148. Weitkamp B, Cullen P, Plenz G, Robenek H, Rauterberg J. Human macrophages synthesize type VIII collagen in vitro and in the atherosclerotic plaque. FASEB J. (1999) 13:1445–57. doi: 10.1096/fasebj.13.11.1445
149. Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. (2008) 180:5707–19. doi: 10.4049/jimmunol.180.8.5707
150. Xia S, Huang Y, Zhang Y, Zhang M, Zhao K, Han P, et al. Role of macrophage-to-myofibroblast transition in chronic liver injury and liver fibrosis. Eur J Med Res. (2023) 28:502. doi: 10.1186/s40001-023-01488-7
151. Little K, Llorián-Salvador M, Tang M, Du X, Marry S, Chen M, et al. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J Neuroinflammation. (2020) 17:355. doi: 10.1186/s12974-020-02033-7
152. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367:eaau6977. doi: 10.1126/science.aau6977
153. Marie PP, Fan SJ, Mason J, Wells A, Mendes CC, Wainwright SM, et al. Accessory ESCRT-III proteins are conserved and selective regulators of Rab11a-exosome formation. J Extracell Vesicles. (2023) 12:e12311. doi: 10.1002/jev2.12311
154. Zhu Z, Chen B, Peng L, Gao S, Guo J, Zhu X. Blockade of LINC01605-enriched exosome generation in M2 macrophages impairs M2 macrophage-induced proliferation, migration, and invasion of human dermal fibroblasts. Int J Immunopathol Pharmacol. (2021) 35:20587384211016724. doi: 10.1177/20587384211016724
155. Chen J, Zhou R, Liang Y, Fu X, Wang D, Wang C. Blockade of lncRNA-ASLNCS5088-enriched exosome generation in M2 macrophages by GW4869 dampens the effect of M2 macrophages on orchestrating fibroblast activation. FASEB J. (2019) 33:12200–12. doi: 10.1096/fj.201901610
156. Zhu X, Zhao Y, Liu Y, Shi W, Yang J, Liu Z, et al. Macrophages release IL11-containing filopodial tip vesicles and contribute to renal interstitial inflammation. Cell Commun Signal. (2023) 21:293. doi: 10.1186/s12964-023-01327-6
157. Khorsandi K, Hosseinzadeh R, Esfahani H, Zandsalimi K, Shahidi FK, Abrahamse H. Accelerating skin regeneration and wound healing by controlled ROS from photodynamic treatment. Inflammation Regen. (2022) 42:40. doi: 10.1186/s41232-022-00226-6
158. Li X, Wang X, Shen T, Xiong J, Ma Q, Guo G, et al. Advances in photodynamic therapy of pathologic scar. Photodiagnosis Photodyn Ther. (2024) 46:104040. doi: 10.1016/j.pdpdt.2024.104040
159. Chen D, Zhang Y, Long W, Chai L, Myint TP, Zhou W, et al. Visible light-driven photodynamic therapy for hypertrophic scars with MOF armored microneedles patch. Front Chem. (2023) 11:1128255. doi: 10.3389/fchem.2023.1128255
160. Huang Y, Peng T, Hu W, Gao X, Chen Y, Zhang Q, et al. Fully armed photodynamic therapy with spear and shear for topical deep hypertrophic scar treatment. J Control Release. (2022) 343:408–19. doi: 10.1016/j.jconrel.2022.01.043
161. Zivari-Ghader T, Hamishehkar H, Shokouhi B, Kosari-Nasab M, Farahpour MR, Memar MY, et al. Chitosan-alginate hydrogel enriched with hypericum perforatum callus extract for improved wound healing and scar inhibition. ACS Appl Mater Interfaces. (2024) 16:67344–61. doi: 10.1021/acsami.4c15091
