 Laura Schuster1,2
Laura Schuster1,2 Marcin Zaradzki3
Marcin Zaradzki3 Henrike Janssen1Nadia Gallenstein1
Henrike Janssen1Nadia Gallenstein1 Melanie Etheredge1,4
Melanie Etheredge1,4 Ilse Hofmann5
Ilse Hofmann5 Markus A. Weigand1
Markus A. Weigand1 Stephan Immenschuh6
Stephan Immenschuh6 Jan Larmann1,4*
Jan Larmann1,4*- 1Department of Anesthesiology, Heidelberg University Hospital, Heidelberg, Germany
- 2Faculty of Biosciences, Heidelberg University, Heidelberg, Germany
- 3Department of Cardiac Surgery, Heidelberg University Hospital, Heidelberg, Germany
- 4Department of Anesthesiology, University Hospital Rheinisch-Westfälische Technische Hochschule (RWTH) Aachen, Aachen, Germany
- 5Division of Vascular Oncology and Metastasis, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 6Department of Transfusion Medicine and Transplant Engineering, Hannover Medical School, Hannover, Germany
The main risk factor for developing transplant vasculopathy (TV) after solid organ transplantation is de-novo production of donor-specific antibodies (DSAs) binding to endothelial cells (ECs) within the graft’s vasculature. Diverse leukocyte populations recruited into the vessel wall via activated ECs contribute to vascular inflammation. Subsequent smooth muscle cell proliferation results in intima hyperplasia, the pathophysiological correlate of TV. We demonstrated that incubating aortic EC with anti-HLA-I antibodies led to increased monocyte adhesion to and transmigration across an EC monolayer. Both occurred in a CD62E-dependent fashion and were sensitive toward the anti-inflammatory enzyme heme oxygenase (HO)-1 modulation. Using a murine heterotopic aortic transplantation model, we demonstrated that anti-MHC I antibody-induced TV is ameliorated by pharmacologically induced HO-1 and the application of anti-CD62E antibodies results in a deceleration of developing TV. HO-1 modulation is a promising therapeutic approach to prevent leukocyte recruitment and subsequent intima hyperplasia in TV and thus precludes organ failure.
1 Introduction
Solid organ transplantation is the treatment of choice for end-stage organ failure. Over recent decades, the 1-year survival after organ transplantation increased to over 90% due to continuously improved surgical techniques and perioperative care as well as increasingly effective immunosuppressive medication (1, 2). To date, the occurrence of chronic rejection determines long-term survival after organ transplantation. Every second patient develops transplant vasculopathy (TV) characterized by intima hyperplasia. The subsequent reduction in tissue perfusion limits organ survival. Preventive or therapeutic options are rare (3–5) and limited to monoclonal antibodies (6) or a combination of plasmaphereses and intravenous application of immunoglobulins (7). As therapeutic success is still limited, TV remains the major obstacle for long-term graft survival.
Owing to immunological differences between the recipient and donor organ, either a cellular or a humoral-driven immune reaction in which both pathways share common features is elicited. In the end, the activated immune system causes tissue injury which in turn leads to organ damage followed by graft failure (8). The main risk factor for developing TV is donor-specific antibodies (DSAs) recognizing MHC class I and II molecules on the cell surface of the donor organ (9). DSAs are associated with a two-fold increased risk for developing chronic rejection (10), and their concentration correlates with reduced survival time of the transplanted organ (11). DSA’s predominant role in TV has been proven by several mouse models where solely the application of anti-MHC I antibodies was capable of evoking TV, even in the absence of T and B cells or a functional complement system (12, 13).
TV particularly affects the blood vessels of the transplanted organ, and accordingly, one hallmark is intimal hyperplasia causing neointima formation and reduced vessel diameter (14). Dysfunctional endothelial cells (ECs) and migration of vascular smooth muscle cells (VSMCs) from the media into the intima as well as accumulation of collagen and elastin are responsible for the phenotypical alteration of transplanted organs undergoing chronic rejection (15, 16). Migrated VSMCs adopt a synthetic phenotype and directly influence the fate of ECs due to paracrine and autocrine effects of proinflammatory cytokines (17). Intima thickening occurs at an early timepoint after the onset of TV (18) and results in altered tissue perfusion (19) and impaired vasodilatation capacity (20) accompanied by reinforced stiffness of the vessel wall (16). ECs are a preferred target for DSA because their location predisposes them to interface between the recipient’s circulation and the graft and because they express all major sets of antigens. Besides complement activation by bound antibodies, the binding of anti-HLA-I antibodies to EC induces complement-independent upregulation of intracellular signaling pathways, such as NF-κB, ERK, and FGF, manifesting itself in phenotypical conversion and consequently in EC activation (21). EC activation results in the reinforced expression of adhesion receptors on the surface as well as intensified cytokine exocytosis (22). Established cytokine gradients and the availability of unbound adhesion receptors lead to the recruitment of leukocytes to the site of inflammation and to their transmigration into the vessel wall. The first loose interactions between ECs and leukocytes are predominantly mediated by integrins and selectins (23), and the resulting transmigration of leukocytes into the vessel wall is achieved through additional binding of specific receptors, such as PECAM, JAM A, and CD99 (24). If the immune system fails to resolve EC activation and the proinflammatory environment of the endothelium persists, chronic inflammatory status—and therefore TV—is established.
Meanwhile, histopathological analysis of transplanted organs has identified macrophages as the predominant cell type forming the mononuclear infiltrate (25). The implication of the amount of transmigrated monocytes and thus the extent of the constituted mononuclear infiltrate within the graft’s vessel wall bears a close negative correlation with organ function (26). A retrospective study revealed that transmigrated monocytes correlate with a serological occurrence of DSA and could be used as a diagnostic marker for early asymptomatic disease (27). This finding would allow for faster therapy initiation and thus prevent progression to fully developed TV. Patients with TV also bear a significantly higher number of transmigrated monocytes at an earlier timepoint after transplantation than patients who do not suffer from chronic rejection (28). Moreover, a cardiac transplantation model in mice showed that monocytes contribute to the progression of organ rejection, whereby systemic monocyte depletion decreases the extent of TV (29). Blocking specific adhesion receptors on the EC’s surface prevents monocyte transmigration and leads to a reduction in experimentally induced chronic rejection (30).
The presence of DSA does not inevitably lead to pathological changes in transplanted tissue. Stable organ function can be maintained over a longer period, a state referred to as accommodation (31). Accommodation is reflected in an anti-inflammatory EC phenotype that is noticeable in the increased expression of anti-apoptotic and antioxidative genes (32) when the expression of adhesion receptors simultaneously decreases (33). The consequences of induced anti-inflammatory pathways have not been fully determined, and accommodation—as well as its underlying mechanisms—has not been characterized in detail. A mouse-to-rat xenotransplantation model has provided evidence that the inducible isoform of heme oxygenase (HO)-1 mediates the main anti-inflammatory effect of the enzyme (34). HO-1 catalyzes the conversion of heme to biliverdin through the release of carbon monoxide (CO) and iron [Fe (II)]. Furthermore, targeted HO-1 induction protects against anti-HLA-I antibody-induced EC activation (22) and diminishes leukocyte adhesion (35). Reduced leukocyte adhesion is conveyed by biliverdin whereby low concentrations of CO impede the expression of proinflammatory cytokines, such as TNF-α and IL-1β (36). The heme derivate cobalt (III) protoporphyrin IX chloride (CoPPIX) has also been identified as an HO-1 expression enhancer that increases the homeostatic-relevant IL-8 in EC (37). Statins also exert anti-inflammatory effects in an HO-1-dependent fashion. Besides reducing cholesterol levels, statin treatment for patients suffering from TV results in declined neointima formation within the graft (38, 39).
Nevertheless, it is essential to understand the underlying mechanisms leading to the formation of a neointima and mononuclear infiltrate in more detail. This study demonstrates the effects of HO-1 expression on anti-HLA-I-induced adhesion and leukocyte transmigration on and across an endothelial monolayer. Moreover, our initial results suggest that HO-1 activity has a protective effect on developing TV in an allogenic aortic transplantation model in mice.
2 Materials and methods
2.1 Reagents
We used an anti-HLA-I antibody (clone: w6/32, isotype: IgG2a) from Thermo Fisher Scientific (Waltham, USA) and an isotype control from BioLegend (San Diego, USA). Recombinant human monocyte chemoattractant protein (MCP)-1 was obtained from PeproTech (Winterhude, GER), CoPPIX was from Frontier Scientific Services (Newark, USA), and CO-releasing molecule (CORM)-401 as well as Cell Tracker Green was from Sigma-Aldrich (St. Louis, USA). For statin treatment, pravastatin tablets from Novartis (Basel, CH) were used, and the pharmacological substances were dissolved in ultrapure H2O. Subsequently, carrier substances were removed and the solution was sterile-filtered. HO-1-specific siRNA (#L-006372-00-0005) and non-target siRNA control were purchased from Dharmacon (Lafayette, USA), and our transfection reagents, OptiMEM-Medium and Lipofectamine RNAi/Max, were from Thermo Fisher Scientific. RNeasy Micro Kit and QuantiTect Rev. Transcription Kit were obtained from Qiagen (Hilden, GER). TaqMan® gene expression assays and TaqMan® Fast Advanced Master Mix were purchased from Thermo Fisher Scientific. Polyclonal anti-human CD62E antibody and isotype control for blocking experiments in vitro were both from Abcam (Cambridge, GB) (mouse anti-human CD62E: ab18981 and mouse IgG isotype control: ab37355). For the blocking experiments, in-vivo mouse anti-mouse CD62E (clone: RME-1, isotype: IgG1, 148802, BioLegend, San Diego, USA) and mouse IgG1 isotype control (clone: MG1-45, 401402, BioLegend, San Diego, USA) were used.
2.2 Cell culture
Primary human aortic endothelial cells and a culture medium consisting of basal medium MV2 supplemented with growth factor MV 2 Kit were obtained from PromoCell (Heidelberg, GER) (40). ECs were cultured until reaching 80% confluency before splitting. For all experiments, ECs were used between passages 4 and 6. Stimulation of ECs with antibodies or HO modulators was conducted in a starvation medium consisting of basal medium and 2% FCS. The human monocytic cell line THP-1 was purchased from Life Technologies (Carlsbad, USA) and cultured in RPMI-1640 medium supplemented with 10% FCS and maintained at a density of 106 cells/mL. ECs were stimulated with different concentrations of anti-HLA-I antibody (0.1 µg/mL or 1 µg/mL) or with the corresponding isotype control (1 µg/mL). Modulation of HO-1 activity in ECs was induced by stimulation with 5 µM of CoPPIX or 1 µM of statin for 24 h followed by further stimulation with the HO-1 modulator alone or in combination with 1 µg/mL of anti-HLA-I for the time period indicated. For siRNA transfection, ECs were cultured until 80% confluency and incubated with siRNA/Lipofectamine complexes in OptiMEM-Medium for 4 h. ECs were further cultivated in a culture medium, and RNA or proteome extraction was performed 24, 36, or 48 h after transfection.
2.3 Adhesion and transmigration assays
For adhesion assays, ECs were cultured in 12-well plates until they reached confluency and then stimulated. Shortly before the assay, 106 THP-1 cells/mL were stained with 1 µg/mL of Cell Tracker Green (CTG) in Hank’s balanced salt solution (HBSS) medium for 30 min, washed, and resuspended in a binding buffer consisting of HBSS, 2 mM of Ca2+, and 2 mM of Mg2+. A concentration of 105 THP-1 cells/mL was adjusted and cells were incubated for 15 min. The stimulation medium was replaced by 500 µL of binding buffer, and the plate was placed on a shaker at a speed of 20 rotations/min. Five hundred microliters of THP-1 suspension was added to every EC cultured well, and the plate was covered with aluminum foil and incubated for 30 min. The supernatant was removed and ECs were washed five times with binding buffer. In the end, ECs and adherent THP-1 were detached and washed, and the number of CTG+ cells out of the total cell count was analyzed with flow cytometry.
For transmigration assays, ECs were cultured in CIM plates until confluency followed by the indicated stimulation. The medium from the lower chamber was replaced by 160 µL of transmigration medium (50% starvation medium + 50% THP-1 culture medium) supplemented with 20 ng/mL of MCP-1. The medium from the upper chamber was aspirated and 50 µL of the transmigration medium was added. The CIM plate was placed in the xCELLigence system (OLS OMNI Life Science) and background measurement was begun. THP-1 cells (2.5 × 104) resuspended in 100 µL of transmigration medium were added to the upper chamber, and the cell index (CI) was measured for 4 h. Readout included the area under the curve (AUC) of the CI.
For CD62E blocking experiments, ECs were incubated with 5 µg/mL of mouse anti-human CD62E antibody or the corresponding isotype control for 1 h prior to beginning the adhesion or transmigration experiment.
