 Lucy G. Fry1
Lucy G. Fry1 Charity L. Washam2,3Hayden Roys1Anne K. Bowlin1Gopinath Venugopal1Jordan T. Bird1,2
Charity L. Washam2,3Hayden Roys1Anne K. Bowlin1Gopinath Venugopal1Jordan T. Bird1,2 Stephanie D. Byrum2,3
Stephanie D. Byrum2,3 Tiffany Weinkopff1*
Tiffany Weinkopff1*- 1Department of Microbiology and Immunology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 2Department of Biochemistry and Molecular Biology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 3Arkansas Children’s Research Institute, Little Rock, AR, United States
Cutaneous leishmaniasis (CL) contributes significantly to the global burden of neglected tropical diseases, with 12 million people currently infected with Leishmania parasites. CL encompasses a range of disease manifestations, from self-healing skin lesions to permanent disfigurations. Currently there is no vaccine available, and many patients are refractory to treatment, emphasizing the need for new therapeutic targets. Previous work demonstrated macrophage HIF-α-mediated lymphangiogenesis is necessary to achieve efficient wound resolution during murine L. major infection. Here, we investigate the role of macrophage HIF-α signaling independent of lymphangiogenesis. We sought to determine the relative contributions of the parasite and the host-mediated inflammation in the lesional microenvironment to myeloid HIF-α signaling. Because HIF-α activation can be detected in infected and bystander macrophages in leishmanial lesions, we hypothesize it is the host’s inflammatory response and microenvironment, rather than the parasite, that triggers HIF-α activation. To address this, macrophages from mice with intact HIF-α signaling (LysMCreARNTf/+) or mice with deleted HIF-α signaling (LysMCreARNTf/f) were subjected to RNASequencing after L. major infection and under pro-inflammatory stimulus. We report that L. major infection alone is enough to induce some minor HIF-α-dependent transcriptomic changes, while infection with L. major in combination with pro-inflammatory stimuli induces numerous transcriptomic changes that are both dependent and independent of HIF-α signaling. Additionally, by coupling transcriptomic analysis with several pathway analyses, we found HIF-α suppresses pathways involved in protein translation during L. major infection in a pro-inflammatory environment. Together these findings show L. major induces a HIF-α-dependent transcriptomic program, but HIF-α only suppresses protein translation in a pro-inflammatory environment. Thus, this work indicates the host inflammatory response, rather than the parasite, largely contributes to myeloid HIF-α signaling during Leishmania infection.
Introduction
Leishmaniasis is the family of diseases caused by infection with protozoan Leishmania parasites. Because pathology depends upon both the species of parasite and the host immune response, leishmaniasis can manifest in three main forms: cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL), and visceral leishmaniasis (VL). Leishmania parasites are transmitted via sandfly bites and are endemic in more than 90 countries across Africa, Asia, and Latin America resulting in 1-2 million new cases of leishmaniasis each year (1, 2). There is currently no human vaccine and existing treatments against Leishmania parasites are toxic to the host, difficult to administer, require a long duration, and are often ineffective (3). The lack of new treatments or vaccines has made global disease control and elimination efforts challenging, reiterating the importance of understanding the host immune response to identify potential therapeutic targets (4).
Upon a sandfly bite, parasites are taken up by macrophages in the skin. After phagocytosis, parasites reside in phagolysosomes in macrophages and begin multiplying. Controlling parasite burden is dependent on a predominant Th1 immune response where CD4+ T cells produce IFNγ which activates macrophages to kill parasites by releasing nitric oxide (NO) and reactive oxygen species (ROS) (5). During CL, neutrophils and inflammatory monocytes are initially recruited to the site of infection (6). Severity of disease is highly dependent upon both parasite burden and the host inflammatory response with excessive inflammation contributing to overall pathology and extending the duration of disease (7). Despite an effective immune response, parasites can persist at low levels in the skin for years even after dermal lesions have resolved (8).
Leishmanial lesions are characterized by hypoxia and the presence of pro-inflammatory cells and cytokines (9–12). During inflammatory hypoxia, transcription factors hypoxia-inducible factor (HIF)-1α and HIF-2α are induced by decreased oxygen availability in tissues (13). HIF-α transcription factors are master regulators of genes involved in metabolism and the cellular response to oxygen deprivation (14, 15). Upon activation, HIF-α subunits bind aryl hydrocarbon receptor nuclear translocator (ARNT; also known as HIF-1β), and ARNT/HIF-α heterodimers translocate to the nucleus where they induce the transcription of HIF-α target genes (16). HIF-α subunits can also be activated by oxygen-independent mechanisms such as TLR ligation, pro-inflammatory cytokines, or ROS stimulation (17, 18). Furthermore, under normoxic conditions LPS induces HIF-1α expression via MyD88/NFκB signaling in macrophages, and mice deficient in HIF-1α are more susceptible to a variety of bacterial and fungal infections (17, 19–22).
During CL, human lesions contain elevated levels of HIF-1α and the HIF-α target, VEGF-A (19, 23, 24). Similarly, HIF-1α and VEGF-A are also elevated in lesions following experimental murine L. major infection (25, 26). Both inflammatory signaling, such as IFNγ production, as well as hypoxia in the skin promote HIF-1α accumulation in L. major-infected macrophages, but which signal occurs first and the relative contributions of each signal to HIF-1α signaling are not known (19, 27, 28). Myeloid-specific HIF-1α-/- mice infected with L. major exhibit increased lesion sizes and parasite burdens due to impaired expression of NOS2, a HIF-1α-specific target gene (19). These data suggest activated HIF-1α in dermal myeloid cells contributes to parasite control through NO production. Additionally, mice deficient in myeloid ARNT/HIF-α signaling (LysMCreARNTf/f; missing both HIF-1α and HIF-2α pathways) infected with L. major exhibit decreased myeloid-derived NOS2 and VEGF-A which impairs lymphangiogenesis at the site of infection, resulting in larger lesion sizes, despite parasites being controlled (26). Altogether, these data suggest myeloid HIF-α signaling plays critical roles in both parasite control and lesion resolution during L. major infection.
HIF-α activation depends on the Leishmania parasite species. In contrast to L. amazonensis and L. donovani parasites, L. major parasites alone do not increase HIF-1α expression or activation under normoxic conditions (11, 19, 27, 29). Additionally, during in vivo L. major infection, both infected and bystander macrophages exhibit HIF-α activation compared to macrophages from naïve skin (28). Based on these findings, we hypothesize that during L. major infection, it is the host’s inflammatory response and microenvironment, rather than the parasite itself, that triggers HIF-α activation. To address this hypothesis, we performed transcriptomic analyses on macrophages from LysMCreARNTf/+ or LysMCreARNTf/f that either exhibit intact or impaired ARNT/HIF-α signaling in myeloid cells, respectively. LysMCreARNTf/+ or LysMCreARNTf/f macrophages were infected or not with L. major parasites and then treated or not with LPS and IFNγ to define the importance of ARNT/HIF-α signaling in response to L. major parasites in the presence or absence of a pro-inflammatory milieu. We find infection with L. major parasites induces transcriptional changes in macrophages and some of these early transcriptomic changes are absent in macrophages without HIF-α signaling. This indicates L. major induces some transcriptomic changes that are HIF-α-dependent, and L. major infection is sufficient to induce HIF-α activation in vitro, albeit minimal compared to pro-inflammatory stimuli. We discovered under inflammatory conditions, HIF-α signaling suppresses transcripts and pathways involved in translation such as ribosomal transcripts, EIF2 signaling and the ribosome pathway during infection with L. major. Additionally, we identified top enriched pathways associated with L. major infection during and apart from inflammatory conditions as well as with and without intact HIF-α signaling.
Materials and methods
Parasites
Leishmania major strain (WHO/MHOM/IL/80/Friedlin) parasites were cultured with Schneider’s insect media (Gibco) supplemented with 20% heat-inactivated fetal bovine serum (FBS) (Invitrogen), 100 U/mL penicillin/streptomycin (Sigma), and 2 mM L-glutamine (Sigma). Metacyclic promastigotes were isolated from 4-5 day old cultures using Ficoll (Sigma) gradient separation for infections (23).
Mice
C57BL/6 mice were purchased from the National Cancer Institute. Mice with a myeloid-specific ARNT conditional knockout were developed by crossing a strain expressing the LysMCre allele with another strain with a floxed ARNT conditional allele and were bred on campus in the vivarium. The LysMCreARNTf/f and LysMCreARNTf/+ mice were a gift from M. Celeste Simon (University of Pennsylvania, Philadelphia, PA). LysMCreARNTf/+ mice were used as controls for LysMCreARNTf/f mice. All animals were housed in the vivarium under pathogen-free conditions at the University of Arkansas for Medical Sciences (UAMS). All mice were infected between 6-8 weeks of age and all procedures were approved by UAMS IACUC and followed institutional guidelines.
Murine infection in vivo
For dermal ear infections in C57BL/6 mice, 2×106 promastigote Leishmania major (WHO/MHOM/IL/80/Friedlin) parasites in 10 µL PBS (Gibco) were injected intradermally into the ear. For analyses, ears were excised, dorsal and ventral sheet were separated. Ear sheets were enzymatically digested for 90 min at 37°C using 0.25 mg/mL Liberase (Roche) and 10 mg/mL DNase I (Sigma) in incomplete RPMI 1640 (Gibco). After digestion, ears were smashed through a filter to obtain a single-cell suspension (28).
Single-cell RNASequencing sample preparation
The scRNASeq samples were prepared and data was acquired as a part of a previous study (28). In short, the Arkansas Children’s Research Institute (ACRI) Genomics and Bioinformatics Core prepared NGS libraries from fresh single-cell suspensions using the 10X Genomics NextGEM 3’ assay for sequencing on the NextSeq 500 platform using Illumina SBS reagents. Trypan Blue exclusion determined cell quantity and viability. Library quality was evaluated with the Advanced Analytical Fragment Analyzer (Agilent) and Qubit (Life Technologies) instruments.
scRNASeq data analysis
Data analysis was performed as a part of a previous study (28). Briefly, the UAMS Genomics Core generated Demultiplexed fastq files which were analyzed using 10X Genomics Cell Ranger alignment and gene counting software, a self-contained scRNASeq pipeline developed by 10X Genomics. The reads were aligned to the mm10 reference transcriptomes using STAR and transcript counts were generated (30, 31). The Seurat R package processed the raw counts generated by cellranger count (32, 33). Potential doublets, low quality cells, and cells with a high percentage of mitochondrial genes were filtered out. Cells that have unique feature counts > 75th percentile plus 1.5 times the interquartile range (IQR) or < 25th percentile minus 1.5 time the IQR were filtered. Similarly, cells with mitochondrial counts falling outside the same range for mitochondrial gene percentage were filtered. After filtering, all 8 sequencing runs were merged. The counts were normalized using the LogNormalize method which log-transforms the results (28). Subsequently, the 2000 highest variable features were selected. The data was scaled, and Principal component analysis (PCA) was performed. A JackStraw procedure was implemented to determine the significant PCA components that have a strong enrichment of low p-value features.
A graph-based clustering strategy embedded cells in graph structure (34) Seurat visualized the results in t-distributed stochastic neighbor embedding (tSNE) and Uniform Manifold Approximation and Projection (UMAP) plots (35). Seurat FindNeighbors and FindClusters functions were optimized to label clusters. Seurat FindAllMarkers function finds markers that identify clusters by differential expression, defining positive markers of a single cluster compared to all other cells and comparing those to known markers of expected cell types from previous single-cell transcriptome studies. Cell type determinations were determined by manually reviewing these results, and some clusters were combined if their expression was found to be similar. From here for this work, we specifically provide Feature maps showing transcript expression of HIF-1α, HIF-2α, and corresponding target genes of these transcription factors amongst all clusters, and particularly in macrophages.
Generation of bone marrow-derived macrophages
Femurs collected from mice were soaked in 70% ethanol for 2 minutes and then flushed with 10 mL of cDMEM to extract bone marrow cells. Bone marrow cells were counted before plating 5x106 cells per 100 mm Petri dish in 10 mL of conditioned macrophage media (cDMEM with 25% L929 cell supernatants). Cells were cultured for 7 days, refreshing media at day 3. To remove the macrophages from the Petri dish, macrophages were washed with ice-cold PBS and gently removed with a cell scraper. The collected macrophages were counted and loaded into 24-well plates with 1x106 cells in 1 mL cDMEM per well.
In vitro infection of BMDM and RNASeq
Bone marrow-derived macrophages (BMDMs) were plated into 24-well plates and allowed to rest overnight. Parasites were added to the wells at a 5:1 multiplicity of infection (MOI). Extracellular parasites were washed away at 2 hours post-infection. After washing, BMDMs were cultured in media with or without 100 ng/mL LPS (Sigma) and 10 ng/mL IFNγ (Peprotech). After 8 hours, the cells washed with PBS, lysed with RLT lysis buffer for RNA extraction, and stored at -80 °C. For transcriptomic RNASeq studies, RNA was extracted following the Qiagen RNEasy Mini-Kit instructions before being subjected to RNASeq analysis. Each experiment group contained 2 or 3 samples for RNASeq analysis.
RNASeq analysis
Following demultiplexing, RNA reads were checked for sequencing quality using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and MultiQC (36)(version 1.6). The raw reads were then processed according to Lexogen’s QuantSeq data analysis pipeline with slight modification. Briefly, residual 3’ adapters, polyA read through sequences, and low quality (Q < 20) bases were trimmed using BBTools BBDuk (version 38.52) (https://sourceforge.net/projects/bbmap/). The first 12 bases were also removed per the manufacture’s recommendation. The cleaned reads (> 20bp) were then mapped to the mouse reference genome (GRCm38/mm10/ensemble release-84.38/GCA_000001635.6) using STAR (30) (version 2.6.1a), allowing up to 2 mismatches depending on the alignment length (e.g. 20-29bp, 0 mismatches; 30-50bp, 1 mismatch; 50–60+bp, 2 mismatches). Reads mapping to > 20 locations were discarded. Gene level counts were quantified using HTSeq (htseq-counts) (37) (version 0.9.1) (mode: intersection-nonempty).