162. Chen Z, Hu X, Lin Z, Mao H, Qiu Z, Xiang K, et al. Layered gelMA/PEGDA hydrogel microneedle patch as an intradermal delivery system for hypertrophic scar treatment. ACS Appl Mater Interfaces. (2023) 15:43309–20. doi: 10.1021/acsami.3c06800
163. Zhang C, Wang T, Zhang L, Chen P, Tang S, Chen A, et al. Combination of lyophilized adipose-derived stem cell concentrated conditioned medium and polysaccharide hydrogel in the inhibition of hypertrophic scarring. Stem Cell Res Ther. (2021) 12:23. doi: 10.1186/s13287-020-02061-3
164. Liu X, Sun Y, Wang J, Kang Y, Wang Z, Cao W, et al. A tough, antibacterial and antioxidant hydrogel dressing accelerates wound healing and suppresses hypertrophic scar formation in infected wounds. Bioact Mater. (2024) 34:269–81. doi: 10.1016/j.bioactmat.2023.12.019
165. Fu D, Huang J, Wu X, Li Y, Zhang Y, Chen L, et al. Shape-fixing hydrogel promotes scarless healing of wounds under tension. Acta Biomater. (2024) 183:173–90. doi: 10.1016/j.actbio.2024.05.036
166. Zhang Z, Fan C, Xu Q, Guo F, Li W, Zeng Z, et al. A new strategy to inhibit scar formation by accelerating normal healing using silicate bioactive materials. Adv Sci (Weinh). (2024) 11:e2407718. doi: 10.1002/advs.202407718
167. Zhang CZ, Niu J, Chong YS, Huang YF, Chu Y, Xie SY, et al. Porous microspheres as promising vehicles for the topical delivery of poorly soluble asiaticoside accelerate wound healing and inhibit scar formation in vitro &in vivo. Eur J Pharm Biopharm. (2016) 109:1–13. doi: 10.1016/j.ejpb.2016.09.005
168. Ouyang XK, Zhao L, Jiang F, Ling J, Yang LY, Wang N. Cellulose nanocrystal/calcium alginate-based porous microspheres for rapid hemostasis and wound healing. Carbohydr Polym. (2022) 293:119688. doi: 10.1016/j.carbpol.2022.119688
169. Kumar PM, Ghosh A. Development and evaluation of silver sulfadiazine loaded microsponge based gel for partial thickness (second degree) burn wounds. Eur J Pharm Sci. (2017) 96:243–54. doi: 10.1016/j.ejps.2016.09.038
170. Patole VC, Awari D, Chaudhari S. Resveratrol-loaded microsponge gel for wound healing: in vitro and in vivo characterization. Turk J Pharm Sci. (2023) 20:23–34. doi: 10.4274/tjps.galenos.2022.93275
171. Shi J, Wu Y, Guo S, Zhang H, Chen G, Xu X. The efficacy of anti-VEGF antibody-modified liposomes loaded with paeonol in the prevention and treatment of hypertrophic scars. Drug Dev Ind Pharm. (2019) 45:439–55. doi: 10.1080/03639045.2018.1546315
172. Xie P, Dolivo DM, Jia S, Cheng X, Salcido J, Galiano RD, et al. Liposome-encapsulated statins reduce hypertrophic scarring through topical application. Wound Repair Regen. (2020) 28:460–9. doi: 10.1111/wrr.12811
173. Zhang Z, Chen J, Huang J, Wo Y, Zhang Y, Chen X. Experimental study of 5-fluorouracil encapsulated ethosomes combined with CO2 fractional laser to treat hypertrophic scar. Nanoscale Res Lett. (2018) 13:26. doi: 10.1186/s11671-017-2425-x
174. Yu Z, Meng X, Zhang S, Wang X, Chen Y, Min P, et al. IR-808 loaded nanoethosomes for aggregation-enhanced synergistic transdermal photodynamic/photothermal treatment of hypertrophic scars. Biomater Sci. (2021) 10:158–66. doi: 10.1039/D1BM01555A