2.4 RT-qPCR
For quantification of mRNA expression analysis, ECs were cultured in 12-well plates until 80% confluency and stimulated either with antibodies alone or in combination with HO-1 modulators. Total RNA extraction was performed at different timepoints and 500 ng of total RNA was used for reverse transcription reaction. qPCR was performed with TaqMan® Fast Advanced Master Mix protocol and TaqMan® gene expression assays specific for specific target sequences. All steps were performed according to the manufacturer’s specifications. The following TaqMan® gene expression assays were used: HMOX-1 (Hs01110250_m1), ESAM (Hs00332781_m1), PECAM-1 (Hs01065282_m1), ICAM-2 (Hs00609563_m1), CD99 (Hs00908458_m1), JAM-1 (Hs00170991_m1), JAM-3 (Hs00230289_m1), P-selectin (Hs00927900_m1), and E-selectin (Hs00174057_m1). GAPDH (Hs03929097_g1) was used as a housekeeping reference.
2.5 Flow cytometry
To assess the amount of CD62E expressed on the surface of ECs, cells were cultivated in 12-well plates until they reached 80% confluency followed by antibody stimulation with or without HO-1 modulation. After stimulation, the cells were washed, detached with Accutase solution (Merck KGaA, Darmstadt, Germany), and centrifuged. Afterward, cells were incubated with 1 µL of PE-coupled mouse anti-human CD62E antibody (322605, BioLegend, San Diego, USA) for 30 min at 4°C in the dark. Readout was the mean fluorescence intensity (MFI) of PE on ECs.
The presence of passively transferred donor-specific antibodies in recipient mice was confirmed using the plasma of Rag2 KO mice collected at the end of the experiment. A single cell suspension of Balb/c splenocytes (from the graft donor) was produced and incubated with 25 µL of plasma for 30 min on ice. The cells were washed and then stained with 1 µL of FITC-conjugated rat anti-mouse CD3 (100204, BioLegend, San Diego, USA) and 1 µL of PE-conjugated goat anti-mouse IgG (405307, BioLegend, San Diego, USA) for 30 min at 4°C in the dark. The readout was MFI of PE on CD3+ Balb/c splenocytes. All measurements were performed using FACSVerse™ Flow Cytometer and FACSuite Software Version 1.0.5.3840 I (both from BD Biosciences, Heidelberg, Germany).
2.6 Semiquantitative analysis of selected mouse and human cytokines
The Proteome Profiler Cytokine Arrays were used for human EC cell culture supernatants (ARY022B, Bio-Techne, Minneapolis, USA) and mouse plasma (ARY028, Bio-Techne, Minneapolis, USA) to measure the relative concentration of various cytokines. ECs were stimulated either with 1 µg/mL of anti-HLA-I antibody or IgG2a isotype control for 24 h in starvation medium. The supernatants were collected and pooled before being transferred onto the membranes. For the mouse cytokine array, plasma from the anti-MHC I antibody or isotype-treated mice was pooled and used for the assay. Both cytokine arrays were run in accordance with the manufacturer’s specifications.
The mean intensity of each spot was determined using ImageJ to evaluate the array. After subtracting the background of each membrane, all values were normalized to the reference spots on the same membrane and compared to the corresponding isotype control membrane.
2.7 Use of the heterotopic aortic transplantation model in mice to induce TV
Infrarenal transplantation of thoracic aorta segments into the abdominal aorta was performed using a protocol originally described by Koulack and colleagues (41). Briefly summarized, Balb/c donor mice, bearing the MHC I haplotype H2-Kd and purchased from Janvier-Labs (FRA), were euthanized using CO2, exsanguinated, and perfused using 0.9% saline. Thoracic aortas were explanted, segmented, and stored in PBS. B6.129S7-Rag2tm1Mom/J (Rag2 KO) mice, carrying a recombination activating gene 2 knockout mutation on a C57BL/6 background (JAX, #008449), were used as graft recipients. Due to their C57BL/6 background, these mice express the MHC I haplotype H2-Kb and represent a full MHC mismatch toward Balb/c mice. Rag2 KO mice are completely T- and B-cell-deficient and have a non-leaky immunodeficiency. At the age of 8 weeks, Rag2 KO mice were anesthetized using isoflurane. Following longitudinal laparotomy, the aorta was clamped and cut transversally. The graft was anastomosed in an end-to-end fashion with 11-0 nylon sutures (Prolene, 11-0, nylon black, S&T AG, Neuhaus, CHE) to the native abdominal aorta. The clamps were removed and the peritoneal and skin incisions were closed with 6-0 nylon sutures. For analgesia, 0.05 mg/kg of body weight (bw) buprenorphine was given immediately after induction and before the end of anesthesia. Analgesia was preserved with intraperitoneal (i.p.) injection of buprenorphine every 6 h for 2 days. Three days postoperatively, non-paralyzed mice were used for the experiments. Weekly i.p. application of 1.5 µg/g of bw anti-H-2Kd antibody (clone: SF1-1.1, isotype: IgG2a, BioLegend, San Diego, USA) induced TV in the transplanted vessel. After 30 days, the mice were euthanized, exsanguinated, and perfused with 0.9% saline to maintain vascular volume. The graft was harvested, embedded in O.C.T. medium (optimal cutting temperature), snap-frozen in liquid nitrogen, and stored at −80°C until ready for processing and histological examination.
Only male mice were used for all of the experiments, handled according to the recommendations of the Society of Laboratory Animal Science, and maintained under controlled SPF conditions. Animal experiments were approved by the Regional Council in Karlsruhe (permission number G222/19, date of approval 10/17/2019) and performed according to national legislation.
2.8 Modulation of HO-1 in vivo
To investigate the influence of HO-1 modulation on developing TV, the HO-1 modulators CoPPIX and statins were used. Additionally, the HO-1 metabolite CO was applied and CD62E blockage was tested. Simultaneous with the anti-MHC I antibody, H2O-solved CoPPIX was injected i.p. at a concentration of 5 µg/g of bw. Statins were dissolved in H2O and 40 µg/g of bw was administered daily in drinking water. CORM-401 was dissolved in DMSO in a nitrogen atmosphere and 30 µg/g of bw was given orally three times a week. Inactive CORM-401 (iCORM) was produced by incubating CORM-401 overnight at 60°C. Mouse anti-mouse CD62E antibody or isotype control was injected i.p. twice a week at the concentration of 3.5 µg/g of bw.
2.9 Morphometric analysis of grafts
Cross-sections of grafts at 5 µm thickness were prepared using a cryomicrotome (Leica Microsystems, Wetzlar, GER), and serial sections were H&E-stained for quantification of the neointima index (NI). Three distinct vascular compartments (lumen, intima, and media) were defined and the corresponding areas were determined. The NI was calculated based on the following formula (42):
The average of all NIs, calculated for every section independently, was assessed as the NI of the graft. For morphometric and immunohistochemical analyses, an Olympus BX63 microscope (Olympus Life Science Solutions, Waltham, USA) and CellSens (Olympus Life Science Solutions, Waltham, USA) were used for image capturing and analyzing, respectively. Besides the grafts, short segments of the naive aorta of Rag2 KO mice were explanted and H&E-stained.
2.10 Immunohistological analysis of grafts
Tissue sections were fixed with ice-cold acetone and blocked with Mouse on Mouse (M.O.M.) blocking reagent (Vector Laboratories, Burlingame, USA) for 30 min. Stainings were performed with the following antibodies: rat anti-mouse CD68 (MCA1957, Bio-Rad Laboratories, Hercules, USA) diluted 1:400 and incubated overnight at 4°C; goat anti-rat F(ab) Alexa Fluor® 555 (ab21434, Abcam, Cambridge, UK) diluted 1:100 and incubated for 2 h at room temperature (RT); rabbit anti-mouse CD62E (ab2497, Abcam, Cambridge, UK) diluted 1:300 and incubated overnight at 4°C; goat anti-rabbit F(ab) Alexa Fluor® 555 (A21428, Thermo Fisher Scientific, Waltham, USA) diluted 1:200 and incubated for 2 h at RT; mouse anti-vWF (CBMAB-V0158-YC, Creative Biolabs, Shirley, USA) diluted 1:50 and incubated overnight at 4°C; goat anti-mouse F(ab) Alexa Fluor® 488 (A11017, Thermo Fisher Scientific, Waltham, USA) diluted 1:100 and incubated for 2 h at RT; and mouse anti-mouse SMA (A2547, Sigma-Aldrich, St. Louis, USA) diluted 1:400 and incubated overnight at 4°C. Nuclei were stained with DAPI and slides were mounted using a fluorescence mounting medium (Dako North America, Carpinteria, USA) and coverslipped. The number of macrophages was calculated in four randomly selected areas on each section and normalized, and the average was calculated. ImageJ was used for quantifying CD62E expression on either EC (vWF/CD62E) or VSMC (SMA/CD62E). The amount of double-positive EC or VSMC cells was also measured.
2.11 Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc.). Data were presented as mean ± standard error of the mean. All data sets were tested for outliers using the ROUT method, and the identified outliers were excluded from further analysis. Data were tested for Gaussian distribution using the Shapiro–Wilk test. The Student’s t-test was used to compare two different groups of normally distributed data. For three or more groups, normally distributed data were tested for significant differences of the means using one-way ANOVA comparing three or more groups. For data sets not following normal distribution, the Kruskal–Wallis test was used. Multifactorial analyses were performed using two-way ANOVA. To adjust for multiple testing, the post-hoc Sidak’s multiple comparison test was employed. Significant differences were assumed if p <0.05.
3 Results
3.1 Stimulation of human ECs with anti-HLA-I antibodies induces adhesion and transmigration of monocytes and is dependent on HO-1 modulation
To examine whether anti-HLA-I antibodies affect EC/monocyte interactions, confluent monolayers of human primary EC were treated either with 0.1 µg/mL or 1 µg/mL of murine pan-HLA-I antibody (w6/32) or with 1 µg/mL of isotype control (IgG2a). After 24 h of stimulation, adhesion and transmigration assays were performed. For the adhesion assays, relative numbers of fluorescent THP-1 monocytes were counted using flow cytometry after adhesion to ECs was allowed for 30 min on a rocking plate (Figure 1A). Stimulation with the anti-HLA-I antibody increased the adhesion of monocytes to 2.65-fold (0.1 µg/mL of anti-HLA-I antibody) and 3.60-fold (1 µg/mL of anti-HLA-I antibody) compared to the IgG2a antibody control-treated EC (Figure 1B). The amount of transmigrated THP-1 was quantified in a commercially available assay by impedance measurement (Figure 1C). In accordance with adhesion, transmigration increased up to 2.80-fold (0.1 µg/mL of anti-HLA-I antibody) and 4.49-fold (1 µg/mL of anti-HLA-I antibody) after antibody stimulation (Figure 1D).
