 Guangqiang Meng
Guangqiang Meng Saran Feng
Saran Feng- Department of Hematology, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
Langerhans cell histiocytosis (LCH) is a disease caused by clonal expansion of CD1a+/CD207+ cells and is characterized by organ involvement and dysfunction. Treatment of LCH in children is risk-adapted, and multisystem LCH requires systemic therapy. Although systemic treatments such as chemotherapy and BRAF/MEK inhibitors have improved the cure rate of LCH, disease reactivation rates remain 30%, and eventually some patients progress to relapse-refractory LCH. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a promising salvage treatment strategy for children with relapse-refractory LCH. However, many questions such as the efficacy and indications of allo-HSCT, as well as suitable conditioning regimen are still undetermined for children with LCH. This review aimed to provide an update on advances in allo-HSCT for LCH in children, including indications, stem cell sources, conditioning regimens, chimerism, transplant-related complications, outcomes, and treatment of relapse.
Introduction
Langerhans cell histiocytosis (LCH) is a rare proliferative disease of the Langerhans cells (1–3). LCH has a wide spectrum of clinical presentations, ranging from solitary benign bony lesions to multi-organ involvement and dysfunction, which pose a high risk of death despite aggressive treatment. Depending on the number of organs or systems involved, LCH can be divided into single system LCH (SS-LCH) and multi-system LCH (MS-LCH). LCH with risk organs (RO-LCH) refers to infiltration of vital risk organs (RO) such as bone marrow, spleen, and liver. MS-LCH is divided into MS-RO+ and MS-RO- according to whether it involves the RO. The treatment of LCH in children varies depending on the severity of the disease (4–6). Poor prognostic factors include age < 2 years, RO involvement, relapse-refractory patients, and complications of hemophagocytic lymphohistiocytosis (HLH) or associated malignancies (7–10). Especially, LCH associated with another hematological neoplasm (LCH-AHN) were reported more frequently. Because LCH may associate in both children and adults to clonally related myeloid malignancies (11). High-risk LCH refers to the presence of the above risk factors. High-risk LCH have poor outcomes, and their 2-year survival rates are < 30% (12). Treatment for these high-risk LCH patients has not yet been established, but allogeneic hematopoietic stem cell transplantation (allo-HSCT) has curative potential for these patients.
Allo-HSCT for LCH treatment has a short history since 1987, with only a limited number of reported cases. The main reason for this is the low incidence of LCH. Approximately 5–6 children per million are diagnosed annually, and more than 50% of the patients are between 1 and 15 years of age, with a peak between 1 and 4 years (13). The reported incidence in adults is lower, approximately 1–2 individuals per million (14). The disease was previously considered non-malignant, and allo-HSCT is controversial. Recent studies have found that LCH is an inflammatory neoplasm of myeloid origin (15–17), and the reduction of transplant-related mortality after reduced intensity conditioning (RIC), more patients with LCH have been treated with allo-HSCT than before. Moreover, with an understanding of the pathogenesis of LCH, patients with BRAF-V600E and MAPK pathway (RAS-RAF-MEK-ERK) mutations accounted for a large proportion of LCH (18–20). Although inhibitors targeting these gene mutations are not curative, and they should be employed for an undetermined time in children, with currently unknown long-term consequences. Inhibitors have been used in clinical practice and have shown satisfactory therapeutic effects (18–20). As a result, the status of allo-HSCT in LCH has been challenged and the number of patients receiving allo-HSCT has decreased, especially in adults with LCH. However, for high-risk and relapse refractory LCH, allo-HSCT remains an effective salvage treatment for LCH.
Indication of allogeneic hematopoietic stem cell transplant
Currently, there are two promising treatment strategies for patients with high-risk and relapse/refractory LCH. The first is a combination of 2-chlorodeoxyadenosine(2-CDA) and cytarabine(Ara-C) (21–23). Another promising approach is allo-HSCT because of its strong immunomodulatory effects and the ability to eradicate tumor cells. MS-RO+ and failure of conventional therapy have very poor outcomes (7, 8). For these patients, salvage approaches based on novel agents such as 2-CDA alone or in combination with Ara-C did not result in improved survival rates. Therefore, allo-HSCT has recently been used as an alternative salvage treatment. In LCH, organ dysfunction including that of the liver and bone marrow, and central nervous system involvement/neurodegeneration are associated with a higher mortality rate. Therefore, for repeated relapses of LCH, it is recommended that patients undergo allo-HSCT early to prevent irreversible organ damage and poor survival. In addition, allo-HSCT is also a promising treatment strategy for patients with LCH and hematologic malignancies, especially acute myeloid leukemia(AML) (24–28).