Genes with unique Entrez IDs and a minimum of ~2 counts-per-million (CPM) in 4 or more samples were selected for statistical testing. This was followed by scaling normalization using the trimmed mean of M-values (TMM) method (38) to correct for compositional differences between sample libraries. Differential expression between naive and infected ears was evaluated using limma voomWithQualityWeights (39) with empirical bayes smoothing. Genes with Benjamini & Hochberg (40) adjusted p-values ≤ 0.05 and absolute fold-changes ≥ 1.5 were considered significant.
Gene Set Enrichment Analysis (GSEA) was carried out using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway databases and for each KEGG pathway, a p-value was calculated using hypergeometric test. Cut-off of both p < 0.05 and adjusted p-value/FDR value < 0.05 was applied to identify enriched KEGG pathways. DEGs that are more than 1.5-fold relative to controls were used as input, with upregulated and downregulated genes considered separately. Subsequently, the heat maps were generated using these genes with complex Heatmap. All analyses and visualizations were carried out using the statistical computing environment R version 3.6.3, RStudio version 1.2.5042, and Bioconductor version 3.11. The raw data from our bulk RNA-Seq analysis were deposited in Gene Expression Omnibus (GEO accession number— GSE273822).
Ingenuity pathway analysis
To categorize the extensive list of differentially expressed genes identified by RNASeq, we performed Ingenuity Pathway Analysis (IPA). IPA allows for the upload and analyzation of high throughput data by placing the data into biological pathways, while also building networks to represent biological systems. To perform the IPA, we inputted our list of DEGs from the RNASeq data into the IPA software (Qiagen). We used a p-value cut-off of <0.05 so that anything below that would be considered for analysis. For the fold change (FC), we used a range of FC -2 to 2 so any values outside of that range would be analyzed by IPA.
In vitro infections and DMOG treatment
Bone marrow-derived macrophages were cultured in cDMEM in polypropylene tubes overnight. Macrophages were then infected with L. major parasites at an MOI of 5:1 and extracellular parasites were washed away at 2 hour post-infection. For HIF-α stabilization, macrophages were cultured with DMOG at a concentration of 0.1 mM.
mRNA extraction and real-time PCR
mRNA was extracted with the RNeasy mini kit (Qiagen). RNA was reverse transcribed with the High-Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using SYBR green PCR Master Mix and a QuantStudio 6 Flex real-time PCR system (Life Technologies). Mouse primer sequences were selected from the PrimerBank (http://pga.mgh.harvard.edu/primerbank/): Rpl4 (forward 5′-CCCCTCATATCGGTGTACTCC-3′ and reverse 5′-ACGGCATAGGGCTGTCTGT-3′), Rpl12 (forward 5′-ACTGGAAGGGTCTCAGAATTACA-3′ and reverse 5′-TGCCGGGCAATGTTGACAA-3′), Rpl23 (forward 5′-GAAGATCCGAACGTCACCCAC -3′ and reverse 5′-GGCCTTGACATCCACAATGAA-3′), and RpsII (forward 5′-CGTGACGAAGATGAAGATGC-3′ and reverse 5′-GCACATTGAATCGCACAGTC-3′). The results were normalized to the housekeeping ribosomal protein S14 gene (RpsII) using the comparative threshold cycle method (2-ΔΔCT) for relative quantification.
Flow cytometry
To assess cell viability, macrophages infected or not with L. major and treated or not with DMOG were incubated with fixable Aqua dye (Invitrogen) for 10 min at room temperature. Cells were treated with FcγR blocking reagent (Bio X Cell) and 0.2% rat IgG for 10 minutes at 4°. Next, macrophages were surface stained with anti-CD45-AF700 (eBioscience, clone 30-F11), anti-CD11b-BV605 (Biolegend, clone M1/70), anti-CD64-BV711 (Biolegend, clone X54-5/7.1), and anti-Ly6C-PerCP-Cy5.5 (eBioscience, clone HK1.4). Surface staining was performed in Super Bright staining buffer (eBiosciences).
In vitro translation analysis
To assess translational activity, puromycin incorporation was measured using flow cytometry as previously described (41). After 24 hours of infection with L. major parasites, macrophages were treated with puromycin at a concentration of 10 μg/mL in PBS. Puromycin was detected by flow cytometry using an anti-puromycin antibody conjugated to AF647 (Sigma, MABE343-AF647) after intracellular staining with the Foxp3 kit (Life technologies).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9. Besides the scRNASeq of leishmanial lesions and total RNASeq of BMDMs where statistics are described above, a t-test was performed with p ≤ 0.05 being considered statistically significant. A Grubbs’ test was used to identify and mathematically remove outlier data points.
Results
HIF-α signaling is a hallmark of lesions following Leishmania infection, but the specific cell types in lesions undergoing HIF-α activation are not known (11, 12, 27). To identify the cell types in leishmanial lesions that express the transcription factors HIF-1α and HIF-2α as well as their transcriptional target genes, we performed scRNASeq on lesions 4 weeks after dermal L. major inoculation (28). Specifically, single cells from the ears of infected and naive mice were bar-coded and sequenced using the droplet-based 10X Genomics Chromium platform. Unbiased hierarchical clustering using Seurat was performed to identify clusters indicative of individual cell types (Figure 1A). Of the 35 distinct cell types, HIF-1α is mainly expressed in keratinocytes, fibroblasts, chondrocytes, and endothelial cells in naïve uninfected skin. In contrast, HIF-1α is predominantly expressed by infiltrating cells including T cells, neutrophils, and monocyte-derived macrophages during L. major infection (Figure 1B). Of the 35 distinct cell types, HIF-2α is mainly expressed in fibroblasts, chondrocytes, and endothelial cells in naïve uninfected skin (Figure 1B). After infection, HIF-2α retains expression in fibroblasts, chondrocytes, and endothelial cells and is additionally expressed in infiltrating T cells and monocyte-derived macrophages during L. major infection (Figure 1B).
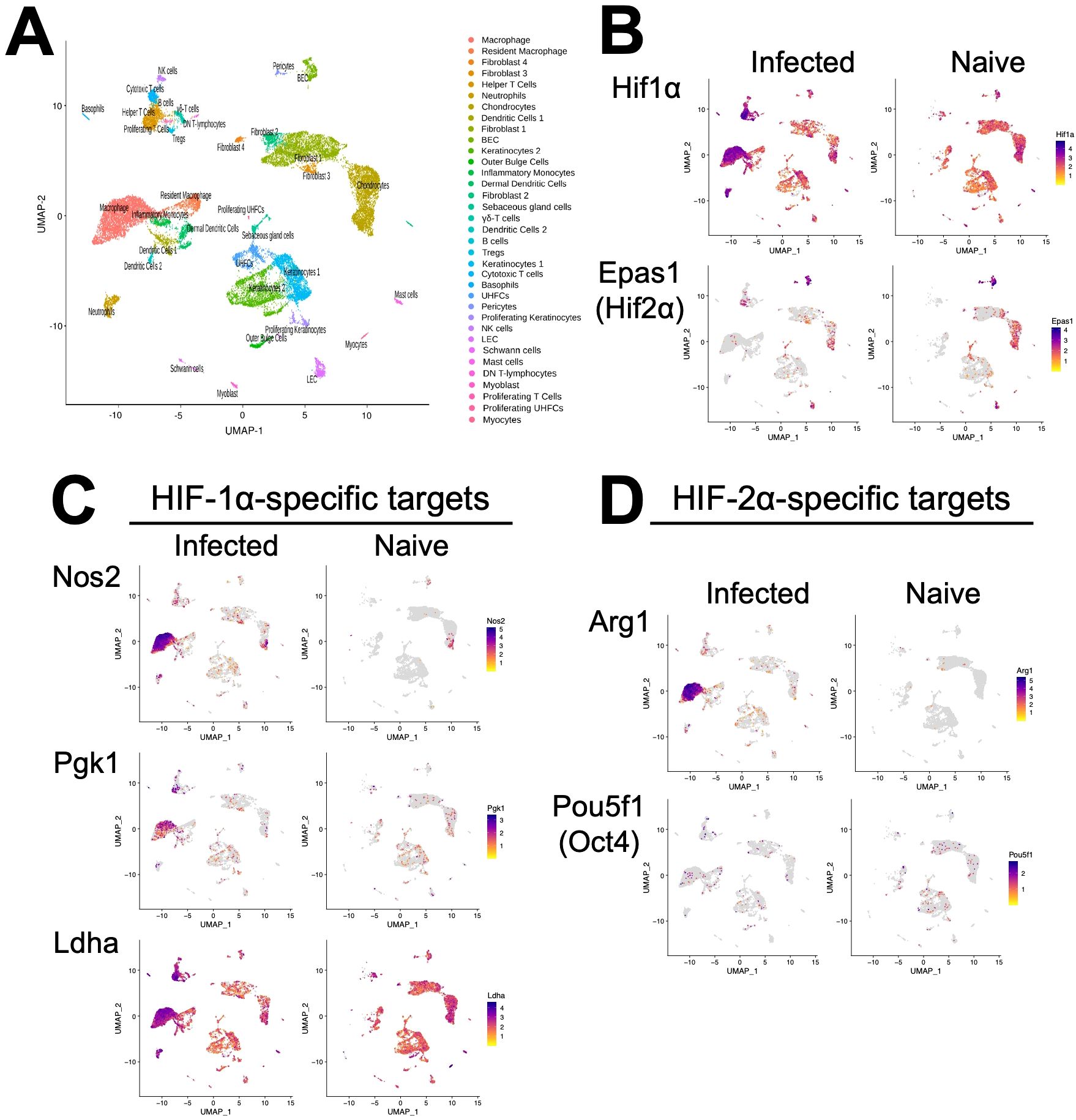
Figure 1. Single-cell RNASequencing (scRNASeq) shows HIF-α transcriptional targets are elevated in murine lesions during L. major infection. C57BL/6 mice were infected or not with 2×106 L. major parasites intradermally in the ear. At 4 weeks, infected ears and naïve uninfected control ears were digested and subjected to scRNASeq as a part of a previous study (28). (A) Uniform Manifold Approximation and Projection (UMAP) plot revealed 35 distinct cell clusters. Seurat’s FindClusters function identified each cell cluster and cell type designation to the right. To initially define cell clusters both naive and infected groups were combined, but here naive and infected groups are shown to specify transcript expression under each condition. (B) Feature plots of expression distribution for HIF-1α and HIF-2α (gene Epas1). (C) Feature plots of expression distribution for HIF-1α-specific target genes (Nos2, Pgk1, and Ldha). (D) Feature plots of expression distribution for HIF-2α-specific target genes (Arg1 and Oct4 (gene Pou5f1)). Expression levels for each gene are color-coded and overlaid onto UMAP plot. Cells with the highest expression level are colored dark purple.
Because HIF-α expression does not always correlate to HIF-α activity, we examined HIF-1α and HIF-2α transcriptional target genes as a surrogate for HIF-α activation. Besides Ldha, overall HIF-1α and HIF-2α target genes are expressed at low levels in naïve skin (Figures 1C, D). In contrast, HIF-1α-specific target genes including Nos2, Pgk1 and Ldha are dramatically increased upon infection, and these are predominantly expressed in monocyte-derived macrophages (Figure 1C). Similarly, the HIF-2α-specific target gene Arg1 is also highly expressed in monocyte-derived macrophages and Arg1 is significantly upregulated during infection (Figure 1D). However, another HIF-2α-specific target gene Pou5f1 (protein name Oct4) was only minorly expressed in monocyte-derived macrophages (Figure 1D). Altogether these transcriptomic data show that monocyte-derived macrophages exhibit HIF-1α and HIF-2α activation following L. major infection.
Given lesional monocyte-derived macrophages exhibited HIF-1α and HIF-2α activation, we evaluated the host macrophage responses during L. major infection using macrophages derived from monocytes from the bone marrow. To investigate the role of HIF-α signaling, we used macrophages from mice missing both HIF-1α and HIF-2α signaling where ARNT is deleted in myeloid cells and compared those responses to macrophages with intact HIF-1α and HIF-2α signaling (11, 27, 42). To first explore the host macrophage response in cells with intact HIF-1α and HIF-2α signaling, differential expression analysis was conducted on infected LysMCreARNTf/+ control macrophages compared to uninfected LysMCreARNTf/+ control macrophages referred to as ARNTf/+ going forward. Macrophages were infected with L. major at an MOI of 5:1. Several differentially expressed genes (DEGs) were upregulated with L. major infection including Gm15564, Gca, Stk35, and Socs1 while only Tcf4 was found to be downregulated with L. major infection (Figure 2A, Table 1).