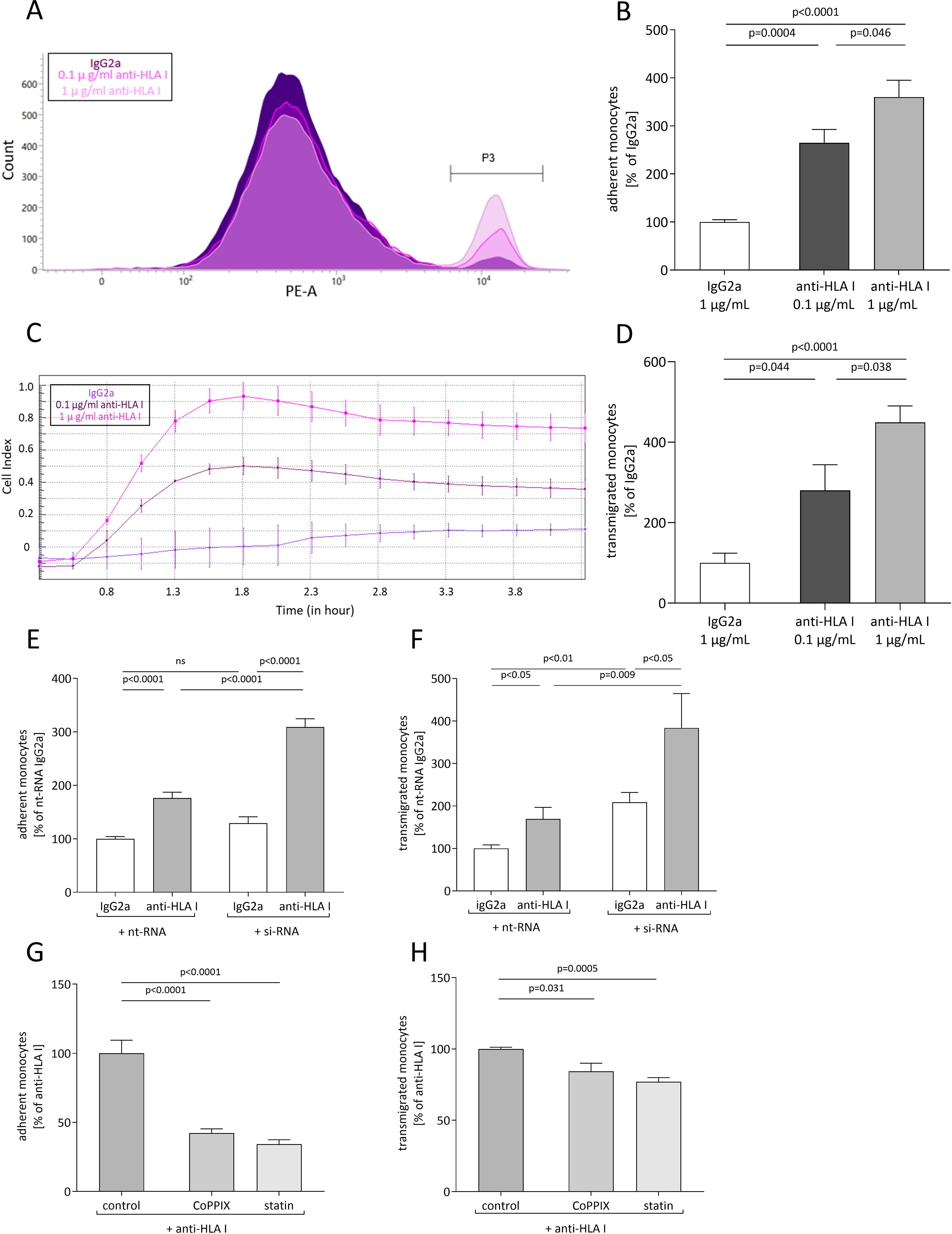
Figure 1. Anti-HLA-I antibody stimulation induces heme oxygenase (HO)-1-dependent adhesion and transmigration of monocytes. Human primary endothelial cells (ECs) were stimulated with 0.1 µg/mL or 1 µg/mL of anti-HLA-I antibodies or isotype control (IgG2a) in a starvation medium for 24 h. Adhesion and transmigration of THP-1 on or across an endothelial monolayer were quantified using flow cytometry and impedance measurement, respectively. (A) Representative flow cytometry graph for adhesion of THP-1 on stimulated ECs. Adherent THP-1 are shown in gate P3. (B) Statistical analysis of flow cytometry data normalized to IgG2a-treated ECs. (C) Representative image of impedance measurement of THP-1 transmigrated across a stimulated endothelial monolayer. (D) Statistical analysis of impedance data normalized to IgG2a-treated ECs. (E, F) HO-1 expression in ECs was suppressed with HO-1-specific siRNA transfection 36 h before anti-HLA-I antibody or IgG2a stimulation. Non-target siRNA (nt-RNA) was used as a control. Adhesion and transmigration of THP-1 were normalized to nt-RNA-transfected and IgG2a-stimulated ECs. (G, H) ECs were incubated with CoPPIX or statins for 48 h to increase HO-1 activity. The last 24 h of incubation occurred in the presence of anti-HLA-I antibody stimulation. Adhesion and transmigration were normalized to ECs stimulated with anti-HLA-I antibodies only. Normally distributed data (mean ± SEM) were analyzed using a one- or two-way ANOVA followed by Sidak’s multiple comparison test to adjust for multiple testing. Differences were considered statistically significant if p <0.05; n = 9.
The effect of diminished HO-1 expression in EC on monocyte adhesion and transmigration was analyzed after experimentally inducing HO-1 suppression. HO-1 knockdown was engendered by siRNA transfection and its efficiency was validated on mRNA as well as on the protein level. Twenty-four hours after transfection, HO-1 mRNA was already significantly reduced by 90% when compared to the nt-RNA-transfected control cells and reached its lowest expression after 48 h (Supplementary Figure S1A). Subsequently, a significant decrease in HO-1 protein concentration was seen 36 h after transfection and remained stable for up to 48 h (Supplementary Figures S1B, C).
After HO-1 knockdown in ECs, anti-HLA-I antibody stimulation resulted in a significant increase in adhesion and transmigration of monocytes compared to antibody-stimulated cells transfected with non-target (nt)-RNA. Adhesion was reinforced by 133% and transmigration by 214% (Figures 1E, F). Within the HO-1 siRNA-transfected groups, adhesion and transmigration were still significantly increased by anti-HLA-I antibody treatment when compared to the IgG2a antibody-treated ECs. As expected, a similar effect was observed in the groups transfected with nt-RNA-treated cells. Interestingly, HO-1 knockdown, when compared to nt-RNA-transfected ECs, also significantly increased transmigration in IgG2a-stimulated cells by 108%, suggesting that sufficient HO-1 expression in ECs is essential to maintaining EC homeostasis.
The effect of anti-HLA-I antibodies in the presence of induced HO-1 activity was further quantified. ECs were incubated with the HO-1 activator CoPPIX or statins that stimulate HO-1 as one of their pleiotropic effects, to increase HO-1 activity for 24 h prior to anti-HLA-I antibody stimulation. Compared to antibody stimulation alone, adhesion and transmigration were significantly decreased in CoPPIX-treated ECs to 42% and 84%, respectively (Figures 1G, H). Statin pretreatment of ECs decreased adhesion to 34% and transmigration to 77%. It can thus be concluded that stimulation of ECs with anti-HLA-I antibodies leads to increased numbers of adherent and transmigrated monocytes in a dose-dependent manner. Furthermore, genetic or pharmacological suppression of HO-1 concentration in EC intensifies anti-HLA-I antibody-driven effects. On the other hand, increasing HO-1 activity with CoPPIX or treatment with statins ameliorates anti-HLA-I antibody effects on adhesion and transmigration.
3.2 Anti-HLA-I antibody-mediated monocyte adhesion and transmigration depend on CD62E
To investigate the underlying mechanism of anti-HLA-I antibody-mediated adhesion and transmigration in more detail, ECs were screened for anti-HLA-I antibody-mediated changes of expression in a variety of established adhesion receptors. ECs were stimulated with two different antibody concentrations, and the transcriptome was collected and analyzed at indicated timepoints. At least a two-fold change of mRNA levels compared to the IgG2a control was considered a relevant change in expression. As confirmed by qPCR analysis, only mRNA levels for CD62E (E-selectin) fulfilled this criterion. A significant increase in CD62E mRNA concentration after 6, 12, and 18 h of anti-HLA-I antibody stimulation was observed (Figure 2A). In contrast, a two-fold mRNA increase was not obtained for JAM1, CD99, JAM3, ICAM2, or P-selectin (Supplementary Figure S2). Therefore, CD62E was the only EC adhesion receptor under investigation with our predefined cutoff value that proved susceptible to anti-HLA-I antibody stimulation. In addition, flow cytometry was used to clarify whether changes in mRNA expression resulted in alterations of CD62E expression on the surface of ECs (Figure 2B). Staining of EC with PE-coupled anti-CD62E antibodies following anti-HLA-I antibody stimulation showed a significant increase in CD62E protein concentration on the cell surface by 1.25-fold for 0.1 µg/mL of anti-HLA-I and by 2.05-fold for 1 µg/mL of anti-HLA-I. For CD62E surface expression, an anti-HLA-I concentration-dependent effect was not detected. To confirm the causal contribution of CD62E expression on the observed adhesion and transmigration results, ECs were incubated with 5 µg/mL of anti-CD62E antibody for 1 h after antibody stimulation. Blocking CD62E resulted in significantly reduced numbers of adherent and transmigrated monocytes compared to anti-HLA-I-activated cells incubated with an IgG control matching the CD62E blocking antibody. Adhesion decreased by 142% and transmigration by 88% (Figures 2C, D). Incubation with anti-CD62E isotype control IgG did not diminish anti-HLA-I-induced EC–monocyte interactions.
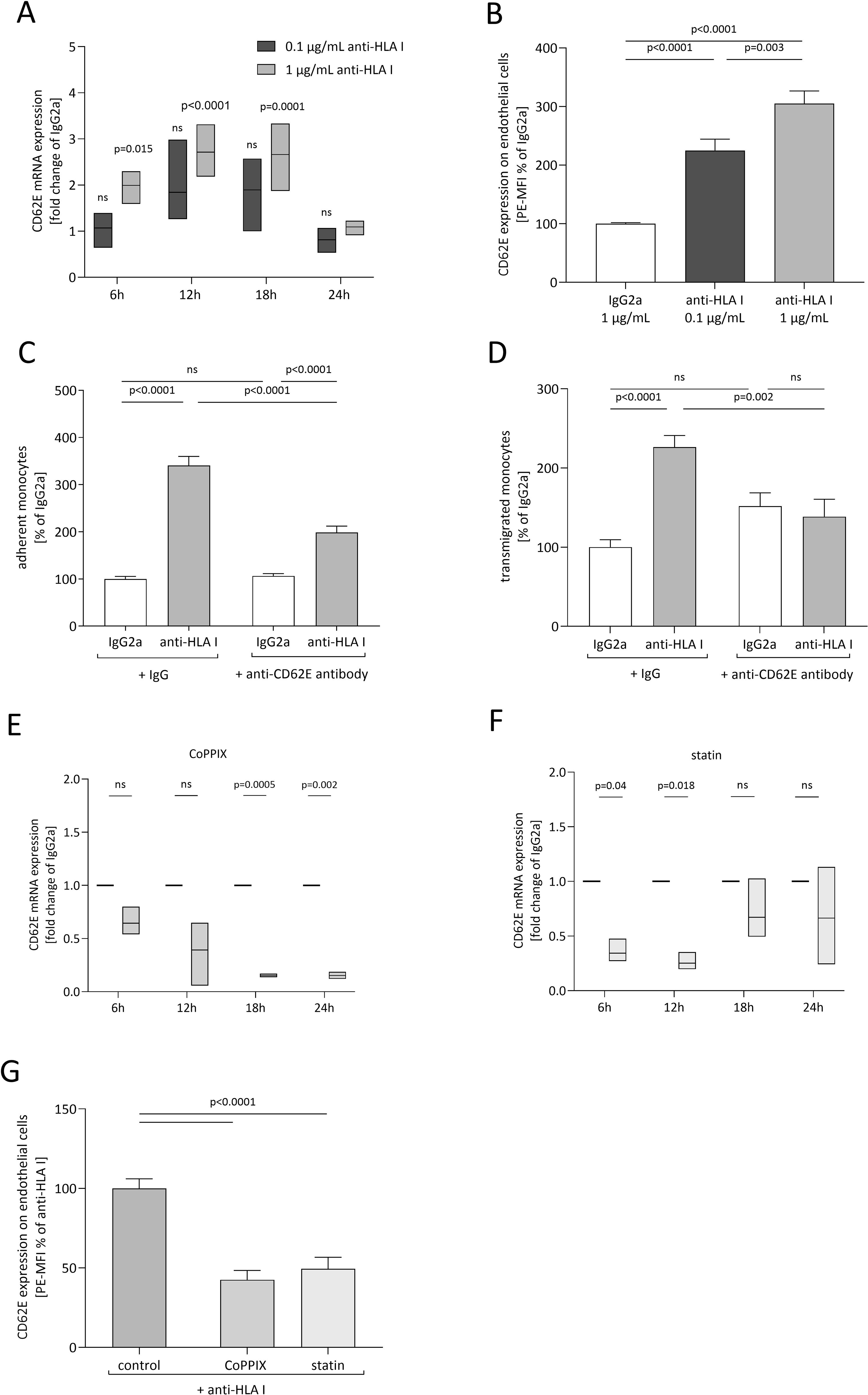
Figure 2. CD62E expression depends on anti-HLA-I stimulation and HO-1 activity, while its blockade decreases adhesion and transmigration. ECs were stimulated with 0.1 µg/mL (dark gray) or 1 µg/mL (light gray) of anti-HLA-I antibodies for 24 h in a starvation medium, and the expression of CD62E was analyzed. (A) At the indicated timepoints, fold change of CD62E mRNA level was measured and normalized to IgG2a (not shown)-treated ECs. (B) Mean fluorescence intensity (MFI) of PE-coupled anti-CD62E antibodies was measured to analyze the surface expression of CD62E on ECs after anti-HLA-I antibody stimulation. Data were normalized to IgG2a-treated ECs. (C, D) After stimulation with 1 µg/mL anti-HLA-I antibodies or IgG2a, ECs were incubated with anti-CD62E antibody or IgG isotype control for receptor blocking before adhesion or transmigration of THP-1 was quantified. (E, F) ECs were incubated with CoPPIX (E) or statins (F) for 24 h to induce HO-1 activity. After incubation, ECs were stimulated with 1 µg/mL of anti-HLA-I antibodies or IgG2a for up to 24 h in the presence of the corresponding HO-1 inducer. At the indicated timepoints, fold change of CD62E mRNA was measured using qPCR and normalized to IgG2a-treated ECs. (G) MFI of PE-coupled anti-CD62E antibodies was measured to analyze surface expression of CD62E on ECs after CoPPIX and statin incubation followed by 24 h of anti-HLA-I antibody stimulation. Data were normalized to anti-HLA-I antibody-treated ECs. Normally distributed data were analyzed using one-way (B–D, n = 9; G) or two-way (A, E, F, n = 3) ANOVA followed by Sidak’s multiple comparison test to adjust for multiple testing; statistically significant differences were assumed if p <0.05. ns, not significant.