Up to 55% of patients with LCH have BRAF mutations, especially in children younger than 2 years (29–31). Therefore, molecular-targeted therapies are likely to provide more opportunities for LCH treatment (32). Vemurafenib (VMF), a BRAF inhibitor, has been used to treat refractory childhood LCH that was BRAFV600E mutation-positive. A total of 54 children participated in the clinical trial (33). At 8 weeks, 38 children achieved complete responses and 16 achieved partial responses. VMF seemed to be effective in children with refractory LCH BRAFV600E-positive. However, discontinuation of VMF in 30 children resulted in 24 cases of LCH reactivation. The median time to reactivation of LCH was 0.9 months, and the reactivation rates at 6 months and 12 months were 72% and 84%, respectively. Therefore, molecular targeted therapies cannot eradicate the BRAFV600E Clone. Molecularly targeted therapy diminishes the status of allo-HSCT; however, allo-HSCT can still be considered for children with gene-negative LCH and repeated relapses. Therefore, molecular targeted therapies can be used as a treatment method for bridging allo-HSCT in LCH patients, and those who fail inhibitor treatment should receive allo-HSCT as soon as possible. In conclusion, allo-HSCT is suitable for relapsed and refractory LCH in children who have failed chemotherapy and targeted therapy (Figure 1).

Figure 1. Treatment algorithm for children with LCH.
Donor and stem cell source
Because the number of reported cases is limited, the best method and optimal time for transplantation are still not defined. Stem cell donors included matched family donors, matched unrelated donors, haploidentical donor, and unrelated cord blood donors. Due to the limited number of cases of children with LCH receiving HSCT and the lack of randomized controlled clinical studies, there is no clear and uniform conclusion on which donor is more beneficial to children. Compared with the stem cell source donor, the current researchers are more concerned about whether the stem cell source is bone marrow, peripheral blood or cord blood, because they are more likely to affect the efficacy and survival after transplantation. In earlier studies, most studies on transplantation for LCH have focused on bone marrow blood as the source of stem cells, whereas only a small proportion of studies have reported the use of peripheral and cord blood; However, in recent years, the number of applications of peripheral blood and cord blood stem cells has gradually increased. For example, of the 87 patients with LCH from the International Blood and Bone Marrow Transplantation Research Center (CIBMTR) and European Blood and Bone Marrow Transplant (EBMT), 20 underwent allo-HSCT before 2000 (34). There were 16 patients underwent bone marrow stem cell transplantation, 2 underwent peripheral blood stem cell transplantation, and 2 underwent cord blood stem cell transplantation. After the year 2000, 67 patients underwent allo-HSCT, 32 patients underwent bone marrow stem cell transplantation, 14 underwent peripheral blood stem cell transplantation, and 23 underwent cord blood stem cell transplantation.
At present, the application of peripheral blood and cord blood stem cells is also more favored, mainly because they have their own advantages. The infusion of peripheral blood stem cells containing a large number of CD34+ cells can result in more rapid hematological recovery (35). In addition, donor T lymphocytes in peripheral blood can have a stronger graft versus tumor (GVT) effect, which is important for patients with advanced-stage hematological malignancy (36). For infants, cord blood has many advantages such as an increased chance of suitable donors, rapid availability from banks, low risk of severe graft-versus-host disease (GVHD), and a low probability of transmitting infectious diseases. Most patients with LCH who require allo-HSCT are younger than 2 years, and cord blood can provide a large number of stem cells, leading to rapid engraftment and hematopoietic reconstitution (37–40). The number of patients eligible for some groups was small; therefore, it was difficult to comment on the efficacy and success of the different sources of hematopoietic stem cells.
Conditioning
The conditioning regimens of allo-HSCT in patients with LCH reported in the current study were mainly myeloablative conditioning (MAC) and reduced intensity conditioning (RIC). The specific drugs for MAC and RIC are consistent with those for other hematological malignancies, and no special new drugs have been added. For example, the MAC regimens mainly contains: busulfan, cyclophosphamide or fludarabine, and some children also choose the total body irradiation (TBI) containing regimens; The RIC mainly contains melphalan and fludarabine containing regimens (34). The choice of conditioning regimens for allo-HSCT in patients with LCH has been an important issue in recent years, and the best choice remains uncertain.