175. Zhang H, Zang C, Zhao W, Zhang L, Liu R, Feng Z, et al. Exosome derived from mesenchymal stem cells alleviates hypertrophic scar by inhibiting the fibroblasts via TNFSF-13/HSPG2 signaling pathway. Int J Nanomed. (2023) 18:7047–63. doi: 10.2147/IJN.S433510
176. Yuan R, Dai X, Li Y, Li C, Liu L. Exosomes from miR-29a-modified adipose-derived mesenchymal stem cells reduce excessive scar formation by inhibiting TGF-β2/Smad3 signaling. Mol Med Rep. (2021) 24:758. doi: 10.3892/mmr.2021.12398
177. Chen J, Yu W, Xiao C, Su N, Han Y, Zhai L, et al. Exosome from adipose-derived mesenchymal stem cells attenuates scar formation through microRNA-181a/SIRT1 axis. Arch Biochem Biophys. (2023) 746:109733. doi: 10.1016/j.abb.2023.109733
178. Cui HS, Joo SY, Lee SY, Cho YS, Kim DH, Seo CH. Effect of hypertrophic scar fibroblast-derived exosomes on keratinocytes of normal human skin. Int J Mol Sci. (2023) 24:6132. doi: 10.3390/ijms24076132
179. Cui HS, Kim DH, Joo SY, Cho YS, Kim JB, Seo CH. Exosomes derived from human hypertrophic scar fibroblasts induces smad and TAK1 signaling in normal dermal fibroblasts. Arch Biochem Biophys. (2022) 722:109215. doi: 10.1016/j.abb.2022.109215
180. Wang P, Peng Z, Yu L, Liu Y, Wang H, Zhou Z, et al. Verteporfin-loaded bioadhesive nanoparticles for the prevention of hypertrophic scar. Small Methods. (2024) 8:e2301295. doi: 10.1002/smtd.202301295
181. Zuo J, Ma S. Resveratrol-laden mesoporous silica nanoparticles regulate the autophagy and apoptosis via ROS-mediated p38-MAPK/HIF-1a/p53 signaling in hypertrophic scar fibroblasts. Heliyon. (2024) 10:e24985. doi: 10.1016/j.heliyon.2024.e24985
182. Jiang K, Chen Y, Zhao D, Cheng J, Mo F, Ji B, et al. A facile and efficient approach for hypertrophic scar therapy via DNA-based transdermal drug delivery. Nanoscale. (2020) 12:18682–91. doi: 10.1039/D0NR04751A
183. Dong Y, Wang H, Zhang Y, Wu Y, Lu L, Yu H, et al. NIR-II light based combinatorial management of hypertrophic scar by inducing autophagy in fibroblasts. J Nanobiotechnol. (2024) 22:625. doi: 10.1186/s12951-024-02876-9
184. Xiao Y, Xu D, Song H, Shu F, Wei P, Yang X, et al. Cuprous oxide nanoparticles reduces hypertrophic scarring by inducing fibroblast apoptosis. Int J Nanomed. (2019) 14:5989–6000. doi: 10.2147/IJN.S196794
185. Jiang Z, Zhao L, He F, Tan H, Li Y, Tang Y, et al. Palmatine-loaded electrospun poly(ϵ-caprolactone)/gelatin nanofibrous scaffolds accelerate wound healing and inhibit hypertrophic scar formation in a rabbit ear model. J Biomater Appl. (2021) 35:869–86. doi: 10.1177/0885328220950060
186. Sun X, Cheng L, Zhu W, Hu C, Jin R, Sun B, et al. Use of ginsenoside Rg3-loaded electrospun PLGA fibrous membranes as wound cover induces healing and inhibits hypertrophic scar formation of the skin. Colloids Surf B Biointerfaces. (2014) 115:61–70. doi: 10.1016/j.colsurfb.2013.11.030
187. Xiang Y, Fan B, Shang P, Ding R, Du J, Zhu T, et al. VR23 and bisdemethoxycurcumin enhanced nanofiber niche with durable bidirectional functions for promoting wound repair and inhibiting scar formation. Small Methods. (2024) 8:e2400273. doi: 10.1002/smtd.202400273