3.3 Target induction of HO-1 activity reduces anti-HLA-I induced CD62E expression
To further characterize the effects of CoPPIX- or statin-mediated HO-1 modulation on adhesion and transmigration, CD62E expression after increased HO-1 activity was quantified. mRNA expression was analyzed after 24 h of stimulation with HO-1 modulators followed by indicated time periods of anti-HLA-I stimulation in the presence of the respective HO-1 modulator. While anti-HLA-I antibody ligation alone significantly increased CD62E mRNA expression compared to IgG2a control antibody (as seen in Figure 2A), EC preincubation with CoPPIX or statins diminished these effects. In CoPPIX-treated ECs, a significant reduction of CD62E expression was observed after 18 and 24 h (Figure 2E), whereas statin-treated ECs showed a significant reduction after 6 and 12 h of anti-HLA-I antibody stimulation (Figure 2F). Alteration of CD62E protein expression on the surface of ECs was measured with flow cytometry, and a significant reduction of mean fluorescence intensity (MFI) of PE on CoPPIX- as well as statin-treated cells could be seen after 24 h (Figure 2G). In summary, CD62E expression is sensitive toward anti-HLA-I antibody ligation, and its expression can be modulated by HO-1 activity. Experiments with CD62E blocking antibodies revealed that the observed effects of anti-HLA-I antibody-induced adhesion and transmigration of monocytes are partially mediated by binding this specific surface receptor.
3.4 Experimental setup of heterotopic aortic transplantation model in mice to investigate the effects of in-vivo HO-1 modulation on TV
To establish an in-vivo model for investigating TV progression, aortic segments from Balb/c mice were transplanted infrarenally into the abdominal aorta of Rag2 KO mice. Combining mouse strains with different genetic backgrounds represents a full MHC mismatch model, and the application of Balb/c matching anti-MHC I antibodies evokes TV in grafts (30). Beginning on postoperative day (POD) 3, anti-MHC I antibody (SF1-1.1) or IgG2a isotype controls were injected weekly (Figure 3A). After 30 days, the mice were euthanized to collect the graft and blood. During the experiment, mice received either weekly intraperitoneal CoPPIX or statins administered continuously via drinking water for HO-1 modulation. To test the effects of the HO-1 downstream metabolite carbon monoxide (CO) on TV development, CORM or its inactive form (iCORM) was applied three times a week. For blocking experiments, anti-CD62E blocking antibodies were injected twice a week (Figure 3B).
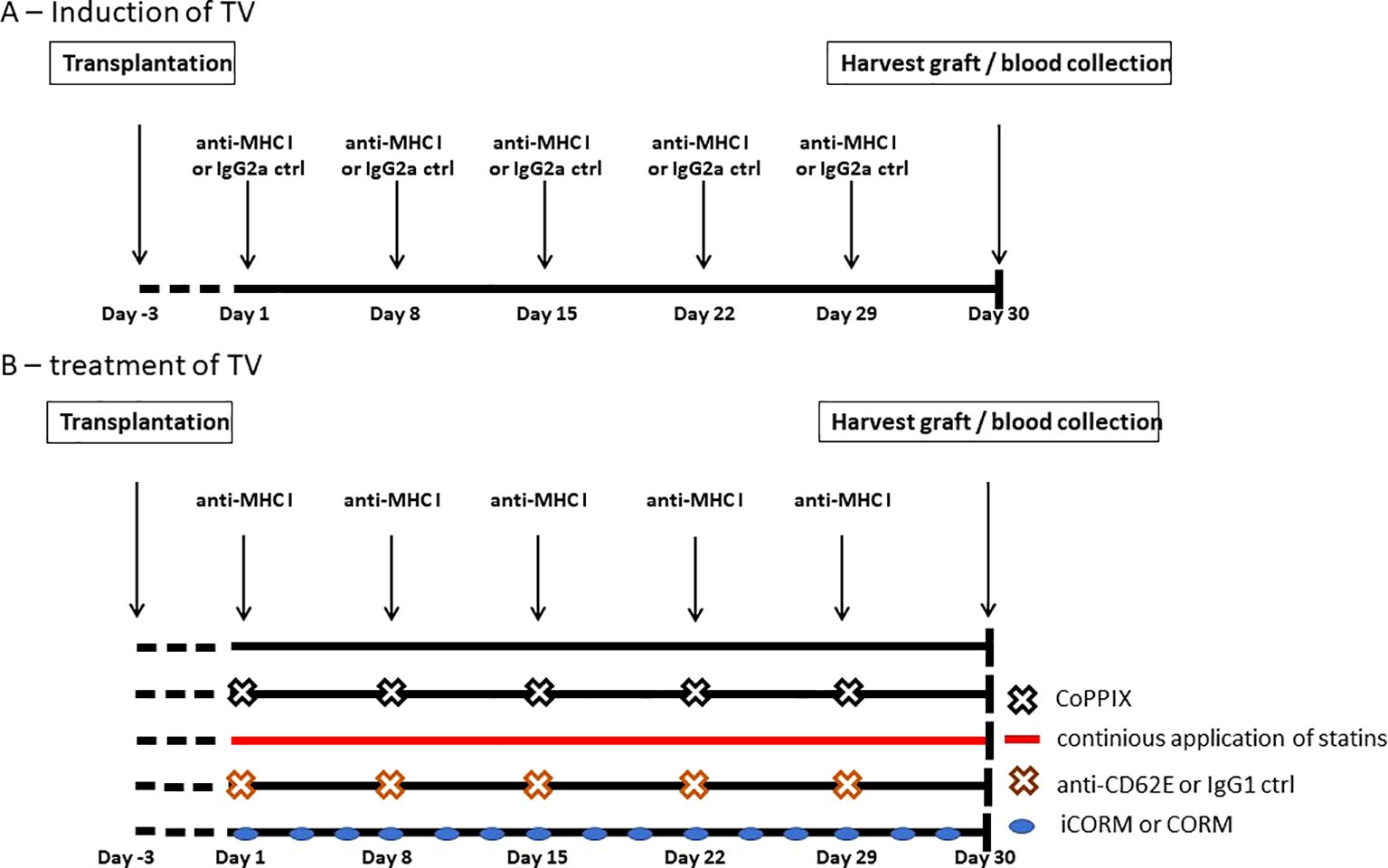
Figure 3. Experimental setup of antibody-induced and HO-1-mediated treatment of TV in Rag2 KO mice. Segments of thoracic aorta from Balb/c mice were transplanted into the abdominal aorta of Rag2 knockout (KO) mice. Experiments were begun on postoperative day 3. (A) Schematic drawing of DSA-induced TV. Mice received i.p. injections of anti-MHC I antibodies or IgG2a isotype control weekly to induce TV in the graft. After 30 days, mice were euthanized and the grafts as well as blood were collected. (B) Schematic drawing of the experimental setup for testing different compounds for reducing TV burden, which were applied in addition to anti-MHC I antibodies. CoPPIX was injected i.p. weekly and statins were continuously administered with the animals’ drinking water. CO-releasing molecule (CORM) or its inactive form (iCORM) was administered enterally, every other day. The CD62E blocking antibody or IgG1 isotype control was injected i.p. biweekly.
3.5 HO-1 modulation minimizes monocytic infiltrates within the vessel wall of anti-MHC I-treated mice
According to in-vitro data where anti-HLA-I antibodies support the adhesion and transmigration of mononuclear cells, one may conclude that monocytes are a driving force in TV development. To prove this assumption, the extent of monocytic infiltrate within the graft was analyzed to investigate whether applying anti-MHC I antibodies promotes monocyte infiltration in vivo, as well as if HO-1 modulation has a preventive effect on monocyte trafficking. Grafts were harvested after 30 days and serial cross-sections were stained for CD68+ monocytes. In addition, the autofluorescence of elastin fibers indicating medial structures was recorded. Grafts from anti-MHC I-treated mice harbor significantly more CD68+ cells in their vessel walls than those of isotype control-treated animals (Figure 4A). On average, the number of CD68+ cells in control mice was 4.2 × 10−4 µm2 and thus significantly lower compared to 10 × 10−4 µm2 CD68+ cells in anti-MHC I-treated mice. In accordance with our in-vitro findings, weekly injections of CoPPIX reduced the number of monocytes significantly to 5.4 × 10−4 µm2, and statin administration lowered the number of CD68+ cells to 6.6 × 10−4 µm2. Analysis of CORM-treated mice revealed a significantly lower number of CD68+ cells compared to the iCORM control group (Figure 4B). Weight gain, documented once a week for general health monitoring, did not differ over time between the experimental groups (Supplementary Figure S3). Furthermore, the hematoxylin and eosin (H&E)-stained segments of Rag2 KO aortas did not show TV or signs of inflammation in the Rag2 KO vasculature, indicating that anti-MHC I antibodies did not induce unspecific vascular inflammation outside of the graft (Figure 4C). Our CD68 staining proves that administering anti-MHC I antibodies is sufficient to induce the formation of a monocytic infiltrate in transplanted aortic segments in the absence of an adaptive immune response. Targeted HO-1 induction attenuates the number of monocytes within the vessel wall, subsequently reducing neointima formation and, ultimately, the burden of TV. Similarly, the HO-1 metabolite CO leads to a reduction of monocyte numbers regardless of its solvent DMSO. H&E staining of the native Rag2 KO aorta does not show TV, demonstrating that DSAs (anti-H2-Kd) specifically affect the graft’s vasculature.
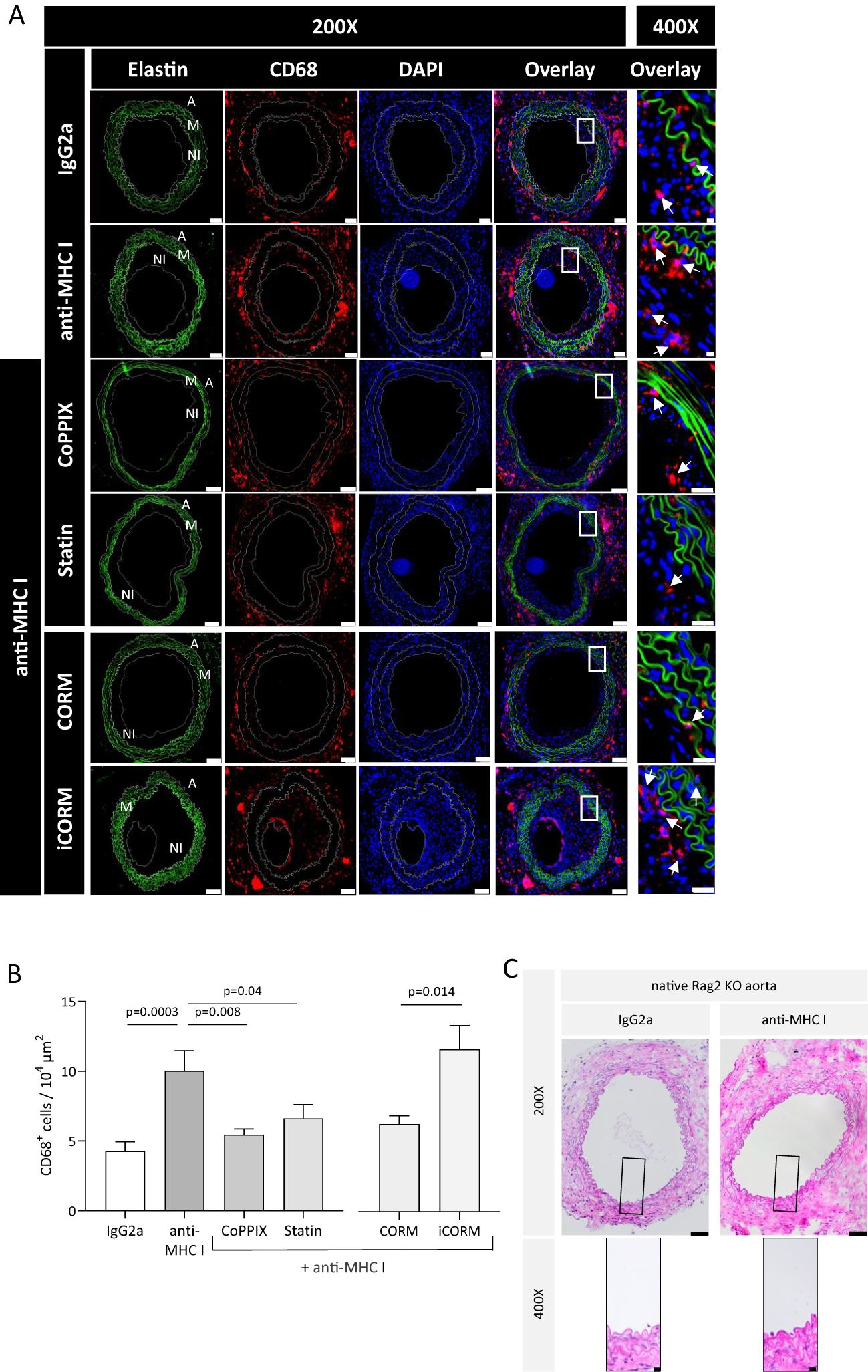
Figure 4. HO-1 modulation diminishes the monocytic infiltrate within the vessel wall of grafts. Thirty days after transplantation, Rag2 KO mice were euthanized, and cross-sections of the explanted grafts for immunohistochemical analysis were prepared. CD68 staining was performed every 75 µm of the graft to quantify the monocytic infiltrate. (A) Representative immunofluorescence images of CD68+ macrophages within the vessel wall (A, adventitia; M, media; NI, neointima). Elastic fibers (green) that made up the media of the vessel wall were pictured using autofluorescence; CD68 (red), DAPI (blue) (scale bar = 50 µm). High-magnification images of the box-indicated regions are shown for all treatment groups (scale bar = 5 µm). Arrows indicate macrophages. (B) Statistical analysis of CD68+ macrophages in the vessel wall. The number of CD68+ macrophages was statistically evaluated using a one-way ANOVA. Parametric data are shown as mean ± SEM and were adjusted for multiple testing; statistically significant differences were assumed if p <0.05. (C) Representative H&E images of Rag2 KO aorta at ×200 (scale bar = 50 µm) and indicated areas at ×400 magnification (scale bar = 10 µm). Grafted Rag2 KO mice were administered with anti-MHC I antibody or IgG2a to induce TV. Graft and native Rag2KO aorta were explanted after 30 days and analyzed. Here, segments of the native Rag2 KO (not the graft) aorta of mice from both groups are shown.