Some studies have found that the advantage of MAC regimens is that it can eliminate LCH cells in the body as much as possible to reduce the recurrence of the disease. For example, Veys et al. ‘s study found that the 3-year disease recurrence rate was only 3% among 41 children who received MAC regimens from 2000 to 2013, while 28% among 25 children who received RIC regimens (34). And the 3-year Disease-free survival of the two regimens was 77% and 51%, respectively (34). However, MAC regimens are often associated with high treatment-related morbidity (TRM) (41). In a retrospective study, Veys et al. summarized 29 pediatric patients with RO-LCH who underwent myeloablative allo-HSCT (34). The overall survival rate was 48%, and TRM was exceedingly high at 45%. Preexisting disease-related organ (the liver, bone marrow, and lungs) dysfunction together with infection in the majority of patients seemed to be the main cause of high TRM. And Kesik et al. reviewed 44 children with refractory LCH who underwent allo-HSCT (42). Of the 18 children with fatal outcomes, 16 received myeloablative allo-HSCT, while only 2 patients who were treated with RIC died. Death was attributed to recurrence, treatment-related toxicity, and GVHD. Owing to the high TRM of MAC transplantation, the number of patients undergoing RIC transplantation for LCH has increased in recent years. Steiner et al. reported nine children with high-risk LCH treated with RIC allo-HSCT (41). The conditioning was well tolerated, and an encouraging result was obtained; seven patients without disease progression were alive at a median follow-up of 390 days after transplantation. Good outcomes after RIC allo-HSCT have also been confirmed in patients with LCH (43, 44). Therefore, in recent years, RIC’s regimens has been gradually recommended more and more. However, Kudo et al. also found that there was no difference in overall survival (OS) or failure-free survival between children with RIC and MAC (45). So, for children with LCH, the choice between MAC and RIC transplantation requires further clinical research data to be confirmed. In clinical studies on HSCT in children with LCH, the conditioning regimens and outcomes are shown in Table 1.
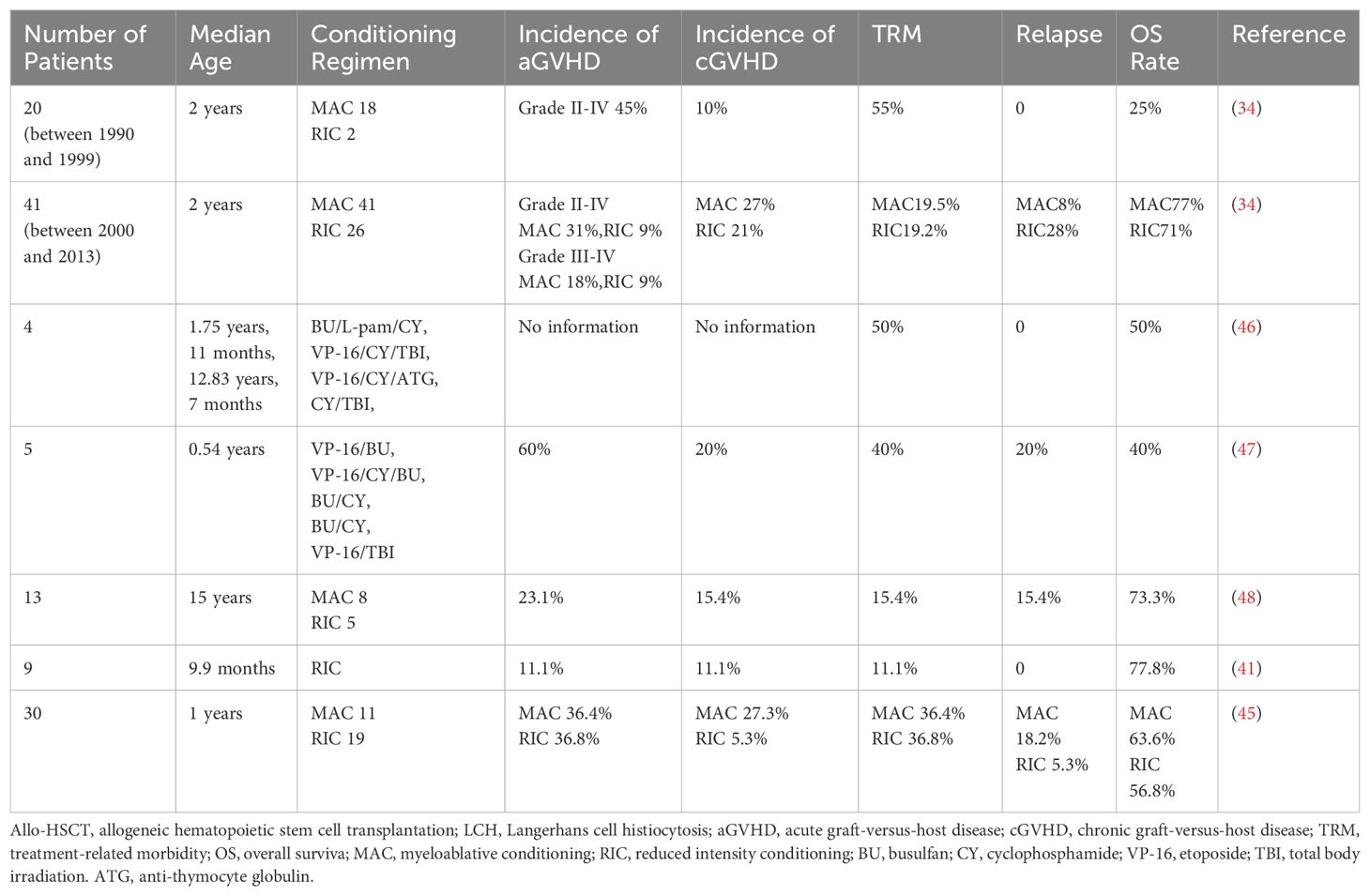
Table 1. Cohort studies reporting allo-HSCT results in the treatment of LCH in children.
Chimerism
Generally, the median time reported for successful neutrophil implantation after RIC allo-HSCT in patients with LCH is 14 days (12–49 days) (34). Successful red blood cell and platelet implantation typically occurs on 38 and 37 days after transplantation, respectively (34). Veys et al. found that hematopoietic recovery rates after MAC transplantation were not significantly different from those after RIC transplantation (34). However, recovery rates were slow for both MAC and RIC transplantations, with most patients undergoing implantation 2 months after transplantation. The disadvantage of RIC allo-HSCT is the increased risk of non-engraftment and graft rejection. In contrast to allo-HSCT after MAC conditioning, RIC allo-HSCT usually leads to mixed chimerism, which is defined as the coexistence of hematopoietic cells from the host and donor. However, that neither