188. Tottoli EM, Benedetti L, Chiesa E, Pisani S, Bruni G, Genta I, et al. Electrospun naringin-loaded fibers for preventing scar formation during wound healing. Pharmaceutics. (2023) 15:747. doi: 10.3390/pharmaceutics15030747
189. Tottoli EM, Benedetti L, Riva F, Chiesa E, Pisani S, Bruni G, et al. Electrospun fibers loaded with pirfenidone: an innovative approach for scar modulation in complex wounds. Polymers (Basel). (2023) 15:4045. doi: 10.3390/polym15204045
190. Ogawa R. Update on hypertrophic scar management in burn patients. Clin Plast Surg. (2024) 51:349–54. doi: 10.1016/j.cps.2024.02.001
191. Bronte J, Zhou C, Vempati A, Tam C, Khong J, Hazany S, et al. A comprehensive review of non-surgical treatments for hypertrophic and keloid scars in skin of color. Clin Cosmet Investig Dermatol. (2024) 17:1459–69. doi: 10.2147/CCID.S470997
192. Chen Y, Chen K, Zhong S, Wang J, Yu Z, Sun X, et al. Transdermal transfersome nanogels control hypertrophic scar formation via synergy of macrophage phenotype-switching and anti-fibrosis effect. Adv Sci (Weinh). (2024) 11:e2305468. doi: 10.1002/advs.202305468
193. Xu Y, Bian Q, Zhang Y, Zhang Y, Li D, Ma X, et al. Single-dose of integrated bilayer microneedles for enhanced hypertrophic scar therapy with rapid anti-inflammatory and sustained inhibition of myofibroblasts. Biomaterials. (2024) 312:122742. doi: 10.1016/j.biomaterials.2024.122742
194. Li T, Chen W, Zhang Q, Deng C. Human-specific gene CHRFAM7A mediates M2 macrophage polarization via the Notch pathway to ameliorate hypertrophic scar formation. BioMed Pharmacother. (2020) 131:110611. doi: 10.1016/j.biopha.2020.110611
195. Zhang J, He Z, Xiong C, Yao Y, Zhang C, Yao W, et al. SHH induces macrophage oxidative phosphorylation and efferocytosis to promote scar formation. Cell Commun Signal. (2024) 22:336. doi: 10.1186/s12964-024-01692-w
196. Rahmani-Neishaboor E, Jallili R, Hartwell R, Leung V, Carr N, Ghahary A. Topical application of a film-forming emulgel dressing that controls the release of stratifin and acetylsalicylic acid and improves/prevents hypertrophic scarring. Wound Repair Regen. (2013) 21:55–65. doi: 10.1111/j.1524-475X.2012.00857.x
197. Song JY, Truong DV, Yang BS. Quercetin shows the pharmacological activity to simultaneously downregulate the inflammatory and fibrotic responses to tissue injury in association with its ability to target multi-kinases. Pharmacology. (2018) 102:142–53. doi: 10.1159/000490417
198. Huang PP, Zhang R, Zhang XF, Xu ZT, Zeng DC, Sun FB, et al. Effects of ultrashort wave diathermy on skin wounds in rabbit ears. Connect Tissue Res. (2023) 64:569–78. doi: 10.1080/03008207.2023.2242655
199. Jun-Zeng, Huang TY, Wang ZZ, Gong YF, Liu XC, Zhang XM, et al. Scar-reducing effects of gambogenic acid on skin wounds in rabbit ears. Int Immunopharmacol. (2021) 90:107200. doi: 10.1016/j.intimp.2020.107200
200. Fei H, Qian Y, Pan T, Wei Y, Hu Y. Curcumin alleviates hypertrophic scarring by inhibiting fibroblast activation and regulating tissue inflammation. J Cosmet Dermatol. (2024) 23:227–35. doi: 10.1111/jocd.15905