3.6 HO-1 modulation reduces anti-MHC I antibody-mediated TV burden in grafts
We analyzed the extent of neointima formation to evaluate whether reduced monocytic infiltrate within the vessel wall correlates with reduced pathophysiological TV manifestation. Serial cross-sections were stained with H&E to quantify the extent of anti-MHC I-induced neointima hyperplasia. Administration of antibodies once a week resulted in extensive neointima formation compared to IgG2a-treated mice. Otherwise, pharmacologically induced HO-1 activity with CoPPIX or statins prevented anti-MHC I antibody-induced neointima formation (Figures 5A, B). The media of the vessel wall was identified by elastin and collagen fibers, which are arranged concentrically within it (Figure 5B, arrows). The effects of the HO-1 downstream metabolite CO on antibody-induced neointima formation were investigated in mice treated with either iCORM or CORM. Grafts of mice treated with anti-MHC I antibodies and CORM revealed less newly formed neointima when compared to mice who received iCORM (Figures 5C, D). For statistical analysis, the NI was calculated and compared between the treatment groups. Mice treated with anti-MHC I antibodies showed a significantly higher NI of 51.27 in comparison to IgG2a control animals, with an NI of 20.23 (Figure 5E). CoPPIX treatment to induce HO-1 limited the NI to 28.3 and continuously applied statins significantly lowered it to 29.97. Additionally, the HO-1 metabolite CO released from CORMs also led to a significant reduction in the calculated NI to 23.6, compared to its iCORM control with an NI of 50.
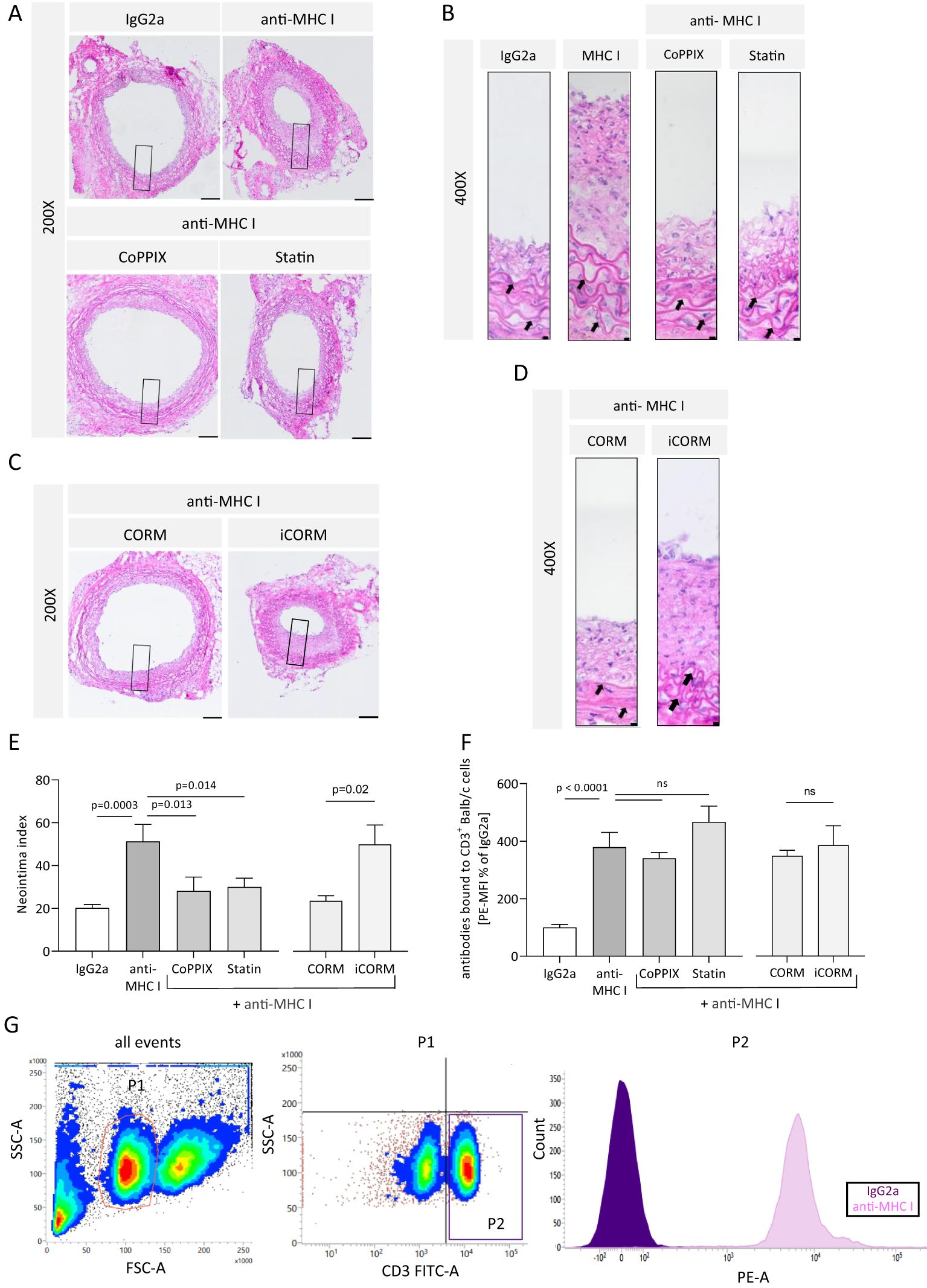
Figure 5. HO-1 modulation mitigates anti-MHC I antibody-induced neointima hyperplasia in vivo. The neointima index was derived from H&E-stained sections every 75 µm of the graft. (A, C) Representative images of different treatment groups at ×200 magnification (scale bar = 100 µm). High-magnification images of the box-indicated regions are shown in (B) and (D) (scale bar = 5 µm). Arrows indicate elastin fibers of the media. (E) Statistical analysis of the neointima index of n = 6–8 mice per group. (F) Mean fluorescence intensity (MFI) of PE on CD3+ cells was determined and normalized to the plasma of IgG2a-treated control mice. Normally distributed data (mean ± SEM) were analyzed using a one-way ANOVA or two-way ANOVA (F) followed by Sidak’s multiple comparison test to adjust for multiple testing; statistically significant differences were assumed if p <0.05. (G) Gating strategy to confirm the presence of anti-MHC I antibodies within the circulation of Rag2 KO mice. Balb/c splenocytes were incubated with plasma from recipient mice and stained with a combination of anti-CD3 antibodies coupled with FITC and PE-coupled anti-mouse IgG. CD3+ cells were gated as seen in the representative scatterplot. ns, not significant.
The presence of anti-MHC I antibodies within the circulation of recipient mice was confirmed using flow cytometry. CD3+ Balb/c splenocytes were incubated with the plasma of Rag2 KO mice collected at the end of the experiment and identified with FITC-coupled anti-CD3 antibodies. Bound anti-MHC I antibodies from the plasma on CD3+ splenocytes were determined using PE-coupled anti-IgG antibodies and quantified by the MFI of PE. The results revealed a significant increase in bound anti-MHC I antibodies to Balb/c CD3+ splenocytes incubated with plasma samples from antibody-treated mice when compared to the control mice. Plasma from CoPPIX-, statin-, CORM-, and iCORM-treated mice, in addition to passively transferred antibodies, showed no significant decrease in bound antibodies compared to the antibody-treated group, thus proving that the observed effects of minimized NI are not dependent on lower antibody concentrations (Figure 5F). The corresponding gating strategy is shown in Figure 5G. Taken together, we were able to successfully induce TV in transplanted aortic segments between genetically different mice strains. Targeted induction of HO-1 activity as well as the administration of the HO-1 downstream metabolite CO resulted in alleviated neointima formation. This finding suggests that the HO-1 pathway plays a causal role in minimizing proinflammatory conditions. Furthermore, we were able to detect circulating anti-MHC I antibodies in the blood of all mice who received this antibody.
3.7 CD62E expression on vascular smooth muscle cells is intensified in grafts after anti-MHC I treatment
Serial cross-sections of grafts of anti-MHC I- or IgG2a-treated mice were double-stained for either CD62E and EC marker von Willebrand factor (vWF) or CD62E and VSMC marker αSMA to quantify the influence of antibody application on CD62E expression on ECs and VSMCs in vivo. The relative amounts of CD62E+ ECs and VSMCs were measured (Figure 6A). Weekly applications of anti-MHC I antibodies did not induce CD62E expression on graft ECs compared to those of the control mice. In both groups, approximately 55% of ECs were stained positive for CD62E (Figure 6B). Nevertheless, immunostaining revealed a profound area of CD62E-positive cells within the vessel wall. In the grafts of IgG2a isotype control mice, 29% of VSMCs stained positive for CD62E; 59% of VSMCs, however, expressed CD62E in the anti-MHC I antibody group (Figure 6C). VSMC rather than EC cells were mainly responsible for CD62E expression in graft vasculature after 30 days of DSA treatment.
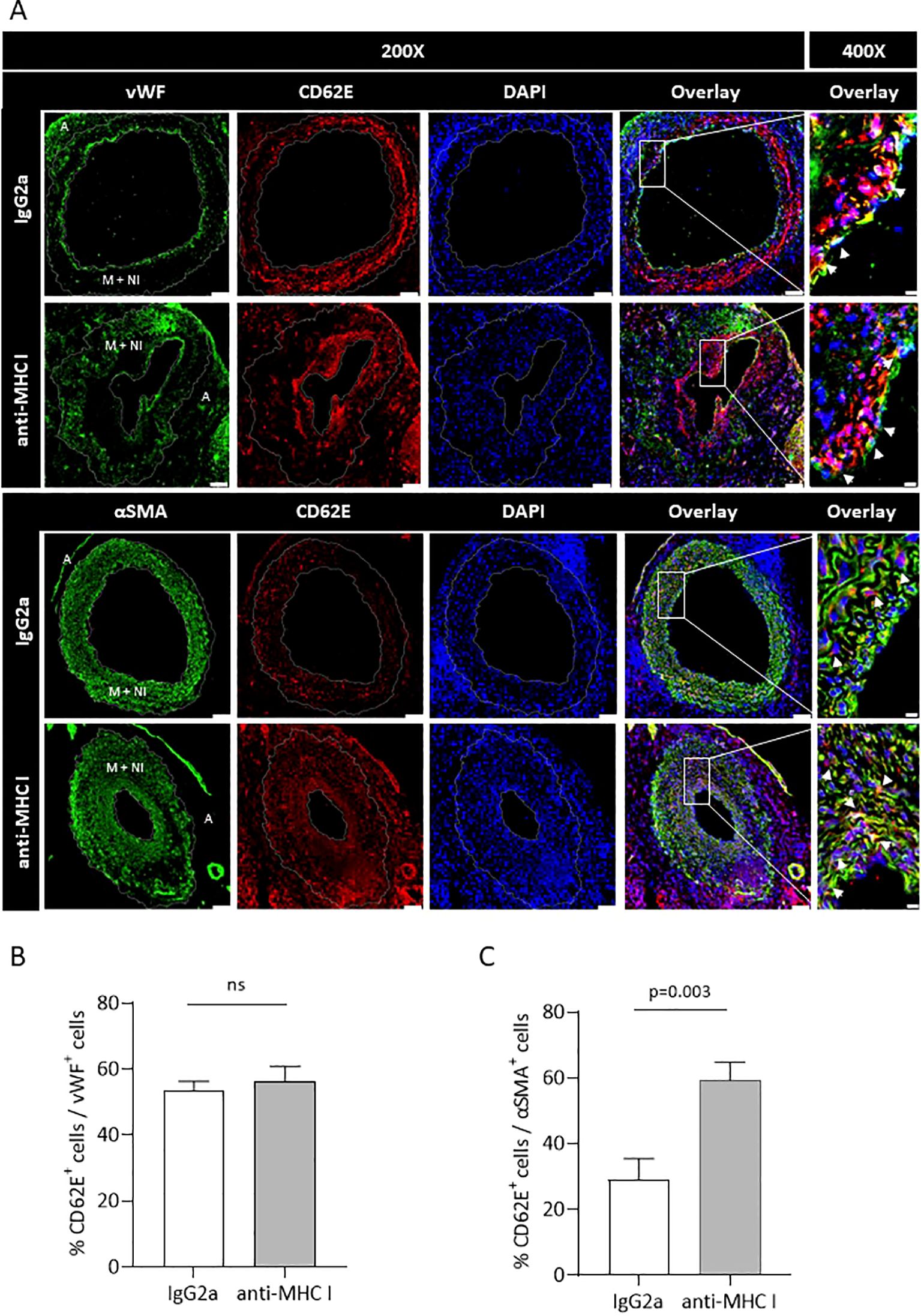
Figure 6. Anti-MHC I antibody induces CD62E expression on VSMCs but not on ECs. Serial cross-sections of grafts after 30 days of anti-MHC I antibody injection were double-stained for either CD62E/EC or CD62E/VSMC to quantify CD62E expression on different cell types of the graft’s vessel wall. (A) Immunofluorescence micrographs of MHC I antibody- or IgG2a-treated mice for ECs (vWF, green) VSMC (αSMA, green), CD62E (red), and DAPI (blue) at ×200 magnification (scale bar = 50 µm). Box-indicated areas are enlarged at ×400 magnification (scale bar = 5 µm), and physiologically different parts of the vessel wall are indicated on vWF- and αSMA-stained sections (A = adventitia, M + NI = media + neointima). (B) Statistical analysis of the relative number of CD62E+ ECs and (C) relative number of CD62E+ VSMCs. Normally distributed data are shown as mean ± SEM and were analyzed using Student’s t-test; statistically significant differences were assumed if p <0.05; n = 8 mice per group. ns, not significant.