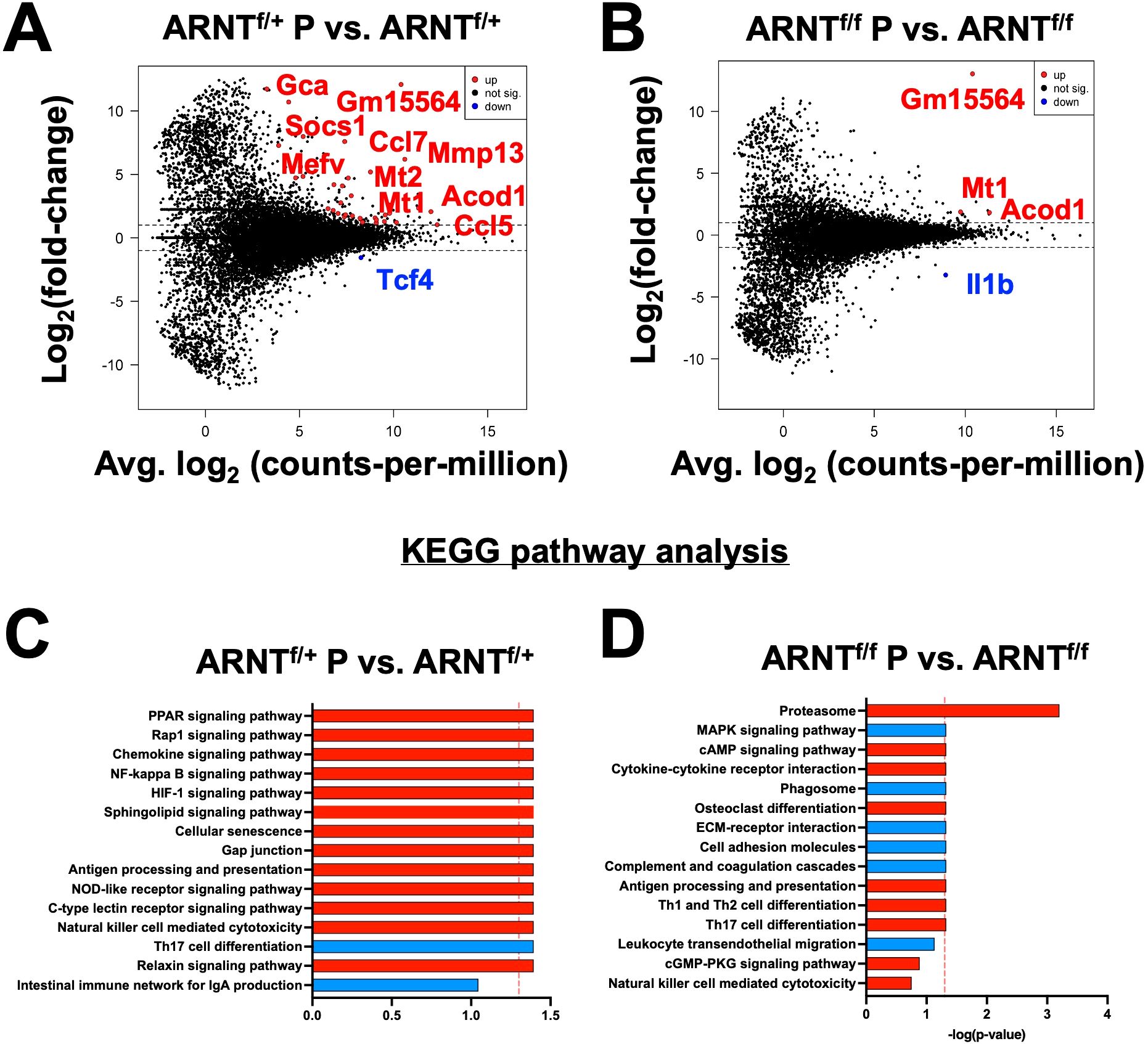
Figure 2. Infection with L. major induces transcriptional changes that are absent in infected macrophages deficient for HIF-α signaling. Macrophages infected with L. major were lysed and prepped for RNASequencing. (A) A mean-difference plot (MD plot) depicts transcripts upregulated (red) and downregulated (blue) with infection in macrophages with intact HIF-α signaling (ARNTf/+ P vs. ARNTf/+) where P indicates parasites. (B) An MD plot shows upregulated and downregulated transcripts in infected macrophages deficient for HIF-α signaling (ARNTf/f P vs. ARNTf/f). (C, D) KEGG analysis was performed to identify the top enriched pathways during infection with L. major in macrophages with or without HIF-α signaling. Upregulated pathways are shown in red and downregulated pathways are shown in blue.
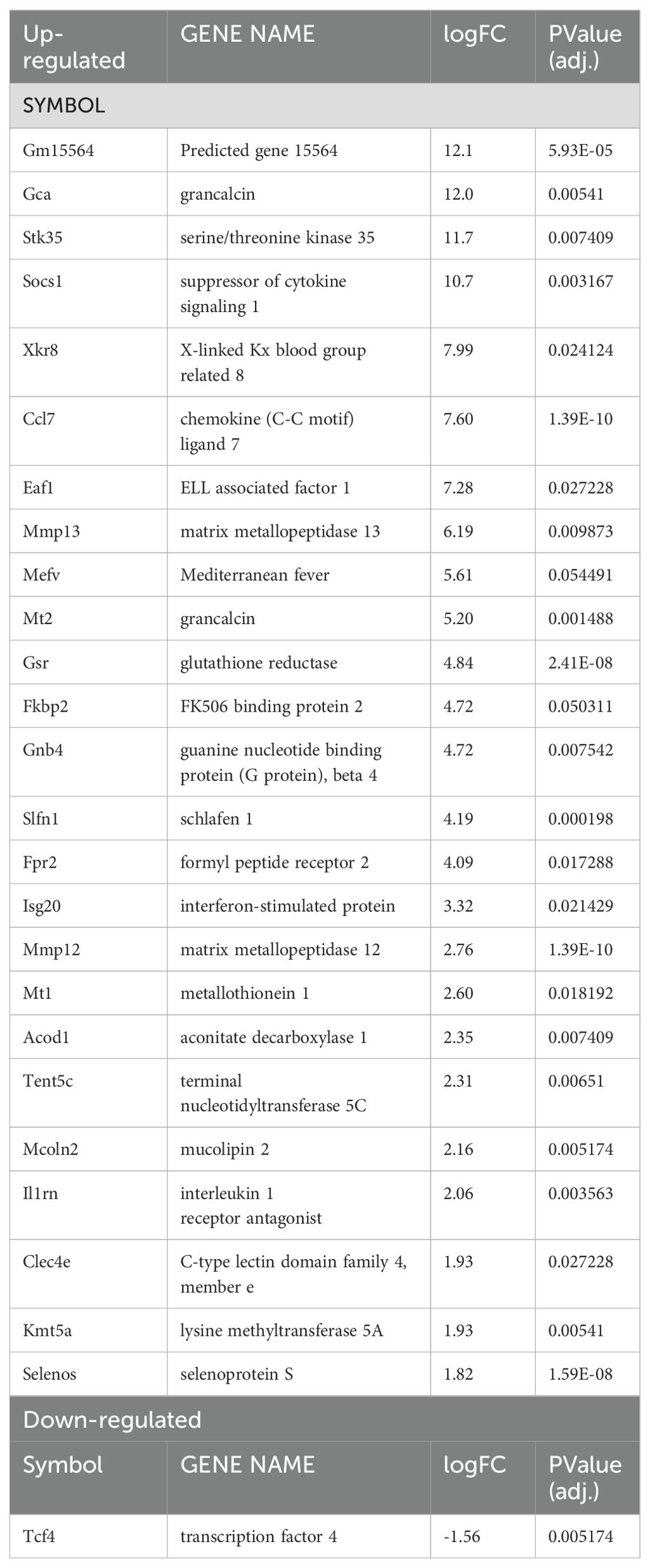
Table 1. Significantly up- or down-regulated DEGs between ARNTf/+ infected macrophages compared to ARNTf/+ uninfected macrophages.
We next investigated transcriptional changes during L. major infection in macrophages devoid of HIF-α signaling by comparing the transcriptome of infected LysMCreARNTf/f macrophages to uninfected LysMCreARNTf/f macrophages referred to as ARNTf/f for the duration of the study. Four genes were differentially expressed, including Mt1, Acod1, Il1β and a predicted gene, gm15564 (Figure 2B, Table 2). Mt1, Acod1, and Gm15564 were also upregulated with infection in HIF-α competent macrophages, indicating these transcriptional changes are independent of HIF-α signaling (Figure 2A, Table 1). Most of the transcriptomic changes seen during L. major infection were ablated in the absence of HIF-α signaling. For instance, 22 DEGs were upregulated in HIF-α competent macrophages with infection, that were not detected during infection in macrophages deficient for HIF-α signaling (Figure 2A, Table 1).
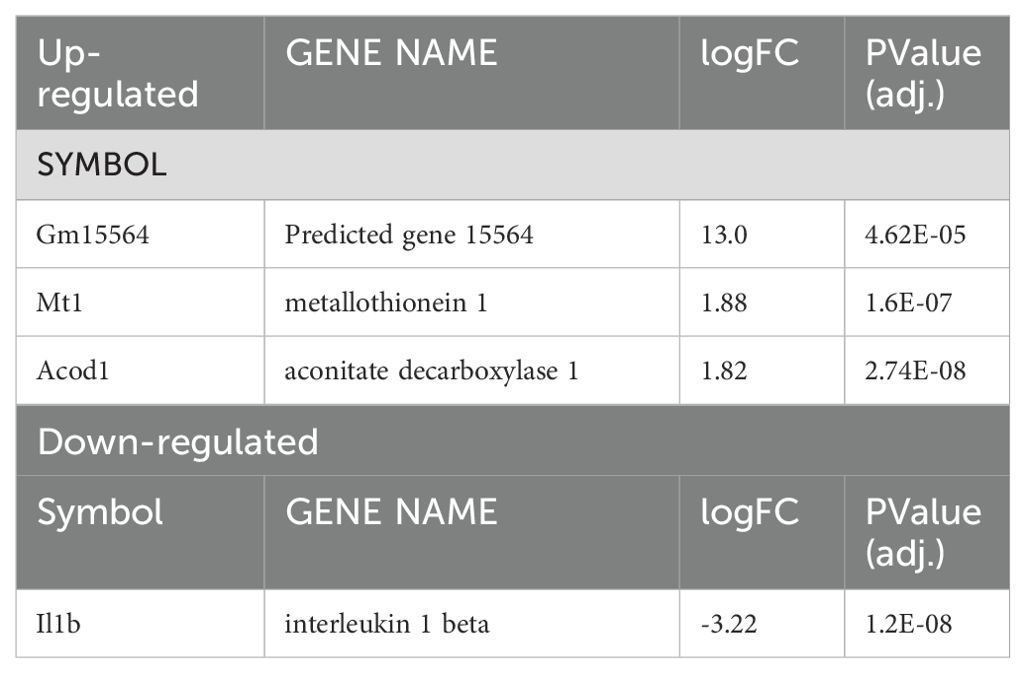
Table 2. Significantly up- or down-regulated DEGs between ARNTf/f infected macrophages compared to ARNTf/f uninfected macrophages.
Next, we analyzed enriched pathways during infection with L. major in HIF-α competent macrophages compared to their uninfected counterparts. KEGG analysis revealed several enriched pathways with L. major infection including the ‘PPAR signaling pathway’, ‘Rap1 signaling pathway’, and ‘Chemokine signaling pathway’ (Figure 2C). Additionally, the ‘Th17 cell differentiation pathway’ was downregulated in infected macrophages compared to uninfected ARNTf/+ macrophages (Figure 2C). Of note, the ‘HIF-1α signaling pathway’ was upregulated with infection (Figure 2C). These data demonstrate that infection with L. major is sufficient to drive transcriptional changes in macrophages and activate HIF-α signaling.
To further characterize the cellular processes most affected by infection in macrophages either with or without HIF-α signaling, we conducted KEGG pathway analyses. The analysis revealed that during infection with L. major, the proteasome pathway is upregulated in the absence of HIF-α signaling (Figure 2D). When we investigated enriched pathways in infected macrophages with intact HIF-α, the proteosome pathway was not upregulated suggesting this pathway is normally suppressed by HIF-α during L. major infection. These data together indicate infection alone induces transcriptional changes that are HIF-α-dependent, suggesting infection with L. major parasites is sufficient to activate HIF-α signaling in vitro contrary to other reports (23, 25, 27).
During infection with L. major parasites, a strong Th1 immune response is formed resulting in the release of a multitude of pro-inflammatory mediators. To identify inflammation-related transcriptomic changes, both HIF-α signaling competent and deficient macrophages were infected with L. major and treated with LPS and IFNγ to mimic the in vivo pro-inflammatory environment. We identified 1,076 genes that were differentially expressed when comparing infected ARNTf/+ macrophages treated with LPS and IFNγ to infected ARNTf/+ macrophages not treated with LPS and IFNγ (Figure 3A, Table 3). The top upregulated DEGs were Gpr18 and Mmp25 (Table 3). Additionally, there were many immune-related transcripts that were upregulated to a lesser extent including Il12b, Cd40, and Nos2, all of which participate in the immune response to Leishmania parasites (Figure 3A, Table 3). The top downregulated DEGs were Arrdc3, Rasgrp3, and Cdca7l comparing infected ARNTf/+ macrophages treated or not with LPS and IFNγ (Table 3). Furthermore, we investigated transcriptional changes in infected ARNTf/f macrophages treated with LPS and IFNγ compared to infected ARNTf/f not treated with LPS and IFNγ. We identified 1,191 DEGs (Figure 3B, Table 4). The top 25 most upregulated genes in response to pro-inflammatory stimuli were the same for infected macrophages with or without competent HIF-α signaling, indicating these DEGs are independent of HIF-α signaling (Figures 3A, B, Table 4). In contrast, there were differences in the top 25 downregulated DEGs in response to LPS and IFNγ stimulation in infected macrophages that are competent or impaired for HIF-α signaling (Figure 3B, Table 4). The top downregulated DEGs in infected HIF-α deficient macrophages treated with LPS and IFNγ compared to infected HIF-α deficient macrophages not treated with LPS and IFNγ were Mdp1, Arap3, and Prmt3 (Table 4).