non-engraftment nor mixed chimerism leads to the deterioration of LCH and a life-threatening state, and does not necessarily implicate an exacerbation of LCH. Kinugawa et al. and Akkari et al. reported that two patients failed engraftment but achieved complete autologous recovery of hematopoiesis without disease activity (46, 47). Both patients were alive and disease-free at 3 and 12 years after transplantation at the time of the last follow-up. Steiner et al. reported five of nine patients had full donor chimerism on day +28 after RIC allo-HSCT, which persisted until the last follow-up (69–390 days after allo-HSCT) (41). Two patients exhibited mixed chimerism until the last follow-up. Steiner et al. also reported that one patient achieved complete remission after RIC allo-HSCT despite mixed chimerism after transplantation, in which only a T-cell subset proved to be of donor origin (41). There are two reasons for this phenomenon. Patients with LCH in a state of engraftment and mixed chimerism are in sustained remission after transplantation. On one hand, a highly immunosuppressive conditioning regimen together with GVHD prophylaxis may decisively contribute to the stabilization of the disease. In contrast, donor T-cells play an important role. The presence of donor T-cells is sufficient to restore normal immunoregulation and prevent hypercytokinemia and macrophage activation, which play important roles in the pathogenesis of LCH (36). Moreover, myeloablation is not critical for the treatment of LCH, although data on this disease are limited.
Transplant-related complication
GVHD remains an important complication of allo-HSCT in patients with LCH. Prophylaxis drugs for GVHD and graft rejection consisted of ciclosporin A (CSA), mycophenolate mofetil (MMF), prednisone, and methotrexate (MTX) (34, 41). Acute GVHD was more common than chronic GVHD in patients with LCH. Veys et al. reported 18 patients who received MAC regimens, with period prior to 2000 (34). Grades II–III acute GVHD occurred in eight patients (two grade II and six grade III), and chronic GVHD occurred in two patients. One of the two patients who received the RIC regimen developed grade II acute GVHD. Akkari et al. reported that three out of five allo-HSCT recipients developed evidence of grade I or II acute GVHD, but transient limited chronic GVHD occurred in only one patient (47). Kudo et al. reported three of ten patients who underwent unrelated cord blood transplantation developed acute GVHD (grades I, III, and IV) (48). One of them and another patient developed chronic GVHD. In patients with LCH, acute GVHD often involves the gastrointestinal mucosa, liver, and skin. Acute GVHD and the subsequent limited chronic GVHD are often responsive to immunosuppressive therapy. Veys et al. retrospectively analyzed patients who underwent allo-HSCT between 2000 and 2013 (34). The incidence of grade II–IV acute GVHD was marginally higher after MAC regimens than after RIC regimens. However, there were no differences in the incidences of grades III–IV acute and chronic GVHD. Other transplant-related complications include infection, mucositis, liver damage, interstitial pneumonia, organ failure, and hypertension (34, 47, 48).