3.8 Application of anti-CD62E antibody diminished anti-MHC I-mediated TV
The in-vitro experiments demonstrated that the application of an anti-CD62E blocking antibody on antibody-stimulated EC causes reduced monocytic adhesion as well as transmigration. We therefore tested the effects of CD62E blockade in our in-vivo mouse model. Mice were injected with DSA once a week to induce TV. They then received an additional anti-CD62E antibody or the corresponding isotype control twice a week. Grafts were harvested after 30 days and immunohistochemically processed for CD68 and H&E staining to quantify the extent of the monocytic infiltrate within the vessel wall as well as the newly formed neointima. Immunofluorescence evaluation revealed that the grafts of mice treated with anti-CD62E antibodies bear significantly less CD68+ monocytes within the vessel wall than those of control animals (Figures 7A, B). The intima index, calculated on chromogen-stained serial cross-sections, significantly decreased from 48.09 in isotype-treated mice to 23.25 in CD62E blocking antibody-treated mice (Figures 7C, D). There were no differences in weight fluctuations between the groups (Supplementary Figure S4). In accordance with prior in-vitro experiments, we proved that CD62E blockade in an allogenic transplantation model is sufficient for reducing the number of monocytes within the vessel wall and lowering the extent of neointima formation.
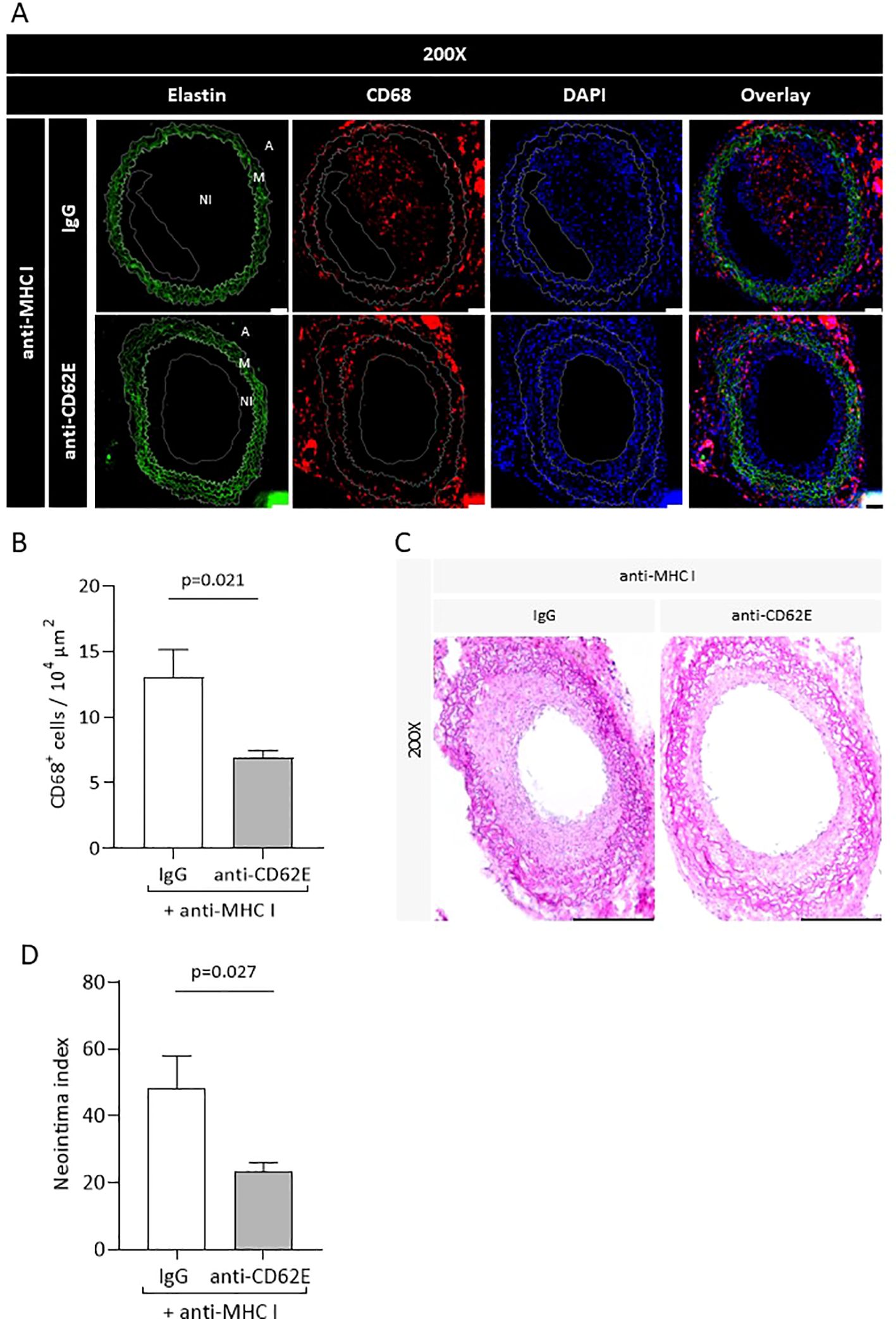
Figure 7. Blockade of CD62E results in reduced monocyte numbers in the vessel wall and attenuates the neointima index. Transplanted mice received CD62E blocking antibody or isotype control (IgG) biweekly in addition to anti-MHC I antibody administration. After 30 days, the grafts were harvested and histologically analyzed. (A) Immunohistological staining for elastin (autofluorescence, green), CD68 (red), and DAPI (blue) to quantify CD68+ monocytes (scale bar = 50 µm). (B) Statistical analysis of CD68+ monocytes. (C) Images of H&E stainings of mice who received a combination of anti-MHCI antibody and CD62E antibody or IgG (scale bar = 50 µm). (D) Statistical analysis of neointima index between groups.
3.9 Anti-MHC I antibodies induce profound changes of soluble cytokine expression in mice
Plasma was analyzed to semiquantify the expression of different cytokines after applying the anti-MHC I antibody and compared to the cytokine profile of control mice. We were able to measure 80 of 111 cytokines that could have been technically detected with the assay used. A reduction of more than 50% or an increase of more than two-fold compared to the control plasma was seen as physiologically relevant. For the sake of clarity, only these 23 physiologically relevant cytokines are shown in Figure 8A. The cytokine CCL22 was subject to the greatest change with a 10-fold increase of expression compared to the expression in control plasma. CXCL1, GDF-15, and DPPIV were more than twice as high as the control, and Gas-6, CXCL16, IL12-p40, CX3CL1, and Chitinase3-like1 were increased by a factor of 3 to 5 (Figure 8B). The remaining cytokines in the table were clearly decreased or no longer detectable at all in the plasma of antibody-treated mice.
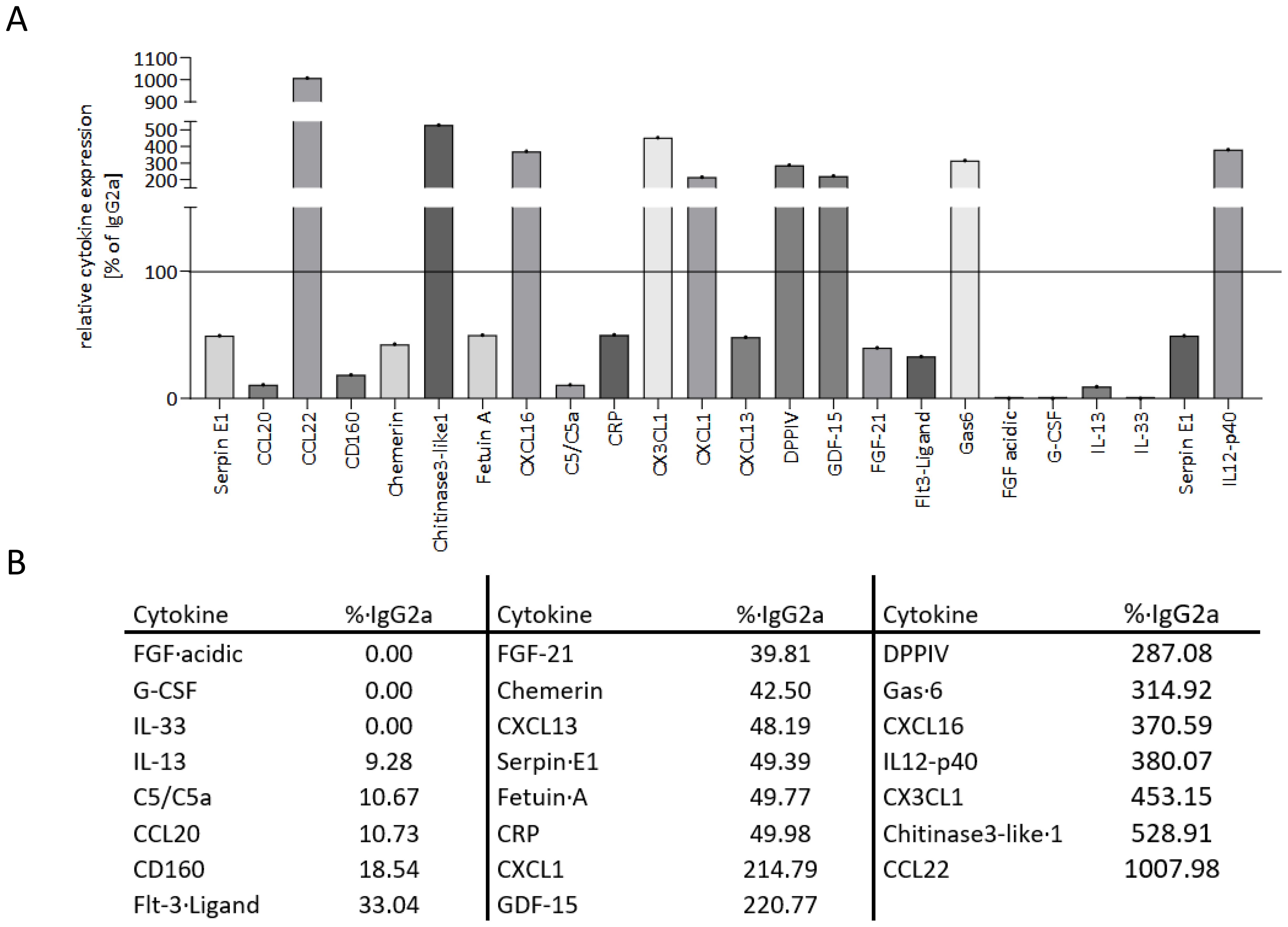
Figure 8. Anti-MHC I antibody induces up- and downregulation of different cytokines in mice. Plasma from transplanted mice was collected after 30 days of anti-MHC I antibody or IgG2a stimulation. With the membrane-based cytokine array, the expression profile of 80 cytokines was determined and compared to IgG2a isotype control. (A) Descriptive presentation of 23 cytokines that revealed a physiologically relevant change. (B) Table of the cytokines from (A) with their corresponding expression normalized to IgG2a.
In the cell culture supernatants, we measured 36 of 105 technically detectable cytokines of which only two had physiologically relevant changes (Supplementary Figures S5A, B).
4 Discussion
The main obstacle to long-term graft survival after solid organ transplantation is DSA-evoked TV. DSAs bind endothelial cells of the vasculature and induce activation of this specific cell population (43). Activation with proinflammatory stimuli results in endothelial dysfunction, further leading to expression changes within adhesion receptors on the surface, subsequent trafficking of immune cells, and chronic inflammation (44). In the context of chronically inflamed vessel walls, activation of different anti-inflammatory pathways, such as the HO-1 pathway, leads to accommodation, a condition in which DSAs do not further harm the graft (45). Here, we demonstrated in vitro and in a murine allogenic aortic transplantation model that stimulation and activation of human primary artery endothelial cells with anti-HLA-I antibodies reinforces CD62E-dependent interactions between ECs and monocytes. Furthermore, we provided evidence that targeted induction of HO-1 diminishes these effects and protects against the development of transplant vasculopathy.