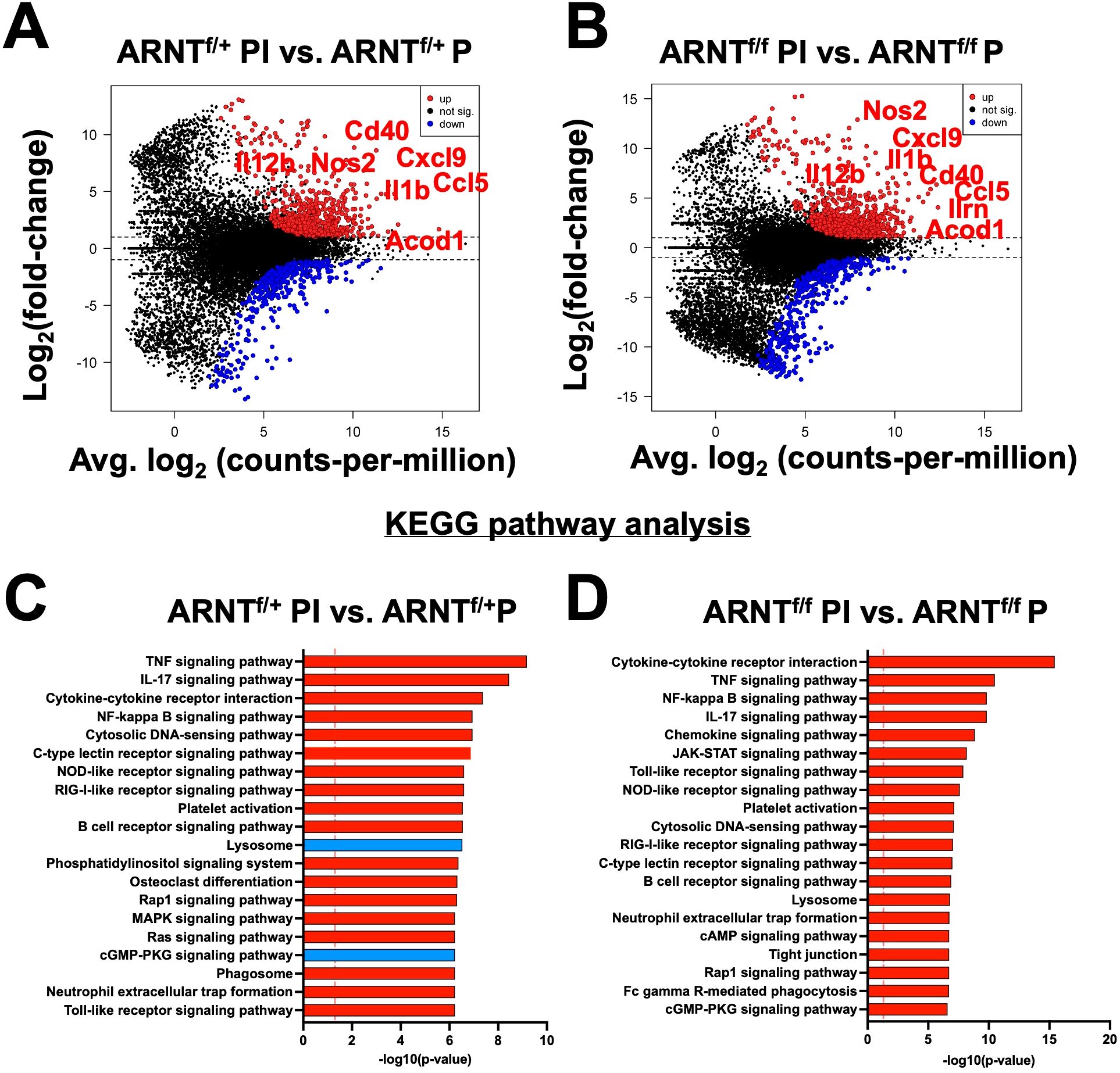
Figure 3. Inflammation related transcriptomic changes are both HIF-α dependent and independent. Macrophages were infected or infected and treated with LPS/IFNγ and prepped for subsequent analysis utilizing RNASeq. (A, B) An MD plot illustrates transcriptomic changes during infection and stimulation with LPS/IFNγ in HIF-α competent (ARNTf/+ PI vs. ARNTf/+P) and HIF-α deficient macrophages (ARNTf/f PI vs. ARNTf/f P). Here the PI indicates parasites and inflammatory stimuli, LPS/IFNγ, and P describes parasite infection alone. Red dots identify upregulated transcripts and blue dots identify downregulated transcripts. (C, D) Enriched pathways were identified using KEGG analysis for both comparisons. Red pathways indicate upregulation while blue pathways are downregulated.
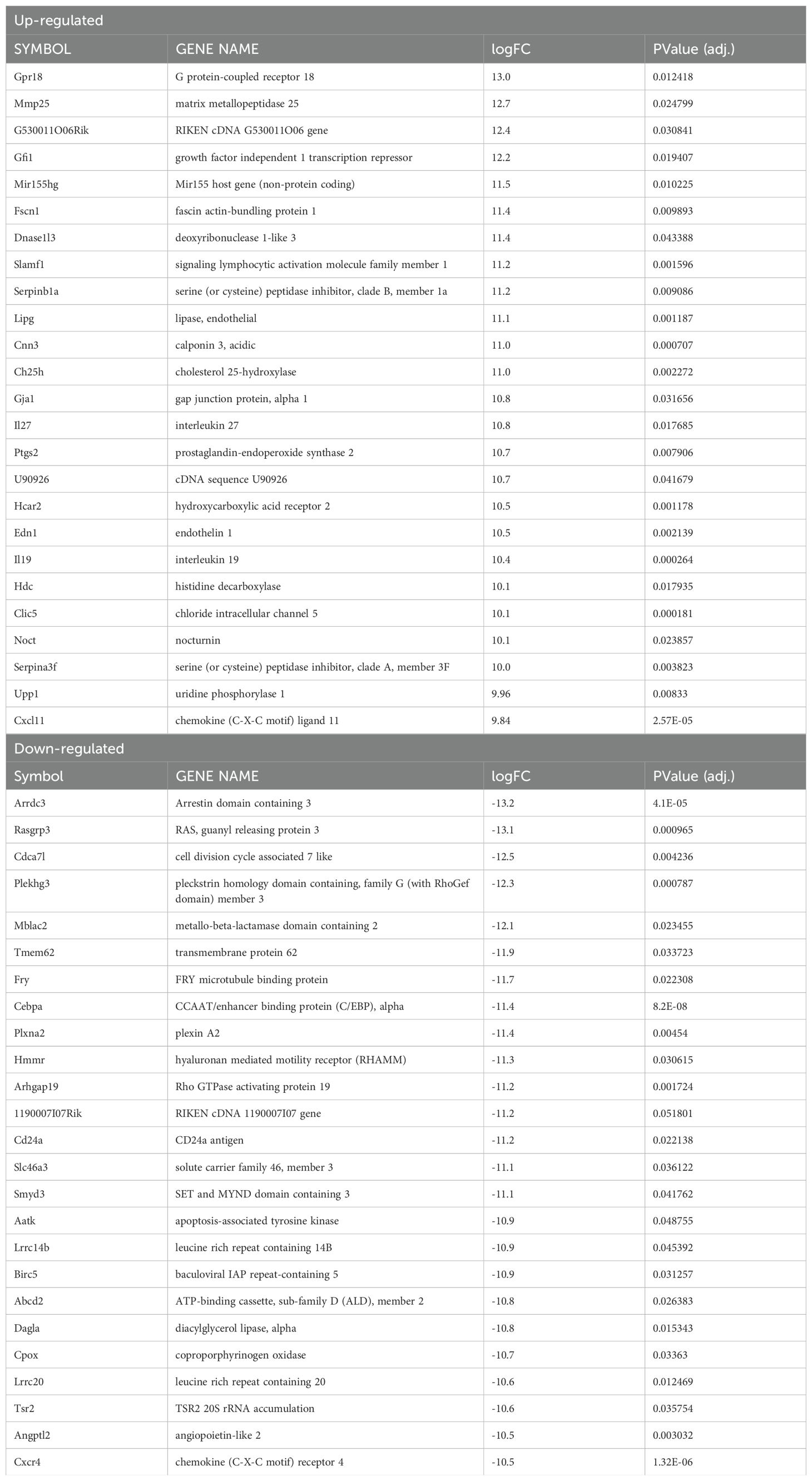
Table 3. Significantly up- or down-regulated DEGs between infected ARNTf/+ macrophages treated with LPS/IFNg compared to infected ARNTf/+ macrophages.
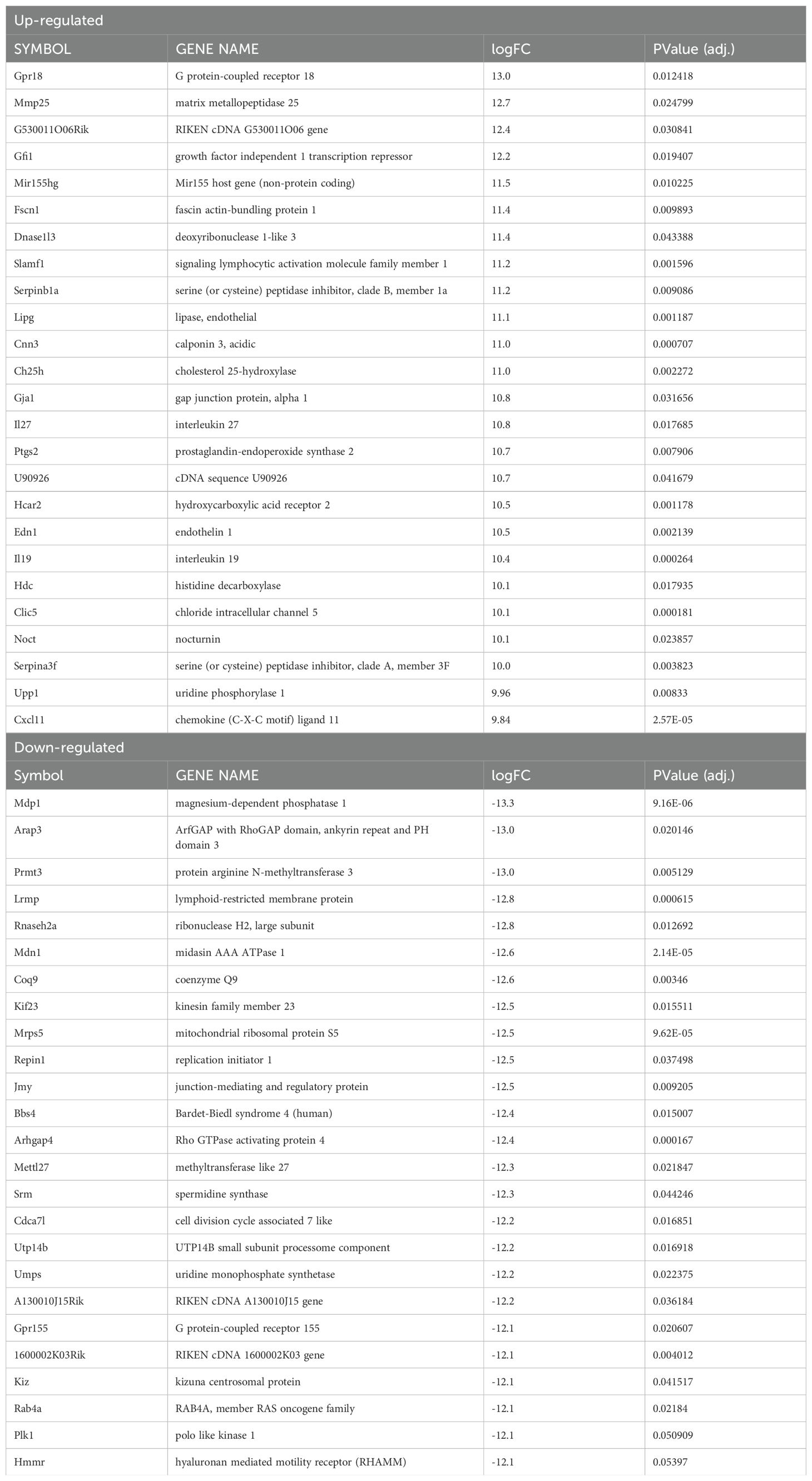
Table 4. Significantly up- or down-regulated DEGs between infected ARNTf/f macrophages treated with LPS/IFNg compared to infected ARNTf/f macrophages.
A functional analysis was performed to identify pathways associated with pro-inflammatory stimulus administration in infected macrophages with or without HIF-α signaling compared to their infected macrophage counterparts with no stimulus. The KEGG analysis revealed pro-inflammatory stimuli upregulated pathways such as ‘TNF signaling receptor’, ‘IL-17 signaling pathway’, and several other inflammatory pathways (Figure 3C). In addition, these infected macrophages downregulated the ‘lysosome’ and ‘cGMP-PKG pathway’ in response to LPS and IFNγ administration (Figure 3C). Next, we compared infected ARNTf/f macrophages treated or not with LPS and IFNγ by KEGG analysis. The results revealed similar upregulated pathways as the pro-inflammatory treated and infected ARNTf/+ macrophages including ‘cytokine-cytokine receptor interaction’ and ‘TNF signaling pathway’ (Figure 3D). Interestingly, there were no significantly downregulated pathways in infected pro-inflammatory stimulated macrophages deficient for HIF-α signaling compared to infected macrophages also deficient for HIF-α signaling. In contrast to the HIF-α competent macrophages, during HIF-α deficiency, the ‘lysosome’ and ‘cGMP-Pk3 signaling pathways’ were upregulated in infected macrophages treated with LPS and IFNγ (Figure 3D). These data suggest in a pro-inflammatory environment, HIF-α suppresses pathways related to L. major infection including those involved in production of the phagolysosome and second messenger signaling.