The incidence of GVHD after LCH transplantation in children is slightly different from that after transplantation for other childhood diseases. Veys et al. ‘s study found that the incidence of grade II-IV acute GVHD after LCH transplantation in 41 children after 2000 was 31%, while that of chronic GVHD was 27% (34). In a transplant study of 618 children with benign and malignant disease, Cumulative incidence of acute GvHD (I-IV) was 31.5% and chronic GvHD was12.8% (49). Thus, the incidence of acute GVHD in children after LCH transplantation is roughly the same as that of other childhood diseases, while the incidence of chronic GVHD is slightly higher. However, the number of cases of LCH transplantation studies in children is still small, and further confirmation is still needed in large sample studies. In terms of TRM of both studies, the incidence of LCH in children within 3 years after transplantation was 15%, compared to 31.2% in the median follow up of 115.1 months after transplantation for other childhood diseases (34, 49). Because of the different follow-up times, it is also difficult to compare the incidence of TRM after transplantation between LCH and other diseases.
Survival and outcome
Chemotherapy failure occurs in approximately 20% of patients with multisystem LCH, with an overall survival of only 20% (7, 50). Allo-HSCT has recently been used in these patients as a salvage treatment to improve prognosis. Kudo et al. reported that 15 children younger than 15 years with refractory LCH underwent allo-HSCT (48). Eleven of the fifteen patients survived without evidence of disease, with a 10-year OS rate of 73.3%. The 10-year OS rate of nine patients with RO at diagnosis was 55.6%, and six patients without RO survived without evidence of disease. These results indicated that allo-HSCT is a promising salvage approach for children with refractory LCH. Three patients with LCH reported by Coope et al. treated with RIC allo-HSCT remained alive and in remission at a median of 5.1 years after allo-HSCT (43). Allo-HSCT can improve the prognosis of relapse and refractory LCH and prolong patient survival.
In addition, studies have found that the disease status of LCH during HSCT can affect a patient’s OS. Kudo et al. retrospectively analyzed 30 children with refractory LCH (45). The 5-year OS of children with no active disease and active disease regression was 100% after HSCT, significantly better than the 54.5% of children with active disease and active disease progression. This study suggests that LCH disease status at HSCT can affect OS. It is important for patients to achieve better remission with active and effective treatments before undergoing HSCT.
Treatment for relapse after allo-HSCT
Compared with other treatments, the rates of relapse or progression were lower in patients with LCH after allo-HSCT. The treatment options for LCH recurrence after allo-HSCT include secondary transplantation and combined chemotherapy. Six patients reported by Veys et al. underwent a second transplantation due to non-engraftment (n=4) or recurrent disease (n=2) (34). Of the patients re-transplanted for non-engraftment, one received MAC and three received RIC regimens for their first transplantation. Two of the three patients who received the RIC regimen for the first transplantation were alive 2 and 9 years after their first transplantation. Of the two patients who received a second transplant for recurrent disease, one was alive 6 years after the second transplantation and the other died 18 months after the second transplant. Therefore, in patients with graft failure and recurrence after allo-HSCT, a second transplant can result in long-term survival. Veys et al. reported that four of six patients who relapsed after RIC allo-HSCT achieved further remission after receiving chemotherapy as a salvage treatment (30). Patients who are BRAF-positive and do not use targeted drugs can also try this targeted therapy.
Conclusion and future prospectives
In summary, allo-HSCT is an important curative method for the relapsed/refractory LCH in children, especially for the failure of chemotherapy and targeted therapy. Due to the fact that LCH is a rare disease, and the research data of allo-HSCT are still few, there are still many problems that are still unclear and worth exploring, such as the indication and timing of allo-HSCT, the selection of preconditioning intensity and the treatment for relapse after allo-HSCT. In the future, we still need large, prospective, multineutral clinical studies to explore the unknown questions of allo-HSCT for LCH in children.