Besides non-immunological factors such as ischemia–reperfusion injury (46), mode of brain death (47), and cold ischemia time (48), several immunological factors initiate EC activation and are reflected in the criteria for chronic antibody-mediated rejection. Binding of DSA on EC induces activation of the complement system, as seen in C4d-positive staining of graft biopsies (44). The presence of DSA and C4d covalently bound to EC does not necessarily lead to pathological graft changes (13). Host-derived immune cell infiltrates within the vessel wall consist predominantly of T cells, macrophages, NK cells, and to a lesser extent, dendritic cells (49). How each of these cell types contributes to TV development, as well as how they influence each other, remains under investigation. Stimulating human EC with anti-HLA-I antibodies induced a concentration-dependent increase in adhesion and transmigration of monocytes as typically seen with other proinflammatory stimuli (50–52). Furthermore, anti-HLA-I antibody stimulation results in an increased expression of the adhesion receptor CD62E on the surface of ECs. CD62E, as a member of the selectin family, provides the initial temporary interaction between ECs and monocytes and is responsible for extracting leukocytes from blood circulation. It is already known that CD62E expression is sensitive toward proinflammatory stimuli, such as TNF-α and LPS (53–55). In contrast to CD62P (P-selectin), which plays a major role in leukocyte homing, CD62E binding activates leukocytes via phosphorylation of the intracellular AKT and NF-κB pathways (56). Furthermore, CD62E binding via its corresponding ligands on leukocytes activates β2-integrins, which in turn results in enhanced transmigration (57). The regulation of CD62E by anti-HLA antibodies, however, has not been shown before. Furthermore, our CD62E blocking experiments have proven the functional relevance of anti-HLA-I-mediated CD62E expression on ECs. Blocking CD62E and therefore reducing the number of available adhesion receptors for monocytes diminish the number of adherent as well as transmigrated immune cells. These observed effects have been intensified by the HO-1-specific siRNA transfection of EC. HO-1 catalyzes the rate-limiting step of heme degradation (58) and, beyond that, plays an important role in anti-inflammatory pathways in order to protect ECs from activation (59, 60). Successfully reduced HO-1 protein expression has a noticeable effect on increased adhesion and monocyte transmigration. Our studies indicate that target induction of HO-1 with CoPPIX as well as incubation with statins results in decreased anti-HLA-I-mediated CD62E expression and minimized adhesion and transmigration of monocytes after anti-HLA-I ligation to ECs. Both reagents induce HO-1 expression and prevent EC activation (37, 61). It is also worth noting that statins exert a plurality of pleiotropic effects independent of the HO-1 pathway. Statin treatment mitigates NF-κB pathway activation, which itself mediates DSA-induced EC activation (62, 63). Nevertheless, sufficient HO-1 expression is mandatory for maintaining EC homeostasis, and its deficiency results in serious chronic inflammation and a shortened life expectancy in mammals (64, 65).
Chronic inflammation of the endothelium due to bound DSAs results in neointima formation and monocytic infiltrate within the vessel wall (44, 66). Both features are hallmarks of chronic transplant rejection, or so-called transplant vasculopathy. For our present study, we adapted the allogenic transplantation mouse model established by Koulack et al. (41) and successfully evoked TV by utilizing anti-MHC I antibodies. Mice treated with anti-MHC I antibodies revealed significantly increased neointima formation compared to isotype-treated control mice. Besides these local effects on the vessel wall, anti-MHC I antibodies induce profound changes in the plasma cytokine profile. The increased expression of soluble factors such as CCL22, CXCL16, CX3CL1, and CXCL1, which all play a crucial role as chemoattractants (67–70), proves the systemic effects of anti-MHC I antibodies in mice. In the experiments carried out here, it is not possible to distinguish whether these soluble factors have been released by ECs or already transmigrated immune cells, serving as a self-amplifying trigger. The HO-1 activators CoPPIX or statins lead to a reduced chronic inflammation of the vessel wall, which was manifested by a decreased neointima index. Upregulated expression of anti-inflammatory genes (such as Bcl-2, Bcl-xL, and HO-1) in the context of accommodation alleviates ICAM-1 and Il-1β expression and protects grafts from rejection (33). Furthermore, target upregulation of HO-1 impairs proinflammatory conditions and protects against viral infections, acute rejection, and cellular infiltration (71). Besides its beneficial effects on reduced neointima formation, CoPPIX and statins also diminish the extent of monocytes within the vessel wall. These observations are not associated with systemic inflammation. Mice treated with CO also benefit from a decreased TV burden. CO, a downstream metabolite within the HO-1 pathway, impedes TNF-α, IL-1, and MIP-1β (36). Beyond that, the application of CO after liver transplantation delays rejection (72). Transmigrated monocytes, whose numbers directly influence fibrosis in transplanted organs (73), are a driving force of neointima formation and therefore TV development. Growing concentrations of IL-12p40 in the periphery might be responsible for the induced recruitment of monocytes (74). Once recruited to the transplant, transmigration of monocytes into the vessel wall might be mediated by binding CD62E. Shortly after CD62E binding, leukocytes paracellularly transmigrate from the vasculature into the vessel wall, where they adopt proinflammatory phenotypes (27, 66). We have proven that VSMC of the graft increases CD62E expression on its surface and that CD62E blocking antibody ameliorates anti-MHC I-induced monocytic infiltrate and neointima formation.
Therapeutical options to treat TV or to decelerate its development are limited and there is an urgent need for improved therapies. Target induction of the anti-inflammatory enzyme HO-1 might be a possible solution, and we have shown that this goal can be achieved by statin therapy. Besides their LDL-lowering effects, statins induce HO-1 upregulation, prevent EC activation, and protect the cardiovascular system (75, 76). In our experiments, statins and CoPPIX were administered systemically to activate HO-1, but it also possible to overexpress HO-1 in the target tissue by viral transfection approaches.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by Regierungspräsidium Karlsruhe- Referat 35 -. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LS: Data curation, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. MZ: Investigation, Methodology, Writing – original draft, Writing – review & editing. HJ: Data curation, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing. NG: Investigation, Methodology, Writing – original draft, Writing – review & editing. ME: Project administration, Resources, Writing – original draft, Writing – review & editing. IH: Data curation, Formal analysis, Supervision, Writing – original draft, Writing – review & editing. MW: Funding acquisition, Resources, Validation, Writing – original draft, Writing – review & editing. SI: Writing – original draft, Writing – review & editing. JL: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the German Research Foundation (LA 2343/7-1) to JL and by the Heidelberger Stiftung Chirurgie (2021/461) to LS. This research did not receive any other specific grants from funding agencies within the public, commercial, or non-profit sectors. Departmental funds were provided from the Department of Anesthesiology, Heidelberg University Hospital, Heidelberg, Germany.
Acknowledgments
We gratefully acknowledge Klaus Stefan for his excellent technical support and would further like to thank the animal caretakers Illona Krämer, Silvio Krasemann, and Ulrike Gärtner.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1447319/full#supplementary-material
References
1. Lund LH, Khush KK, Cherikh WS, Goldfarb S, Kucheryavaya AY, Levvey BJ, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-fourth Adult Heart Transplantation Report-2017; Focus Theme: Allograft ischemic time. J Heart Lung Transplant. (2017) 36:1037–46. doi: 10.1016/j.healun.2017.07.019
2. Rana A, Ackah RL, Webb GJ, Halazun KJ, Vierling JM, Liu H, et al. No gains in long-term survival after liver transplantation over the past three decades. Annals Surgery. (2019) 269:20–7. doi: 10.1097/SLA.0000000000002650
3. Wilhelm MJ. Long-term outcome following heart transplantation: current perspective. J Thorac Dis. (2015) 7:549–51. doi: 10.3978/j.issn.2072-1439.2015.01.46
4. Royer PJ, Olivera-Botello G, Koutsokera A, Aubert JD, Bernasconi E, Tissot A, et al. Chronic lung allograft dysfunction: A systematic review of mechanisms. Transplantation. (2016) 100:1803–14. doi: 10.1097/TP.0000000000001215
5. Wolfe RA, Roys EC, Merion RM. Trends in organ donation and transplantation in the United States, 1999-2008. Am J Transplant. (2010) 10:961–72. doi: 10.1111/j.1600-6143.2010.03021.x
6. Gheith O, Al-Otaibi T, Halim MA, Mahmoud T, Nair P, Abdel Monem M, et al. Early versus late acute antibody-mediated rejection among renal transplant recipients in terms of response to rituximab therapy: A single center experience. Exp Clin Transplant. (2017) 15:150–5. doi: 10.6002/ect.mesot2016.P32
7. Lefaucheur C, Nochy D, Andrade J, Verine J, Gautreau C, Charron D, et al. Comparison of combination Plasmapheresis/IVIg/anti-CD20 versus high-dose IVIg in the treatment of antibody-mediated rejection. Am J Transplant. (2009) 9:1099–107. doi: 10.1111/j.1600-6143.2009.02591.x
8. Taylor AL, Negus SL, Negus M, Bolton EM, Bradley JA, Pettigrew GJ, et al. Pathways of helper CD4 T cell allorecognition in generating alloantibody and CD8 T cell alloimmunity. Transplantation. (2007) 83:931–7. doi: 10.1097/01.tp.0000257960.07783.e3
9. Smith JD, Banner NR, Hamour IM, Ozawa M, Goh A, Robinson D, et al. De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant. (2011) 11:312–9. doi: 10.1111/j.1600-6143.2010.03383.x
10. Tikkanen JM, Singer LG, Kim SJ, Li Y, Binnie M, Chaparro C, et al. De novo DQ donor-specific antibodies are associated with chronic lung allograft dysfunction after lung transplantation. Am J Respir Crit Care Med. (2016) 194:596–606. doi: 10.1164/rccm.201509-1857OC
11. Mao Q, Terasaki PI, Cai J, Briley K, Catrou P, Haisch C, et al. Extremely high association between appearance of HLA antibodies and failure of kidney grafts in a five-year longitudinal study. Am J Transplant. (2007) 7:864–71. doi: 10.1111/j.1600-6143.2006.01711.x
12. Uehara S, Chase CM, Cornell LD, Madsen JC, Russell PS, Colvin RB, et al. Chronic cardiac transplant arteriopathy in mice: relationship of alloantibody, C4d deposition and neointimal fibrosis. Am J Transplant. (2007) 7:57–65. doi: 10.1111/j.1600-6143.2006.01599.x
13. Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, et al. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. (2010) 10:510–7. doi: 10.1111/j.1600-6143.2009.02958.x
14. Chenzbraun A, Pinto FJ, Alderman EL, Botas J, Oesterle SN, Schroeder JS, et al. Distribution and morphologic features of coronary artery disease in cardiac allografts: an intracoronary ultrasound study. J Am Soc Echocardiogr. (1995) 8:1–8. doi: 10.1016/S0894-7317(05)80351-7
15. Lin H, Wilson JE, Roberts CR, Horley KJ, Winters GL, Costanzo MR, et al. Biglycan, decorin, and versican protein expression patterns in coronary arteriopathy of human cardiac allograft: distinctness as compared to native atherosclerosis. J Heart Lung Transplant. (1996) 15:1233–47.