After investigating changes involved with L. major infection alone and with pro-inflammatory stimulus in macrophages with and without competent HIF-α signaling, we directly compared the gene expression profiles of macrophages without HIF-α signaling to macrophages with HIF-α signaling under each condition. First, we compared ARNTf/f to ARNTf/+ under basal conditions. We identified two upregulated DEGs in the ARNTf/f macrophages compared to ARNTf/+ macrophages including, Isg20 and Spp1 suggesting HIF-α inhibits these genes during homeostasis (Figure 4A, Table 5). Next, we analyzed differences between ARNTf/f to ARNTf/+ during infection with L. major parasites. When comparing macrophages without or with HIF-α signaling during L. major infection, we found several downregulated DEGs in macrophages with impaired HIF-α signaling which suggests under normal conditions these DEGs are mediated by HIF-α (Figure 4B, Table 6). These DEGs include Il1β, Ccl5, Mcoln2, Mevf, and Socs1 (Figure 4B, Table 6). In line with these results, Socs1, Mcoln2, Mevf were upregulated during infection with L. major parasites in HIF-α competent macrophages compared to uninfected HIF-α competent macrophages further suggesting these specific genes are dependent on HIF-α during infection with L. major (Figure 2A). By KEGG analysis, we found under basal conditions macrophages without HIF-α signaling upregulate the ‘ribosome’ and ‘DNA replication pathways’ compared to macrophages with HIF-α signaling (Figure 4C). This finding suggests that HIF-α restricts cell processes in the absence of infection in steady state. Furthermore, when we analyzed enriched pathways in infected macrophages without HIF-α signaling compared to infected macrophages with HIF-α signaling, we found there were minimal significantly enriched pathways (Figure 4D). Together, this dictates there are HIF-α-dependent transcriptomic changes during homeostasis and in response to L. major infection supporting previous data depicting infection activates HIF-α.
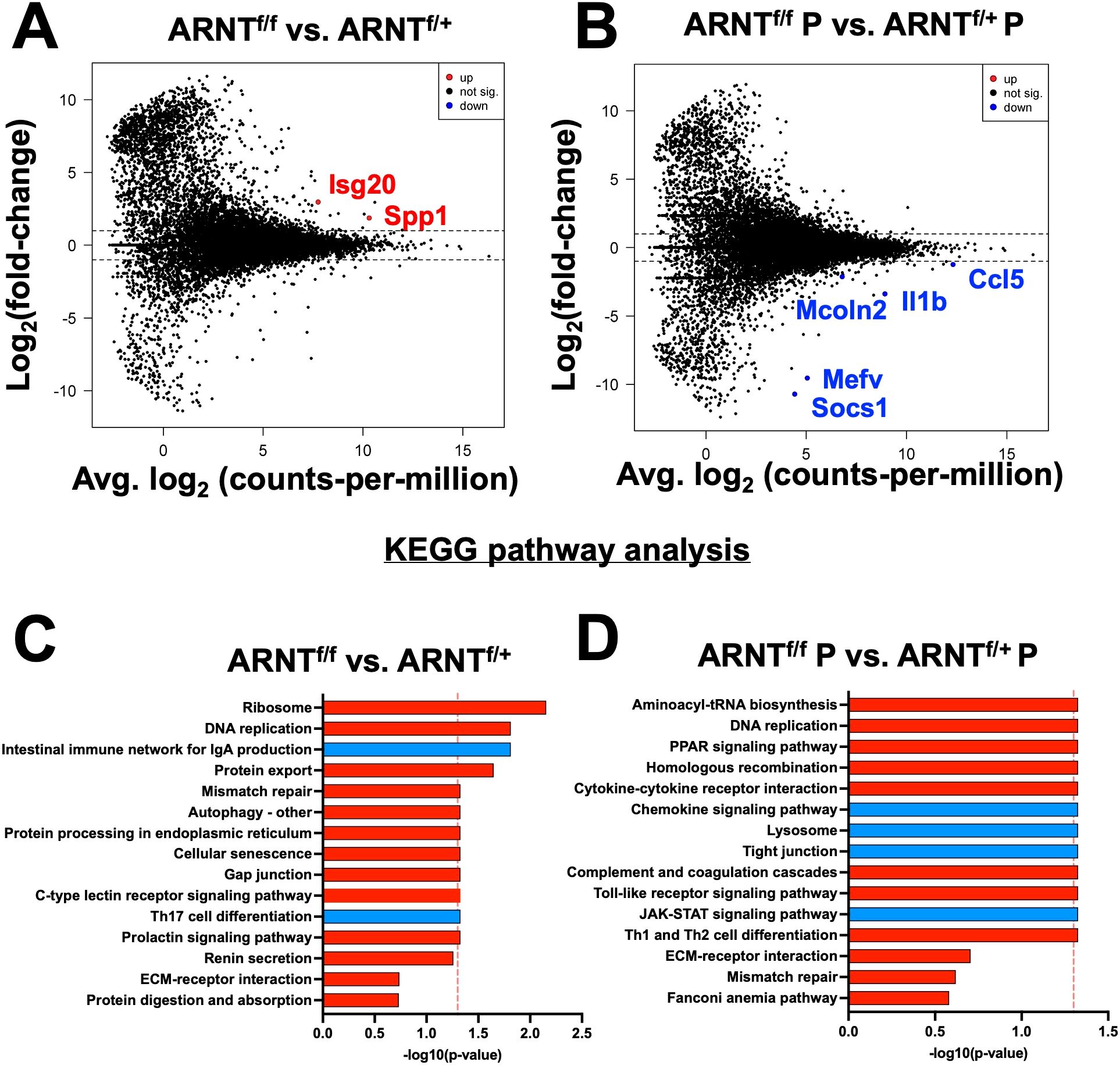
Figure 4. HIF-α mediates DEGs induced by L. major parasites. Macrophages both with and without HIF-α signaling were cultured in media alone or infected with L. major parasites for 8 hours before being prepped for RNASequencing. (A) An MD plot illustrates transcripts inhibited by HIF-α under basal conditions. (B) An MD plot shows transcripts mediated by HIF-α during L. major infection. (C) KEGG pathway analysis identified enriched pathways in macrophages without HIF-α signaling under basal conditions. (D) KEGG pathway analysis identified enriched pathways in macrophages without HIF-α signaling during infection. Red pathways are upregulated and blue pathways are downregulated.

Table 5. Significantly up- or down-regulated DEGs between ARNTf/f compared to ARNTf/+ uninfected macrophages.
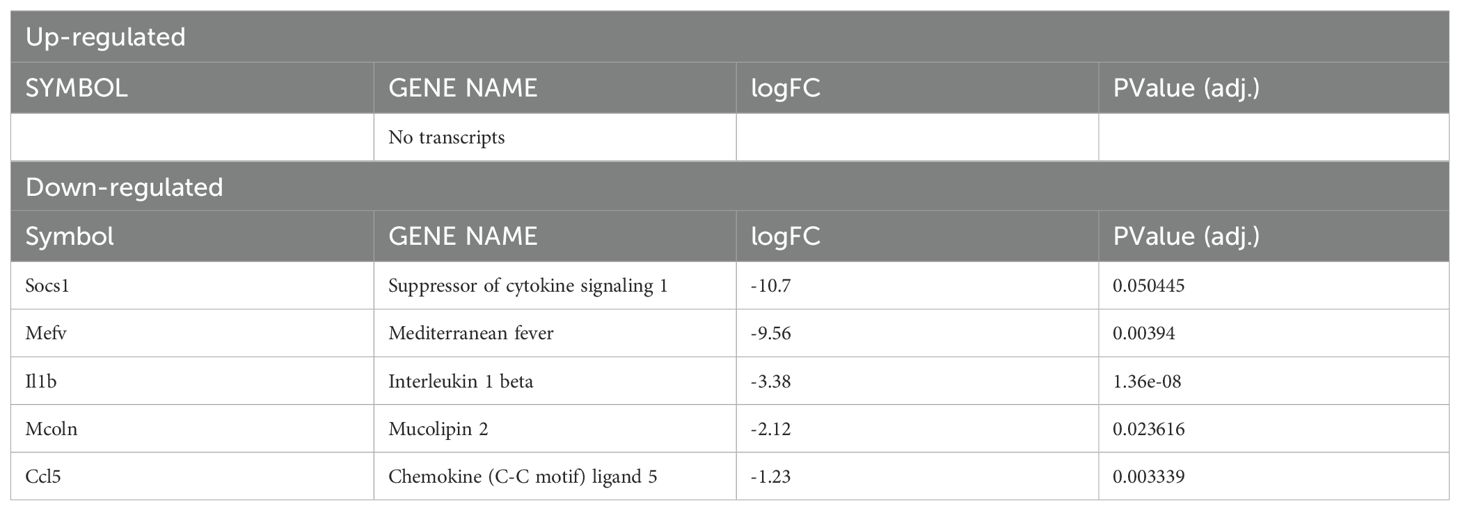
Table 6. Significantly up- or down-regulated DEGs between ARNTf/f compared to ARNTf/+ infected macrophages.
Finally, to further characterize the role of HIF-α signaling in infected macrophages under pro-inflammatory conditions, we compared the gene expression profile of infected macrophages deficient for HIF-α signaling stimulated with LPS/IFNγ to infected macrophages with intact HIF-α signaling under the same conditions. There were 102 DEGs between infected and stimulated macrophage without and with HIF-α signaling, 63 being upregulated and 39 downregulated (Figure 5A, Table 7). Of note, the top upregulated DEGs included Slc26a11, Agap1, and Cxcr4 and the top downregulated DEGs contained Mmgt1, Arhgap4, and Mdn1 (Figure 5A, Table 7). We predict the upregulated genes are inhibited by HIF-α signaling (Slc26a11, Agap1, and Cxcr4) while the downregulated DEGs are mediated by HIF-α signaling (Mmgt1, Arhgap4, and Mdn1). Interestingly, Cxcr4 expression is downregulated during infection and pro-inflammatory stimulation in HIF-α competent macrophages compared to HIF-α competent infected macrophages (Table 3). This indicates HIF-α inhibits Cxcr4 during L. major infection under inflammatory conditions. To identify pathways enriched for our DEGs, we performed a KEGG analysis. The KEGG pathway analysis revealed the DEGs clustered into pathways related to translation and protein production (‘Ribosomes’ and ‘Protein export’) (Figure 5B). Upregulation of the ‘ribosome’ and ‘protein export’ pathways in macrophages without HIF-α signaling suggests that these pathways are suppressed by HIF-α. To further conduct gene set enrichment analysis we utilized the molecular signature database (MSigDB). The MSigDB analysis revealed that the ‘interferon gamma response pathway’ was significantly upregulated in stimulated and infected macrophages without HIF-α signaling indicating this pathway is inhibited by HIF-α signaling (Figure 5C). Additionally, the ‘oxidative phosphorylation pathway’ was found to be upregulated in HIF-α deficient macrophages indicating HIF-α signaling suppresses this pathway in infected macrophages in response to pro-inflammatory stimuli (Figure 5C). This suggests that HIF-α deficient macrophages are shunted towards a predominant oxidative phosphorylation profile rather than a dominant metabolic glycolytic profile.
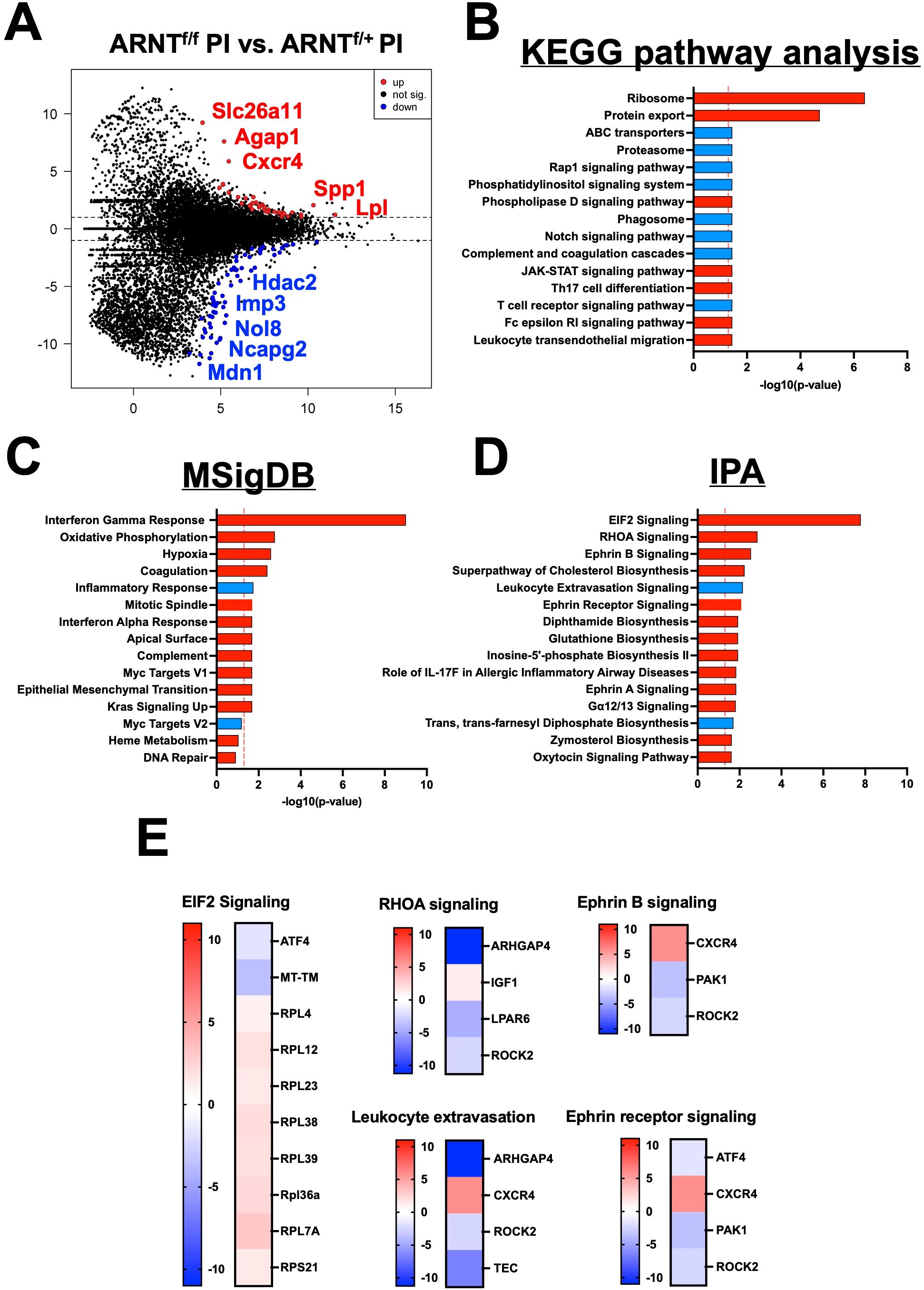
Figure 5. HIF-α signaling suppresses translational pathways under inflammatory conditions. RNASeq and pathway analysis on macrophages with and without intact HIF-α signaling infected with L. major and treated with pro-inflammatory stimuli. (A) DEGs upregulated (red) and downregulated (blue) in infected macrophages treated with LPS/IFNγ without HIF-α signaling compared to macrophages with intact HIF-α signaling under the same conditions. (B) KEGG analysis identified enriched pathways in infected macrophages without HIF-α signaling stimulated with LPS/IFNγ. (C) MSigDB pathway analysis defined upregulated pathways in red and downregulated pathways in blue in the infected macrophages without HIF-α signaling compared to macrophages with intact HIF-α signaling. (D) Ingenuity pathway analysis (IPA) was run to determine upregulated and downregulated pathways (red and blue respectively). (E) Heatmap plots of each upregulated or downregulated pathway defined by the IPA with individual altered DEGs represented in each pathway.