Author contributions
GM: Writing – original draft. SF: Validation, Writing – review & editing. YW: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. (2010) 16:S82–9. doi: 10.1016/j.bbmt.2009.11.014
2. Berres ML, Lim KP, Peters T, Price J, Takizawa H, Salmon H, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. (2014) 211:669–83. doi: 10.1084/jem.20130977
3. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. (2020) 135:1319–31. doi: 10.1182/blood.2019000934
4. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): guidelines fordiagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. (2013) 60:175–84. doi: 10.1002/pbc.24367
5. Rigaud C, Barkaoui MA, Thomas C, Bertrand Y, Lambilliotte A, Miron J, et al. Langerhans cellhistiocytosis: therapeutic strategy and outcome in a 30-year nationwide cohort of 1478 patients under 18 years of age. Br J Haematol. (2016) 174:887–98. doi: 10.1111/bjh.2016.174.issue-6
6. Rodriguez-Galindo C. Clinical features and treatment of Langerhans cell histiocytosis. Acta Paediatr. (2021) 110:2892–902. doi: 10.1111/apa.v110.11
7. Minkov M, Grois N, Heitger A, Pötschger U, Westermeier T, Gadner H, et al. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator. Med Pediatr Oncol. (2002) 39:581–5. doi: 10.1002/mpo.10166
8. Gadner H, Grois N, Pötschger U, Minkov M, Aricò M, Braier J, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. (2008) 111:2556–62. doi: 10.1182/blood-2007-08-106211
9. Morimoto A, Ikushima S, Kinugawa N, Ishii E, Kohdera U, Sako M, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. (2006) 107:613–9. doi: 10.1002/cncr.v107:3
10. Salama HA, Jazieh AR, Alhejazi AY, Absi A, Alshieban S, Alzahrani M, et al. Highlights of the management of adult histiocytic disorders: langerhans cell histiocytosis, erdheim-chester disease, rosai-dorfman disease, and hemophagocytic lymphohistiocytosis. Clin Lymph Myeloma Leuk. (2021) 21:e66–75. doi: 10.1016/j.clml.2020.08.007
11. Bonometti A, Bagnoli F, Fanoni D, Venegoni L, Corti L, Bianchi P, et al. JAK2-mutated Langerhans cell histiocytosis associated with primary myelofibrosis treated with ruxolitinib. Hum Pathol. (2018) 73:171–5. doi: 10.1016/j.humpath.2017.10.017
12. Maria Postini A, del Prever AB, Pagano M, Rivetti E, Berger M, Asaftei SD, et al. Langerhans cell histiocytosis: 40 years' experience. J Pediatr Hematol Oncol. (2012) 34:353–8. doi: 10.1097/MPH.0b013e318257a6ea
13. Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. (2007) 48:555–60. doi: 10.1002/pbc.20884
14. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans'-cell histiocytosis in adults. Med Pediatr Oncol. (1997) 28:9–14. doi: 10.1002/(SICI)1096-911X(199701)28:1<9::AID-MPO3>3.0.CO;2-P
15. Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. (2018) 379:856–68. doi: 10.1056/NEJMra1607548
16. Suh JK, Kang S, Kim H, Im HJ, Koh KN. Recent advances in the understanding of the molecular pathogenesis and targeted therapy options in Langerhans cell histiocytosis. Blood Res. (2021) 56:S65–9. doi: 10.5045/br.2021.2021013
17. Bagnasco F, Zimmermann SY, Egeler RM, Nanduri VR, Cammarata B, Donadieu J, et al. Langerhans cell histiocytosis and associated Malignancies: A retrospective analysis of 270 patients. Eur J Cancer. (2022) 172:138–45. doi: 10.1016/j.ejca.2022.03.036
18. Gao XM, Li J, Cao XX. Signaling pathways, microenvironment, and targeted treatments in Langerhans cell histiocytosis. Cell Commun Signal. (2022) 20:195. doi: 10.1186/s12964-022-00917-0
19. Geerlinks AV, Abla O. Treatment of langerhans cell histiocytosis and histiocytic disorders: A focus on MAPK pathway inhibitors. Paediatr Drugs. (2023) 25:399–409. doi: 10.1007/s40272-023-00569-8