201. Dolivo D, Rodrigues A, Sun L, Hou C, Li Y, Chung E, et al. Simvastatin cream alleviates dermal fibrosis in a rabbit ear hypertrophic scar model. J Cosmet Dermatol. (2023) 22:534–41. doi: 10.1111/jocd.15142
202. Sun X, Phan TN, Jung SH, Kim SY, Cho JU, Lee H, et al. LCB 03-0110, a novel pan-discoidin domain receptor/c-Src family tyrosine kinase inhibitor, suppresses scar formation by inhibiting fibroblast and macrophage activation. J Pharmacol Exp Ther. (2012) 340:510–9. doi: 10.1124/jpet.111.187328
203. Aoki M, Kondo A, Matsunaga N, Honda A, Okubo Y, Takabe K, et al. The immunosuppressant fingolimod (FTY720) for the treatment of mechanical force-induced abnormal scars. J Immunol Res. (2020) 2020:7057195. doi: 10.1155/2020/7057195
204. Liu XJ, Xu MJ, Fan ST, Wu Z, Li J, Yang XM, et al. Xiamenmycin attenuates hypertrophic scars by suppressing local inflammation and the effects of mechanical stress. J Invest Dermatol. (2013) 133:1351–60. doi: 10.1038/jid.2012.486
205. Ding J, Ma Z, Liu H, Kwan P, Iwashina T, Shankowsky HA, et al. The therapeutic potential of a C-X-C chemokine receptor type 4 (CXCR-4) antagonist on hypertrophic scarring in vivo. Wound Repair Regener. (2014) 22:622–30. doi: 10.1111/wrr.2014.22.issue-5
206. Condorelli AG, El Hachem M, Zambruno G, Nystrom A, Candi E, Castiglia D. Notch-ing up knowledge on molecular mechanisms of skin fibrosis: focus on the multifaceted Notch signaling pathway. J BioMed Sci. (2021) 28:36. doi: 10.1186/s12929-021-00732-8
207. Xia Z, Wang J, Yang S, Liu C, Qin S, Li W, et al. Emodin alleviates hypertrophic scar formation by suppressing macrophage polarization and inhibiting the Notch and TGF-β pathways in macrophages. Braz J Med Biol Res. (2021) 54:e11184. doi: 10.1590/1414-431x2021e11184
208. Marjani AA, Nader ND, Aghanejad A. Exosomes as targeted diagnostic biomarkers: Recent studies and trends. Life Sci. (2024) 354:122985. doi: 10.1016/j.lfs.2024.122985
209. Boussios S, Ovsepian SV. Exosomes in renal cell cancer: diagnostic and therapeutic nanovehicles. Technol Cancer Res Treat. (2024) 23:15330338241275403. doi: 10.1177/15330338241275403
210. Shao Y, Jiang Y, Wang J, Li H, Li C, Zhang D. Inhibition of circulating exosomes release with GW4869 mitigates severe acute pancreatitis-stimulated intestinal barrier damage through suppressing NLRP3 inflammasome-mediated pyroptosis. Int Immunopharmacol. (2024) 126:111301. doi: 10.1016/j.intimp.2023.111301
Keywords: hypertrophic scar, macrophages, inflammation, signaling molecules, M1, M2
Citation: Shen L, Zhou Y, Gong J, Fan H and Liu L (2025) The role of macrophages in hypertrophic scarring: molecular to therapeutic insights. Front. Immunol. 16:1503985. doi: 10.3389/fimmu.2025.1503985
Received: 30 September 2024; Accepted: 14 March 2025;
Published: 28 March 2025.
Edited by:
Diana Crisan, University Hospital Ulm, GermanyReviewed by:
William D. Shipman, Skin & Beauty Center- Board Certified Dermatologist, United StatesCopyright © 2025 Shen, Zhou, Gong, Fan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongqiao Fan, MzEwMTAxQGhudWNtLmVkdS5jbg==; Lifang Liu, bGlmYW5nX2xpdTIwMjRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.