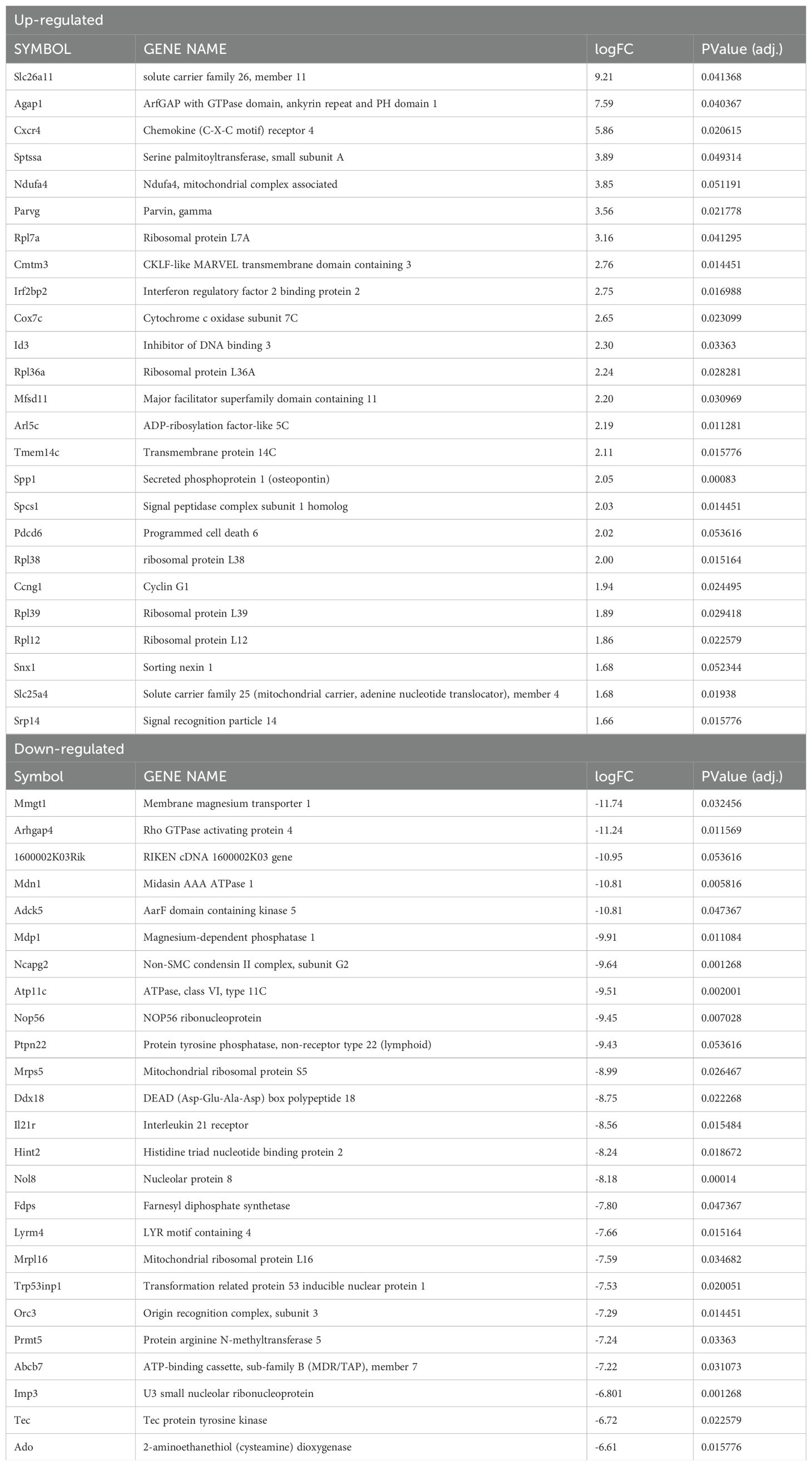
Table 7. Top 25 significantly up- or down-regulated DEGs between ARNTf/f compared to ARNTf/+ infected macrophages treated with LPS/IFNγ.
Furthermore, many ribosomal related transcripts were enriched in the HIF-α deficient infected macrophages treated with pro-inflammatory stimuli such as Rpl4, Rpl7a, Rpl12, Rpl23, Rpl38, Rpl39, and Rps21. To further investigate these altered ribosomal transcripts, we conducted an ingenuity pathway analysis (IPA) comparing infected macrophages treated with LPS and IFNγ with or without intact HIF-α signaling (Figures 5D, E). We pinpointed ‘EIF2 signaling’ as the top upregulated pathway in macrophages without HIF-α signaling, proposing that in a scenario with both infection and inflammatory stimuli, HIF-α inhibits EIF2 signaling. This data is consistent with the KEGG pathway analysis indicating HIF-α signaling suppresses protein translation during inflammatory conditions. Of note, several other enriched pathways were identified by the IPA including ‘RhoA signaling’ and ‘Ephrin B signaling’ suggesting these pathways are inhibited by HIF-α (Figures 5D, E). Finally, ‘Ephrin Receptor Signaling’ and ‘Leukocyte Extravasation’ were downregulated in infected macrophages stimulated with LPS and IFNγ without HIF-α, again suggesting this pathway is mediated by HIF-α (Figures 5D, E).
To validate the transcriptomic findings, we employed quantitative PCR (qPCR). To directly investigate HIF-α dependent transcriptomic changes during L. major infection, we designed an assay to selectively stabilize HIF-α by utilizing dimethyloxallyl glycine (DMOG), a prolyl hydroxylase inhibitor that prevents HIF-α from being targeted for degradation by the proteosome (43). Briefly, macrophages were derived from C57BL/6 mice and 1) cultured in media, 2) infected with L. major, 3) treated with DMOG, or 4) infected and treated with DMOG. We analyzed the relative expression of ribosomal transcripts upregulated in response to HIF-α deletion in our transcriptomic data, suggesting DMOG administration should decrease the relative expression of these transcripts. These selected transcripts were contained within the EIF2 signaling pathway which was the top hit of differentially regulated pathways during L. major infection and pro-inflammatory stimulus administration, suggesting HIF-α suppresses this pathway. In confirmation, we found the expression of Rpl4 was significantly decreased in L. major-infected macrophages treated with DMOG compared to infected macrophages without DMOG suggesting HIF-α stabilization results in downregulation of Rpl4, consistent with our transcriptomic data (Figure 6A). Additionally, the expression of two other ribosomal transcripts, Rpl12 and Rpl23, were additionally decreased in infected macrophages treated with DMOG compared to infected macrophages treated with DMOG (Figure 6A).
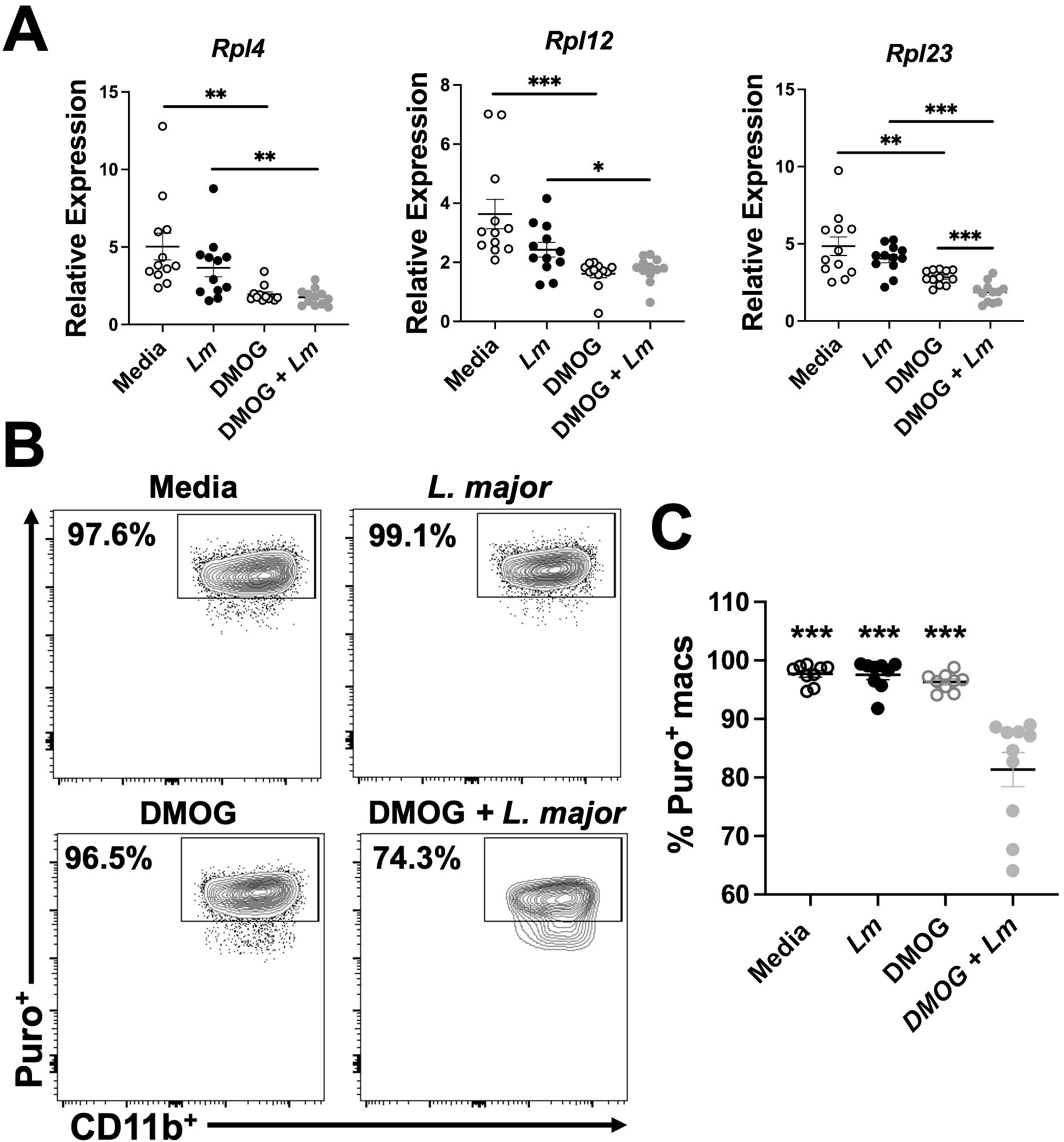
Figure 6. HIF-α stabilization decreases macrophage translation during L. major infection. Macrophages derived from C57BL/6 mice were cultured in media, infected with L. major, treated with DMOG, or infected and treated with DMOG before RNA was isolated and prepped for quantitative PCR. (A) Relative expression of ribosomal protein transcripts is shown for macrophages infected or not and treated or not with DMOG. Data is pooled from two independent experiments. (B) Macrophages were cultured in media, with or without L. major, and with or without DMOG and labeled with puromycin to assess translation activity via flow cytometry. Representative flow plots of Puro+ macrophages. Macrophages were gated as CD45+CD11b+CD64+Ly6G-. (C) Quantification of (B). Data is pooled from two experiments where n=10. Significance was determined using a student’s unpaired t-test *p<0.05, **p<0.01, ***p<0.001 and for (C) significance is relative to the DMOG + L. major group.