20. Rees MJ, Dickinson M, Paterson J, Ng TF, Grigg A, Moore J, et al. Targeting the BRAF pathway in haematological diseases. Intern Med J. (2023) 53:845–9. doi: 10.1111/imj.16091
21. Weitzman S, Braier J, Donadieu J, Egeler RM, Grois N, Ladisch S, et al. 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer. (2009) 53:1271–6. doi: 10.1002/pbc.22229
22. Bernard F, Thomas C, Bertrand Y, Munzer M, Landman Parker J, Ouache M, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. (2005) 41:2682–9. doi: 10.1016/j.ejca.2005.02.007
23. Donadieu J, Bernard F, van Noesel M, Barkaoui M, Bardet O, Mura R, et al. Cladribine and cytarabine in refractory multisystem Langerhans cellhistiocytosis: results of an international phase 2 study. Blood. (2015) 126:1415–23. doi: 10.1182/blood-2015-03-635151
24. Jeong TD, Jang S, Park CJ, Chi HS, Lee JH. A case of Langerhans cellhistiocytosis following acute basophilic leukemia. Ann Hematol. (2013) 92:137–9. doi: 10.1007/s00277-012-1542-y
25. Xu G, Yang M, Huang J, Jin J. Successful treatment of a case of acute myeloid leukemia following Langerhans cell histiocytosis in an adolescent: a case report and review of the literature. Int J Clin Exp Med. (2015) 8:3024–6.
26. Ma J, Laird JH, Chau KW, Chelius MR, Lok BH, Yahalom J. Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid Malignancies. Cancer Med. (2019) 8:58–66. doi: 10.1002/cam4.2019.8.issue-1
27. Hostettler KE, Casañas Quintana E, Tamm M, Savic Prince S, Sommer G, Chen WC, et al. Case report: opposite effects of BRAF inhibition on closely related clonal myeloid disorders. Front Oncol. (2021) 11:779523. doi: 10.3389/fonc.2021.779523
28. Blakley MP, Dutcher JP, Wiernik PH. Pulmonary Langerhans cell histiocytosis, acute myeloid leukemia, and myelofibrosis in a large family and review of the literature. Leuk Res. (2018) 67:39–44. doi: 10.1016/j.leukres.2018.01.011
29. Nelson DS, Quispel W, Badalian-Very G, van Halteren AG, van den Bos C, Bovée JV, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. (2014) 123:3152–5. doi: 10.1182/blood-2013-06-511139
30. Brown NA, Furtado LV, Betz BL, Kiel MJ, Weigelin HC, Lim MS, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. (2014) 124:1655–8. doi: 10.1182/blood-2014-05-577361
31. Nann D, Schneckenburger P, Steinhilber J, Metzler G, Beschorner R, Schwarze CP, et al. Pediatric Langerhans cell histiocytosis: the impact of mutational profile on clinical progression and late sequelae. Ann Hematol. (2019) 98:1617–26. doi: 10.1007/s00277-019-03678-y
32. Egeler RM, Katewa S, Leenen PJ, Beverley P, Collin M, Ginhoux F, et al. Langerhans cell histiocytosis is a neoplasm and consequently its recurrence is a relapse: In memory of Bob Arceci. Pediatr Blood Cancer. (2016) 63:1704–12. doi: 10.1002/pbc.v63.10
33. Donadieu J, Larabi IA, Tardieu M, Visser J, Hutter C, Sieni E, et al. Vemurafenib for refractory multisystem langerhans cell histiocytosis in children: an international observational study. J Clin Oncol. (2019) 37:2857–65. doi: 10.1200/JCO.19.00456
34. Veys PA, Nanduri V, Baker KS, He W, Bandini G, Biondi A, et al. Haematopoietic stem cell transplantation for refractory Langerhans cell histiocytosis: outcome by intensity of conditioning. Br J Haematol. (2015) 169:711–8. doi: 10.1111/bjh.2015.169.issue-5
35. Körbling M, Anderlini P. Peripheral blood stem cell versus bone marrow allotransplantation: does the source of hematopoietic stem cells matter? Blood. (2001) 98:2900–8. doi: 10.1182/blood.V98.10.2900
36. Bensinger WI, Martin PJ, Storer B, Clift R, Forman SJ, Negrin R, et al. Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med. (2001) 344:175–81. doi: 10.1056/NEJM200101183440303
37. Rubinstein P, Carrier C, Scaradavou A, Kurtzberg J, Adamson J, Migliaccio AR, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med. (1998) 339:1565–77. doi: 10.1056/NEJM199811263392201