16. Rahmani M, Cruz RP, Granville DJ, McManus BM. Allograft vasculopathy versus atherosclerosis. Circ Res. (2006) 99:801–15. doi: 10.1161/01.RES.0000246086.93555.f3
17. Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, et al. Mechanisms of induction of endothelial cell E-selectin expression by smooth muscle cells and its inhibition by shear stress. Blood. (2007) 110:519–28. doi: 10.1182/blood-2006-08-040097
18. Tsutsui H, Ziada KM, Schoenhagen P, Iyisoy A, Magyar WA, Crowe TD, et al. Lumen loss in transplant coronary artery disease is a biphasic process involving early intimal thickening and late constrictive remodeling: results from a 5-year serial intravascular ultrasound study. Circulation. (2001) 104:653–7. doi: 10.1161/hc3101.093867
19. Chih S, Chong AY, Mielniczuk LM, Bhatt DL, Beanlands RSB, et al. Allograft vasculopathy: The Achilles’ Heel of heart transplantation. J Am Coll Cardiol. (2016) 68:80–91. doi: 10.1016/j.jacc.2016.04.033
20. Hollenberg SM, Klein LW, Parrillo JE, Scherer M, Burns D, Tamburro P, et al. Changes in coronary endothelial function predict progression of allograft vasculopathy after heart transplantation. J Heart Lung Transplant. (2004) 23:265–71. doi: 10.1016/S1053-2498(03)00150-5
21. Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends Mol Med. (2015) 21:319–29. doi: 10.1016/j.molmed.2015.02.004
22. Zilian E, Saragih H, Vijayan V, Hiller O, Figueiredo C, Aljabri A, et al. Heme oxygenase-1 inhibits HLA class I antibody-dependent endothelial cell activation. PloS One. (2015) 10:e0145306. doi: 10.1371/journal.pone.0145306
23. Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. (2003) 24:327–34. doi: 10.1016/S1471-4906(03)00117-0
24. Mamdouh Z, Mikhailov A, Muller WA. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med. (2009) 206:2795–808. doi: 10.1084/jem.20082745
25. Grandaliano G, Gesualdo L, Ranieri E, Monno R, Stallone G, Schena FP, et al. Monocyte chemotactic peptide-1 expression and monocyte infiltration in acute renal transplant rejection. Transplantation. (1997) 63:414–20. doi: 10.1097/00007890-199702150-00015
26. Girlanda R, Kleiner DE, Duan Z, Ford EAS, Wright EC, Mannon RB, et al. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant. (2008) 8:600–7. doi: 10.1111/j.1600-6143.2007.02109.x
27. Xu L, Collins J, Drachenberg C, Kukuruga D, Burke A. Increased macrophage density of cardiac allograft biopsies is associated with antibody-mediated rejection and alloantibodies to HLA antigens. Clin Transplant. (2014) 28:554–60. doi: 10.1111/ctr.2014.28.issue-5
28. Pilmore HL, Painter DM, Bishop GA, McCaughan GW, Eris JM. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation. (2000) 69:2658–62. doi: 10.1097/00007890-200006270-00028
29. Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, et al. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest. (2014) 124:3579–89. doi: 10.1172/JCI74370
30. Valenzuela NM, Hong L, Shen XD, Gao F, Young SH, Rozengurt E, et al. Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo. Am J Transplant. (2013) 13:299–311. doi: 10.1111/ajt.12016
31. Smith RN, Colvin RB. Chronic alloantibody mediated rejection. Semin Immunol. (2012) 24:115–21. doi: 10.1016/j.smim.2011.09.002
32. Bach FH, Hancock WW, Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today. (1997) 18:483–6. doi: 10.1016/S0167-5699(97)01129-8
33. Fukami N, Ramachandran S, Narayanan K, Liu W, Nath DS, Jendrisak M, et al. Mechanism of accommodation in a sensitized human leukocyte antigen transgenic murine cardiac transplant model. Transplantation. (2012) 93:364–72. doi: 10.1097/TP.0b013e3182406a6b
34. Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. (1998) 4:1073–7. doi: 10.1038/2063
35. Keshavan P, Deem TL, Schwemberger SJ, Babcock GF, CookMills JM, Zucker SD, et al. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J Immunol. (2005) 174:3709–18. doi: 10.4049/jimmunol.174.6.3709
36. Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6:422–8. doi: 10.1038/74680
37. Loboda A, Jazwa A, Wegiel B, Józkowicz J, Dulak J. Heme oxygenase-1-dependent and -independent regulation of angiogenic genes expression: effect of cobalt protoporphyrin and cobalt chloride on VEGF and IL-8 synthesis in human microvascular endothelial cells. Cell Mol Biol (Noisy-le-grand). (2005) 51:347–55.
38. Ellimuttil TM, Harrison K, Rollins AT, Feurer ID, Rega SA, Gray J, et al. Effect of statin intensity on the progression of cardiac allograft vasculopathy. Card Fail Rev. (2021) 7:e15. doi: 10.15420/cfr
39. Jang HJ, Balcells M, Alegret MC, Santacana M, Molins B, Hamik A, et al. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget. (2016) 7:46219–29. doi: 10.18632/oncotarget.10078
40. Methe H, Balcells M, Alegret MC, Santacana M, Molins B, Hamik A, et al. Vascular bed origin dictates flow pattern regulation of endothelial adhesion molecule expression. Am J Physiol Heart Circ Physiol. (2007) 292:H2167–75. doi: 10.1152/ajpheart.00403.2006
41. Koulack J, McAlister VC, Giacomantonio CA, BitterSuermann H. Development of a mouse aortic transplant model of chronic rejection. Microsurgery. (1995) 16:110–3. doi: 10.1002/micr.1920160213
42. Armstrong AT, Strauch AR, Starling RC, Sedmak DD, Orosz CG. Morphometric analysis of neointimal formation in murine cardiac allografts: II. Rate location lesion Dev Transplant. (1997) 64:322–8. doi: 10.1097/00007890-199707270-00025
43. Walsh RC, Brailey P, Girnita A, Alloway RR, Shields AR, Wall GE, et al. Early and late acute antibody-mediated rejection differ immunologically and in response to proteasome inhibition. Transplantation. (2011) 91:1218–26. doi: 10.1097/TP.0b013e318218e901
44. Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. (2005) 5:807–17. doi: 10.1038/nri1702
45. Kenta I, Takaaki K. Molecular mechanisms of antibody-mediated rejection and accommodation in organ transplantation. Nephron. (2020) 144 Suppl 1:2–6. doi: 10.1159/000510747
46. Bharat A, Kreisel D. Immunopathogenesis of primary graft dysfunction after lung transplantation. Ann Thorac Surg. (2018) 105:671–4. doi: 10.1016/j.athoracsur.2017.11.007
47. Floerchinger B, Oberhuber R, Tullius SG. Effects of brain death on organ quality and transplant outcome. Transplant Rev (Orlando). (2012) 26:54–9. doi: 10.1016/j.trre.2011.10.001
48. Kayler LK, Srinivas TR, Schold JD. Influence of CIT-induced DGF on kidney transplant outcomes. Am J Transplant. (2011) 11:2657–64. doi: 10.1111/j.1600-6143.2011.03817.x
49. Merola J, Jane-Wit DD, Pober JS. Recent advances in allograft vasculopathy. Curr Opin Organ Transplant. (2017) 22:1–7. doi: 10.1097/MOT.0000000000000370
50. Lee BK, Lee WJ, Jung YS. Chrysin attenuates VCAM-1 expression and monocyte adhesion in lipopolysaccharide-stimulated brain endothelial cells by preventing NF-kappaB signaling. Int J Mol Sci. (2017) 18:1424. doi: 10.3390/ijms18071424
51. Li M, van Esch BCAM, Henricks PAJ, Folkerts G, Garssen J. The Anti-inflammatory Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor alpha-Stimulated Endothelial Cells via Activation of GPR41/43 and Inhibition of HDACs. Front Pharmacol. (2018) 9:533. doi: 10.3389/fphar.2018.00533
52. Murphy JM, Jeong K, Rodriguez YAR, Kim JH, Ahn EYE, Lim STS, et al. FAK and Pyk2 activity promote TNF-alpha and IL-1beta-mediated pro-inflammatory gene expression and vascular inflammation. Sci Rep. (2019) 9:7617. doi: 10.1038/s41598-019-44098-2
53. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. (2011) 118:6743–51. doi: 10.1182/blood-2011-07-343566
54. Yao Y, Jia H, Wang G, Ma Y, Sun W, Li P. miR-297 protects human umbilical vein endothelial cells against LPS-induced inflammatory response and apoptosis. Cell Physiol Biochem. (2019) 52:696–707. doi: 10.33594/000000049
55. Weishaupt C, Steinert M, Brunner G, Schulze HJ, Fuhlbrigge RC, Goerge T, et al. Activation of human vascular endothelium in melanoma metastases induces ICAM-1 and E-selectin expression and results in increased infiltration with effector lymphocytes. Exp Dermatol. (2019) 28:1258–69. doi: 10.1111/exd.v28.11
56. Davies JM, Radford KJ, Begun J, Levesque JP, Winkler IG. Adhesion to E-selectin primes macrophages for activation through AKT and mTOR. Immunol Cell Biol. (2021) 99:622–39. doi: 10.1111/imcb.12447
57. Gong Y, Zhang Y, Feng S, Liu X, Lü S, Long M, et al. Dynamic contributions of P- and E-selectins to beta2-integrin-induced neutrophil transmigration. FASEB J. (2017) 31:212–23. doi: 10.1096/fj.201600398RRR
58. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U.S.A. (1968) 61:748–55. doi: 10.1073/pnas.61.2.748
59. Lee T, Park HS, Jeong JH, Jung TW. Kynurenic acid attenuates pro-inflammatory reactions in lipopolysaccharide-stimulated endothelial cells through the PPARdelta/HO-1-dependent pathway. Mol Cell Endocrinol. (2019) 495:110510. doi: 10.1016/j.mce.2019.110510
60. Soares MP, Seldon MP, Pombo Gregoire I, Vassilevskaia T, Berberat PO, Yu J, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. (2004) 172:3553–63. doi: 10.4049/jimmunol.172.6.3553
61. Grosser N, Hemmerle A, Berndt G, Erdmann K, Hinkelmann U, Schürger S, et al. The antioxidant defense protein heme oxygenase 1 is a novel target for statins in endothelial cells. Free Radic Biol Med. (2004) 37:2064–71. doi: 10.1016/j.freeradbiomed.2004.09.009
62. Prasad R, Giri S, Nath N, Singh I, Singh AK. Inhibition of phosphoinositide 3 kinase-Akt (protein kinase B)-nuclear factor-kappa B pathway by lovastatin limits endothelial-monocyte cell interaction. J Neurochem. (2005) 94:204–14. doi: 10.1111/j.1471-4159.2005.03182.x
63. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. (2015) 35:1306–16. doi: 10.1161/ATVBAHA.114.304650
64. Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U.S.A. (1997) 94:10925–30. doi: 10.1073/pnas.94.20.10925
65. Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. (1999) 103:129–35. doi: 10.1172/JCI4165
66. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. (2015) 15:692–704. doi: 10.1038/nri3908
67. Ushio A, Arakaki R, Otsuka K, Yamada A, Tsunematsu T, Kudo Y, et al. CCL22-producing resident macrophages enhance T cell response in Sjogren’s syndrome. Front Immunol. (2018) 9:2594. doi: 10.3389/fimmu.2018.02594
68. Korbecki J, BajdakRusinek K, Kupnicka P, Kapczuk P, Simińska D, Chlubek D, et al. The role of CXCL16 in the pathogenesis of cancer and other diseases. Int J Mol Sci. (2021) 22:3490. doi: 10.3390/ijms22073490
69. Rivas-Fuentes S, SalgadoAguayo A, ArratiaQuijada J, GorocicaRosete P. Regulation and biological functions of the CX3CL1-CX3CR1 axis and its relevance in solid cancer: A mini-review. J Cancer. (2021) 12:571–83. doi: 10.7150/jca.47022
70. Wu CL, Yin R, Wang SN, Ying R. A review of CXCL1 in cardiac fibrosis. Front Cardiovasc Med. (2021) 8:674498. doi: 10.3389/fcvm.2021.674498
71. Blancou P, Tardif V, Simon T, Rémy S, Carreño L, Kalergis A, et al. Immunoregulatory properties of heme oxygenase-1. Methods Mol Biol. (2011) 677:247–68. doi: 10.1007/978-1-60761-869-0_18
72. Ikeda A, Ueki S, Nakao A, Tomiyama K, Ross MA, Stolz DB, et al. Liver graft exposure to carbon monoxide during cold storage protects sinusoidal endothelial cells and ameliorates reperfusion injury in rats. Liver Transpl. (2009) 15:1458–68. doi: 10.1002/lt.21918
73. Toki D, Zhang W, Hor KLM, Liuwantara D, Alexander SI, Yi Z, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. (2014) 14:2126–36. doi: 10.1111/ajt.12803
74. Ha SJ, Lee CH, Lee SB, Kim CM, Jang KL, Shin HS, et al. A novel function of IL-12p40 as a chemotactic molecule for macrophages. J Immunol. (1999) 163:2902–8. doi: 10.4049/jimmunol.163.5.2902
75. Chen JC, Huang KC, Lin WW. HMG-CoA reductase inhibitors upregulate heme oxygenase-1 expression in murine RAW264.7 macrophages via ERK, p38 MAPK and protein kinase G pathways. Cell Signal. (2006) 18:32–9. doi: 10.1016/j.cellsig.2005.03.016
Keywords: transplantation, chronic rejection, heme oxygenase-1, anti-HLA-1 antibodies, accommodation, endothelial cells, adhesion, monocyte transmigration
Citation: Schuster L, Zaradzki M, Janssen H, Gallenstein N, Etheredge M, Hofmann I, Weigand MA, Immenschuh S and Larmann J (2025) Heme oxygenase-1 modulates CD62E-dependent endothelial cell–monocyte interactions and mitigates HLA-I-induced transplant vasculopathy in mice. Front. Immunol. 16:1447319. doi: 10.3389/fimmu.2025.1447319
Received: 11 June 2024; Accepted: 11 February 2025;
Published: 07 March 2025.
Edited by:
Sina Naserian, Hôpital Paul Brousse, FranceReviewed by:
Sudipta Tripathi, University of Massachusetts Medical School, United StatesMichael Adu Gyamfi, Charité University Medicine Berlin, Germany
Copyright © 2025 Schuster, Zaradzki, Janssen, Gallenstein, Etheredge, Hofmann, Weigand, Immenschuh and Larmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Larmann, YW4tZGlyZWt0aW9uQHVrYWFjaGVuLmRl