To further validate our transcriptomic findings and investigate the functional impact of HIF-α stabilization during L. major infection, we designed an in vitro experiment to assess translational activity with or without HIF-α stabilization. We used puromycin (puro), a tyrosyl-tRNA mimic that inhibits translation and labels active ribosomes, to determine if HIF-α suppresses translation as suggested by our IPA analysis (Figure 5 and Figure 6). HIF-α stabilization was achieved using DMOG. Macrophages were derived from C57BL/6 mice and cultured in four conditions: media alone, L. major parasites, DMOG alone, or both L. major and DMOG. Previously, we showed that lesional macrophages exhibit the highest puro signal during L. major infection compared to other cell types within the lesion, demonstrating lesional macrophages exhibit high translational activity in vivo (206). In line with this, macrophages cultured with media, L. major, or DMOG alone had 90-95% of cells positive for puro (Figures 6B, C). However, when macrophages were treated with both L. major and DMOG, the percentage of puro+ macrophages significantly decreased (Figures 6B, C). These results support that HIF-α stabilization during infection inhibits translation. Notably, this effect was specific to L. major infection, as macrophages treated with DMOG alone showed similar puro levels to those cultured with media or L. major alone (Figures 6B, C). Overall, these findings indicate that HIF-α suppresses translation during L. major infection, but this effect requires a pro-inflammatory environment potentially to allow for maximal HIF-α stabilization.
Discussion
HIF-α activation is a hallmark of both CL and VL occurring in response to tissue hypoxia, TLR activation, ROS and cytokines like TNFα and IL-1β, all of which are present during Leishmania infection (9, 12, 29, 44–46). However, the direct contribution of the parasite versus the host response/microenvironment to HIF-α activation is not clear. Leishmania parasites can directly activate HIF-1α in macrophages, but the direct activation of macrophage HIF-1α is context dependent with the parasite species playing a major role. For instance, L. amazonensis parasites, which cause CL in South America, directly induce the expression of HIF-1α in human and mouse macrophages in vitro under normoxic conditions (10, 47). HIF-1α is also present in L. amazonensis-infected skin (10). While L. amazonensis parasites can drive HIF-1α expression on their own, HIF-1α also promotes L. amazonensis killing by macrophages under hypoxic conditions (47). Similar to L. amazonensis, L. donovani parasites, which cause VL in Africa and Asia, directly activate HIF-1α in macrophages in vitro under normoxic conditions (48, 49). L. donovani parasites increase HIF-1α expression, nuclear translocation and activity in a variety of macrophages including J774 cells, peritoneal macrophages and splenic-derived macrophages from BALB/c mice (48). To stabilize HIF-1α, L. donovani parasites use an array of mechanisms including depleting host iron pools to modulate prolyl hydroxylase activity and inducing microRNAs to limit NF-κB activation which establishes a suitable environment for parasite survival (48, 49). In vitro, HIF-1α blockade inhibits L. donovani intracellular growth and HIF-1α stabilization promotes L. donovani growth inside macrophages (48, 49). However, myeloid-specific HIF-1α-/- mice infected with L. donovani and humans with a loss-of-function HIF1A gene polymorphism are more susceptible to infection (50). The role of HIF-2α in L. amazonensis and L. donovani infection has not been investigated.
Although L. amazonensis and L. donovani parasites can activate HIF-α directly, previous work shows that L. major parasites do not increase HIF-1α expression or activation under normoxic conditions in macrophages (11, 23, 27, 29). For example, HIF-1α and HIF-2α as well as HIF-1α-specific and HIF-2α-specific target genes are increased at the site of murine L. major infection, but in vitro infection of macrophages with L. major does not induce HIF-1α expression (11, 25). Rather L. major parasites require additional inflammatory signals such as LPS and/or IFNγ to induce HIF-1α accumulation and subsequent HIF-1α target expression like NOS2 and VEGF-A in macrophages (23, 27, 29). While HIF-α stabilization promotes L. donovani survival in macrophages, previous work has shown HIF-α stabilization does not impact L. major parasite growth in macrophages and may be why L. major parasites alone do not induce significant HIF-α protein accumulation (11, 27, 29). However, previous work from our laboratory found that macrophages derived from mice deficient in HIF-α signaling possess higher parasite burdens at 2 and 72 hours post-infection compared to macrophages derived from HIF-α competent mice (27). In support of this data, through pathway analysis, we have shown that in vitro, the HIF-1α signaling pathway is enriched during infection with L. major and many initial transcriptomic changes are HIF-α-dependent suggesting infection with L. major initiates the HIF-α transcriptional program (Figure 2). This is consistent with an additional study investigating initial transcriptomic changes after in vitro L. major infection, reporting HIF-1α signaling is enriched in murine macrophages at 4 hours post-infection (51). Despite these transcriptomic indications, it is possible pro-inflammatory stimuli are required for optimal HIF-α activation and subsequent target gene activation. It is important to note that in the above-mentioned study, L. major infection was not associated with changes specifically in HIF-1α accumulation. In the present study we have investigated changes in the absence of both HIF-1α and HIF-2α signaling which could account for the discrepancies.
Among the transcripts involved in the subtle HIF-α program activated during infection with L. major were Socs1 and Mevf (Figure 2A). Here, we show that during infection these transcripts are upregulated in HIF-α competent macrophages (Figure 2A). Additionally, when we compared infection in HIF-α deficient macrophages compared to HIF-α competent infected macrophages, Socs1 and Mevf were strongly downregulated suggesting that HIF-α mediates the expression of Socs1 and Mevf during L. major infection (Figure 4B). Interestingly, Socs1 is involved in immune regulation suggesting that mediation of this transcript by HIF-α is another mechanism to limit excess energetic use during conditions of low oxygen availability.
HIF-α activation occurs in a wide variety of circumstances playing a central role in tissue adaptation to low oxygen tensions (52, 53). Namely, hypoxia can be a characteristic of both tissue injury and subsequent inflammation, where infiltrating cells increase the demand for nutrients and oxygen, further depleting the tissue stores (54, 55). Protein translation is an energetically demanding process and during hypoxia, inhibition of translation supports energy homeostasis and possibly promotes survival when energy stores are insufficient (56, 57). Therefore, translation during hypoxic conditions becomes selective; coordinating adaptation to promote cell survival under low oxygen and energy conditions (58). Specifically, hypoxic conditions have been shown to stifle protein translation through downregulation of EIF2α signaling which we have shown is directly suppressed by HIF-α signaling in macrophages during inflammatory conditions (Figure 5) (59). Phosphorylation of eIF2a is necessary for mRNA translation inhibition during hypoxia and may be coordinated by HIF-α based on the current findings (57, 60).
In addition to acclimating tissue to low oxygen availability, HIF-α is also a master regulator of macrophage inflammatory and innate immune function (15, 61, 62). Inhibition of protein translation coupled with a shift in metabolism to glycolysis during hypoxia are both mechanisms to conserve energy directly manipulated by HIF-α (63–65). Previous reports have demonstrated that HIF-α is capable of shunting macrophages towards a M1 dominant phenotype by targeting glucose metabolism (66, 67). Elevated glucose metabolism coupled with HIF-α-induced ATP production are two major cellular mechanisms of overcoming low oxygen tension. As a result, macrophage-specific deletion of HIF-1α leads to impaired macrophage responses including lower glycolytic rates, lower energy generation, and impaired motility (68–70). Here we have shown that macrophages deficient for HIF-α signaling are predisposed to a dominant oxidative phosphorylation profile in comparison to HIF-α competent macrophages (Figure 5). Our study confirms that HIF-α reprograms macrophages during L. major infection to cope with the energetic demand. We have also shown through IPA analysis that pathways associated with macrophage motility are dysregulated during genetic deletion of HIF-α signaling including RhoA signaling and leukocyte extravasation consistent with what is reported in the literature (71–73). Although we investigated the impact of HIF-α signaling in resting M0 macrophages and M1 polarized macrophages (through LPS/IFNγ administration), a limitation of our study is that we have not considered the importance of HIF-α signaling in M2 polarized macrophages. Because M2 macrophages serve as a permissive niche during Leishmania infection, future work will investigate the role of HIF-α signaling in M2 macrophages during L. major infection (74, 75).
In summary, we showed L. major infection elicits a subtle macrophage HIF-α program, but major transcriptomic changes dependent on HIF-α are only present in a pro-inflammatory environment. This supports our hypothesis that during in vivo L. major infection, HIF-α stabilization is dependent on the pro-inflammatory milieu and not L. major directly, which is in contrast to L. donovani infection where the parasite alone can stabilize HIF-α (48). Additionally, we have evidence suggesting HIF-α suppresses protein translation in response to L. major infection and pro-inflammatory stimulus. However, a limitation of our study is that we have not determined if HIF-α suppresses protein translation during infection of primary macrophages, human macrophages, or following in vivo infection with L. major. So, future work will assess the extent to which protein translation occurs in a HIF-α dependent manner, and if this is unique to L. major or if it is conserved in other skin infections and diseases. We hypothesize suppression of translation is a mechanism of cellular adaptation to the pro-inflammatory response and subsequent hypoxic conditions from infiltrating cells and their high energetic demand during infection. A complete understanding of HIF-α during inflammation is vital in developing targeted therapeutics not only for CL, but also for other inflammatory skin diseases psoriasis (76). These results are also broadly relevant to diseases where HIF-α is highly expressed such as metabolic disorders including obesity and diabetes and inflammatory conditions like rheumatoid arthritis (77–79).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE273822 (GEO).
Ethics statement
The animal study was approved by IACUC of the University of Arkansas for Medical Sciences. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LF: Conceptualization, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing, Data curation. CW: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. HR: Formal analysis, Investigation, Methodology, Writing – review & editing, Writing – original draft. AB: Investigation, Methodology, Writing – original draft. GV: Investigation, Methodology, Writing – original draft, Formal analysis. JB: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. SB: Investigation, Methodology, Writing – review & editing, Conceptualization, Funding acquisition, Supervision. TW: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Writing – review & editing, Formal analysis, Project administration, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Center for Microbial Pathogenesis and Host Inflammatory Responses (funded by NIH NIGMS Centers of Biomedical Research Excellence Grant P20-GM103625). This publication was also supported in part by funds provided by the National Center for Advancing Translational Sciences of the NIH under awards TL1 TR003109 and UL1 TR003107 for the Systems Pharmacology and Therapeutics (SPaT) NIH T32 training grant GM106999 to LF. This study was supported by the Arkansas Children’s Research Institute, the Arkansas Biosciences Institute, and the Center for Translational Pediatric Research funded under the National Institutes of Health National Institute of General Medical Sciences (NIH/NIGMS) grant P20-GM121293 and the National Science Foundation Award No. OIA-1946391. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data analysis, decision to publish or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Desjeux P. Leishmaniasis: Current situation and new perspectives. Comp Immunol Microbiol Infect Dis. (2004) 27:305–18. doi: 10.1016/j.cimid.2004.03.004
2. Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PloS One. (2012) 7. doi: 10.1371/journal.pone.0035671
3. Sundar S, Singh B. Emerging therapeutic targets for treatment of leishmaniasis. Expert Opin Ther Targets. (2018) 22:467–86. doi: 10.1080/14728222.2018.1472241
4. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol. (2016) 16:581–92. doi: 10.1038/nri.2016.72
5. Horta MF, Mendes BP, Roma EH, Noronha FSM, MacDo JP, Oliveira LS, et al. Reactive oxygen species and nitric oxide in cutaneous leishmaniasis. J Parasitol Res. (2012) 2012. doi: 10.1155/2012/203818
6. Glennie ND, Volk SW, Scott P. Skin-resident CD4+T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PloS Pathog. (2017) 13. doi: 10.1371/journal.ppat.1006349
7. Pacheco-Fernandez T, Volpedo G, Verma C, Satoskar AR. Understanding the immune responses involved in mediating protection or immunopathology during leishmaniasis. Biochem Soc Trans. (2021) 49:297–311. doi: 10.1042/BST20200606
8. Volpedo G, Pacheco-Fernandez T, Holcomb EA, Cipriano N, Cox B, Satoskar AR. Mechanisms of immunopathogenesis in cutaneous leishmaniasis and Post Kala-azar Dermal Leishmaniasis (PKDL). Front Cell Infection Microbiol. (2021) 11:685296. doi: 10.3389/fcimb.2021.685296
9. Mahnke A, Meier RJ, Schatz V, Hofmann J, Castiglione K, Schleicher U, et al. Hypoxia in Leishmania major Skin Lesions Impairs the NO-Dependent Leishmanicidal Activity of Macrophages. J Invest Dermatol. (2014) 134:2339–46. Available online at: https://www.sciencedirect.com/science/article/pii/S0022202X15369542 (Accessed April 4, 2023).