38. Meyer-Wentrup F, Foell J, Wawer A, Burdach S. Unrelated cord blood transplantation in an infant with severe multisystem Langerhans cell histiocytosis: clinical outcome, engraftment and culture of monocyte-derived dendritic cells. Bone Marrow Transplant. (2004) 33:875–6. doi: 10.1038/sj.bmt.1704441
39. Suminoe A, Matsuzaki A, Hattori H, Ishii S, Hara T. Unrelated cord blood transplantation for an infant with chemotherapy-resistant progressive Langerhans cell histiocytosis. J Pediatr Hematol Oncol. (2001) 23:633–6. doi: 10.1097/00043426-200112000-00018
40. Nagarajan R, Neglia J, Ramsay N, Baker KS. Successful treatment of refractory Langerhans cell histiocytosis with unrelated cord blood transplantation. J Pediatr Hematol Oncol. (2001) 23:629–32. doi: 10.1097/00043426-200112000-00017
41. Steiner M, Matthes-Martin S, Attarbaschi A, Minkov M, Grois N, Unger E, et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant. (2005) 36:215–25. doi: 10.1038/sj.bmt.1705015
42. Kesik V, Citak C, Kismet E, Koseoglu V, Akyuz C. Hematopoietic stem cell transplantation in Langerhans cell histiocytosis: case report and review of the literature. Pediatr Transplant. (2009) 13:371–4. doi: 10.1111/j.1399-3046.2008.00989.x
43. Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. (2008) 42 Suppl 2:S47–50. doi: 10.1038/bmt.2008.283
44. Hatakeyama N, Hori T, Yamamoto M, Inazawa N, Hirako Y, Tsutsumi H, et al. Successful treatment of refractory Langerhans cell histiocytosis with pulmonary aspergillosis by reduced-intensity conditioning cord blood transplantation. Pediatr Transplant. (2010) 14:E4–10. doi: 10.1111/j.1399-3046.2008.01124.x
45. Kudo K, Maeda M, Suzuki N, Kanegane H, Ohga S, Ishii E, et al. Nationwide retrospective review of hematopoietic stem cell transplantation in children with refractory Langerhans cell histiocytosis. Int J Hematol. (2020) 111:137–48. doi: 10.1007/s12185-019-02760-5
46. Kinugawa N, Imashuku S, Hirota Y, Yamada K, Yamamoto A, Akazai A, et al. Hematopoietic stem cell transplantation (HSCT) for Langerhans cell histiocytosis (LCH) in Japan. Bone Marrow Transplant. (1999) 24:935–8. doi: 10.1038/sj.bmt.1702001
47. Akkari V, Donadieu J, Piguet C, Bordigoni P, Michel G, Blanche S, et al. Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group. Bone Marrow Transplant. (2003) 31:1097–103. doi: 10.1038/sj.bmt.1704065
48. Kudo K, Ohga S, Morimoto A, Ishida Y, Suzuki N, Hasegawa D, et al. Improved outcome of refractory Langerhans cell histiocytosis in children with hematopoietic stem cell transplantation in Japan. Bone Marrow Transplant. (2010) 45:901–6. doi: 10.1038/bmt.2009.245
49. Khan S, Siddiqui K, ElSolh H, AlJefri A, AlAhmari A, Ghemlas I, et al. Outcomes of blood and marrow transplantation in children less than 2-years of age: 23 years of experience at a single center. Int J Pediatr Adolesc Med. (2022) 9:190–5. doi: 10.1016/j.ijpam.2022.09.002
Keywords: Langerhans cell histiocytosis, allogeneic hematopoietic stem cell transplantation, myeloablative conditioning, reduced intensity conditioning, chimerism
Citation: Meng G, Feng S and Wang Y (2025) Advances in allogeneic hematopoietic stem cell transplantation for Langerhans cell histiocytosis in children. Front. Immunol. 16:1345855. doi: 10.3389/fimmu.2025.1345855
Received: 28 November 2023; Accepted: 13 January 2025;
Published: 28 January 2025.
Edited by:
Rémy Dulery, Hôpital Saint-Antoine, FranceReviewed by:
Pranali Nitin Shah, Brigham and Women’s Hospital and Harvard Medical School, United StatesArturo Bonometti, Humanitas University, Italy
Copyright © 2025 Meng, Feng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangqiang Meng, bWVuZ2dxMjAwMUAxNjMuY29t; Yan Wang, cWZzeXl3eUAxNjMuY29t