10. Arrais-Silva WW, Paffaro VAJ, Yamada AT, Giorgio S. Expression of hypoxia-inducible factor-1alpha in the cutaneous lesions of BALB/c mice infected with Leishmania amazonensis. Exp Mol Pathol. (2005) 78:49–54. doi: 10.1016/j.yexmp.2004.09.002
11. Bowlin A, Roys H, Wanjala H, Bettadapura M, Venugopal G, Surma J, et al. Hypoxia-inducible factor signaling in macrophages promotes lymphangiogenesis in Leishmania major infection. Infect Immun. (2021) 89:e00124-21. doi: 10.1128/IAI.00124-21
12. Fowler EA, Farias Amorim C, Mostacada K, Yan A, Amorim Sacramento L, Stanco RA, et al. Neutrophil-mediated hypoxia drives pathogenic CD8+ T cell responses in cutaneous leishmaniasis. J Clin Invest. (2024) 134(14):e177992. doi: 10.1172/JCI177992
13. Cimmino F, Avitabile M, Lasorsa VA, Montella A, Pezone L, Cantalupo S, et al. HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC Med Genet. (2019) 20(37). doi: 10.1186/s12881-019-0767-1
14. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. (2013) 123:3664–71. doi: 10.1172/JCI67230
15. Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol Nat Res. (2022) 18:573–87. doi: 10.1038/s41581-022-00587-8
16. Lin N, Simon MC. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J Clin Invest. (2016) 126:3661–71. doi: 10.1172/JCI84426
17. Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage- differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. (2008) 10:983–95. doi: 10.1089/ars.2007.1825
18. Miyata T, Van Ypersele De Strihou C. Diabetic nephropathy: A disorder of oxygen metabolism? Nat Rev Nephrol. (2010) 6:83–95. doi: 10.1038/nrneph.2009.211
19. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. (2008) 453:807–11. doi: 10.1038/nature06905
20. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor-1α in development of lipopolysaccharide-induced sepsis. J Immunol. (2007) 178:7516–9. doi: 10.4049/jimmunol.178.12.7516
21. Shepardson KM, Jhingran A, Caffrey A, Obar JJ, Suratt BT, Berwin BL, et al. Myeloid Derived Hypoxia Inducible Factor 1-alpha Is Required for Protection against Pulmonary Aspergillus fumigatus Infection. PloS Pathog. (2014) 10(9):e1004378. doi: 10.1371/journal.ppat.1004378
22. Cramer T, Yamanishi Y, Clausen BE, Fö rster I, Pawlinski R, Mackman N, et al. HIF-1 Is Essential for Myeloid Cell-Mediated Inflammation profound impairment of myeloid cell aggregation, mo-tility, invasiveness, and bacterial killing. This role for HIF-1 demonstrates its direct regulation of survival and function in the inflammatory microenvironment. Cell. (2003) 112(5):645–57. doi: 10.1016/S0092-8674(03)00154-5
23. Schatz V, Strüssmann Y, Mahnke A, Schley G, Waldner M, Ritter U, et al. Myeloid cell–derived HIF-1α Promotes control of leishmania major. J Immunol. (2016) 197:4034–41. doi: 10.4049/jimmunol.1601080
24. de Carvalho Fraga CA, de Oliveira MVM, Alves LR, Viana AG, de Sousa AA, Carvalho SFG, et al. Immunohistochemical profile of HIF-1α, VEGF-A, VEGFR2 and MMP9 proteins in tegumentary leishmaniasis. Bras Dermatol. (2012) 87:709–13. Available online at: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0365-05962012000500006&lng=en&tlng=en (Accessed August 15, 2023).
25. Weinkopff T, Konradt C, Christian DA, Discher DE, Hunter CA, Scott P. Leishmania major infection–induced VEGF-A/VEGFR-2 signaling promotes lymphangiogenesis that controls disease. J Immunol. (2016) 197:1823–31. doi: 10.4049/jimmunol.1600717
26. Weinkopff T, Roys H, Bowlin A, Scott P. Leishmania infection induces macrophage vascular endothelial growth factor A production in an ARNT/HIF-dependent manner. Infect Immun. (2019) 87(11):e00088-19. doi: 10.1128/IAI.00088-19
27. Bettadapura M, Roys H, Bowlin A, Venugopal G, Washam CL, Fry L, et al. Hif-α activation impacts macrophage function during murine leishmania major infection. Pathogens. (2021) 10(12):1584. doi: 10.3390/pathogens10121584
28. Venugopal G, Bird JT, Washam CL, Roys H, Bowlin A, Byrum SD, et al. In vivo transcriptional analysis of mice infected with Leishmania major unveils cellular heterogeneity and altered transcriptomic profiling at single-cell resolution. PloS Negl Trop Dis. (2022) 16(7):e0010518. doi: 10.1371/journal.pntd.0010518
29. Schatz V, Neubert P, Rieger F, Jantsch J. Hypoxia, hypoxia-inducible factor-1α, and innate antileishmanial immune responses. Front Immunol. (2018) 9:216. doi: 10.3389/fimmu.2018.00216
30. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
31. Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. (2017) 8:14049. doi: 10.1038/ncomms14049
32. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. (2018) 36:411–20. doi: 10.1038/nbt.4096
33. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. (2019) 177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031
34. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. (2015) 161:1202–14. doi: 10.1016/j.cell.2015.05.002
35. McInnes L, Healy J, Melville J. UMAP: uniform manifold approximation and projection for dimension reduction(2018). Available online at: http://arxiv.org/abs/1802.03426 (Accessed August 1, 2024).
36. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. (2016) 32:3047–8. doi: 10.1093/bioinformatics/btw354
37. Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
38. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data(2010). Available online at: http://genomebiology.com/2010/11/3/R25 (Accessed August 1, 2024).
39. Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. (2015) 43(15):e97. doi: 10.1093/nar/gkv412
40. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Society: Ser B (Methodological). (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
41. Fry L, Roys H, Bowlin A, Venugopal G, Bird JT, Weaver A, et al. Enhanced translational activity is linked to lymphatic endothelial cell activation in cutaneous leishmaniasis. bioRxiv. (2024). doi: 10.1101/2024.08.07.605632.
42. Lin N, Shay JES, Xie H, Lee DSM, Skuli N, Tang Q, et al. Myeloid cell hypoxia-inducible factors promote resolution of inflammation in experimental colitis. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.02565
43. Duscher D, Januszyk M, Maan ZN, Whittam AJ, Hu MS, Walmsley GG, et al. Comparison of the hydroxylase inhibitor dimethyloxalylglycine and the iron chelator deferoxamine in diabetic and aged wound healing. Plast Reconstr Surg. (2017) 139:695e–706e. doi: 10.1097/PRS.0000000000003072
44. Kropf P, Freudenberg MA, Modolell M, Price HP, Herath S, Antoniazi S, et al. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite leishmania major. Infect Immun. (2004) 72:1920–8. doi: 10.1128/IAI.72.4.1920-1928.2004
45. Van Assche T, Deschacht M, Da Luz RAI, Maes L, Cos P. Leishmania-macrophage interactions: Insights into the redox biology. Free Radical Biol Med. (2011) 51:337–51. doi: 10.1016/j.freeradbiomed.2011.05.011
46. Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasitol. (2003) 130:65–74. doi: 10.1016/S0166-6851(03)00160-9
47. Alonso D, Serrano E, Bermejo FJ, Corral RS. HIF-1α-regulated MIF activation and Nox2-dependent ROS generation promote Leishmania amazonensis killing by macrophages under hypoxia. Cell Immunol. (2019) 335:15–21. doi: 10.1016/j.cellimm.2018.10.007
48. Singh AK, Mukhopadhyay C, Biswas S, Singh VK, Mukhopadhyay CK. Intracellular pathogen Leishmania donovani activates hypoxia inducible factor-1 by dual mechanism for survival advantage within macrophage. PloS One. (2012) 7(6):e38489. doi: 10.1371/journal.pone.0038489
49. Kumar V, Kumar A, Das S, Kumar A, Abhishek K, Verma S, et al. Leishmania donovani activates hypoxia inducible factor-1α and miR-210 for survival in macrophages by downregulation of NF-κB mediated pro-inflammatory immune respons. Front Microbiol. (2018) 9. doi: 10.3389/fmicb.2018.00385
50. Mesquita I, Ferreira C, Moreira D, Kluck GEG, Barbosa AM, Torrado E, et al. The Absence of HIF-1α Increases Susceptibility to Leishmania donovani Infection via Activation of BNIP3/mTOR/SREBP-1c Axis. Cell Rep. (2020) 30:4052–4064.e7. doi: 10.1016/j.celrep.2020.02.098
51. Dillon LAL, Suresh R, Okrah K, Corrada Bravo H, Mosser DM, El-Sayed NM. Simultaneous transcriptional profiling of Leishmania major and its murine macrophage host cell reveals insights into host-pathogen interactions. BMC Genomics. (2015) 16:1108. doi: 10.1186/s12864-015-2237-2
52. Ziello JE, Jovin IS, Huang Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in Malignancy and ischemia. Yale J OF Biol AND Med. (2007) 80(2):51–60.
53. Ramakrishnan S, Anand V, Roy S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol. (2014) 9:142–60. doi: 10.1007/s11481-014-9531-7
54. Amin N, Chen S, Ren Q, Tan X, Botchway BOA, Hu Z, et al. Hypoxia inducible factor-1α attenuates ischemic brain damage by modulating inflammatory response and glial activity. Cells. (2021) 10(6):1359. doi: 10.3390/cells10061359
55. Lokmic Z, Musyoka J, Hewitson TD, Darby IA. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol. (2012) 296:139–85. doi: 10.1016/B978-0-12-394307-1.00003-5
56. Casey TM, Pakay JL, Guppy M, Arthur PG. Hypoxia causes downregulation of protein and RNA synthesis in noncontracting mammalian cardiomyocytes. Circ Res. (2002) 90:777–83. doi: 10.1161/01.RES.0000015592.95986.03
57. Koritzinsky M, Magagnin MG, Van Den Beucken T, Seigneuric R, Savelkouls K, Dostie J, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. (2006) 25:1114–25. doi: 10.1038/sj.emboj.7600998
58. Chee NT, Lohse I, Brothers SP. MRNA-to-protein translation in hypoxia. Mol Cancer. (2019) 18(49). doi: 10.1186/s12943-019-0968-4
59. Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of Protein Synthesis by Hypoxia via Activation of the Endoplasmic Reticulum Kinase PERK and Phosphorylation of the Translation Initiation Factor eIF2α. Mol Cell Biol. (2002) 22:7405–16. doi: 10.1128/MCB.22.21.7405-7416.2002
60. Mulroney TE, Pöyry T, Willis AE. Hypoxia: uncharged tRNA to the rescue! Curr Biol. (2021) 31:R25–7. doi: 10.1016/j.cub.2020.10.067
61. D’Ignazio L, Bandarra D, Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J. (2016) 283:413–24. doi: 10.1111/febs.13578
62. Murdoch C, Muthana M, Lewis CE. IMMUNOLOGY hypoxia regulates macrophage functions in inflammation 1(2005). Available online at: http://journals.aai.org/jimmunol/article-pdf/175/10/6257/1203360/6257.pdf (Accessed August 1, 2024).
63. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
64. Flood D, Lee ES, Taylor CT. Intracellular energy production and distribution in hypoxia. J Biol Chem. (2023) 299(9):105103. doi: 10.1016/j.jbc.2023.105103
65. Boutilier RG, St-Pierre J. Surviving hypoxia without really dying. Comp Biochem Physiol Part A. (2000) 126,4:481–90. Available online at: www.elsevier.com/locate/cbpa (Accessed August 1, 2024).
66. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, et al. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. (2010) 31:1863–72. doi: 10.1093/carcin/bgq088
67. Ferraro E, Germanò M, Mollace R, Mollace V, Malara N. HIF-1, the warburg effect, and macrophage/microglia polarization potential role in COVID-19 pathogenesis. Oxid Med Cell Longev. (2021) 2021:. doi: 10.1155/2021/8841911
68. Corcoran SE, O’Neill LAJ. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
69. Stothers CL, Luan L, Fensterheim BA, Bohannon JK. Hypoxia-inducible factor-1α regulation of myeloid cells. J Mol Med. (2018) 96:1293–306. doi: 10.1007/s00109-018-1710-1
70. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. (2016) 7:11635. doi: 10.1038/ncomms11635
71. Chen J, Chen J, Tan J, Li J, Cheng W, Ke L, et al. HIF-1α dependent RhoA as a novel therapeutic target to regulate rheumatoid arthritis fibroblast-like synoviocytes migration in vitro and in vivo. J Orthop Translat. (2023) 40:49–57. doi: 10.1016/j.jot.2023.05.004
72. Hutami IR, Izawa T, Khurel-ochir T, Sakamaki T, Iwasa A, Tanaka E. Macrophage motility in wound healing is regulated by hif-1α via s1p signaling. Int J Mol Sci. (2021) 22(166):8992. doi: 10.3390/ijms22168992
73. Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation - A potential mechanism. Eur J Immunol. (2001) 31:480–9. doi: 10.1002/1521-4141(200102)31:2<480::AID-IMMU480>3.0.CO;2-L
74. Carneiro MB, Lopes ME, Hohman LS, Romano A, David BA, Kratofil R, et al. Th1-th2 cross-regulation controls early leishmania infection in the skin by modulating the size of the permissive monocytic host cell reservoir. Cell Host Microbe. (2020) 27:752–768.e7. doi: 10.1016/j.chom.2020.03.011
75. Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. (2018) 215:357–75. doi: 10.1084/jem.20171389
76. Subudhi I, Konieczny P, Prystupa A, Castillo RL, Sze-Tu E, Xing Y, et al. Metabolic coordination between skin epithelium and type 17 immunity sustains chronic skin inflammation. Immunity. (2024) 57:1665–1680.e7. doi: 10.1016/j.immuni.2024.04.022
77. AbdelMassih A, Yacoub E, Husseiny RJ, Kamel A, Hozaien R, El Shershaby M, et al. Hypoxia-inducible factor (HIF): The link between obesity and COVID-19. Obes Med. (2021) 22. doi: 10.1016/j.obmed.2020.100317
78. Zhu D, Wei W, Zhang J, Zhao B, Li Q, Jin P. Mechanism of damage of HIF-1 signaling in chronic diabetic foot ulcers and its related therapeutic perspectives. Heliyon. (2024) 10(3):324656. doi: 10.1016/j.heliyon.2024.e24656
Keywords: leishmania, leishmaniasis, macrophages, HIF - 1α, translation
Citation: Fry LG, Washam CL, Roys H, Bowlin AK, Venugopal G, Bird JT, Byrum SD and Weinkopff T (2025) HIF-α signaling regulates the macrophage inflammatory response during Leishmania major infection. Front. Immunol. 16:1487311. doi: 10.3389/fimmu.2025.1487311
Received: 27 August 2024; Accepted: 03 March 2025;
Published: 21 March 2025.
Edited by:
Ramona Hurdayal, University of Cape Town, South AfricaReviewed by:
Pedro Cecílio, National Institute of Allergy and Infectious Diseases (NIH), United StatesChengxian Xu, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2025 Fry, Washam, Roys, Bowlin, Venugopal, Bird, Byrum and Weinkopff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiffany Weinkopff, dHdlaW5rb3BmZkB1YW1zLmVkdQ==